

Abstract
Nucleic acid amplification test (NAAT)-based point-of-care (POC) devices are rapidly growing for use in low-resource settings. However, key challenges are the ability to store the enzyme-based reagents in dry form in the device and the long-term stability of those reagents at elevated temperatures, especially where ambient temperatures could be as high as 45 °C. Here, we describe a set of excipients including a combination of trehalose, polyethylene glycol and dextran, and a method for using them that allows long-term dry storage of enzyme-based reagents for an isothermal strand displacement amplification (iSDA) reaction in a porous matrix. Various porous materials, including nitrocellulose, cellulose, and glass fiber, were tested. Co-dried reagents for iSDA always included those that amplified the ldh1 gene in Staphylococcus aureus (a polymerase and a nicking enzyme, 4 primers, dNTPs and a buffer). Reagents also either included a capture probe and a streptavidin-Au label required for lateral flow (LF) detection after amplification, or a fluorescent probe used for real-time detection. The reagents showed the best stability in a glass fiber matrix when stored in the presence of 10% trehalose and 2.5% dextran. The reagents were stable for over a year at ~22 °C as determined by lateral flow detection and gel electrophoresis. The reagents also exhibited excellent stability after 360 h at 45 °C; the assay still detected as few as 10 copies of ldh1 gene target by lateral flow detection, and 50 copies with real-time fluorescence detection. These results demonstrate the potential for incorporation of amplification reagents in dry form in point-of-care devices for use in a wide range of settings.
Introduction
Point-of-care (POC) devices that are accurate, robust, low cost, rapid, easy-to-use, equipment-free and disposable are in great demand for the diagnosis of diseases, especially in low-resource settings (LRS).1,2 However, the COVID-19 crisis has brought into sharp focus a great need to universally apply our tremendous gains in knowledge in molecular methods toward better POC diagnostics within the developed as well as developing worlds with high disease burden. Nucleic acid amplification tests (NAATs) are extremely sensitive methods for detecting DNA or RNA from pathogens and have relatively rapid turnaround times (getting results on the same day) compared to microbiological cultures or ELISA.3–8 Unfortunately, while NAATs aimed at POC for bacterial and viral infectious disease diagnosis have been developed, they are instrumented and expensive because they either require thermal cycling and/or complicated optics for fluorescence readout.9–12 Examples of commercial POC instrumented diagnostic NAAT platforms include Cepheid GeneXpert, Abbott ID NOW, Credo Biomedical VitaPCR, Mesa Biotech Accula system, and Rapid Diagnostics Rapid MiniLab. They provide dry storage of reagents in cartridges, are rapid, and have high sensitivity but require trained users, need electricity, and are not affordable, limiting their use in low-resource settings.
There has been progress towards developing isothermal amplification systems in miniaturized microfluidic devices that are easy to use and suitable for LRS, but often at high prices and with varying levels of automation.13–16 A low-cost cartridge system with fluidic controls that executes isothermal amplification with lateral flow (LF) detection has recently been reported, but still requires a (portable) instrument.17 Paper fluidic devices have also been used for NAAT based testing as an inexpensive alternative to instrumented systems.18–20 All these systems, however, required fresh NAAT reagents to demonstrate the capability and did not address the dry storage of the reagents.
One of the key technical requirements for truly LRS-compatible NAAT-based POC systems is the ability to store dry reagents on the device. There are two main challenges. The first is the long-term stability of enzymes at elevated temperatures, especially for use in places where the ambient temperature could be as high as 45 °C. The most promising approach is to store all reagents except simple buffers in dry form in a way that limits exposure to oxygen. The subsequent challenge is uniform rehydration of the reagents within the device to achieve optimal performance. Overcoming these challenges would circumvent the need for the “cold chain” (continuous refrigeration from the point of manufacture to the point of use), which is inconvenient in any diagnostic product, and maybe too expensive and even unavailable in many LRS settings.
Methods for stabilizing proteins and enzymes in dry form using sugars and sugar alcohols are widely used in the pharmaceutical industry. The most common method of making solid proteins is lyophilization.21 Trehalose, a non-reducing disaccharide, can form protein-stabilizing glass and is the most common sugar used in the dry preservation of biomolecules.22–24 There have been a few reports on dry storage of PCR and isothermal amplification reagents, but they did not address long-term stability.25–30 A lyophilization method using Biolyph’s patented technology for long-term dry storage for loop-mediated isothermal amplification was reported but did not include the primers.31 A direct blood dry LAMP system (CZC-LAMP) was applied to the diagnosis of malaria with different reagents dried separately in the lid and tube.32 In all the above studies, the dry reagents were in a tube, and the reactions carried out using an instrument. Short-term stability studies were reported for helicase dependent amplification (HDA) reagents in Whatman 1 chromatography paper stored at room temperature.33 Similarly, reagents for isothermal reverse transcriptase recombinase polymerase amplification (RT-RPA) were freeze-dried onto cellulose pads and stored at room temperature.34 Recently an autonomous microfluidic device with vitrified reverse transcription loop-mediated isothermal amplification reagents was reported with reagents stored dry up to three weeks at room temperature.35 A sample-to-result integrated paper-based device with short-term reagent stability was also reported.36 However, the stability of the reagents at elevated temperatures was not undertaken in any of these studies.
We previously reported successful long-term dry storage and stability of various reagents such as horseradish peroxidase (HRP)37 and gold enhancement38 reagents in a porous matrix and incorporated into an automated two-dimensional paper network (2DPN) fluidic devices for malarial antigen detection. An autonomous device with a combination of isothermal amplification and paper fluidics for NAAT with dry reagents stable at elevated temperatures holds great promise for an inexpensive POC diagnosis, especially for LRS settings. We reported an isothermal strand displacement amplification (iSDA) method for a Staphylococcus aureus DNA target that uses two enzymes, namely a nicking enzyme and a polymerase, in combination with high specificity probes either for real-time fluorescence measurement or twin probes for lateral flow detection.39 We have shown that speed, sensitivity, and specificity of iSDA make it a powerful method for point-of-care molecular diagnosis.
In this report, we describe a method for the long-term dry storage of the iSDA reagents, including probes with either a gold nanoparticle label for downstream lateral flow detection or a fluorescent probe for real-time detection in a porous matrix. The dry preservation of amplification reagents in a solid porous matrix could have high value for several applications with fluid connectivity especially in paper-based POC devices and applicable to low-resource settings, especially in hot climates where the ambient temperatures can be in the 40–45 °C range. We have also embodied this amplification method in an integrated 2-dimensional paper network (2DPN) sample-to-result device for methicillin resistant Staphylococcus aureus (MRSA) detection, the use of which could reduce the cost of diagnosis by eliminating the need for expensive instruments.40
Experimental
Isothermal amplification in porous matrices
Porous matrices tested for iSDA compatibility included nitrocellulose FF80HP, cellulose CF5, Quartz QMA, Fusion 5, and Standard 17 Glass fiber (Std 17 GF). All these materials were purchased from GE Healthcare Life Sciences, Pittsburgh, PA. Each material was cut to size with a hole-puncher to hold 25 μl of fluid when saturated. Reagents specific for detection of the Staphylococcus aureus ldh1 gene using iSDA (as noted in Toley et al.39) contained 50 mM potassium phosphate buffer pH 7.6, 3.8 mM magnesium sulfate, 100 μM of each dNTP, 250 nM extension primer E1, 500 nM extension primer E2, 50 nM each of bumper primers B1 and B2, 8 units of WarmStart Bst 2.0 polymerase (New England Biolabs, Ipswich, MA) and 1.6 units of nicking enzyme Nt.BbvCI (New England Biolabs, Ipswich, MA). Primer and probe sequences for ldh1 amplification have been previously published.39
To test the compatibility of the porous matrices, about 20 μl of iSDA reagent solution containing 100 copies of purified methicillin-resistant S. aureus (MRSA) genomic DNA (ATCC, BAA-1556) was added to the different porous matrices; the porous matrix was placed in a Secure-Seal hybridization chamber (Electron Microscopy Sciences, Hatfield, PA). DNA solutions were replaced with pure water for no-template controls (NTCs) for each of the materials tested. The samples were incubated in a custom electric oven at 49 °C for 30 min, which has been shown to be sufficient for amplification of even single copies of the target sequence using iSDA (although a custom oven was used, any device capable of holding 49 °C for 30 min could be substituted). After amplification, the pads were placed in a 0.2 ml tube with a hole at the bottom, which in turn was placed in a 0.5 ml tube, and centrifuged to collect the solution containing free amplicons. The amplicons were run on a gel electrophoresis system as described below.
Effect of leachates from porous matrices on iSDA
The effect of possible inhibitors contained in the leachates from the different matrices (listed in the previous section) on iSDA was tested. The materials were first incubated in clean water at 49 °C for 30 minutes and then centrifuged in a 0.45 μM filter spin column. The extracted fluid was used as a replacement for water in the iSDA reaction. The reactions were carried out in a tube in a thermocycler (CFX96, Bio-Rad Laboratories, Hercules, CA) with 100 copies of the MRSA genomic DNA. The amplicons were run on a gel electrophoresis system, as described below.
