

Significance
Milk is fundamental for early life in mammals, providing nutrients and immunoregulatory compounds to developing young. Our study represents the largest mammalian milk microbiome survey to date. Across the mammalian tree of life, we show that lactating female mammals host a diverse collection of bacterial and archaeal taxa in their milk, representing one of the earliest exposures of mammalian young to the microbial world. The communities of microbes found within milks were largely a reflection of random, stochastic process. Nonetheless, milk microbiomes still showed signatures of selection that reflected the mother’s evolutionary history and ecology, including environment and diet. Milk microbiomes warrant study as the microbial species in milk likely colonize offspring guts and impact offspring physiology and behavior.
Keywords: microbiota, symbiosis, neonate, bacteria, breast milk
Abstract
Milk production is an ancient adaptation that unites all mammals. Milk contains a microbiome that can contribute to offspring health and microbial-immunological development. We generated a comprehensive milk microbiome dataset (16S rRNA gene) for the class Mammalia, representing 47 species from all placental superorders, to determine processes structuring milk microbiomes. We show that across Mammalia, milk exposes offspring to maternal bacterial and archaeal symbionts throughout lactation. Deterministic processes of environmental selection accounted for 20% of milk microbiome assembly processes; milk microbiomes were similar from mammals with the same host superorder (Afrotheria, Laurasiathera, Euarchontoglires, and Xenarthra: 6%), environment (marine captive, marine wild, terrestrial captive, and terrestrial wild: 6%), diet (carnivore, omnivore, herbivore, and insectivore: 5%), and milk nutrient content (sugar, fat, and protein: 3%). We found that diet directly and indirectly impacted milk microbiomes, with indirect effects being mediated by milk sugar content. Stochastic processes, such as ecological drift, accounted for 80% of milk microbiome assembly processes, which was high compared to mammalian gut and mammalian skin microbiomes (69% and 45%, respectively). Even amid high stochasticity and indirect effects, our results of direct dietary effects on milk microbiomes provide support for enteromammary trafficking, representing a mechanism by which bacteria are transferred from the mother’s gut to mammary gland and then to offspring postnatally. The microbial species present in milk reflect both selective pressures and stochastic processes at the host level, exemplifying various ecological and evolutionary factors acting on milk microbiomes, which, in turn, set the stage for offspring health and development.
The production of milk and nursing of offspring is unique to mammals. Milk is a complex bioactive substance of nutritional and immunoregulatory compounds that has far-reaching impacts on developing mammalian offsprings’ growth and development (1, 2). For instance, milk macronutrient content is a primary factor guiding development and varies widely across Mammalia according to phylogeny, maternal diet, nursing frequency, litter size, and neonatal growth rate, among others (3, 4). Over 100 million years of adaptive evolution has shaped the composition and diversity of milk nutrient and immunological profiles across placental mammals.
Milk was historically believed to be sterile. We now know milk harbors a microbiome, a collection of diverse bacteria, fungi, and archaea (5–7). Early exposure of mammalian young to microbiota occurs through the birthing process, the external environment, and maternal milk (8). Microbes may colonize milk from the maternal gut via the entero-mammary trafficking pathway (9), maternal and infant skin by passive exposure [as reviewed by Fernandez et al. (10)], and the infant oral cavity by retrograde flow during nursing (11). Yet, milk microbiomes are compositionally unique from these other niches (12), indicating that milk microbiomes are unique ecological communities. Understanding factors underlying why certain microbes are present in milk is important as the milk microbiome can colonize neonates’ guts (13) and is likely a mechanism by which milk affects neonatal growth and microbial–immunological development (10, 14).
The adaptive functions of the milk microbiome remain an open question, however, several reasonable hypotheses have been suggested. The microbes in milk are among the first to reach the neonatal gut and may shape the early gut microbiome if they are able to establish residence (13). Microbial metabolic products may have profound effects on the neonates developing gut and immune system. For example, the microbial breakdown of certain milk oligosaccharides has been shown to produce bioactive compounds which have beneficial effects on gut and immune function development (15). In addition, the development of tolerance to benign antigens is as critical as the development of active immune responses to potential pathogens. Exposure to antigenic signals from maternal milk microbes may be a mechanism to develop immune tolerance to these benign and sometimes beneficial microbes (16). These suggested functions of milk microbes are not exclusive; all could be important, and other potential functions may yet be discovered.
Host-associated microbiomes are ecological systems, and like any multispecies assemblage, they are structured by some combination of deterministic and stochastic processes. Deterministic processes are generally considered those that involve nonrandom, niche-based mechanisms such as environmental filtering (e.g., pH, moisture) and species interactions (microbe–microbe, host–microbe). Stochastic processes are generally considered those that involve ecological drift and probabilistic dispersal (e.g., random changes in communities due to birth, death, and reproduction and random chance of colonization, respectively), and are indistinguishable from random, neutral-based processes (17). It is now generally accepted that both deterministic and stochastic processes simultaneously structure ecological communities but vary in their relative importance depending on the system. In host-associated microbial communities, community assembly processes are generally understudied (18), but are gaining attention (17, 19). By applying ecological theory to host microbiomes, we can increase our ability to explain inter- and intraspecies variation (18, 20).
Deterministic processes such as host evolutionary history and environment can shape mammalian host-associated microbial communities, as has been shown for skin (21), oral (22), and gut microbiomes (23, 24). Host species generally differ in their microbiomes and sometimes these differences parallel differences in host evolutionary history. Phylosymbiosis describes when more closely related hosts have more similar microbiomes (25) and has been observed in mammalian gut microbiomes (24) and skin microbiomes (21). However, ecological traits that mirror evolutionary history may explain phylosymbiosis (20), such as when host diet co-varies with phylogeny (26). For instance, diet can act as an ecological filter on the gut microbiome likely through filtering dietary-associated microbes for species that aid in digestion (26). Additionally, the environment has long been regarded as a strong ecological filter, selecting specific ecological communities from the regional pool of species. Host-associated microbial communities are unique in that these filters are acting at both the host level and the host body site level, such that the environment of the host affects the regional pool of microbial species (e.g., in a forest) and the environment of the host’s body site for which the microbes will colonize acts as a secondary ecological filter (e.g., gut pH). For instance, we have previously shown that where an individual lives influences their milk microbiome (27) and that milk nutrient content (milk fat, milk sugar, and milk protein) is related to species differences in milk microbiome composition in primates (5).
Our objective was to generate a comprehensive milk microbiome dataset for class Mammalia to determine the relative contribution of host evolutionary history, diet, environment, milk nutrient content and lactation stage on structuring milk microbiomes. We utilized samples from the world’s largest collection of mammalian milks housed at the Smithsonian National Zoo and Conservation Biology Institute (NZCBI) Milk Repository. We selected samples in a manner to reduce bias by selecting species within the four superorders of placental mammals (Placentalia) that represented different environments, diets, milk nutrient contents, and that were collected, when possible, at both early and mid-lactation stages (Fig. 1).
Fig. 1.
