

Abstract
The ability to successfully predict the three-dimensional structure of a protein from its amino acid sequence has made considerable progress in the recent past. The progress is propelled by the improved accuracy of deep learning-based inter-residue contact map predictors coupled with the rising growth of protein sequence databases. Contact map encodes interatomic interaction information that can be exploited for highly accurate prediction of protein structures via contact map threading even for the query proteins that are not amenable to direct homology modeling. As such, contact-assisted threading has garnered considerable research effort. In this chapter, we provide an overview of existing contact-assisted threading methods while highlighting the recent advances and discussing some of the current limitations and future prospects in the application of contact-assisted threading for improving the accuracy of low-homology protein modeling.
Keywords: Protein threading, Residue-residue contact, Contact-assisted threading, Template-based modeling, Protein structure prediction
1. Introduction
The computational prediction of the three-dimensional (3D) structure of a protein from its amino acid sequence remains elusive [1–4]. Despite the encouraging recent progress in ab initio protein structure prediction [5–12], template-based modeling (TBM) [13] remains one of the most reliable approaches in protein structure prediction [14–21], especially when homologous templates are available in the Protein Data Bank (PDB) [22]. TBM approaches can be broadly classified into homology modeling and protein threading based on the degree of homology. Homology modeling or comparative modeling is the process of building a structure of a query protein from a homologous template with a high degree of sequence similarity [23], whereas threading or fold recognition corresponds to an advanced template identification strategy where only distant homologs are available in the PDB but are not easily identifiable [3, 24, 25]. The primary objective of threading is to recognize one or more templates that are consistent with the query sequence, that is, existing folds that might be potentially analogous to the query sequence. Since its inception at the beginning of the 1990s [3, 24], threading remains an active area of research. The general principle behind protein threading is that there exists a finite number of unique folds in nature and many proteins (~90% [14]) share the same folds [26, 27], even though their sequences differ, illustrating that in theory the structure of most proteins can be successfully predicted by threading a query protein sequence onto a library of structural templates [14].
Current threading strategies are based on various techniques ranging from dynamic programming to profile-profile comparison based on hidden Markov models to more advanced machine learning approaches [18, 21, 28–49]. Some of these methods use only sequence-based features, while others [14, 18, 19, 31] use sequence and structure-based features for calculating the fitness score between the query and template. With the recent advances in residue-residue contact prediction [50–61] driven by sequence coevolution and deep learning, predicted contact information has become an additional structural feature in protein threading, leading to the development of numerous contact-assisted threading methods in the last few years [16, 20, 62–65]. The usefulness of these cutting-edge contact-assisted threading methods are particularly noteworthy in low-homology (see Note 1) protein modeling scenarios [63, 64, 66]. Here, we provide an overview of existing contact-assisted threading methods, highlighting some of the recent advances in low-homology protein modeling. We also discuss some of the current limitations and future prospects in contact-assisted threading.
2. Materials
Most threading methods have certain aspects in common. Here, we provide a brief overview of the common methodologies used in threading.
2.1. Template Library
Template library is a collection of representative protein structures (aka templates) from the PDB. A query protein sequence is threaded (or aligned) across each template in the library. Therefore, in order to minimize the time to search the whole template library, it is a common practice to make the library nonredundant by considering a small fraction of representative templates from a group of highly similar templates [14].
2.2. Query and Template Feature Set
Threading approaches use different sequential and structural features for the query protein and the templates. Below, we briefly discuss various common features used in threading.
2.2.1. Sequence Profiles
Sequence profile contains the evolutionary information as well as the sequence diversity among homologous sequences of the query protein. A sequence profile is considered as a rich source of information in threading because homologous proteins tend to have similar sequence profiles. Programs such as PSI-BLAST [17] and HHblits [67] can be used to generate sequence profiles.
2.2.2. Secondary Structures
A protein’s local conformation may be defined in terms of its secondary structure and using the secondary structure as a feature in threading has attracted much attention since the early days of threading approaches. The secondary structure of a query protein can be predicted using secondary structure predictors such as PSIPRED [68], SPIDER3 [69], and RaptorX Property [70]. Most of these methods predict the likelihood of various secondary structure types. Most popular secondary structure predictors [16, 18, 71] use the three-class secondary structures (alpha helices, beta strands, and loop), even though some of the recent threading methods [72] use both three-class and eight-class secondary structure types. While the secondary structure of a query protein is typically predicted from its sequence information, the secondary structures of the template proteins are calculated directly from the PDB structures using programs such as DSSP [73] and STRIDE [74].
2.2.3. Solvent Accessibility
Solvent accessibility is related to the spatial organization and packing of residues and is therefore considered as an important feature for threading. Solvent accessibility can be categorized using binary classification (buried or exposed) or using a three-class classification (buried, intermediate, and exposed). While solvent accessibility predictors such as PSIPRED, SPIDER3, and RaptorX Property are typically used to predict the solvent accessibility of each residue in the query protein, DSSP and STRIDE can be used for calculating that of the template.
2.2.4. Backbone Dihedral Angles
A protein’s dihedral angle is the angle of the polypeptide backbone where two neighboring planes meet. The dihedral angles for the query protein can be predicted using predictors such as PSIPRED and SPIDER3.
2.2.5. Additional Features
In addition to these features, structure profiles, hydrophobicity, and amino acid substitution matrix such as BLOSUM are also considered as features for threading [71, 72]. Contact-assisted threading methods use the pairwise predicted (or native) contact information for a query (or template) protein because contact information is considered as a rich source of information for threading. Contact-assisted threading methods use contact information either implicitly such as in PROSPECT [46], PROSPECTOR [75, 76], and RAPTOR [14] or explicitly such as in EigenTHREADER [20], map_align [62], CEthreader [63], CATHER [64], ThreaderAI [65], and our in-house threading method [16].
2.3. Threading Performance Measure
Measuring the structural similarity between the predicted and the native protein 3D structure is critically important for objectively evaluating the performance of a threading method. Some most commonly used scores are the template modeling score (TM-score) [77], the root-mean-square deviation (RMSD) [78], the global distance test (GDT) [79], and the local distance difference test (lDDT) [80]. TM-score is one of the most widely used scoring metrics having scores in the range (0, 1) with higher scores indicating better similarities. A TM-score >0.5 typically indicates the correct overall fold [81].
3. Methods
3.1. Overview of Protein Threading
The goal of protein threading is to optimally align a query sequence to a known structural template [82]. This requires identifying the correct or best-fit template from a library of templates and the optimal query-template alignment from the space of all possible query-template alignments. The query-template alignment represents a correspondence between each query residue and the spatial positioning of the aligned template residues. Overall, protein threading can be mainly considered to involving three components: (1) a threading scoring function that evaluates the fitness of query-template alignments, (2) identification of the best-fit structural template from the library of templates, and (3) an optimal alignment of the query sequence to the template. In the following, we discuss each component in more details.
3.1.1. Threading Scoring Function
The scoring function plays an important role to quantitatively assess the fitness of query-template alignments [14]. The scoring function normally consists of the profile similarity score, the structural consistency score, and the gap penalty. The profile similarity score can be calculated by comparing the query and template profiles. It quantifies how the query is evolutionary related to the template. The structural consistency score contains two components: consistency of local structures such as secondary structure and solvent accessibility compatibility and consistency of global structures or pairwise interatomic interactions. Weights can be used in the scoring function to control the relative importance of different scoring terms.