Lyophilization and long-term storage of iSDA and detection reagents
Several combinations of the formulation containing trehalose (Life Sciences Advanced Technologies, St Petersburg, FL), polyethylene glycol (PEG) (Hampton Research, Aliso Viejo, CA) and dextran (Sigma-Aldrich, St Louis, MO) were used for preservation of iSDA reagents (Table 1). The effect of the combinations of the excipients on the iSDA was first determined in a conventional tube reaction at 49 °C with MRSA genomic DNA using the thermal cycler. For lyophilization, the iSDA reagents specific to the S. aureus ldh1 target were mixed with the different formulation given in Table 1. The mixtures also contained the lateral flow (LF) detection twin probes, including 10 nM of biotin probe (ELITechGroup, Bothell, WA) (5′-Biotin Phosphoramidite 10–5950-95, Glen Research, Sterling, Virginia, USA), and 20 nM of capture probe (ELITechGroup, Bothell, WA). We also included a 40 nm streptavidin-coated gold (Au) detection label (Innova Biosciences, Cambridge, UK) into the iSDA reagent mix by premixing one microliter of Au label (OD 10) with 10 nM of biotin detection probe for 5 min before adding to the iSDA reagents.
Table 1.
List of preservation mix used for dry storage and stabilization of iSDA reagents
| Preservation mix for dry storage (w/v) | |
|---|---|
|
| |
| T | 10% trehalose |
| TP | 10% trehalose |
| 1% PEG 8000 | |
| TD 70 | 10% trehalose |
| 2.5% dextran (~70 kDa) | |
| TD 500 | 10% trehalose |
| 2.5% dextran (500 kDa) | |
Standard 17 glass fiber (Std 17 GF) was laser-cut (VLS3.60, Universal Laser Systems, Scottsdale, AZ) to 20 mm × 5 mm strips having a fluid capacity of 40 μl. The Std 17 GF pads were incubated in 1% bovine serum albumin (BSA) containing 0.1% Tween 20 for one hour, drained of the solution, and dried overnight at 45 °C. An experimental setup for the lyophilization procedure is shown in ESI Fig. S1.† The BSA-coated Std 17 GF pads were placed in a sterile 12-well polystyrene plate. The outer four wells contained a neodymium magnet. An aluminum plate with neodymium magnets embedded at the four corners was placed below the 12 well-plate, serving as a thermal mass to ensure the reagent pads in the well-plate remain frozen during lyophilization. The magnets held the aluminum and well plates together (ESI Fig. S1a†). About 35 μl of iSDA reagent containing Au label and the preservation mix was pipetted onto the pads. The entire apparatus, with samples loaded, was flash-frozen in liquid nitrogen, placed in a fast-freeze flask (Labconco, Kansas City, MO), and lyophilized overnight in a freeze-drying system (FreeZone 2.5 liter benchtop, Labconco, Kansas City, MO) operated at a temperature of −51 °C and vacuum pressure of 0.018 mbar (ESI Fig. S1b†). After lyophilization, each pad was individually transfered to 0.5 ml sterile tubes with a hole in the cap. Sets of four tubes along with 1 gram of silica desiccant (Delta Absorbents, Roselle, IL) were placed in a moisture barrier foil pouch (Ted Pella Inc., Redding, CA) and heat-sealed. The samples were stored at 22, 35, 40, or 45 °C for 360 h or stored at 22 °C for a year.
Amplification assay for iSDA preservation
The Std 17 GF pads containing the dry iSDA and detection reagents stored at varying temperatures for 360 h were taken out of dry storage and placed in a Secure-Seal hybridization chamber (Electron Microscopy Sciences, Hatfield, PA), rehydrated with 33 μl of water containing the MRSA genomic DNA template (10–200 copies) and incubated at 49 °C for 30 min in the aforementioned custom oven (ESI Fig. S1c†). For long-term storage study at 22 °C, the pads were periodically taken out of dry storage at 1, 2, 3, 6, and 12 months and iSDA performed with 100 copies of the genomic DNA template. After amplification, the pads were placed in a 0.2 ml tube with a hole at the bottom, which in turn was placed in a 0.5 ml tube and centrifuged to collect the amplicons and processed for lateral flow detection or gel electrophoresis.
Lateral flow detection of amplicons
Lateral flow detection of iSDA amplicons was by a twin probe method co-developed with our colleagues at ELITechGroup.39 In short, a 3′-biotinylated detection probe hybridizes to the amplicon and binds a streptavidin-coated Au nanoparticle label. A chimeric pyranosyl DNA (pDNA)–DNA capture probe hybridizes by DNA independently to the amplicon and hybridizes by pDNA to an immobilized pDNA complement. A dipstick-style LF assay format was used for demonstrating the dry preservation of the iSDA reagents. Cardboard-backed nitrocellulose FF80HP (GE Healthcare, Waukesha, WI) striped with (1) a pDNA complement linked to T20 (twenty repeats of thymidine) and (2) a T20-biotin control line (provided by ELITechGroup, Bothell, WA), and attached to a cellulose wicking pad, was cut into ~5 mm-wide strips. For detection, 28 μl of the iSDA reaction mix containing amplicons was mixed with a solution of NaCl and Triton-X100 to give final concentrations of 0.6 M and 0.8%, respectively. The mixture was added into a 96-well deep well plate (VWR, Radnor, PA) and a detection strip placed into each well for 20 minutes. The LF strips were then imaged using the procedure described below.
Lateral flow image capture and quantification
Lateral flow strips were imaged wet using a flatbed scanner (Epson Perfection V700, CA, USA) in 48-bit RGB mode at a resolution of 600 dpi. The intensities in the region of interest (ROI) were quantified using ImageJ41 by measuring the mean green-channel intensity of each signal band and a background region within each strip. The assay signal was calculated as the background-subtracted intensity normalized to the full intensity range of the image.42
Gel electrophoresis
PAGE analysis was conducted under denaturing conditions. A 2.2 μl sample (from a 35 μl reaction) was mixed with 6.8 μl gel loading buffer II (Life Technologies, Carlsbad, CA) heated to 95 °C for 5 minutes, then kept on a cold block. About 7 μl of this sample mix was loaded into pre-cast 15% Novex® TBE-Urea gels (Life Technologies). A 10 bp ladder (Life Technologies) was used as a marker. Electrophoresis was conducted in an XCell SureLock™ Mini-Cell Electrophoresis System (Life Technologies) at 160 volts for ~40 minutes using a 1× TBE running buffer. The electrophoresis cell was kept in a pre-warmed water bath at 70 °C to ensure that denaturing conditions were maintained throughout. After electrophoresis, gels were stained with 2× SYBR Gold® Nucleic Acid Gel Stain (Life Technologies) in 1× TBE for 20 minutes. Stained gels were imaged using a Gel Doc™ EZ System (Bio-Rad, Hercules, CA).
Real time fluorescence in porous matrix
An in-house method to monitor iSDA amplification in real time in the porous matrix was developed using a standard microplate reader (SpectraMax ID3, Molecular Device). Circular Std 17 GF pads with a fluid capacity of 13 μl were laser cut to fit the wells of a black flat-bottomed 96 well plate (Greiner Bio-One). A minor-groove-binding (MGB) fluorescent Pleiades probe,43 which has previously been described,39 was used to monitor amplification in real time. In short, the DNA probe has fluorophore and a minor groove binder at the 5′ end and a quencher at the 3′ end. The fluorophore is quenched in the unbound state and fluoresces when hybridized to the target amplicon. About 200 nM red-emitting Pleiades probe (ELITechGroup, Bothell, WA) was mixed to the iSDA reagents with the excipients (Table 1) and added to the Std 17 GF pads placed in a black flat-bottomed 96 well-plate and lyophilized as previously described. The samples were stored at 22 or 45 °C for 360 h and reagents were rehydrated with water containing the MRSA genomic DNA (50–1000 copies). The well-plate was sealed with a PCR plate sealer (Bio-Rad, Hercules CA) and placed in the microplate reader set at 50 °C. Real-time kinetic measurement used the 593/650 nm excitation/emission setting, and fluorescence signals were acquired at 2-minute intervals.
Enzyme activity assay
The nicking enzyme Nt.BbvCI and WarmStart Bst polymerase 2.0 were separately lyophilized with the same formulations listed in Table 1, and tested for their activity after dry storage at different temperatures using enzyme-specific assays. The assays use reagent mixes of two components: an enzyme mix and a substrate/probe mix.