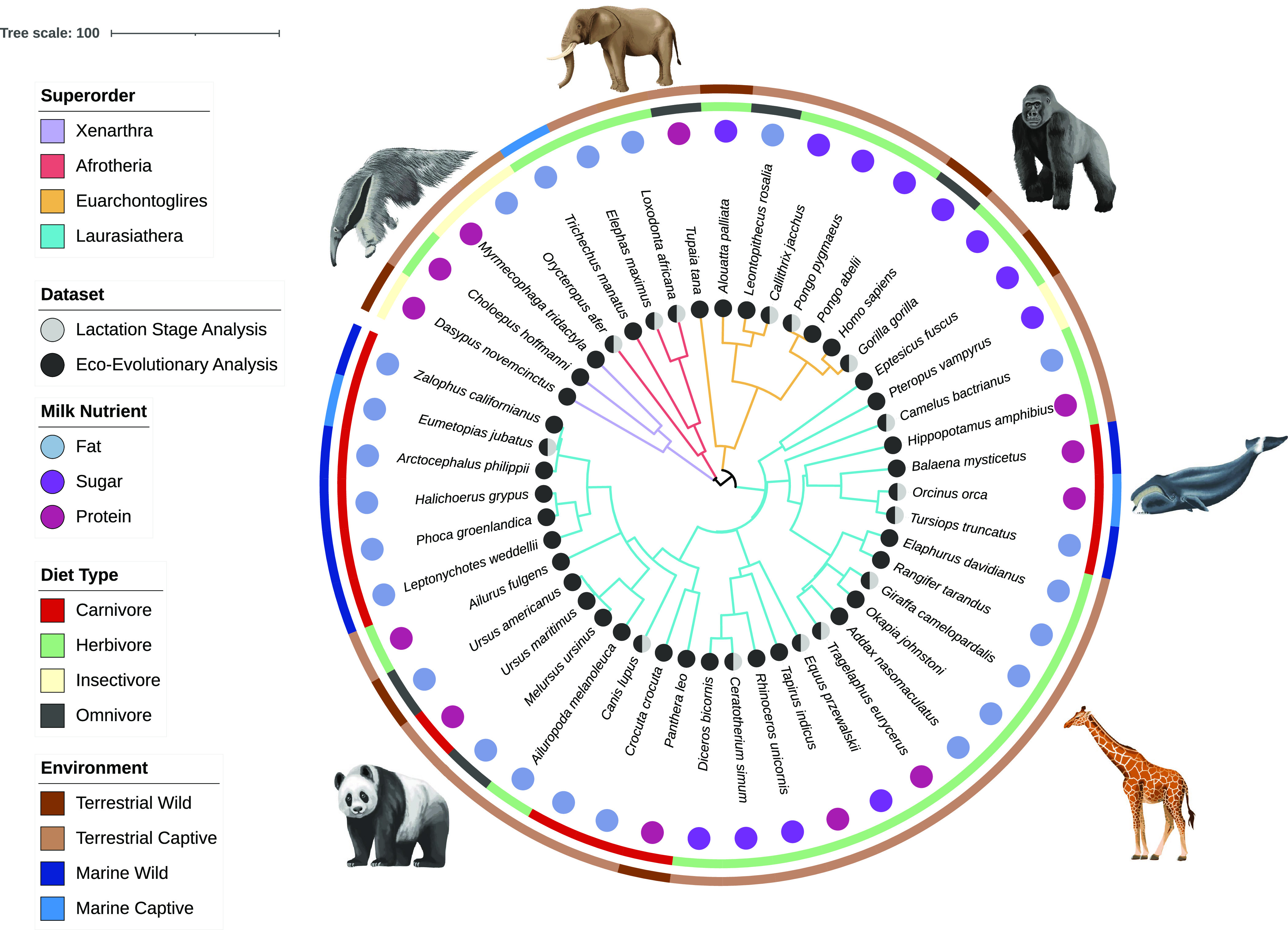
Host phylogenetic tree representing our experimental design. Host environment is shown in the outer ring, diet type in the inner ring, milk nutrient category as letters and branches colored by superorder. Two data subsets (represented by circles) were analyzed: i) a repeated measures (lactation stage) dataset of 21 females from 15 species sampled at an early and a mature lactation stage to quantify the effect of lactation stage on milk microbiomes, and ii) an independent measures (ecoevolutionary) dataset of 83 females from 47 species to examine host superorder, diet, environment and milk nutrient content impact on milk microbiomes. The host phylogenetic tree (rooted and ultra-metric) was created using TimeTree (28) and visualized with Interactive Tree of Life (iTol) v6 (29).
Results
Overview.
We generated 317,041 high-quality bacterial and archaeal 16S sequences (V3-V5 region) from 107 mammalian milk samples housed in the NZCBI’s Milk Repository (SI Appendix, Table S1). An additional 16 samples were processed, but did not yield sufficient sequences for analyses. The complete mammal milk microbiome dataset consisted of 13,413 microbial amplicon sequence variants (ASVs) from 26 bacterial phyla and 2 archaeal phyla. The dominant bacterial phyla were Actinobacteria (18.5%, 2,409 ASVs, 106 samples), Bacteroidetes (9.8%, 2,501 ASVs, 104 samples), Firmicutes (23.3%, 2,648 ASVs, 102 samples), and Proteobacteria (41.3%, 3,323 ASVs, 106 samples). Archaea were rarer, but detected in individuals from all superorders, diet types and environments; the archaeal phyla found were Crenarchaeota (0.6%, 151 ASVs, 33 samples) and Euryarchaeota (0.02%, 12 ASVs, 6 samples). The distribution of microbial phyla (SI Appendix, Fig. S1A) and genera (SI Appendix, Fig. S1B) differed greatly across host taxa. No microbial ASVs were identified as being core mammal milk microbes (in 90% of samples); only one ASV was found in at least 40% of samples, which was a Staphylococcus sp. (ASV16). This was consistent with no core bacteria found within superorders and diet types, other than ASV16 (Staphylococcus sp., core in Xenartha) and ASV141 (Lactococcus sp., core in Insectivores).
We found that microbiome structure was similar between early and mature lactation stages in the repeated measures dataset of 21 females from 15 species (SI Appendix, Table S2). Lactation stage did not influence bacterial ASV richness or phylogenetic diversity (SI Appendix, Fig. S2A; Linear Mixed Model Wald X2 P = 0.62 and P = 0.3) or bacterial composition (SI Appendix, Fig. S2B; Bray–Curtis PERMANOVA: Pseudo F1,41 = 0.58, P = 0.81). We then identified the effect of host superorder, diet, environment and milk nutrient content on milk microbiomes (SI Appendix, Table S3) with an independent-measures dataset of 83 females from 47 species. For all composition analyses, we report Bray–Curtis distances only, but found similar results with Jaccard and unweighted UniFrac unless otherwise noted.
Ecoevolutionary Factors Shape Milk Microbiome Structure.
We first assessed the effects of host superorder, diet, environment and milk nutrient categories on milk microbiome structure. Milk microbiome structure (alpha and beta diversity) consistently differed among environments and diet types, while milk microbiome composition (beta diversity) also differed among superorders and milk nutrient categories. Among all independent females sampled (n = 83), the average number of microbial ASVs present in mammalian milk was 210 (SD ± 152). Among environments, captive marine mammals had the lowest microbial richness and phylogenetic diversity in their milk, which was significantly lower than captive terrestrial mammals (Fig. 2A; richness ANOVA F3,71 = 3.1, P = 0.032, post hoc P = 0.08; phylogenetic diversity ANOVA F3,71 = 3.67, P = 0.016, post hoc P = 0.024). Among diet types, herbivores had the highest microbial richness and phylogenetic diversity in their milk, which was significantly higher than carnivores (Fig. 2A; richness ANOVA F3,71 = 9.18, P < 0.001, post hoc P < 0.001; phylogenetic diversity ANOVA F3,71 = 4.49, P = 0.006; post hoc P = 0.013). Milk microbiome composition differed among all superorders, environments, diet types, and milk nutrient categories, which in total accounted for explaining 20.3% in microbiome variation (Fig. 2 B and C and SI Appendix, Fig. S3; Bray–Curtis PERMANOVA: superorder Pseudo F3,71 = 1.78, R2 = 6.0%, P = 0.001; environment Pseudo F3,71 = 1.96, R2 = 6.6%, P = 0.001; diet type Pseudo F3,71 = 1.38, R2 = 4.7%, P = 0.001; milk nutrient category Pseudo F3,71 = 1.34, R2 = 3.0%, P = 0.001; all pairwise < 0.05). We then analyzed how ecoevolutionary factors affected milk microbiomes individually and through interactions.
Fig. 2.