3.1.2. Template Selection
Identifying the best-fit template inevitably requires using the alignment score of query-template alignments. The raw query-template alignment score cannot be directly used to rank templates due to the biases introduced by the protein length [14]. Both machine learning-based methods and Z-score are used to mitigate the bias. Several protein threading methods [40, 46, 83–85] use machine learning models such as the neural network for the template ranking by formulating the template selection as a classification problem, even though a majority of the threading methods [18, 63, 64] rely on Z-score for the template selection. Z-scores of the query-template pair are computed from the means and standard deviations of the scores of the query sequence with all templates of the template library. However, it cannot cancel out all the biases introduced by the protein length. A large protein appears to have a high Z-score. It is also difficult to interpret the Z-score, particularly when the scoring function is the weighted sum of different scoring terms [14].
3.1.3. Optimal Query-Template Alignment
The optimal query-template alignment is the alignment that optimally aligns residues in the query sequence homologous to residues in the template. It is often the case that a threading scoring function is effective in selecting the homologous template, but the query-template alignment is significantly weak [25, 86]. In such cases, the alignment may be suboptimal, which might result in less accurate template-based models built from such an alignment, that is, the sensitivity of query-template alignment directly affects the overall performance of template-based modeling.
3.2. Contact-Assisted Protein Threading
3.2.1. Residue-Residue Contact Map
A contact map of a protein is a binary, square, symmetric matrix with vertices corresponding to residues of the protein, and a contact edge indicates that the distance between a residue pair is smaller than a given threshold. Typically, this distance threshold is considered 8 Å between the Cα and Cβ atoms of the residue pairs [16, 20]. Here, the set of contacts between residue pair (i, j) is defined as:
| (1) |
where dij is the distance between the residue pair (i, j). Figure 1 shows a representative protein 3D structure and its corresponding 2D residue-residue contact map.
Fig. 1.
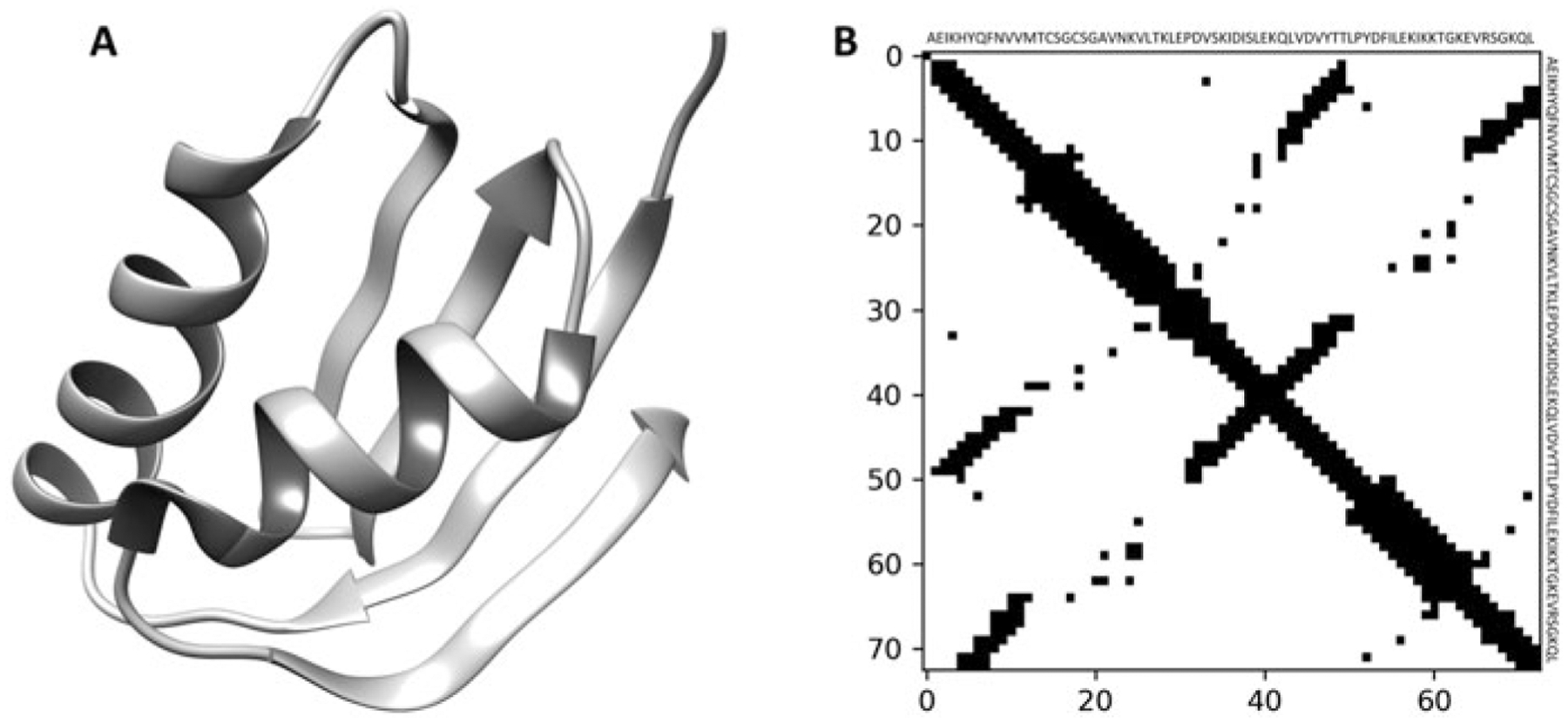
A representative protein 3D structure and its corresponding 2D binary contact map. (a) 3D structure of a representative protein (PDB ID 1cc8A), (b) the corresponding 2D residue-residue contact map, considering Cα atoms and a distance threshold of 8 Å
3.2.2. Contact Map Alignment
Contact map alignment is a way of measuring the similarity between two contact maps. The maximum contact map overlap problem tries to evaluate the similarity of the two proteins by calculating the maximum overlap between their contact maps while preserving the ordering of residues of both sequences, leading to a pairwise sequence alignment as illustrated in Fig. 2. Since direct contact map alignment is computationally expensive [63], several approximation algorithms [62, 87–92] have been developed to address the contact map alignment problem including the eigendecomposition-based strategy, graphlet degree-based approach, and iterative double dynamic programming-based approach. Eigendecomposition decomposes a contact map into eigenvectors and corresponding eigenvalues. This approach compares two proteins by comparing their contact map eigenvectors, which can be performed in polynomial time. For example, approaches such as EIGAs [87], SABERTOOTH [89], and Al-Eigen [90] use the eigendecomposition to approximate contact maps using the top eigenvectors and use the global alignment of key eigenvectors to find the similarity between two contact maps. GR-Align [92] is a fast contact map alignment approach based on graphlet degree distribution. Moreover, [93] proposes a contact map alignment algorithm C-Align based on Cα atoms using dynamic programming. Recent methods such as map_align [62] employ iterative double dynamic programming to calculate contact map alignment, with the goal of optimizing the number of contact overlaps while minimizing the number of gaps.
Fig. 2.
