

Abstract
With the advent and widespread adoption of high-throughput DNA sequencing, genetic discoveries in neurodevelopmental disorders are advancing very rapidly. Recent findings include the identification of previously unknown neurodevelopmental disorder genes, as well as multiple examples of rare, highly-penetrant variants in specific genes, which in turns is leading to improved understanding of genotype-phenotype correlations. Here we emphasize the importance of large-scale, reference databases such as gnomAD to determine gene and variant level constraints and facilitate gene discovery, variant interpretation, and genotype-phenotype correlations. While the majority of dominant NDD genes are highly intolerant to variation, some apparent exceptions in reference databases are related to the presence of variants in transcripts that are not brain expressed and/or genes that show acquired somatic mosaicism in blood. Multiple NDD genes are being identified where varying phenotypes depend on either the mode of inheritance (e.g., dominant or recessive), or the nature (e.g., missense or nonsense) and location of the mutation. Ongoing genome-wide analyses and targeted functional studies provide enhancements to the annotation of genes, gene products and variants, which will continue to facilitate gene and variant discovery and variant interpretation.
Keywords: autism, intellectual disability, mutation, constraint score, regional constrain
Introduction
As clinical and research sequencing studies get ever larger, we are seeing unprecedented progress in gene discovery for a broad group of neurodevelopmental disorders (NDDs), including developmental delay (DD), intellectual disability (ID), autism spectrum disorder (ASD), and epilepsy. For all of these disorders, genes carrying highly penetrant rare variants are being identified, and these variants account for a significant proportion of the risk in the individual. Here we provide some background around gene discovery in these NDDs, with a focus on gene and variant level constraints and how they play out in the genetics of NDDs and then take examples from recent literature, focusing on informative genes as well as emerging genotype-phenotype correlations.
Gene and variant level constraints
No discussion of the genetics of NDDs can ignore the profound impact of large-scale population sequencing studies and the accumulation of these data in publicly available databases. The Genome Aggregation Database (gnomAD) is the best example of such an effort, and currently includes over 120,000 whole exome sequences and over 70,000 whole genome sequences [1]. Within gnomAD, which is a follow on of the Exome Aggregation Consortium (ExAC) [2], individuals with severe pediatric diseases and their first-degree relatives have been removed, and there are subsets of individuals who have been identified as free of neuropsychiatric disorders, so this cohort serves as a useful reference set of allele frequencies for NDDs, which in turn advancing gene discovery and facilitates the interpretation of variant pathogenicity in NDDs.
Some of the key parameters that can be derived from gnomAD are the (i) frequency of specific variants in controls, (ii) tolerance of a given gene to heterozygous protein truncating variants (PTVs) predicted to result in loss-of-function (although PTVs are often referred to as loss-of-function variants, the term ‘loss of function’ should be reserved to the function of the protein, as both PTVs and missense variants can result in loss of function), and (iii) tolerance of a gene to missense variation. The probability of being loss-of-function intolerant (pLI) is a metric that compares the expected number of PTVs in a given gene to the observed number of PTVs in gnomAD [2]. The closer pLI is to 1, the more intolerant a gene is to PTVs. Not surprisingly, the vast majority of the genes implicated in autosomal dominant and X-linked forms of NDDs have very high pLI (>0.9), reflecting selective pressure at these loci (Figure 1). In a first study of pLI in NDDs, Kosmicki et al. showed that individuals with ASD have a 3.24-fold increase in PTVs in genes that have high pLI (>0.9) and where PTVs are not seen in ExAC [3]. For individuals with ID/DD, this excess increases to 6.70, as compared to controls. There are, however, two caveats to the very high pLI finding in NDDs; first, we are most powered to identify highly penetrant genes and, second, some of the newest approaches to gene discovery focus on high pLI genes. But, while these points may bias the discovery to the genes with the highest pLI, there is no question that pLI is a very strong predictor of haploinsufficient genes that are likely to lead to severe, early-onset phenotypes when mutated with a PTV. On other hand, genes with many PTVs in controls are much less likely to be associated with phenotypes impacting development under a dominant mode of inheritance.
Figure 1. gnomAD constraint metrics of NDD genes.