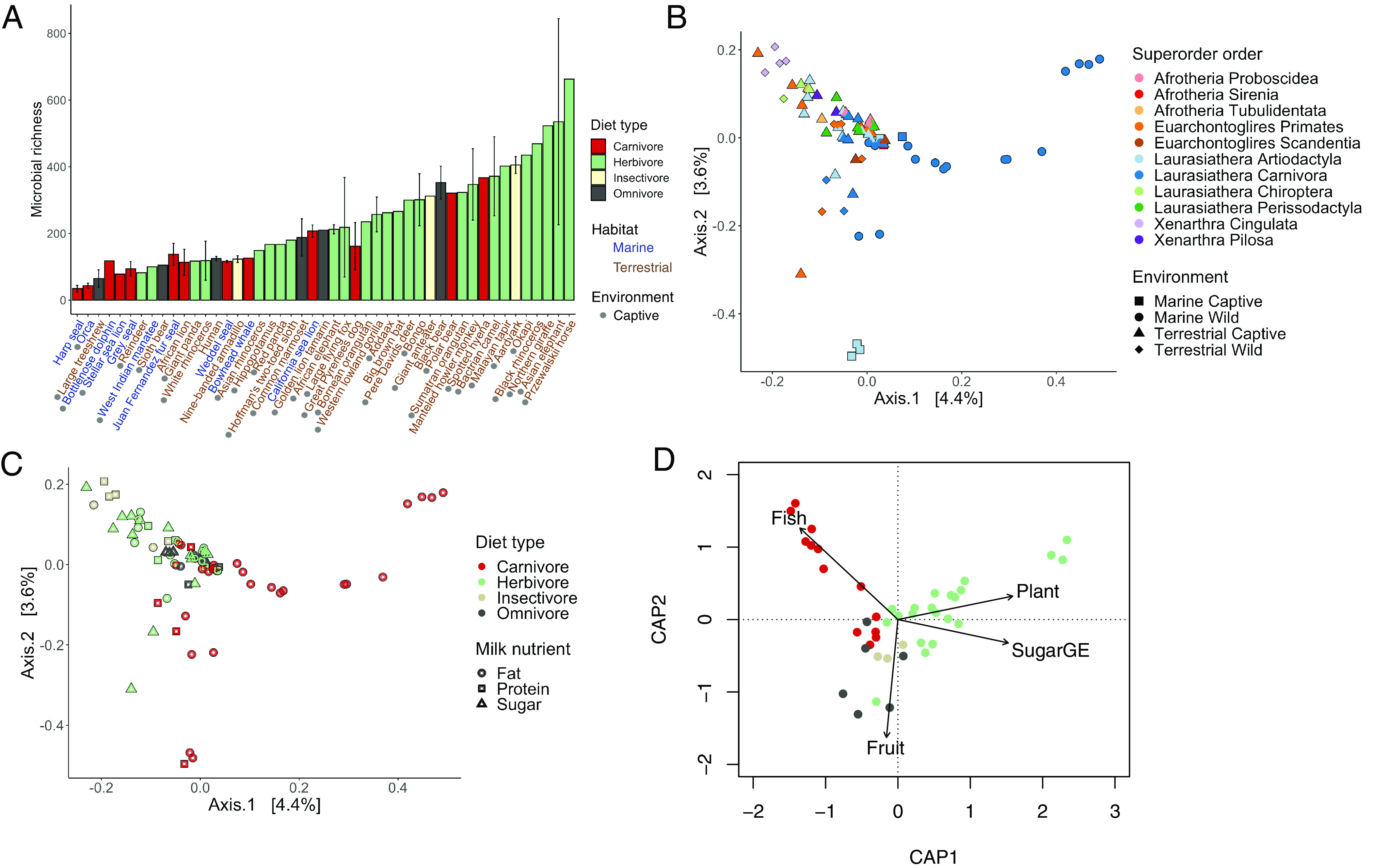
Milk microbiome structure explained by deterministic processes. (A) Captive terrestrial mammals had greater bacterial richness than captive marine mammals. Bottlenose dolphins displayed in figure as captive (n = 2 captive, n = 1 wild). Herbivores had greater bacterial richness and phylogenetic diversity than carnivores. Mammalian milk microbiome composition covaries with (B) superorder and environment, as well as (C) diet type and milk nutrient category as show with principal coordinate analysis of Bray–Curtis distances. (D) Milk microbiome composition changed in a linear manner with host consumption of fish, plants, and fruit, and the amount of sugar GE in milk (based on Bray–Curtis composition measure and distance-based linear models).
Individual and Interactive Effects of Ecoevolutionary Factors on Milk Microbiomes.
We examined if there was a relationship between host phylogeny and changes in microbial composition using phylosymbiosis analyses. Although closely related species tended to have more similar microbiomes, there was no evidence for phylosymbiosis (SI Appendix, Fig. S4). We found no evidence of phylosymbiosis between milk microbiomes and host phylogeny (Robinson–Foulds = 0.91) or host divergence time (Jaccard Mantel test, P = 0.086). We also verified within superorders and diet types with sufficient sample size (n > 7: Euarchontroglires, Laurasiathera, Herbivore, and Carnivore) that there was no indication of phylosymbiosis (Jaccard Mantel test, P = 0.074, P = 0.39, P = 0.33, P = 0.34).
Then, we assessed the individual and collective contributions of host phylogeny and quantitative traits of diet items (based on Elton dietary traits) and milk nutrient content [milk sugar gross energy (GE) in mg/kcal (GE) and milk protein GE] on milk microbiome composition using multiple regression on dissimilarity matrices (MRM) (30–32). It is important to note that we did not directly quantify maternal diet, but used Elton traits to represent dietary components of each species. All three variables were significant (Bray–Curtis: P = 0.001), with their shared variance explaining 2.6% of variation and their individual variance explaining 5.4% (Bray–Curtis: dietary items 3.2%, phylogeny 1.7%, milk nutrient content 0.5%; SI Appendix, Table S4 and Fig. S5 Venn diagram). We found similar results for Jaccard measures, but not for UniFrac (only diet was significant and explained 1.3%).
To uncover direct and indirect effects of diet and milk nutrient content on milk microbiome composition we used quantitative traits of dietary items and milk nutrient content in distance-based linear modelling (DBLM) and structural equation modeling (SEMs). In DBLMs, we determined whether individual milk nutrient content (sugar GE or protein GE) or individual dietary items (invertebrate, mammal/bird, fish, scavenged, fruit and plant) were linked to milk microbiome compositional changes. Milk microbiome composition consistently changed in a linear relationship as mammalian hosts ate more fish and plants (Fig. 2D and SI Appendix, Fig. S6; Jaccard DBLM: fish P = 0.002, plant P = 0.002; Bray–Curtis DBLM: fish P = 0.004, plant P = 0.04). Likewise, changes in the amount of milk protein GE, milk sugar GE and fruit consumption showed linear relationships with milk microbiome composition, but this depended on the composition measure (SI Appendix, Fig. S6; Jaccard DBLM: milk protein GE P = 0.002; Bray–Curtis DBLM: milk sugar GE P = 0.002, fruit P = 0.004). Dietary items of invertebrates, mammals/birds, and scavenged did not show any indication of linear relationships with composition. In SEMs, we tested whether diet (principal component axis 1 of Elton traits as used in DBLM) directly impacted microbiome structure (species richness, phylogenetic diversity, Bray–Curtis, UniFrac) or indirectly impacted microbiome structure via individual milk nutrient components (milk sugar GE or milk protein GE). We found diet to significantly influence milk sugar GE (standardized coefficient = ± 0.52, P < 0.001), but not milk protein GE (Fig. 3); reflecting that herbivores generally have more sugar GE in their milk and also higher species richness (SI Appendix, Fig. S7). Diet directly influenced microbial species richness, a pattern that was observed in earlier analyses with herbivores having the highest microbial richness and carnivores the lowest (Fig. 2A). Diet indirectly influenced microbial composition through milk sugar GE (UniFrac: standardized coefficient = ± 0.22, P = 0.03; Bray–Curtis: standardized coefficient = ± 0.46, P < 0.001). Protein had a direct effect on milk microbial phylogenetic diversity and composition (Faith’s phylogenetic diversity: standardized coefficient = ± 0.285, P = 0.01; Unifrac: standardized coefficient = ± 0.447, P < 0.001; Bray–Curtis: standardized coefficient = ± 0.268, P = 0.007).
Fig. 3.