For the nicking enzyme activity assay, the 20 μL enzyme mix contained 1.6 units of Nt.BbvCI, 100 μM of each dNTPs, 50 mM potassium phosphate buffer pH 7.6, 3.75 mM magnesium sulfate, and one of the preservative mix (Table 1); and the 20 μL substrate/probe mix contained 50 mM potassium phosphate buffer, the same preservative mix, and 188 nM substrate/probe. For this assay, the substrate/probe was a DNA hairpin labeled with a quencher and a fluorophore with a minor groove binder (MGB), provided courtesy of ELITechGroup. The substrate/probe mix, always freshly prepared, and enzyme mix, either freshly prepared or extracted from pads as described below, were added to alternating rows of a 96-well microplate. The plate was loaded onto a real-time thermal cycler (CFX96, Bio-Rad Laboratories, Hercules, CA) and equilibrated to 49 °C for 10 min. Each row of enzyme mixes was combined with the adjacent row of substrate/probe mixes using a twelve-channel pipette, and then the plate was immediately read for fluorescence in the FAM channel about every 17 s for ~25 min.
For the polymerase activity assay, the 20 μL enzyme mix contained 8 units of WarmStart Bst 2.0 polymerase, 50 mM potassium phosphate buffer pH 7.6, 3.75 mM magnesium sulfate, and one of the preservative mix (Table 1); and the 40 μL substrate/probe mix contained 50 mM potassium phosphate buffer pH 7.6, the same preservative mix, and 333 nM substrate, 333 μM dNTPs, 1.25 μM probes. For the polymerase activity assay, the substrate was a 90-nt, single-stranded DNA template (GCA CCG ATT TCC ACA GTT CTC CCG ACA CGC CCC TCA TAA ACA CAA TAC CAC CCA TTC ATT CCA AGC CAT ACC GAT TCC CAC AAA GCA TCT) with a 25-nt DNA primer (AGA TGC TTT GTG GGA ATC GGT ATG G), which were synthesized by Integrated DNA Technologies (Coralville, IA), and the probe was EvaGreen (Biotium). The substrate/probe mix was added to every other row of a 96-well microplate. The plate was loaded onto a real-time thermal cycler (CFX96, Bio-Rad Laboratories, Hercules, CA), and the template/primer substrate was annealed by heating to 95 °C for 2 min, then cooling to 25 °C. The enzyme was then added to interstitial rows, and the plate was equilibrated to 50 °C for 10 min. Each row of enzyme mixes was combined with the adjacent row of substrate/probe mixes using a twelve-channel pipette, and then the plate was immediately read for fluorescence in the FAM channel about every 22 s for ~25 min.
Enzyme mixes were either freshly prepared or rehydrated from lyophilized Std 17 pads with water and extracted by centrifugation in a 0.45 μM filter spin column at 10 000g for 8 min. Enzyme activities were assayed in each preservative mix (Table 1); with fresh reagents, from pads with dry reagents on the day of preparation; and from pads with dry storage at RT, 35 °C, 40 °C, and 45 °C after day 1 and day 15.
For the nicking activity assay, freshly prepared standard conditions used enzyme concentrations of 0×, 0.1×, 0.5×, 1.0×, and 2.0× the nominal amount of enzyme. For the polymerase activity assay, freshly prepared standard conditions used enzyme concentrations of 0×, 0.2×, 0.5×, 1.0×, and 2.0× the nominal amount of enzyme.
For analysis, a line was fit to the initial linear region of each curve while maintaining an R2 value of greater than 0.985. The slope of this fit line was plotted vs. concentration to construct a kcatvs. concentration calibration curve. A line was fitted to each data point in the same manner, and the slope was used to calculate the kcat using the calibration curve. The 100% activity was determined by the calibration curve, which uses fresh reactions in the plate. The relative activity for each condition was calculated using three technical replicates, each from 5 dried pads.
Imaging of dried reagents in porous matrix
Std 17 GF pads containing iSDA reagents, 10% trehalose and 2.5% dextran and Au label were lyophilized as previously described. The lyophilized pads were set on a flatbed scanner (Epson Perfection V700, CA, USA) either dry or rehydrated with water, and scanned in 48-bit RGB mode at a resolution of 600 dpi.
For scanning electron microscopy, Std 17 GF pads with iSDA reagents containing 10% trehalose were either lyophilized or dried under vacuum using a centrifugal evaporator for 4 h (Genevac Inc., Gardiner, NY). All samples were Au/Pd sputtered (SPI Module Control, Structure Probe, Inc., West Chester, PA, USA) for 103 seconds, at a ~7 nm per minute deposition rate, leaving an estimated 12 nm Au/Pd coating. A FEI Sirion scanning electron microscope with a 5 kV beam and a spot size of 3 was used for imaging.
Results and discussion
The goal of this study was to develop a method for the long-term dry preservation of amplification reagents used in iSDA in a porous matrix that could be easily implemented in a POC NAAT platform for LRS settings. Porous materials were chosen as a matrix for drying the reagents since they are convenient to handle during drying and provided a format appropriate for integration with our 2D paper network-based POC MAD NAAT device, which has porous material connectivity for fluidic operations,40 and a fluorescence-based MD NAAT device (Shah et al., submitted). We included LF detection probes, and the Au-label in the iSDA reagent dry-down matrix itself, which could be applicable for an inexpensive colorimetric readout in POC device.40 We also used real-time fluorescence kinetics as a tool to monitor iSDA performance after dry storage. Reagents for an iSDA reaction that targeted the S. aureus ldh1 gene were selected for dry storage, and the limit of detection determined after rehydration.
We first tested iSDA assay compatibility with a variety of porous matrices. After the selection of a porous matrix, we tested a suitable drying and storage method. We then tested the stability of dry iSDA reagents, including the detection labels, in various preservation formulations by performing amplification in the porous matrix itself, followed by dipstick-style LF detection and gel electrophoresis. We also used a real-time fluorescence measurement in the porous matrix to assess the iSDA reagent stability. Furthermore, we separately tested the stability of the two enzymes (nicking enzyme and the polymerase) in dry storage in the porous matrix by using enzyme-specific activity assays. A range of temperatures of storage (22–45 °C) was studied for applicability to LRS, especially in hot climates where the ambient temperatures can be in the 40–45 °C range.
Porous matrix selection for iSDA assay
To select porous matrices that are compatible with iSDA, we first added the iSDA reagents directly to the native porous materials without any pre-treatment. We used a variety of porous matrices, including nitrocellulose (FF80), cellulose (CF5), quartz (QMA), Fusion 5, and Std 17 GF, and tested for the ldh1 iSDA with 100 MRSA genomic copies in a custom oven. We found that, of this set, only Std 17 GF supported iSDA, and had sensitivity comparable to in-tube assay, as seen by gel electrophoresis (Fig. 1a). The target amplification products (100 and 120 bp) and the primer-dimer side products, as seen on the gel, are an indication of a successful iSDA. The other materials (nitrocellulose, cellulose, Fusion 5, and quartz) inhibited iSDA, as neither the ldh1-specific products nor the inevitable primer-dimer side products could be seen on the gel. We speculated two reasons for the failure of amplification in these materials; (1) that the materials had assay inhibiting compounds, or (2) the interaction of the enzymes with the surface of the materials rendered them inactive. To investigate the failure of amplification with those matrices, we tested the leachate from incubation of these materials in water in an in-tube iSDA with 100 MRSA genomic copies. We found that the leachate from the nitrocellulose, cellulose, and Fusion 5 significantly inhibited the assay, as seen by the very faint product-specific bands, compared to the strong control bands in the condition with clean water (Fig. 1b). Interestingly, for the quartz material, the leachate did not inhibit iSDA in the in-tube assay (Fig. 1b) compared to the in-material testing in Fig. 1a, possibly indicating that the surface of the quartz material inactivated or immobilized some or all of the reagents. These porous materials could support iSDA after washing away the inhibitors and/or blocking surfaces with protein coating, although we did not pursue these efforts. We used Std 17 GF as our material of choice for all the reagent dry storage studies.
Fig. 1.
