

Abstract
This review uses the National Cancer Institute (NCI) COMPARE program to establish an extensive list of heterocyclic iminoquinones and quinones with similarities in differential growth inhibition patterns across the 60-cell line panel of the NCI Developmental Therapeutics Program (DTP). Many natural products and synthetic analogues are revealed as potential NAD(P)H:quinone oxidoreductase 1 (NQO1) substrates, through correlations to dipyridoimidazo[5,4-f]benzimidazoleiminoquinone (DPIQ), and as potential thioredoxin reductase (TrxR) inhibitors, through correlations to benzo[1,2,4]triazin-7-ones and pleurotin. The strong correlation to NQO1 infers the enzyme has a major influence on the amount of the active compound with benzo[e]perimidines, phenoxazinones, benz[f]pyrido[1,2-a]indole-6,11-quinones, seriniquinones, kalasinamide, indolequinones, and furano[2,3-b]naphthoquinones, hypothesised as prodrugs. Compounds with very strong correlations to known TrxR inhibitors had inverse correlations to the expression of both reductase enzymes, NQO1 and TrxR, including naphtho[2,3-b][1,4]oxazepane-6,11-diones, benzo[a]carbazole-1,4-diones, pyranonaphthoquinones (including kalafungin, nanaomycin A, and analogues of griseusin A), and discorhabdin C. Quinoline-5,8-dione scaffolds based on streptonigrin and lavendamycin can correlate to either reductase. Inhibitors of TrxR are not necessarily (imino)quinones, e.g., parthenolides, while oxidising moieties are essential for correlations to NQO1, as with the mitosenes. Herein, an overview of synthetic methods and biological activity of each family of heterocyclic imino(quinone) is provided.
Keywords: bioreduction, heterocycles, natural products, NQO1, reductases, synthesis, thioredoxin
1. Introduction
The National Cancer Institute’s (NCI, USA) Developmental Therapeutics Program (DTP) has supported the development of numerous clinical anticancer agents, including synthetic and natural compounds, vaccines, and antibodies [1,2,3,4]. The NCI human cancer 60-cell line panel consists of the nine major histological tissue types, allowing high throughput screening of thousands of compounds and natural product extracts each month, using the same assay under strictly identical conditions. Compounds showing high toxicity and variable patterns at an initial single dose (10 µM) may be selected for five dose in vitro testing. The in vitro mean growth (inhibition) data against each cell line represents a pattern, or “fingerprint”, for the evaluated compound (the seed). The seed is identified using a designated NCI accession number (the Cancer Chemotherapy National Service Center number, NSC number). Paull et al. transformed numerical cell line response data into the mean graph format, for visualising differential growth inhibition [5]. After five dose testing, differential growth inhibition is depicted on a log scale by bars (in delta units), which project either side of the mean (e.g., Figures S1–S82, in Supplementary Materials). The COMPARE algorithm is used to rank, in order of similarity to the seed, the activity of compounds in the huge NCI-DTP database, as well as the similarity of the activity of the seed to the expression of key cancer molecular targets in the panel of cell lines [1,2,3,5]. A molecular target is a protein, enzyme, gene, or any other cellular molecule whose presence within the 60-cell line panel has been identified and quantified by the NCI. For some test compounds, a single cellular component may determine activity, while in most cases, cell sensitivity is complex and determined by gene expression, cell signalling, and repair pathways. The similarity of anti-cancer activity patterns to the seed is expressed quantitatively as a Pearson correlation coefficient (PCC). PCCs are between −1 and +1, with −1 indicating a perfect inverse correlation, zero indicating no correlation, and +1 indicating a perfect direct correlation. A PCC of 0.3–0.5 is generally accepted as weak to moderate, 0.5–0.7 as being moderate to strong, and above 0.7 as strong to very strong [1,5,6]. The toxicity evaluation service and COMPARE is available to the public, free of charge, from the NCI website (https://dtp.cancer.gov/, (accessed on 20 October 2022 until 12 May 2023)), and as a new technology platform. Herein, we continue to assess similarities using the 50% growth inhibition (GI50) parameter. GI50 is defined as the concentration that prevents half the proliferation from baseline compared to the maximal value of the untreated cells. Strong correlations may indicate similarities in the mechanisms of the action of anti-cancer compounds, as well as structure–activity relationships. Two previous drug discovery investigations are revisited that established two diverse heterocyclic iminoquinone scaffolds as potent anti-cancer agents [7,8,9,10,11]. Among the tools utilised to ascertain molecular targets was the NCI COMPARE program [7,10,11], which now reveals a more extensive list of correlated compounds. Other high-throughput compound screening programs are not utilised in this review [3,4].
Aldabbagh and co-workers introduced ring-fused imidazo[5,4-f]benzimidazolequinones, e.g., 2a and 2b, with iminoquinones 1a and 1b isolated from the Frémy oxidation of the amine intermediate in the presence of KH2PO4 acidic buffer (Scheme 1) [7,9].
Scheme 1.

Iminoquinones via the synthesis of imidazo[5,4-f]benzimidazolequinones [7,9].
The dipyrido-fused iminoquinone 1a (DPIQ) was isolated in high yield (91%). DPIQ exhibited significant and variable cell growth inhibitory activity against the NCI-60 cell lines, and was selected for five-dose testing [7]. The seven-membered (azepino) analogue 1b is inactive, and the hydrolysed quinones 2a and 2b were less potent than DPIQ [7]. Notably, the isomeric iminoquinone of DPIQ, the imidazo[4,5-f]benzimidazole also exhibited low toxicity at the NCI [12]. The COMPARE analysis gave a moderate correlation for DPIQ (PCC = 0.51) to NAD(P)H:quinone oxidoreductase 1 (NQO1, formerly known as DT-diaphorase) [7], an enzyme with heterogeneity of expression across the NCI-60 panel (Figure 1) [13]. The COMPARE analysis of DPIQ against the huge library of synthetic compounds in the NCI-DTP database gave strong correlations in anti-cancer activity to two other heterocyclic iminoquinones 3a (PCC = 0.87) and 4 (0.77), indicating that the compounds possibly possess similar mechanisms of action. Iminoquinone 3a (PCC = 0.64) gave a stronger correlation than DPIQ and 4 (0.47) to NQO1 expression in the NCI-DTP 60 cell lines. The higher PCC for 3a was supported by computational docking at the NQO1 active site, which predicted a higher affinity and a shorter distance for hydride reduction from the isoalloxazine ring of FADH2 [7].
Figure 1.

6-Imino-1,2,3,4,8,9,10,11-octahydropyrido[1,2-a]pyrido[1’,2’:1,2]imidazo[5,4-f]benzimidazole-13-one (DPIQ), 3a and 4, and PCCs from COMPARE analysis [7].
A collaboration between the groups of Aldabbagh and Koutentis led to the discovery of anti-cancer activity for the benzo[e][1,2,4]triazin-7-ones, e.g., 5a–d, (Figure 2) based on inhibition of the growth of cancer cell lines [10,11]. An earlier report demonstrated the potency of 6-substituted 1,3-diphenylbenzo[1,2,4]triazinones, and 5-substituted [1,2,4]triazino[5,6,1-jk]carbazol-6-one derivatives 6, as multi-target inhibitors for Alzheimer’s disease (AD) [14].
Figure 2.

Benzo[1,2,4]triazin-7-ones 5a–d and PCCs to pleurotin [10,11], and anti-AD [1,2,4]triazino[5,6,1-jk]carbazol-6-one 6 [14].
The anti-cancer evaluation studies were stimulated by our interest in the iminoquinone motif, which is common to both DPIQ and the benzo[1,2,4]triazinones (Figure 1 and Figure 2). The benzotriazinones 5a–d showed sufficient potency for selection by the NCI for five-dose testing. The five-dose testing gave the GI50 parameter used in the COMPARE analysis. For 5a–d, the PCCs of ~0.8 indicate close to perfect direct correlations to the naturally occurring saturated benzoquinone, pleurotin [10,11]. Pleurotin possesses antibiotic and anti-cancer activities, including the inhibiting of hypoxia-induced factor 1α (HIF-1α) [15]. Pleurotin is a potent irreversible inhibitor of thioredoxin reductase (TrxR), and compounds 5a and 5b are reversible inhibitors with Ki values of 3.90 and 0.78 µM, respectively [10]. The greater enzyme inhibition by 5b was assumed to be related to the highly electron-withdrawing 3-CF3 group increasing the susceptibility to reductive activation and thereby increasing the specificity towards cancer cell lines.
NQO1 and TrxR are obligatory 2-electron reductases involved in regulating the production of toxic reactive oxygen species (ROS), including superoxide (O2●−) (Scheme 2). Although O2●− is used in cells of the immune system and provides a defence against pathogens, excessive amounts are implicated in many physiological disorders, including cancer [16]. The 2-electron reduction overrides the single electron transfer (SET) by detoxifying the quinone, through the formation of a relatively stable aromatic hydroquinone, which is eliminated through the conjugation to glutathione, sulfate, or glucose [17].
Scheme 2.

General mechanism for NQO1 and TrxR-Trx quinone detoxification (hydrolysis required for X = NH).
NQO1 and TrxR, however, are over-expressed in many solid tumours and, thus, are viable molecular targets for cancer therapy [18,19]. In this context, the heterocyclic (imino)quinone is designed as a prodrug to form cytotoxic hydroquinone. For instance, in the case of the archetypical bioreductive quinone prodrug, mitomycin C, cytotoxicity occurs through quinone methide formation at C-1 and C-10, which react with the nucleophilic DNA bases [20] (Figure 3). However, in the case of heterocyclic quinones and iminoquinones, such as DPIQ, which lack obvious reactive sites upon reduction, cell death is likely attributed to ROS formation with simultaneous reoxidation of the hydroquinone to the quinone, as observed in deoxynyboquinone (Figure 3) [21].
Figure 3.

