

Abstract
The effect of cytokines on non-traditional immunological targets under conditions of chronic inflammation is an ongoing subject of study. Fatigue is a symptom often associated with autoimmune diseases. Chronic inflammatory response and activated cell-mediated immunity are associated with cardiovascular myopathies which can be driven by muscle weakness and fatigue. Thus, we hypothesize that immune dysfunction-driven changes in myocyte mitochondria may play a critical role in fatigue-related pathogenesis. We show that persistent low-level expression of in designated AU-Rich Element deletion mice (ARE mice) under androgen exposure resulted in mitochondrial and metabolic deficiencies in myocytes from male or castrated ARE mice. Most notably, echocardiography unveiled that low ejection fraction in the left ventricle post-stress correlated with mitochondrial deficiencies, explaining how heart function decreases under stress. We report that inefficiencies and structural changes in mitochondria, with changes to expression of mitochondrial genes, are linked to male-biased fatigue and acute cardiomyopathy under stress. Our work highlights how male androgen hormone backgrounds and active autoimmunity reduce mitochondrial function and the ability to cope with stress and how pharmacological blockade of stress signal protects heart function. These studies provide new insight into the diverse actions of in fatigue, energy metabolism, and autoimmunity.
Keywords: mitochondria, inflammation, autoimmune disease, systemic lupus erythematosus (SLE), cardiomyopathy, IFN-γ
Introduction
Recent studies estimate that about80% of the 23.5 million Americans suffering an autoimmune disease describe long-term muscle weakness or fatigue as the most debilitating symptom [1–3]. Heart failure associated with exercise intolerance related to acute fatigue carries high mortality rates in male patients [4,5]. Cardiovascular disease and fatigue are ubiquitous complaints in male patients with systemic lupus erythematosus (SLE) [6–8]. A multicenter study found that cardiac dysfunction preceded or appeared very early in SLE, supporting a strong association between disease status and cardiomyopathy [9]. Additionally, postmortem examination found that myocarditis in 40% of lupus cases [6] and cardiomyopathy in male SLE patients have two common risk factors, the cardiotoxic effects of interferons (IFNs) and androgenic hormones [10]. However, the mechanisms of how those factors affect the heart are not well understood.
Generally, male patients present a bias towards developing myocarditis, pericarditis, and/or congestive heart failure (CHF) [11–13]. Notably, sustained high levels of have been associated with acute severe myocarditis in mice [13,14]. Interestingly, heart immune cell infiltration correlates with heart wall weakness, a factor associated with 25% of all cases of sudden cardiac death in young adults [15,16]. During infections, IFNs can regulate mitochondrial fission/fusion proteins [17], while androgens regulate mitochondrial respiratory elements [12,18,19]. Inefficiencies, in the conversion of carbohydrate molecules aerobically through the mitochondrial TCA cycle to charge internal membranes (cristae), disrupt the creation of high-energy phosphates from polarization of cristae, leading to fatigue [20,21]. As mitochondria modify energy management and adapt internal structures under stress [22], the lack of energy drives poor muscle mechanical action in chronic inflammatory conditions [23].
Here, we report how chronic inflammation and autoimmunity induce fatigue under acute stress by affecting mitochondrial function in myocytes, particularly in the left ventricle. Importantly, this stress-driven condition significantly decreases muscle heart function in lupus-prone male mice overexpressing as a result of replacement of the AU-rich element (ARE) with random nucleotides [24]. Specifically, we confirmed that the actions of under androgen-driven conditions limit mitochondrial recovery and metabolic efficiency, resulting in disruption of mitochondrial structures and energy management, respectively. Thus, our data revealed a novel underlying mechanism of association that may be responsible for causing cardiovascular disease in male SLE patients.
Materials and methods
Mice
C57BL/6 wildtype (WT), , , B6.129S7 () mice and relevant genetic crosses have been described previously [24,25]. All mice were age-matched (20–25 weeks). Mice were housed at animal facilities at the National Institutes of Health, Bethesda and Frederick, MD, USA. Animal handling was conducted in accordance with the Animal Care and Use Committee, under protocol number 21–018 [26].
Pathology
Pathological evaluation was conducted on serially sectioned slides stained with hematoxylin and eosin (H&E), phosphotungstic acid hematoxylin (PTAH), and immunohistochemistry (IHC). All slides were evaluated by a board-certified veterinary pathologist (EE). Tissue sections were fixed in 10% neutral-buffered formalin and routinely processed. IHC staining was performed using Leica Biosystems’ (Richmond, IL, USA) BondMax autostainer with the following conditions: F4/80, proteinase K for 5 min (S3020; Dako, Santa Clara, CA, USA), followed by anti-F4/80 (14–4801; eBioscience, San Diego, CA, USA) at a 1:200 dilution; CD3, HIER (heat-induced epitope retrieval) for 20 min, anti-CD3 (MCA1477; Bio-Rad, Tucson, AR, USA) at a 1:100 dilution with a bond polymer refine detection. Isotype control reagents were used for negative controls. Slides were digitized using an Aperio ScanScope XT (Leica Biosystems) at 200× in a single z-plane. Myocardial lesions, including inflammatory infiltrates, myocardial degeneration, and myocardial mineralization were assessed and quantified using semi-quantitative histopathological grades (supplementary material, Table S1). Intestinal smooth muscle and skeletal muscle were also evaluated. Myocarditis was evaluated by the Dallas criteria, which account for cell type and lesion distribution [27].
Mobility and fatigue testing
Forced swim testing (FST) was employed to examine exercise stress response as described elsewhere [28], in both a preliminary unmodified and a modified form with differences listed below. Tests failed if a mouse’s nose dipped below the water line (> 4 s). FST was modified as follows: the procedure was reduced to 90 s to accommodate testing tolerance for ARE mice. After testing, mice were removed from the water (maintained at 21–22 °C), dried, and allowed to recover for subsequent echocardiography or other tests.
The RotaRod test assesses coordination of rodents. Animals were placed with both hind legs and forearms situated facing away from rotation, requiring them to walk forward to remain upright (start speed of 4 rpm accelerating to 20 rpm after 10 s). Fall time indicates completion of the test. Average time trials were calculated from three consecutive and independent trials.
Open field testing (OFT) was employed on mice acclimated to the testing room in their home cages for 15 min. Test chamber activity was recorded for a duration of 300 s using automated detectors with three replicates and no experimenter present.
