

SUMMARY
Regulation of RNA substrate selectivity of m6A demethylase ALKBH5 remains elusive. Here, we identify RNA binding motif protein 33 (RBM33) as a previously unrecognized m6A binding protein that plays a critical role in ALKBH5-mediated mRNA m6A demethylation of a subset of mRNA transcripts by forming a complex with ALKBH5. RBM33 recruits ALKBH5 to its m6A-marked substrate and activates ALKBH5 demethylase activity through removal of its SUMOylation. We further demonstrate that RBM33 is critical for the tumorigenesis of Head-Neck Squamous Cell Carcinoma (HNSCC). RBM33 promotes autophagy by recruiting ALKBH5 to demethylate and stabilize DDIT4 mRNA, which is responsible for the oncogenic function of RBM33 in HNSCC cells. Collectively, our study uncovers the mechanism of selectively demethylate m6A methylation of a subset of transcripts during tumorigenesis that may explain demethylation selectivity in other cellular processes, and we showed its importance in the maintenance of tumorigenesis of HNSCC.
Graphical Abstract
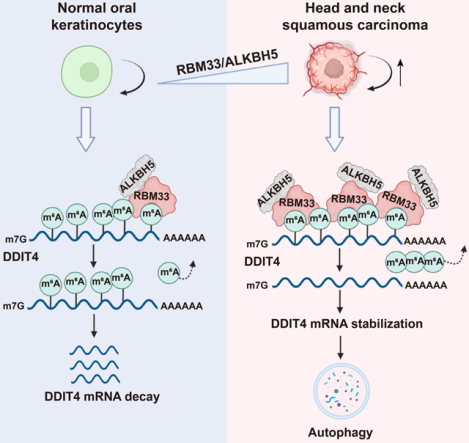
eToc Blurb
Yu et al. identify RBM33 as a new m6A binding protein that forms a complex with ALKBH5 and mediates m6A demethylation of selected transcripts by regulating ALKBH5 substrate accessibility and activity. In HNSCC cells, highly expressed RBM33 promotes HNSCC tumorigenesis by stabilizing DDIT4 in an m6A-dependent manner, thereby inducing autophagy.
INTRODUCTION
RNA binding proteins (RBPs) are a structurally and functionally diverse class of proteins, which are involved in a variety of biological processes. Accumulating evidence has shown that RBPs play a key role in post-transcriptional regulation of gene expression by controlling the alternative splicing, transport, stability, degradation, and translation of coding or non-coding RNAs.1–3 RBPs directly interact with RNAs in a sequence-specific manner through RNA recognition motifs (RRMs), which are one of multiple common RNA binding domains; other domains include K-homology (KH) domains, DEAD-box domains, and double-stranded RBD (dsRBD). Most RBPs are conserved across species and ubiquitously expressed in human tissues.1 Given the importance of RBPs in the post-transcriptional regulation of gene expression, dysregulation of RBPs contributes to various human diseases, including cancer.4–7 Approximately 1542 RBPs have been identified using bioinformatic and experimental methods, accounting for 7.5% of protein-coding genes. However, the biological functions of one-third of RBPs remain unknown.1 RBM33 is one such member of these RBPs, and to date, the biological function of RBM33 is largely unknown. A very recent study suggests that RBM33 predominantly binds to the intron-less RNA and guide its nuclear export.8
N6-methyladenosine (m6A) methylation, the most abundant internal modification in eukaryotic mRNA, is a dynamic and reversible process. m6A is deposited by the m6A methyltransferase complex, composed of core subunits METTL39,10 and METTL1411 as well as additional adaptor proteins such as WTAP,12 VIRMA (KIAA1429),13RBM15/15B,14 and ZC3H13.15 m6A methylation can be removed by m6A demethylases such as FTO16 and ALKBH5.17 The crucial roles of RNA m6A methylation in diverse biological processes are mediated by many RNA binding proteins (RBPs) with ‘reader’ or ‘nonreader’ functions including the YTH domain-containing proteins (YTHDF1/2/3 and YTHDC1/2),18,19 and hnRNPs (hnRNPG and hnRNPC).20 A previous study suggests that RNA-binding protein, HNRNPA2B1 binds to m6A-marked RNA substrate presumably through its RRM domains,21 but later a subsequent study shows that the RRMs in HNRNPA2B1 protein doesn’t bind m6A directly.22 Additionally, a group of RNA binding proteins including eIF3, IGF2BPs, Prrc2, and FMR1 has been newly identified as m6A readers.23–26 Adaptor proteins RBM15 and RBM15B, which are components of the m6A methyltransferase complex (MTC), are necessary for the recruitment of the MTC complex to its RNA substrate to trigger m6A methylation.14 While MTC can be recruited by various cellular components to target transcript, we currently do not have a clear understanding of how demethylation selectivity is achieved. We do not know whether similar adaptor proteins exist to facilitate m6A demethylation of selected transcripts.
In this study, we uncovered a previously unnoticed function and the underlying mechanism of RBM33 in facilitating ALKBH5-mediated m6A demethylation of selected mRNA transcripts, and elucidate the importance of RBM33-mediated selectivity of m6A demethylation of specific transcripts during tumorigenesis in human HNSCC cells. Our data suggest that selective demethylation mediated by RBM33 may play a critical role in diverse of biological process.
RESULTS
RBM33 forms a complex with ALKBH5 and regulates mRNA m6A methylation dependent on ALKBH5
RBM33 is a known RBP, whereas little is known about the biological function of RBM33. To glean any potential role in cellular metabolism or oncogenesis, we analyzed The Cancer Genome Atlas (TCGA) database. Notably, RBM33 is commonly up-regulated in diverse solid tumors including HNSCC, kidney renal clear cell carcinoma (KIRC), bladder urothelial carcinoma (BLCA), cervical squamous cell carcinoma(CESC), colon adenocarcinoma (COAD), liver hepatocellular carcinoma (LIHC), and adrenocortical carcinoma (ACC) while its expression has no changes or down-regulation in glioblastoma multiforme (GBM), lung adenocarcinoma (LUAD), prostate adenocarcinoma (PRAD), and breast invasive carcinoma (BRCA) (Figure S1), suggesting that RBM33 may have oncogenic functions in diverse cancers. To explore the function of RBM33, we employed the Strep-Tactin XT system,27,28 a protein pull-down-based approach, to search for the potential binding partners of RBM33 in HNSCC cells. We then employed liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis of the proteins pulled down by RBM33 co-immunoprecipitation (Co-IP), and found that ALKBH5 is one of the most abundant proteins that potentially interact with RBM33 in UM-SCC-1 cells (Figure 1A). However, none of the other known regulators of m6A RNA methylation including METTL3, METTL14, and FTO was identified in the RBM33 associated complex. ALKBH5 plays an important role in the maintenance of cancer stem cells.29 Thus, we hypothesize that ALKBH5 is a key partner of RBM33, and mediates the function of RBM33 in cancers. To verify the interaction between ALKBH5 and RBM33, we performed the Strep-Tactin pull-down assay. Consistent with the LC-MS/MS result, endogenous ALKBH5 could co-immunoprecipitate with Strep-tagged RBM33 (Figure S2A) and endogenous RBM33 in UM-SCC-1 cells (Figure 1B). As shown by immunostaining analysis, RBM33 colocalizes with ALKBH5 (Figure 1C) in the cells. However, RBM33 does not interact with Strep-tagged METTL3, METTL14, and FTO (Figure 1D and Figure S2B). These results provide compelling evidence that RBM33 forms a complex with ALKBH5 but is not associated with the m6A MTC or FTO demethylase, suggesting a unique role of RBM33 in ALKBH5-mediated RNA m6A demethylation. Additionally, we observed that RNAase treatment significantly disrupted the interaction between ALKBH5 and RBM33, suggesting that their interaction depends on the presence of RNA substrates (Figure S2C).
Figure. 1.
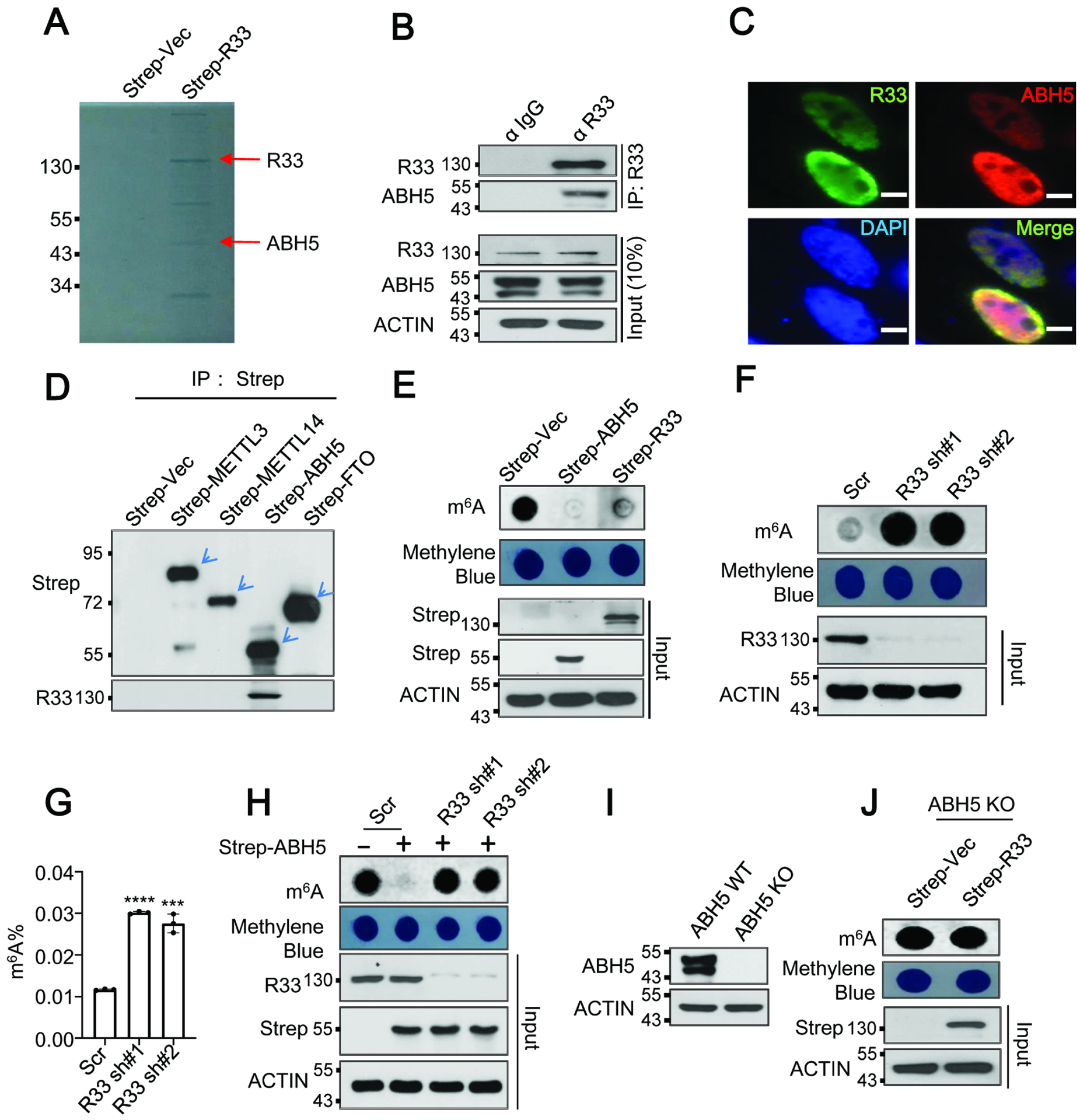
RBM33 forms a complex with ALKBH5, and regulates mRNA m6A methylation dependent on ALKBH5. (A) Coomassie staining of co-immunoprecipitants of RBM33. (B) Co-IP analysis was performed as indicated. (C) Immunostaining analysis of RBM33 and ALKBH5. Scale bars: 39.5 μm (D) Strep-Tactin pull-down assay was performed as indicated. (E) Dot-blot analysis of mRNA m6A methylation in UM-SCC-1 cells. (F and G) RNA m6A methylation analysis as determined by dot-blot and RNA m6A methylation quantification kit respectively, G shown as mean ± SEM. (H) Dot-blot analysis as indicated. (I) Western blot analysis of ALKBH5 protein levels. (J) Dot-blot analysis of global mRNA m6A methylation as indicated.
Next, we determined whether RBM33 regulates mRNA m6A methylation. Similar to ALKBH5 overexpression-induced m6A demethylation, ectopic expression of RBM33 resulted in a substantial decrease in mRNA m6A methylation (Figure 1E and Figure S2D). Conversely, knockdown (KD) of endogenous RBM33 by two RBM33-specific shRNAs dramatically increased global mRNA m6A methylation in UM-SCC-1 cells, as determined by dot-blot and RNA m6A methylation quantification analyses, suggesting that RBM33 negatively regulates RNA m6A methylation in UM-SCC-1 cells (Figure 1F and 1G). Similarly, RBM33 interacted with ALKBH5 and RBM33 KD led to an increase in global RNA m6A methylation in 293T cells, suggesting that the regulation of RBM33 towards mRNA m6A methylation is likely a general-phenomenon (Figure S2E–S2G). To examine the underlying mechanism for RBM33-mediated down-regulation of global RNA m6A methylation, we analyzed the effect of RBM33 depletion on both protein levels and enzymatic activities of m6A methylation writers, METTL3 and METTL14 and m6A methylation erasers, ALKBH5 and FTO. We observed that neither protein levels of m6A methylation writers nor m6A erasers could be affected by RBM33 depletion (Figure S2H). However, RBM33 depletion selectively inhibited ALKBH5 m6A demethylase activity (Figure S2I–S2L). Consistently, RBM33 KD by two specific shRNAs also prevented ALKBH5-mediated mRNA m6A demethylation (Figure 1H and Figure S2M). To determine whether regulation of global mRNA m6A methylation by RBM33 relies on ALKBH5, we overexpressed strep-vector and strep-RBM33 in an ALKBH5 knockout (KO) HEK293T cell generated by the CRISPR-Cas9 system30 (Figure 1I). Surprisingly, RBM33-mediated down-regulation of mRNA m6A methylation was rescued by ALKBH5 KO and this phenomenon could be further reversed by co-expression RBM33 with wild-type but not enzymatic mutant ALKBH5, suggesting that RBM33 negatively regulates global RNA m6A methylation dependent on ALKBH5 (Figure 1J and Figure S2N). Taken together, these results suggest that RBM33 forms a complex with ALKBH5 and that RBM33 and ALKBH5 mutually depend on one another to mediate mRNA m6A demethylation.