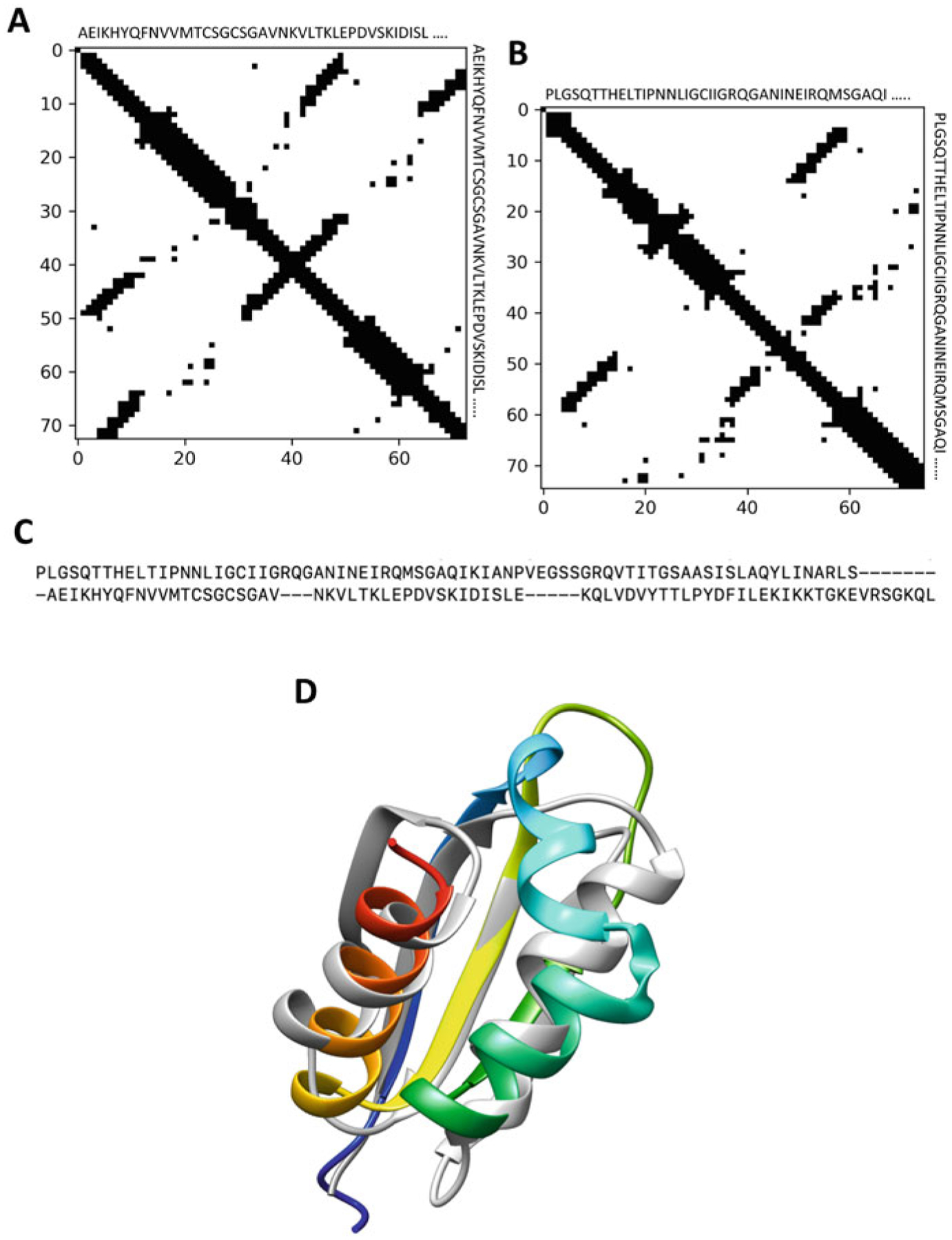
Contact map alignment. (a) contact map of a representative protein (PDB ID 1cc8A), (b) contact map of another representative protein (PDB ID 1wvnA), (c) sequence alignment of 1cc8A and 1wvnA using Al-Eigen. In both cases, Cα atoms and the distance threshold of 8 Å are considered. (d) 1wvnA (in rainbow) is structurally superimposed on 1cc8A (in gray)
3.3. Overview of Existing Contact-Assisted Threading Methods
Table 1 shows several publicly available contact-assisted threading methods. These approaches can be broadly subdivided into two classes: (1) methods that implicitly use contact information via pairwise contact potential such as PROSPECT [46], PROSPECTOR [75, 76], and RAPTOR [14]; and (2) methods that explicitly use contact information via predicted residue-residue contacts including the current state-of-the-art contact-assisted threading methods such as EigenTHREADER [20], map_align [62], CEthreader [63], CATHER [64], ThreaderAI [65], and our in-house threading method [16]. We briefly discuss them below.
Table 1.
Selected publicly accessible threading methods that implicitly or explicitly use contact information
| Name (reference) | Method | Availability |
|---|---|---|
| PROSPECT (Xu and coworkers [46]) | Divide-and-conquer algorithm | http://compbio.ornl.gov/structure/prospect/ |
| PROSPECTOR (Skolnick and coworkers [75, 76]) | Hierarchical approach | http://bioinformatics.danforthceneter.org/services/threading.html |
| RAPTOR (Xu and coworkers [14]) | Linear programming | http://www.cs.uwaterloo.ca/~j3xu/RAPTOR_form.htm |
| EigenTHREADER (Jones and coworkers [20]) | Dynamic programming and eigendecomposition | http://bioinfadmin.cs.ucl.ac.uk/downloads/eigenTHREADER/ |
| map_align (Baker and coworkers [62]) | Iterative double dynamic programming | https://github.com/sokrypton/map_align |
| CEthreader (Zhang and coworkers [63]) | Dynamic programming and eigendecomposition | https://zhanglab.ccmb.med.umich.edu/CEthreader/ |
| CATHER (Yang and coworkers [64]) | Iterative double dynamic programming | https://yanglab.nankai.edu.cn/CATHER/ |
| ThreaderAI (Shen and coworkers [65]) | Deep residual neural network and dynamic programming | https://github.com/ShenLab/ThreaderAI |
3.3.1. Threading Methods That Implicitly Use Contact Information via Pairwise Contact Potential
PROSPECT (PROtein Structure Prediction and Evaluation Computer Toolkit) [46] is one of the earliest protein threading methods, which makes use of pairwise contact potential by introducing a contact term into its scoring function. This study considers that pairwise contact potentials are measured only between core secondary structures. The contact cutoff is set at 7 Å between the Cβ atoms. Additionally, the method uses a divide-and-conquer algorithm for the alignment searching procedure. Another method, PROSPECTOR (PROtein Structure Predictor Employing Combined Threading to Optimize Results) [75, 76], uses a “partly thawed” technique to assess the contact potential based on the previous alignment iterations. RAPTOR (RApid Protein Threading by Operation Research technique) [14] is another protein threading method that introduces contact capacity score. It considers only contacts between two core residues where the spatial distance between Cα atoms is 7 Å with a sequence separation of 4. It addresses threading as a problem of wide-scale integer programming, relaxes it to a problem of linear programming, and uses a branch-and-bound approach to solve the integer program. However, the performance contribution of pairwise contact potential in the above methods is not significant compared to that of sequence profile, particularly for distantly related proteins. The underlying reason may be noisy contacts that do not hold any extra signal, yielding just modest improvement.