Echocardiography
Two-dimensional images of in vivo lightly anesthetized hearts were acquired using the Vevo 2100 ultrasound system (VisualSonics, Toronto, Canada) with a 40 MHz probe (MS550D, VisualSonics). The probe has a focal length of 7 mm, a lateral resolution of 90 μm, and an axial resolution of 40 μm. Mice were anesthetized with isoflurane and placed in the supine position on a heated platform with electrocardiogram (ECG) leads during the examination. Data evaluation included the assessment of heart wall thickness, velocities, specific dimensions, and ejection fraction (EF%), which expresses the difference between diastole and systole expressed as a percentage. Tests were repeated at least twice.
Modifying levels
The XMG-6 monoclonal antibody, an anti- antibody that neutralizes murine (a gift from Dr Giorgio Trinchieri, NCI), or isotype (IgG2a) was administered intraperitoneally (i.p.) 0.2 mg every other day for 3 weeks. Tests were repeated at least twice.
Adoptive cell transfer of immune cells from ARE mice and WT controls to irradiated mice was performed with both Jackson Laboratories B6.129S7- and WT recipients. Recipient mice were treated with an antibiotic (enrofloxacin, Baytril®; Bayer, Philadelphia, PA, USA) 1 week prior to whole body irradiation (900 rad). Each animal received 1 × 107 donor splenic cells followed by post-irradiation support; blood samples were taken at 1 week post-treatment, and at the end of 3 weeks, the mice were given final monitoring and sacrificed. Adoptive transfers were done at least twice.
Structural and functional mitochondrial assessment
For transmission electron microscopy (TEM) analysis of fixed cell structure, animals were perfused with 4% paraformaldehyde (PFA) and hearts were harvested, post-fixed in 1% osmium tetroxide, and embedded in Epon resin. A Leica UC6 ultramicrotome cut 0.6-μm sections, stained with toluidine blue for areas of interest, and 90–100 nm sections were cut, stained with 4% methanolic uranyl acetate and Reynold’s lead citrate, and examined on a Hitachi H7650 transmission electron microscope (Hitachi, Stoke Poges, UK).
Live mitochondria were extracted from myocytes as described by Das et al [29]. A Pierce BCA Protein Assay Kit was used to measure protein concentration (Thermo Fisher Scientific, Philadelphia, IL, USA). A Complex I Enzyme Activity Microplate Assay Kit (ab109721; Abcam, Cambridge, MA, USA) or Mitochondrial Complex 1 Activity Colorimetric Assay Kit (K968; Biovision, San Francisco, CA, USA) was used following the manufacturer’s instructions, without detergent. Mitochondrial Complex 1 (MC1) activity was measured by NAD/NADH turnover. Plates were incubated for 3 h at room temperature and measured at 450 nm at 1-min intervals for 31 min.
The immortalized mouse cardiomyocyte cell line HL-1, obtained from Sigma-Aldrich Inc. (Darmstadt, Hesse, Germany; SCC065), was grown as reported previously [30]. 4’,6-Diamidino-2-phenylindole (DAPI) and MitoTracker Deep Red (M22426, Thermo Fisher Scientific) were used as recommended by the manufacturer. Cells were stained with tetramethylrhodamine methyl ester perchlorate (TMRM) (T668, Thermo Fisher Scientific), a rhodamine-based dye showing charged cristae (Invitrogen, Carlsbad, CA, USA). Mitochondrial density is displayed as a percentage of control (MitoTracker Deep Red/DAPI). Mitochondrial competence is displayed as (TMRM/Mitochondrial density as a percentage of control).
Metabolic serum analysis
For myocyte damage markers in the blood, serum Creatine Kinase-Muscular Isotype (CK-MM) expression was determined using a colorimetric assay (MBS261027; MyBiosource, San Diego, CA, USA). Absorbance was measured at 450 nm, with the correction wavelength set at 570 nm. Cardiac troponin (cTNNT) in serum was measured using an ELISA (ab285235; Abcam) with secondary incubation for 25 min and reading at 450 nm. Experiments were repeated at least twice. For measuring lactic acid concentrations, experimental animals underwent the modified FST. Blood was collected in serum-separator tubes, and lactic acid was measured in fresh serum using the MAK064 assay kit and following the manufacturer’s instructions (Sigma Aldrich, St Louis, MO, USA). Experiments were repeated at least thrice. To measure glucose for glucose tolerance testing, 12 h prior to testing, all animals were fasted with ad libitum access to distilled water and then given × μl (= 7.5 × body weight of the mouse in grams) of 20% glucose stock solution. Intraperitoneal blood glucose levels were measured using an AccuChek Glucose Monitor (Roche Diagnostics, Indianapolis, IN, USA) following the manufacturer’s instructions, in three experiments.
Castration
Mice were castrated at 13 weeks of age using a standard surgical protocol [31]. Three serum blood samples were taken from experimental mice: one prior to surgery, one 3 weeks after surgery, and one 7 weeks post-castration. Mice were terminally bled, and tissues fixed in 4% PFA. Serum testosterone was measured using a Mouse Testosterone ELISA Kit from MyBiosource (MBS494055) following the manufacturer’s instructions, and this was done in triplicate.
Beta-antagonist treatments
Propranolol (P0884; Sigma Aldrich Inc., Darmstadt, Germany) and metoprolol (1441301, Sigma Aldrich Inc.) treatment (1 mg/kg in HBSS), or placebo was administered by i.p. injection daily for 5 days. Mice were examined for functional changes as listed above. The test was repeated twice for each treatment.
RNA sequencing and RT-qPCR
Mice underwent FST and subsequently hearts were excised and placed in RNAlater (AM7021, Thermo Fisher Scientific). RNA isolation was performed using a Qiagen (Bethesda, MD, USA) Tissue RNA isolation kit, then assessed using an Agilent (Santa Clara, CA, USA) BioAnalyzer. RNA samples were sequenced and analyzed commercially (Novogene, Chaoyang District, Beijing, PRC) using Illumina paired-end sequencing. In brief, alignment to mm10 used STAR; Differential Expression of Genes (DEG) analysis used DEseq2 (p adj ≤ 0.05); and Gene Ontology (GO) enrichment analysis used ClusterProfiler [32]. Validation was performed using cDNA from the Qiagen RT2 Easy First Strand Kit, with RT-qPCR completed using the Qiagen Fast SYBR Green Master Mix (330404, 4385610, Thermo Fisher Scientific). Supplementary material, Table S2 lists the primers used.
Statistical analysis
Alpha values of 0.05 or less were considered significant; all asterisks reflect levels of significance. Statistical analysis was performed using the GraphPad Prism software (Version 7.01; GraphPad Software Inc., San Diego, CA, USA) Parametric t-tests, ANOVA, or the non-parametric Mann–Whitney U-tests significance at 0.05. Data are presented as mean ± SEM unless otherwise stated. The indicated statistical tests to compare datasets were performed using GraphPad Prism 8 (Dotmatics, San Diego, CA, USA).