RBM33 preferentially binds to m6A-modified RNA substrate
Since RBM33 was recognized as a potential RNA binding protein according to its protein sequence, we next examined its ability to bind to m6A modified or unmodified RNA oligos with a biotinylated RNA pull-down assay. As shown in Figure S2O and Figure 2A, both exogenous RBM33 and endogenous RBM33 had a higher binding affinity with the m6A modified RNA substrate than they had with the unmodified RNA oligos, suggesting that RBM33 is a previously unrecognized m6A binding protein. RBM33 protein contains an RNA recognition motif (RRM) at its carboxyl-terminal region (Figure 2B). We found that removal of the RRM of RBM33 completely abrogated its ability to bind to the m6A modified RNA oligos (Figure 2C), suggesting the RRM domain of RBM33 is required for its binding ability to the m6A modified substrate. Meanwhile, depletion of the RRM domain of RBM33 also blocked RBM33-mediated down-regulation of mRNA m6A methylation as determined by both dot-blot and RNA m6A methylation quantification analyses (Figure 2D and Figure S2P). In addition, RBM33 overexpression significantly facilitated ALKBH5 binding affinity towards the m6A modified RNA substrate (Figure S3A). In contrast, ALKBH5 substrate accessibility was dramatically blocked by RBM33 depletion (Figure 2E and Figure S3B). We next determined whether RBM33 regulates ALKBH5 m6A-binding affinity in vitro. As shown in Figure S3C–S3D, ALKBH5 purified from RBM33 KO cells exhibited a significantly lower m6A-binding affinity. However, RBM33 m6A-binding affinity was not affected by ALKBH5 overexpression or KD (Figure.S3E–S3F). Together, these results suggest that RBM33 regulates ALKBH5 m6A-binding ability in vivo and in vitro.
Figure. 2.
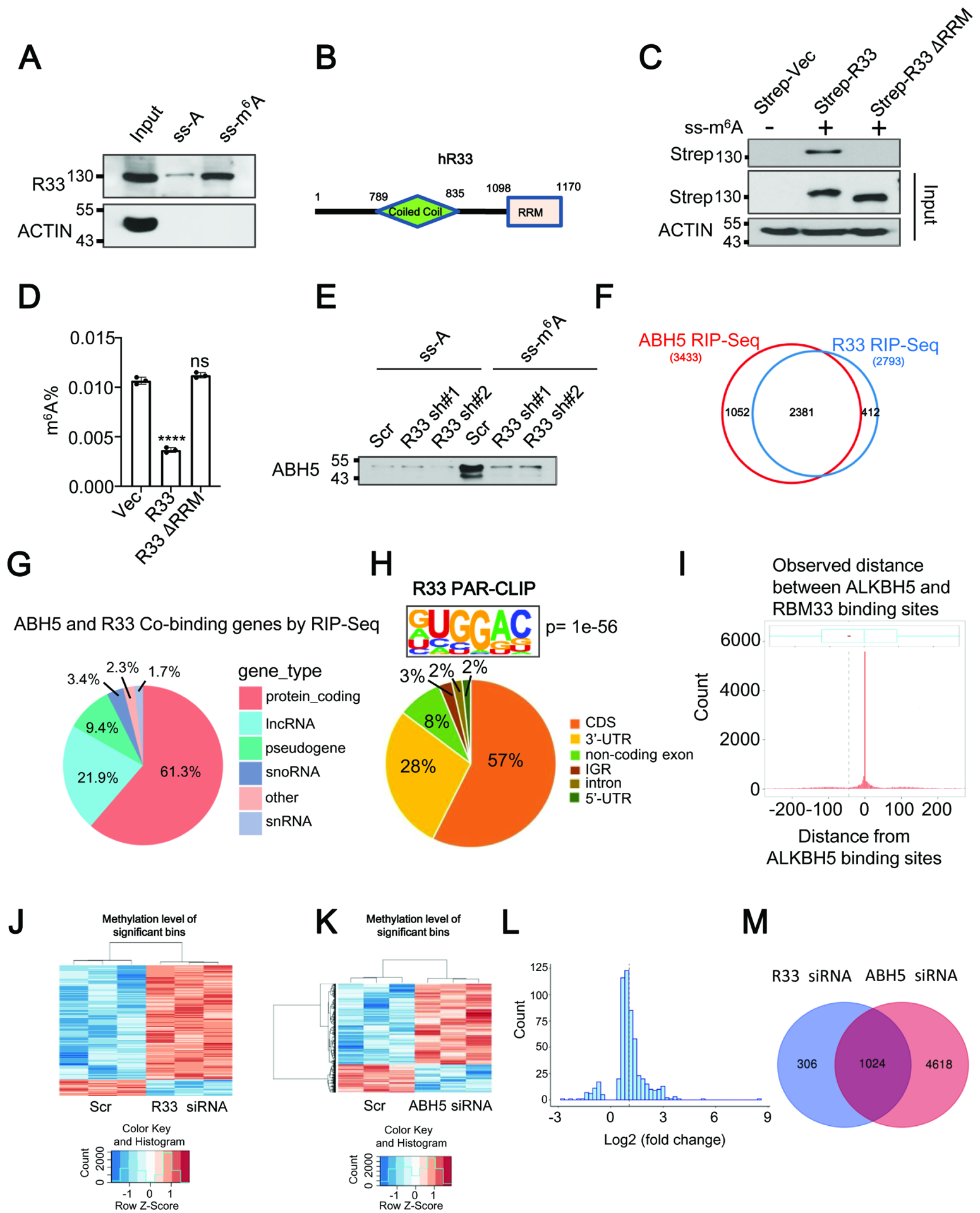
RBM33 acts as a novel m6A binding protein. (A) Substrate pull-down analyses of endogenous RBM33. (B) Domain structures of human RBM33. (C) Substrate pull-down analysis as indicated. (D) RNA m6A methylation quantification analysis as indicated. Data shown as medians with SEM. (E) Substrate pull-down analysis showing ALKBH5 substrate accessibility. (F) RIP-Seq analyses of RBM33 and ALKBH5 binding transcripts. (G) Pie charts showing the distribution of the overlapped RBM33 and ALKBH5 RIP-seq reads in RNA classes. (H) RBM33 PAR-CLIP-seq analysis showing the distribution of RBM33 binding sites and consensus binding motif of RBM33 in 293 T cells. (I) Histogram plot showing the observed distance between ALKBH5 and RBM33 binding sites in 293T cells. The black dashed line represents the average distance (−49 nt) between them. (J and K) Heat map showing mRNA transcripts with significant m6A alterations by m6A-Seq. (L) The histogram showing the majority of genes co-regulated by RBM33 and ALKBH5 exhibit an increased m6A methylation in RBM33 KD cells. (M) The Venn diagram showing the genes with a significant m6A increase upon RBM33 and ALKBH5 KD.
To determine RNA transcripts that are directly bound by RBM33 and ALKBH5 in HNSCC cells at a global level, we performed RBM33 and ALKBH5 RIP-seq analysis. The results showed that 2793 genes contain RBM33 binding sites while 3433 genes contain ALKBH5 binding sites in HNSCC cells. Interestingly, 2381 transcripts contain both ALKBH5 and RBM33 binding sites (Figure 2F), indicating that nearly 70% of ALKBH5 target genes are bound by RBM33. Notably, the vast majority of RBM33 and ALKBH5 common binding sites are located in protein-coding transcripts and long non-coding RNAs (LncRNAs) (Figure2G). To precisely identify the transcripts co-bound by RBM33 and ALKBH5, we performed the photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation followed by next-generation sequencing (PAR-CLIP-seq) analysis for both RBM33 and ALKBH5 in HEK 293T cells. In contrast to a recent study showing that RBM33 predominantly binds to the GC-rich and intron-less RNA, we observed that RBM33 predominantly binds to the coding sequencing region of protein coding genes, and that the canonical m6A methylation consensus motif RRACH (R = G or A; H = A, C, or U; where A is converted to m6A) appears at RBM33 binding sites, suggesting that RBM33 is a potential m6A-binding protein (Figure2H and Figure S3G). Consistent with our RIP-seq data, the PAR-CLIP-seq data analysis suggests that the majority of RBM33 binding sites were also bound by ALKBH5 (Figure S3H). Interestingly, 2672 out of 8639 transcripts that are bound by ALKBH5 showed significant reduced ALKBH5 binding peaks upon RBM33 knockout in 293T cells (Figure S3I). ALKBH5 and RBM33 predominantly bind to the coding sequences of protein coding genes. The genes co-bound by RBM33 and ALKBH5 are mainly involved in regulating RNA splicing, protein processing in endoplasmic reticulum, mRNA stability and mRNA catabolic process (Figure S3J–S3L). More importantly, RBM33 binding sites are very close to ALKBH5 binding sites and more than 50% of RBM33 binding sites locate at 100-nt upstream of ALKBH5 binding sites, as determined by the metagene analysis of the distance between RBM33 and ALKBH5 binding sites (Figure 2I). Integrative analysis of RBM33 and ALKBH5 PAR-CLIP-seq results in wild-type and RBM33 KO cells, we showed that the several ALKBH5 and RBM33 binding sites are co-localized in three representative genes including HSPA5, DDIT4, MAT2A in the control and RBM33 KO cells (Figure S3M–S3O). Interestingly, RBM33 KO significantly inhibits the binding abilities of ALKBH5 to these three transcripts.
To further determine the effect of RBM33 and ALKBH5 depletion on mRNA m6A methylation transcriptome-wide, we performed m6A-Seq analyses. Consistent with previous studies,31,32 our m6A-Seq data showed that the majority of m6A peaks are distributed in the protein-coding region (CDS) and 3’ untranslated region (3’UTR) of mRNA transcripts in UM-SCC-1 cells, and also contain a GGACU consensus motif (Figure S3P). Consistent with the m6A quantification analysis and dot blot results, KD of either RBM33 or ALKBH5 induced a significant increase in mRNA m6A levels, as determined by m6A-seq analysis (Figure 2J–2K). Meanwhile, the majority of genes regulated by RBM33 and ALKBH5 exhibited a significant increase in mRNA m6A methylation in RBM33 or ALKBH5 KD cells (Figure 2L and Figure S3Q). RBM33 KD led to an increase of m6A methylation in 1330 RNA transcripts. More than 65% of these transcripts had an increased m6A methylation upon ALKBH5 KD (Figure. 2M). Additionally, 458 out of 2672 transcripts, in which ALKBH5 binding peaks were significantly reduced upon RBM33 KO in 293T cells, showed increased m6A levels upon RBM33 KD in UM-SCC-1 cells (Figure S3I). Together, these results suggest that RBM33 acts as a novel m6A binding protein, which recognizes m6A RNA substrate through its RNA recognition motif and recruits ALKBH5 to demethylate its m6A RNA substrates.
Characterization of the RBM33 and ALKBH5 interaction domain.
To further determine how RBM33 interacts with ALKBH5, we used the HDOCK server (http://hdock.phys.hust.edu.cn/) to predict the interaction between RBM33 and ALKBH5. As shown in Figure 3A and 3B, the predicted interaction surface between RBM33 and ALKBH5 involves amino acids, specifically S1101, S1108, T1110, R1134, V1169 in RBM33, and T123, P128, K132, G151, E153, D206, T265, C267 in ALKBH5. By performing site-directed mutagenesis followed by Co-IP analyses in HNSCC cells, we showed that the replacement of amino acids Serine 1101 (S1101) or Arginine 1134 (R1134) with Alanine in RBM33 protein or amino acids Lysine 132 (K132), Glutamine153 (E153) or Threonine 265 (T265) with Alanine in ALKBH5 significantly disrupted the interaction between ALKBH5 and RBM33 (Figure 3C, 3D), suggesting that these amino acids in RBM33 or ALKBH5 are critical for maintaining the interaction between them. Interestingly, the binding affinities of RBM33S1101A and RBM33R1134A mutants and wild-type RBM33 to the m6A-modified substrate were quite comparable (Figure 3E). However, both RBM33S1101A and RBM33R1134A mutants lost the ability to regulate global mRNA m6A methylation (Figure 3F, 3G), indicating that the function of RBM33 in regulation of mRNA m6A methylation depends on its association with ALKBH5. On the other hand, ALKBH5K132A, ALKBH5E153A, and ALKBH5T265A mutants lost substrate accessibility as well as mRNA m6A demethylase activity (Figure 3H, 3I and 3J), suggesting that the binding ability of ALKBH5 to m6A RNA substrate heavily relies on RBM33. Taken together, these results further support the hypothesis that ALKBH5 binds to mRNA m6A substrates through RBM33 by forming a complex with RBM33 to regulate mRNA m6A methylation in HNSCC cells.
Figure. 3.