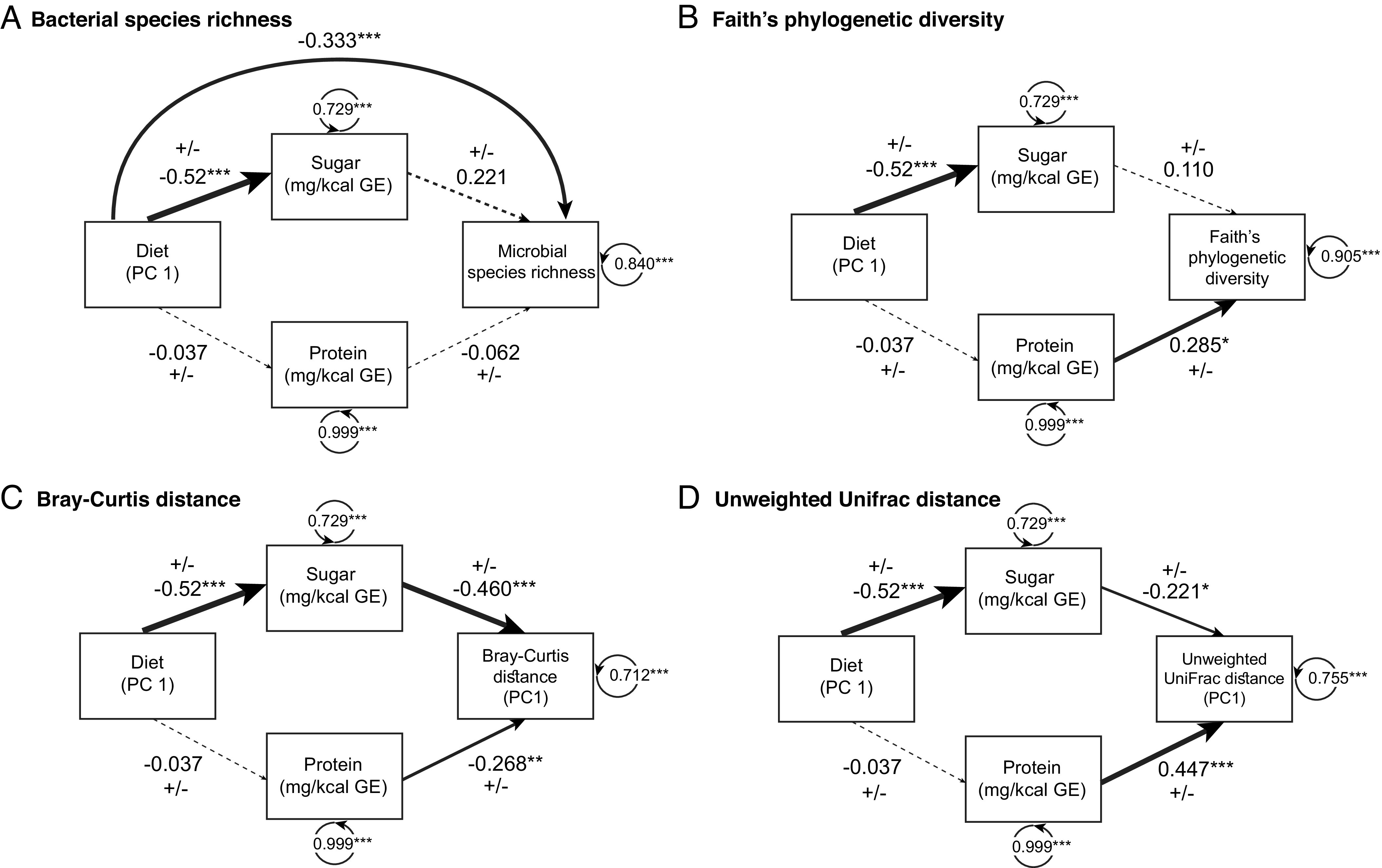
Direct and indirect effects of diet and milk nutrient content on milk microbiome structure using structural equation modeling. Diagrams represent models used in structural equation modeling. In all models, diet influences milk sugar GE. (A) Diet directly influenced microbial species richness. (B–D) Milk protein GE influenced microbial diversity and composition (Faith’s phylogenetic diversity, Bray–Curtis, and UniFrac) and (C and D) diet indirectly influenced microbial composition via milk sugar GE (Bray–Curtis and UniFrac). Principal coordinate axis 1 (PC1) values were analyzed for both metrics of composition (Bray–Curtis and UniFrac) and principal component axis 1 (PC1) for Elton dietary traits. Line thickness is scaled in proportion to the associated effect size, with dashed lines representing non-significant paths. ± symbols indicated that at one variable in the path is represented by a principal coordinate axis, which has positive and negative values that are reversible. Residual variance of observed variables listed within circular arrow.
We determined whether any abundant microbial ASVs (n = 135 ASVs) were related to milk sugar GE and milk protein GE amount. We found six bacterial ASVs (Rothia sp. ASV 154, Rothia sp. ASV2, Streptococcus sp. ASV31, Acinetobacter sp. ASV 259, Actinobacillus sp. ASV 126, Streptococcus sp. ASV 45) whose abundance positively correlated with milk sugar GE and one bacterial ASV (Solibacillus silvestris ASV 347) positively correlated with milk protein GE (SI Appendix, Table S5 and Fig. S8), after adjusting for multiple comparisons.
Community Assembly Processes in Mammalian Milk, Gut, and Skin Microbiomes.
We assessed microbial community assembly processes using a null model-based approach (33, 34). We found that ecological drift, dispersal limitation, and environmental selection-guided assembly of milk microbiomes, creating dissimilarity between individuals’ milk microbiomes. Processes generally considered stochastic (17, 33, 34) had the largest influence on milk microbiome assembly: Drift accounted for 53% of all pairwise community comparisons; dispersal limitation accounted for 26% and homogenizing dispersal 1%. We accessed previously published datasets on mammalian gut (24) and skin (21) microbiomes, subset to similar number of samples from similar number of taxonomic groups (SI Appendix, Table S7), and used the same modeling approach to estimate their community assembly processes. Mammalian milk microbiomes experienced the highest ecological stochasticity, accounting for 80% of all pairwise community comparisons, in community assembly mechanisms compared to 69% for mammalian gut microbiomes and 45% for mammalian skin microbiomes (Fig. 4A). In contrast, deterministic processes of species sorting or environmental selection accounted for 20% in milk microbiomes, followed by gut microbiomes at 40% and skin microbiomes at 55%. The high ecological stochasticity in milk microbiomes may be a reflection of their lower microbial diversity (Kruskal-Wallis, X2 = 55.9, df = 2, P < 0.001) compared to gut and skin microbiomes (pairwise Wilcoxon rank sum test P < 0.001; Fig. 4B). Collectively, we explained all of the deterministic processes occurring in mammal milk microbiome composition with superorder, environment, diet type and milk nutrients explaining 20.3% of the variation in microbiomes among individuals (Figs. 2 and 5) as reported earlier.
Fig. 4.

Comparative analysis of milk microbiomes versus mammalian gut (24) and skin (21) microbiomes. (A) Relative influence of assembly mechanisms shaping mammalian milk, gut, and skin microbiomes. We used null model analysis to compare phylogenetic beta diversity distances from each bacterial community to random phylogenetic trees (33, 34). Comparison calculations indicated how community assembly processes influenced microbial variation. (B) Milk microbiomes had the lowest microbial diversity, which may partially explain high ecological stochasticity in milk microbiomes, while skin and gut microbiomes did not differ from one another. *** indicates p < 0.001 from pairwise Wilcoxon rank sum test.
Fig. 5.

Host evolutionary history, environment, diet, and milk nutrient content contribute to milk microbiome structure, yet much of milk microbial assembly is due to random, stochastic processes. Null model analysis assigned 80% of variation to stochastic processes and 20% to deterministic processes. We collectively explained all deterministic processes in milk composition with phylogeny, diet, milk nutrient content, and lactation stage (tested via PERMANOVA). Modifying diet and environmental conditions can affect milk composition, thus impacting offspring gut microbiome colonization and health.
Discussion
We demonstrate that stochastic and deterministic processes influence milk microbiome community assembly. Ecological drift had a large influence on milk microbiomes. Microbial taxa in milk microbiomes likely originate from gut, skin, and oral microbiomes through enteromammary trafficking (9), skin contact (10) and retrograde flow from infant oral cavity (11). Ecological stochasticity may be compounded in milk microbiomes given the multiple contributing sources of microbial taxa. Nonetheless, milk microbiomes still showed signatures of environmental selection, accounting for 20% of variance, that reflected the mother’s evolutionary history and ecology. We also observed similar microbiomes at early and mature lactation stages, suggesting similar ecoevolutionary processes are controlling milk microbiome composition across lactation. Previous studies have found variation in milk microbiomes across lactation in some species, namely primates (5, 12, 27), suggesting that temporal variation in milk microbiomes occurs in some species and may best be detected with greater longitudinal sampling resolution within specific species or animal groups. Our study encompasses hosts across the mammalian tree and lends itself to broad comparisons across lactation, in which we found that factors such as evolutionary history, external and internal environments, and stochasticity shape the mammalian milk microbiome throughout lactation.
