

Abstract
The human 3-methyladenine DNA glycosylase [alkyladenine DNA glycosylase (AAG)] catalyzes the first step of base excision repair by cleaving damaged bases from DNA. Unlike other DNA glycosylases that are specific for a particular type of damaged base, AAG excises a chemically diverse selection of substrate bases damaged by alkylation or deamination. The 2.1-Å crystal structure of AAG complexed to DNA containing 1,N6-ethenoadenine suggests how modified bases can be distinguished from normal DNA bases in the enzyme active site. Mutational analyses of residues contacting the alkylated base in the crystal structures suggest that the shape of the damaged base, its hydrogen-bonding characteristics, and its aromaticity all contribute to the selective recognition of damage by AAG.
DNA bases are chemically reactive and readily undergo deamination and alkylation on the inevitable exposure to reactive cellular metabolites and environmental toxicants (1–4). Alkylation occurs at many different positions of DNA, producing a variety of lesioned bases (4, 5) that can block replication or interfere with other enzymatic activities templated by DNA. Hypoxanthine is an abundant deaminated base, and it too corrupts the DNA template. Remarkably, human cells appear to produce a single enzyme, alkyladenine DNA glycosylase [AAG (3-methyladenine DNA glycosylase, ANPG, or MPG)], which recognizes and removes hypoxanthine plus a variety of alkylated bases that include 3-methyladenine, 7-methylguanine, and 1,N6-ethenoadenine (ɛA; refs. 6–13). AAG cleaves the N-glycosylic bond joining the damaged base to the DNA backbone, and the resulting abasic nucleotide is excised and replaced with a normal nucleotide by the sequential action of an endonuclease, a polymerase, and DNA ligase (14). The high selectivity for damaged vs. normal bases is essential because normal bases are present in vast excess. AAG can distinguish alternations in both adenine and guanine and can recognize changes present in both the major and minor grooves of DNA. We set out to determine how AAG achieves selectivity for chemically diverse substrates.
We previously reported a 2.7-Å crystal structure of AAG complexed to DNA containing a transition-state mimic of the glycosylase reaction, the pyrrolidine abasic nucleotide (pyr; PDB ID code 1bnk; refs. 15 and 16). In the AAG/pyr-DNA complex, the pyr ring is flipped into the proposed active site by intercalation of the Tyr-162 side chain into the minor groove of the DNA (15). A bound water molecule in the active site is aligned for a back-side attack of the abasic sugar, but the pyr inhibitor lacks a base, and we could not deduce how AAG recognizes alkylated bases in preference to normal bases. Structures of several other DNA N-glycosylases complexed to their DNA substrates have been reported (17–20). These enzymes are selective for one type of damaged DNA base and, correspondingly, their active site structures are tailor made for specific interactions with these substrates. For example, uracil DNA glycosylase flips uracil bases out of DNA and into a pocket that is too small to bind purine bases or a thymine with its bulky C5 methyl group, and cytosine is excluded by unfavorable interactions with its exocyclic amine (N4). Thus, catalytic selectivity is achieved by selective binding of the flipped-out uridine nucleotide. Substrate recognition by AAG is more puzzling, because its active site must accommodate a wide variety of differently shaped alkylated bases while excluding normal purine bases. The alkylated base ɛA is generated endogenously by lipid peroxidation (21) or by exposure of cells to vinyl chloride or chloroacetaldehyde (ref. 22; reviewed in ref. 3). ɛA lesions are efficiently excised from DNA by AAG (11, 13, 23). As a first step toward identifying the basis of AAG's catalytic specificity, we determined crystal structures of AAG bound to DNA containing ɛA and performed mutational analyses of residues that contact the DNA substrate. The structures and related functional studies identify key determinants for selecting damaged bases for excision.
Materials and Methods
Mutagenesis and Methylmethane Sulfonate (MMS) Resistance.
Site-specific mutants of full-length AAG (residues 1–298) were constructed in the yeast expression vector pYes (24) by using Stratagene's QuikChange kit, and the mutated genes were sequenced in their entirety. Wild-type and mutant AAG proteins were expressed in a Saccharomyces cerevisiae strain (BGY148) lacking the endogenous yeast 3-methyladenine DNA glycosylase [Mag1 (25)]. The transformed cells were assayed for resistance to the alkylating agent MMS by growth on a concentration gradient of MMS in yeast/peptone/dextrose medium containing either 2% glucose (basal expression of AAG) or 2% galactose (inducing condition; ref. 24).