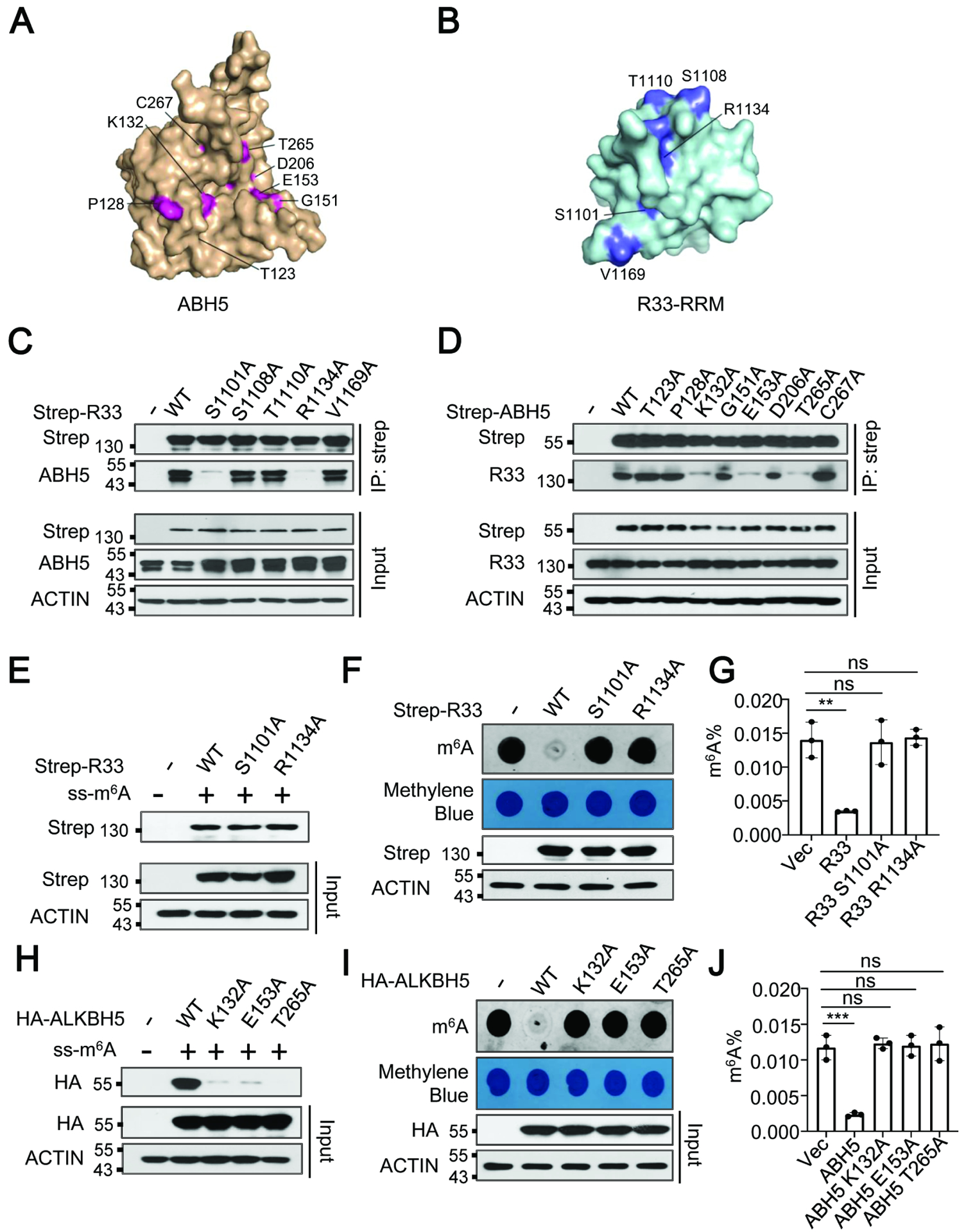
RBM33 plays an essential role in ALKBH5 loading to the substrate. (A and B) Amino acids in the interaction interface between RBM33 and ALKBH5 based on the HDOCK prediction. (C and D) Co-IP analysis as indicated. (E) m6A-modified RNA oligos pull-down analysis as indicated. (F and G) Dot-blot and RNA m6A methylation quantification analysis of global mRNA m6A methylation as indicated, G shown as mean ± SEM. (H) m6A-modified RNA oligos pull-down analysis as indicated. (I and J) Dot-blot and RNA m6A methylation quantification analysis of mRNA m6A demethylase activity of ALKBH5 K132A, E153A, and T265A mutants, J shown as mean ± SEM.
RBM33 activates ALKBH5 demethylase activity by recruiting deSUMOylase SENP1
Our previous study demonstrated that ALKBH5 SUMOylation significantly inhibited its m6A demethylase activity and that ALKBH5 SUMOylation is dynamically controlled by SUMO E3 ligase PIAS4 and desumoylase SENP1.30 Thus, we determined the role of RBM33 in the SUMOylation of ALKBH5. Denaturing IP analysis revealed that RBM33 KD dramatically induced ALKBH5 SUMOylation (Figure 4A). Interestingly, RBM33 KD did not affect the interaction between ALKBH5 and PIAS4, but it did significantly disrupt the association between ALKBH5 and SENP1 (Figure 4B–4D). Conversely, the interaction between ALKBH5 and SENP1 was markedly strengthened by RBM33 overexpression (Figure 4E and 4F). In contrast, RBM33 interacts with SENP1, which is independent of ALKBH5 (Figure 4G). Furthermore, the interaction between ALKBH5 and RBM33 is not affected by SENP1 KD (Figure S4A), suggesting that their interaction is independent of SENP1. Interestingly, compared with wild-type RBM33, RBM33 mutants (RBM33 S1101A and RBM33 R1134A) defective for interaction with ALKBH5, have a similar binding ability towards SENP1 (Figure S4B). However, SUMOylation of ALKBH5 was significantly enhanced by overexpression of RBM33 S1101A and RBM33 R1134A mutants (Figure. S4C). Consistently, ALKBH5 K132A, ALKBH5 E153A and ALKBH5T265A mutants defective for interaction with RBM33 failed to interact with SENP1 but induced SUMOylation of ALKBH5 at the same time (Figure S4D–S4E). More importantly, endogenous RBM33 interacted with endogenous SENP1 (Figure S4F). The Co-IP assays with a series of truncation mutants for both RBM33 and SENP1 (Figure S4G) showed that the coiled coil domain of RBM33 interacted with the N-terminal of SENP1 (Figure S4H–S4I). Collectively, these results provide compelling evidence that RBM33 also plays an important role in the activation of ALKBH5 demethylase activity by inhibiting ALKBH5 SUMOylation through the recruitment of deSUMOylase SENP1 (Figure 4H).
Figure. 4.
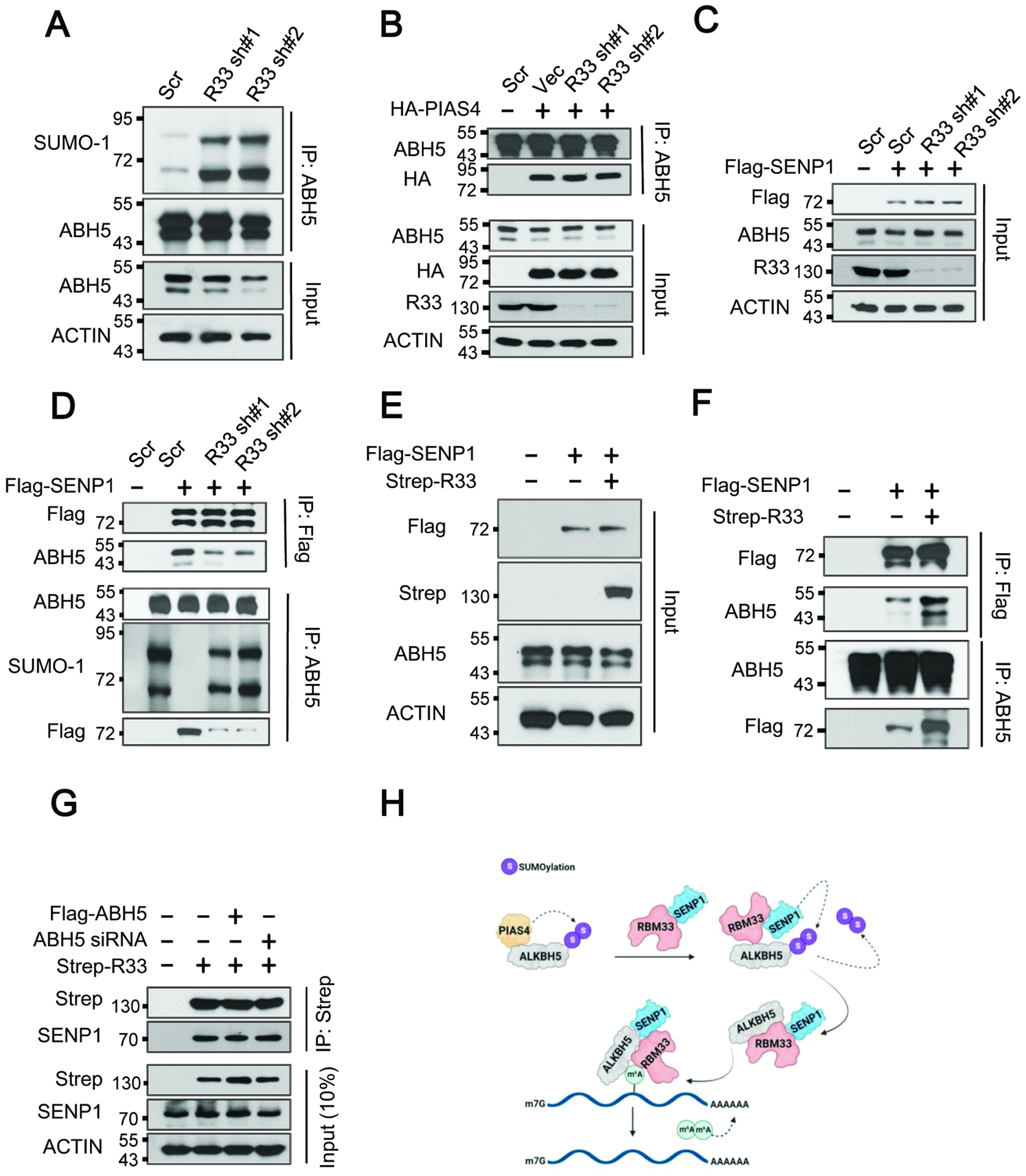
RBM33 activates ALKBH5 demethylase activity by recruitment of deSUMOylase SENP1. (A) Denaturing IP analysis as indicated. (B) Co-IP analysis of ALKBH5 and PIAS4. (C, D, E and F) Co-IP analysis of ALKBH5 and SENP1 as indicated. (G) Co-IP analysis of RBM33 and SENP1 as indicated. (H) Carton showing that the molecular mechanisms by which RBM33 regulates ALKBH5 substrate accessibility and its demethylase activity.
RBM33/ALKBH5 regulates tumorigenesis of HNSCC in an m6A-dependent manner
Next, we determined whether RBM33 plays an oncogenic role in HNSCC. Consistent with the TCGA database findings, Western blot analysis revealed that RBM33 was barely detectable in normal human oral keratinocytes (HOK) cells, whereas it was highly expressed in the HNSCC cell lines (UM-SCC-1, UM-SCC-2, SCC090, JHU022, 93-VU-147T, SqCC/Y1, 1483) (Figure 5A). As determined by immunohistochemistry (IHC) staining, RBM33 exhibited much higher expression in primary tumors from HNSCC patients at different disease stages, compared to the normal adjacent tissues (Figure 5B). The expression of RBM33 was significantly increased in the stage IV HNSCC TNM compared to the earlier stages of HNSCC TNM (I, II, and III) (Figure 5C, 5D). To further investigate the role of RBM33 in HNSCC cells, we knocked down RBM33 via RBM33-specific shRNAs in normal control HOK cells and three HNSCC cell lines including UM-SCC-1, UM-SCC-2, and SCC090, as confirmed by Western Blot (Figure 5E, and Figure S4J). RBM33 KD specifically induced apoptosis and inhibited the colony-forming ability of HNSCC cells (UM-SCC-1, UM-SCC-2, and SCC090), but not the normal control HOK cells (Figure 5F–5I). In addition, RBM33 KD also significantly suppressed HNSCC growth in vivo in xenograft mice (Figure 5J–5L). Together, these data indicate that RBM33 is highly expressed in HNSCC and is required for the growth and survival of human HNSCC but not its normal human oral keratinocyte counterpart.
Figure. 5.
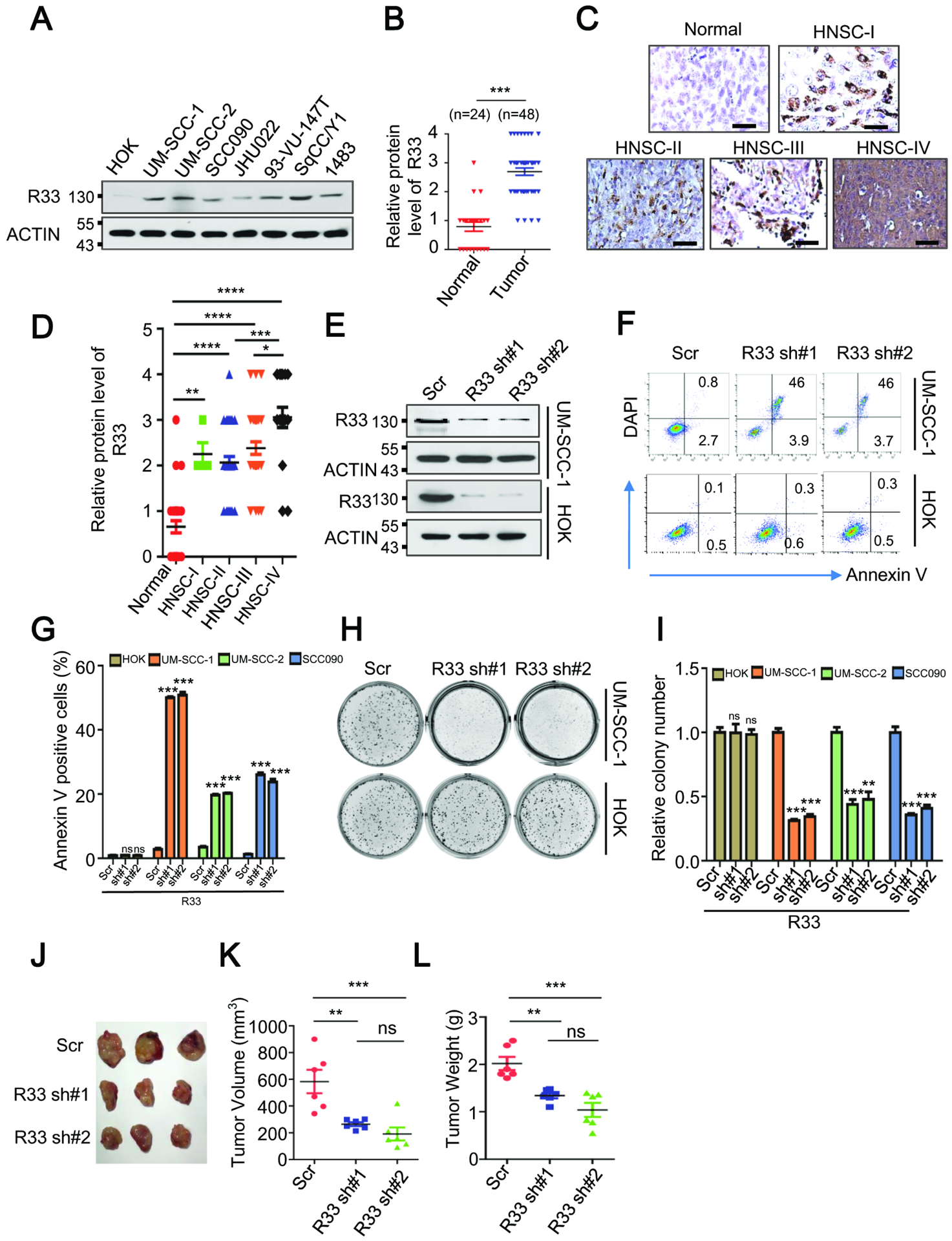
RBM33 is commonly up-regulated in HNSCC and is essential for tumorigenesis. (A) Western blot analysis showing protein levels of RBM33 in cell lines as indicated. (B) IHC analyses showing protein levels of RBM33 in normal adjacent tissues and HNSCC tumors. Data shown as medians with SEM. (C) IHC analyses showing protein levels of RBM33 across HNSCC TNM clinical stages. Scale bars: 124.5 μm (D) Summary and statistical analysis of Figure 5C. Data shown as medians with SEM. (E) Western blot analysis of RBM33 expression in UM-SCC-1 and HOK cells. (F) Annexin V staining showing cell apoptosis. (G) Summary and statistical analysis of Figure 5F. (H) Colony-forming analyses. Data shown as medians with SEM. (I) Summary and statistical analysis of Figure. 5H. Data shown as medians with SEM. (J-L) Effect of RBM33 KD on tumor growth of UM-SCC-1 xenograft mice. Data shown as medians with SEM.