Results
Exercise exacerbated left ventricle functional deficiency in male mice
Generally, fatigue-like states are associated with systemic structural muscle abnormalities [33]. To determine whether ARE mice experience fatigue, mice were subjected to a battery of standard motility tests including the open field test (OFT), RotaRod examination, and forced swim test (FST) (Figure 1A). OFT involves passive observation in an open grid for a set time (supplementary material, Figure S1A) [34]. The RotaRod test distinguishes neurological effects from muscle fatigue by monitoring coordination (supplementary material, Figure S1C) [35]. OFTs showed that ARE male mice exhibit a decline in motility compared with male WT controls under steady conditions (257.6 ± 7.63 s versus 217.0 ± 12.22 s) (Figure 1B). No motility difference was observed in female mice (supplementary material, Figure S1B). While neurological causes were dismissed due to no significant changes in RotaRod testing in ARE male mice (supplementary material, Figure S1D), the FST uncovered a potential muscle compromise, as ARE male mice ( 122.1 ± 10.33 s and average 164.9 ± 14.29 s) failed the swim test compared with WT mice (Figure 1C).
Figure 1.
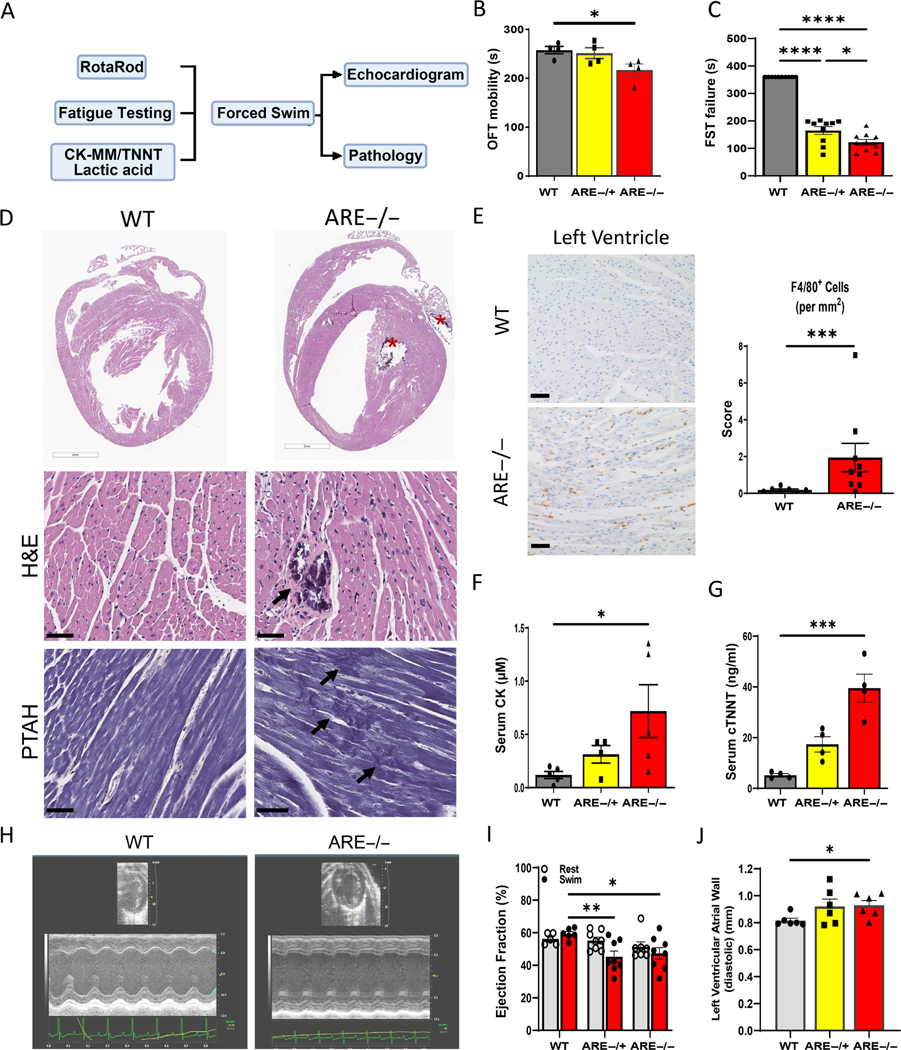
Heart function post-exercise decreases in male ARE mice. (A) Graphical representation of the methodologies and data collection process employed to evaluate heart function. (B) Open field test (OFT) outcomes in WT male mice and ARE −/− male mice (n = 10; p = 0.0135). (C) Low FST time denotes need of animal recovery from water (failure) prior to test limit (600 s). n = 10. Experiments were repeated thrice. (D) Representative myocardial images showing multifocal cardiac mineralization (*) within the left ventricle and left atria in male mice (top, right). Different from H&E, PTAH staining reveals disruption and fragmentation of cardiomyocyte myofibrils in the left ventricle of mice (arrows, bottom). (E) Representative images and bar graph showing infiltration of F4/80+ macrophages in the ventricular wall. (F, G) Graphs show significantly high levels of (F) creatine kinases (CK) and (G) cardiac troponin T (cTNNT) in mice (n = 5 animals per group). (H–J) In vivo assessment of heart function. (H) Representative echocardiograms post-swimming showing abnormal wall contraction in male mice. From H, quantitation of left ventricle anterior wall thickness in mm at diastole (J) and EF% values post-exercise (I) (n = 6 animals per group). All experiments were repeated at least twice, unless otherwise indicated. *p < 0.05, **p < 0.01, ***p < 0.001, ****p ≤ 0.0001.
To determine the extent of muscle compromise, histopathology with serum biomarkers was used to investigate the status of different muscle types in ARE mice. Histopathology revealed lesions primarily within the heart, followed by skeletal muscle (left hindlimb) and intestinal smooth muscle (supplementary material, Table S1). Cardiac histopathology included cardiomyocyte degeneration, characterized by myofibril fragmentation and cardiomyocyte necrosis and mineralization in mice (Figure 1D). Based on the Dallas criteria [27,36], myocarditis was not shown due to minimal T-cell infiltration; however, significant F4/80+ macrophage infiltration was found in ventricle walls of ARE male mice compared with WT counterparts (1.95 ± 0.77 and 0.18 ± 0.05 cells per mm2, respectively) (Figure 1E). However, a lower frequency of inducible nitric oxide (iNOS)-positive cells (~27 per mm2) was found in ARE males than in ARE female (~40 cells per mm2) mice (supplementary material, Figure S2A,B), indicating that iNOS from activated macrophages did not drive myocyte changes [37,38]. These data together with cumulative pathology scoring show a male predominance (supplementary material, Figure S1E), which led us to work herein on male mice only.