Crystal Growth and X-Ray Data Collection.
Wild-type AAG and the inactive E125Q mutant protein were overproduced from the T7 expression vector pLM1 (26) in BL21(DE3) Escherichia coli (Novagen) and purified as previously described (15). Oligonucleotides used for crystallization were purified by anion exchange HPLC (Poros HQ medium, PE Biosystems). Crystallization of the wild-type AAG/pyr-DNA complex has been previously described (15). For the ɛA complex, ɛA (Glen Research, Sterling, VA) was incorporated into one DNA strand [5′-GACATG(ɛA)TTGCCT-3′] and annealed to a complementary strand with “T” opposite the lesion (5′-GGCAATCATGTCA-3′). Equimolar amounts of duplex ɛA DNA and the wild-type or E125Q AAG protein were mixed together (final complex concentration of 0.3 mM) in 100 mM sodium chloride/20 mM Hepes (pH 7.5)/0.1 mM EDTA/5% glycerol. Crystals of the ɛA complexes grew overnight in hanging drops maintained at 22°C after equilibration against an equal volume of a reservoir solution containing 200 mM magnesium chloride, 100 mM Tris⋅HCl (pH 8.5), 24% (wt/vol) polyethylene glycol 4000, and 10% glycerol. The flash-frozen crystals belong to space group P212121 and have unit cell dimensions of a = 42.1 Å, b = 57.3 Å, and c = 125.5 Å. One AAG/ɛA-DNA complex (Mr = 33,000) occupies the crystallographic asymmetric unit. Native x-ray data from crystals of the ɛA complex were collected at beamline X-25 of the National Synchrotron Light Source (NSLS, Upton, NY) by using a 4-module Brandeis charge-coupled device detector (W. Phillips and M. Stanton, personal communication; Table 1). Native data from crystals of the pyr complex were collected at beamline X-12C of the NSLS by using the same detector.
Table 1.
Data collection and refinement statistics
| Data collection complex | E125Q–ɛA | Wild-type ɛA | Wild-type pyr |
|---|---|---|---|
| Wavelength, Å | 1.01 | 1.01 | 1.00 |
| Resolution limit, Å | 2.1 | 2.4 | 2.4 |
| Total observations | 247,464 | 167,384 | 100,868 |
| Unique observations | 18,163 | 12,133 | 13,252 |
| Rsym | 0.042 | 0.043 | 0.071 |
| Rsym (last shell) | 0.201 | 0.137 | 0.253 |
| |/sigma (last shell) | 6.4 | 12.1 | 3.5 |
| Completeness (overall) | 0.993 | 0.994 | 0.861 |
| Completeness (last shell) | 0.978 | 0.997 | 0.756 |
| Model refinement | |||
| Rwork/Rfree | 0.230/0.259 | 0.239/0.276 | 0.219/0.282 |
| Resolution: | 500–2.1 Å | 500–2.4 Å | 500–2.4 Å |
| rmsd from stereochemical target values: | |||
| rmsd, bond length, Å | 0.006 | 0.006 | 0.006 |
| rmsd, bond angles, ° | 1.22 | 1.20 | 1.20 |
Rsym = ∑j|/j − </j>|/∑j/j, where </j> is the average intensity of reflection j for its symmetry equivalents. Rwork and Rfree = ∑|Fo − kFc/∑Fo, where Fo and Fc are the observed and calculated structure factor amplitudes. Rfree was calculated with 10% of reflections against which the model was not refined. The Rfree test set for each complex contained the same reflections. rmsd, root-mean-square deviation.
Phasing, Model Building, and Refinement.
X-ray intensity data were processed with denzo/scalepack (27), and the structures of the ɛA complexes were determined by molecular replacement by using the software suite crystallography and nmr system cns (ref. 28; Table 1), by using the AAG/pyr-DNA complex (PDB ID code 1bnk) as a search mode after omitting the pyrrolidine abasic nucleotide. The packing arrangement of DNAs in the ɛA complex crystals is different from that of the pyr complex. The DNAs in the ɛA complexes pack end to end by a mispairing of C12 and A26 (Fig. 1b) that is stabilized by a water molecule bridging N4 of C12 and N1 of A26. Nucleotides T13 and G14 are apparently disordered in the ɛA/DNA complex crystals. After rotation and translation, initial models of the AAG/ɛA-DNA complexes were subjected to rigid-body and positional refinement in cns. 2Fo − Fc, Fo − Fc, and Fc − Fo difference electron density maps were used to guide the fitting of the model during manual rebuilding. Model rebuilding was performed with the program o (see http://kaktus.kemi.aau.dk). The model was further refined by Powell conjugate gradient minimization and torsion angle-restrained molecular dynamics by using cns. The success of model refinement was evaluated at each stage by the change in the free R factor (29) and inspection of stereochemical parameters with the program procheck (ref. 30; Table 1).