Bioreductive anti-cancer agents.
Similar to the NQO1 reductive activation mechanism [22], NAD(P)H provides hydride reducing equivalents for transfer via a cascade in the TrxR-Trx (thioredoxin protein) system to the (imino)quinone [19]. Consequently, inhibition of TrxR leads to the accumulation of Trx protein and the activation of ROS-induced apoptosis pathways [16].
We now employ the NCI COMPARE analysis to examine heterocyclic quinones and iminoquinones, specifically focusing on their strong correlations with DPIQ, benzo[1,2,4]triazinone 5a and pleurotin. The objective of this analysis is to determine whether these correlations in anti-cancer activity patterns reflect actual similarities in mechanisms of action and structure-activity relationships. In addition to assessing the biological activities, we provide descriptions of selected syntheses utilised to obtain each heterocyclic iminoquinone and quinone scaffold. We believe that this review represents the first compilation of heterocyclic compounds derived from a COMPARE analysis.
2. Compound Search Methods
Our previous COMPARE analyses were repeated [6,7,10,11,23], and new analyses were carried out under identical conditions to allow comparisons of Pearson correlation coefficients (PCCs). The public COMPARE (https://dtp.cancer.gov/public_compare/, (accessed on 20 October 2022 until 12 May 2023)) was used to access the NCI-60 data, which did not include one-dose experiments. The mean graphs generated are listed along with the PCCs in the Supplementary Materials document (Figures S1–S82). Scifindern (https://scifinder-n.cas.org, https://dtp.cancer.gov/public_compare/, (accessed on 20 October 2022 until 12 May 2023)) was used to find the relevant literature using chemical structure and citation searches with, where applicable, publication data refinements.
2.1. COMPARE Using DPIQ and 5a as the Seed
The seed NSC numbers for DPIQ (NCS753790) and benzo[1,2,4]triazinone 5a (NCS768093) were used to search using the COMPARE algorithm screening data limited to GI50 end points. A user’s guide is provided below (Scheme 3). The mean graph data was subjected to a standard COMPARE using GI50 of the synthetic compound as the target set, with compounds ranked according to the magnitude of the PCCs. The process was repeated using pleurotin (NSC401005) as the seed to confirm similarities in PCCs with 5a.
Scheme 3.

Workflow for COMPARE analysis on DPIQ, as the seed.
2.2. COMPARE Using Molecular Target Expression
A COMPARE analysis was carried out for the selected compound NSC numbers against the molecular target expression in the NCI-60 (Scheme 4). NAD(P)H:quinone oxidoreductase 1 (NQO1) used the MT22 MOLTID.MT.SERIES, and thioredoxin reductase (TrxR) used the MT143 TXNRD1 MOLTID.MT.SERIES. A PCC was obtained using a matrix COMPARE by appending the GI50 cell viability profile of a given NSC number against an appended molecular target expression across the NCI-60.
Scheme 4.

Workflow for COMPARE analysis using NQO1 as the molecular target.
The strongest correlations to NQO1 (MT22 MOLTID.MT.SERIES) were found by submitting the molecular target expression data to a standard COMPARE using screening data limited to GI50 end points. Compounds were listed according to the magnitude of the PCCs.
3. Discussion
3.1. COMPARE Analysis: Strong Correlations to DPIQ as the Seed
This section reviews compounds 3a, 4, 7–12 with strong to very strong correlations (PCC = 0.72–0.87) in anti-cancer activity to DPIQ (Figure 1 and Figure 4). COMPARE gave planar fused heterocyclic compounds containing the oxidising quinone or iminoquinone motif, apart from azaanthracenone 11b, which is the para-dimethoxybenzene (bio)synthetic precursor (see Section 3.1.5). Weak to moderate correlations (PCC = 0.38–0.64) to NQO1 expression across the NCI-60 cell line panel were demonstrated, although benzo[e]perimidine-4-carboxylic acid 3a is found to have a stronger correlation to NQO1 (PCC = 0.64, Figure 1). The following sub-sections review each heterocyclic scaffold, apart from quinoline-5,8-dione 9a, since quinoline-5,8-diones also have strong correlations to the benzo[1,2,4]triazinone 5a (reviewed in Section 3.3).
Figure 4.

The strongest COMPARE correlations to DPIQ, excluding 3a and 4.
3.1.1. Benzo[e]perimidines
Commercially available benzo[e]perimidine-4-carboxylic acid 3a displays the greatest similarity in anti-cancer activity to DPIQ, with a PCC of 0.87, and a moderate to strong PCC of 0.64 to NQO1 expression across the NCI-60 cell line panel (Figure 1). Recently, iminoquinone 3a was shown to induce apoptosis in NCI cell lines with high nuclear factor erythroid 2-related factor 2 (NRF2) activation [24]. NRF2 is a transcription factor over-expressed in pancreatic adenocarcinomas, which regulates expression of many redox enzymes, including NQO1 [25]. Compound 3a induced apoptosis in ex vivo cultures of pancreatic cancer xenografts with a high NQO1/NRF2 activation, and inhibited the biosynthesis of amino acids, including asparagine and methionine [24].
The benzo[e]perimidine scaffold is obtained using the condensation of acetamide with l,4-diamino-9,10-anthracenedione in molten phenol, with transamination providing 6-[(aminoalkyl)amino]-substituted examples 3c and 3d (Scheme 5) [26]. Substitutions mimic the well-known clinical anti-cancer agent mitoxantrone, originally used to treat leukaemia. Borowski and co-workers demonstrated the requirement of the 6-[(aminoalkyl)amino]-substituent in antileukemic activity, e.g., 3c, with the synthetic precursor 3b inactive [26], and 8,11-dihydroxybenzo[e]perimidin-7-one 3d displaying significant toxicity against NCI leukemic and solid tumour cell lines [27]. Compound 3d displayed in vivo toxicity and overcomes multi-drug resistance tumour cells and, like mitoxantrone, induces cell cycle accumulation in the G2/M phase. The same group evaluated guanidine condensation adducts, with 3e displaying the greatest in vitro toxicity [28].
Scheme 5.

Synthesis and anti-cancer evaluation of benzo[e]perimidines 3b–e [26,27,28].
3.1.2. Phenoxazinones
DPIQ displays a strong similarity in anti-cancer activity to the extensively studied and abundant natural chromophore, 2-aminophenoxazin-3-one 4 (PCC = 0.77, Figure 1), also known as the antibiotic questiomycin A [29,30,31,32,33]. Iminoquinone 4 is isolated from the 6-electron oxidative coupling of two molecules of 2-aminophenol [33,34], mediated by the enzyme phenoxazinone synthase [35]. The antibiotic has a weak to moderate correlation to NQO1 expression (PCC = 0.47) [7]. Anti-cancer activity of 4 includes cytotoxicity against a variety of human cancer cells lines, with induction of apoptosis [31,36,37] and suppression of NO and prostaglandin E2 production leading to anti-inflammatory effects [38]. Cancer spread was monitored in vivo in mice, where the number of pulmonary metastasis of B6 melanoma cells remained the same [37]. The first reported antibiotic with anti-cancer activity is actinomycin D [39], containing 2-aminophenoxazin-3-one 4 at its core (Figure 5) [34]. Actinomycin D (also known as dactinomycin) has the trade name Cosmegen and is clinically used to treat a variety of solid tumours.
Figure 5.

Actinomycin D, showing 2-aminophenoxazin-3-one 4 core in bold.
The phenoxazinone scaffold is contained in heterocyclic iminoquinone 7, which also displays a strong PCC of 0.74 to DPIQ activity (Figure 4). Benzo[a]phenoxazin-5-one 7 was assembled by condensation of 1-nitroso-2-naphthol with L-tyrosine [40]. The iminoquinone was identified from the NCI diversity set of 2000 compounds as an inhibitor of HIF-1α induced by insulin-like growth factor-1 [41]. HIF-1α is one of the sub-units of HIF-1, the transcription factor over-expressed in hypoxic tumour cells, and responsible for regulating anaerobic metabolism that leads to tumour progression and angiogenesis [42]. SET, which is reversible by oxygen and mediated by NADPH-cytochrome (P450) reductase, is associated with tumour hypoxia [16,43,44], and is prevented by the NQO1-mediated two-electron reduction (Scheme 2) [7,18,21,45]. This specificity towards hypoxia provides a rationale for the weak correlation of iminoquinone 7 (0.38) to NQO1 expression across the NCI 60 cell lines.
Increased cell signalling mediated by reversible protein phosphorylation supports cancer cell proliferation. The human cytoplasmic protein tyrosine phosphatases (HCPTPs) are overexpressed in hypophosphorylated breast cancer cells [46]. Benzo[a]phenoxazin-5-one 7 was identified as the third-most potent inhibitor of HCPTP isoform B (IC50 = 31 µM), after in silico docking of the compounds of the NCI Diversity Set I and enzyme inhibition assays on five selected compounds [47].
3.1.3. Benz[f]pyrido[1,2-a]indole-6,11-quinone
Updating the 2012 COMPARE analysis on DPIQ [7] revealed that the carboxylic acid 8a of benz[f]pyrido[1,2-a]indole-6,11-quinone (Figure 4) has the second-highest PCC of 0.81. Testing against the NCI-DTP cell panel revealed the N,N-dimethylethyl carboxamide 8b to be the most potent amongst carboxamide derivatives evaluated (Figure 6), with cytotoxicity against the adriamycin-resistant breast tumour cell line (NCI/ADR-RES) at a concentration lower than clinical anticancer agents, daunorubicin and mitoxantrone [48]. More recently, methylester 8c was found to inhibit indoleamine 2,3-dioxygenase 1 (IDO1) [49]. IDO1 depletes tryptophan, with deficiencies in this essential amino acid leading to suppression of immune response to tumours [50]. 12-Unsubstituted and carboxylate ester derivatives of 8a, including ethylester 8d, exhibit anti-fungal activities [51]. Carboxylates 8e–h display micromolar anti-bactericidal activity against the Erdman strain of Mycobacterium tuberculosis, with the molecular target being a membrane-bound, iron−thiol reductase (IspQ) [52].
Figure 6.