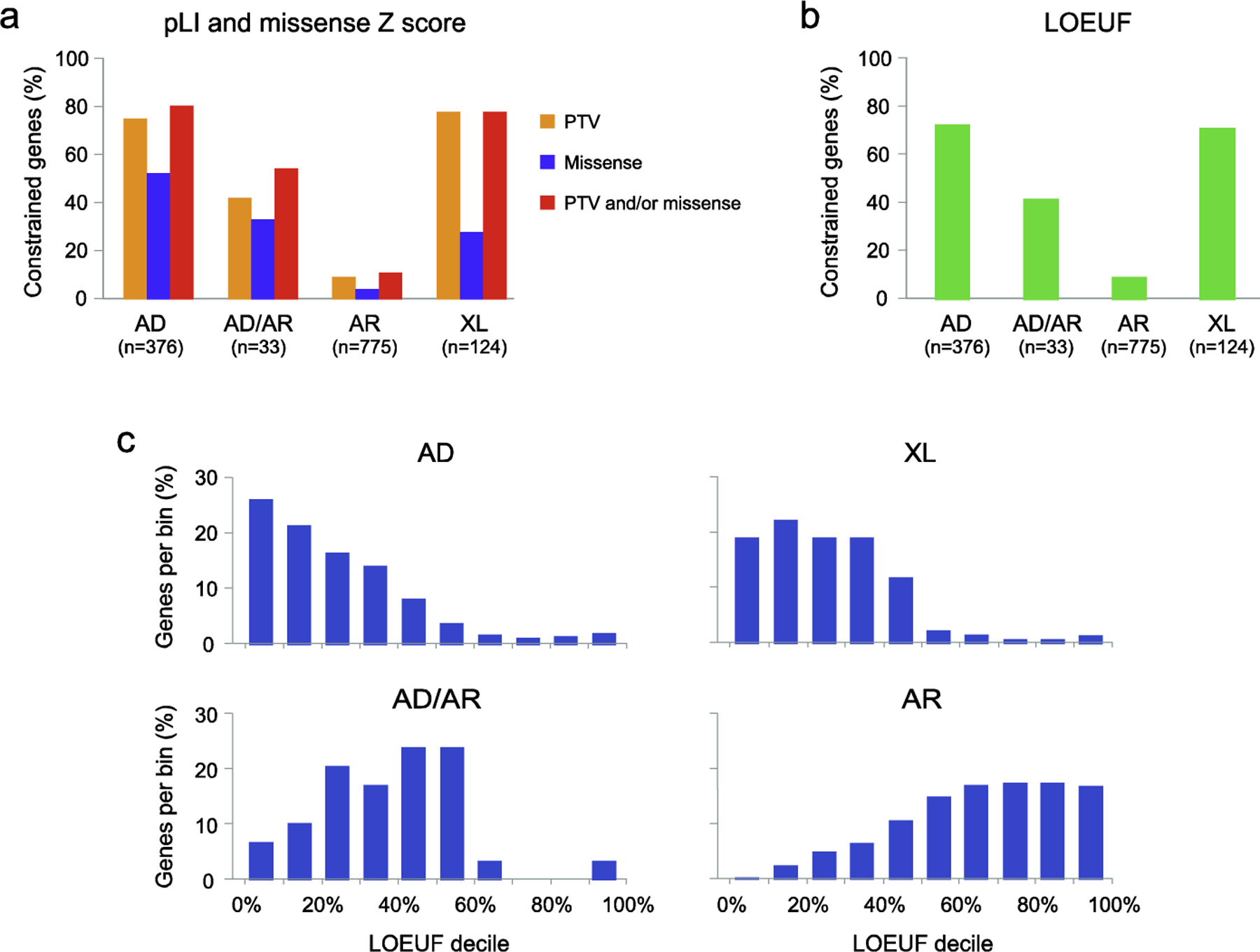
The majority of the genes implicated in autosomal dominant (AD) and X-linked (XL) forms of NDDs are highly constrained for PTVs and/or missense variants. In contrast, genes involved in autosomal recessive (AR) disorders are usually not constrained. This is illustrated using curated sets of autosomal dominant, autosomal recessive, and X-linked genes, as well as autosomal genes showing NDD phenotypes with either monoallelic or biallelic mutations (AD/AR). a, The percentage of genes showing evidence of constraint for PTVs (pLI ≥0.9) and/or missense variants (missense Z score ≥3.09). b, The percentage of genes showing evidence of constraint for PTVs using the LOEUF metric (< 0.35). The proportion of constrained genes is similar to that obtained using the pLI score. c, The distribution of LOEUF scores is shown for each of the 4 gene sets. LOEUF, loss-of-function observed/expected upper bound fraction; pLI, probability of loss-of-function intolerant; PTV, protein truncating variant.
The gnomAD gene constraint metric for missense variants is the missense Z-score [2]. Here too, the comparison is made between the expected and the observed number of missense variants in a given gene. The expected number of missense variants is derived by sequence-specific mutational models with corrections using synonymous variation. A high missense Z-score (≥3.09) indicates that a gene is intolerant to missense variation, and is common in genes involved in autosomal dominant or X-linked NDDs, as opposed to recessive disorders (Figure 1).