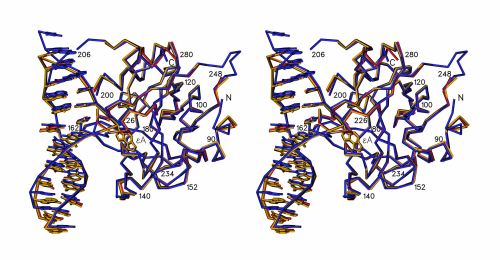
Figure 1.

Crystal structure of the E125Q AAG/ɛA-DNA complex. (a) The ɛA base (black) is flipped into the protein active site to stack between Tyr-127 on one side and His-136 and Tyr-159 on the other (shown in purple). Tyr-162 intercalates between the bases that flank the flipped-out ɛA, filling the abasic gap in the DNA. (b) Schematic diagram of contacts between AAG and the ɛA–DNA. The flipped-out ɛA base (labeled ɛA7) participates in a hydrogen-bonding interaction with the main chain amide of His-136 (solid line labeled “mc136”) and many van der Waals interactions (wavy lines) with residues of the active site (see Fig. 4). Hydrogen bonds and salt bridges (solid lines) with the DNA backbone anchor the protein to DNA. The nucleotides T13 and G14 (dashed outlines) are not visible in the electron density.
The model of the E125Q AAG/ɛA-DNA complex consists of 200 residues, 24 nucleotides, 80 molecules of water that have refined temperature factors of less than 50 Å2, and one Na+ molecule. Density is not seen for protein residues 80–81, 200–207, 249–254, and 296–298, and these disordered segments are omitted from the model. Density is also not seen for nucleotides T13 and G14, so they have been omitted from the model. Electron density is lacking for the side chains of residues His-82, Glu-131, Thr-199, Val-208, Lys-210, Gln-238, Glu-240, and Glu-269, and these residues are modeled as alanines. The model of the wild-type AAG/ɛA-DNA complex includes 199 residues, 24 nucleotides, 48 molecules of water that have refined temperature factors of less than 50 Å2, and one Na+ molecule. Density is not seen for protein residues 80–81, 200–207, 249–254, and 295–298. These disordered segments are omitted from the model. Density is also not seen for nucleotides T13 and G14, so they have been omitted from the model. Interpretable electron density is lacking for the side chains of residues His-82, Glu-131, Thr-199, Val-208, Lys-210, Gln-238, Glu-240, and Glu-269, and Gln-294, which are modeled as alanines. The model of the 2.4-Å resolution wild-type AAG/pyr-DNA complex includes 211 residues, 26 nucleotides, 134 molecules of water that have refined temperature factors of less than 50 Å2, and one Na+ molecule. Density is not seen for protein residues 201–205 and 296–298. These disordered segments are omitted from the model. Interpretable electron density is lacking for the side chains of residues Arg-207, Lys-210, and Gln-294, and these residues are modeled as alanines. The atomic coordinates of the E125Q–ɛA, wild-type ɛA, and wild-type pyr complexes have been deposited in PDB (ID codes are 1ewn, 1f4r, and 1f6o, respectively).
Results and Discussion
Overview of the Structures.
Crystal structures of wild-type AAG and a catalytically inactive mutant (E125Q) complexed to DNA containing the alkylated base ɛA were determined by molecular replacement and refined to 2.4-Å and 2.1-Å resolution, respectively (Table 1; Figs. 1 and 2). To our surprise, the glycosylic bond is uncleaved in the wild-type enzyme/DNA complex, despite growing the crystals at room temperature over a period of days. The difference electron density calculated after omitting the ɛA nucleotide from the model and performing a limited refinement by simulated annealing and conjugate gradient minimization (28) clearly shows that the glycosylic bond is intact. We subsequently confirmed that the purified AAG protein is enzymatically active but found that 0.2 M MgCl2 present during crystallization inhibits glycosylase activity for unknown reasons. No electron density that could be ascribed to a bound magnesium ion was seen. A similar concentration of NaCl or KCl does not affect AAG's activity. We have also refined the previously reported AAG–(pyr-DNA) structure to a resolution limit of 2.4 Å, and we compare the three AAG/DNA complexes below.