Biologically active benz[f]pyrido[1,2-a]indole-6,11-quinones [48,49,51,52].
For the biological evaluation purposes described above, the benz[f]pyrido[1,2-a]indole-6,11-quinone scaffold was obtained using adaptations of the traditional three-component, one-pot condensation of 2,3-dichloro-1,4-naphthoquinone, an active methylene dicarbonyl and pyridine [48,49,51,52]. Yields are improved when using three-component reactions with 1,4-naphthoquinone and pyridine catalysed by CuCl2 with acyl bromides [53], or cleavage of butynedioates [54] (Scheme 6).
Scheme 6.

Synthesis of benz[f]pyrido[1,2-a]indole-6,11-quinones [53,54].
3.1.4. Analogues of Seriniquinone
Dibenzo[b,i]thianthrene-5,7,12,14-tetrone was first synthesized in 1991 [55], and identified in 2014 as seriniquinone (Figure 7), derived from the marine bacterium Serinicoccus marinus [56].
Figure 7.
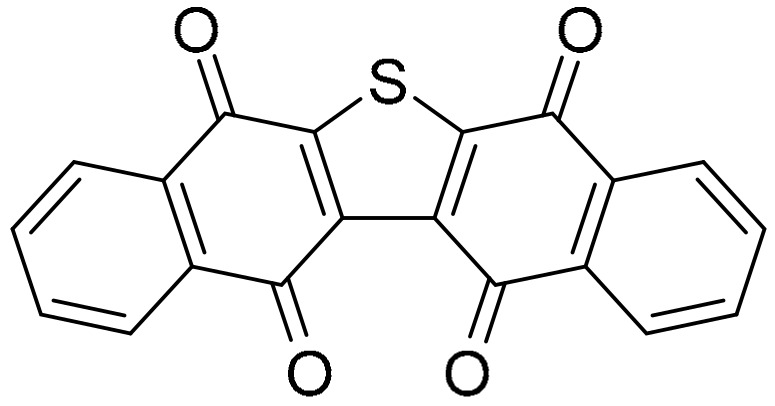
Seriniquinone.
Synthetic analogues of seriniquinone are required due to its poor water solubility, with 10a and 10b showing strong PCCs of 0.75 and 0.72 to DPIQ anti-cancer activity, respectively, but weak to moderate PCCs of 0.47 and 0.48 to NQO1 expression, respectively (Figure 4). Phenol 10a and carbamate 10b were prepared on a multi-gram scale via naphthalene-1,4-dione coupling, thiophene ring-formation, and Pd-induced deprotection (Scheme 7) [57]. Carbamate 10b was shown to hydrolyse at room temperature to 10a under phosphate-buffered saline (PBS) at pH 7.2. Analogues 10a and 10b, like DPIQ [7], possess specificity towards the NCI melanoma cell lines. Cytotoxicity of seriniquinones was demonstrated through binding to dermcidin [56,57], a small protein involved in cancer cell proliferation [58] and induction of cell death via autophagocytosis [56]. More recently, seriniquinones were shown to induce elevated levels of intracellular ROS, which trigger apoptotic events [59], supporting the relatively weak correlations to NQO1. ROS is related to the SET radical producing processes (Scheme 2), which are not mediated by NQO1.
Scheme 7.

Synthesis of seriniquinone analogues [57].
3.1.5. Azaanthracenone
Kalasinamide (11a) is an azaanthracenone first isolated from the tree Polyalthia suberosa (Figure 4) [60]. The unreported analogue 11b without the 4-methyl substituent was found by the COMPARE analysis to have a strong PCC to DPIQ of 0.73 and a weak to moderate PCC to NQO1 of 0.44. Kalasinamide is a photosensitiser thought to prevent the invasion of pathogens by generating singlet oxygen, and/or through conversion to the quinone marcanine A [61]. Kalasinamide is prepared through acid-mediated substitutions onto the activated p-dimethoxybenzene [61,62], including using Knorr cyclisation, which gives a mixture of 11a and marcanine A, in the presence of oxygen and light (Scheme 8). Marcanine A is a plant natural product [61,63], also prepared through photochemical cyclisation onto the naphthoquinone acrylamide substituent [64]. The potent anti-bacterial marcanine A [63] has cytotoxicity comparable to adriamycin, using five different solid tumour cell lines [65].
Scheme 8.

Syntheses of kalasinamide and marcanine A with yields [61,64].
3.1.6. Furano[2,3-b]naphthoquinone (FNQ)
The polyketide-isoprenoid 12a was isolated from Streptomyces cinnamonensis (Figure 4) [66]. The biosynthesis of furano[2,3-b]naphthoquinone (FNQ) 12a has attracted considerable attention [67]; however, there is no literature on antimicrobial and anti-cancer studies. The COMPARE analysis shows that 12a exhibits a strong PCC to DPIQ of 0.72, and a moderate PCC of 0.55 to NQO1 expression across the NCI-60, which is comparable with DPIQ (0.51) (Figure 1). Related synthetic structures (FNQ13, Figure 8) induce mitochondrial swelling and apoptosis, due to induction of ROS including H2O2 [68]. Furano[2,3-b]naphtho-4,9-dione 12b, isolated from Tabebuia avellanedae, shows a preference for inhibition of cancer cell growth over normal cells, with STAT3 inhibitory mechanisms proposed [69], while other FNQ natural products inhibit IDO1 (see Section 3.1.3) [70]. Inhibition of STAT3 may be important because the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway plays a key role in membrane-to-nucleus signalling, which is critical in mediation of cancer and inflammation [71].
Figure 8.

Anti-cancer FNQs.
Most syntheses of FNQ involve forming the furan ring from 2-hydroxy-1,4-naphthoquinone [70,72,73], including by an acetyl chloride/Et3N-mediated Wittig reaction (Scheme 9A) [72,73]. The product (RJ-LC-07-48) is potent against drug-resistant non-small-cell lung cancer (NSCLC) cells by interaction with the minichromosomal maintenance protein MCM2, disrupting the formation of the MCM complex that is required for the initiation of DNA replication [73]. Similar specificity towards non-small cell lung cancer cell lines was shown by DPIQ [7]. Wu and co-workers transformed 2-(5-hydroxy-1-pentynyl)benzonitriles to FNQ via a NaOMe-mediated ring-closure, followed by Frémy’s salt oxidation (Scheme 9B) [74].
Scheme 9.

3.2. COMPARE Analysis: Strong Correlations to Benzo[1,2,4]triazin-7-one 5a as the Seed
The NCI COMPARE analysis using benzo[1,2,4]triazin-7-ones 5a and pleurotin, as the seed revealed very strong correlations to synthetic heterocyclic quinone scaffolds 13a–d and 14a–f, as well as to the natural products pyranonaphthoquinones 15 and discorhabdin C (PCC = 0.68–0.90, Figure 9). However, unlike DPIQ (above), very strong PCCs of ~0.85 (to 5a) are also found to non-quinone structures, notably synthetic derivatives of the anti-cancer sesquiterpene lactone melampomagnolide B (MMB), isolated from Magnolia grandiflora [75,76]. MMB derivatives show specificity towards leukemic cell lines [76,77], with dimeric examples showing nanomolar activity against solid tumour cell lines in the NCI-60 panel [77]. Similarly, benzotriazin-7-ones 5a–d also showed potent anti-proliferative effects against most NCI leukemic cell lines [10,11]. MMB is an extensively investigated scaffold that is thought to target the NFkB pathway through inhibition of the IkBa/p65/p50 kinase complex (IKK) [78]. The following section reviews the heterocyclic quinones and iminoquinones that have strong to very strong correlations to the anti-cancer activity of TrxR inhibitors 5a and pleurotin. Quinoline-5,8-diones (e.g., 9b) are reviewed separately in Section 3.3.
Figure 9.

Very strong COMPARE correlations to benzo[1,2,4]triazinone 5a and pleurotin.
3.2.1. Naphtho[2,3-b][1,4]oxazepine-6,11-dione
COMPARE gave an almost perfect PCC of ~0.90 for the anti-cancer activity of benzo[1,2,4]triazinone 5a to 1,4-benzoxazepine derivatives of 5,8-dihydroxy-1,4-naphthoquinones 13a–d (related to Echinamines A and B) (Figure 10). Echinamines A and B are antioxidants isolated from the sea urchin Scaphechinus mirabilis [79], which suppress herpes simplex virus type 1 infection in the Vero monkey kidney cell line [80]. COMPARE analysis-derived 1,4-benzoxazepines 13a–d are unreported, giving (Figure 3) very strong correlations to pleurotin anticancer activity (PCC = 0.8–0.9), analogous to 5a–d. The almost perfect correlations of 13a–d to pleurotin are perhaps unsurprising, given the common 1,4-naphthoquinone motif.
Figure 10.

1,4-Benzoxazepine derivatives of 1,4-naphthoquinones with PCCs and the related natural products Echinamine A and B.
You et al. used an intramolecular copper-catalysed hydroxyl displacement of iodides (16a–d) to synthesize homomorpholine 17b-fused and morpholine-fused, e.g., 17a, 17c and 17d, naphthoquinone adducts with compounds giving moderate inhibition of lung A549 cell growth (Scheme 10) [81].
Scheme 10.