To confirm the above findings, we evaluated the serum levels of lactic acid, CK-MM, and cTNNT under basal conditions. Lactic acid, a common marker of fatigue, is produced by anaerobic respiratory pathways [39]. Consistently, ARE mice had lactic acid serum levels significantly higher than those of WT mice (45.13 ± 6.23 and 13.68 ± 2.82 μM in males, respectively; supplementary material, Figure S1F). Moreover, the levels of CK-MM and cTNNT were elevated seven and eight times compared with WT counterparts (0.718 ± 0.2476 μM and 39.50 ± 5.533 ng/ml versus 0.1192 ± 0.03316 μM and 5.20 ± 0.6014 ng/ml, respectively) (Figure 1F,G). It should be noted that cTNNT levels are significantly present only in males (supplementary material, Figure S1G). Thus, these data indicate that heart muscle is primarily affected in ARE male mice.
To determine if heart muscle fatigue worsens under stress in ARE male mice, heart function was assessed in vivo using ultrasound imaging before and after swim exercise (Figure 1H, supplementary material, Table S3). Notably, stress exercise-driven cardiac function, denoted by ejection fraction percentage (EF%) post-exercise, declined in and mice (47.33 ± 3.58% and 45.29 ± 3.39%, respectively) compared with WT counterparts (56.2 ± 1.26%; Figure 1I). Consequently, mild compensatory hypertrophy of the left ventricular anterior wall appeared in mice (Figure 1J). Further histological examination of the liver, lungs, and pericardium found no instances of congestion or pericarditis in ARE mice (not shown). Without distinct focal inflammatory locations or congestive heart disease, we conclude that heart muscle fatigue worsens under stress in ARE male mice.
Myocyte mitochondria exhibit abnormal metabolism
To investigate the causes of poor myocyte performance under stress, TEM was used to examine myocyte internal architecture in ARE and WT male mice. Surprisingly, mitochondria in ARE myocytes featured variable sizes and shapes, with poorly defined outer membranes, and internal gaps in cristae and matrix (Figure 2A). Consistently, ARE hearts exhibited greater mitochondrial density (65.90 per 10 μm2 ± 1.634) compared with WT counterparts (49.65 per 10 μm2 ± 1.432; Figure 2B). To assess the functional status of mitochondria, energy generation via MC1 was assessed in mitochondria isolated from the left ventricle. As expected, NADH was oxidized at one third of the rate in mice compared with WT controls. (Figure 2C). These data indicate loss of energy efficiency in ARE myocyte mitochondria.
Figure 2.

Myocyte mitochondrial dysfunction in ARE male mice. (A) Representative ultrastructural images from left ventricle showing abnormal mitochondria (10,000 × original magnification). (B) Number of mitochondria per 10 μm2 from A (n = 10). (C) Measurement of NAD/NADH turnover over time (minutes) representing mitochondrial complex 1 (MC1) activity in myocytes (n = 4). (D) ATP levels post-exercise assayed in male heart lysates (n = 4). Experiments were repeated thrice. (E) Graph depicts top downregulated mitochondrial gene pathways post-exercise in male heart. Red boxes denote pathways associated with mitochondrial energy generation. (F, G) Validation of RNAseq data. Fold-change of gene expression for the indicated genes involved in (F) mitochondrial fusion/fission or (G) metabolism. Experiments were repeated twice, unless otherwise indicated. ‘n’ denotes animals per group. **p < 0.01, ****p ≤ 0.0001.
Next, the mechanisms of energy metabolism were examined by assessing ATP generation in heart lysates and global glucose consumption. The latter measured muscular consumption after 12 h fasting [40,41]. Consistent with low metabolic outputs, ATP generation from ARE heart lysates was lower (6.73 ± 0.8711 μM) than in WT counterparts (14.83 ± 1.45 μM) (Figure 2D). Moreover, ARE mice utilized blood glucose fuel faster than WT mice, who may retain elevated glucose for up to 3 h (supplementary material, Figure S3A) [40]. Considered together, these data revealed that ARE myocytes feature abnormal mitochondrial structure and function.
Mitochondrial functional and structural genes are downregulated in ARE myocytes
To understand mitochondrial genetic changes, bulk RNAseq was performed on hearts of male (n = 4) and WT animals (n = 4) post-exercise. Consistent with our data, gene ontology (GO) analysis showed that 14 of the top 20 downregulated pathways corresponded to mitochondrial functions (i.e. mitochondrial inner matrix, cellular respiration, ATP synthesis coupled electron transport) (Figure 2E, red boxes). By contrast, the 20 highest upregulated GO pathways corresponded to immune responses, including innate immune activation and signatures (i.e. T-cell activation and cytokine production, respectively) (supplementary material, Figure S4A, Table S4). As expected, RT-qPCR confirmed downregulation of a key gene for mitochondrial fusion, Mfn2 [42] (0.0147 fold-change compared with WT), and strong upregulation of Stat1 (2.6732-fold), a direct signature gene in ARE myocytes, while no changes were observed in expression of the key mitochondrial fission gene Drp1 [43] (Figure 2F). Moreover, transcripts of lactate dehydrogenase B (Ldhb), encoding a critical enzyme regulating anaerobic fermentation [44], and pyruvate dehydrogenase subunit E1 alpha (Pdha1), encoding a ratelimiting enzyme for mitochondrial aerobic respiration [45], were significantly decreased in ARE myocytes (Figure 2G). Of note, ARE myocytes also exhibited upregulation of lactate dehydrogenase A (Ldha;1.67-fold), the subunit with higher binding affinity in the conversion of pyruvate to lactate [46]. In contrast, no changes were seen for Pdhb, another component of the pyruvate dehydrogenase complex (Figure 2G and supplementary material, Figure S4). Thus, these data confirmed that myocyte mitochondrial function is primarily affected in the -driven autoimmune landscape of ARE male mice.
Elevated , not type 1 IFNs, affects myocyte function and energy metabolism
To confirm that altersmyocyte mitochondrial function, WT and spleen cells were transferred into male WT and receptor knockout mice () (Figure 3A). As reported, successful adoptive transfer of spleen cells increased serum levels of (from 1.6 ± 0.24 to 19 ± 4.28 pg/ml; Figure 3B) which disorganized germinal centers (supplementary material, Figure S5A) [47]. As expected, adoptive transfer of cells to WT mice decreased global cardiovascular health and endurance, as reflected by decreased swim time in FST and decreased MC1 rate (Figure 3C,D). By contrast, despite high levels (Figure 3E), no changes to FST (Figure 3F) and mitochondrial metabolism (Figure 3G) happened without Ifngr signaling.
Figure 3.