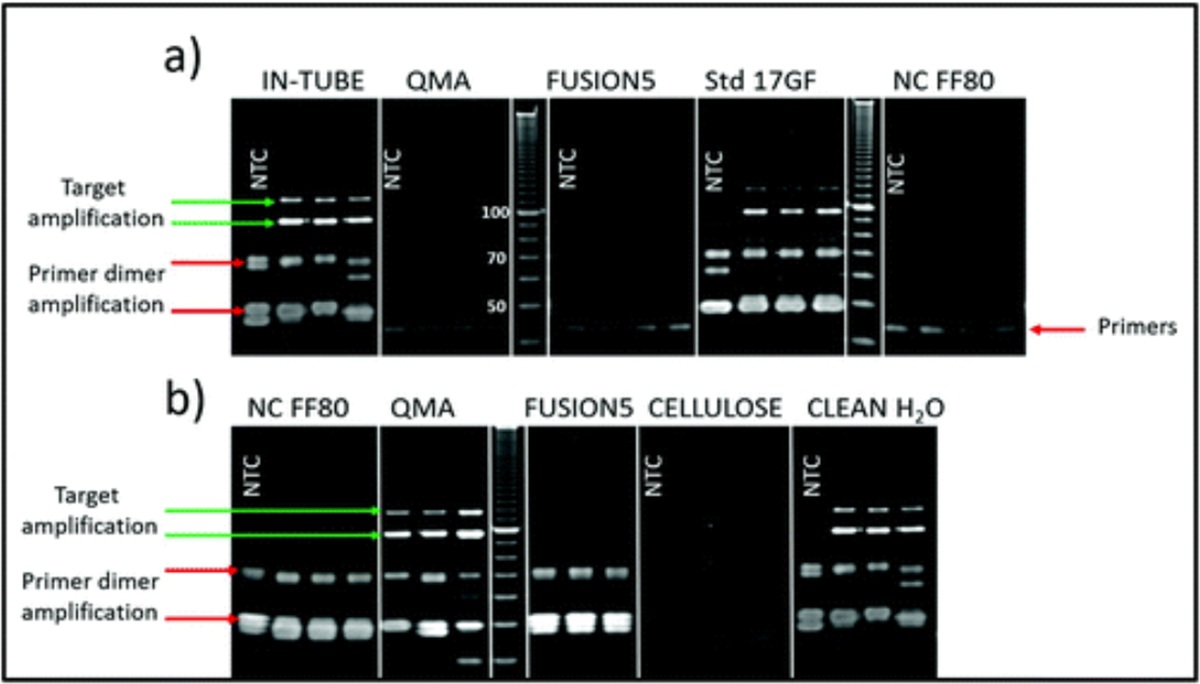
(a) Denaturing PAGE image showing products of the ldh1 iSDA assay in a few selected porous materials. Only Std 17 GF showed the targeted amplification product bands at ~100 and 120 nucleotides (green arrow) comparable to the assay as performed in-tube (conventional polypropylene tube). The inevitable primer dimer side products are indicated with a red arrow. Quartz (QMA), nitrocellulose (NC-FF80), and Fusion 5 totally inhibited amplification, including the primer-dimer side reactions. (b) Gel image showing the effect of leachate from porous materials on iSDA as performed in a tube. Leachate from NC-FF80, Fusion 5, and cellulose significantly inhibited iSDA as seen by very faint product-specific products, whereas the leachate from QMA did not affect the iSDA assay.
Dry storage of iSDA reagents
Several formulations of preservatives involving combinations of trehalose, PEG, and dextrans (Table 1) were considered for stabilizing the iSDA reagents for dry storage. It was assumed that the most unstable components of the amplification “master mix” would be the proteins: the nicking enzyme and the polymerase. The preservative formulations were initially tested for any adverse effect on iSDA by real-time amplification using a red-emitting fluorescent target-specific MGB probe in an in-tube assay with fresh reagents. The lift-off time of the reaction and the fluorescence levels serve as an indication of the performance of iSDA.39 In the literature, PEG and dextran have been reported to act as crowding agents that enhance loop-mediated isothermal amplification (LAMP)44,45 and also to provide protein stabilization by volume exclusion.46 We found that neither trehalose alone, nor in combination with moderate concentrations of the polymers, dextran and PEG, had any negative effect on iSDA when tested at 250 input copies of MRSA genomic DNA (Fig. 2). The lift-off time of the reaction (~7 minutes) and the fluorescence levels were comparable to the control sample without preservatives.
Fig. 2.
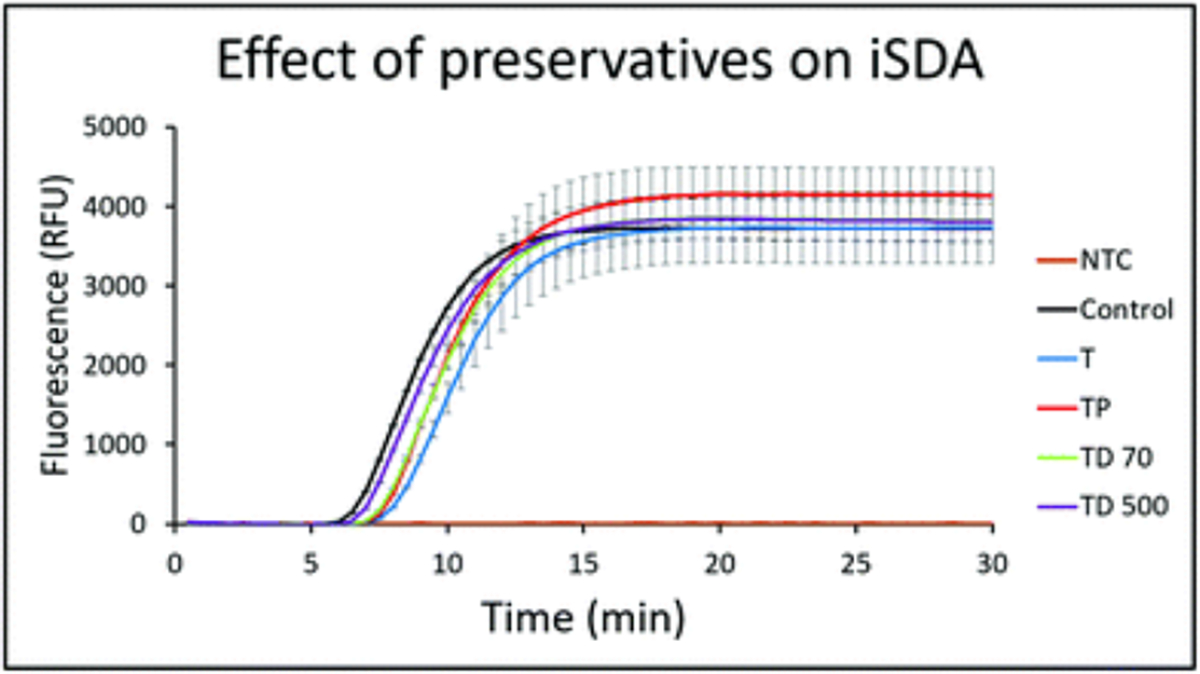
Effect of combinations of preservatives on ldh1 iSDA with 250 copies of MRSA genomic DNA measured by real-time fluorescence using the target-specific probe. The control sample was without any preservatives. Curves are mean of 3 replicates, and error bars are standard deviation. The preservatives did not have any negative effect on iSDA. The lift-off times of the reactions were nearly identical (~7 min), and the peak fluorescence levels were similar for all the formulations (T = 10% trehalose, TP = 10% trehalose + 1% PEG, TD70 = 10% trehalose + 2.5% dextran (~70 kDa), TD500 = 10% trehalose + 2.5% dextran (500 kDa) and NTC was a no-template control).
Next, we developed a freeze-drying method to store all the iSDA reagents in a Std 17 GF porous matrix. In addition, we were able to include the target-specific detection probes and the streptavidin-coated Au label into the iSDA reagent mix itself. The iSDA reagents, the detection probes, and the Au label in various preservation formulations (Table 1) were lyophilized in 5 mm × 20 mm Std 17 GF pads and stored at a range of temperatures (22–45 °C) for 360 hours. The pads were rehydrated, and iSDA assay performed within the matrix with 100 copies of MRSA genomic DNA followed by dip-stick style LF detection. The lateral flow detection of the iSDA amplicons was by a twin probe method co-developed with our colleagues at ELITechGroup and previously published.39 In this paper, we used Au label instead of the blue latex beads for colorimetric detection. In this LF detection method, the twin probes hybridize with the amplicon and are captured by the test line via the complementary pDNA on the nitrocellulose. Any free probe will flow downstream and bind to a biotin control line via streptavidin on the Au label.
An example of an LF detection strip with either a strong, medium, weak or negative amplification signal at the test line for the dry storage conditions with 100 genomic copy numbers is shown in Fig. 3a. Under the condition of the biotin probe and the streptavidin-Au label concentrations we used, a robust amplification gave us an intense test line signal with a very weak control line, and a negative amplification showed only an intense control line. Based on the stability of the iSDA reagents in different dry storage conditions, we saw a range of varying intensities for the test and the control line in an inverse relationship.
Fig. 3.