Synthesis (with yields) of naphtho[2,3-b][1,4]-oxazine-1,4-diones 17a–d and IC50 against A549 cells [81].
3.2.2. Benzo[a]carbazole-1,4-dione
Benzo[a]carbazole-1,4-dione 14c and others appear in the patent literature as inhibitors for the thioredoxin TrxR-Trx system (Figure 11) [82]. This supports the very strong anti-cancer activity correlations with TrxR inhibitors, benzo[e][1,2,4]triazin-7-one 5a (~0.74–0.82) and pleurotin (0.78–0.87). Benzo[a]carbazole-1,4-dione 14a was one of 6 hits out of 2000 in the NCI library to inhibit Plasmodium kinesin-5 from both human and malaria vivax [83]. Compounds 14a and 14d–f were amongst 35 hits out of 1597 compounds in the NCI Diversity SET III screen with >50% inhibitory activity, at concentrations of 20 µM of the immunosuppressive enzyme IDO1 [84]. Benzo[a]carbazole-1,4-dione 14a is effective against vancomycin-resistant Staphylococcus aureus by targeting the cysteine thiol of bacterial MarR transcription factors [85].
Figure 11.

Benzo[a]carbazole-1,4-dione antibiotics and anti-cancer agents [82,83,84,85].
The traditional synthesis of the benzo[a]carbazole-1,4-dione scaffold (Figure 11) involves a Diels–Alder cycloaddition of 3-vinylindole with benzoquinone and oxidative dehydrogenation [86,87]. The 3-vinylindole can be generated in situ from 3-ethylindole using benzoquinone as the oxidising agent, and the benzoquinone then acts as the dienophile to give the benzo[a]carbazole (Scheme 11) [87].
Scheme 11.

Synthesis of benzo[a]carbazole-1,4-dione [87].
3.2.3. Pyranonaphthoquinones
Pyranonaphthoquinones are natural products possessing the naphtho[2,3-c]pyran-5,10-dione ring system (Figure 12) [88,89]. Benzotriazinone 5a and pleurotin show strong to very strong PCCs of 0.72–0.83 to the antibiotics kalafungin and nanaomycin A first isolated from the Streptomyces tanashiensis strain Kala [90,91]. Nanaomycin D isolated from Streptomyces rosa is enantiomeric to kalafungin with nanaomycin A, the cleaved lactone [92]. Kalafungin and nanaomycin analogues show specificity towards breast cancer cell lines over-expressing cytochrome P450 oxidoreductase with cytotoxicity diminished under anoxia, where reactive oxygen production is inhibited [93]. The SET recycling mechanism is also important in antibacterial activity [94]. Conversely, kalafungin and analogues were reported inhibitors of the serine-threonine kinase AKT via a proposed two-electron reduction to the hydroquinone with alkylation of the quinone methide by a cysteine in the activation loop (T-loop) of the kinase domain of AKT [95]. Nanaomycin A was identified in a screening program as a DNA methyltransferase inhibitor, with biochemical assays revealing specificity towards DNMT3B with docking hypothesising a reduction in nanaomycin A by the sulfur atom of a cysteine in the catalytic site [96].
Figure 12.

The pyranonaphthoquinones with COMPARE analysis PCCs.
Griseusin A, isolated from Streptomyces griseus, is a pyranonaphthoquinone antibiotic [97], that bears structural similarity to kalafungin, but possesses a 1,7-dioxaspiro[5,5]-undecane ring system. Synthetic griseusin 15a exhibits a strong correlation in anti-cancer activity to benzotriazinone 5a (PCC = 0.78) and pleurotin (0.68) (Figure 12). The multi-step synthesis of griseusin 15a involves a cerium(IV) ammonium nitrate-mediated oxidative rearrangement and acid-mediated cyclisation to the spiroacetal [98,99,100,101]. Brimble and co-workers have reported the separation of isomers 15a and 15b using flash chromatography (Scheme 12) [99,100,101].
Scheme 12.

Brimble and co-workers synthesis of the griseusin scaffold [99,100].
3.2.4. Discorhabdin C
The pyrroloiminoquinone alkaloids, discorhabdins, are isolated from numerous cold water marine sponges, with cytotoxic discorhabdin C from the New Zealand sponge, Latrunculia Bocage [102,103]. Discorhabdin C exhibits strong to very strong PCCs to benzotriazinone 5a (0.78) and pleurotin (0.84) (Figure 9). Munro and co-workers reported selectivity towards the NCI colon and leukaemia subpanels [104], while others reported in vitro anti-hepatitis virus C, antimalarial and antimicrobial activities [105]. Figg and co-workers performed high-throughput screens on crude natural product extracts and identified 3-dihydro-discorhabdin C (Figure 13), as a HIF-1α/p300 inhibitor [106], which interferes with the HIF-1α and p300 protein−protein interaction to decrease HIF-1α-dependent transcription [43].
Figure 13.
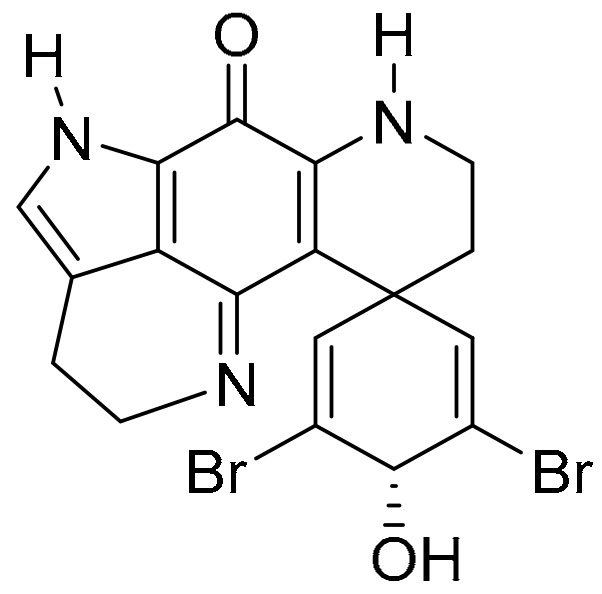
3-Dihydrodiscorhabdin C.
In the 1990s, many multi-step total syntheses of discorhabdin C were reported, with approaches starting from the quinoline with the formation of the fused pyrrole [107,108], and from the indole with the formation of the six-membered imino-ring through condensation with the quinone [109,110,111]. One approach involves oxidation of the tyramine-substituted indoloquinonimine 18 in a nucleophilic addition onto the iminoquinone (Scheme 13) [111]. Kublak and Confalone had earlier used the para-phenoxide approach for alkylation of an adjacent naphthoquinone ethylamino-substituent by displacement of a mesylate [112].
Scheme 13.

Aubart and Heathcock synthesis of discorhabdin C [111].
3.3. COMPARE Analysis: Strong Correlations to DPIQ and Benzo[1,2,4]triazin-7-one 5a as the Seed: Quinoline-5,8-diones
Synthetic quinoline-5,8-diones scaffolds correlate strongly with the anti-cancer activities of DPIQ, benzotriazinone 5a, and pleurotin. 6-Aminoethyl substituted derivative 9a shows one of the strongest PCC to DPIQ of 0.80, with a relatively weak PCC to NQO1 expression of 0.42 (Figure 4). 2-Methyl substituted derivative 9b shows a very strong PCC to pleurotin of 0.89, and a strong correlation to benzotriazinone 5a of 0.71 (Figure 9). There is an abundance of synthetic and anti-cancer studies on quinoline-5,8-diones [113], with most stimulated by the broad range of cytotoxicity against solid tumours displayed by streptonigrin, a recognised substrate for NQO1 (Figure 14) [114]. Streptonigrin was isolated from Streptomyces flocculus [115], with lavendamycin a biosynthetically related antibiotic isolated from Streptomyces lavendulae [116]. Unfortunately, both streptonegrin and lavendamycin have proved too toxic for clinical use, although analogues of lavendamycin display potent HIV reverse-transcription inhibition [117], and improved specificity for NQO1 [118].
Figure 14.

Popular quinoline-5,8-dione scaffolds for biological activity investigations.
N-Alkylamino compounds, analogous to dione 9a, also possess antimalarial activity [119], while compound 9b is a reported inhibitor of Cdc25B (IC50 = 4.6 µM), a protein phosphatase involved in regulating cyclin-dependent kinase activity during the cell cycle [120]. Recent studies have reported 6-N-arylquinoline-5,8-diones 9c (Figure 14) as inhibitors of Mycobacterium tuberculosis [121], as well as of Gram-negative and Gram-positive bacteria [122]. 6-N-Arylquinoline-5,8-diones 9c are also reported to cleave DNA as an underlying mechanism for apoptosis induction in leukemic cell lines [123]. Nevertheless, the consensus is that there is a strong correlation between NQO1 bioreduction and anticancer activity for many quinoline-5,8-diones [118,124,125], given that NQO1 is strongly over-expressed in solid tumours [13] relative to normal tissues [126].
3.4. COMPARE Analysis Using Molecular Target Expression
In this section, the COMPARE algorithm was used to derive PCCs for the similarity in expression of chosen cancer molecular targets, NQO1 and TrxR, to compound growth inhibition across the NCI-60 cell line panel. Section 3.4.1 deals with NQO1 expression and establishes the strongest correlations to compound cytotoxicity. Section 3.4.2 reveals the PCCs of compounds with almost perfect direct correlations to the anti-cancer activity of known TrxR inhibitors, benzo[1,2,4]triazinones 5a–d and pleurotin.
3.4.1. Compound Correlations to NQO1 Expression
Since most compounds that correlated strongly with DPIQ anti-cancer activity gave modest PCCs of 0.38–0.55 to NQO1 expression, except for benzo[e]perimidine 3a, which was noticeably stronger (PCC = 0.64, Figure 1 and Figure 4), we searched for compounds with the strongest PCCs to NQO1 expression. The strongest compound correlations to NQO1 were of similar magnitude to the PCC for benzo[e]perimidine 3a (Figure 15). Phenazine-5,10-dioxide 19, 5-hydroxy-6-methoxy-8-nitroquinoline 20, and indolequinones 21a and 21b, gave PCCs of ~0.6–0.7 to NQO1 and DPIQ. Phenazine-5,10-dioxide 19 is unreported, and has a marginally lower PCC of 0.51 than the other NQO1 substrates to DPIQ.
Figure 15.