Recent enhancements to the pLI and missense Z-score metrics include loss-of-function and missense observed/expected (o/e) scores, loss-of-function o/e upper bound fraction (LOEUF), as well as Missense badness, PolyPhen-2, Constraint (MPC) [1, 4]. Low values of the o/e and LOEUF metrics are indicative of strong intolerance. Importantly, the o/e metric includes estimates of confidence intervals, taking into account the gene size and numbers of samples, as well as providing a continuous measure across the constraint. LOEUF incorporates the upper bound of the confidence interval of the o/e ratio, and is thus a more reliable metric than pLI. MPC incorporates multiple metrics to better predict the impact of specific deleterious missense variants within a given gene, in contrast to missense Z-score, which looks at o/e for missense variation at the gene level. A recent study showed that the odds ratios associated with a de novo missense variant with high MPC were as high as the odds ratios associated with de novo PTVs in highly constrained genes (pLI > 0.9) [4]. Specifically, the study showed that there is a 5.79-fold increase in de novo variants with MPC > 2 in NDD cases, as compared to controls.
These constraint metrics are being used to facilitate gene discovery. For example, the most recent implementation of TADA (Transmitted and De Novo Association test), an approach to identify genes significantly associated with NDDs through ultra-rare variation, included pLI and MPC scores to greatly increase power to identify ASD genes [5, 6]. Since there is a strong bias for dominant highly penetrant NDD genes to have high pLI scores and/or to harbor missense variants with high MPC, incorporating these metrics enhances gene discovery [6].
Given the very strong association between autosomal dominant NDD genes with high constraint scores, great care should be taken before suggesting a causal implication of a heterozygous truncating or missense variant in a gene that is not constrained for that type of variant. To highlight just how powerful these approaches are, it is interesting to look at what appear to be exceptions to the above rules.
Transcript level constraints
Isoform diversity through alternative mRNA splicing across various tissues can sometimes explain the paradoxical finding of PTVs in known dosage-sensitive NDD genes in apparently healthy individuals in gnomAD [7]. SHANK2, encoding a synaptic scaffolding protein, is known to be involved in ID and ASD, based on both targeted and genome-wide studies [8, 9]. However, the gene has a pLI of 0 in gnomAD as of this writing, which is highly unusual for a dominant disease gene presumed to act through haploinsufficiency. A careful review of the data resolved this apparent conflict. gnomAD uses the longest transcript in GENCODE as the canonical transcript (even when a different transcript is used as the reference sequence in medical genetics). However, when looking at the exon distribution of SHANK2 in various tissues, there is little or no expression from over half the exons in this transcript in most tissues, including the brain, and the PTVs identified in gnomAD are primarily in these exons (Figure 2). Selecting the brain-specific transcript of SHANK2 in gnomAD resulted in a pLI of 1. MEF2C, a gene encoding a transcription factor and involved in ID, ASD, epilepsy, and cerebral malformations, and responsible for the neurologic features of the 5q14.3 deletion syndrome [10], is another example, where the pLI for the default transcript in gnomAD, which is not expressed in the brain, is 0.02, but the pLI for the brain-expressed transcript is 0.97. For a more subtle example, CAMK2B, encoding a calcium/calmodulin-dependent protein kinase involved in ID [11], has a pLI of 0.74 for the reference transcript, but the truncating variants reported in gnomAD map to a small number of exons, which are not expressed in brain. pLI in the two brain-specific transcripts is 1.
Figure 2. Distribution of truncating variants in gnomAD across SHANK2 transcripts.