3.3.2. Threading Methods That Explicitly Use Contact Information via Predicted Residue-Residue Contacts
Recent successful applications of deep learning have resulted in significantly improved inter-residue contact prediction methods [53, 56, 60, 94]. As such, the newest contact-assisted threading methods have been explicitly integrating predicted residue-residue contact information to improve threading performance. EigenTHREADER [20], developed in 2017, extends Al-Eigen [90] to enable threading by predicting a protein’s contact map using classical neural network-based predictor MetaPSICOV [53] and then searching a library of templates’ contact maps. Despite the superior performance of EigenTHREADER over other profile-based threading methods for low-homology threading, it can be further improved by integrating other linear features such as sequence profiles along with inter-residue contact maps. map_align [62], developed in 2017, proposes an iterative double dynamic programming algorithm [95] that aligns contact maps, predicted by pure coevolutionary-based predictor GREMLIN [96], in combination with metagenomic sequences of microbial DNA [97]. The elevated performance of map_align can be attributed to the contribution of contact maps in low-homology threading. However, considering that the outcomes rely on the initial estimate of the similarity matrix, which is not always optimal, this approach does not necessarily guarantee optimal solutions. CEthreader [63] (Contact Eigenvector-based threader), developed in 2019, uses contact maps predicted from deep residual neural-network-based predictor ResPRE [94]. Similar to Al-Eigen, this work uses the eigendecom-position technique to approximate contact maps by the cross product of single-body eigenvectors. CEthreader introduces a dot-product scoring function by incorporating contact information along with secondary structures and sequence profiles to align contact eigenvectors and uses dynamic programming to generate the query-template alignments. However, the method can be further strengthened by considering negative eigenvalues in addition to positive eigenvalues, since the incorporation of both positive and negative eigenvalues restores the contact map. Another new contact-assisted threading algorithm CATHER [64] (contact-assisted THreadER), developed in 2020, uses both conventional sequential profiles and contact maps predicted by a deep learning-based method MapPred [98]. A very recent method ThreaderAI [65] integrates deep learning-based contact information with traditional sequential and structural features by formulating the task of threading as the classical computer vision’s classification problem. This work introduces a deep residual neural network to predict query-template alignments. Based on the reported results of the above methods, contact-assisted threading methods significantly outperform profile-based threading methods by a large margin, particularly for low-homology targets.
Our in-house threading method [16], developed in 2019, integrates the standard threading technique along with inter-residue contact information predicted by the state-of-the-art ultra-deep learning-based method RaptorX [56]. First, our method applies the standard threading technique to select the top templates based on the Z-score and then applies the contact map overlap score using Al-Eigen along with the Z-score to calculate the final score for selecting the best-fit template. Based on large-scale bench-marking results, this method outperforms profile-based threading method MUSTER as well as other contact-assisted threading methods EigenTHREADER and map_align.
3.4. Significance of Contact Maps Quality in Threading
While incorporating contact information into threading is highly effective, our recent study [99] shows the impact of diverse quality of contact maps on contact-assisted threading performance in that integrating high-quality contacts having the Matthews correlation coefficient (MCC) ≥0.5 results in improved threading performance for ~30% of the cases, while low-quality contacts having MCC <0.35 degrade the threading performance for 50% of the cases. The results reveal the reciprocal coupling between the quality of predicted contact maps and contact-assisted threading performance and indicate that the rapid advancement in contact prediction methods powered by deep learning can synergistically assist contact-assisted threading, leading to improved low-homology protein modeling.
3.5. Growth of Protein Sequence Databases and Its Implication in Threading
Since most contact map predictions, secondary structure predictions, and sequence profiles depend on the evolutionary signal derived from multiple sequence alignments (MSA) (see Note 2), the adequate number of homologous sequences is critical to the success of these approaches. This limitation can be largely overcome by taking advantage of the fast-paced growth of whole-genome sequence databases such as the nr database compiled by the National Center for Biotechnology Information (NCBI), UniRef [100], UniProt [101], Uniclust [102], as well as metagenome databases from the European Bioinformatics Institute (EBI) Meta-genomics [103, 104] and Metaclust [105]. For instance, with the addition of two billion metagenomic protein sequences, there is a significant increase in the number of families of unknown structures, which can now be reliably modeled by using the coevolutionary information [62]. A recent paper [106] demonstrates improved protein structure prediction through marine metagenomics for low-homology proteins, illustrating the potential usefulness of growing sequence databases on protein structure prediction. Two newest emerging sequence databases, BFD [107] and MGnify [108], may further enrich the evolutionary information. Recently, DeepMSA [109] method for generating multiple sequence alignment information shows the benefit of generating deep multiple sequence alignment by combining the multiple sequence databases for threading as well as contact predictions.
3.6. Discussion
The improved performance of contact-assisted threading methods is attributed to successfully integrating inter-residue contact information along with traditional linear and nonlinear threading features. Although contact-assisted threading approaches have witnessed promising progress so far, but there is still room for improvement with the advancement of deep learning-based inter-residue distance prediction [6, 7, 110–113] instead of binary contacts (see Note 3). A protein can be represented by a 2D inter-residue distance map, where a distance map is a square, symmetric matrix with vertices corresponding to residues of the protein and an edge indicates the distance between a residue pair. As distances carry more information than contacts [85], recent distance-based threading method DeepThreader [85] shows further improvement, particularly for low-homology threading, by outperforming existing contact-assisted threading approaches. Inspired by the promising results, CEthreader method is extended to distance-guided threading method DEthreader in the recently concluded 14th critical assessment of protein structure prediction (CASP14) experiment (see Note 4) by adding a distance map-based energy term in the threading scoring function. Similarly, CATHER has also replaced contacts with distances in CASP14. Our most recent threading method DisCovER [71] (distance- and orientation-based Covariational threadER) goes one step further by effectively integrating information from inter-residue distance and orientation along with the topological network neighborhood (see Note 5) of a query-template alignment. DisCovER shows the usefulness of incorporating inter-residue orientation along with distance information together with the neighborhood effect induced by the query-template alignment, leading to improved threading performance.
While no single-template threading method works well for all types of targets [13], multiple-template approaches as well as meta-approaches work better in protein structure prediction [41, 45, 66]. For instance, previous multiple-template approaches [45, 114–119] demonstrate their superior performance over the best single-template threading method by attaining better alignments. Moreover, meta-approaches [41, 66, 120] show promising results over individual approaches, particularly for distantly homologous proteins. In the case of meta-servers, there is a need to select the top model based on various scoring functions by scoring predicted 3D models using model quality assessment programs (MQAPs), including single-model [121–130] and consensus [131–134] methods. Furthermore, even when using the most advanced template-based modeling pipeline, predicted models often fail to reach near-native accuracy. Protein structure refinement methods [135–144] are needed to bring these moderately accurate predicted models closer to the native state.
Acknowledgments
This work was supported in part by the National Science Foundation (IIS2030722, DBI1942692 to DB) and the National Institute of General Medical Sciences (R35GM138146 to DB).
Footnotes
Low homology refers to the lack of available sufficient homologous information for the query sequence.
Multiple sequence alignment refers to the alignment of evolutionary-related protein sequences.
A binary contact indicates that the distance between a residue pair is smaller than a given distance threshold, typically 8 Å.
CASP is a community-wide blind assessment of protein structure prediction, taking place in each alternative year since 1994.
Network neighborhood attempts to capture the similarity between the neighboring residues. It works on the assumption that a pair of query-template residues are likely to be aligned if their adjacent residues are also aligned.