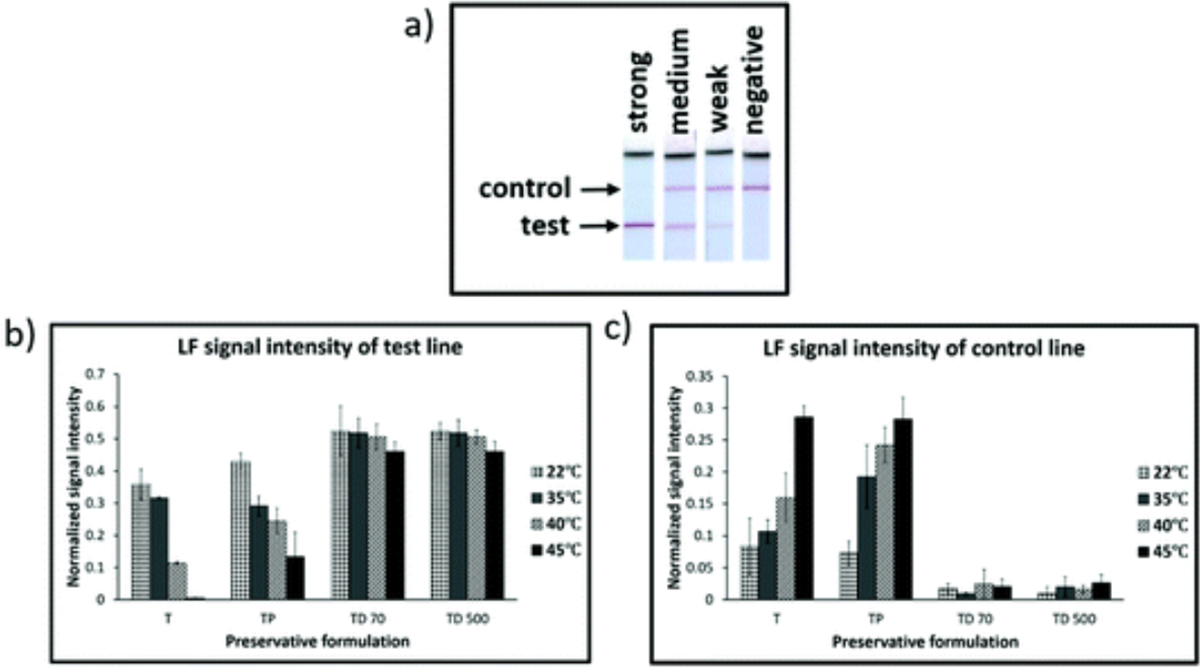
Performance of ldh1 iSDA of reagents stored dry in Std 17 GF for 360 h with different preservative formulations (Table 1), and range of temperatures. (a) Example of images of lateral flow (LF) detection strips showing strong, medium, weak, and negative amplification signal for ldh1 iSDA with 100 genomic copies. (b) and (c) Normalized intensities of LF test line and control line, respectively. Excellent stability was achieved for formulations that contained both trehalose and dextran (TD 70 and TD 500), and at all temperatures of storage as indicated by strong signal intensities for the test line and a weaker control line. With trehalose alone (T), target amplification failed with storage at 45 °C, and amplification was weaker for 22, 35, and 40 °C (when compared to TD 70 and TD 500) as indicated by weaker signal intensity for the test line and stronger intensity for the control lines. Samples with both trehalose and PEG (TP) performed only slightly better than trehalose alone.
The signal intensities for the test and control line were measured for all conditions of dry storage (Fig. 3b and c). For formulations in which both trehalose and dextran were included, excellent stability was achieved at all temperatures of storage, including 45 °C, as indicated by strong signal intensities of the test line and a very weak control line. The test line signal intensity was comparable to iSDA with a fresh reagent for the 100 genomic copy number (ESI Fig. S2†). With trehalose alone, target amplification failed for storage at 45 °C, and amplification was weaker for 22, 35, and 40 °C (when compared to samples with trehalose and dextran) as indicated by weaker signal intensity for the test line and stronger intensity for the control lines. Samples with both trehalose and PEG performed only slightly better than trehalose. This inverse relationship of the control and test line intensities served as a qualitative measure for screening for iSDA reagent stability under different storage conditions. A similar relationship was seen when a LOD for ldh1 iSDA using fresh reagents was performed with varying genomic copy numbers (ESI Fig. S2b†). At lower copy numbers, the signal intensity of the control line is stronger than the test line, and vice versa with increasing copy number. While the method for LF detection is very robust and sensitive, it gives us only a qualitative measure of reagent stability. Testing the stability of the reagents with real-time kinetics in terms of lift-off time and fluorescence levels gave us more quantitative results without the influence of excipients in lateral flow detection.
We included a target-specific red-emitting fluorescent probe instead of LF probes to monitor iSDA in real time after rehydration in the porous matrix using a plate reader to compare the performance of different formulations at 22 °C and 45 °C storage. We found that the lift-off time of the iSDA assay for almost all conditions was within 15–18 minutes and comparable to fresh reagents in the porous matrix using 250 copies of MRSA genomic DNA (Fig. 4).
Fig. 4.
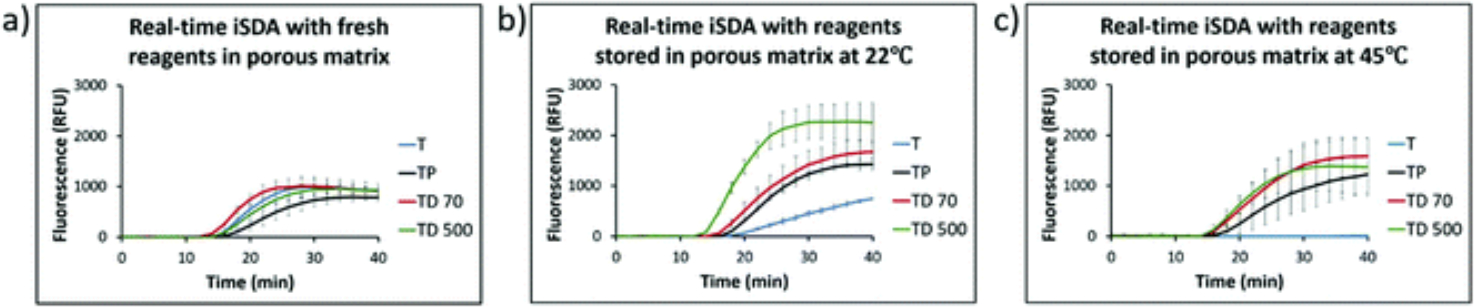
Real-time fluorescence of iSDA in Std 17 GF for the ldh1 target measured by plate-reader after rehydration in the porous matrix with different preservative formulations stored for 360 h. (a) With fresh reagents (b) with reagents stored at 22 °C and (c) with reagents stored at 45 °C. Curves are mean of 3 replicates, and error bars are standard deviation. The lift-off time of the reaction was within 15–18 minutes for all combinations of the preservatives. The peak fluorescence levels varied for different formulations with the highest signal for the formulation with trehalose and dextran. Trehalose only formulation did not amplify at 45 °C and performed poorly at 22 °C (T = 10% trehalose, TP = 10% trehalose + 1% PEG, TD70 = 10% trehalose + 2.5% dextran (~70 kDa), TD500 = 10% trehalose + 2.5% dextran (500 kDa)).
The peak fluorescence levels, however, varied for different formulations with the highest signal for formulations with trehalose and dextran. The trehalose only formulation did not amplify at 45 °C storage and performed poorly at 22 °C. Our results with fluorescence measurement directly matched the LF detection data presented in Fig. 3 for the formulations used for dry storage of iSDA reagents.
Interestingly, the peak fluorescence levels for dry stored conditions, especially for TP TD 70 and TD 500 (Fig. 4b and c), were higher than for iSDA with fresh reagents (Fig. 4a) in the porous matrix. We believe that this is due to non-uniform rehydration of reagents in the porous matrix, causing a concentration gradient. As a result, some areas in the matrix might have had robust amplification resulting in higher fluorescence measured locally, compared to the fresh reagents which are homogenous throughout the matrix. The result also agrees with additional unpublished data which suggests that the iSDA reaction is tolerant of a wide concentration range (0.5×–2.5× fold). While we do not get the spatial information of the amplification using the plate reader, support for this hypothesis has been addressed by fluorescence imaging in a submitted publication (Shah et al.).
Based on these results, we conclude that the addition of dextran to trehalose preserved the iSDA reagents better than other formulations at elevated temperatures in the Std 17 GF. In the literature, dextran has been reported to provide protein stability during freeze-drying.47
Enzyme activity
Another way to address the performance of iSDA at different storage conditions is to assess the stability of the two enzymes individually because they are likely the components of the amplification mixture most vulnerable to degradation. We, therefore, stored the nicking and the polymerase enzymes separately in Std 17 GF porous matrix under the same dry storage conditions as the iSDA reagents and used enzyme-specific assays to test the stability.
The results of the enzyme activity assays are shown in Fig. 5. The activity of polymerase does not appear to decrease appreciably over storage temperatures for all formulations except trehalose + PEG. The activity of the nicking enzyme does appear to decrease significantly over time at elevated temperatures. PEG was observed to be an overall detriment to storage for enzymes. The preservation of polymerase activity at 45 °C over time was expected as the optimal working temperature for the enzyme is listed as 65 °C. Loss of nicking enzyme activity during storage at 45 °C appears the likely candidate for the decrease of performance of iSDA over time. The decrease in activity at the elevated temperature over time of nicking enzyme was expected because the reported optimal temperature of the enzyme is 37 °C.
Fig. 5.