The three strongest PCCs to NQO1.
Analogues of phenazine 19 are reported as π-stacking DNA intercalators with differential toxicity through DNA-damaging ●OH release under hypoxic conditions [127,128]. Phenazine-5,10-dioxides are designed to model 3-amino-1,2,4-benzotriazine-1,4-dioxide (tirapazamine, TPZ, Figure 16), which reached advanced clinical trials as a hypoxia-activated prodrug [129]. However, although there are successful Phase I and Phase II trials, Phase III randomised controlled trials showed no benefit of TPZ in chemotherapy without using an approach to ensure sustained tumour hypoxia [130]. Quinoline 20 is a synthetic precursor for 5-alkoxy derivatives of the clinical anti-malarial primaquine [131]. The biological activity of 20 is unreported, and, tentatively, the compound is metabolised to a quinoline-5,8-dione antibiotic upon reductive activation-oxidation (Section 3.3).
Figure 16.

TPZ and primaquine related to 19 and 20, respectively.
Pyrrolo[1,2-a]indoles are highly pursued synthetic targets [132], because they form the core of mitomycins, in particular MMC (Figure 3) [133]. The 7-methoxymitosene skeleton of 21a and 21b was accessed via a Pt-promoted cyclisation of a β-lactam onto an internal acetylene (Scheme 14) [134]. Triphosgene and triethylamine preferentially chlorinate the primary alcohol over the secondary alcohol in 22 to give 21a [135], with the substitution reported to be driven by steric demand [136].
Scheme 14.

Cycloisomerisation to the 7-methoxymitosene skeleton [134,135].
7-Methoxymitosenes 21a and 22b are designed to form an electrophilic spiro-cyclopropane intermediate upon reductive activation, which enables crosslinking, possibly within the same DNA molecule (Scheme 15) [135]. Indolequinones 21a and 21b were evaluated using the prostate cancer cell line PPC-1 and the normal prostate cell line RWPE-1, with alcohol 21a exhibiting the greater selectivity towards the cancer cell line [135].
Scheme 15.