To test our hypothesis that the oncogenic function of RBM33 is mediated by ALKBH5, we examined the expression of ALKBH5 in primary tissues from HNSCC patients and determined the effects of ALKBH5 KD in HNSCC cells. Similar to RBM33, ALKBH5 expression was much higher in the HNSCC primary tumors compared with the normal adjacent tissues (Figure 6A and 6B). Similarly, transcription levels of both RBM33 and ALKBH5 are higher in HNSCC cell lines as compared to the normal control HOK cells (Figure S4K–S4L). However, protein turn-over rates of RBM33 and ALKBH5 were comparable in HNSCC cells and the normal control HOK cells (Figure S4M–S4P), suggesting that the elevated protein level of RBM33 and ALKBH5 in HNSCC cells is likely due to the deregulation of transcription of RBM33 and ALKBH5. IHC analysis of the protein levels of both ALKBH5 and RBM33 in different HNSCC TNM stages further revealed that both ALKBH5 and RBM33 are simultaneously overexpressed in the majority of HNSCC primary tumors, especially at more advanced stages (Figure S4Q). Additionally, ALKBH5 KD by ALKBH5-specific shRNAs selectively induced apoptosis and significantly inhibited the colony-forming ability of HNSCC cells but not normal control HOK cells (Figure 6C, 6D, Figure S4R). In addition, the ALKBH5 overexpression-induced cell proliferation increase in UM-SCC-1 cells was blocked by RBM33 KD and vice versa (Figure 6E and 6F). More importantly, ALKBH5 KD had a slightly more inhibitory effect on HNSCC in vivo cell growth in xenograft mice than RBM33 KD (Figure 6G–6I). Together, these data suggest that RBM33 and ALKBH5 are dependent on each other to maintain HNSCC tumorigenicity.
Figure. 6.
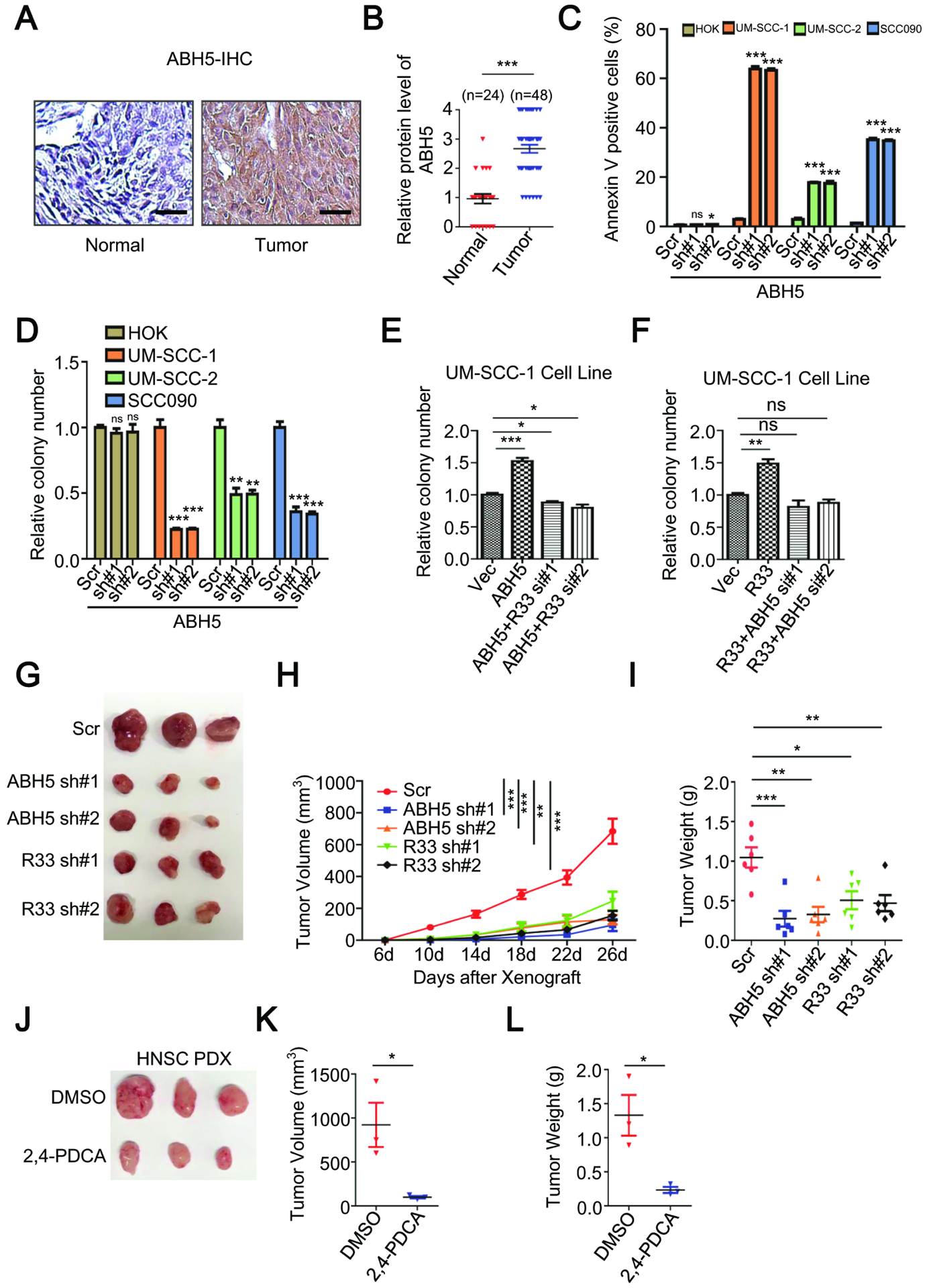
ALKBH5 is highly expressed in HNSCC patients and its depletion leads to HNSCC growth inhibition in vivo. (A) IHC analysis of ALKBH5 protein level in HNSCC primary tumors and normal adjacent tissues. Scale bars: 124.5 μm (B) Summary and statistical analysis of Figure. 6A. Data shown as medians with SEM. (C) Annexin V staining analysis as indicated. Data shown as medians with SEM. (D, E and F) Colony-forming analysis, shown as medians with SEM. (G, H and I) UM-SCC-1 xenograft assay, shown as medians with SEM. (J, K and L) HNSCC PDX assay, shown as medians with SEM.
We next investigate whether ALKBH5 and RBM33 regulate HNSCC tumorigenesis in an m6A-dependent manner. Consistent with the observation that RBM33 and ALKBH5 are highly expressed in HNSCC cell lines, RNA m6A methylation levels in HNSCC cells are lower as compared to the normal control HOK cells (Figure S4S). Additionally, ALKBH5 KD-mediated UM-SCC-1 cell proliferation inhibition was only rescued by wild-type but not enzymatic mutant ALKBH5 overexpression (Figure S4T). Consistently, RBM33 KD-mediated UM-SCC-1 cell proliferation inhibition was rescued by wild-type but not ALKBH5 interaction defective mutant (RBM33 S1101A) in vitro and in vivo, suggesting that the regulation of ALKBH5/RBM33 towards HNSCC growth is m6A-dependent (Figure S4U–S4X).
Pharmacological inhibition of ALKBH5 markedly suppresses primary HNSCC tumor cell growth in vivo
Given its role in cancer proliferation, we next sought to determine whether the RBM33/ALKBH5 complex could be therapeutically targeted. Pyridine-2,4-dicarboxylate (2,4-PDCA) is the first reported ALKBH5 small-molecule inhibitor.33 We next determined whether inhibition of ALKBH5 by 2,4-PDCA affected the growth of HNSCC cell lines as well as human patient primary HNSCC tumor cells in vitro and in vivo. As shown in Figure S4Y–S4Z, 2,4-PDCA treatment significantly inhibited the colony-forming ability and induced apoptosis of UM-SCC-1 cells. Additionally, the 2,4-PDCA treatment also significantly inhibited UM-SCC-1 xenograft growth in vivo (Figure S5A–S5C). Dot-blot analysis revealed that global mRNA m6A methylation was markedly increased as a consequence of ALKBH5 inhibition either by ALKBH5-specific shRNA or 2,4-PDCA inhibitor in UM-SCC-1 cells in vivo (Figure S5D). To exclude the possibility that the inhibitory effect of 2,4-PDCA inhibitor in HNSCC cells was due to any off-target effects of 2,4-PDCA, we established UM-SCC-1 cells stably expressing wild-type and ALKBH5 enzymatic mutant (ALKBH5 H204A) (Figure S5E). As shown in Figure S5F–S5G, the 2,4-PDCA-induced apoptosis and growth inhibition in UM-SCC-1 cells was almost completely reversed by overexpression of wild-type ALKBH5 but not the enzymatic mutant ALKBH5, suggesting that 2–4-PDCA inhibits HNSCC cell growth and survival mainly through suppression of ALKBH5 demethylase activity. Finally, we examined the effect of 2,4-PDCA treatment on the patient primary HNSCC tumor in xenograft mice in vivo. Strikingly, the growth of human HNSCC patient-derived xenograft (PDX) was significantly inhibited by 2,4-PDCA (Figure 6J–6L). Taken together, these results suggest that the ALKBH5/RBM33 complex is a potential therapeutic target for the treatment of HNSCC patients.
Identification of DDIT4 as a key downstream target of RBM33 in HNSCC
To further understand the underlying mechanisms that mediate the function of RBM33 in HNSCC tumorigenicity maintenance, we performed RNA sequencing (RNA-Seq) analysis of UM-SCC-1 cells with or without RBM33 or ALKBH5 KD. As shown in Figure 7A and Figure S5H, both RBM33 and ALKBH5 KD led to significant alterations in gene expression of 2544 and 2418 genes respectively (Log2FC > 0.5, or Log2FC< −0.5, p < 0.05). Gene set enrichment analysis (GSEA) analyses reveal that the down-regulated genes upon RBM33 or ALKBH5 KD are enriched in pathways involved in hypoxia, RAS, and RAF pathways (Figure 7B). These data suggest that suppression of RBM33/ALKBBH5 complex activity inhibits hypoxia and RAS/RAF pathways, which play significant roles in the process of cancer progression, cancer cell migration, and therapy resistance in head and neck cancer patients.34,35 Additionally, Gene Ontology (GO) analysis of 609 genes, which were identified as significantly differentially expressed genes in both RBM33 and ALKBH5 KD HNSCC cells showed that the commonly regulated genes by both RBM33 and ALKBH5 are enriched in the pathways involved in the unfolded protein response, protein localization to the endoplasmic reticulum (ER), and spliceosome (Figure 7C–7D). Notably, the transcripts with up-regulation of m6A methylation upon RBM33 or ALKBH5 KD are also involved in protein processing in ER and spliceosome (Figure 7E). It was previously reported that ALKBH5 controls the splicing of 3’-UTR mRNA in male germ cells.36 Likewise, these data support that the RBM33/ALKBH5 complex controls mRNA splicing in HNSCC cells. Additionally, the RBM33/ALKBH5 complex may have a pivotal role in the regulation of protein processing in ER. Deregulation of protein modification and folding in ER provokes ER stress, which significantly contributes to tumor initiation, progression, and therapy resistance.37 To further analyze the m6A methylation level changes of differentially expressed genes upon ALKBH5 or RBM33 KD, we identified 36 differentially expressed genes with upregulation of m6A methylation (Figure S5I). Among the 36 genes, DDIT4 DNA damage-inducible transcript 4 (DDIT4) stood out due to its important role in various cellular stresses as well as multiple pathways that regulate protein folding in the ER, hypoxia, and the RAS pathway.38 Notably, DDIT4 is one of the most significantly repressed genes by RBM33 or ALKBH5 KD (Figure 7A and Figure S5H), and it is also differentially expressed with a significant increase of m6A methylation upon RBM33 or ALKBH5 KD (Figure 7F, 7G, and Figure S5J–S5K). Additionally, RBM33 and ALKBH5 PAR-CLIP-seq data revealed that the binding sites of RBM33 and ALKBH5 on DDIT4 transcripts are significantly overlapped (Figure. S3N), suggesting that both ALKBH5 and DDIT4 directly bind DDIT4 transcripts. Interestingly, by analysis of the TCGA database, we found that DDIT4 exhibits a higher expression in HNSCC primary tumors compared to the normal adjacent tissues and that high DDIT4 expression is associated with a poor prognosis in HNSCC patients (Figure 7H, 7I). Consistently, DDIT4 was highly expressed in HNSCC cell lines as compared to the normal control HOK cells, as determined by Western Blot analysis (Figure 7J). We showed that the transcription level of DDIT4 was only promoted by wild-type RBM33 and ALKBH5, but not by the RRM-deleted RBM33 (RBM33 ΔRRM) or ALKBH5 enzymatic mutant (ALKBH5 H204A) (Figure 7K and Figure S5L), suggesting that RBM33 and ALKBH5 both regulate DDIT4 gene expression in an m6A-dependent manner. Consistent with the PAR-CLIP-seq data, RBM33 and ALKBH5 RNA immunoprecipitation (RIP)-qPCR assays indicate that both RBM33 and ALKBH5 directly bind to DDIT4 mRNA transcripts (Figure 7L and Figure S5M) in UM-SCC-1 cells. The methylated RNA immunoprecipitation (MeRIP) followed by RT-PCR analysis suggests that mRNA m6A methylation of DDIT4 could be significantly increased by either RBM33 or ALKBH5 KD (Figure S5N–S5O). Furthermore, ALKBH5 enrichment at transcripts of DDIT4 was markedly disrupted by RBM33 KD, suggesting that the binding ability of ALKBH5 to DDIT4 was dependent on the presence of RBM33 (Figure S5P). To determine how the RBM33/ALKBH5 complex regulates DDIT4 expression, we measured the half-life of DDIT4 mRNAs by RT-qPCR analysis upon RBM33 or ALKBH5 KD by blocking nascent RNA synthesis with Actinomycin D. As shown in Figure S5Q–S5R, either RBM33 or ALKBH5 KD promoted DDIT4 mRNA decay. Additionally, only wild-type but not enzymatic mutant ALKBH5 overexpression can rescue the altered DDIT4 transcription, mRNA m6A methylation and stability (Figure S5S–S5U), providing direct evidence that ALKBH5 regulates DDIT4 transcription, mRNA m6A methylation, and RNA stability in an m6A-dependent manner. Meanwhile, RBM33 KD-mediated alterations in DDIT4 transcription, ALKBH5 enrichment at DDIT4 transcripts, and DDIT4 mRNA m6A methylation levels were reversed by wild-type but not ALKBH5 interaction-defective mutant RBM33, suggesting the regulation of RBM33 towards DDIT4 relies on ALKBH5 and m6A (Figure S5V-S5X). Collectively, these results suggest that RBM33 and ALKBH5 stabilize DDIT4 mRNA in an m6A-dependent manner.