Figure 2.

AAG active-site structure. (a) The 2Fo − Fc electron OMIT density contoured at 2σ above the mean (purple) clearly shows the position of the flipped-out ɛA and a bound water molecule in the active site of the E125Q/ɛA complex. The OMIT density for the wild-type AAG/ɛA-DNA complex has a similar appearance. (b) A superposition of the active sites of the E125Q/ɛA-DNA (green), wild-type/ɛA DNA (red), and wild-type/pyr-DNA (blue) complexes shows these DNAs and the active-site water are bound in similar orientations. However, the pyr ring has rotated to optimize the geometry of a hydrogen bond between pyr N4′ and the bound water. (c) The main chain amide of His-136 makes a key hydrogen-bonding interaction with N6 of ɛA. The N6 of an unmodified adenine would be protonated and repelled by the His-136 amide nitrogen. The side chain of His-136 bridges between the Tyr-157 side chain and the phosphate of the ɛA nucleotide. This fixes the position of the imidazole ring, which stacks against the alkylated base. (d) A guanosine modeled in the active site by superposition on the ɛA nucleotide reveals a clash between N2 of guanine and the Asn-169 side chain (arrow). This steric clash and the conformational constraints on the Asn-169 side chain are best visualized by examining the atomic coordinates of the crystallized complexes (PDB ID codes 1ewn, 1f4r, and 1f6o).
The conformation of the AAG protein is unchanged in DNA complexes with either the pyr-abasic inhibitor or the ɛA substrate. The root-mean-square deviation (rmsd) of all protein atoms is 0.4 Å for the wild-type and E125Q mutant complexes with ɛA-DNA. The pyr and ɛA complexes with wild-type AAG have an rmsd of 1.3 Å (see supplemental data, www.pnas.org). Furthermore, the side chains lining the pocket that accepts the ɛA base have identical orientations in the presence or absence of a bound base. A feature of the higher-resolution pyr complex, now refined to 2.4 Å, is clear electron density for a loop consisting of residues 247–254 that was poorly ordered in the 2.7-Å structure (15). The loop might be stabilized by a slight shift in crystal packing that apparently improved the diffraction quality of the crystals. We also see evidence in the 2Fo − Fc difference electron density of a bound monovalent metal that is octahedrally coordinated by the side chain of Ser-171, a water molecule, and the main chain carbonyls of Met-149, Ser-172, Gly-174, and Ala-177. The distance between the proposed metal and the oxygen ligands ranges from 2.4 to 2.6 Å, consistent with a bound Na+ ion (31). The bound metal could add to the structural integrity of the floor of the active site, but it is unlikely to participate directly in glycosylic bond cleavage.
DNA Binding and Base Flipping.
The DNA in the AAG/ɛA-DNA complexes is bent away from the protein by about 20°, as in the pyr complex (15). The center of the bend is located at Tyr-162 (Fig. 1), where the width of the minor groove is increased by more than 2 Å. The distortion of DNA structure in the AAG complex resembles that caused by other DNA glycosylases (17, 18, 20, 32). The side chain of Tyr-162 projects from a β-hairpin on the surface of AAG and inserts into the minor groove DNA, flipping the nucleotide targeted for cleavage into the enzyme active site (Fig. 1). We tested the functional significance of Tyr-162 by expressing mutant and wild-type AAG proteins in a S. cerevisiae strain lacking the endogenous yeast Mag1 glycosylase. Expression of basal levels of wild-type AAG in the mag1 yeast cells confers more than 5-fold resistance to the alkylating agent MMS (Fig. 3). The true extent of resistance has not been determined and would require testing cell growth at higher concentrations of MMS. In contrast to wild-type AAG, cells expressing the Y162A mutant are very sensitive to MMS, and induction of Y162A protein expression by growth on 2% galactose does not increase resistance. We conclude that the Y162A mutant has minimal glycosylase activity. Consistent with this interpretation, the Y162A AAG protein binds weakly to ɛA–DNA and pyr–DNA in vitro (not shown). The loss of the Tyr-162 side chain probably hinders the ability to extrude target nucleotides out of duplex DNA by base flipping. Two neighboring residues, Met-164 and Tyr-165, assist in base flipping by destabilizing the base pair next to the flipped-out nucleotide (15). When expressed at high levels, the M164A and Y165A mutants confer significant resistance to MMS (Fig. 3), suggesting that alanine substitutions at these flanking positions have only modest effects on AAG function.