References
- 1.Dill KA, MacCallum JL (2012) The protein-folding problem, 50 years on. Science 338: 1042–1046. 10.1126/science.1219021 [DOI] [PubMed] [Google Scholar]
- 2.Baker D, Sali A (2001) Protein structure prediction and structural genomics. Science 294: 93–96. 10.1126/science.1065659 [DOI] [PubMed] [Google Scholar]
- 3.Jones DT, Taylor WR, Thornton JM (1992) A new approach to protein fold recognition. Nature 358:86–89. 10.1038/358086a0 [DOI] [PubMed] [Google Scholar]
- 4.Moult J, Fidelis K, Kryshtafovych A, Schwede T, Tramontano A (2014) Critical assessment of methods of protein structure prediction (CASP) — round x. Proteins 82: 1–6. 10.1002/prot.24452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang S, Li W, Zhang R, Liu S, Xu J (2016) CoinFold: a web server for protein contact prediction and contact-assisted protein folding. Nucleic Acids Res 44:W361–W366. 10.1093/nar/gkw307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D (2020) Improved protein structure prediction using predicted interresidue orientations. PNAS 117:1496–1503. 10.1073/pnas.1914677117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greener JG, Kandathil SM, Jones DT (2019) Deep learning extends de novo protein modelling coverage of genomes using iteratively predicted structural constraints. Nat Commun 10:1–13. 10.1038/s41467-019-11994-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adhikari B, Bhattacharya D, Cao R, Cheng J (2015) CONFOLD: residue-residue contact-guided ab initio protein folding. Proteins 83: 1436–1449. 10.1002/prot.24829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adhikari B, Cheng J (2018) CONFOLD2: improved contact-driven ab initio protein structure modeling. BMC Bioinformatics 19: 22. 10.1186/s12859-018-2032-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marks DS, Colwell LJ, Sheridan R, Hopf TA, Pagnani A, Zecchina R, Sander C (2011) Protein 3D structure computed from evolutionary sequence variation. PLoS One 6:e28766. 10.1371/journal.pone.0028766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roche R, Bhattacharya S, Bhattacharya D (2020) Hybridized distance- and contact-based hierarchical structure modeling for folding soluble and membrane proteins. PLoS Comput Biol 17:e1008753. 10.1371/journal.pcbi.1008753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J (2019) Distance-based protein folding powered by deep learning. PNAS 116: 16856–16865. 10.1073/pnas.1821309116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y (2008) Progress and challenges in protein structure prediction. Curr Opin Struct Biol 18:342–348. 10.1016/j.sbi.2008.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu J, Li M, Kim D, Xu Y (2003) Raptor: optimal protein threading by linear programming. J Bioinforma Comput Biol 01:95–117. 10.1142/S0219720003000186 [DOI] [PubMed] [Google Scholar]
- 15.Källberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J (2012) Template-based protein structure modeling using the RaptorX web server. Nat Protoc 7:1511–1522. 10.1038/nprot.2012.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhattacharya S, Bhattacharya D (2019) Does inclusion of residue-residue contact information boost protein threading? Proteins 87: 596–606. 10.1002/prot.25684 [DOI] [PubMed] [Google Scholar]
- 17.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu S, Zhang Y (2008) MUSTER: improving protein sequence profile–profile alignments by using multiple sources of structure information. Proteins 72:547–556. 10.1002/prot.21945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu S, Zhang Y (2010) Recognizing protein substructure similarity using segmental threading. Structure 18:858–867. 10.1016/j.str.2010.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buchan DWA, Jones DT (2017) EigenTHREADER: analogous protein fold recognition by efficient contact map threading. Bioinformatics 33:2684–2690. 10.1093/bioinformatics/btx217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lobley A, Sadowski MI, Jones DT (2009) pGenTHREADER and pDomTHREADER: new methods for improved protein fold recognition and superfamily discrimination. Bioinformatics 25:1761–1767. 10.1093/bioinformatics/btp302 [DOI] [PubMed] [Google Scholar]
- 22.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–242. 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moult J (1996) The current state of the art in protein structure prediction. Curr Opin Biotechnol 7:422–427. 10.1016/S0958-1669(96)80118-2 [DOI] [PubMed] [Google Scholar]
- 24.Bowie JU, Luthy R, Eisenberg D (1991) A method to identify protein sequences that fold into a known three-dimensional structure. Science 253:164–170. 10.1126/science.1853201 [DOI] [PubMed] [Google Scholar]
- 25.Petrey D, Honig B (2005) Protein structure prediction: inroads to biology. Mol Cell 20: 811–819. 10.1016/j.molcel.2005.12.005 [DOI] [PubMed] [Google Scholar]
- 26.Kinch LN, Grishin NV (2002) Evolution of protein structures and functions. Curr Opin Struct Biol 12:400–408. 10.1016/S0959-440X(02)00338-X [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Skolnick J (2005) The protein structure prediction problem could be solved using the current PDB library. PNAS 102: 1029–1034. 10.1073/pnas.0407152101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Y, Faraggi E, Zhao H, Zhou Y (2011) Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics 27:2076–2082. 10.1093/bioinformatics/btr350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma J, Wang S, Zhao F, Xu J (2013) Protein threading using context-specific alignment potential. Bioinformatics 29:i257–i265. 10.1093/bioinformatics/btt210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng J, Xu J (2010) Low-homology protein threading. Bioinformatics 26:i294–i300. 10.1093/bioinformatics/btq192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Söding J (2005) Protein homology detection by HMM–HMM comparison. Bioinformatics 21:951–960. 10.1093/bioinformatics/bti125 [DOI] [PubMed] [Google Scholar]
- 32.Peng J, Xu J (2009) Boosting protein threading accuracy. In: Batzoglou S (ed) Research in computational molecular biology. Springer, Berlin Heidelberg, pp 31–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma J, Peng J, Wang S, Xu J (2012) A conditional neural fields model for protein threading. Bioinformatics 28:i59–i66. 10.1093/bioinformatics/bts213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaroszewski L, Rychlewski L, Li Z, Li W, Godzik A (2005) FFAS03: a server for profile–profile sequence alignments. Nucleic Acids Res 33:W284–W288. 10.1093/nar/gki418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rychlewski L, Li W, Jaroszewski L, Godzik A (2000) Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci 9:232–241. 10.1110/ps.9.2.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng J, Baldi P (2006) A machine learning information retrieval approach to protein fold recognition. Bioinformatics 22:1456–1463. 10.1093/bioinformatics/btl102 [DOI] [PubMed] [Google Scholar]
- 37.Marti-Renom MA, Madhusudhan MS, Sali A (2004) Alignment of protein sequences by their profiles. Protein Sci 13:1071–1087. 10.1110/ps.03379804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ginalski K, Pas J, Wyrwicz LS, Grotthuss M v, Bujnicki JM, Rychlewski L (2003) ORFeus: detection of distant homology using sequence profiles and predicted secondary structure. Nucleic Acids Res 31:3804–3807. 10.1093/nar/gkg504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou H, Zhou Y (2005) Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments. Proteins 58: 321–328. 10.1002/prot.20308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones DT (1999) GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences 11 Edited by B. Honig. J Mol Biol 287:797–815. 10.1006/jmbi.1999.2583 [DOI] [PubMed] [Google Scholar]