Hypothesis for cytotoxicity of 7-methoxymitosenes [135].
Interestingly, known TrxR inhibitors, benzo[1,2,4]triazinones 5a, 5b and pleurotin, as well as compounds that correlate very strongly to their anti-cancer activity, benzo[a]carbazole-1,4-dione 14b, kalafungin, and discorhabdin C, gave negative PCCs to NQO1 expression across the NCI-60 panel (PCC = −0.27 to −0.48, Table 1). This suggests that TrxR inhibitors may also act as inhibitors of other two-electron reductases over-expressed in solid tumours, namely NQO1 (see discussion below).
Table 1.
COMPARE analysis-derived PCCs to NQO1 and TrxR expression across the NCI-60 panel for TrxR inhibitors 1.
| Compound | NQO1 | TrxR |
|---|---|---|
| 5a | −0.37 | −0.24 |
| 5b | −0.30 | −0.12 |
| Pleurotin | −0.27 | −0.21 |
| 14b | −0.48 | −0.22 |
| Kalafungin | −0.27 | −0.24 |
| Discorhabdin C | −0.29 | −0.37 |
3.4.2. Compound Correlations to TrxR
The negative correlations to TrxR (TXNRD1) expression by reported TrxR-Trx inhibitory compounds [10,11,15,19,137], as well as by compounds with very strong similarities in anti-cancer activity, suggest that the cytotoxicity of these compounds is greater when TrxR levels are low (Table 1). Increased inhibition may, thus, prevent the reducing system’s essential role in tumour development, such as inhibiting apoptosis, and angiogenesis promotion [19,137]. The TrxR-Trx system modulates cell signalling, in particular through interactions with the tumour suppressor protein PTEN (protein tyrosine phosphatase and tensin homolog). The TrxR-Trx system activates PTEN through reduction [138], with oxidation of PTEN leading to inactivation [138,139]. Trx-1–PTEN interactions through disulfide bond formation between Cys 32 in the Trx-1 active site and Cys 212 in the lipid membrane binding domain of PTEN inhibits PTEN phosphatase activity [140]. The inactivation of PTEN leads to activation of tumour propagating PI3K-AKT kinase signalling pathways [138]. The naturally occurring sesquiterpene lactone, parthenolide (structurally similar to MMB, Figure 9), is reported to inhibit TrxR by shifting the enzyme from antioxidant activity to ROS generation, leading to promotion of apoptosis in HeLa cells (Figure 17) [141]. Cell death through ROS generation seems a common mode of action for most classes of compounds inhibiting TrxR [19,93,137,141,142]. Alternatively, the greater potency of compounds as cytotoxins when reductase expression is low may be due to the TrxR-Trx or NQO1 systems detoxifying the heterocyclic (imino)quinones through bioreduction (Scheme 2). Further, COMPARE reveals a diverse range of chemical structures, including many structures without the (imino)quinone moiety, that have strong PCCs to the anti-cancer activity of TrxR inhibitors, 5a, 5b and pleurotin. Since benzotriazinones 5a and 5b exhibit reversible mixed and uncompetitive inhibition of TrxR, respectively [10,137], with binding to positions other than the active site likely, it seems that bioreduction may not be directly involved in the inhibition or inactivation of TrxR by many compounds.
Figure 17.
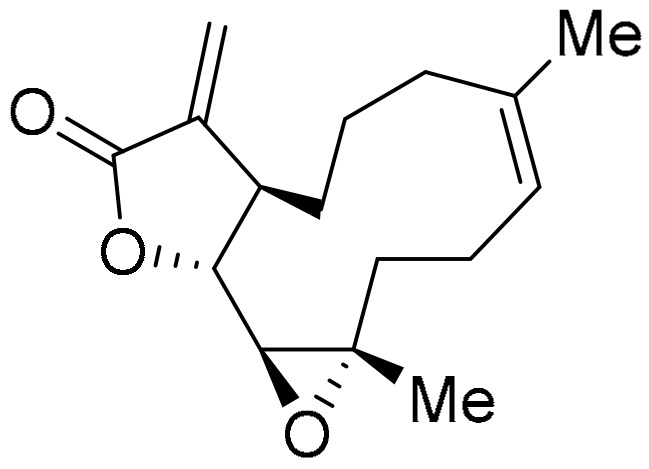
Parthenolide.
Importantly, specificity for NQO1 was demonstrated by DPIQ, benzo[e]perimidine 3a, 2-aminophenoxazinone 4 and phenazine 19, which have no correlations to TrxR (TXNRD1: PCC = −0.15 to 0.12), but moderate to strong correlations to NQO1 expression across the NCI-60 cell line panel (PCC = 0.47–0.67, Table 2). This supports the premise that these compounds are specifically substrates for cellular NQO1, which facilitates reductively-activated cytotoxicity. Further, MMC and other recognised indolequinone NQO1 substrates [7,13,143,144,145], are shown to irreversibly inhibit TrxR through formation of covalently bound adducts (Figure 18) [135,143,145]. Comparing mean growth (inhibition) graphs of NQO1 substrates, the greatest cytotoxicity is towards solid tumour cell lines, including most melanoma, non-small cell lung, and colon cancer cell lines, with negligible toxicity towards leukemic cell lines, in contrast with the strong anti-leukemic activity of TrxR inhibitors (see Supplementary Materials).
Table 2.
COMPARE analysis-derived PCCs to NQO1 and TrxR expression across the NCI-60 panel for NQO1 substrates 1.
| Compound | NQO1 | TrxR |
|---|---|---|
| DPIQ | 0.51 | 0.12 |
| 3a | 0.64 | 0.11 |
| 4 | 0.47 | −0.15 |
| 19 | 0.67 | 0.10 |
Figure 18.
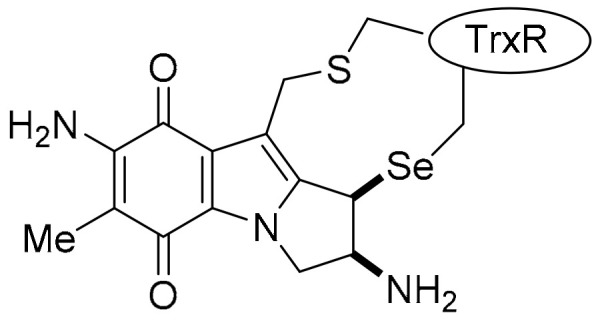
Inactivation of TrxR: MMC-TrxR adduct [143].
4. Conclusions
The NCI COMPARE program has enabled the composition of this review on the synthesis and biological activity of heterocyclic iminoquinones and quinones, where scaffolds are categorised according to similarities in their anti-cancer activity. Although it is important to emphasise that all correlations herein need to be verified experimentally [2], as with many literature studies [1,2,3,4,5,146], COMPARE has enabled hypotheses of mechanisms of actions to be made. We reveal several natural products with strong to very strong correlations in patterns of anti-cancer activity to DPIQ, benzo[1,2,4]triazin-7-one 5a, and pleurotin. Most heterocyclic scaffolds exhibit significant biological activity to warrant extensive synthetic investigations, while others are yet to be synthesized and are revealed as important future targets for medicinal chemists.
Compounds strongly correlating to DPIQ, and showing specificity to NQO1 expression, have no correlation to the alternative two-electron reductase, TrxR. These “NQO1 specific prodrugs” are flat aromatic heterocycles with fused oxidizable, e.g., quinone or iminoquinone, moieties. The correlations to NQO1 expression, however, are modest, suggesting that NQO1 is not the sole cellular molecular component that influences cell sensitivity. NQO1 has a “Janus” effect in cancer biology [18], where it behaves as either a tumour suppressor or a tumour promotor, with the former based on the prevention of SET processes that lead to an accumulation of harmful ROS. However, the redox appears interchangeable, and NQO1-activated pathways can lead to ROS-induced apoptosis [18,21]. Ultimately, designed prodrugs for NQO1 may also effectively act as inhibitors of redox defensive signalling pathways that contribute to carcinogenesis, including the transcriptional regulators Nrf2 [25], HIF-1α [43], and JAK/STAT [71].
There is a diverse range of compounds with almost direct correlations to the anti-cancer activity patterns of the TrxR inhibitors, benzotriazinone 5a, and pleurotin. These compounds do not always possess an oxidising (iminoquinone or quinone) group, suggesting bioreduction is unnecessary, and compounds are more cytotoxic in the absence of the reductase. Among these compounds, there are similarities in biological activity, with many possessing antiviral, antimicrobial (including antimalarial), as well as anti-cancer activity. These compounds tend to be cytotoxic towards leukemic as well as solid tumour cell lines, unlike NQO1 substrates, which show specificity towards solid tumours over-expressing NQO1. There are similarities in the mechanism of action, with compounds correlating to NQO1 expression or acting as TrxR inhibitors, both showing inhibition of the monomeric oxidase, IDO1 [50]. 1,3-Diphenylbenzo[1,2,4]triazinones are inhibitors in AD [14], as well as cancer [10,11,137]. Reduced Trx protein binds to apoptosis signal-regulating kinase 1 (ASK1), thus inhibition of TrxR leads to oxidised Trx, which cannot bind to ASK1. The outcome is mitochondrial apoptosis through activation of ASK1, downstream JNK and mitogen-activated protein kinase (MAPK14) signalling pathways [145], which lead to reduced inflammatory response and tumorigenesis or the initiating of neurodegenerative disorders, such as AD [137,142].
Acknowledgments
F.A. is grateful to Vincent Fagan [6], Robert Coyle [23], and Martin Sweeney [137] for preliminary data. This manuscript was completed as part of the MPharm. dissertation of N.H. and M.F. at Kingston University. F.A. dedicates this review to William J. Spillane (University of Galway) on his 80th birthday.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28135202/s1.
Author Contributions
Investigation, methodology, and writing: N.H. and M.F.; Review and editing: P.A.K. and M.P.C.; Conceptualisation, supervision, writing, review, and editing: F.A. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This research received no external funding.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Shoemaker R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 2.Holbeck S.L. Update on NCI in vitro drug screen utilities. Eur. J. Cancer. 2004;40:785–793. doi: 10.1016/j.ejca.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 3.Sharma S.V., Haber D.A., Settleman J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Rev. Cancer. 2010;10:241–253. doi: 10.1038/nrc2820. [DOI] [PubMed] [Google Scholar]
- 4.Holbeck S.L., Camalier R., Crowell J.A., Govindharajulu J.P., Hollingshead M., Anderson L.W., Polley E., Rubinstein L., Srivastava A., Wilsker D., et al. The National Cancer Institute ALMANAC: A Comprehensive screening resource for the detection of anticancer drug pairs with enhanced therapeutic activity. Cancer Res. 2017;77:3564–3576. doi: 10.1158/0008-5472.CAN-17-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paull K.D., Shoemaker R.H., Hodes L., Monks A., Scudiero D.A., Rubinstein L., Plowman J., Boyd M.R. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: Development of mean graph and COMPARE algorithm. J. Natl. Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- 6.Fagan V. Ph.D. Thesis. National University of Ireland Galway; Galway, Ireland: 2011. Synthesis and Anti-Cancer Activity of Novel Imidazo[5,4-f]benzimidazolequinones. [Google Scholar]