Deterministic processes guiding milk composition included the superorder of the host species. Yet, we found no evidence of phylosymbiosis in milk microbiomes, in contrary to patterns seen in mammalian gut (23, 24) and skin microbiomes (21). The lack of phylosymbiosis may be a result of the low microbial diversity in milk and the increased stochasticity in the assembly of complex microbial communities (35) or it may reflect our sampling design, which we strategically picked samples to represent multiple environments, diet types and milk nutrient contents to limit ecological filtering covariation with evolutionary history (20). We conclude that host evolutionary history is a selective force on which microbes exist in milk, but it is not solely based on how distantly related species are. Diet, environment, and high levels of stochasticity/drift were likely mechanisms decoupling host evolutionary change from milk microbiome change.
The external environment of the host and the internal nutritional environment of the host’s mammary gland were linked to milk microbiome structure. Regional microbial pools and niche preference or nutritional constraint may explain the mechanism behind environmental conditions shaping milk microbial structure. We found that marine mammals had the lowest milk microbial diversity. The marine environment is microbially diverse, consisting of a few abundant taxa and many rare, locally endemic taxa (36). Limited dispersal of marine microbes due to seawater stratification (37) may contribute to restricted microbial pools and thus low milk microbial richness and high dissimilarity among marine mammal milk microbial composition. Milk microbial composition varied by all environments (captive, wild, marine, and terrestrial) highlighting the role of regional microbial pools on microbiome structure. Mammalian gut and skin microbiomes are influenced by the environment, including captivity (38), geographic location (39), and co-habitation (40), suggesting that microbial symbionts are acquired from the external environment such as community contact (number of individuals housed together) and contact with novel substances (dietary diversity, antibiotics, medical care, and human contact). Our results indicate milk microbiomes are similarly influenced by environmental conditions.
Mammalian hosts also vary in milk nutrient content (milk fat, milk sugar, and milk protein), which may lead to filtering of microbes in milk via nutritional constraints. Milk microbes may be selected on their ability to feed off dominant milk nutrients or incorporate nutrients into their cellular structure (41). Milk sugar and milk protein had linear relationships with milk microbial composition, indicating that mammals with similar milk nutrient profiles harbor more similar milk microbiomes. It is important to note that milk fat was correlated with milk sugar and milk protein in our dataset, and we cannot differentiate the effect of milk protein and milk sugar from milk fat. Additionally, we found six bacterial taxa whose abundance correlated with milk sugar GE and one with milk protein GE that merit further study. Milk nutrient content may reflect ecological filtering via nutrient utilization and constraints. Previous studies have demonstrated milk microbial composition to be associated with milk fatty acid profiles in humans (41) and milk fat and milk protein profiles in humans and other primates (5). Our results provide additional evidence for bacterial selection processes by milk nutrient content and that this likely occurs widely across the mammalian tree of life.
Our analytic method to measure milk sugar content measures total sugar and cannot distinguish between free glucose and galactose, lactose, and other larger oligosaccharides. The proportion of sugar as oligosaccharides as opposed to lactose varies widely in mammals (42). Milk oligosaccharides vary substantially in molecular weight, carbohydrate composition and structure and can have antibiotic, prebiotic and osmotic functions (43, 44). Many pathogenic microbes obtain access to cells by attaching to oligosaccharide residues on cell membranes. When these microbes attach to free oligosaccharides in milk, they remain bound and will transit the neonatal gut until being excreted in the feces. Some oligosaccharides can be metabolized by select microbes. This has been shown in detail for human milk oligosaccharides (43). Thus, some milk oligosaccharides serve as food for specific benign or even beneficial microbes giving them a boost in colonizing the neonatal gut. Metabolic products from the breakdown of oligosaccharides have been shown to have important beneficial effects on the neonate (45, 46). Finally, low molecular weight oligosaccharides produce an osmotic gradient, in the same manner as does lactose, which draws intracellular water into the milk. In species such as sea lions, which produce no lactose (47), small oligosaccharides likely serve as the main mechanism to bring water into the milk. Future research on the mechanisms structuring milk microbiomes should consider investigating the potential roles of milk oligosaccharides.
Diet directly and indirectly influenced milk microbial diversity and composition. Diet directly impacted the number of microbial species in milk, with herbivores having higher bacterial richness and diversity than carnivores. We also observed that diet uniquely explained up to 3% of variance in microbial composition in MRMs, and particularly that the amount of plant and fish (and fruit to a lesser extent) in diet was directly linked to turnover in communities in DBLMs. Diet had an indirect effect on milk microbiome structure via milk sugar GE content. Maternal diet is one component by which milk nutrient composition varies (3, 4), and in turn we found milk sugar GE content influenced milk microbiome composition. Our findings suggest that particular microbial taxa may colonize milk based on maternal dietary choice and on their ability to utilize dominant milk nutrients (48).
One direct mechanism by which microbes may colonize milk and feed on milk nutrients includes enteromammary trafficking. The enteromammary pathway is hypothesized to be a mode by which specific bacterial taxa move from the maternal gut to the mammary gland and then to infant gut postnatally (49–51). Evidence from mouse models suggests that live commensal bacteria coated with immunoglobulin A reside in dendritic cells in gut-associated lymphoid tissue, particularly the mesenteric lymph node and Peyer’s patch (50, 52), and they are trafficked from these lymphoid tissues through the bloodstream to the mammary gland (9). Evidence for this pathway also exists in a human study in which lactating women given oral supplements of Lactobacillus had increases in the same strain in their milk (53). We found 50 Lactobacillus ASVs in 24 mammalian species and 28 Bifidobacterium ASVs in 17 mammalian species, indicating that these probiotics could be similarly examined in future experimental studies; specifically in host species with both probiotic genera, which in our dataset included: Aardvark, Nine-banded armadillo, Bongo, Bactrian camel, Great Pyrenees dog, Western lowland gorilla, Golden lion tamarin, humans, Common marmoset, Bornean orangutan, and Hoffman’s two-toed sloth. On contrary to the enteromammary pathway, Petrullo et al. (54) found that vervet monkey gut microbiomes are no more similar to the milk microbiome within individuals as between individuals (54). We hypothesize that this result may be a reflection of high stochasticity in milk microbiomes. Our study shows that while gut and milk microbiomes may differ, broad dietary categories (omnivore, carnivore, and herbivore) and specific dietary items (fish and plants, and fruit to a lesser extent) directly impacted milk microbiome composition. Together, these results are consistent with an entero-mammary pathway and suggest gut microbes associated with specific diets may be passed from the gut to the mammary gland. The enteromammary pathway is one of a series of milk inoculation pathways, with others including passive transfer from maternal and infant skin microbiomes and retrograde flow transfer from infant oral microbiomes. These pathways are likely common across mammalian taxa, but likely differ in contribution to seeding mammary glands based on nursing frequency and duration, fur type, and infant oral cavity structure.
We demonstrate that stochastic and deterministic processes influence milk microbiome community assembly. Ecological drift, reflecting the random chance of microbial birth, death, and reproduction rather than niche preference, had the greatest influence on milk microbiomes compared to mammalian gut and skin microbiomes. Milk microbiomes had the lowest microbial diversity, which may partially explain the high ecological drift (35, 55). We also found that gut microbiomes experienced greater drift than skin microbiomes, and this may reflect transient dietary associated microbes in gut microbiomes (56). All the host-associated microbial communities examined (milk, gut, and skin) had low homogenizing dispersal as seen in other gut metacommunities (34), but contrasting to free-living bacterial metacommunities (33). This suggests hosts limit mass homogenizing effects. Microbial taxa in milk microbiomes likely originate from gut, skin, and oral microbiomes, which may compound ecological stochasticity given the multiple contributing sources of microbial taxa.
When working with lower microbial biomass samples, such as milk, steps to limit contamination are important. Contrasting findings have motivated best practices when analyzing microbial communities of extremely low to non-existent microbial biomass samples such as in placenta and brain tissue (57). However, milk samples are much higher in microbial quantity than such extreme cases and estimates in humans and livestock estimate microbial quantities of approximately 1 × 105 to 1 × 107 colony-forming units/mL of focal bacterial taxa (58). Regardless, we followed best practices throughout laboratory and postsequencing analyses, including working in clean, controlled laboratory spaces, sample randomization, negative controls in extraction and in PCR, mock communities in extraction, and postsequencing techniques that identify contamination such as the R package decontam (16, 57). We could not control for variation in sampling across time and space given that these samples were often collected by independent researchers to deposit in the Smithsonian Milk Repository for a multitude of different projects over many years. We acknowledge that this variation in sample collection (e.g., multiple collectors, different years) may have increased the stochasticity observed in milk microbiomes, but would be highly improbable to contribute to the deterministic processes we observed (e.g., effects of diet). We encourage future work to adhere to recommendations put forth as techniques (e.g., ref. (57)) to improve characterization of reproductive microbiomes (16).