- 41.Wu S, Zhang Y (2007) LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res 35:3375–3382. 10.1093/nar/gkm251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gniewek P, Kolinski A, Kloczkowski A, Gront D (2014) BioShell-threading: versatile Monte Carlo package for protein 3D threading. BMC Bioinformatics 15:22. 10.1186/1471-2105-15-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rost B, Schneider R, Sander C (1997) Protein fold recognition by prediction-based threading 11 Edited by F. E. Cohen. J Mol Biol 270: 471–480. 10.1006/jmbi.1997.1101 [DOI] [PubMed] [Google Scholar]
- 44.Olmea O, Rost B, Valencia A (1999) Effective use of sequence correlation and conservation in fold recognition 11 Edited by J. M. Thornton. J Mol Biol 293:1221–1239. 10.1006/jmbi.1999.3208 [DOI] [PubMed] [Google Scholar]
- 45.Peng J, Xu J (2011) A multiple-template approach to protein threading. Proteins 79: 1930–1939. 10.1002/prot.23016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Y, Xu D (2000) Protein threading using PROSPECT: design and evaluation. Proteins 40:343–354. [DOI] [PubMed] [Google Scholar]
- 47.Ma J, Wang S, Wang Z, Xu J (2014) MRFalign: protein homology detection through alignment of Markov random fields. PLoS Comput Biol 10:e1003500. 10.1371/journal.pcbi.1003500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan R, Xu D, Yang J, Walker S, Zhang Y (2013) A comparative assessment and analysis of 20 representative sequence alignment methods for protein structure prediction. Sci Rep 3:2619. 10.1038/srep02619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee SY, Skolnick J (2010) TASSER_WT: a protein structure prediction algorithm with accurate predicted contact restraints for difficult protein targets. Biophys J 99:3066–3075. 10.1016/j.bpj.2010.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones DT, Buchan DWA, Cozzetto D, Pontil M (2012) PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics 28:184–190. 10.1093/bioinformatics/btr638 [DOI] [PubMed] [Google Scholar]
- 51.Seemayer S, Gruber M, Söding J (2014) CCMpred—fast and precise prediction of protein residue–residue contacts from correlated mutations. Bioinformatics 30:3128–3130. 10.1093/bioinformatics/btu500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaján L, Hopf TA, Kalaš M, Marks DS, Rost B (2014) FreeContact: fast and free software for protein contact prediction from residue co-evolution. BMC Bioinformatics 15:85. 10.1186/1471-2105-15-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones DT, Singh T, Kosciolek T, Tetchner S (2015) MetaPSICOV: combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics 31:999–1006. 10.1093/bioinformatics/btu791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adhikari B, Hou J, Cheng J (2018) DNCON2: improved protein contact prediction using two-level deep convolutional neural networks. Bioinformatics 34:1466–1472. 10.1093/bioinformatics/btx781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanson J, Paliwal K, Litfin T, Yang Y, Zhou Y (2018) Accurate prediction of protein contact maps by coupling residual two-dimensional bidirectional long short-term memory with convolutional neural networks. Bioinformatics 34:4039–4045. 10.1093/bioinformatics/bty481 [DOI] [PubMed] [Google Scholar]
- 56.Wang S, Sun S, Li Z, Zhang R, Xu J (2017) Accurate De novo prediction of protein contact map by ultra-deep learning model. PLoS Comput Biol 13:e1005324. 10.1371/journal.pcbi.1005324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ovchinnikov S, Kamisetty H, Baker D (2014) Robust and accurate prediction of residue–residue interactions across protein interfaces using evolutionary information. eLife 3: e02030. 10.7554/eLife.02030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang S, Li Z, Yu Y, Xu J (2017) Folding membrane proteins by deep transfer learning. Cell Syst 5:202–211.e3. 10.1016/j.cels.2017.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morcos F, Pagnani A, Lunt B, Bertolino A, Marks DS, Sander C, Zecchina R, Onuchic JN, Hwa T, Weigt M (2011) Direct-coupling analysis of residue coevolution captures native contacts across many protein families. PNAS 108:E1293–E1301. 10.1073/pnas.1111471108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kandathil SM, Greener JG, Jones DT (2019) Prediction of interresidue contacts with DeepMetaPSICOV in CASP13. Proteins 87: 1092–1099. 10.1002/prot.25779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He B, Mortuza SM, Wang Y, Shen H-B, Zhang Y (2017) NeBcon: protein contact map prediction using neural network training coupled with naı¨ve Bayes classifiers. Bioinformatics 33:2296–2306. 10.1093/bioinformatics/btx164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ovchinnikov S, Park H, Varghese N, Huang P-S, Pavlopoulos GA, Kim DE, Kamisetty H, Kyrpides NC, Baker D (2017) Protein structure determination using metagenome sequence data. Science 355:294–298. 10.1126/science.aah4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zheng W, Wuyun Q, Li Y, Mortuza SM, Zhang C, Pearce R, Ruan J, Zhang Y (2019) Detecting distant-homology protein structures by aligning deep neural-network based contact maps. PLoS Comput Biol 15: e1007411. 10.1371/journal.pcbi.1007411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Du Z, Pan S, Wu Q, Peng Z, Yang J (2020) CATHER: a novel threading algorithm with predicted contacts. Bioinformatics 36:2119–2125. 10.1093/bioinformatics/btz876 [DOI] [PubMed] [Google Scholar]
- 65.Zhang H, Shen Y (2020) Template-based prediction of protein structure with deep learning. BMC Genomics 21:878. 10.1186/s12864-020-07249-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng W, Zhang C, Wuyun Q, Pearce R, Li Y, Zhang Y (2019) LOMETS2: improved meta-threading server for fold-recognition and structure-based function annotation for distant-homology proteins. Nucleic Acids Res 47:W429–W436. 10.1093/nar/gkz384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Remmert M, Biegert A, Hauser A, Söding J (2012) HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods 9:173–175. 10.1038/nmeth.1818 [DOI] [PubMed] [Google Scholar]
- 68.McGuffin LJ, Bryson K, Jones DT (2000) The PSIPRED protein structure prediction server. Bioinformatics 16:404–405. 10.1093/bioinformatics/16.4.404 [DOI] [PubMed] [Google Scholar]
- 69.Heffernan R, Yang Y, Paliwal K, Zhou Y (2017) Capturing non-local interactions by long short-term memory bidirectional recurrent neural networks for improving prediction of protein secondary structure, backbone angles, contact numbers and solvent accessibility. Bioinformatics 33:2842–2849. 10.1093/bioinformatics/btx218 [DOI] [PubMed] [Google Scholar]
- 70.Wang S, Peng J, Ma J, Xu J (2016) Protein secondary structure prediction using deep convolutional neural fields. Sci Rep 6:18962. 10.1038/srep18962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bhattacharya S, Roche R, Bhattacharya D (2020) DisCovER: distance- and orientation-based covariational threading for weakly homologous proteins. bioRxiv. 2020.01.31.923409 10.1101/2020.01.31.923409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu F, Xu J (2021) Deep template-based protein structure prediction. PLoS Comput Biol 17:e1008954. 10.1371/journal.pcbi.1008954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22:2577–2637. 10.1002/bip.360221211 [DOI] [PubMed] [Google Scholar]
- 74.Heinig M, Frishman D (2004) STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res 32:W500–W502. 10.1093/nar/gkh429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Skolnick J, Kihara D (2001) Defrosting the frozen approximation: PROSPECTOR— a new approach to threading. Proteins 42: 319–331. [DOI] [PubMed] [Google Scholar]
- 76.Skolnick J, Kihara D, Zhang Y (2004) Development and large scale benchmark testing of the PROSPECTOR_3 threading algorithm. Proteins 56:502–518. 10.1002/prot.20106 [DOI] [PubMed] [Google Scholar]