Figure. 7.
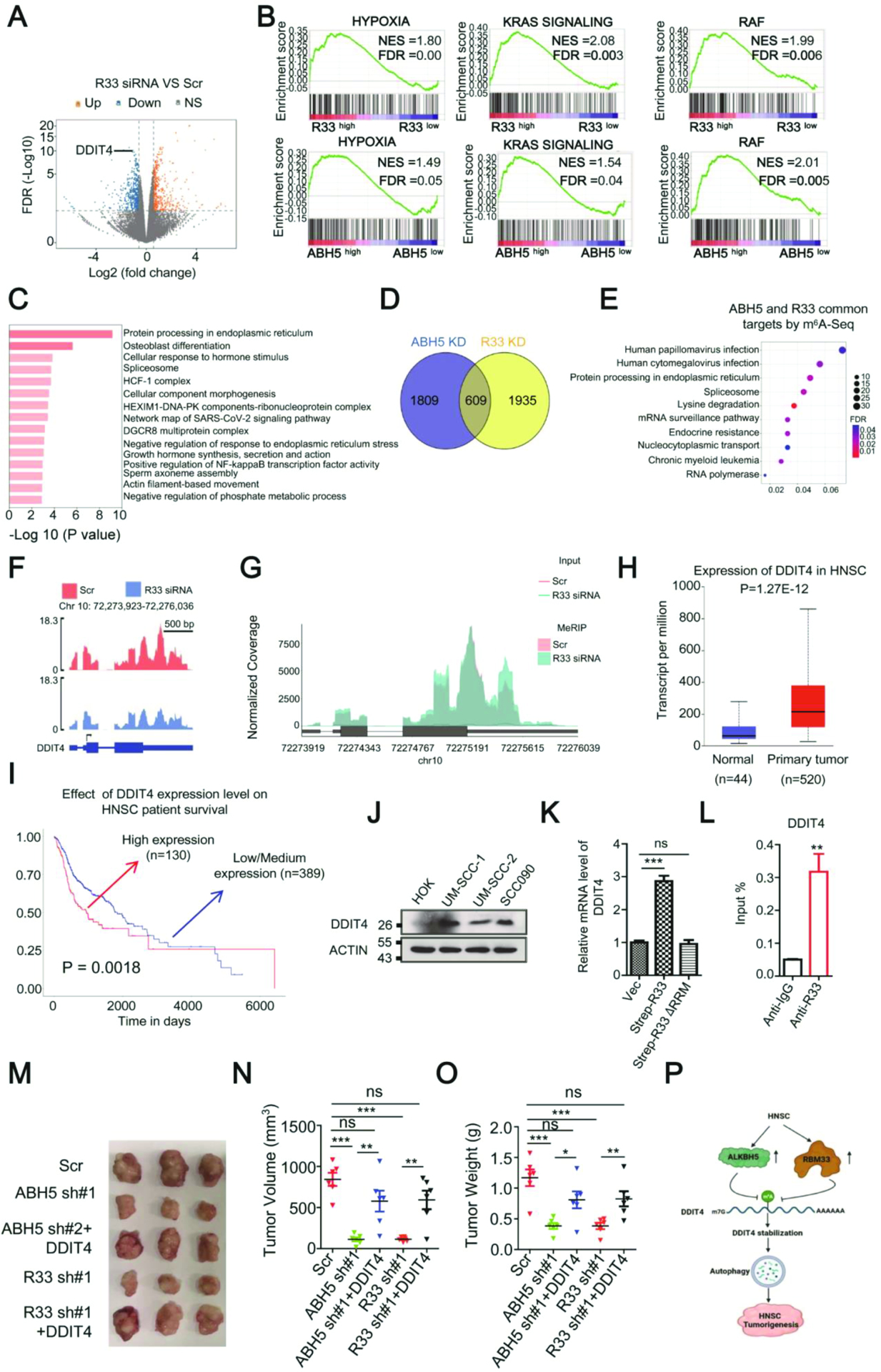
RBM33 maintains HNSCC tumorigenicity by regulating DDIT4-mediated ROS and autophagy. (A)Volcano figure showing differentially expressed genes in wild-type and RBM33 KD cells. (B) GSEA plot showing enrichment of gene sets of hypoxia, KRAS, and RAF signaling in RBM33 and ALKBH5 KD UM-SCC-1 cells respectively. (C) Gene Ontology (GO) analysis for the RBM33 and ALKBH5 common downstream target genes, as determined by RNA-Seq analysis. (D) The Venn diagram shows many genes with differential gene expression upon RBM33 or ALKBH5 KD. (E) KEGG analysis for the genes co-regulated by ALKBH5 and RBM33 by the m6A-Seq analysis. (F) RNA-Seq peak visualization of DDIT4 in wild-type and RBM33 KD cells. (G) m6A-Seq peak visualization of DDIT4 in wild-type and RBM33 KD cells. (H) TCGA database analysis of DDIT4 transcripts in normal adjacent tissues and HNSCC primary tumors. Data shown as medians with SEM. (I) Association of DDIT4 expression and HNSCC patient survival, shown as medians with SEM. (J) Western blot analysis as indicated. (K) RT-PCR analysis as indicated. (L) RBM33 RIP analysis, shown as medians with SEM. (M, N and O) UM-SCC-1 xenograft assay, shown as medians with SEM. (P) Working model showing that RBM33 and ALKBH5-mediated HNSCC tumorigenesis are through m6A demethylation-mediated stabilization of DDIT4 transcripts, thereby inducing autophagy.
DDIT4-mediated autophagy is responsible for the oncogenic function of RBM33 in HNSCC.
To determine the function of DDIT4 in HNSCC cells, we knocked down DDIT4 using two specific shRNAs (Figure S6A). Similar to the effect of RBM33 or ALKBH5 KD, DDIT4 KD significantly induced UM-SCC-1 cell apoptosis and at the same time markedly inhibited the colony-forming ability of UM-SCC-1 cells (Figure S6B–S6C). Consistent with the RT-qPCR results, RBM33 or ALKBH5 KD led to a significant decrease in DDIT4 protein expression (Figure S6D–S6E). In contrast, overexpression of wild-type, but not RRM-deleted RBM33 markedly increased the protein expression of DDIT4, suggesting that RBM33 requires its RNA binding ability to regulate DDIT4 expression (Figure S6F). Numerous studies suggest that DDIT4 forms a complex with TXNIP, and DDIT4/TXNIP complex promotes autophagy by facilitating autophagosome formation via increasing reactive oxygen species (ROS) level.39,40 Thus, we next investigated whether DDIT4-induced autophagy is critical for RBM33/ALKBH5 complex-mediated HNSCC tumorigenesis. As shown in Figure S6G–S6J , either RBM33 or ALKBH5 KD significantly inhibited autophagy. In addition, we found that HNSCC cells are more sensitive to autophagy inhibitor-induced autophagy suppression and cell apoptosis compared to the normal control HOK cells (Figure S6K–S6M). Finally, RBM33 or ALKBH5 KD-mediated HNSCC growth inhibition in xenograft mice was reversed by DDIT4 overexpression (Figure 7M–7O). Collectively, our results imply that DDIT4 is the functional mediator of the RBM33/ALKBH5 complex in HNSCC tumorigenesis by modulating DDIT4 mRNA stability in an m6A-dependent manner, thereby controlling DDIT4-mediated autophagy (Figure 7P).
DISCUSSION
RNA m6A methylation can be guided by different adaptors,12–14,41 transcription factors,42,43 and histone methylation.44 How demethylation selectivity is achieved is not clear. In this manuscript, we report a first clear example that a large portion of m6A demethylation in HNSCC and 293T cells requires RBM33, a previously unrecognized m6A binding protein. The binding protein recognizes m6A-marked RNA substrates through its RNA recognition motif (RRM) and then recruits ALKBH5 for transcript-specific demethylation. We find that RBM33 forms a complex with ALKBH5, and inhibits ALKBH5 SUMOylation, leading to activation of ALKBH5 demethylase activity by recruitment of deSUMOylase SENP1. As determined by site-directed mutagenesis analysis, ALKBH5 K132, E153, and T265 were identified as critical amino acids required for the interaction between ALKBH5 and RBM33 as well as ALKBH5 demethylase activity in vivo. Of note, the crystal structure of ALKBH5 in a complex with an m6A-ssRNA 8-mer substrate has been published recently, suggesting ALKBH5 can directly bind to 8-mer m6A RNA in vitro.45 Analysis of the electrostatic surfaces among ALKBH5 and other AlkB family demethylases such as FTO, ALKBH1, and AlkB revealed that ALKBH5 is less positively charged compared to other AlkB demethylases, whereas the predicted RBM33 protein structure suggests that the electrostatic surface of RBM33 is more positively charged (Figure S6N–S6R). This suggests that a complex formed by RBM33 and ALKBH5 provides a structure with a more positive charge than ALKBH5 alone, thereby enhancing the binding affinity of ALKBH5 with its RNA substrates. RIP-seq and PAR-CLIP-seq analyses reveal that the RNA transcripts bound by RBM33 or ALKBH5 are significantly overlapped in different cell types. Additionally, the transcripts displaying an increase of m6A methylation upon ALKBH5 or RBM33 KD are also substantially overlapped. These results imply that RBM33 and ALKBH5 act on the same substrates of the transcriptome, further suggesting that the two proteins are mutually dependent on one another in the regulation of m6A RNA demethylation of selected genes, involved in diverse biological processes. Thus, we conclude that RBM33 is required for ALKBH5-mediated m6A demethylation of selected mRNA transcripts.
Importantly, we further demonstrate that RBM33 mediated selectivity of ALKBH5-induced demethylation of specific transcripts is critical for tumorigenesis in HNSCC cells. We find that both RBM33 and ALKBH5 are simultaneously overexpressed in the majority of HNSCC primary tumors, especially at more advanced stages. RBM33 and ALKBH5 function as a complex to maintain the tumorigenesis of HNSCC by directly binding to DDIT4 transcripts and regulating DDIT4 expression in an m6A-dependent manner. Consistent with previously published results,46–48 we demonstrate that DDIT4 transcripts harbor m6A methylation and DDIT4 m6A methylation lead to its mRNA decay in HNSCC cells. Notably, re-expression of DDIT4 reverses the negative impact of RBM33 and ALKBH5 on the growth and survival of HNSCC cells in vivo, suggesting that DDIT4 is a key functional mediator of the RBM33/ALKBH5 complex in HNSCC. The role of DDIT4 in cancer is controversial.38 DDIT4 was reported to function as a tumor suppressor through inhibiting mTOR pathways49,50 but numerous studies suggest that DDIT4 plays an oncogenic role in multiple solid tumors including Ovarian Cancer (OC),51 Bladder Urothelial Carcinoma (BUC),52 and Colorectal Cancer (CRC).53We find that DDIT4 is significantly up-regulated in HNSCC patients and high expression of DDIT4 is associated with a poor prognosis in HNSCC patients. DDIT4 KD substantially inhibits growth and induces apoptosis of HNSCC cells, implying that DDIT4 plays an oncogenic role in HNSCC. Interestingly, RBM33, ALKBH5 and DDIT4 are upregulated in HNSCC, KIRC, LIHC and ACC (Figure S1 and Figure S7), suggesting that the RBM33/ALKBH5/m6A demethylation/DDIT4 signaling axis maybe deregulated in a wide range of solid tumors. Consistent with RNA-seq data showing that RBM33 and ALKBH5 KD both significantly affect the expression of genes related to hypoxia and ER stress response pathways, which regulate autophagy, either ALKBH5 or RBM33 KD reduces autophagy in HNSCC cells. Autophagy has a dynamic role in malignancy. Despite autophagy limiting tumor initiation, it can play a positive role in malignant progression and tumor maintenance.54 We show that inhibition of autophagy substantially suppresses growth and inhibits apoptosis of HNSCC cells. Together, these results support our model that significant up-regulation of both RBM33 and ALKBH5 in HNSCC, particularly at the late stage of HSNCs, promotes tumorigenesis at least partially through DDIT4-mediated autophagy. In summary, our study reveals a unique mechanism underlying the selective demethylation of m6A transcripts mediated by ALKBH5 in HNSCC and 293T cells. This process of selectively demethylate of m6A-marked transcripts such as DDIT4 by RBM33/ALKBH5 is critical for tumorigenesis in HNSCC. This process might be important for tumorigenesis in other cancers as well as other biological processes involved ALKBH5.
Limitations of the study
Here we demonstrated that RBM33 mediates substrate selectivity of ALKBH5 in a significant number of ALKBH5 targeting transcripts. It remains unclear whether there are other RNPs or adaptor proteins are involved in ALKBH5 targeting transcripts that are not recognized by RBM33. Although we showed both ALKBH5 and RBM33 were upregulated in HSNCC, the mechanism that leads to upregulation of ALKBH5 and RBM33 in HSNCC remains elusive. Additional studies are required to address these questions in the future.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Dr. Zhijian Qian (Zhijian.Qian@medicine.ufl.edu).