Figure 3.

Functional analyses of residues in the AAG active site. Wild-type and mutant AAG proteins were expressed in a S. cerevisiae strain lacking the endogeneous Mag1 glycosylase activity, and resistance to the alkylating agent methylmethane sulfonate was determined by growth on a gradient of MMS. The extent of growth on MMS is shown for cells expressing a basal level of AAG (gray) and induced cells expressing a higher level of AAG protein (black). Immunoblots indicated that all of the AAG mutant proteins are expressed at the same level as the wild-type AAG protein (not shown). Compared with the null cells (−AAG), cells expressing wild-type AAG (wt) are resistant to growth inhibition by MMS. Substitution of Glu-125 with alanine (E125A) or glutamine (E125Q) eliminates detectable MMS resistance. The functional significance of these residues is discussed in the text.
The Glycosylase Active Site.
Fig. 2b shows the superimposed active sites of wild-type AAG complexed to pyr (blue) and ɛA (red) DNAs, and the E125Q mutant complexed to ɛA (yellow). AAG binds the pyr abasic inhibitor and the ɛA substrate in a similar manner, but the pyr ring has rotated to allow nitrogen N4′ to donate a hydrogen bond to a water molecule bound in the active site. In this orientation, N4′ of pyr is nearly superimposed on the anomeric C1′ of the ɛA substrate. We previously proposed that Glu-125 deprotonates the bound water, forming the hydroxyl nucleophile for glycosylic bond cleavage (15). Consistent with this suggestion, the substitution of Glu-125 with alanine or glutamine eliminates detectable glycosylase activity in vitro (not shown) and abrogates resistance to MMS (Fig. 3). Arg-182 donates hydrogen bonds to the active-site water and to the 3′ phosphate of the flipped-out nucleotide (Fig. 2). A lysine substitution at this position eliminates one of the two contacts made by Arg-182 in the wild-type enzyme, and the R182K mutant provides less resistance to MMS than wild-type AAG (Fig. 3).
The location of the active-site water is similar in all three AAG/DNA complexes (Fig. 2b), but its hydrogen-bonding partners are different. In the pyr complex, the water interacts with the pyr N4′, the side chains of Glu-125 and Arg-182, and the main chain carbonyl of Val-262. In the E125Q/ɛA complex, the same Arg-182 and Val-262 contacts are made, but the amide nitrogen of Gln-125 donates a hydrogen bond, and the O3′ of ɛA accepts a hydrogen bond. Although the Gln-125 side chain could be modeled into the electron density in a flipped orientation (180° rotation about χ3), the chosen orientation satisfies the hydrogen-bonding requirements with neighboring groups. In the wild-type ɛA complex, the water interacts only with the side chains of Glu-125 and Arg-182 and the Val-262 carbonyl oxygen, leaving one hydrogen without an interaction. It is inevitable that some movement of the bound water and the associated protein side chains accompanies cleavage of the glycosylic bond.
The added rotation of the bound pyr ring, which allows for interaction with nitrogen N4′, previously caused us to incorrectly model a substrate base stacked between Tyr-127 and Tyr-159 (15). Tyr-159 does not stack face to face with a substrate base as previously suggested, but it instead makes an edge-to-face packing interaction with the flipped-out ɛA (Fig. 4), which has rotated about its glycosylic bond 85° away from its B-DNA anti conformation (χ = −94°) to a high-anti conformation (χ = −179°). The sugar pucker, which was not restrained during refinement, appears to be C2′-endo. It is notable that an unstacked deoxyadenosine nucleoside with a high-anti conformation and C2′-endo sugar puckering has been observed in duplex DNA by NMR (33).
Figure 4.