Milk is essential to neonatal health and development, providing nutrients and immune factors. We show that across Mammalia, milk is providing bacterial and archaeal symbionts to offspring. Our study is the first to uncover the processes involved in structuring milk microbiomes across the mammalian tree of life. We were able to identify factors contributing to deterministic processes including host evolutionary history, environment, diet, and milk nutrition. Stochastic processes drive much of the milk microbiome composition, as well as gut and skin microbiomes, albeit to a lesser extent. The strong influence of stochastic processes explains why manipulating microbiomes is challenging and often unsuccessful. Notably, our results are consistent with entero-mammary trafficking, representing a mechanism by which microbes are transferred from the mother’s gut to mammary gland and then to offspring postnatally. Mediating the milk microbiome through the maternal diet may offer subtle effects that influence offspring gut microbiomes and health (48). Together, we demonstrate that applying community ecological theories to microbiome science yields valuable information on how microbial communities assemble and provides perspective on developing therapeutics, such as probiotics, which could be used for mammals in human care (16).
Material and Methods
Data Overview.
Milk samples were used from the Department of Nutrition Science’s Milk Repository at NZCBI. Our study comprised 107 milk samples from 47 species (SI Appendix, Table S1) representing four mammalian superorders (Afrotheria, Euarchontoglires, Laurasiathera, and Xenarthra), four diet types (carnivore, herbivore, insectivore, and omnivore), four environments (marine captive, terrestrial captive, marine wild, and terrestrial wild) and three nutrient content categories (protein, fat, and sugar; SI Appendix, Fig. S9). We defined early lactation samples as coming from the first quarter of the average length of lactation, and mature lactation samples as the second quarter prior to weaning, declining milk yields, or offspring primarily consuming food as supplement (59). Researchers that provided samples to the milk repository were provided general guidelines on milk collection, which included following general standard operating procedures for handling animals (e.g., wearing gloves), washing the nipple area with sterile water, using oxytocin for animals that require anesthetization for sample collection, not collecting any milk that was in contact with skin, and freezing milk samples following collection. All samples were maintained in a −20 °C freezer since deposition into the milk repository, with limited freeze–thaws.
We analyzed two data subsets to properly account for pseudoreplication in the complete dataset: i) a repeated measures dataset of 21 females from 15 species sampled at an early and a mature lactation stage to quantify the effect of lactation stage on milk microbiomes (SI Appendix, Table S2) and i) an independent measures dataset of 83 females from 47 species to identify the effect of host superorder, diet, environment and milk nutrient content on milk microbiomes (SI Appendix, Table S3).
Molecular Methods and Sequencing.
We extracted DNA from 300 µL of milk using Qiagen ProFecal DNA Extraction Kit. Prior to following the manufacturer’s instructions, milk samples were centrifuged at 10,000 g for 10 min, the supernatant discarded and the remaining pellet extracted (12). Negative extraction controls (n = 6), negative PCR controls (n = 3) and one positive mock control (ZymoBIOMICS D6300) were included. We targeted the 16S rRNA gene with primers 515F-Y and 939R, performed a two-step PCR library prep, pooled clean libraries in equimolar amounts, and size selected our desired amplicon size following methods outlined in Keady et al. (39), with one modification: increase in annealing temp to 68 °C, which improved amplicon DNA yields. We sequenced the pooled library on an Illumina MiSeq (2 × 300 bp kit) at the Center for Conservation Genomics at NZCBI.
Sequence Data Processing.
Data processing and subsequent analyses was conducted in R (v 4.0.3). We quality filtered sequence data using dada2 package and identified bacterial and archaeal ASVs (60, 61). Taxonomy was assigned to ASVs using the Ribosomal Database Project classifier (62). We built a phylogenetic tree using FastTree (63) in QIIME2 (64), and combined data files for further analysis using the phyloseq package (65). Contaminants were filtered with the decontam package using the “either” method to identify contaminants (66). We filtered singletons, ASVs not assigned to Bacteria or Archaea, and ASVs in class Chloroplast. We rarified samples to 3,000 total reads per sample given high variation in sequencing depth (SI Appendix, Fig. S10).
Milk Nutrient Content.
We assayed milk samples for nutrient content at the NZCBI Nutrition Laboratory. Samples with sufficient volume and part of the independent measures dataset (n = 64) were assayed for macronutrient composition (% crude protein, % fat, % sugar) using standard methods developed at NZCBI (67) and detailed in Power et al. (68). Protein GE, fat GE, and sugar GE were calculated in mg/kcal GE from those percentages and were used in cluster analysis to create milk nutrient categories; see SI Appendix for additional detail.
Measures of Microbiome Structure.
We used alpha diversity (ASV richness and Faith’s phylogenetic diversity) (69) and beta diversity measures (Bray Curtis, Jaccard, and unweighted UniFrac distances) to quantify milk microbiome structure. We performed square root and log transformations on alpha diversity measures when appropriate to improve normality. We report Bray–Curtis distances (except for phylosymbiosis analyses; see below), but found similar results with Jaccard and unweighted UniFrac unless otherwise noted. We verified that our explanatory variables were not confounded by examining generalized variance inflation factors (70) using the package car. We were unable to use host order as a factor as it was confounded with diet type in our ecoevolutionary analysis.
Lactation Stage Analysis.
We quantified the effect of lactation stage on milk microbiome structure using a repeated measures dataset of 21 independent females (15 unique species) sampled at early and mature lactation stages that represented a diversity of individuals from superorders, environments, and diets (n = 42; SI Appendix, Table S2). For alpha and beta diversity, we used linear mixed models and PERMANOVAs, respectively with animal ID as a random effect.
Ecoevolutionary Analysis.
We quantified the effect of superorder, environment, diet type, and milk nutrient content on milk microbiome structure. For alpha diversity, we used ANOVAs and conducted post hoc analyses using Tukey Honest Significant Differences method. For beta diversity, we used PERMANOVAs. PERMANOVA results are sensitive to unbalanced designs (71, 72); we conducted a subsequent analysis using a balanced dataset, which supported all of our findings presented herein. We tested dispersion for explanatory variables for each microbial measure and in both datasets using function betadisper (package vegan) (73) (SI Appendix, Table S6). We then examined individual and combined effects of ecoevolutionary factors on milk microbiomes.
Individual and Interactive Effects of Ecoevolutionary Factors.
We quantified phylosymbiosis between host phylogeny and microbial dendrograms using two methods: topological congruence between host phylogenies and milk microbiome dendrograms using normalized Robinson–Foulds (21) and Mantel tests with Pearson correlation between matrices of host divergence time and Jaccard microbiome dissimilarities (24). We built a rooted and ultra-metric host phylogenetic tree (newick format) using TimeTree (28). We randomly selected a single sample per host species (n = 47) and created microbiome dendrograms and distance matrices with Jaccard distances as in Song et al. (24). We also subset within superorder and diet to test for phylosymbiosis at lower taxonomic scales as in Ross et al. (21).
We assessed diet and milk nutrient effects on milk microbiomes, using quantitative traits as opposed to categories used in the prior analyses. We quantified the effects of milk nutrient content and dietary items (n = 45) on milk microbiome composition. For milk nutrient content, we used raw sugar GE and protein GE values for 73 individuals for which we had these data. For dietary items, we identified percent dietary content from the EltonTraits 1.0 database (74); we only included dietary categories with at least five data points in our dataset (Invertebrates, Mammals/birds, Fish, Scavenged, Fruit, and Plant) and one individual per species (n = 47) following Song et al. (24). The EltonTrait database did not include Przewalski horse (Equus ferus przewalskii), however we verified its diet composition with the NZCBI’s senior nutritionist M. Maslanka.