Materials availability
All the materials generated in this study are accessible upon request.
Data and code availability
Data have been deposited at GEO database, ProteomeXchange Consortium, and the flow cytometry repository and are publicly available as of the date of publication.
Accession numbers are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | ||
|---|---|---|---|---|
| Antibodies | ||||
| α-RBM33 | Sigma | CAT #HPA021768 | ||
| α- m6A | Synaptic Systems | CAT #202003 | ||
| α- ALKBH5 | Sigma | CAT #HPA007196 | ||
| α-Strep | Sigma | CAT #SAB2702216 | ||
| α- Annexin V | Thermo Fisher | CAT #17800774 | ||
| α- SENP1 | Cell Signaling Technology | CAT #11929S | ||
| α- Actin | Cell Signaling Technology | CAT #8457S | ||
| α- Flag | Sigma | CAT #F1804 | ||
| α- SUMO-1 | Thermo Fisher | CAT #33–2400 | ||
| α- DDIT4 | Cell Signaling Technology | CAT #2516 | ||
| Alexa Fluor 594 goat anti-rabbit IgG | Jackson immunoResearch | CAT #111–585-003 | ||
| Alexa Fluor 488 goat anti-mouse IgG | Jackson immunoResearch | CAT #115–545-003 | ||
| ImmPRESS® HRP Horse Anti-Rabbit IgG Polymer Detection Kit, Peroxidase | Vector Laboratories Inc | CAT #MP-7401 | ||
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, HRP | Thermo Fisher | CAT #G21234 | ||
| Chemicals, peptides, and recombinant proteins | ||||
| SuperScript III Reverse Transcriptase | Thermo Fisher | CAT #18080044 | ||
| RNaseOUT Recombinant Ribonuclease Inhibitor | Thermo Fisher | CAT #10777019 | ||
| dNTP Mix | Thermo Fisher | CAT # R0192 | ||
| TRIzol™ Reagent | Thermo Fisher | CAT #15596018 | ||
| ACTINOMYCIN D | Sigma | CAT #A1410 | ||
| ECL Western Blotting Detection Kit | LAMDA BIOTECH | CAT #G075 | ||
| 4-Thiouridine | Sigma | CAT #T4509 | ||
| GLYCOGEN | Sigma | CAT #G1767 | ||
| FBS | Thermo Fisher | CAT #16140071 | ||
| Protein G Agarose/Salmon Sperm DNA, 2.5 mL | Sigma | CAT #16–201 | ||
| Critical commercial assays | ||||
| FITC Annexin V Apoptosis Detection Kit I | BD Pharmingen | CAT #556547 | ||
| Dynabeads mRNA Purification Kit | Thermo Fisher | CAT #61006 | ||
| EpiQuik m6A RNA Methylation Quantification Kit | epigentek | CAT # P-9008–48 | ||
| NEBNext® Small RNA Library Prep Set for Illumina® | NEB | CAT # E7330S | ||
| Monarch® PCR & DNA Cleanup Kit | NEB | CAT #T1030L | ||
| Deposited data | ||||
| The raw and processed sequencing data | This paper | GEO: GSE202878 | ||
| The raw data and the analyzed data of spectrometry proteomics data | This paper | ProteomeXchange Consortium: PXD033861 | ||
| The raw data for FACS | This paper | the flow cytometry repository:http://flowrepository.org/id/RvFrqmygFTFYfcUcMryb0KueJ8wKpvgyEki6PXvB6HaI40zqK9fCXJcFDBT6buOV http://flowrepository.org/id/RvFriG3gY7h6XPt2gugIUmmw1vzSe1Gvt03h2jc2euZZfEaRREZzf8lrYCadvLFf http://flowrepository.org/id/RvFrmftoEHSGaS0skVN2AhpzyuDdEC0HeG4OGIXRwucrZGop4UldROZZ82qWgCPw http://flowrepository.org/id/RvFrRxAK0ZGiqGZsSC08zL9tmesGvqLDl72VutMbi9SicrLBxYWiWU3AwjQhv35MVw http://flowrepository.org/id/RvFrltp8tLuTzHOdz9OhyeCWDy9ZJZ5UTimYscHtvDO9lrSguFXqVU5jbpgwsXxk http://flowrepository.org/id/RvFrKDtRbfCXZiIaCXUNuNrxvNC5J7UxHHLQZf294tN9j1ULVyHmuc7T22GvQfkZ |
||
| Experimental models: Cell lines | ||||
| HEK293T | ATCC | Cat# CRL-11268 | ||
| HOK | ScienCell | 2610 | ||
| UM-SCC-1 | Gift from Dr. Lingtao Jin’s lab | RRID: CVCL_7707 | ||
| UM-SCC-2 | Gift from Dr. Lingtao Jin’s lab | N/A | ||
| SCC090 | Gift from Dr. Lingtao Jin’s lab | N/A | ||
| JHU022 | Gift from Dr. Lingtao Jin’s lab | RRID: CVCL_5991 | ||
| 93-VU-147T | Gift from Dr. Lingtao Jin’s lab | COSS2296308 | ||
| SqCC/Y1 | Gift from Dr. Lingtao Jin’s lab | RRID: CVCL_0551 | ||
| 1483 | Gift from Dr. Lingtao Jin’s lab | RRID: CVCL_6980 | ||
| Oligonucleotides | ||||
| Table S1 | This paper | N/A | ||
| Recombinant DNA | ||||
| pCDH- Strep-RBM33 | This paper | N/A | ||
| p3XFlag-RBM33 | This paper | N/A | ||
| pMSCV-HA-ALKBH5 | This paper | N/A | ||
| pCDH-Strep-ALKBH5 | This paper | N/A | ||
| p3XFlag-SENP1 | This paper | N/A | ||
| pCDH-Strep-DDIT4 | This paper | N/A | ||
| Software and algorithms | ||||
| Cutadapt (v1.18) | Martin, 2011 | N/A | ||
| HISAT2 (v2.1.0) | Kim et al., 2015 | N/A | ||
| featureCounts (v1.6.4) | Liao et al., 2014 | N/A | ||
| edgeR (v3.24.3) | Robinson et al., 2010 | PMID: 19910308 | ||
| DESeq2 | Love et al., 2014 | N/A | ||
| RADAR (v1.0) | Zhang et al., 2019 | N/A | ||
| MeRIPtools (v1.0) | Zhang et al., 2020 | N/A | ||
| Bowtie2 (v2.4.5) | Langmead and Salzberg, 2012 | N/A | ||
| STAR (v 2.7.9a) | Dobin et al., 2013 | N/A | ||
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HOK cell line was purchased from ScienCell (Cat#2610). For rountine maintenance, HOK cells were cultured at 37 0C with 5% CO2 in Oral Keratinocyte Medium (OKM), when used with Oral Keratinocyte Growth Supplement (OKGS, Cat #2652). HNSCC cell lines are kindly provided by Dr. Lingtao Jin’s lab. UM-SCC-1, UM-SCC-2 were cultured in DMEM containing 10% FBS and 1% penicillin/streptomycin. SCC090 was cultured in DMEM-F12 medium containing 10% FBS and 1% penicillin/streptomycin. JHU022, 1483, 93-VU-147T, SqCC/Y1 were cultured in DMEM-F12 medium with 10% FBS plus 1% penicillin/streptomycin.
METHOD DETAILS
Western blot analysis and Co-immunoprecipitation
The western blot and co-immunoprecipitation analyses were performed according to standard protocols as described previously,57 using the indicated antibodies. For examining protein and protein interactions, cells were lysed in IP lysis buffer (50 mM Tris-HCl pH7.5 , 150 mM NaCl, 0.5% NP-40, 1mM EDTA, 8% glycerol, 50mM NaF, 1 mM DTT, 1mM PMSF) supplemented with protein inhibitors. For immunoprecipitation assays, the lysates were further diluted to 0.1% NP-40 and immunoprecipitated with antibodies against target proteins at 40C overnight. Next, the IP proteins were tested by western blotting.
Streptavidin pull-down analysis
To determine whether RBM33 has a binding preference towards m6A-modified RNA oligos than unmodified RNA oligos, biotin-labeled m6A-modified and unmodified RNA oligonucleotide baits were synthesized at GE Dharmacon, USA (ss-A: 5’biotin-CGUCUCGGACUCGGACUGCU-3’ and ss-m6A: 5’biotin-CGUCUCGG(m6A)CUCGG(m6A)CUGCU-3’). The pull-down assay was performed according to what was previously described.30 Briefly, infected UM-SCC1 cells were collected, washed with PBS, and lysed in lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5% NP-40, 1 mM EDTA, 8% glycerol supplemented with protease inhibitor mixture, phosphatase inhibitors, and 1 mM DTT). 10% of whole cell lysate was used as input and 90% of the whole cell lysate was used for the following Streptavidin sepharose beads pull-down. Next, 2 μg of biotinylated RNA baits were incubated with the above-mentioned whole cell lysate, diluted with a binding buffer containing 10 mM Tris-HCl pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 0.05% NP-40 and subjected to rotation at 4 °C for 2 hours. The resulting beads were washed three times with washing buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.05% NP-40, 1 mM EDTA). The enriched proteins were subjected to Western blotting analysis.
HNSCC Xenograft studies
Animal experiments were performed according to animal protocols approved by the animal care facility of the University of Florida. Briefly, two million UM-SCC-1 cells stably expressing shRNA scramble or shRNAs against RBM33 and ALKBH5 were subcutaneously injected into two flanks of each NSGS mice. Tumor volume and mice weight measurements were taken every 4 days and 7 days respectively. For the in vivo ALKBH5 inhibition assay, the HNSCC PDX tumors TM01145 and TM01420 were purchased from Jackson Laboratory. 15 mg/kg 2,4-PDCA was given i.p. every four days when tumor volume reaches ~100 mm3. And, tumor volume was calculated according to the formula: [D×(d2)] /2 where D represents the large diameter of the tumor and d represents the small diameter of the tumor. Animals were individually monitored throughout the experiment.
Immunohistochemical Staining (IHC)
All the formalin-fixed, paraffin-embedded tissue specimens from human HNSCC patients were purchased from US Biomax, Inc. IHC analyses of RBM33 and ALKBH5 were performed according to the protocol as previously described.58 Briefly, all the tissue microarray slices went through deparaffinization, rehydration, suppressing endogenous peroxidase activity, antigen retrieval, blocking, primary and secondary antibodies incubation, staining & counterstaining, and finally dehydration and mounting. Positive staining of RBM33 and ALKBH5 was identified by using IHC signal intensity, scored as 0 to 4.
shRNA, siRNA-mediated target genes depletion, and quantitative RT-PCR
Deletion of target genes by shRNAs was done as described previously.57 The vector for shRNAs was pLKO.1. For siRNA-mediated target genes’ depletion, siRNAs against RBM33 and ALKBH5 were inserted into the pSiEB vector according to the published paper59 with minor changes. siRNA scrambles or siRNAs against RBM33 and ALKBH5 were packaged into a retrovirus. After retrovirus infection and selected by blasticidin, UM-SCC-1 cells stably expressing scrambled or siRNAs against RBM33 and ALKBH5 were subjected to RT-qPCR analysis. The sequences for shRNAs and siRNAs are listed in Table S1. For qRT-PCR analysis, total RNA was extracted from various cells as indicated and reverses transcribed using kits purchased from Thermo Fisher. The primer sequences used in the qRT-PCR are listed in Table S1.
RNA immunoprecipitation (RIP) and RIP-seq
RNA immunoprecipitation was performed as previously described,31 with some modifications. Briefly, after formaldehyde crosslinking, 25 million cells per sample were harvested and washed with PBS. Cells were lysed with 500 μL (approximately 3 volumes) of lysis buffer consisting of 10 mM HEPES pH 7.6, 150 mM KCl, 2 mM EDTA, 0.5% NP-40, 0.5 mM DTT, 1X complete Protease Inhibitor (Roche), and 400 U/mL SUPERase-In RNase Inhibitor (Thermo). To fragment RNA, the lysate was also subject to sonication with the Bioruptor Pico sonication device (Diagenode) for 30 cycles consisting of 30 seconds ON and 30 seconds OFF. Sample lysate was then centrifuged at 15k × g for 15 minutes. Input sample for RNA sequencing was prepared by saving 5% of lysate and adding 500 μL TRIzol reagent. Samples were subjected to immunoprecipitation using MagStrep type3 XT magnetic beads (IBA Life Sciences). Beads were washed 3 times and re-suspended with cold NT2 buffer (50 mM HEPES pH 7.6, 200 mM NaCl, 2 mM EDTA, 0.05% NP-40, 0.5 mM DTT, and 200 U/mL RNase inhibitor). Sample lysates were immunoprecipitated with orbital rotation at 4°C overnight. Afterward, beads were washed 6 times with cold NT2 buffer. The samples of immunoprecipitated beads were subjected to Proteinase K digestion in NT2 buffer supplemented with 1% SDS and 1.2 mg/mL Proteinase K (ThermoFisher) incubated with shaking at 1200 rpm at 55°C for 1 hour. Afterward, 500 μL TRIzol reagent was added. RNA was extracted from both input and immunoprecipitated RNA through the Direct-zol RNA Miniprep Kit (Zymo) including DNase I digestion and subsequently used for high-throughput sequencing. For RIP-seq, libraries for input and RIP RNA samples were constructed using the SMARTer Stranded Total RNA-seq Kit v2 (Takara) and were quantified by the Bioanalyzer High Sensitivity DNA chip. RIP-seq libraries were sequenced on the Illumina NovaSeq 6000 according to the manufacturer’s instructions with paired-end 50-bp reads.
PAR-CLIP-seq
PAR-CLIP-seq was performed according to a previous published study60 with minor changes. Briefly, 200 uM 4SU was firstly incorporated into 50 million of HEK293T cells stably expressing 3XFlag-ALKBH5 and 3XFlag-RBM33. Then the cells was UV-crosslinked twice at 1500*100 uJ/cm2 by using the 365 nm UV cross-linker. Cultures were then washed, lysed, and partially digested by RNase T1 (Final concentration, 0.2U/uL). Lysates were used for immunoprecipitation of Flag-ALKBH5 and Flag-RBM33 by using a monoclonal anti-Flag tag antibody (Sigma, # F1804–5 mg) and ProteinG beads (Sigma, Cat #16–201). 10 mg of antibody was used in 100 mL bead slurry per 1mg of input lysate. Save 5% of cell lysate as Input. After IP enrichment and three times washes, the samples were subjected to the second time RNase T1 (1000 U/uL, Thermo Fisher) digestion (Final concentration, 10 U/uL). After that, all the samples were subjected to dephosphorylation, end-repair/radioactivity-labeling and SDS-PAGE protein size selection. After the enriched RNA fragments were collected by the phenol/choloroform, the library was constructed with NEBNext multiplex small RNA library prep set for illumine (Set1) (NEB, Cat #E7300S). Finally, purified cDNA libraries were submitted to the next-generation sequencing service at University of Florida for sequencing. All libraries were processed on a NovaSeq S4 2X150 platform (Illumina) and 50 M reads per sample was required.