drives mitochondrial dysfunction in cardiac muscle. (A) Experimental design scheme for adoptive transfer experiments. (B) Serum levels of (pg/ml) at indicated days pre- and post-adoptive transfer to WT mice. (C) Bar chart showing failure time for FST post-adoptive transfer of WT (dWT) or (dARE) cells into WT mice (p = 0.0003). (D) NAD/NADH slope showing generation of substrate in dWT or dARE mice. (E–G) Data obtained after transfer of immune cells from WT and mice (adoptive transfer) into irradiated Ifngr−/− mice. (E) Serum levels of (pg/ml) taken at indicated days pre- and post-adoptive transfer to Ifngr−/− mice. (F) EF% measurements post-stress. (G) Consumption of MC1 fuel shown as NAD/NADH slope of mice engrafted with WT (n = 4) or spleen cells (n = 3). (H) Scheme for the use of anti- antibody. (I) Time to FST failure before and after treatment with anti- (n = 4; ****p ≤ 0.0001). (J) MC1 rates from treated and untreated ARE−/− mice. (K) FST for WT, , and mice (n = 10; ****p ≤ 0.0001, **p = 0.0082). (L) Measurement of EF% of WT, , and ARE mice (n = 4; p = 0.0090). (M, N) HL-1 cells demonstrate altered mitochondrial density and function after (M) 5 days’ treatment with 100 IU of and/or 1,000 IU of . (N) Quantitation of mitochondrial stain in HL-1 cells. Normalized TMRM signal values expressed as a percentage of control (n = 16; ****p ≤ 0.0001). All experiments were repeated at least twice. ‘n’ denotes animals per group. **p < 0.01, ****p ≤ 0.0001.
To investigate if therapeutic blockade of could prevent cardiovascular and mitochondrial metabolic damage, male animals received anti- antibody prior to echocardiogram evaluation post-swim (Figure 3H). As expected, global cardiovascular improvement post- blockade allowed for mice completion of FST compared with isotype-treated controls (Figure 3I). This resulted from a 4-fold increase in mitochondrial NAD/NADPH conversion in isolated mitochondria from anti- -treated WT myocytes; we have also shown restoration of germinal centers (Figure 3J and supplementary material, Figures S3B and S5B). These data indicate that overexpression of induces abnormal mitochondrial metabolic outcomes in myocytes.
Like , SLE patients exhibit abnormally high type 1 IFN signaling [48]. To determine whether could drive myocyte mitochondrial dysfunction in the absence of type 1 IFN signaling, cardiac performance was tested in type 1 interferon receptor-deficient ARE male mice ( ). These type 1 Ifnar-deficient ARE mice produced constitutively high serum levels but unlike ARE mice exhibited mild autoimmunity due to lack of the common type 1 IFN receptor (Ifnar) [49]. Surprisingly, like mice, failed the FST and had a similarly low EF% post-swim stress to mice (49.2 ± 1.53) compared with WT controls (56.0 ± 1.26) (Figure 3K,L). Pathological evaluation of left ventricular heart walls showed that and mice exhibited no evidence of necrosis or mineralization. However, minimal immune infiltrates and mild myocyte disorganization were observed (supplementary material, Figure S5C). To support these findings, mitochondrial density was assessed in murine cardiac HL-1 cells grown in vitro with either alone, alone, or their combination for 5 days. Consistent with in vivo data, alone failed to increase mitochondrial density or affect mitochondrial polarization compared with significant changes by alone or in combination (Figure 3M,N). Thus, these data demonstrate a minimal role for type 1 IFN in global myocyte organization, supporting a model where directly induces mitochondrial dysfunction in the myocytes of ARE male mice.
Androgen suppression improves cardiac function
We asked whether androgens together with the IFN signature modify heart and myocyte mitochondrial function. To examine this possibility, heart function was assessed in vivo in castrated male mice (Figure 4A). Successful castration significantly lowered testosterone levels after 8 weeks compared with controls (Figure 4B). Surprisingly, no differences in EF% were obtained between castrated ARE−/− and WT mice (Figure 4C). Histological analysis of hearts revealed no substantial immune infiltrate or calcifications in left ventricles of castrated mice (Figure 4D). However, TEM revealed mild to moderate disruption of mitochondrial size and cristae in myocytes (Figure 4D). Assessment of NADH oxidation from isolated ARE myocytes was no different from that of WT controls (Figure 4E), suggesting that androgens affect mitochondrial internal structures in the presence of elevated .
Figure 4.

Castration improves cardiac outcomes in male mice. (A) Scheme detailing sample collection from mice prior to or after castration. (B) Serum levels of testosterone at indicated time points post-castration (n = 6). (C) Ejection fraction (%) in castrated mice before and after swim test (n = 3). (D) Representative H&E staining (top; 20× original magnification) and ultrastructural images (10,000× original magnification) from left ventricle post-exercise in WT and castrated mice. (E) Measurement of myocyte mitochondrial complex 1 activity after 8 weeks post-recovery from castration surgery in WT and mice. (F, G) In vitro responses of the myocyte cell line (HL-1) to testosterone (T; 3,000 pg/ml) and (10 IU/ml) after 5 days of treatment (n = 16). (F) Mitochondrial density (mitodensity). (G) Note that combined treatment with T and decreases mitochondrial function as assessed by TMRM signal normalized by mitodensity. Values are expressed as a percentage of control. Experiments were repeated twice, unless otherwise indicated. ‘n’ denotes animals per group. *p < 0.05, **p < 0.01, ****p ≤ 0.0001.
To confirm the association between androgens and on myocyte mitochondrial metabolism, mitochondrial population and function were assessed in vitro using murine HL-1 cardiac cells after 7 days’ treatment with alone, testosterone (T) alone, or their combination () [30]. As expected, mitochondrial density in HL-1 cells increased after treatment with alone and was independent of testosterone (Figure 4F). However, and testosterone significantly decreased the charge potential of mitochondrial membranes via TMRM signaling by lowering competence 40% compared with controls (Figure 4G). These data suggest that the association of and androgens disrupts mitochondrial metabolic functions in ARE myocytes.
Beta-adrenergic receptor antagonists normalize ARE mice
Catecholamines (adrenaline and norepinephrine) drive muscle stress signals via structurally different beta-adrenergic receptors, 1 and 2 [50–52]. We asked whether beta adrenergic receptor blockade lessens the detrimental impact of stress on myocytes in male ARE mice. Mice were treated for 5 days with either a general or a specific beta-adrenergic receptor 1 antagonist, propranolol and metoprolol, respectively (Figure 5A,B). Assessment of EF% post-swim showed that both propranolol and metoprolol increased EF% in mice. However, only metoprolol preserved EF% without altering myocardial pathology both in WT and in mice (Figure 5C,D and supplementary material, Figure S6). Collectively, these data demonstrate that therapeutic managing of stress helps to prevent the deleterious consequences of myocyte dysfunction in male mice.