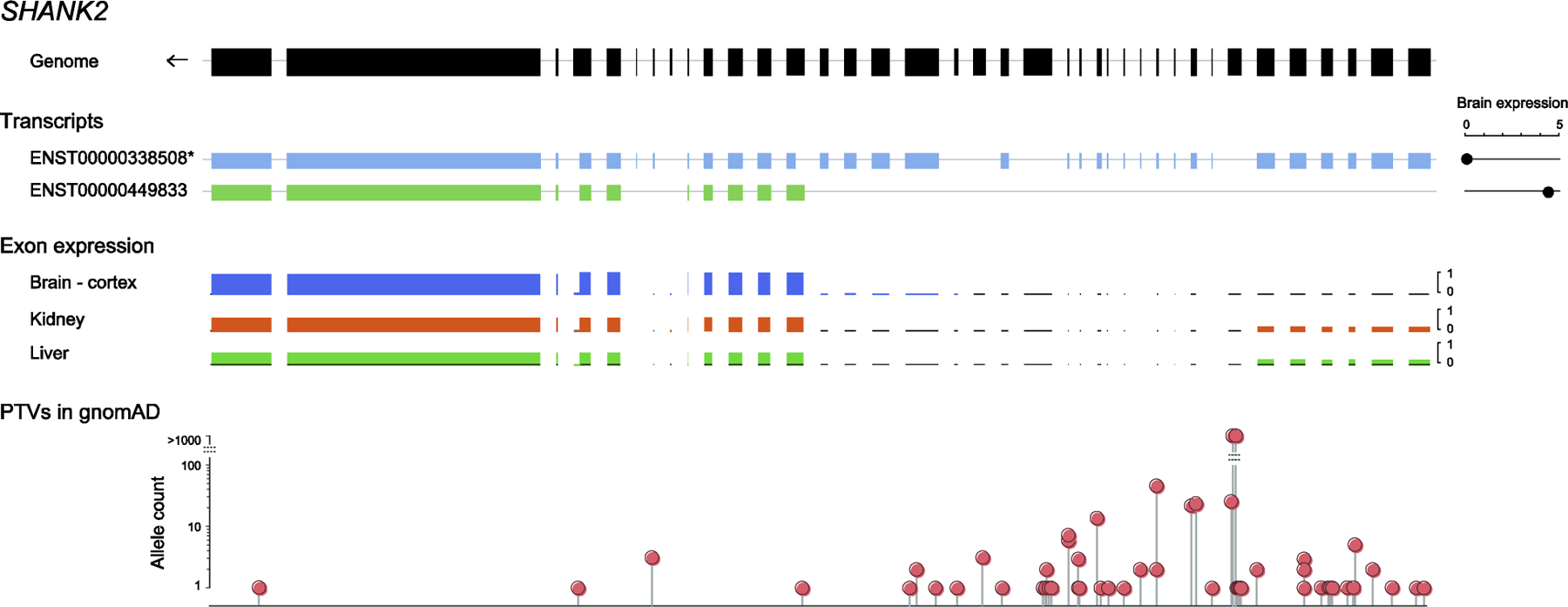
The figure shows (i) the genomic structure of SHANK2, (ii) two SHANK2 transcripts, including the longest transcript selected as canonical in gnomAD ([1], noted with an asterisk), and the brain-specific transcript, with brain expression of the transcripts (read counts) shown on the right, based on the Genotype Tissue Expression (GTEx) dataset [39], (iii) ratios of exon expression in three example tissues, and (iv) the position and numbers of PTVs in gnomAD. There is no brain expression from over half the exons of SHANK2 in the canonical transcript, where most of the PTVs are found. As a result, the pLI is 0 in the canonical transcript, but increases to 1 in the brain-specific transcript.
Hence, careful attention needs to be paid to brain-expressed transcripts when attempting to assign clinical significance to a novel variant. In gene discovery studies, a focus on constraint scores from brain-expressed transcripts would enhance NDD gene discovery in genes where the constraint metrics of the reference sequence diverge significantly from those of the brain-expressed sequence. A recent ‘transcript-expression aware’ metric has been developed, called pext (proportion expressed across transcripts) [7]. Using pext it was observed that, for NDDs, de novo PTVs in low-expressed exons have effect sizes similar to those of synonymous variants (rate ratio ~1), while de novo PTVs in highly expressed exons have much larger effect sizes (rate ratio 4.64 for ID/DD and 2.11 for ASD). In addition, the metric proved useful for filtering PTVs in NDD genes.