We evaluated individual and shared variance of host phylogeny, dietary traits (Elton traits), and milk nutrient content on milk microbiome structure using variance partitioning (ecodist package) (30–32). We analyzed samples with complete nutrient content data for milk sugar GE and milk protein GE (n = 73). Results were visualized with package eulerr (75). We also assessed the relationship between individual milk nutrient components and dietary items (Elton Traits) with microbiome composition using DBLMs (package vegan, function capscale) (73). The dataset included one individual per species with complete milk nutrient data (n = 45). We selected our final model using forward model selection (package vegan, function ordiR2step) (73).
We tested direct and indirect effects of quantitative dietary items (Elton Traits) on milk nutrient content and microbial structure with structural equation modeling (package lavaan) (76). We tested three a priori models with four metrics of microbial structure (species richness, Faith’s phylogenetic diversity, Bray–Curtis, and unweighted UniFrac) (SI Appendix, Fig. S11). Briefly, our three models are i) a full model representing all possible relationships between variables, ii) model 2 without a bidirectional relationship between milk sugar GE and milk protein GE, and iii) model 3 assumes no direct relationship between diet and milk microbiome structure (SI Appendix, Fig. S11). Samples with milk sugar GE and milk protein GE were analyzed (n = 73). Milk fat GE was not included due to covariance with milk protein and milk sugar (SI Appendix, Supporting Text). Distance matrices were created for microbial composition (Bray–Curtis and unweighted UniFrac) (package phyloseq, function distance) (65) and principal coordinate axis 1 (PC1) values were analyzed for both metrics of composition and from principal component axis 1 for Elton dietary traits (package stats, function princomp). Response variables were log10 transformed as needed to meet test assumptions and models were compared using ANOVA P-values and Akaike Information Criterion. If no significant difference was observed between two models, the most parsimonious model was interpreted as the final model.
We assessed relationships between milk nutrient content and microbial abundance using linear regressions. All samples with complete milk nutritional data (milk sugar GE, milk protein GE, and milk fat GE) were analyzed (n = 67). We log transformed microbial taxa abundance and only included ASVs with at least 5% abundance and presence in a minimum of five samples (n = 135 ASVs). P values were adjusted for multiple comparisons with Benjamini and Hochberg correction.
Community Assembly Processes.
We performed a null model analysis to estimate the relative influence of ecological processes on the assembly of three distinct microbial communities from mammals (i.e., subset data from gut, milk and skin bacterial communities). Gut and skin microbiome data was downloaded from Song et al. (24, 77) and Ross et al. (21) respectively, and were subset to similar number of samples from similar number of taxonomic groups (SI Appendix, Table S7). Null model analysis was performed separately on each dataset. We followed the framework developed by Stegen et al. (33) and methodology from Zha et al. (34), by comparing observed and null-predicted bacterial communities using two metrics: the β-nearest taxon index (βNTI) and a modified version of Raup–Crick distances (RCBray) (78) based on relative abundance data according to Chase et al. (79). The workflow of the analysis was performed as described by Zha et al. (34). See SI Appendix for additional detail.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Acknowledgments
We graciously thank all of the researchers who contributed milk samples to the Smithsonian Milk Repository. Funding was provided by Smithsonian Scholarly Study grants to C.R.M.-W. and M.L.P. We thank Nancy McInerney for advice and assistance with lab work and sequencing. Some of the null model analyses were conducted on the Smithsonian High Performance Cluster, Smithsonian Institution. https://doi.org/10.25572/SIHPC.
Author contributions
M.L.P. and C.R.M.-W. designed research; M.M.K., M.B., J.C.P.W., and C.R.M.-W. performed research; M.M.K., R.R.J., S.L.B., and C.R.M.-W. analyzed data; and M.M.K., R.R.J., S.L.B., M.L.P., and C.R.M.-W. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
The data generated and analyzed during this study are available in the National Center for Biotechnology Information Sequence Read Archive repository (BioProject ID: PRJNA843071) (80). Final files for analyses (feature table, taxonomy table, phylogenetic tree, and metadata file) and R code are available on figshare: https://doi.org/10.6084/m9.figshare.21482829 (81). Previously published data were used for this work (21, 24).
Supporting Information
References
- 1.Bartol F., Wiley A., Bagnell C., Epigenetic programming of porcine endometrial function and the lactocrine hypothesis. Reprod. Domest. Anim. 43, 273–279 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Power M. L., Schulkin J., Maternal regulation of offspring development in mammals is an ancient adaptation tied to lactation. Appl. Transl. Genomics 2, 55–63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skibiel A. L., Downing L. M., Orr T. J., Hood W. R., The evolution of the nutrient composition of mammalian milks. J. Anim. Ecol. 82, 1254–1264 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Power M. L., Schulkin J., Milk: The Biology of Lactation (Johns Hopkins University Press, 2016). [Google Scholar]
- 5.Muletz-Wolz C. R., et al. , Diversity and temporal dynamics of primate milk microbiomes. Am. J. Primatol. 81, e22994 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Togo A., et al. , Repertoire of human breast and milk microbiota: A systematic review. Future Microbiol. 14, 623–641 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Boix-Amorós A., et al. , Mycobiome profiles in breast milk from healthy women depend on mode of delivery, geographic location, and interaction with bacteria. Appl. Environ. Microbiol. 85, e02994–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Penders J., et al. , Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118, 511–521 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Perez P. F., et al. , Bacterial imprinting of the neonatal immune system: Lessons from maternal cells? Pediatrics 119, e724–e732 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Fernández L., Pannaraj P. S., Rautava S., Rodríguez J. M., The microbiota of the human mammary ecosystem. Front Cell Infect. Microbiol. 10, 586667 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramsay D. T., Kent J. C., Owens R. A., Hartmann P. E., Ultrasound imaging of milk ejection in the breast of lactating women. Pediatrics 113, 361–367 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Cabrera-Rubio R., et al. , The human milk microbiome changes over lactation and is shaped by maternal weight and mode of delivery. Am. J. Clin. Nutr. 96, 544–551 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Asnicar F., et al. , Studying vertical microbiome transmission from mothers to infants by strain-level metagenomic profiling. mSystems 2, e00164–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrullo L., et al. , The early life microbiota mediates maternal effects on offspring growth in a nonhuman primate. iScience 25, 103948 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nunez N., Réot L., Menu E., Neonatal immune system ontogeny: The role of maternal microbiota and associated factors. How might the non-human primate model enlighten the path? Vaccines 9, 584 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Comizzoli P., Power M. L., Bornbusch S. L., Muletz-Wolz C. R., Interactions between reproductive biology and microbiomes in wild animal species. Anim. Microbiome 3, 87 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou J., Ning D., Stochastic community assembly: Does it matter in microbial ecology? Microbiol. Mol. Biol. Rev. 81, e00002-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller E. T., Svanbäck R., Bohannan B. J. M., Microbiomes as metacommunities: Understanding host-associated microbes through metacommunity ecology. Trends Ecol. Evol. 33, 926–935 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Grisnik M., Grinath J. B., Walker D. M., The presence of Pseudogymnoascus destructans, a fungal pathogen of bats, correlates with changes in microbial metacommunity structure. Sci. Rep. 11, 11685 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazel F., et al. , Is host filtering the main driver of phylosymbiosis across the tree of life? mSystems 3, e00097-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ross A. A., Müller K. M., Weese J. S., Neufeld J. D., Comprehensive skin microbiome analysis reveals the uniqueness of human skin and evidence for phylosymbiosis within the class Mammalia. Proc. Natl. Acad. Sci. U.S.A. 115, E5786–E5795 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lutz H. L., et al. , Ecology and host identity outweigh evolutionary history in shaping the bat microbiome. mSystems 4, e00511–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Groussin M., et al. , Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat. Commun. 8, 1–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song S. J., et al. , Comparative analyses of vertebrate gut microbiomes reveal convergence between birds and bats. mBio 11, e02901-19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brooks A. W., Kohl K. D., Brucker R. M., van Opstal E. J., Bordenstein S. R., Phylosymbiosis: Relationships and functional effects of microbial communities across host evolutionary history. PLOS Biol. 14, e2000225 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ingala M. R., et al. , You are more than what you eat: Potentially adaptive enrichment of microbiome functions across bat dietary niches. Anim. Microbiome 3, 82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bornbusch S. L., Keady M. M., Power M. L., Muletz-Wolz C. R., Milk microbiomes of three great ape species vary among host species and over time. Sci. Rep. 12, 11017 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar S., Stecher G., Suleski M., Hedges S. B., Timetree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Letunic I., Bork P., Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goslee S. C., Urban D. L., The ecodist package for dissimilarity-based analysis of ecological data. J. Stat. Softw. 22 (2007). [Google Scholar]