Figure 5.

Beta-antagonist therapy improves response to exercise stress in ARE mice. (A) Timeline for treatments with beta-blockers. (B) Diagram of the structures for beta receptors 1 and 2 produced using BioRender.com. (C, D) Heart EF% after 5 days’ treatment with the indicated beta-blocker (n = 4). (C) EF% in propranolol-treated mice. (D) EF% in metoprolol-treated mice. Values expressed as a percentage of control. ‘n’ denotes animals per group. *p < 0.05. Experiments were repeated twice, unless otherwise indicated.
Discussion
Cardiovascular disease is a major cause of death in SLE patients, yet is often poorly addressed and reported postmortem [53]. We have uncovered a hidden, stress-triggered, heart impairment that relates to underlying autoimmune conditions in lupus-prone ARE male mice. Deprivation and transcriptional experiments after light exercise show detrimental results from an associative effect between and androgens, an age-dependent event that eventually affects both male and female SLE patients [10,54]. Based on our data, the use of FDA-approved beta-adrenergic antagonists to relieve muscular stress response could provide a therapeutic course of treatment to prevent heart failure, the leading cause of death in male SLE patients [10]. To our knowledge, this is the first report that addresses myocyte mitochondrial dysfunction as a precursor mechanism of cardiovascular disease and proposes a therapeutic intervention to treat this condition in male lupus-prone mice or SLE patients.
Different from myocarditis or pericarditis with inflammatory foci [27], the existence of structural and functional abnormalities affecting mitochondria in myocytes highlights underlying processes connected to global SLE status. Specifically, we uncovered that a decreased expression of essential mitochondrial genes, e.g. pyruvate dehydrogenase (Pdha), in myocytes was associated with heart dysfunction and fatigue only in male hearts [55]. Conversely, other mitochondrial genes associated with muscle stress and lactate metabolism, such as ldha which was upregulated with associated high serum levels of lactic acid, provide a broader view of the global impact of SLE [56]. Importantly, our finding that measurement of EF% after stress correlated with ultrastructural mitochondrial abnormalities associated with dysfunctional fuel utilization in myocytes [57] provides a potential tool to evaluate these processes in SLE patients [24,58]. Thus, with the downregulation of metabolic markers for efficient mitochondrial energy generation, our data demonstrate that echocardiography becomes a reliable screen tool to assess cardiac mitochondrial disease in males with underlying autoimmunity.
Generally, type 1 interferons are considered the major driver of SLE and other lupus-like diseases [59]. Surprisingly, our in vivo studies with mice, Ifnar-deficient ARE mice, and blockade of with antibodies in ARE mice demonstrate that deprivation of , not type 1 IFNs, prevents the deleterious mitochondrial functional loss in male myocytes by downregulating genes controlling mitochondrial function and fusion. Poor fusion causes accumulation of deficient myocyte mitochondria incapable of handling stress and demonstrating potentially poor energy generation, features that may suggest mitochondrial immaturity or defects [60,61]. Similarly, it is also well known that testosterone protects mitochondria from oxidative stress by upregulating genes associated with radical scavenging [62]. Again, adoptive transfer data into castrated mice showed that loss of androgen mitochondrial protection enabled to drastically disrupt myocyte mitochondrial function under stress. This associative mechanism could explain, in part, how pre-existent autoimmune conditions could predispose male cancer patients undergoing immunotherapy to develop idiopathic cardiomyopathy [63,64]. Together, these findings lead us to conclude that drives myocyte mitochondrial dysfunction in autoimmune landscapes.
Finally, we provide proof of principle that effective treatment of cardiac stress with targeted beta-blocking medication may offer a viable therapeutic option to potentially decrease cardiovascular death risk for SLE patients. Particularly, the specific use of beta-adrenergic receptor 1 antagonists such as metoprolol could selectively protect the mitochondrial myocyte from high energy demands, improve the heart’s response to stress, and limit heart damage [65]. Thus, our work highlights how androgens and signatures reduce mitochondrial function in myocytes, limiting the heart’s ability to cope with stress in autoimmunity.
Supplementary Material
Figure S1. Functional and pathological scoring of muscular fatigue in ARE mice
Figure S2. Inducible nitric oxide synthase (iNOS) expression in left ventricles from female and male mice
Figure S3. Additional metabolic evaluations
Figure S4. Gene expression (bulk RNAseq) in male ARE hearts
Figure S5. Overexpression of modifies cellular organization in organs
Figure S6. Heart pathology of metoprolol-treated mice
Table S1. Pathology scoring scale from differing muscle groups in male mice
Table S2. Primers used for RT-qPCR validation
Table S3. Echocardiogram data post-swimming
Table S4. Differentially expressed gene (DEG) list. Excel file providing complete DEG listing from RNAseq (Sheet 1), upregulated genes (Sheet 2), and downregulated genes (Sheet 3)
Acknowledgements
We acknowledge technical assistance from the Electron Microscopy Core Imaging Facility (EMCIF), Center for Innovative Biomedical Resources, University of Maryland, Baltimore for the embedding of the samples. We also acknowledge the electron microscopy expertise of Dr Kunio Nagashima from the Advanced Technology Research Facility in Frederick, Maryland, and the technical assistance of Lisa Riffle from the Small Animal Imaging Program at the Laboratory Animal Sciences Program at the National Cancer Institute in Frederick. We would also like to acknowledge the advice of Dr Sam Das, Dr Michael N Sack, Dr Erika M Palmieri, Dr Enitome E Bafur, Dr Javier Rivera-Guzman, and Dr Javid J Moslehi for providing expertise and assistance with our work. We gratefully acknowledge the skills of Mrs Megan Karwan, Ms Heather Potts, Mr Tim Back, and Mrs Loretta Smith for assistance with the animal studies; and The Scientific Publications, Graphics and Media Department; Frederick National Laboratory for Cancer Research, National Cancer Institute for assistance with figure finishing. This project was funded in whole or in part by Federal funds directly from the intramural research programs of the NCI, Center for Cancer Research, Cancer Innovation Laboratories, under Contract No. HHSN261200800001E and the iCure grant (JMF). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Footnotes
No conflicts of interest were declared.
Data availability statement
The raw data and processed files pertaining to the RNAseq results presented in this article are made available in NCBI’s GEO under accession number GSE211217 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE211217).