The substrate-binding pocket of AAG. The ɛA base (purple surface) fits snugly into a pocket next to the enzyme active site. The base-binding pocket is viewed from the perspective of the protein, with the DNA helix oriented almost vertically behind the plane of the diagram. Met-149 and Cys-178 make additional van der Waals contacts to the ɛA base that are not shown.
The Alkylbase-Binding Pocket.
The flipped-out ɛA is fully inserted into a deep pocket next to the enzyme active site (Figs. 1a and 4), where the base stacks between the aromatic side chains of Tyr-127 on one side and His-136 and Tyr-159 on the other (Fig. 2). These side chains are in the same orientations in the abasic pyr complex and the ɛA bound complex, suggesting that the shape of the pocket is predetermined rather than being induced by substrate binding. Tyr-127 also donates a hydrogen bond to the catalytic residue Glu-125, stabilizing it in the active site (Fig. 2). This interaction with Glu-125 is eliminated by the substitution of Tyr-127 with phenylalanine, which can still stack against the flipped-out ɛA base. Yeast expressing a basal or induced level of Y127F AAG are sensitive to MMS (Fig. 3). In addition to stacking against ɛA, the His-136 side-chain hydrogen bonds with the 5′ phosphate of the flipped-out base and with Tyr-157 (Fig. 2c). The H136Q AAG mutant was engineered to eliminate aromatic stacking interactions with ɛA while retaining the ability to donate a hydrogen bond to the DNA backbone. Although basal expression of H136Q confers little resistance to MMS, high-level expression results in significant resistance (Fig. 3). We infer that the H136Q substitution only partly compromises AAG's glycosylase activity, consistent with this mutant's low activity in vitro (data not shown). The imidazole ring of His-136 primarily stacks against the aberrant ɛA moiety of the ɛA base (Fig. 2c). His-136 might be expected to play less of a role in the excision of monomethylated bases, which lack this alkyl moiety. One face of the ɛA base is contacted by the hydroxyl group of Tyr-159 (Figs. 2 and 4). A conservative substitution of phenylalanine at this position, which eliminates the hydroxyl group (Y159F mutant; Fig. 3), has only a modest effect on resistance to alkylation damage produced by MMS.
Achieving Catalytic Specificity.
The widely different shapes of the alkylated bases that are substrates for AAG suggest that shape complementarity between the substrate base and the enzyme active site cannot completely account for catalytic selectivity. Some alkylation-damaged bases are electron deficient and have a delocalized positive charge. These positively charged alkylated bases could be selectively recognized by tight-binding interactions with an aromatic side chain(s) of the glycosylase active site (15, 34), which would constitute a π-electron donor–acceptor pair with considerably more potential binding energy than a neutral π-electron stacking interaction (35, 36). A second unique feature is that a positively charged alkylated base is a good leaving group with a weakened glycosylic bond. With minimal catalytic assistance, these destabilized bases could be readily excised by a glycosylase lacking the catalytic strength to efficiently excise normal bases (37). Thus, catalytic selectivity for the electropositive alkylation adducts, like 3-methyladenine and 7-methylguanine, could be achieved by enhanced binding of these substrates coupled with their chemical instability in comparison to unmodified bases. However, these features are not present in the neutral alkylated substrates that are efficiently cleaved by AAG, like hypoxanthine (23) and ɛA (ref. 7; Figs. 1 and 2).
Features of the ɛA-binding site seen in the crystal structure suggest several additional means for achieving selective binding of substrates. The ɛA-binding pocket snugly accommodates the flipped-out base by a combination of aromatic stacking interactions and a hydrogen bond between the main chain amide of His-136 and N6 of ɛA, which offers an acceptor lone pair that is unique to the alkylated adduct (Fig. 2c). The N6 nitrogen of a normal adenine is protonated and would instead be repelled by the main chain amide. The etheno adduct contributes an additional 17 Å2 of van der Waals surface area that contacts the sandwiching residues of the binding site and probably favors tighter binding of ɛA. The structure of the base-binding pocket also suggests why inosine is a preferred substrate and guanosine is a poor substrate. Superposition of inosine or guanosine on the ɛA in the crystal structure shows that O6 of these bases can accept a hydrogen bond from the His-136 main chain amide like N6 of ɛA. However, the exocyclic amino group of guanine (N2) clashes with the side chain of Asn-169, which is highly constrained in conformation, creating a repulsive interaction that disfavors binding of guanosine (Fig. 2d). Inosine lacks a N2 amino group and fits nicely. The alkylated substrate base 7-methylguanine has a N2 amino group, but it is positively charged and might be pulled into the active site to satisfy cation–π interactions with Tyr-127 and His-136 strongly enough to push aside Asn-169 (38). This hypothesis remains to be tested.