Influence of somatic mosaicism on constraint metrics
Another explanation for the presence of potentially deleterious variants in NDD genes in population databases is somatic mosaicism. Although it is generally assumed that all variants present in ExAC and gnomAD are germline events, this is not always the case. DNMT3A, encoding a DNA methyltransferase involved in epigenetic regulation, is an interesting example. Germline mutations in DNMT3A result in Tatton-Brown-Rahman syndrome, an autosomal dominant disorder characterized by ID, tall stature, and a distinctive facies; many individuals also have ASD [12]. Missense and truncating variants, as well as whole gene deletions, have been reported, suggesting a loss-of-function mechanism. However, DNMT3A has dozens of PTVs reported in gnomAD and hence has a pLI of 0. The question then is how are the many PTVs in gnomAD consistent with the findings in Tatton-Brown-Rahman syndrome. While somatic mutations in DNMT3A are known to be common in acute myeloid leukemia [13], recent, large-scale, exome sequencing studies identified somatic mutations in this gene in tissue from older, healthy individuals [14]. Importantly, the mutations, although mosaic, are highly represented in the blood samples because of clonal hematopoiesis [15], a process where a substantial proportion of mature blood cells is derived from a single dominant hematopoietic stem cell lineage. Many of the DNMT3A truncating variants reported in ExAC and gnomAD exhibit allelic imbalance, consistent with somatic mosaicism [16]. In addition, numerous missense variants associated with Tatton-Brown-Rahman syndrome are present in gnomAD due to clonal hematopoiesis, limiting the usefulness of population databases in the interpretation of DNMT3A variant pathogenicity [12, 16]. Interestingly, while germline loss-of-function variants in DNMT3A cause macrocephalic overgrowth in Tatton-Brown-Rahman syndrome, gain-of-function missense substitutions are associated with the reciprocal phenotype, microcephalic dwarfism (Heyn-Sproul-Jackson syndrome) [17]. Another autosomal dominant NDD gene that is frequently mutated in blood in older individuals is ASXL1 [14]. ASXL1 is associated with Bohring-Opitz syndrome and, like DNMT3A, encodes an epigenetic modifier [18]; it has a pLI of 0 in gnomAD because of the large number of cases with acquired hematopoietic mosaicism in the database [16]. PPM1D, involved in Jansen-de Vries syndrome [19] and encoding a protein phosphatase, is also enriched for truncating somatic mutations in the blood of cancer patients, asymptomatic individuals and the elderly [20–22], and has a pLI of 0. Of note, the majority of the somatic mutations occur in the C-terminal domain encoded by the two last exons, which is also the site of the truncating mutations reported in Jansen-de Vries syndrome; these PTVs escape nonsense-mediated decay and result in a truncated protein that retains the phosphatase domain [19, 20]. Taken together, these findings indicate that somatic mosaicism in blood should be considered to avoid misinterpreting the pathogenicity of germline variants or assume inaccurately that neurodevelopmental syndromes associated with genes affected by clonal hematopoiesis have reduced penetrance. Variant interpretation in clinical and research settings would be greatly facilitated by the systematic annotation in gnomAD of variants with decreased allele balance that are likely due to somatic mosaicism.
Complex genotype-phenotype correlations depending on mutation type and location
As the numbers of examples of mutations in NDD genes are increasing, we are beginning to find genes that show exclusively or primarily missense mutations, others that show primarily or exclusively truncating mutations, and others that show both types of variants. One gene with a preponderance of missense mutations is DEAF1. De novo heterozygous missense variants in DEAF1, encoding a transcription factor involved in embryonic and neuronal development, have been implicated in autosomal dominant ID and behavioral problems, including ASD [23]. Consistent with these findings, in a recent, large ASD study, five deleterious missense substitutions were observed in DEAF1, with no PTVs contributing to risk [6]. DEAF1 shows a pLI of 0, and appears to be completely tolerant to truncating variation, hence the missense variants are likely acting through a dominant-negative or gain-of-function mechanism. In fact, deleterious missense variants cluster in the SAND domain, which is critical for dimerization and DNA binding , or the adjacent zinc binding motif [24, 25]. Of note, because only missense variants in specific functional domains are deleterious, the gene is not constrained for missense variants overall (Z-score=1.5). For some de novo missense variants in the SAND domain, it has been shown experimentally that they lead to a loss of transcriptional repression [23–25], suggesting a dominant-negative mechanism. Interestingly, biallelic recessive variants in DEAF1 have been reported in several mostly consanguineous families with ID, ASD, epilepsy, microcephaly, and dyskinesia [25, 26]. The biallelic variants, inherited from unaffected parents, include nonsense, frameshift and splice variants predicted to result in loss of function due to nonsense mediated decay, as well as three missense variants in the SAND domain. Functional analysis of these inherited missense variants showed that, in contrast to de novo variants, they do not affect transcriptional regulation or DNA binding affinity [24, 25]. While the pathobiology of recessive missense variants has not been elucidated, it is likely that they are hypomorphic compared to dominant alleles, and result in partial loss of function. Thus, these findings provide evidence for distinct underlying mechanisms in dominant and recessive DEAF1-associated NDDs.
Like DEAF1, an increasing number of NDD genes with autosomal dominant inheritance are being implicated in recessive disorders. For instance, de novo variants in CAMK2A, GRIN1, and EEF1A2, cause dominant forms of ID and epilepsy [11, 27, 28]. The variants reported are most often dominant-negative or gain-of-function missense variants, which tend to cluster in functionally important coding regions. Considering that these three genes are highly intolerant to variation, they are a priori not expected to be involved in recessive disorders. However, recent studies have reported novel recessive NDDs associated with homozygous missense variants in these genes resulting in hypomorphic alleles, which become pathogenic when recessively inherited [27, 29, 30]. Functional studies were necessary to demonstrate the pathogenicity of these variants. It is expected that as functional studies progress, it will be possible to categorize additional pathogenic variants as dominant-negative, gain-of-function and hypomorphic alleles, to improve our understanding of disease mechanisms, expand the disease spectrum, and better predict outcomes.