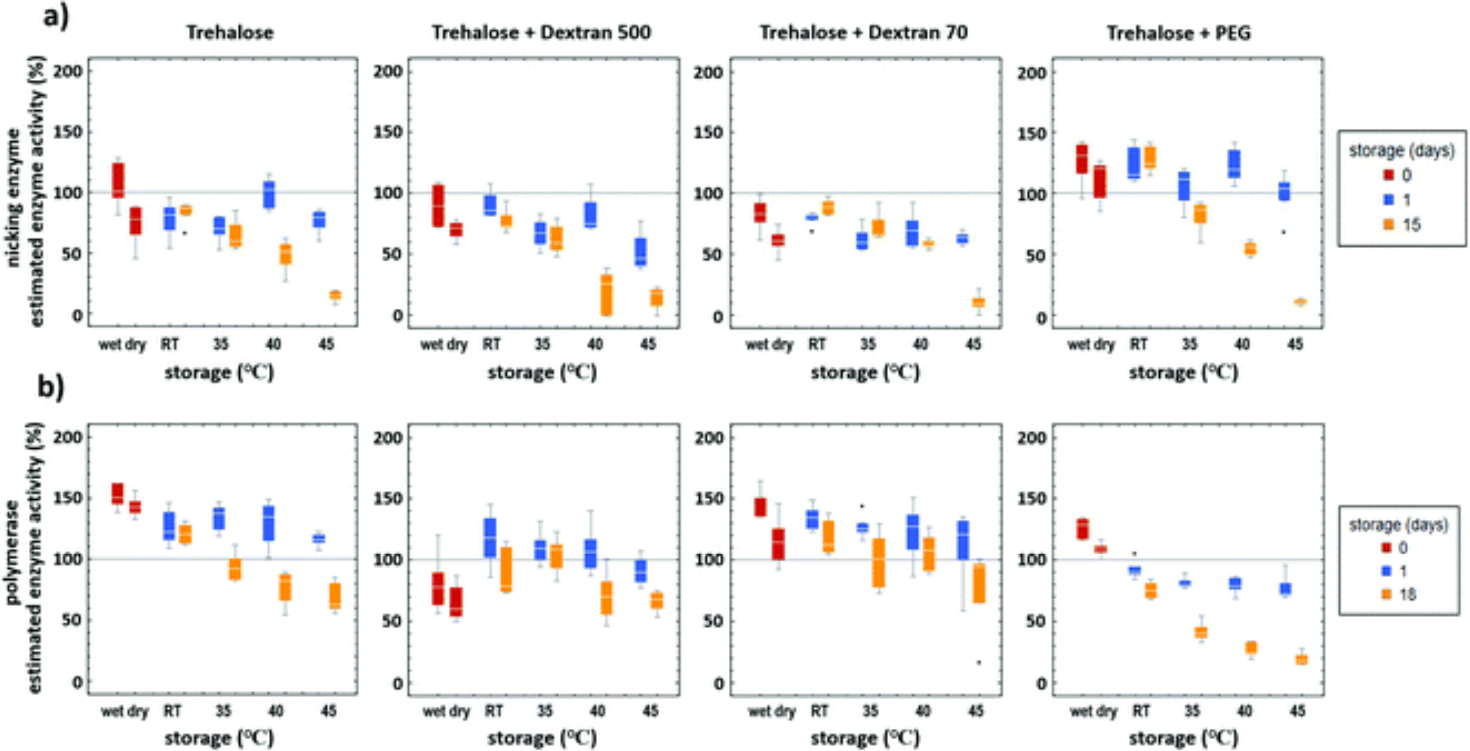
Enzyme activity after dry storage in Std 17 GF with different preservation formulations (Table 1) and temperatures. The results are mean of 5 replicates, and error bars are standard deviation. (a) Nicking enzyme assay showed decreased activity at elevated temperatures of storage after 15 days. (b) Polymerase activity does not seem to decrease appreciably over storage temperatures except for formulation with trehalose and PEG. PEG was overall detrimental to the storage of both the enzymes.
Limit of detection with dry stored iSDA reagents
Next, the limit of detection (LOD) for the iSDA reagents stored dry in Std 17 GF at 22 °C and 45 °C for 360 h with various formulations was determined by LF detection. From the LOD with fresh iSDA reagents (ESI Fig. S2a†), we determined that the linear range of the normalized signal intensity of the test line was below 100 copies. We, therefore, tested the ldh1 iSDA performance in the range of 10–200 genomic copies for all the formulations stored at 22 °C and 45 °C. The normalized test line signal intensities for varying copy numbers are given in Fig. 6. We found that the samples stored in the presence of both trehalose and dextran showed excellent stability down to 10 copies of genomic DNA for both at 22 °C and 45 °C and comparable to fresh reagents (ESI Fig S2a†). For samples with trehalose only, or with trehalose and PEG, the test line signal intensities were lower below 50 genomic copies at 22 °C storage compared to TD 70 and TD 500 (Fig. 6a), and near zero for samples stored at 45 °C indicating poor stability (Fig. 6b).
Fig. 6.
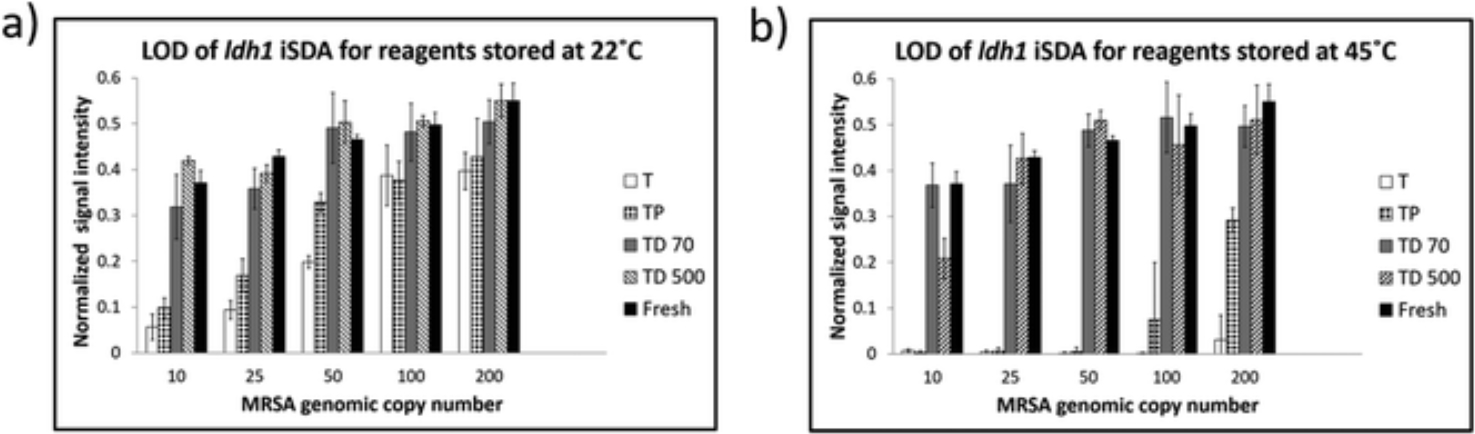
LOD of ldh1 iSDA with reagents stored dry in Std 17 GF at 22 °C and 45 °C for 360 h in different formulations of the preservatives. (a) Normalized intensity of the LF strip test line for samples stored at 22 °C. Samples with trehalose and dextran (TD 70 & TD 500) in the iSDA mix showed excellent stability down to 10 genomic copies. Samples with either trehalose only (T) or trehalose and PEG (TP) in the iSDA mix had diminishing signal intensity with decreasing genomic copy numbers. (b) Normalized intensity of the LF strip test line for samples stored at 45 °C. Excellent stability down to 10 copies was achieved for samples with trehalose and dextran (TD 70 & TD 500), whereas samples with trehalose (T) only or trehalose and PEG (TP) showed poor stability.
We also tested the LOD for the ldh1 iSDA by real-time fluorescence for the reagents stored in Std 17 GF with both trehalose and dextran (TD 500) at 22 °C and 45 °C for 360 h. We found that the lift-off time for all copy numbers (50–1000) was ~15 min (ESI Fig. S3†). The fluorescence peak intensities were a little higher for samples stored at 22 °C than 45 °C indicating slightly lower amplification efficiency at the higher storage temperature.
Long-term stability of iSDA reagents
To study the long-term stability in dry storage, the iSDA reagent mix and the Au label with 10% trehalose and 2.5% dextran (~70 kDa) were stored in Std 17 GF at room temperature (~22 °C) for one year. They were periodically tested by performing iSDA assay within the porous matrix with 100 copies of MRSA genomic DNA, and the amplicons detected by LF and gel electrophoresis. We found excellent stability of the iSDA reagents after one year of dry storage by LF detection and by gel electrophoresis (Fig. 7). The test line signal intensities on the LF strips were comparable to those with fresh reagents.
Fig. 7.