The selective recognition of alkylation-damaged bases could occur at several points along the pathway of base excision repair. Substrate nucleotides are exposed to the enzyme active site by base flipping, which might be energetically more favorable for damaged nucleotides than for unmodified nucleotides in DNA. The fit of the damaged nucleotide into the enzyme active site is crucial for the formation of the Michaelis complex that leads to glycosylic bond cleavage. The combination of stacking interactions involving the additional surface area of the ɛA adduct and changes in the hydrogen-bonding potential of the alkylated purine plays a role in stabilizing the substrate nucleotide in the enzyme active site. Although the structure of AAG's nucleotide-binding pocket changes little in the unbound and ɛA-bound structures, some rearrangements might be required to bind other alkylated substrates like 7-methylguanine. As previously noted by Seeberg and coworkers (37), the chemical instability of the glycosylic bond might be an additional selectivity factor for some alkyl–purine substrates. It is likely that these factors are used to varying extents for the recognition of different damaged bases.
Supplementary Material
Acknowledgments
We thank Robert Sweet and the staff of beamline X-12C and Lonnie Berman, Hal Lewis, and the staff of beamline X-25 at the National Synchrotron Light Source (Upton, NY) for their expert assistance with x-ray data collection. Orlando Schärer and Gregory Verdine (Harvard University) kindly provided reagents for the synthesis of pyrrolidine-containing DNAs. We appreciate the help and advice of Hyock Joo Kwon, Anang Shelat, Tom Hollis, and other members of the Ellenberger and Samson groups. This work was supported by research grants from the National Institutes of Health (T.E., L.D.S.), by a training grant from the National Institute of Environmental Health Sciences (A.Y.L.), and by the Harvard–Armenise Center for Structural Biology at Harvard Medical School.
Abbreviations
- AAG
alkyladenine DNA glycosylase
- ɛA
1,N6-ethenoadenine
- MMS
methylmethane sulfonate
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1ewn, 1f4r, and 1f6o).
References
- 1.Rydberg B, Lindahl T. EMBO J. 1982;1:211–216. doi: 10.1002/j.1460-2075.1982.tb01149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindahl T. Nature (London) 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 3.Nair J, Barbin A, Velic I, Bartsch H. Mutat Res. 1999;424:59–69. doi: 10.1016/s0027-5107(99)00008-1. [DOI] [PubMed] [Google Scholar]
- 4.Singer B, Hang B. Chem Res Toxicol. 1997;10:713–732. doi: 10.1021/tx970011e. [DOI] [PubMed] [Google Scholar]
- 5.Wyatt M D, Allan J M, Lau A Y, Ellenberger T E, Samson L D. BioEssays. 1999;21:668–676. doi: 10.1002/(SICI)1521-1878(199908)21:8<668::AID-BIES6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 6.O'Connor T R, Laval J. Biochem Biophys Res Commun. 1991;176:1170–1177. doi: 10.1016/0006-291x(91)90408-y. [DOI] [PubMed] [Google Scholar]
- 7.Dosanjh M K, Roy R, Mitra S, Singer B. Biochemistry. 1994;33:1624–1628. doi: 10.1021/bi00173a002. [DOI] [PubMed] [Google Scholar]
- 8.Mattes W B, Lee C S, Laval J, O'Connor T R. Carcinogenesis. 1996;17:643–648. doi: 10.1093/carcin/17.4.643. [DOI] [PubMed] [Google Scholar]
- 9.Roy R, Kennel S J, Mitra S. Carcinogenesis. 1996;17:2177–2182. doi: 10.1093/carcin/17.10.2177. [DOI] [PubMed] [Google Scholar]
- 10.Saparbaev M, Laval J. Proc Natl Acad Sci USA. 1994;91:5873–5877. doi: 10.1073/pnas.91.13.5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saparbaev M, Kleibl K, Laval J. Nucleic Acids Res. 1995;23:3750–3755. doi: 10.1093/nar/23.18.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouziane M, Miao F, Ye N, Holmquist G, Chyzak G, O'Connor T R. Acta Biochim Pol. 1998;45:191–202. [PubMed] [Google Scholar]