Many disease-associated genes (including DEAF1 as noted above) show clustering of mutations within functional domains; these domain can also show evidence for regionalized constraint on deleterious variants [31–33]. Recent studies in ID, ASD and epilepsy identified deleterious, dominant missense variants in KCNQ3, a gene which encodes a subunit of the KV7potassium channel [6, 34–36]. The missense variants cluster in transmembrane regions involved in voltage sensing and appear to be gain-of-function affecting the neurophysiological properties of the channel. Similarly, NDD-associated mutations in DDX3X cluster around the helicase domain. DDX3X is emerging as the cause of 1–3% of unexplained ID and DD in girls and is implicated in ASD as well [37]. Dominant-negative missense mutations are associated with the most severe developmental and neurological phenotypes, while PTVs and hypomorphic missense alleles are associated with milder phenotypes [38]. Methods incorporating structural information and spatial constraints will enhance gene and variant discovery and variant interpretation.
Discovery of novel recessive NDD genes
For all of the success of genome-wide discovery for dominant NDD disorders, recessive variation still proves challenging in genome-wide analyses. Much of what we currently know about recessive genes is derived from traditional clinical genetic studies where multiply affected families were ascertained and sequenced. In large-scale, whole-exome and whole-genome sequencing studies, families typically have just one affected individual, or are case-control cohorts. The presence of heterozygous variants in unaffected individuals, and the relative paucity of de novo variation in recessive genes, decreases power to identify such NDD genes. A metric termed pREC, for probability of being recessive, which was developed alongside pLI [2], has failed to gain traction because it is very hard to show convincing evidence of true recessive constraint (i.e., a specific deficit of individuals with biallelic variants, given the rates of heterozygous individuals) with sample sizes even at the level of gnomAD. In addition, phasing of rare variants (unless they are near each other) is very difficult with short read sequencing, hence distinguishing compound heterozygotes from two variants on a single haplotype is challenging. New approaches to address these issues are being developed now.
Conclusions
Large-scale sequence datasets from cohorts with and without NDDs are leading to enhanced gene discovery, variant interpretation, and etiologic diagnoses. Rates of different types of variation in such cohorts can also begin to illuminate mechanism and explain genotype-phenotype correlations. While interpretation of PTVs is most straightforward, careful attention to the localization and consequences of missense variation within genes can begin to shed light on hypomorphic, loss-of-function, gain-of-function, and dominant-negative alleles. The incorporation of improved annotation, as well as a focus on brain-expressed transcripts and critical domains within encoded proteins will enhance both gene discovery and variant interpretation.
Acknowledgements
We thank Dr. Mark Daly for discussions concerning pREC.
Funding
This work was supported by the Beatrice and Samuel A. Seaver Foundation (to JDB) and by the National Institute of Mental Health (MH111661, to CB and JDB, and, MH097849 to JDB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
Nothing declared.
Papers of particular interest, published within the period of review, have been highlighted as:
of special interest
of outstanding interest
References and recommended reading
- 1. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP et al. : The mutational constraint spectrum quantified from variation in 141,456 humans. bioRxiv 2020. ** This is the flagship paper for gnomAD. By careful and in depth analysis of over 140,000 sequenced samples, the authors provide a detailed look the tolerance of human genes to truncating mutations, which can enhance gene discovery for human disease. The authors also introduce LOEUF, an enhanced metric for categorizing genes for tolerance to truncation mutations.
- 2.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB et al. : Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536(7616):285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kosmicki JA, Samocha KE, Howrigan DP, Sanders SJ, Slowikowski K, Lek M, Karczewski KJ, Cutler DJ, Devlin B, Roeder K et al. : Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat Genet 2017, 49(4):504–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samocha KE, Kosmicki JA, Karczewski KJ, O’Donnell-Luria AH, Pierce-Hoffman E, MacArthur DG, Neale BM, Daly MJ: Regional missense constraint improves variant deleteriousness prediction. bioRxiv 2017.
- 5.He X, Sanders SJ, Liu L, De Rubeis S, Lim ET, Sutcliffe JS, Schellenberg GD, Gibbs RA, Daly MJ, Buxbaum JD et al. : Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet 2013, 9(8):e1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, Peng M, Collins R, Grove J, Klei L et al. : Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180(3):568–584. ** This study identifies genes for autism spectrum disorder from over 535,000 sequenced samples. The authors show that gene discovery is enhanced using constraint metrics, and highlight examples of genotype-phenotype correlations.