- 77.Zhang Y, Skolnick J (2004) Scoring function for automated assessment of protein structure template quality. Proteins 57:702–710. 10.1002/prot.20264 [DOI] [PubMed] [Google Scholar]
- 78.Kabsch W (1976) A solution for the best rotation to relate two sets of vectors. Acta Crystallogr A 32:922–923. 10.1107/S0567739476001873 [DOI] [Google Scholar]
- 79.Zemla A (2003) LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res 31:3370–3374. 10.1093/nar/gkg571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mariani V, Biasini M, Barbato A, Schwede T (2013) lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 29:2722–2728. 10.1093/bioinformatics/btt473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu J, Zhang Y (2010) How significant is a protein structure similarity with TM-score =0.5? Bioinformatics 26:889–895. 10.1093/bioinformatics/btq066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bienkowska J, Lathrop R (2005) Threading algorithms. In: Encyclopedia of genetics, genomics, proteomics and bioinformatics. American Cancer Society [Google Scholar]
- 83.Xu Y, Xu D, Uberbacher EC (1998) An efficient computational method for globally optimal threading. J Comput Biol 5:597–614. 10.1089/cmb.1998.5.597 [DOI] [PubMed] [Google Scholar]
- 84.Akutsu T, Miyano S (1999) On the approximation of protein threading. Theor Comput Sci 210:261–275. 10.1016/S0304-3975(98)00089-9 [DOI] [Google Scholar]
- 85.Zhu J, Wang S, Bu D, Xu J (2018) Protein threading using residue co-variation and deep learning. Bioinformatics 34:i263–i273. 10.1093/bioinformatics/bty278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Venclovas Č (2003) Comparative modeling in CASP5: Progress is evident, but alignment errors remain a significant hindrance. Proteins 53:380–388. 10.1002/prot.10591 [DOI] [PubMed] [Google Scholar]
- 87.Shibberu Y, Holder A, Lutz K (2010) Fast protein structure alignment. In: Borodovsky M, Gogarten JP, Przytycka TM, Rajasekaran S (eds) Bioinformatics research and applications. Springer, Berlin, Heidelberg, pp 152–165 [Google Scholar]
- 88.Shibberu Y, Holder A (2011) A spectral approach to protein structure alignment. IEEE/ACM Trans Comput Biol Bioinform 8:867–875. 10.1109/TCBB.2011.24 [DOI] [PubMed] [Google Scholar]
- 89.Teichert F, Bastolla U, Porto M (2007) SABERTOOTH: protein structural alignment based on a vectorial structure representation. BMC Bioinformatics 8:425. 10.1186/1471-2105-8-425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Di Lena P, Fariselli P, Margara L, Vassura M, Casadio R (2010) Fast overlapping of protein contact maps by alignment of eigenvectors. Bioinformatics 26:2250–2258. 10.1093/bioinformatics/btq402 [DOI] [PubMed] [Google Scholar]
- 91.Teichert F, Minning J, Bastolla U, Porto M (2010) High quality protein sequence alignment by combining structural profile prediction and profile alignment using SABERTOOTH. BMC Bioinformatics 11: 251. 10.1186/1471-2105-11-251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Malod-Dognin N, Pržulj N (2014) GR-align: fast and flexible alignment of protein 3D structures using graphlet degree similarity. Bioinformatics 30:1259–1265. 10.1093/bioinformatics/btu020 [DOI] [PubMed] [Google Scholar]
- 93.Skolnick J, Zhou H (2017) Why is there a glass ceiling for threading based protein structure prediction methods? J Phys Chem B 121: 3546–3554. 10.1021/acs.jpcb.6b09517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li Y, Hu J, Zhang C, Yu D-J, Zhang Y (2019) ResPRE: high-accuracy protein contact prediction by coupling precision matrix with deep residual neural networks. Bioinformatics 35:4647–4655. 10.1093/bioinformatics/btz291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Taylor WR (1999) Protein structure comparison using iterated double dynamic programming. Protein Sci 8:654–665. 10.1110/ps.8.3.654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kamisetty H, Ovchinnikov S, Baker D (2013) Assessing the utility of coevolution-based residue–residue contact predictions in a sequence- and structure-rich era. PNAS 110: 15674–15679. 10.1073/pnas.1314045110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Söding J (2017) Big-data approaches to protein structure prediction. Science 355:248–249. 10.1126/science.aal4512 [DOI] [PubMed] [Google Scholar]
- 98.Wu Q, Peng Z, Anishchenko I, Cong Q, Baker D, Yang J (2020) Protein contact prediction using metagenome sequence data and residual neural networks. Bioinformatics 36: 41–48. 10.1093/bioinformatics/btz477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bhattacharya S, Bhattacharya D (2020) Evaluating the significance of contact maps in low-homology protein modeling using contact-assisted threading. Sci Rep 10:2908. 10.1038/s41598-020-59834-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Suzek BE, Wang Y, Huang H, PB MG, Wu CH, The UniProt Consortium (2015) Uni-Ref clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31:926–932. 10.1093/bioinformatics/btu739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.The UniProt Consortium (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47:D506–D515. 10.1093/nar/gky1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mirdita M, von den Driesch L, Galiez C, Martin MJ, Söding J, Steinegger M (2017) Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res 45:D170–D176. 10.1093/nar/gkw1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mitchell AL, Scheremetjew M, Denise H, Potter S, Tarkowska A, Qureshi M, Salazar GA, Pesseat S, Boland MA, Hunter FMI, ten Hoopen P, Alako B, Amid C, Wilkinson DJ, Curtis TP, Cochrane G, Finn RD (2018) EBI metagenomics in 2017: enriching the analysis of microbial communities, from sequence reads to assemblies. Nucleic Acids Res 46: D726–D735. 10.1093/nar/gkx967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Markowitz VM, Chen I- MA, Chu K, Szeto E, Palaniappan K, Pillay M, Ratner A, Huang J, Pagani I, Tringe S, Huntemann M, Billis K, Varghese N, Tennessen K, Mavromatis K, Pati A, Ivanova NN, Kyrpides NC (2014) IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res 42:D568–D573. 10.1093/nar/gkt919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Steinegger M, Söding J (2018) Clustering huge protein sequence sets in linear time. Nat Commun 9:2542. 10.1038/s41467-018-04964-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang Y, Shi Q, Yang P, Zhang C, Mortuza SM, Xue Z, Ning K, Zhang Y (2019) Fueling ab initio folding with marine metagenomics enables structure and function predictions of new protein families. Genome Biol 20:229. 10.1186/s13059-019-1823-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Steinegger M, Mirdita M, Söding J (2019) Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nat Methods 16:603–606. 10.1038/s41592-019-0437-4 [DOI] [PubMed] [Google Scholar]