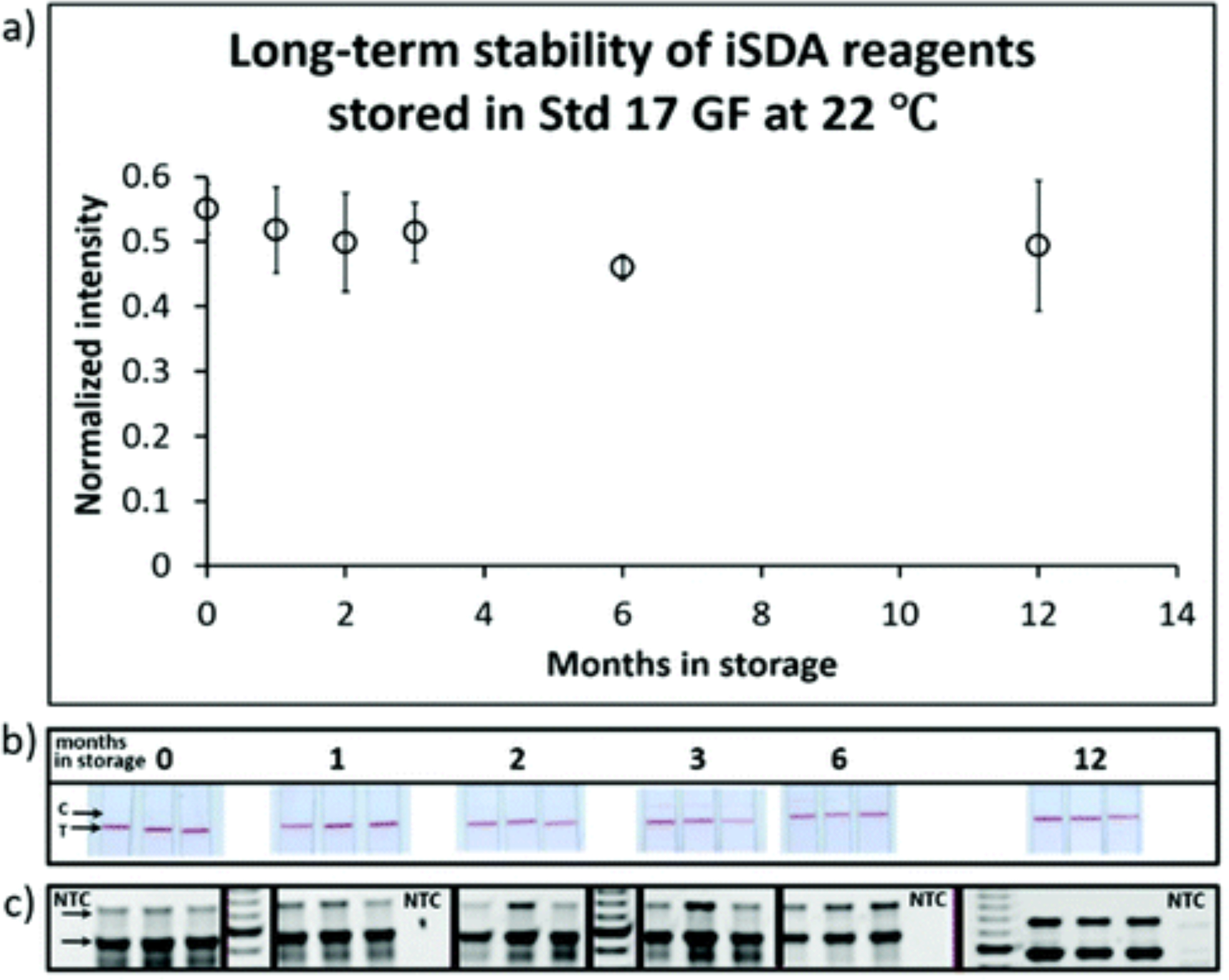
Long-term iSDA reagent stability in the Std 17 GF. (a) Normalized intensity of the LF test line after dry storage at 22 °C at different times. (b) Images of lateral flow detection strips at various time points. “C” indicates the control line, and “T” is the test line for ldh1 assay on the detection strips. (c) Corresponding gel images of the ldh1 amplicon products (indicated by arrow) at various time points. NTC is no template control. The iSDA reagents in the presence of 10% trehalose and 2.5% dextran performed well after 12 months of dry storage, with signal intensities similar to that observed with fresh reagents.
Our method of dry storage of isothermal amplification reagents in the porous matrix has several useful features. First, reagents stored in a glass fiber pad can be easily implemented for several applications with porous material fluid connectivity, especially in paper-based POC integrated systems. An image of the Std 17 GF pad with iSDA reagents lyophilized in the presence of 10% trehalose and 2.5% dextran before and after rehydration is shown in ESI Fig. S4a.† The glass fiber provides physical support during lyophilization, allowing passive spreading of the reagents across the matrix that allows rehydration and amplification function without excessive variation in reagent concentration throughout the volume of the pad. We also compared scanning electron micrographs of reagents dried in Std 17 GF in the presence of 10% trehalose, either by vacuum drying or by lyophilization. In the lyophilized sample, the reagents migrate to the small features of the Std 17 GF and appear as dry sheets stretched across the void with high surface-area-to-volume ratio compared to the vacuum dried samples, were reagents appear as large clumps (ESI Fig. S4b†). Differences in freezing rate, handling time, vacuum, and time-temperature profiles could all contribute to the differential distribution of the reagents once dry. These two different drying conditions could be expected to perform differently in both how well reagents are preserved, along with how the dry structures enable imbibition and eventual dissolution once wet. We observed differences in wetting: the lyophilized pads tended to wet faster and more uniformly than the vacuum-dried pads (data not shown).
Second, we included all the detection reagents, including LF probes and Au label, with the amplification reagents mix for dry storage. This is an advantage when implemented in an inexpensive POC device with colorimetric LF readout, as it alleviates the need to store the detection label separately, and the complexity necessitated by subsequent rehydration and mixing detection chemistry with the amplicon.
Third, we were also able to include the target-specific fluorescent probe in the dry pads in our study: this facilitates real-time fluorescence imaging of amplification using mobile phone in POC integrated systems (Shah et al., submitted). One potential disadvantage of our method is that scale-up requires creation of a specialized production facility for a specialized lyophilization procedure for the dry pads.
This method of dry storage of iSDA reagents along with the LF detection probes in the same mix has been incorporated into a sample-to-result instrument-free MAD NAAT device for the detection of MRSA from nasal swabs.40 A modification of this device with a USB-powered printed circuit board, the MD NAAT device, with dry reagents including fluorescence detection with mobile phone imaging has been described in a submitted publication (Shah et al.). Our future publications will describe our progress in adapting this dry-down method to include reverse transcriptase for isothermal amplification methods for detection of RNA viruses such as SARS-CoV-2.
Conclusions
We developed a method for dry preservation of all reagents necessary for iSDA amplification and detection of DNA targets in a porous matrix. The reagents also included either fluorescence probes or Au-label probes for detection, thus eliminating the need to store them separately. We showed that the iSDA reagents retained a high level of activity after dry storage in a glass fiber matrix with trehalose and dextran at temperatures up to 45 °C. Lateral flow readout of iSDA produced consistently detectable colorimetric signals at 10 copies or above (visual readout by eye or by scanner), while fluorescence readout was reliably measurable at 50 copies or greater (fluorescence measurement in a plate reader). We demonstrated long-term stability of reagents up to one year at 22 °C. Our method for drying the reagents onto a glass fiber pad has the benefit of easy incorporation into POC devices, including conventional microfluidic or paper-based devices, especially in places where the ambient temperatures are in the 40–45 °C range. Further, we have demonstrated iSDA in a fully integrated 2DPN MAD NAAT device with dry reagents and successfully validated with real patient samples.40 Dry preservation of amplification reagents in porous matrices could be used for a variety of applications and has particular advantages for use in POC devices with LF detection or real-time fluorescence readout, portability, and ease-of-use in low resource settings.
Supplementary Material
Acknowledgements
Support for this project was provided through funding from DARPA DSO grant number HR0011–11-2–0007. We wish to acknowledge ELITechGroup, for collaboration on this grant and providing us with the iSDA assay reagents and probes, and their twin probe lateral flow detection technology. The authors thank Peter Kaufman for building the custom electric oven. SEM imaging and sputter coating work was performed at the University of Washington Nanotech User Facility (NTUF), a member of the NSF-sponsored National Nanotechnology Infrastructure Network (NNIN).
Footnotes
Electronic supplementary information (ESI) available: Fig. S1–S4. See DOI: 10.1039/d0an01098g
Conflicts of interest
As of the date of this publication, Paul Yager has a nonpaying appointment as CSO of the UbiDX corporation. This work was performed before his association with the company.