- 7.Fagan V., Bonham S., Carty M.P., Saenz-Méndez P., Eriksson L.A., Aldabbagh F. COMPARE analysis of the toxicity of an iminoquinone derivative of the imidazo[5,4-f]benzimidazoles with NAD(P)H:quinone oxidoreductase 1 (NQO1) activity and computational docking of quinones as NQO1 substrates. Bioorganic Med. Chem. 2012;20:3223–3232. doi: 10.1016/j.bmc.2012.03.063. [DOI] [PubMed] [Google Scholar]
- 8.Fagan V., Bonham S., McArdle P., Carty M.P., Aldabbagh F. Synthesis and toxicity of new ring-fused imidazo[5,4-f]benzimidazolequinones and mechanism using amine N-oxide cyclizations. Eur. J. Org. Chem. 2012;2012:1967–1975. doi: 10.1002/ejoc.201101687. [DOI] [Google Scholar]
- 9.Fagan V., Bonham S., Carty M.P., Aldabbagh F. One-pot double intramolecular homolytic aromatic substitution routes to dialicyclic ring fused imidazobenzimidazolequinones and preliminary analysis of anticancer activity. Org. Biomol. Chem. 2010;8:3149–3156. doi: 10.1039/c003511d. [DOI] [PubMed] [Google Scholar]
- 10.Sweeney M., Coyle R., Kavanagh P., Berezin A.A., Re D.L., Zissimou G.A., Koutentis P.A., Carty M.P., Aldabbagh F. Discovery of anti-cancer activity for benzo[1,2,4]triazin-7-ones: Very strong correlation to pleurotin and thioredoxin reductase inhibition. Bioorganic Med. Chem. 2016;24:3565–3570. doi: 10.1016/j.bmc.2016.05.066. [DOI] [PubMed] [Google Scholar]
- 11.Keane L.-A.J., Mirallai S.I., Sweeney M., Carty M.P., Zissimou G.A., Berezin A.A., Koutentis P.A., Aldabbagh F. Anti-cancer activity of phenyl and pyrid-2-yl 1,3-substituted benzo[1,2,4]triazin-7-ones and stable free radical precursors. Molecules. 2018;23:574. doi: 10.3390/molecules23030574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conboy D., Aldabbagh F. 6-Imino-1,2,3,4,8,9,10,11-octahydropyrido[1,2-a]pyridoimidazo[4,5-f]benzimidazole-13-one: Synthesis and cytotoxicity evaluation. Molbank. 2020;2020:M1118. doi: 10.3390/M1118. [DOI] [Google Scholar]
- 13.Fitzsimmons S.A., Workman P., Grever M., Paull K., Camalier R., Lewis A.D. Reductase enzyme expression across the National Cancer Institute tumor cell line panel: Correlation with sensitivity to mitomycin C and EO9. J. Natl. Cancer Inst. 1996;88:259–269. doi: 10.1093/jnci/88.5.259. [DOI] [PubMed] [Google Scholar]
- 14.Catto M., Berezin A.A., Re D.L., Loizou G., Demetriades M., De Stradis A., Campagna F., Koutentis P.A., Carotti A. Design, synthesis and biological evaluation of benzo[e][1,2,4]triazin-7(1H)-one and [1,2,4]-triazino[5,6,1-jk]carbazol-6-one derivatives as dual inhibitors of beta-amyloid aggregation and acetyl/butyryl cholinesterase. Eur. J. Med. Chem. 2012;58:84–97. doi: 10.1016/j.ejmech.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Welsh S.J., Williams R.R., Birmingham A., Newman D.J., Kirkpatrick D.L., Powis G. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1α and vascular endothelial growth factor formation. Mol. Cancer Ther. 2003;2:235–243. [PubMed] [Google Scholar]
- 16.Saikolappan S., Kumar B., Shishodia G., Koul S., Koul H.K. Reactive oxygen species and cancer: A complex interaction. Cancer Lett. 2019;452:132–143. doi: 10.1016/j.canlet.2019.03.020. [DOI] [PubMed] [Google Scholar]
- 17.Bolton J.L., Dunlap T. Formation and biological targets of quinones: Cytotoxic versus cytoprotective effects. Chem. Res. Toxicol. 2017;30:13–37. doi: 10.1021/acs.chemrestox.6b00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang K., Chen D., Ma K., Wu X., Hao H., Jiang S. NAD(P)H:quinone oxidoreductase 1 (NQO1) as a therapeutic and diagnostic target in cancer. J. Med. Chem. 2018;61:6983–7003. doi: 10.1021/acs.jmedchem.8b00124. [DOI] [PubMed] [Google Scholar]
- 19.Bian M., Fan R., Zhao S., Liu W. Targeting the thioredoxin system as a strategy for cancer therapy. J. Med. Chem. 2019;62:7309–7321. doi: 10.1021/acs.jmedchem.8b01595. [DOI] [PubMed] [Google Scholar]
- 20.Tomasz M., Lipman R., Chowdary D., Pawlak J., Verdine G.L., Nakanishi K. Isolation and structure of a covalent cross-link adduct between mitomycin C and DNA. Science. 1987;235:1204–1208. doi: 10.1126/science.3103215. [DOI] [PubMed] [Google Scholar]
- 21.Parkinson E.I., Bair J.S., Cismesia M., Hergenrother P.J. Efficient NQO1 substrates are potent and selective anticancer agents. ACS Chem. Biol. 2013;8:2173–2183. doi: 10.1021/cb4005832. [DOI] [PubMed] [Google Scholar]
- 22.Li R., Bianchet M.A., Talalay P., Amzel L.M. The three-dimensional structure of NAD(P)H:quinone reductase, a flavoprotein involved in cancer chemoprotection and chemotherapy: Mechanism of the two-electron reduction. Proc. Natl. Acad. Sci. USA. 1995;92:8846–8850. doi: 10.1073/pnas.92.19.8846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coyle R. Ph.D. Thesis. National University of Ireland Galway; Galway, Ireland: 2014. Barton Esters for Initiator-Free Radical Cyclisation with Heteroaromatic Substitution and Anti-Cancer Evaluation of Benzo[e][1,2,4]triazin-7-ones. [Google Scholar]
- 24.Dai B., Augustine J.J., Kang Y., Roife D., Li X., Deng J., Tan L., Rusling L.A., Weinstein J.N., Lorenzi P.L., et al. Compound NSC84167 selectively targets NRF2-activated pancreatic cancer by inhibiting asparagine synthesis pathway. Cell Death Dis. 2021;12:693. doi: 10.1038/s41419-021-03970-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lister A., NedjadI T., Kitteringham N.R., Campbell F., Costello E., Lloyd B., Copple I.M., Williams S., Owen A., Neoptolemos J.P., et al. Nrf2 is overexpressed in pancreatic cancer: Implications for cell proliferation and therapy. Mol. Cancer. 2011;10:37. doi: 10.1186/1476-4598-10-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stefañska B., Dzieduszycka M., Martelli S., Tarasiuk J., Bontemps-Gracz M., Borowski E. 6-[(Aminoalkyl)amino]-substituted 7H-benzo[e]perimidin-7-ones as novel antineoplastic agents. Synthesis and biological evaluation. J. Med. Chem. 1993;36:38–41. doi: 10.1021/jm00053a005. [DOI] [PubMed] [Google Scholar]
- 27.Stefañska B., Dzieduszycka M., Bontemps-Gracz M.M., Borowski E., Martelli S., Supino R., Pratesi G., De Cesare M., Zunino F., Kuśnierczyk H., et al. 8,11-Dihydroxy-6-[(aminoalkyl)amino]-7H-benzo[e]perimidin-7-ones with activity in multidrug-resistant cell lines: Synthesis and antitumor evaluation. J. Med. Chem. 1999;42:3494–3501. doi: 10.1021/jm980682p. [DOI] [PubMed] [Google Scholar]
- 28.Dzieduszycka M., Martelli S., Arciemiuk M., Bontemps-Gracz M.M., Kupiec A., Borowski E. Effect of modification of 6-[(Aminoalkyl)amino]-7H-benzo[e]perimidin-7-ones on their cytotoxic activity toward sensitive and multidrug resistant tumor cell lines. Synthesis and biological evaluation. Bioorganic Med. Chem. 2002;10:1025–1035. doi: 10.1016/S0968-0896(01)00358-3. [DOI] [PubMed] [Google Scholar]
- 29.Anzai K., Isono K., Okuma K., Suzuki S. The new antibiotics, questiomycins A and B. J. Antibiot. 1960;13:125–132. [PubMed] [Google Scholar]
- 30.Kozlovsky A.G., Zhelifonova V.P., Antipova T.V., Adanin V.M., Novikova N.D., Deshevaya E.A., Schlegel B., Dahse H.M., Gollmik F., Grafe U. Penicillium expansum, a resident fungal strain of the orbital complex Mir, producing xanthocyllin X and questiomycin A. Appl. Biochem. Microbiol. 2004;40:344–349. doi: 10.1023/B:ABIM.0000025954.82316.b4. [DOI] [PubMed] [Google Scholar]
- 31.Bitzer J., Große T., Wang L., Lang S., Beil W., Zeeck A. New aminophenoxazinones from a marine Halomonas sp.: Fermentation, structure elucidation, and biological activity. J. Antibiot. 2006;59:86–92. doi: 10.1038/ja.2006.12. [DOI] [PubMed] [Google Scholar]
- 32.Hanawa T., Osaki T., Manzoku T., Fukuda M., Kawakami H., Tomoda A., Kamiya S. In vitro antibacterial activity of Phx-3 against Helicobacter pylori. Biol. Pharm. Bull. 2010;33:188–191. doi: 10.1248/bpb.33.188. [DOI] [PubMed] [Google Scholar]
- 33.Guo S., Hu H., Bilal M., Zhang X. Production of antibacterial questiomycin A in metabolically engineered Pseudomonas chlororaphis HT66. J. Agric. Food Chem. 2022;70:7742–7750. doi: 10.1021/acs.jafc.2c03216. [DOI] [PubMed] [Google Scholar]
- 34.Barry C.E., III, Nayar P.G., Begley T.P. Phenoxazinone synthase: Mechanism for the formation of the phenoxazinone chromophore of actinomycin. Biochemistry. 1989;28:6323–6333. doi: 10.1021/bi00441a026. [DOI] [PubMed] [Google Scholar]
- 35.Le Roes-Hill M., Goodwin C., Burton S. Phenoxazinone synthase: What’s in a name? Trends Biotechnol. 2009;27:248–258. doi: 10.1016/j.tibtech.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 36.Nakachi T., Tabuchi T., Takasaki A., Arai S., Miyazawa K., Tomoda A. Anticancer activity of phenoxazines produced by bovine erythrocytes on colon cancer cells. Oncol. Rep. 2010;23:1517–1522. doi: 10.3892/or_00000790. [DOI] [PubMed] [Google Scholar]
- 37.Hongo T., Miyano-Kurosaki N., Kurosaki K., Hata A., Harigae S., Tomoda A. 2-Aminophenoxazine-3-one prevents pulmonary metastasis of mouse B16 melanoma cells in mice. J. Pharmacol. Sci. 2010;114:63–68. doi: 10.1254/jphs.10023FP. [DOI] [PubMed] [Google Scholar]
- 38.Kohno K., Miyake M., Sano O., Tanaka-Kataoka M., Yamamoto S., Koya-Miyata S., Arai N., Fujii M., Watanabe H., Ushio S., et al. Anti-inflammatory and immunomodulatory properties of 2-amino-3H-phenoxazin-3-one. Biol. Pharm. Bull. 2008;31:1938–1945. doi: 10.1248/bpb.31.1938. [DOI] [PubMed] [Google Scholar]
- 39.Baindara P., Mandal S.M. Bacteria and bacterial anticancer agents as a promising alternative for cancer therapeutics. Biochimie. 2020;177:164–189. doi: 10.1016/j.biochi.2020.07.020. [DOI] [PubMed] [Google Scholar]
- 40.Bhansali K.G., Kook A.M. Synthesis and characterization of a series of 5H-benzo[a]phenoxazin-5-one derivatives as potential antiviral/antitumor agents. Heterocycles. 1993;36:1239–1251. doi: 10.3987/COM-92-6170. [DOI] [Google Scholar]