References
- 1.Dinse G, Parks C, Weinberg C, et al. Increasing prevalence of antinuclear antibodies in the United States. Arthritis Rheumatol 2020; 74: 2032–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tench CM, McCurdie I, White PD, et al. The prevalence and associations of fatigue in systemic lupus erythematosus. Rheumatology 2000; 39: 1249–1254. [DOI] [PubMed] [Google Scholar]
- 3.Roberts MH, Erdei E. Comparative United States autoimmune disease rates for 2010–2016 by sex, geographic region, and race. Autoimmun Rev 2020; 19: 102423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Myers J, Prakash M, Froelicher V, et al. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med 2002; 346: 793–801. [DOI] [PubMed] [Google Scholar]
- 5.Craig JC, Colburn TD, Caldwell JT, et al. Central and peripheral factors mechanistically linked to exercise intolerance in heart failure with reduced ejection fraction. AmJPhysiolHeartCirc Physiol 2019; 317: H434–H444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doherty NE, Siegal RJ. Cardiovascular manifestations of systemiclupus erythematosus. Am Heart J 1985; 110: 1257–1265. [DOI] [PubMed] [Google Scholar]
- 7.Doria A, Iaccarino L, Sarzi-Puttini P, et al. Cardiac involvement in systemic lupus erythematosus. Lupus 2005; 14: 683–686. [DOI] [PubMed] [Google Scholar]
- 8.Lee YH, Choi SJ, Ji JD, et al. Overall and cause-specific mortality in systemiclupus erythematosus: an updatedmeta-analysis. Lupus 2016; 25: 727–734. [DOI] [PubMed] [Google Scholar]
- 9.Urowitz MB, Gladman DD, Anderson NM, et al. Cardiovascular events prior to or early after diagnosis of systemic lupus erythematosus in the systemic lupus international collaborating clinics cohort. Lupus Sci Med 2016; 3: e000143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falasinnu T, Chaichian Y, Simard JF. Impact of sex on systemic lupuserythematosus-related causes of premature mortality in the United States. J Womens Health 2017; 26: 1214–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amaya-Amaya J, Montoya-Sánchez L, Rojas-Villarraga A. Cardiovascular involvement in autoimmune diseases. Biomed Res Int 2014; 2014: 367359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopes RA, Neves KB, Pestana CR, et al. Testosterone induces apoptosis in vascular smooth muscle cells via extrinsic apoptotic pathway with mitochondria-generated reactive oxygen species involvement. Am J Physiol Heart Circ Physiol 2014; 306: H1485–H1494. [DOI] [PubMed] [Google Scholar]
- 13.Reifenberg K, Lehr HA, Torzewski M, et al. Interferon-gamma induces chronic active myocarditis and cardiomyopathy in transgenic mice. Am J Pathol 2007; 171: 463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torzewski M, Wenzel P, Kleinert H, et al. Chronic inflammatory cardiomyopathy of interferon γ-overexpressing transgenic mice is mediated by tumor necrosis factor-α. Am J Pathol 2012; 180: 73–81. [DOI] [PubMed] [Google Scholar]
- 15.Lynge TH, Nielsen TS, Gregers Winkel B, et al. Sudden cardiac death causedby myocarditis in persons aged1–49 years: a nationwide study of 14 294 deaths in Denmark. Forensic Sci Res 2019; 4: 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ha FJ, Han HC, Sanders P, et al. Sudden cardiac death in the young: incidence, trends, and risk factors in a nationwide study. Circ Cardiovasc Qual Outcomes 2020; 13: e006470. [DOI] [PubMed] [Google Scholar]
- 17.Batista AF, Rody T, Forny-Germano L, et al. Interleukin-1β mediates alterations in mitochondrial fusion/fission proteins and memory impairment induced by amyloid-β oligomers. J Neuroinflammation 2021; 18: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pronsato L, Milanesi L, Vasconsuelo A. Testosterone induces upregulation of mitochondrial gene expression in murine C2C12 skeletal muscle cells accompanied by an increase of nuclear respiratory factor-1 and its downstream effectors. Mol Cell Endocrinol 2020; 500: 110631. [DOI] [PubMed] [Google Scholar]
- 19.Diaconu R, Donoiu I, Mirea O, et al. Testosterone, cardiomyopathies, and heart failure: a narrative review. Asian J Androl 2021; 23: 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Q,Liu R, Yu Q,et al. Metabolic regulation of inflammasomesin inflammation. Immunology 2019; 157: 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sundberg CW,Fitts RH.Bioenergetic basis of skeletal musclefatigue. Curr Opin Physiol 2019; 10: 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eisner V, Picard M, Hajnoczky G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol 2018; 20: 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huss JM, Kelly DP. Mitochondrial energy metabolism in heartfailure: a question of balance. J Clin Invest 2005; 115: 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hodge DL, Berthet C, Coppola V, et al. IFN-gamma AU-rich element removal promotes chronic IFN-gamma expression and autoimmunity in mice. J Autoimmun 2014; 53: 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bae HR, Leung PS, Tsuneyama K, et al. Chronic expression of interferon-gamma leads to murine autoimmune cholangitis with a female predominance. Hepatology 2016; 64: 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.National Research Council (USA) Committee for the Update of theGuide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals (8th edn). National Academies Press (USA): Washington, DC, 2011. [Google Scholar]
- 27.Aretz HT. Myocarditis: the Dallas criteria. Hum Pathol 1987; 18: 619–624. [DOI] [PubMed] [Google Scholar]
- 28.Commons KG, Cholanians AB, Babb JA, et al. The rodent forced swim test measures stress-coping strategy, not depression-like behavior. ACS Chem Neurosci 2017; 8: 955–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das S, Bedja D, Campbell N, et al. miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS One 2014; 9: e96820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Claycomb WC, Lanson NA, Stallworth BS, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A 1998; 95: 2979–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valkenburg KC, Amend SR, Pienta KJ. Murine prostate microdissection and surgical castration. J Vis Exp 2016; 11: 53984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.The Gene Ontology Consortium. The Gene Ontology resource: 20 yearsand still GOing strong. Nucleic Acids Res 2019; 47: D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan TC, Fang H, Magder LS, et al. Differences between male and female systemic lupus erythematosus in a multiethnic population. J Rheumatol 2012; 39: 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dougherty J, Springer DA, Cullen MJ, et al. Evaluation of the effects of chemotherapy-induced fatigue and pharmacological interventions in multiple mouse behavioral assays. Behav Brain Res 2019; 360: 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pugsley MK, Authier S, Curtis MJ. Principles of safety pharmacology. Br J Pharmacol 2008; 154: 1382–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Błyszczuk P. Myocarditis in humans and in experimental animal models. Front Cardiovasc Med 2019; 6: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia L, Wang Y, Wang Y, et al. Heme oxygenase-1 in macrophages drives septic cardiac dysfunction via suppressing lysosomal degradation of inducible nitric oxide synthase. Circ Res 2018; 122: 1532–1544. [DOI] [PubMed] [Google Scholar]