References
- 1.Yager P, Domingo GJ and Gerdes J, Annu. Rev. Biomed. Eng, 2008, 10, 107–144. [DOI] [PubMed] [Google Scholar]
- 2.Derda R, Gitaka J, Klapperich CM, Mace CR, Kumar AA, Lieberman M, Linnes JC, Jores J, Nasimolo J, Ndung’u J, Taracha E, Weaver A, Weibel DB, Kariuki TM and Yager P, PLoS Neglected Trop. Dis, 2015, 9(5), e0003676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mothershed EA and Whitney AM, Clin. Chim. Acta, 2006, 363(1–2), 206–220. [DOI] [PubMed] [Google Scholar]
- 4.Ince J and McNally A, Expert Rev. Med. Devices, 2009, 6(6), 641–651. [DOI] [PubMed] [Google Scholar]
- 5.Mahony JB, Clin. Microbiol. Rev, 2008, 21(4), 716–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mori Y, Kanda H and Notomi T, J. Infect. Chemother, 2013, 19(3), 404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piepenburg O, Williams CH, Stemple DL and Armes NA, PLoS Biol, 2006, 4(7), e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker GT, Fraiser MS, Schram JL, Little MC, Nadeau JG and Malinowski DP, Nucleic Acids Res, 1992, 20(7), 1691–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helb D, Jones M, Story E, Boehme C, Wallace E, Ho K, Kop J, Owens MR, Rodgers R, Banada P, Safi H, Blakemore R, Ngoc Lan NT, Jones-López EC, Levi M, Burday M, Ayakaka I, Mugerwa RD, McMillan B, Winn-Deen E, Christel L, Dailey P, Perkins MD, Persing DH and Alland D, J. Clin. Microbiol, 2010, 48(1), 229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanriverdi S, Chen L and Chen S, J. Infect. Dis, 2010, 201(1), S52–S58. [DOI] [PubMed] [Google Scholar]
- 11.Ritchie AV, Ushiro-Lumb I, Edemaga D, Joshi HA, De Ruiter A, Szumilin E, Jendrulek I, McGuire M, Goel N, Sharma PI, Allain JP and Lee HH, J. Clin. Microbiol, 2014, 52(9), 3377–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Douthwaite ST, Walker C, Adams EJ, Mak C, Ortiz AV, Martinez-Alier N and Goldenberg SD, J. Clin. Microbiol, 2016, 54(1), 212–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niemz A, Ferguson TM and Boyle DS, Trends Biotechnol, 2011, 29(5), 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asiello PJ and Baeumner AJ, Lab Chip, 2011, 11, 1420–1430. [DOI] [PubMed] [Google Scholar]
- 15.Mahalanabis M, Do J, Almuayad H, Zhang JY and Klapperich CM, Biomed. Microdevices, 2010, 12(2), 353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang CH, Lien KY, Wang TY, Chen TY and Bin Lee G, Biosens. Bioelectron, 2011, 26(5), 2045–2052. [DOI] [PubMed] [Google Scholar]
- 17.Roskos K, Hickerson AI, Lu HW, Ferguson TM, Shinde DN, Klaue Y and Niemz A, PLoS One, 2013, 8(7), e69355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Magro L, Escadafal C, Garneret P, Jacquelin B, Kwasiborski A, Manuguerra JC, Monti F, Sakuntabhai A, Vanhomwegen J, Lafaye P and Tabeling P, Lab Chip, 2017, 17(14), 2347–2371. [DOI] [PubMed] [Google Scholar]
- 19.Choi JR, Hu J, Tang R, Gong Y, Feng S, Ren H, Wen T, Li XJ, Bakar WA, Pingguan-Murphy B and Xu F, Lab Chip, 2016, 16, 611–621. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez NM, Wong WS, Liu L, Dewar R and Klapperich CM, Lab Chip, 2016, 16(4), 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang W, Int. J. Pharm, 2000, 203(1–2), 1–60. [DOI] [PubMed] [Google Scholar]
- 22.Crowe LM, Reid DS and Crowe JH, Biophys. J, 1996, 71(4), 2087–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazzobre MF, Buera MDP and Chirife J, Biotechnol. Prog, 1997, 13(2), 195–199. [Google Scholar]
- 24.Ohtake S and Wang YJ, J. Pharm. Sci, 2011, 100(6), 2020–2053. [DOI] [PubMed] [Google Scholar]
- 25.Lutz S, Weber P, Focke M, Faltin B, Hoffmann J, Müller C, Mark D, Roth G, Munday P, Armes N, Piepenburg O, Zengerle R and Von Stetten F, Lab Chip, 2010, 10(7), 887–893. [DOI] [PubMed] [Google Scholar]
- 26.Al-Talib H, Yean CY, Al-Khateeb A, Hasan H and Ravichandran M, J. Microbiol., Immunol. Infect, 2014, 47(6), 484–490. [DOI] [PubMed] [Google Scholar]
- 27.Chua AL, Elina HT, Lim BH, Yean CY, Ravichandran M and Lalitha P, J. Med. Microbiol, 2011, 60(4), 481–485. [DOI] [PubMed] [Google Scholar]
- 28.Qu S, Shi Q, Zhou L, Guo Z, Zhou D, Zhai J and Yang R, PLoS Neglected Trop. Dis, 2010, 4(3), e629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bell J, Bonner A, Cohen DM, Birkhahn R, Yogev R, Triner W, Cohen J, Palavecino E and Selvarangan R, J. Clin. Virol, 2014, 61(1), 81–86. [DOI] [PubMed] [Google Scholar]
- 30.Ang GY, Yu CY, Chan KG, Singh KKB and Yean YC, J. Microbiol. Methods, 2015, 118, 99–105. [DOI] [PubMed] [Google Scholar]
- 31.Chander Y, Koelbl J, Puckett J, Moser MJ, Klingele AJ, Liles MR, Carrias A, Mead DA and Schoenfeld TW, Front. Microbiol, 2014, 5, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayashida K, Kajino K, Simukoko H, Simuunza M, Ndebe J, Chota A, Namangala B and Sugimoto C, Parasites Vectors, 2017, 10, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shetty P, Ghosh D, Singh M, Tripathi A and Paul D, RSC Adv, 2016, 6, 56205–56212. [Google Scholar]
- 34.Magro L, Jacquelin B, Escadafal C, Garneret P, Kwasiborski A, Manuguerra JC, Monti F, Sakuntabhai A, Vanhomwegen J, Lafaye P and Tabeling P, Sci. Rep, 2017, 7(1), 1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phillips EA, Moehling TJ, Ejendal KFK, Hoilett OS, Byers KM, Basing LA, Jankowski LA, Bennett JB, Lin LK, Stanciu LA and Linnes JC, Lab Chip, 2019, 19, 3375–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang R, Yang H, Gong Y, You ML, Liu Z, Choi JR, Wen T, Qu Z, Mei Q and Xu F, Lab Chip, 2017, 17, 1270–1279. [DOI] [PubMed] [Google Scholar]
- 37.Ramachandran S, Fu E, Lutz B and Yager P, Analyst, 2014, 139(6), 1456–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fu E, Liang T, Spicar-Mihalic P, Houghtaling J, Ramachandran S and Yager P, Anal. Chem, 2012, 84(10), 4574–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toley BJ, Covelli I, Belousov Y, Ramachandran S, Kline E, Scarr N, Vermeulen N, Mahoney W, Lutz BR and Yager P, Analyst, 2015, 140(22), 7540–7549. [DOI] [PubMed] [Google Scholar]
- 40.Lafleur LK, Bishop JD, Heiniger EK, Gallagher RP, Wheeler MD, Kauffman P, Zhang X, Kline EC, Buser JR, Kumar S, Byrnes SA, Vermeulen NMJ, Scarr NK, Belousov Y, Mahoney W, Toley BJ, Ladd PD, Lutz BR and Yager P, Lab Chip, 2016, 16, 3777–3787. [DOI] [PubMed] [Google Scholar]
- 41.Rasband WS, ImageJ: Image processing and analysis in Java, Astrophys. Source code Libr, 2012. [Google Scholar]
- 42.Stevens DY, Petri CR, Osborn JL, Spicar-Mihalic P, McKenzie KG and Yager P, Lab Chip, 2008, 12, 2038–2045. [DOI] [PubMed] [Google Scholar]
- 43.Lukhtanov EA, Lokhov SG, Gorn VV, Podyminogin MA and Mahoney W, Nucleic Acids Res, 2007, 35(5), e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nose K, Nagamine K, Tokuda J, Takino J and Hori T, Yakugaku Zasshi, 2013, 133(10), 1121–1126. [DOI] [PubMed] [Google Scholar]
- 45.Sasahara K, McPhie P and Minton AP, J. Mol. Biol, 2003, 326(4), 1227–1237. [DOI] [PubMed] [Google Scholar]
- 46.Christiansen A, Wang Q, Samiotakis A, Cheung M and Wittung-Stafshede P, Biochem, 2010, 49(31), 6519–6530. [DOI] [PubMed] [Google Scholar]
- 47.Sun WQ and Davidson P, CryoLetters, 2001, 22(5), 285–292. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.