- 108.Mitchell AL, Almeida A, Beracochea M, Boland M, Burgin J, Cochrane G, Crusoe MR, Kale V, Potter SC, Richardson LJ, Sakharova E, Scheremetjew M, Korobeynikov A, Shlemov A, Kunyavskaya O, Lapidus A, Finn RD (2020) MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res 48:D570–D578. 10.1093/nar/gkz1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang C, Zheng W, Mortuza SM, Li Y, Zhang Y (2020) DeepMSA: constructing deep multiple sequence alignment to improve contact prediction and fold-recognition for distant-homology proteins. Bioinformatics 36:2105–2112. 10.1093/bioinformatics/btz863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ding W, Gong H (2020) Predicting the real-valued inter-residue distances for proteins. Adv Sci 7:2001314. 10.1002/advs.202001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Adhikari B (2020) A fully open-source framework for deep learning protein real-valued distances. Sci Rep 10:13374. 10.1038/s41598-020-70181-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wu T, Guo Z, Hou J, Cheng J (2020) Deep-Dist: real-value inter-residue distance prediction with deep residual convolutional network. bioRxiv. 2020.03.17.995910 10.1101/2020.03.17.995910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kukic P, Mirabello C, Tradigo G, Walsh I, Veltri P, Pollastri G (2014) Toward an accurate prediction of inter-residue distances in proteins using 2D recursive neural networks. BMC Bioinformatics 15:6. 10.1186/1471-2105-15-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Larsson P, Wallner B, Lindahl E, Elofsson A (2008) Using multiple templates to improve quality of homology models in automated homology modeling. Protein Sci 17:990–1002. 10.1110/ps.073344908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cheng J (2008) A multi-template combination algorithm for protein comparative modeling. BMC Struct Biol 8:18. 10.1186/1472-6807-8-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fernandez-Fuentes N, Madrid-Aliste CJ, Rai BK, Fajardo JE, Fiser A (2007) M4T: a comparative protein structure modeling server. Nucleic Acids Res 35:W363–W368. 10.1093/nar/gkm341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rykunov D, Steinberger E, Madrid-Aliste CJ, Fiser A (2009) Improved scoring function for comparative modeling using the M4T method. J Struct Funct Genom 10:95–99. 10.1007/s10969-008-9044-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Joo K, Lee J, Lee S, Seo J-H, Lee SJ, Lee J (2007) High accuracy template based modeling by global optimization. Proteins 69:83–89. 10.1002/prot.21628 [DOI] [PubMed] [Google Scholar]
- 119.Meier A, Söding J (2015) Automatic prediction of protein 3D structures by probabilistic multi-template homology modeling. PLoS Comput Biol 11:e1004343. 10.1371/journal.pcbi.1004343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ginalski K, Elofsson A, Fischer D, Rychlewski L (2003) 3D-jury: a simple approach to improve protein structure predictions. Bioinformatics 19:1015–1018. 10.1093/bioinformatics/btg124 [DOI] [PubMed] [Google Scholar]
- 121.Derevyanko G, Grudinin S, Bengio Y, Lamoureux G (2018) Deep convolutional networks for quality assessment of protein folds. Bioinformatics 34:4046–4053. 10.1093/bioinformatics/bty494 [DOI] [PubMed] [Google Scholar]
- 122.Karasikov M, Pagès G, Grudinin S (2019) Smooth orientation-dependent scoring function for coarse-grained protein quality assessment. Bioinformatics 35:2801–2808. 10.1093/bioinformatics/bty1037 [DOI] [PubMed] [Google Scholar]
- 123.Olechnovič K, Venclovas Č (2017) Voro-MQA: assessment of protein structure quality using interatomic contact areas. Proteins 85:1131–1145. 10.1002/prot.25278 [DOI] [PubMed] [Google Scholar]
- 124.Ray A, Lindahl E, Wallner B (2012) Improved model quality assessment using ProQ2. BMC Bioinformatics 13:224. 10.1186/1471-2105-13-224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Uziela K, Shu N, Wallner B, Elofsson A (2016) ProQ3: Improved model quality assessments using Rosetta energy terms. Sci Rep 6:33509. 10.1038/srep33509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Uziela K, Menéndez Hurtado D, Shu N, Wallner B, Elofsson A (2017) ProQ3D: improved model quality assessments using deep learning. Bioinformatics 33:1578–1580. 10.1093/bioinformatics/btw819 [DOI] [PubMed] [Google Scholar]
- 127.Sato R, Ishida T (2019) Protein model accuracy estimation based on local structure quality assessment using 3D convolutional neural network. PLoS One 14:e0221347. 10.1371/journal.pone.0221347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pagès G, Charmettant B, Grudinin S (2019) Protein model quality assessment using 3D oriented convolutional neural networks. Bioinformatics 35:3313–3319. 10.1093/bioinformatics/btz122 [DOI] [PubMed] [Google Scholar]
- 129.Shuvo MH, Bhattacharya S, Bhattacharya D (2020) QDeep: distance-based protein model quality estimation by residue-level ensemble error classifications using stacked deep residual neural networks. Bioinformatics 36:i285–i291. 10.1093/bioinformatics/btaa455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Baldassarre F, Menéndez Hurtado D, Elofsson A, Azizpour H (2020) GraphQA: protein model quality assessment using graph convolutional networks. Bioinformatics 37:360. 10.1093/bioinformatics/btaa714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Alapati R, Bhattacharya D (2018) clustQ: efficient protein decoy clustering using superposition-free weighted internal distance comparisons. In: Proceedings of the 2018 ACM international conference on bioinformatics, computational biology, and health informatics. Association for Computing Machinery, New York, pp 307–314 [Google Scholar]
- 132.Benkert P, Tosatto SCE, Schwede T (2009) Global and local model quality estimation at CASP8 using the scoring functions QMEAN and QMEANclust. Proteins 77:173–180. 10.1002/prot.22532 [DOI] [PubMed] [Google Scholar]
- 133.Cheng J, Wang Z, Tegge AN, Eickholt J (2009) Prediction of global and local quality of CASP8 models by MULTICOM series. Proteins 77:181–184. 10.1002/prot.22487 [DOI] [PubMed] [Google Scholar]
- 134.McGuffin LJ, Roche DB (2010) Rapid model quality assessment for protein structure predictions using the comparison of multiple models without structural alignments. Bioinformatics 26:182–188. 10.1093/bioinformatics/btp629 [DOI] [PubMed] [Google Scholar]
- 135.Bhattacharya D (2019) refineD: improved protein structure refinement using machine learning based restrained relaxation. Bioinformatics 35:3320–3328. 10.1093/bioinformatics/btz101 [DOI] [PubMed] [Google Scholar]
- 136.Wang D, Geng L, Zhao Y-J, Yang Y, Huang Y, Zhang Y, Shen H-B (2020) Artificial intelligence-based multi-objective optimization protocol for protein structure refinement. Bioinformatics 36:437–448. 10.1093/bioinformatics/btz544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lee GR, Won J, Heo L, Seok C (2019) GalaxyRefine2: simultaneous refinement of inaccurate local regions and overall protein structure. Nucleic Acids Res 47:W451–W455. 10.1093/nar/gkz288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Heo L, Feig M (2020) High-accuracy protein structures by combining machine-learning with physics-based refinement. Proteins 88: 637–642. 10.1002/prot.25847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Park H, Lee GR, Kim DE, Anishchenko I, Cong Q, Baker D (2019) High-accuracy refinement using Rosetta in CASP13. Proteins 87:1276–1282. 10.1002/prot.25784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Heo L, Arbour CF, Feig M (2019) Driven to near-experimental accuracy by refinement via molecular dynamics simulations. Proteins 87: 1263–1275. 10.1002/prot.25759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bhattacharya D, Cheng J (2013) 3Drefine: consistent protein structure refinement by optimizing hydrogen bonding network and atomic-level energy minimization. Proteins 81:119–131. 10.1002/prot.24167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bhattacharya D, Nowotny J, Cao R, Cheng J (2016) 3Drefine: an interactive web server for efficient protein structure refinement. Nucleic Acids Res 44:W406–W409. 10.1093/nar/gkw336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bhattacharya D, Cheng J (2013) i3Drefine software for protein 3D structure refinement and its assessment in CASP10. PLoS One 8: e69648. 10.1371/journal.pone.0069648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bhattacharya D, Cheng J (2013) Protein structure refinement by iterative fragment exchange. In: Proceedings of the international conference on bioinformatics, computational biology and biomedical informatics. Association for Computing Machinery, New York, pp 106–114. 10.1145/2506583.2506601 [DOI] [Google Scholar]