- 38.Nussler AK, Billiar TR, Liu ZZ, et al. Coinduction of nitric oxide synthase and argininosuccinate synthetase in a murine macrophage cell line. Implications for regulation of nitric oxide production. J Biol Chem 1994; 269: 1257–1261. [PubMed] [Google Scholar]
- 39.Lumb AB. Hypoxia. In Nunn’s Applied Respiratory Physiology (8th edn). Elsevier: Amsterdam, Netherlands, 2017; 327–334. [Google Scholar]
- 40.Goren HJ, Kulkarni RN, Kahn CR. Glucose homeostasis and tissuetranscript content of insulin signaling intermediates in four inbred strains of mice: C57BL/6, C57BLKS/6, DBA/2, and 129X1. Endocrinology 2004; 145: 3307–3323. [DOI] [PubMed] [Google Scholar]
- 41.Benedé-Ubieto R, Estévez-Vázquez O, Ramadori P, et al. Guidelines and considerations for metabolic tolerance tests in mice. Diabetes Metab Syndr Obes 2020; 13: 439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen H, Detmer SA, Ewald AJ, et al. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 2003; 160: 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao F, Reynolds MB, Passalacqua KD, et al. The mitochondrial fission regulator DRP1 controls post-transcriptional regulation of TNF-α. Front Cell Infect Microbiol 2021; 10: 593805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim J, Kim E, Lee Y, et al. Decreased lactate dehydrogenase B expression enhances claudin 1-mediated hepatoma cell invasiveness via mitochondrial defects. Exp Cell Res 2011; 317: 1108–1118. [DOI] [PubMed] [Google Scholar]
- 45.Jaiswal M, Haelterman NA, Sandoval H, et al. Correction: impaired mitochondrial energy production causes light-induced photoreceptor degeneration independent of oxidative stress. PLoS Biol 2018; 16: e1002622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Urbańska K, Orzechowski A. Unappreciated role of LDHA and LDHB to control apoptosis and autophagy in tumor cells. Int J Mol Sci 2019; 20: 2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Domeier PP, Chodisetti SB, Soni C, et al. receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J Exp Med 2016; 213: 715–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oke V, Gunnarsson I, Dorschner J, et al. High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res Ther 2019; 21: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Valencia JC, Erwin-Cohen RA, Clavijo PE, et al. Myeloid-derived suppressive cell expansion promotes melanoma growth and autoimmunity by inhibiting CD40/IL27 regulation in macrophages. Cancer Res 2021; 81: 5977–5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Lucia C, Eguchi A, Koch WJ. New insights in cardiac β-adrenergic signaling during heart failure and aging. Front Pharmacol 2018; 9: 904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barth E, Albuszies G, Baumgart K, et al. Glucose metabolism and catecholamines. Crit Care Med 2007; 35: S508–S518. [DOI] [PubMed] [Google Scholar]
- 52.Taylor MR. Pharmacogenetics of the human beta-adrenergic receptors. Pharmacogenomics J 2007; 7: 29–37. [DOI] [PubMed] [Google Scholar]
- 53.Panchal L, Divate S, Vaideeswar P, et al. Cardiovascular involvement in systemic lupus erythematosus: an autopsy study of 27 patients in India. J Postgrad Med 2006; 52: 5–10 discussion 10. [PubMed] [Google Scholar]
- 54.Fogle RH, Stanczyk FZ, Zhang X, et al. Ovarian androgen production in postmenopausal women. J Clin Endocrinol Metab 2007; 92: 3040–3043. [DOI] [PubMed] [Google Scholar]
- 55.Shin HK, Grahame G, McCandless SE, et al. Enzymatic testing sensitivity, variability and practical diagnostic algorithm for pyruvate dehydrogenase complex (PDC) deficiency. Mol Genet Metab 2017; 122: 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lund J, Aas V, Tingstad RH, et al. Utilization of lactic acid in human myotubes and interplay with glucose and fatty acid metabolism. Sci Rep 2018; 8: 9814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bugger H, Chen D, Riehle C, et al. Tissue-specific remodeling of the mitochondrial proteome in type 1 diabetic Akita mice. Diabetes 2009; 58: 1986–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology ofheart failure. J Clin Invest 2018; 128: 3716–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Akiyama C, Tsumiyama K, Uchimura C, et al. Conditional upregulation of alone is sufficient to induce systemic lupus erythematosus. J Immunol 2019; 203: 835–843. [DOI] [PubMed] [Google Scholar]
- 60.Morita Y, Tohyama S. Metabolic regulation of cardiac differentiation and maturation in pluripotent stem cells: a lesson from heart development. JMA J 2020; 3: 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang YF, Lin HL, Wang LJ, et al. Coordinated organization of mitochondrial lamellar cristae and gain of COX function during mitochondrial maturation in Drosophila. Mol Biol Cell 2020; 31: 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yan W, Kang Y, Ji X, et al. Testosterone upregulates the expression of mitochondrial ND1 and ND4 and alleviates the oxidative damage to the nigrostriatal dopaminergic system in orchiectomized rats. Oxid Med Cell Longev 2017; 2017: 1202459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dal’bo N, Patel R, Parikh R, et al. Cardiotoxicity of contemporary anticancer immunotherapy. Curr Treat Options Cardiovasc Med 2020; 22: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anquetil C, Salem JE, Lebrun-Vignes B, et al. Immune checkpoint inhibitor-associated myositis: expanding the spectrum of cardiac complications of the immunotherapy revolution. Circulation 2018; 138: 743–745. [DOI] [PubMed] [Google Scholar]
- 65.Sandroni PB, Fisher-Wellman KH, Jensen BC. Adrenergic receptor regulation of mitochondrial function in cardiomyocytes. J Cardiovasc Pharmacol 2022; 80: 364–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Functional and pathological scoring of muscular fatigue in ARE mice
Figure S2. Inducible nitric oxide synthase (iNOS) expression in left ventricles from female and male mice
Figure S3. Additional metabolic evaluations
Figure S4. Gene expression (bulk RNAseq) in male ARE hearts
Figure S5. Overexpression of modifies cellular organization in organs
Figure S6. Heart pathology of metoprolol-treated mice
Table S1. Pathology scoring scale from differing muscle groups in male mice
Table S2. Primers used for RT-qPCR validation
Table S3. Echocardiogram data post-swimming
Table S4. Differentially expressed gene (DEG) list. Excel file providing complete DEG listing from RNAseq (Sheet 1), upregulated genes (Sheet 2), and downregulated genes (Sheet 3)
Data Availability Statement
The raw data and processed files pertaining to the RNAseq results presented in this article are made available in NCBI’s GEO under accession number GSE211217 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE211217).