RNA-seq
Total RNA was isolated from cells with Direct-zol RNA Miniprep Kit (Zymo) including DNase I digestion (Zymo). 2–4 μg total RNA was then sonicated with the Bioruptor Pico sonication device (Diagenode) for 30 cycles consisting of 30 seconds ON and 30 seconds OFF. Library construction starting from 100 ng total RNA for each sample was made using the SMARTer Stranded Total RNA-seq Kit v2. Each library was quantified using a Qubit fluorometer (Thermo Fisher Scientific) and the size distribution was assessed using the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA). All samples were sequenced by the Illumina NovaSeq 6000 with a single-end 100-base pair read length.
m6A-seq
Total RNA was isolated from cells with Direct-zol RNA Miniprep Kit (Zymo) including DNase I digestion (Zymo). RNA fragmentation was performed by sonicating 2–4 μg total RNA in 100 μl RNase-free water using the Bioruptor Pico (Diagenode) with the 30s on/30s off for 30 cycles at 4°C. m6A-IP and library preparation was performed per the reported protocol61 with some modified instructions based on the EpiMark N6-Methyladenosine Enrichment Kit. Briefly, 25 μL Pierce Protein A/G Magnetic Beads (Thermo Fisher) were washed twice with 1x IP buffer and mixed with 1 μL m6A antibody from the EpiMark N6-Methyladenosine Enrichment Kit (New England Biolabs, E1610S) and incubated with orbital rotation at 4°C for 30 min. The beads were washed twice with 1x IP buffer, and immunoprecipitation was performed by adding 1 μg sonicated RNA and mixing with orbital rotation for 3 hr at 4°C. The beads were then separated and washed twice with 1x IP buffer, twice with low salt reaction buffer (50 mM NaCl, 0.1% NP-40, 10 mM Tris-HCl, pH 7.4), and twice with high salt reaction buffer (500 mM NaCl, 0.1% NP-40, 10 mM Tris-HCl, pH 7.4) before elution with Buffer RLT (Qiagen). The eluate was purified with the RNA Clean and Concentrator kit (Zymo, Orange, CA). The purified RNA fragments were then used to construct libraries with the SMARTer Stranded Total RNA-seq Kit v2 (Takara). Sequencing was carried out on the Illumina NovaSeq 6000 according to the manufacturer’s instructions with a single-end 100-bp read length.
Sequencing data analysis
(1) RNA-seq data.
m6A-seq input samples were used for gene expression analysis. Briefly, adapters of all raw m6A-seq input data sets were removed by Cutadapt62 v1.18. Then reads longer than 19 nt were mapped to the human genome (hg38) and the GENCODE v36 gene annotation using HISAT263 v2.1.0 with default parameters. Raw reads on each gene were counted by featureCounts64 (-t exon -g gene_id -M -O --fraction -T 24 -s 2) from Subread v1.6.4. The trimmed mean of M-values (TMM) normalization method from edgeR55 v3.24.3 package was used for RNA abundance estimation. Gene expression levels are represented by the Counts Per Million (CPM) values. DEGs were determined with the cutoff (|log2FCshRNA/shCtrl| > 0.5849625 and FDR < 0.05). DESeq2 was used for differential gene expression (DEG) analysis. A transcription database was generated:
gtffile <- file.path(“hg38_UCSC.gtf”) txdb <- makeTxDbFromGFF(gtffile, format = “gtf”, circ_seqs = character()) A summarized experiment object was then generated. se <- summarizeOverlaps(features=ebg, reads=bamfiles, mode=“Union”, singleEnd=TRUE, ignore.strand=TRUE, fragments=FALSE)
The dataset was pre-filtered by removing genes that had zero read counts. After running the DESeq command, the results were contrasted based on their genetic condition profile. For example: res = DESeq2∷results (dds, contrast = c(“condition”,“shALKBH5”, “shScramble”) Genes were considered differentially expressed if the adjusted p-value of the logarithm of the fold-change was less than or equal to 0.05.
(2) RIP-seq data.
Adapters of all raw RIP-seq data sets were removed by Cutadapt62 v1.18. Then reads longer than 19 nt were mapped to the human genome (hg38) and the GENCODE v36 gene annotation using HISAT263 v2.1.0 with default parameters. Raw reads on each gene were counted by featureCounts64 (-t exon -g gene_id -M -O --fraction -T 24 -s 2) from Subread v1.6.4. For each gene, the differential enrichment of reads between IP and input samples were calculated by the edgeR55 v3.24.3 package. Genes with the cutoff (log2FCIP/input > 1 and FDR < 0.05) were determined as candidate targets and used for further analysis.
(3) m6A-seq data.
Sequencing was carried out on the Illumina NovaSeq 6000 according to the manufacturer’s instructions with a single-end 50-bp read length. Reads were mapped to human genome version GRCh38 by HiSat2 version 2.2.165. Differentially m6A methylated peaks between different sample conditions were identified using RADAR version 1.066, or MeRIPtools version 1.067 (adjusted p < 0.1). For preprocessing details, trim_galore, a wrapper script that automates quality and adapter trimming as well as quality control was used with the following command and parameters:
trim_galore file.fastq.gz --length 20 –fastqc. The MeRIPtools package was used for analysis of m6A-methylated genes and peaks, as well as differential methylation analysis. First, the package’s countReads tool command was used as shown below:
MeRIP <- countReads(samplenames = samplenames, gtf = gtf_human, bamFolder = bam_dir, modification = “m6A”, strandToKeep = “opposite”,fragmentLength = 150, outputDir = NA, threads = 48, binSize = 50, saveOutput = FALSE). Peaks were then called using Fisher’s exact test with the callPeakFisher command, as shown below. A region was defined as an m6A peak if it had a minimum read count of 15, a false discovery rate of less than 0.05, and was found in a minimum of two replicates.
MeRIP <- callPeakFisher(MeRIP = MeRIP, min_counts = 15, peak_cutoff_fdr = 0.05, peak_cutoff_oddRatio = 1) MeRIP <- reportJointPeak(MeRIPdata = MeRIP, joint_threshold = 2) Joint peaks of reads between replicate samples were then counted with the jointPeakCount command: MeRIP <- jointPeakCount(MeRIP)
In order to assess differentially methylated peaks, a genotype was assigned to each variable with a dataframe using the variable() command, and a quadratic negative binomial model was used to find differentially methylated peaks.
MeRIPshALKBH51 <- MeRIPtools∷QNBtest(MeRIPshALKBH51) allPeakResult <- MeRIPtools∷results(MeRIPshALKBH51)
Peaks were considered differentially methylated if they had an adjusted p-value of less than 0.1.
diff_peaks <- allPeakResult[allPeakResult$padj <0.1, ]
The bedtools software was used in conjunction with the hg38 genome, using the bedtools getfasta command in order to convert the peaks to be analyzed into fasta format. The Homer software was then implemented with the following command.
findMotifs.pl Joint_peak.fa fasta homer_motif -fasta hg38_200bp_randomPeak.fa -rna -p 10 -len 5,6,7
(4) PAR-CLIP-seq data
Adapters of all raw PAR-CLIP data sets were removed by Cutadapt62 v1.18. Reads longer than 17 were first mapped to the human rRNAs and small RNAs with Bowtie268 v2.4.5. Then unmapped reads were aligned to the human genome (hg38) and the GENCODE v36 gene annotation using STAR69 v 2.7.9a with parameters according to the ENCODE RNA-seq pipeline, except that the option (--outFilterMismatchNoverReadLmax 0.06) was modified in order to capture more potential T to C mutations. Only reads with proper pair were remained. Reads with mate unmapped and not in primary alignment were discarded. MarkDuplicates was used to remove PCR duplicates. To get aggregated T to C mutation counts, alignment files of two replicates were merged. T positions with coverage >= 10 were selected and they were concatenated as clusters if they were next to each other within 20 nt. Clusters with total mutation counts >= 3 were considered as potential binding regions. De novo motif enrichment analysis was performed in the binding regions using HOMER70 v4.9. Differentially ALKBH5 binding regions before and after RBM33 knock out were defined according to the following steps: 1) T to C mutation rates of each selected T positions in the clusters were calculated and summed up as the mutation levels of the clusters. 2) Total mutation counts >= 3 in the clusters of the control group, the RBM33 knock out group or both were required. 3) clusters with the mutation level difference >= 20% between the two groups were considered as differentially binding regions.
Quantification and Statistical Analysis
Experiments were performed at least three times, and the representative data were shown. All statistical tests were performed using the unpaired Student’s t-test by GraphPad Prism 5 software. A value of lossless<0.05 was considered statistically significant. In all the results, “*” denotes p<0.05, “**” denotes p<0.01, “***” denotes p<0.001, and “ns” denotes no significant difference.
Supplementary Material
SUPPLEMENTAL INFORMATION
Table S1. 00000000000.... for RT-qPCR primers, shRNAs and siRNAs, related to STAR Method.
Highlights.
RBM33 binds m6A and regulates ALKBH5-mediated m6A demethylation selectivity.
RBM33 activates ALKBH5 demethylase activity via suppression of ALKBH5 SUMOylation.
RBM33/ALKBH5 modulates m6A-mediated DDIT4 mRNA decay and DDIT4/TXNIP-based autophagy.
Suppression of RBM33/ALKBH5 activity inhibits HNSCC and primary patient cells growth.
ACKNOWLEDGMENTS
We thank all members of Dr. Qian’s laboratory for their valuable discussions. This work was partially supported by National Institutes of Health (NIH) RO1 Grants R01 HL157539–01(Z.Q.), DK129489 (ZQ), CA259576–01(Z.Q.), R01CA266659 (Z.Q.) and RM1 HG008935 (C.H.), Z.Q is Leukemia & Lymphoma Society (LLS) Scholars. C.H. is an Investigator of the Howard Hughes Medical Institute (HHMI).