- 31.Legendre P., Studying beta diversity: Ecological variation partitioning by multiple regression and canonical analysis. J. Plant Ecol. 1, 3–8 (2008). [Google Scholar]
- 32.Weinstein S. B., et al. , Microbiome stability and structure is governed by host phylogeny over diet and geography in woodrats (Neotoma spp.). Proc. Natl. Acad. Sci. U.S.A. 118, e2108787118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stegen J. C., et al. , Quantifying community assembly processes and identifying features that impose them. ISME J. 7, 2069–2079 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zha Y., Lindström E. S., Eiler A., Svanbäck R., Different roles of environmental selection, dispersal, and drift in the assembly of intestinal microbial communities of freshwater fish with and without a stomach. Front. Ecol. Evol. 8, 1–15 (2020). [Google Scholar]
- 35.Mallott E. K., Amato K. R., Host specificity of the gut microbiome. Nat. Rev. Microbiol. 19, 639–653 (2021). [DOI] [PubMed] [Google Scholar]
- 36.Sogin M. L., et al. , Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. U.S.A. 103, 12115–12120 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galand P. E., Casamayor E. O., Kirchman D. L., Lovejoy C., Ecology of the rare microbial biosphere of the Arctic Ocean. Proc. Natl. Acad. Sci. U.S.A. 106, 22427–22432 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McKenzie V. J., et al. , The effects of captivity on the mammalian gut microbiome. Integr. Comp. Biol. 57, 690–704 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keady M. M., et al. , Clinical health issues, reproductive hormones, and metabolic hormones associated with gut microbiome structure in African and Asian elephants. Anim. Microbiome 3, 85 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song S. J., et al. , Cohabiting family members share microbiota with one another and with their dogs. eLife 2, e00458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar H., et al. , Distinct patterns in human milk microbiota and fatty acid profiles across specific geographic locations. Front. Microbiol. 7, 1619 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Urashima T., Mineguchi Y., Fukuda K., Whitehouse-Tedd K., Oftedal O. T., “Evolution of milk oligosaccharides of carnivora and artiodactyla: Significance of the ratio of oligosaccharides to lactose in milk” in Evolutionary Biology A Transdisciplinary Approach, Pontarotti P., Ed. (Springer International Publishing, 2020), pp. 359–377. [Google Scholar]
- 43.Bode L., The functional biology of human milk oligosaccharides. Early Hum. Dev. 91, 619–622 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Oliveira D. L., Wilbey R. A., Grandison A. S., Roseiro L. B., Milk oligosaccharides: A review. Int. J. Dairy Technol. 68, 305–321 (2015). [Google Scholar]
- 45.Al Nabhani Z., Eberl G., Imprinting of the immune system by the microbiota early in life. Mucosal Immunol. 13, 183–189 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Berger P. K., Ong M. L., Bode L., Belfort M. B., Human Milk Oligosaccharides and Infant Neurodevelopment: A Narrative Review. Nutrients 15, 719 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reich C. M., Arnould J. P. Y., Evolution of Pinnipedia lactation strategies: A potential role for α-lactalbumin? Biol. Lett. 3, 546–549 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Al Rubaye H., Adamson C. C., Jadavji N. M., The role of maternal diet on offspring gut microbiota development: A review. J. Neurosci. Res. 99, 284–293 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Rodríguez J. M., The origin of human milk bacteria: Is there a bacterial entero-mammary pathway during late pregnancy and lactation? Adv. Nutr. 5, 779–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernández L., et al. , The human milk microbiota: Origin and potential roles in health and disease. Pharmacol. Res. 69, 1–10 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Mulligan C. M., Friedman J. E., Maternal modifiers of the infant gut microbiota: metabolic consequences. J. Endocrinol. 235, R1–R12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Macpherson A. J., Uhr T., Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 303, 1662–1665 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Arroyo R., et al. , Treatment of infectious mastitis during lactation: Antibiotics versus oral administration of lactobacilli isolated from breast milk. Clin. Infect. Dis. 50, 1551–1558 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Petrullo L., Jorgensen M. J., Snyder-Mackler N., Lu A., Composition and stability of the vervet monkey milk microbiome. Am. J. Primatol. 81, e22982. (2019). [DOI] [PubMed] [Google Scholar]
- 55.Gilbert B., Levine J. M., Ecological drift and the distribution of species diversity. Proc. R. Soc. B: Biol. Sci. 284, 20170507 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.David L. A., et al. , Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eisenhofer R., et al. , Contamination in low microbial biomass microbiome studies: Issues and recommendations. Trend Microbiol. 27, 105–117 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Quigley L., et al. , The complex microbiota of raw milk. FEMS Microbiol. Rev. 37, 664–698 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Oftedal O., Milk composition, milk yield and energy output at peak lactation: A comparative review. Symposia Zool. Soc. L. 51, 33–58 (1984). [Google Scholar]
- 60.Callahan B. J., McMurdie P. J., Holmes S. P., Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11, 2639–2643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Callahan B. J., et al. , DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Q., Garrity G. M., Tiedje J. M., Cole J. R., Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Price M. N., Dehal P. S., Arkin A. P., FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bolyen E., et al. , Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McMurdie P. J., Holmes S., phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLOS One 8, e61217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Davis N. M., Proctor D. M., Holmes S. P., Relman D. A., Callahan B. J., Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hood W. R., Oftedal O. T., “Methods of measuring milk composition and yield in small mammals” in Ecological and Behavioral Methods for the Study of Bats (Johns Hopkins University Press, 2009), pp. 539–553. [Google Scholar]
- 68.Power M. L., et al. , Patterns of milk macronutrients and bioactive molecules across lactation in a western lowland gorilla (Gorilla gorilla) and a Sumatran orangutan (Pongo abelii). Am. J. Primatol. 79, e22609 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Faith D. P., Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61, 1–10 (1992). [Google Scholar]
- 70.Fox J., Monette G., Generalized collinearity diagnostics. J. Am. Stat. Assoc. 87, 178–183 (1992). [Google Scholar]
- 71.Anderson M. J., Walsh D. C. I., PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing? Ecol. Monographs 83, 557–574 (2013). [Google Scholar]
- 72.Anderson M. J., Permutational multivariate analysis of variance (PERMANOVA). Wiley StatsRef: Statistics Reference Online 1–15 (2017). [Google Scholar]
- 73.Oksanen J., et al. , vegan: Community ecology package (2019).
- 74.Wilman H., et al. , EltonTraits 1.0: Species-level foraging attributes of the world’s birds and mammals. Ecology 95, 2027–2027 (2014). [Google Scholar]
- 75.Micallef L., Rodgers P., eulerAPE: Drawing area-proportional 3-venn diagrams using ellipses. PLOS One 9, e101717 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rosseel Y., lavaan: An R package for structural equation modeling. J. Stat. Softw. 48, 1–36 (2012). [Google Scholar]
- 77.R Core Team, R: A language and environment for statistical computing (2020).
- 78.Raup D. M., Crick R. E., Measurement of faunal similarity in paleontology. J. Paleontol. 53, 1213–1227 (1979). [Google Scholar]
- 79.Chase J. M., Kraft N. J. B., Smith K. G., Vellend M., Inouye B. D., Using null models to disentangle variation in community dissimilarity from variation in α-diversity. Ecosphere 2, art24 (2011). [Google Scholar]
- 80.Keady M.. Data from “Mammal Milk Microbiome”. NCBI SRA. Available at https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA843071. Deposited 27 May 2022. [Google Scholar]
- 81.Keady M.. Data from “Mammal Milk Microbiome Files”. Figshare. Available at https://figshare.com/articles/dataset/Mammal_Milk_Microbiome_files/21482829. Deposited 15 June 2023. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Data Availability Statement
The data generated and analyzed during this study are available in the National Center for Biotechnology Information Sequence Read Archive repository (BioProject ID: PRJNA843071) (80). Final files for analyses (feature table, taxonomy table, phylogenetic tree, and metadata file) and R code are available on figshare: https://doi.org/10.6084/m9.figshare.21482829 (81). Previously published data were used for this work (21, 24).