- 7. Cummings BB, Karczewski KJ, Kosmicki JA, Seaby EG, Watts NA, Singer-Berk M, Mudge JM, Karjalainen J, Satterstrom FK, O’Donnell-Luria A et al. : Transcript expression-aware annotation improves rare variant discovery and interpretation. bioRxiv 2020. * By leveraging gene expression data across tissues, the authors show that attention to transcripts expressed in disease-relevant tissues and organs provides improved gene discovery and overcomes limitations that can arise when constraint metrics are applied to just a single, canonical transcript. .
- 8.Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U, Endris V, Roberts W, Szatmari P, Pinto D et al. : Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet 2010, 42(6):489–491. [DOI] [PubMed] [Google Scholar]
- 9.Leblond CS, Nava C, Polge A, Gauthier J, Huguet G, Lumbroso S, Giuliano F, Stordeur C, Depienne C, Mouzat K et al. : Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: a gradient of severity in cognitive impairments. PLoS Genet 2014, 10(9):e1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zweier M, Gregor A, Zweier C, Engels H, Sticht H, Wohlleber E, Bijlsma EK, Holder SE, Zenker M, Rossier E et al. : Mutations in MEF2C from the 5q14.3q15 microdeletion syndrome region are a frequent cause of severe mental retardation and diminish MECP2 and CDKL5 expression. Hum Mutat 2010, 31(6):722–733. [DOI] [PubMed] [Google Scholar]
- 11.Kury S, van Woerden GM, Besnard T, Proietti Onori M, Latypova X, Towne MC, Cho MT, Prescott TE, Ploeg MA, Sanders S et al. : De Novo Mutations in Protein Kinase Genes CAMK2A and CAMK2B Cause Intellectual Disability. Am J Hum Genet 2017, 101(5):768–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tatton-Brown K, Zachariou A, Loveday C, Renwick A, Mahamdallie S, Aksglaede L, Baralle D, Barge-Schaapveld D, Blyth M, Bouma M et al. : The Tatton-Brown-Rahman Syndrome: A clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open Res 2018, 3:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bullinger L, Dohner K, Dohner H: Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J Clin Oncol 2017, 35(9):934–946. [DOI] [PubMed] [Google Scholar]
- 14.Risques RA, Kennedy SR: Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet 2018, 14(1):e1007108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayachi S, Buscarlet M, Busque L: 60 Years of clonal hematopoiesis research: From X-chromosome inactivation studies to the identification of driver mutations. Exp Hematol 2020, 83:2–11. [DOI] [PubMed] [Google Scholar]
- 16.Carlston CM, O’Donnell-Luria AH, Underhill HR, Cummings BB, Weisburd B, Minikel EV, Birnbaum DP, Exome Aggregation C, Tvrdik T, MacArthur DG et al. : Pathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz Syndrome. Hum Mutat 2017, 38(5):517–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heyn P, Logan CV, Fluteau A, Challis RC, Auchynnikava T, Martin CA, Marsh JA, Taglini F, Kilanowski F, Parry DA et al. : Gain-of-function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb-regulated regions. Nat Genet 2019, 51(1):96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russell B, Tan WH, Graham JM Jr..: Bohring-Opitz Syndrome. In: GeneReviews Edited by Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A. Seattle (WA); 2018. [PubMed] [Google Scholar]
- 19.Jansen S, Geuer S, Pfundt R, Brough R, Ghongane P, Herkert JC, Marco EJ, Willemsen MH, Kleefstra T, Hannibal M et al. : De Novo Truncating Mutations in the Last and Penultimate Exons of PPM1D Cause an Intellectual Disability Syndrome. Am J Hum Genet 2017, 100(4):650–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M et al. : Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014, 371(26):2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, Hyman DM, Solit DB, Robson ME, Baselga J et al. : Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017, 21(3):374–382 e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, Thorgeirsson TE, Sigurdsson A, Gudjonsson SA, Gudmundsson J et al. : Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130(6):742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vulto-van Silfhout AT, Rajamanickam S, Jensik PJ, Vergult S, de Rocker N, Newhall KJ, Raghavan R, Reardon SN, Jarrett K, McIntyre T et al. : Mutations affecting the SAND domain of DEAF1 cause intellectual disability with severe speech impairment and behavioral problems. Am J Hum Genet 2014, 94(5):649–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Jensik PJ, Alaimo JT, Walkiewicz M, Berger S, Roeder E, Faqeih EA, Bernstein JA, Smith ACM, Mullegama SV et al. : Functional analysis of novel DEAF1 variants identified through clinical exome sequencing expands DEAF1-associated neurodevelopmental disorder (DAND) phenotype. Hum Mutat 2017, 38(12):1774–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nabais Sa MJ, Jensik PJ, McGee SR, Parker MJ, Lahiri N, McNeil EP, Kroes HY, Hagerman RJ, Harrison RE, Montgomery T et al. : De novo and biallelic DEAF1 variants cause a phenotypic spectrum. Genet Med 2019, 21(9):2059–2069. [DOI] [PubMed] [Google Scholar]