- 13.Asaeda A, Ide H, Asagoshi K, Matsuyama S, Tano K, Murakami A, Takamori Y, Kubo K. Biochemistry. 2000;39:1959–1965. doi: 10.1021/bi9917075. [DOI] [PubMed] [Google Scholar]
- 14.Friedberg E C, Walker G C, Siede W. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol.; 1995. [Google Scholar]
- 15.Lau A Y, Scharer O D, Samson L, Verdine G L, Ellenberger T. Cell. 1998;95:249–258. doi: 10.1016/s0092-8674(00)81755-9. [DOI] [PubMed] [Google Scholar]
- 16.Scharer O D, Nash H M, Jiricny J, Laval J, Verdine G L. J Biol Chem. 1998;273:8592–8597. doi: 10.1074/jbc.273.15.8592. [DOI] [PubMed] [Google Scholar]
- 17.Vassylyev D G, Kashiwagi T, Mikami Y, Ariyoshi M, Iwai S, Ohtsuka E, Morikawa K. Cell. 1995;83:773–782. doi: 10.1016/0092-8674(95)90190-6. [DOI] [PubMed] [Google Scholar]
- 18.Slupphaug G, Mol. C D, Kavli B, Arvai A S, Krokan H E, Tainer J A. Nature (London) 1996;384:87–92. doi: 10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- 19.Parikh S S, Mol. C D, Slupphaug G, Bharati S, Krokan H E, Tainer J A. EMBO J. 1998;17:5214–5226. doi: 10.1093/emboj/17.17.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bruner S D, Norman D P, Verdine G L. Nature (London) 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 21.el Ghissassi F, Barbin A, Nair J, Bartsch H. Chem Res Toxicol. 1995;8:278–283. doi: 10.1021/tx00044a013. [DOI] [PubMed] [Google Scholar]
- 22.Matijasevic Z, Sekiguchi M, Ludlum D B. Proc Natl Acad Sci USA. 1992;89:9331–9334. doi: 10.1073/pnas.89.19.9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wyatt M D, Samson L D. Carcinogenesis. 2000;21:901–908. doi: 10.1093/carcin/21.5.901. [DOI] [PubMed] [Google Scholar]
- 24.Glassner B J, Rasmussen L J, Najarian M T, Posnick L M, Samson L D. Proc Natl Acad Sci USA. 1998;95:9997–10002. doi: 10.1073/pnas.95.17.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Derfler B, Samson L. EMBO J. 1990;9:4569–4575. doi: 10.1002/j.1460-2075.1990.tb07910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sodeoka M, Larson C J, Chen L, Leclair K P, Verdine G L. Bioorg Med Chem Lett. 1993;3:1089–1094. [Google Scholar]
- 27.Otwinowski Z, Minor V. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 28.Brunger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 29.Brunger A. Nature (London) 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 30.Laskowski R A, McArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 31.Lide D R. CRC Handbook of Chemistry and Physics1998–1999. Boca Raton, FL: CRC; 1998. [Google Scholar]
- 32.Barrett T E, Savva R, Panayotou G, Barlow T, Brown T, Jiricny J, Pearl L H. Cell. 1998;92:117–129. doi: 10.1016/s0092-8674(00)80904-6. [DOI] [PubMed] [Google Scholar]
- 33.Weber D J, Mullen G P, Mildvan A S. Biochemistry. 1991;30:7425–7437. doi: 10.1021/bi00244a009. [DOI] [PubMed] [Google Scholar]
- 34.Labahn J, Scharer O D, Long A, Ezaz-Nikpay K, Verdine G L, Ellenberger T E. Cell. 1996;86:321–329. doi: 10.1016/s0092-8674(00)80103-8. [DOI] [PubMed] [Google Scholar]
- 35.Gallivan J P, Dougherty D A. Proc Natl Acad Sci USA. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu G, Gershon P D, Hodel A E, Quiocho F A. Proc Natl Acad Sci USA. 1999;96:7149–7154. doi: 10.1073/pnas.96.13.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berdal K G, Johansen R F, Seeberg E. EMBO J. 1998;17:363–367. doi: 10.1093/emboj/17.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leonard G A, McAuley-Hecht K E, Gibson N J, Brown T, Watson W P, Hunter W N. Biochemistry. 1994;33:4755–4761. doi: 10.1021/bi00182a002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}