INCLUSION AND DIVERSITY
We support inclusive, diverse and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Gerstberger S, Hafner M, and Tuschl T (2014). A census of human RNA-binding proteins. Nat Rev Genet 15, 829–845. 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matia-Gonzalez AM, Laing EE, and Gerber AP (2015). Conserved mRNA-binding proteomes in eukaryotic organisms. Nat Struct Mol Biol 22, 1027–1033. 10.1038/nsmb.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitchell SF, and Parker R (2014). Principles and properties of eukaryotic mRNPs. Mol Cell 54, 547–558. 10.1016/j.molcel.2014.04.033. [DOI] [PubMed] [Google Scholar]
- 4.Neelamraju Y, Gonzalez-Perez A, Bhat-Nakshatri P, Nakshatri H, and Janga SC (2018). Mutational landscape of RNA-binding proteins in human cancers. RNA Biol 15, 115–129. 10.1080/15476286.2017.1391436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin H, Ni H, Liu Y, Yuan Y, Xi T, Li X, and Zheng L (2020). RNA-binding proteins in tumor progression. J Hematol Oncol 13, 90. 10.1186/s13045-020-00927-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kapeli K, Martinez FJ, and Yeo GW (2017). Genetic mutations in RNA-binding proteins and their roles in ALS. Hum Genet 136, 1193–1214. 10.1007/s00439-017-1830-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper TA, Wan L, and Dreyfuss G (2009). RNA and disease. Cell 136, 777–793. 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas A, Rehfeld F, Zhang H, Chang T-C, Goodarzi M, Gillet F, and Mendell JT (2022). RBM33 directs the nuclear export of transcripts containing GC-rich elements. Genes & Development 36, 550–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, and Rottman F (1994). Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. Journal of Biological Chemistry 269, 17697–17704. [PubMed] [Google Scholar]
- 10.Bokar J, Shambaugh M, Polayes D, Matera A, and Rottman F (1997). Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. Rna 3, 1233–1247. [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, and Deng X (2014). A METTL3–METTL14 complex mediates mammalian nuclear RNA N 6-adenosine methylation. Nature chemical biology 10, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ping X-L, Sun B-F, Wang L, Xiao W, Yang X, Wang W-J, Adhikari S, Shi Y, Lv Y, and Chen Y-S (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell research 24, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, and Cacchiarelli D (2014). Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell reports 8, 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patil DP, Chen C-K, Pickering BF, Chow A, Jackson C, Guttman M, and Jaffrey SR (2016). m 6 A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, Masiello I, Hares T, Villaseñor R, and Hess D (2018). Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m6A machinery component Wtap/Fl (2) d. Genes & development 32, 415–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, and Yang Y-G (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nature chemical biology 7, 885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ, Vågbø CB, Shi Y, Wang W-L, and Song S-H (2013). ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Molecular cell 49, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao S, Sun H, and Xu C (2018). YTH domain: a family of N6-methyladenosine (m6A) readers. Genomics, proteomics & bioinformatics 16, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patil DP, Pickering BF, and Jaffrey SR (2018). Reading m6A in the transcriptome: m6A-binding proteins. Trends in cell biology 28, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J, Fang X, Zhong P, Song Z, and Hu X (2019). N6-methyladenosine modifications: interactions with novel RNA-binding proteins and roles in signal transduction. RNA biology 16, 991–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, and Tavazoie SF (2015). HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell 162, 1299–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu B, Su S, Patil DP, Liu H, Gan J, Jaffrey SR, and Ma J (2018). Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nature communications 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian S-B, and Jaffrey SR (2015). 5′ UTR m6A promotes cap-independent translation. Cell 163, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, and Yuan CL (2018). Recognition of RNA N 6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nature cell biology 20, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arguello AE, DeLiberto AN, and Kleiner RE (2017). RNA chemical proteomics reveals the N6-methyladenosine (m6A)-regulated protein–RNA interactome. Journal of the American Chemical Society 139, 17249–17252. [DOI] [PubMed] [Google Scholar]
- 26.Wu R, Li A, Sun B, Sun J-G, Zhang J, Zhang T, Chen Y, Xiao Y, Gao Y, and Zhang Q (2019). A novel m 6 A reader Prrc2a controls oligodendroglial specification and myelination. Cell research 29, 23–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt TG, and Skerra A (2007). The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nature protocols 2, 1528–1535. 10.1038/nprot.2007.209. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt T, and Skerra A (2015). The Strep-tag system for one-step affinity purification of proteins from mammalian cell culture. Methods Mol Biol 1286, 83–95. 10.1007/978-1-4939-2447-9_8. [DOI] [PubMed] [Google Scholar]
- 29.Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L, Chen H, Su R, Yin Z, and Li W (2020). RNA demethylase ALKBH5 selectively promotes tumorigenesis and cancer stem cell self-renewal in acute myeloid leukemia. Cell Stem Cell 27, 64–80. e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu F, Wei J, Cui X, Yu C, Ni W, Bungert J, Wu L, He C, and Qian Z (2021). Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic acids research 49, 5779–5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L, Chen H, Su R, Yin Z, and Li W (2020). RNA demethylase ALKBH5 selectively promotes tumorigenesis and cancer stem cell self-renewal in acute myeloid leukemia. Cell Stem Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J, Gao M, He J, Wu K, Lin S, Jin L, Chen Y, Liu H, Shi J, and Wang X (2021). The RNA m6A reader YTHDC1 silences retrotransposons and guards ES cell identity. Nature 591, 322–326. [DOI] [PubMed] [Google Scholar]
- 33.Feng C, Liu Y, Wang G, Deng Z, Zhang Q, Wu W, Tong Y, Cheng C, and Chen Z (2014). Crystal structures of the human RNA demethylase Alkbh5 reveal basis for substrate recognition. Journal of Biological Chemistry 289, 11571–11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li JZ, Gao W, Chan JY, Ho WK, and Wong TS (2012). Hypoxia in head and neck squamous cell carcinoma. ISRN Otolaryngol 2012, 708974. 10.5402/2012/708974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cancer Genome Atlas N (2015). Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582. 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang C, Klukovich R, Peng H, Wang Z, Yu T, Zhang Y, Zheng H, Klungland A, and Yan W (2018). ALKBH5-dependent m6A demethylation controls splicing and stability of long 3′-UTR mRNAs in male germ cells. Proc Natl Acad Sci U S A 115, E325–E333. 10.1073/pnas.1717794115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, and Cubillos-Ruiz JR (2021). Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer 21, 71–88. 10.1038/s41568-020-00312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tirado-Hurtado I, Fajardo W, and Pinto JA (2018). DNA Damage Inducible Transcript 4 Gene: The Switch of the Metabolism as Potential Target in Cancer. Front Oncol 8, 106. 10.3389/fonc.2018.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qiao S, Dennis M, Song X, Vadysirisack DD, Salunke D, Nash Z, Yang Z, Liesa M, Yoshioka J, Matsuzawa S, et al. (2015). A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nature communications 6, 7014. 10.1038/ncomms8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao C, Wang R, Li B, Guo Y, Yin T, Xia Y, Zhang F, Lian K, Liu Y, and Wang H (2020). TXNIP/Redd1 signalling and excessive autophagy: a novel mechanism of myocardial ischaemia/reperfusion injury in mice. Cardiovascular Research 116, 645–657. [DOI] [PubMed] [Google Scholar]
- 41.Wen J, Lv R, Ma H, Shen H, He C, Wang J, Jiao F, Liu H, Yang P, and Tan L (2018). Zc3h13 regulates nuclear RNA m6A methylation and mouse embryonic stem cell self-renewal. Molecular cell 69, 1028–1038. e1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, and Han N (2017). Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 552, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertero A, Brown S, Madrigal P, Osnato A, Ortmann D, Yiangou L, Kadiwala J, Hubner NC, de Los Mozos IR, and Sadée C (2018). The SMAD2/3 interactome reveals that TGFβ controls m6A mRNA methylation in pluripotency. Nature 555, 256–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, Chen Z, Deng X, Xiao G, and Auer F (2019). Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature 567, 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaur S, Tam NY, McDonough MA, Schofield CJ, and Aik WS (2022). Mechanisms of substrate recognition and N 6-methyladenosine demethylation revealed by crystal structures of ALKBH5–RNA complexes. Nucleic Acids Research 50, 4148–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, Song C, Wang N, Li S, Liu Q, Sun Z, Wang K, Yu S-C, and Yang Q (2020). NADP modulates RNA m6A methylation and adipogenesis via enhancing FTO activity. Nature chemical biology 16, 1394–1402. [DOI] [PubMed] [Google Scholar]
- 47.Qin Y, Li B, Arumugam S, Lu Q, Mankash SM, Li J, Sun B, Li J, Flavell RA, and Li H-B (2021). m6A mRNA methylation-directed myeloid cell activation controls progression of NAFLD and obesity. Cell reports 37, 109968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Liu X, Wang Y, Lai S, Wang Z, Yang Y, Liu W, Wang H, and Tang B (2022). The m6A demethylase ALKBH5-mediated upregulation of DDIT4-AS1 maintains pancreatic cancer stemness and suppresses chemosensitivity by activating the mTOR pathway. Molecular Cancer 21, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, and Kaelin WG Jr. (2004). Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 18, 2893–2904. 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeYoung MP, Horak P, Sofer A, Sgroi D, and Ellisen LW (2008). Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev 22, 239–251. 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang B, Meng J, Zhu H, Du X, Sun L, Wang L, Li S, and Yang G (2018). Overexpression of the recently identified oncogene REDD1 correlates with tumor progression and is an independent unfavorable prognostic factor for ovarian carcinoma. Diagnostic pathology 13, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang Y, Xia Z, Wang X, Zhao X, Sheng Z, Ye Y, He G, Zhou L, Zhu H, and Xu N (2018). Small-molecule inhibitors targeting protein sumoylation as novel anticancer compounds. Molecular pharmacology 94, 885–894. [DOI] [PubMed] [Google Scholar]
- 53.Fattahi F, Zanjani LS, Shams ZH, Kiani J, Mehrazma M, Najafi M, and Madjd Z (2021). High expression of DNA damage-inducible transcript 4 (DDIT4) is associated with advanced pathological features in the patients with colorectal cancer. Scientific Reports 11, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kimmelman AC (2011). The dynamic nature of autophagy in cancer. Genes Dev 25, 1999–2010. 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perez-Riverol Y, Bai J, Bandla C, García-Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks-Zipper A, and Eisenacher M (2022). The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic acids research 50, D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu F, Shi G, Cheng S, Chen J, Wu S-Y, Wang Z, Xia N, Zhai Y, Wang Z, and Peng Y (2018). SUMO suppresses and MYC amplifies transcription globally by regulating CDK9 sumoylation. Cell research, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jin L, Chun J, Pan C, Li D, Lin R, Alesi GN, Wang X, Kang H-B, Song L, and Wang D (2018). MAST1 drives cisplatin resistance in human cancers by rewiring cRaf-independent MEK activation. Cancer cell 34, 315–330. e317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He F, Ni N, Zeng Z, Wu D, Feng Y, Li AJ, Luu B, Li AF, Qin K, Wang E, et al. (2020). FAMSi: A Synthetic Biology Approach to the Fast Assembly of Multiplex siRNAs for Silencing Gene Expression in Mammalian Cells. Mol Ther Nucleic Acids 22, 885–899. 10.1016/j.omtn.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp A-C, and Munschauer M (2010). PAR-CliP-a method to identify transcriptome-wide the binding sites of RNA binding proteins. JoVE (Journal of Visualized Experiments), e2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 62.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 3. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 63.Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 65.Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Z, Zhan Q, Eckert M, Zhu A, Chryplewicz A, De Jesus DF, Ren D, Kulkarni RN, Lengyel E, and He C (2019). RADAR: differential analysis of MeRIP-seq data with a random effect model. Genome biology 20, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Z, Luo K, Zou Z, Qiu M, Tian J, Sieh L, Shi H, Zou Y, Wang G, and Morrison J (2020). Genetic analyses support the contribution of mRNA N6-methyladenosine (m6A) modification to human disease heritability. Nature genetics 52, 939–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTAL INFORMATION
Table S1. 00000000000.... for RT-qPCR primers, shRNAs and siRNAs, related to STAR Method.
Data Availability Statement
Data have been deposited at GEO database, ProteomeXchange Consortium, and the flow cytometry repository and are publicly available as of the date of publication.
Accession numbers are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | ||
|---|---|---|---|---|
| Antibodies | ||||
| α-RBM33 | Sigma | CAT #HPA021768 | ||
| α- m6A | Synaptic Systems | CAT #202003 | ||
| α- ALKBH5 | Sigma | CAT #HPA007196 | ||
| α-Strep | Sigma | CAT #SAB2702216 | ||
| α- Annexin V | Thermo Fisher | CAT #17800774 | ||
| α- SENP1 | Cell Signaling Technology | CAT #11929S | ||
| α- Actin | Cell Signaling Technology | CAT #8457S | ||
| α- Flag | Sigma | CAT #F1804 | ||
| α- SUMO-1 | Thermo Fisher | CAT #33–2400 | ||
| α- DDIT4 | Cell Signaling Technology | CAT #2516 | ||
| Alexa Fluor 594 goat anti-rabbit IgG | Jackson immunoResearch | CAT #111–585-003 | ||
| Alexa Fluor 488 goat anti-mouse IgG | Jackson immunoResearch | CAT #115–545-003 | ||
| ImmPRESS® HRP Horse Anti-Rabbit IgG Polymer Detection Kit, Peroxidase | Vector Laboratories Inc | CAT #MP-7401 | ||
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, HRP | Thermo Fisher | CAT #G21234 | ||
| Chemicals, peptides, and recombinant proteins | ||||
| SuperScript III Reverse Transcriptase | Thermo Fisher | CAT #18080044 | ||
| RNaseOUT Recombinant Ribonuclease Inhibitor | Thermo Fisher | CAT #10777019 | ||
| dNTP Mix | Thermo Fisher | CAT # R0192 | ||
| TRIzol™ Reagent | Thermo Fisher | CAT #15596018 | ||
| ACTINOMYCIN D | Sigma | CAT #A1410 | ||
| ECL Western Blotting Detection Kit | LAMDA BIOTECH | CAT #G075 | ||
| 4-Thiouridine | Sigma | CAT #T4509 | ||
| GLYCOGEN | Sigma | CAT #G1767 | ||
| FBS | Thermo Fisher | CAT #16140071 | ||
| Protein G Agarose/Salmon Sperm DNA, 2.5 mL | Sigma | CAT #16–201 | ||
| Critical commercial assays | ||||
| FITC Annexin V Apoptosis Detection Kit I | BD Pharmingen | CAT #556547 | ||
| Dynabeads mRNA Purification Kit | Thermo Fisher | CAT #61006 | ||
| EpiQuik m6A RNA Methylation Quantification Kit | epigentek | CAT # P-9008–48 | ||
| NEBNext® Small RNA Library Prep Set for Illumina® | NEB | CAT # E7330S | ||
| Monarch® PCR & DNA Cleanup Kit | NEB | CAT #T1030L | ||
| Deposited data | ||||
| The raw and processed sequencing data | This paper | GEO: GSE202878 | ||
| The raw data and the analyzed data of spectrometry proteomics data | This paper | ProteomeXchange Consortium: PXD033861 | ||
| The raw data for FACS | This paper | the flow cytometry repository:http://flowrepository.org/id/RvFrqmygFTFYfcUcMryb0KueJ8wKpvgyEki6PXvB6HaI40zqK9fCXJcFDBT6buOV http://flowrepository.org/id/RvFriG3gY7h6XPt2gugIUmmw1vzSe1Gvt03h2jc2euZZfEaRREZzf8lrYCadvLFf http://flowrepository.org/id/RvFrmftoEHSGaS0skVN2AhpzyuDdEC0HeG4OGIXRwucrZGop4UldROZZ82qWgCPw http://flowrepository.org/id/RvFrRxAK0ZGiqGZsSC08zL9tmesGvqLDl72VutMbi9SicrLBxYWiWU3AwjQhv35MVw http://flowrepository.org/id/RvFrltp8tLuTzHOdz9OhyeCWDy9ZJZ5UTimYscHtvDO9lrSguFXqVU5jbpgwsXxk http://flowrepository.org/id/RvFrKDtRbfCXZiIaCXUNuNrxvNC5J7UxHHLQZf294tN9j1ULVyHmuc7T22GvQfkZ |
||
| Experimental models: Cell lines | ||||
| HEK293T | ATCC | Cat# CRL-11268 | ||
| HOK | ScienCell | 2610 | ||
| UM-SCC-1 | Gift from Dr. Lingtao Jin’s lab | RRID: CVCL_7707 | ||
| UM-SCC-2 | Gift from Dr. Lingtao Jin’s lab | N/A | ||
| SCC090 | Gift from Dr. Lingtao Jin’s lab | N/A | ||
| JHU022 | Gift from Dr. Lingtao Jin’s lab | RRID: CVCL_5991 | ||
| 93-VU-147T | Gift from Dr. Lingtao Jin’s lab | COSS2296308 | ||
| SqCC/Y1 | Gift from Dr. Lingtao Jin’s lab | RRID: CVCL_0551 | ||
| 1483 | Gift from Dr. Lingtao Jin’s lab | RRID: CVCL_6980 | ||
| Oligonucleotides | ||||
| Table S1 | This paper | N/A | ||
| Recombinant DNA | ||||
| pCDH- Strep-RBM33 | This paper | N/A | ||
| p3XFlag-RBM33 | This paper | N/A | ||
| pMSCV-HA-ALKBH5 | This paper | N/A | ||
| pCDH-Strep-ALKBH5 | This paper | N/A | ||
| p3XFlag-SENP1 | This paper | N/A | ||
| pCDH-Strep-DDIT4 | This paper | N/A | ||
| Software and algorithms | ||||
| Cutadapt (v1.18) | Martin, 2011 | N/A | ||
| HISAT2 (v2.1.0) | Kim et al., 2015 | N/A | ||
| featureCounts (v1.6.4) | Liao et al., 2014 | N/A | ||
| edgeR (v3.24.3) | Robinson et al., 2010 | PMID: 19910308 | ||
| DESeq2 | Love et al., 2014 | N/A | ||
| RADAR (v1.0) | Zhang et al., 2019 | N/A | ||
| MeRIPtools (v1.0) | Zhang et al., 2020 | N/A | ||
| Bowtie2 (v2.4.5) | Langmead and Salzberg, 2012 | N/A | ||
| STAR (v 2.7.9a) | Dobin et al., 2013 | N/A | ||
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.