- 26.Rajab A, Schuelke M, Gill E, Zwirner A, Seifert F, Morales Gonzalez S, Knierim E: Recessive DEAF1 mutation associates with autism, intellectual disability, basal ganglia dysfunction and epilepsy. J Med Genet 2015, 52(9):607–611. [DOI] [PubMed] [Google Scholar]
- 27.Lemke JR, Geider K, Helbig KL, Heyne HO, Schutz H, Hentschel J, Courage C, Depienne C, Nava C, Heron D et al. : Delineating the GRIN1 phenotypic spectrum: A distinct genetic NMDA receptor encephalopathy. Neurology 2016, 86(23):2171–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carvill GL, Helbig KL, Myers CT, Scala M, Huether R, Lewis S, Kruer TN, Guida BS, Bakhtiari S, Sebe J et al. : Damaging de novo missense variants in EEF1A2 lead to a developmental and degenerative epileptic-dyskinetic encephalopathy. Hum Mutat 2020. [DOI] [PMC free article] [PubMed]
- 29.Chia PH, Zhong FL, Niwa S, Bonnard C, Utami KH, Zeng R, Lee H, Eskin A, Nelson SF, Xie WH et al. : A homozygous loss-of-function CAMK2A mutation causes growth delay, frequent seizures and severe intellectual disability. Elife 2018, 7:e32451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao S, Smith LL, Padilla-Lopez SR, Guida BS, Blume E, Shi J, Morton SU, Brownstein CA, Beggs AH, Kruer MC et al. : Homozygous EEF1A2 mutation causes dilated cardiomyopathy, failure to thrive, global developmental delay, epilepsy and early death. Hum Mol Genet 2017, 26(18):3545–3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Havrilla JM, Pedersen BS, Layer RM, Quinlan AR: A map of constrained coding regions in the human genome. Nat Genet 2019, 51(1):88–95 . ** The authors generated a detailed map of constrained coding regions using the gnomAD databases. Constrained regions are enriched for mutations underlying neurodevelopmental disorders and also provide evidence for protein domains that are as yet unannotated. .
- 32.Turner TN, Douville C, Kim D, Stenson PD, Cooper DN, Chakravarti A, Karchin R: Proteins linked to autosomal dominant and autosomal recessive disorders harbor characteristic rare missense mutation distribution patterns. Hum Mol Genet 2015, 24(21):5995–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sivley RM, Dou X, Meiler J, Bush WS, Capra JA: Comprehensive Analysis of Constraint on the Spatial Distribution of Missense Variants in Human Protein Structures. Am J Hum Genet 2018, 102(3):415–426. ** The authors use three-dimensional structural information available for most of the protein coding genes to identify genes that show clustering of deleterious germline or somatic mutations in protein domains. .
- 34.Sands TT, Miceli F, Lesca G, Beck AE, Sadleir LG, Arrington DK, Schonewolf-Greulich B, Moutton S, Lauritano A, Nappi P et al. : Autism and developmental disability caused by KCNQ3 gain-of-function variants. Ann Neurol 2019, 86(2):181–192. [DOI] [PubMed] [Google Scholar]
- 35.Miceli F, Striano P, Soldovieri MV, Fontana A, Nardello R, Robbiano A, Bellini G, Elia M, Zara F, Taglialatela M et al. : A novel KCNQ3 mutation in familial epilepsy with focal seizures and intellectual disability. Epilepsia 2015, 56(2):e15–20. [DOI] [PubMed] [Google Scholar]
- 36.Orhan G, Bock M, Schepers D, Ilina EI, Reichel SN, Loffler H, Jezutkovic N, Weckhuysen S, Mandelstam S, Suls A et al. : Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann Neurol 2014, 75(3):382–394. [DOI] [PubMed] [Google Scholar]
- 37.Snijders Blok L, Madsen E, Juusola J, Gilissen C, Baralle D, Reijnders MR, Venselaar H, Helsmoortel C, Cho MT, Hoischen A et al. : Mutations in DDX3X Are a Common Cause of Unexplained Intellectual Disability with Gender-Specific Effects on Wnt Signaling. Am J Hum Genet 2015, 97(2):343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lennox AL, Hoye ML, Jiang R, Johnson-Kerner BL, Suit LA, Venkataramanan S, Sheehan CJ, Alsina FC, Fregeau B, Aldinger KA et al. : Pathogenic DDX3X Mutations Impair RNA Metabolism and Neurogenesis during Fetal Cortical Development. Neuron 2020, 106:404–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.GTEX Consortium: Genetic effects on gene expression across human tissues. Nature 2017, 550(7675):204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]