- 41.Chau N.-M., Rogers P., Aherne W., Carroll V., Collins I., McDonald E., Workman P., Ashcroft M. Identification of novel small molecule inhibitors of hypoxia-inducible factor-1 that differentially block hypoxia-inducible factor-1 activity and hypoxia-inducible factor-1α induction in response to hypoxic stress and growth factors. Cancer Res. 2005;65:4918–4928. doi: 10.1158/0008-5472.CAN-04-4453. [DOI] [PubMed] [Google Scholar]
- 42.Lv X., Li J., Zhang C., Hu T., Li S., He S., Yan H., Tan Y., Lei M., Wen M., et al. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017;4:19–24. doi: 10.1016/j.gendis.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Z., You Q., Zhang X. Small-Molecule modulators of the hypoxia-inducible factor pathway: Development and therapeutic applications. J. Med. Chem. 2019;62:5725–5749. doi: 10.1021/acs.jmedchem.8b01596. [DOI] [PubMed] [Google Scholar]
- 44.Sharma A., Arambula J.F., Koo S., Kumar R., Singh H., Sessler J.L., Kim J.-S. Hypoxia-targeted drug delivery. Chem. Soc. Rev. 2019;48:771–813. doi: 10.1039/C8CS00304A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faig M., Bianchet M.A., Talalay P., Chen S., Winski S., Ross D., Amzel L.M. Structures of recombinant human and mouseNAD(P)H:quinone oxidoreductases: Species comparison and structural changes with substrate binding and release. Proc. Natl. Acad. Sci. USA. 2000;97:3177–3182. doi: 10.1073/pnas.97.7.3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song Y., Wang S., Zhao M., Yang X., Yu B. Strategies targeting protein tyrosine phosphatase SHP2 for cancer therapy. J. Med. Chem. 2022;65:3066–3079. doi: 10.1021/acs.jmedchem.1c02008. [DOI] [PubMed] [Google Scholar]
- 47.Homan K.T., Balasubramaniam D., Zabell A.P.R., Wiest O., Helquist P., Stauffacher C.V. Identification of novel inhibitors for a low molecular weight protein tyrosine phosphatase via virtual screening. Bioorganic Med. Chem. 2010;18:5449–5456. doi: 10.1016/j.bmc.2010.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Defant A., Guella G., Mancini I. Synthesis and in vitro cytotoxicity evaluation of novel naphthindolizinedione derivatives. Arch. Pharm. Chem. Life Sci. 2007;340:147–153. doi: 10.1002/ardp.200600160. [DOI] [PubMed] [Google Scholar]
- 49.Yang R., Chen Y., Pan L., Yang Y., Zheng Q., Hu Y., Wang Y., Zhang L., Sun Y., Li Z., et al. Design, synthesis and structure-activity relationship study of novel naphthoindolizine and indolizinoquinoline-5,12-dione derivatives as IDO1 inhibitors. Bioorganic Med. Chem. 2018;26:4886–4897. doi: 10.1016/j.bmc.2018.08.028. [DOI] [PubMed] [Google Scholar]
- 50.Feng X., Liao D., Liu D., Ping A., Li Z., Bian J. Development of indoleamine 2,3-dioxygenase 1 inhibitors for cancer therapy and beyond: A recent perspective. J. Med. Chem. 2020;63:15115–15139. doi: 10.1021/acs.jmedchem.0c00925. [DOI] [PubMed] [Google Scholar]
- 51.Ryu C.-K., Yoon J.-H., Song A.L., Im H.A., Kim J.Y., Kim A. Synthesis and antifungal evaluation of pyrido[1,2-a]indole-1,4-diones and benzo[f]pyrido[1,2-a]indole-6,11-diones. Bioorganic Med. Chem. 2012;22:497–499. doi: 10.1016/j.bmcl.2011.10.092. [DOI] [PubMed] [Google Scholar]
- 52.Székely R., Rengifo-Gonzalez M., Singh V., Riabova O., Benjak A., Piton J., Gimino M., Kornobus E., Mizrahi V., Johnsson K., et al. 6,11-Dioxobenzo[f]pyrido[1,2-a]indoles kill mycobacterium tuberculosis by targeting iron−sulfur protein Rv0338c (IspQ), a putative redox sensor. ACS Infect. Dis. 2020;6:3015–3025. doi: 10.1021/acsinfecdis.0c00531. [DOI] [PubMed] [Google Scholar]
- 53.Liu Y., Sun J.-W. Copper(II)-catalyzed synthesis of benzo[f]pyrido[1,2-a]indole-6,11-dione derivatives via naphthoquinone difunctionalization reaction. J. Org. Chem. 2012;77:1191–1197. doi: 10.1021/jo2023312. [DOI] [PubMed] [Google Scholar]
- 54.Sun J., Wang F., Hu H., Wang X., Wu H., Liu Y. Copper(II)-catalyzed carbon−carbon triple bond cleavage of internal alkynes for the synthesis of annulated indolizines. J. Org. Chem. 2014;79:3992–3998. doi: 10.1021/jo500456d. [DOI] [PubMed] [Google Scholar]
- 55.Matsuoka M., Iwamoto A., Kitao T. Reaction of 2,3-dichloro-1,4-naphthoquinone with dithiooxamide. Synthesis of dibenzo[b,i]thianthrene-5,7,12,14-tetrone. J. Heterocycl. Chem. 1991;28:1445–1447. doi: 10.1002/jhet.5570280543. [DOI] [Google Scholar]
- 56.Trzoss L., Fukuda T., Costa-Lotufo L.V., Jimenez P., La Clair J.J., Fenical W. Seriniquinone, a selective anticancer agent, induces cell death by autophagocytosis, targeting the cancer-protective protein dermcidin. Proc. Natl. Acad. Sci. USA. 2014;111:14687–14692. doi: 10.1073/pnas.1410932111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hammons J.C., Trzoss L., Jimenez P.C., Hirata A.S., Costa-Lotufo L.V., La Clair J.J., Fenical W. Advance of seriniquinone analogues as melanoma agents. ACS Med. Chem. Lett. 2019;10:186–190. doi: 10.1021/acsmedchemlett.8b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burian M., Schittek B. The secrets of dermcidin action. Int. J. Med. Microbiol. 2015;305:283–286. doi: 10.1016/j.ijmm.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 59.Nagao H., Ninomiya M., Sugiyama H., Itabashi A., Uno K., Tanaka K., Koketsu M. Comparative analysis of p-terphenylquinone and seriniquinone derivatives as reactive oxygen species-modulating agents. Bioorganic Med. Chem. Lett. 2022;76:128992. doi: 10.1016/j.bmcl.2022.128992. [DOI] [PubMed] [Google Scholar]
- 60.Tuchinda P., Pohmakotr M., Munyoo B., Reutrakul V., Santisuk T. An azaanthracene alkaloid from Polyalthia suberosa. Phytochemistry. 2000;53:1079–1082. doi: 10.1016/S0031-9422(99)00535-X. [DOI] [PubMed] [Google Scholar]
- 61.Gandy M.N., Piggott M.J. Synthesis of kalasinamide. A putative plant defense photoxin. J. Nat. Prod. 2008;71:866–868. doi: 10.1021/np070582z. [DOI] [PubMed] [Google Scholar]
- 62.Lang S., Groth U. Total syntheses of cytotoxic, naturally occurring kalasinamide, geovanine, and marcanine A. Angew. Chem. Int. Ed. 2009;48:911–913. doi: 10.1002/anie.200804388. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Y.-J., Kong M., Chen R.-Y., Yu D.Q. Alkaloids from the roots of Goniothalamus griffithii. J. Nat. Prod. 1999;62:1050–1052. doi: 10.1021/np990004l. [DOI] [PubMed] [Google Scholar]
- 64.Yin J., Landward M.B., Rainier J.D. Photoelectrocyclization reactions of amidonaphthoquinones. J. Org. Chem. 2020;85:4298–4311. doi: 10.1021/acs.joc.9b03417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soonthornchareonnon N., Suwanborirux K., Bavovada R., Patarapanich C., Cassady J.M. New cytotoxic 1-azaanthraquinones and 3-aminonaphthoquinone from the stem bark of Goniothalamus marcanii. J. Nat. Prod. 1999;62:1390–1394. doi: 10.1021/np990197c. [DOI] [PubMed] [Google Scholar]
- 66.Sedmera P., Pospisil S., Novak J. New furanonaphthoquinone from Streptomyces cinnamonensis. J. Nat. Prod. 1991;54:870–872. doi: 10.1021/np50075a022. [DOI] [Google Scholar]
- 67.Bringmann G., Haagen Y., Gulder T.A.M., Gulder T., Heide L. Biosynthesis of the isoprenoid moieties of furanonaphthoquinone I and endophenazine A in Streptomyces cinnamonensis DSM 1042. J. Org. Chem. 2007;72:4198–4204. doi: 10.1021/jo0703404. [DOI] [PubMed] [Google Scholar]
- 68.Simamura E., Hirai K.-I., Shimada H., Koyama J., Niwa Y., Shimizu S. Furanonaphthoquinones cause apoptosis of cancer cells by inducing the production of reactive oxygen species by the mitochondrial voltage-dependent anion channel. Cancer Biol. Ther. 2006;5:1523–1529. doi: 10.4161/cbt.5.11.3302. [DOI] [PubMed] [Google Scholar]
- 69.Tahara T., Watanabe A., Yutani M., Yamano Y., Sagara M., Nagai S., Saito K., Yamashita M., Ihara M., Iida A. STAT3 inhibitory activity of naphthoquinones isolated from Tabebuia avellanedae. Bioorganic Med. Chem. 2020;28:115347. doi: 10.1016/j.bmc.2020.115347. [DOI] [PubMed] [Google Scholar]
- 70.Feng X., Qiu X., Huang H., Wang J., Xu X., Xu P., Ge R., Liu X., Li Z., Bian J. Palladium(II)-catalyzed reaction of Lawsones and propargyl carbonates: Construction of 2,3-furanonaphthoquinones and evaluation as potential indoleamine 2,3-dioxygenase inhibitors. J. Org. Chem. 2018;83:8003–8010. doi: 10.1021/acs.joc.8b00872. [DOI] [PubMed] [Google Scholar]
- 71.Hu X., Li J., Fu M., Zhao X., Wang W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021;6:402. doi: 10.1038/s41392-021-00791-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu Z.-Z., Jang Y.-J., Lee C.-J., Lin W. A versatile and practical method for the regioselective synthesis of polysubstituted furanonapthoquinones. Org. Biomol. Chem. 2013;11:828–834. doi: 10.1039/C2OB26986D. [DOI] [PubMed] [Google Scholar]
- 73.Lin C.-Y., Wu H.-Y., Hsu Y.-L., Cheng T.-J.R., Liu J.-H., Huang R.-J., Hsiao T.-H., Wang C.-J., Hung P.-F., Lan A., et al. Suppression of drug-resistant non-small-cell lung cancer with inhibitors targeting minichromosomal maintenance protein. J. Med. Chem. 2020;63:3172–3187. doi: 10.1021/acs.jmedchem.9b01783. [DOI] [PubMed] [Google Scholar]
- 74.Tsai C.-J., Chen C.-C., Tsai C.-W., Wu M.J. Base-mediated cyclization reaction of 2-(5-hydroxy-1-pentynyl)benzonitriles to 4-amino-2,3-dihydronaphtho[2,3-b]furanes and synthesis of furanonaphthoquinones. J. Org. Chem. 2016;81:3882–3889. doi: 10.1021/acs.joc.5b02514. [DOI] [PubMed] [Google Scholar]
- 75.El-Feraly F.S. Melampolides from Magnolia grandiflora. Phytochemistry. 1984;23:2372–2374. doi: 10.1016/S0031-9422(00)80557-9. [DOI] [Google Scholar]
- 76.Nasim S., Pei S., Hagen F.K., Jordan C.T., Crooks P.A. Melampomagnolide B: A new antileukemic sesquiterpene. Bioorganic Med. Chem. 2011;19:1515–1519. doi: 10.1016/j.bmc.2010.12.045. [DOI] [PubMed] [Google Scholar]
- 77.Janganati V., Ponder J., Jordan C.T., Borrelli M.J., Penthala N.R., Crooks P.A. Dimers of melampomagnolide B exhibit potent anticancer activity against hematological and solid tumor cells. J. Med. Chem. 2015;58:8896–8906. doi: 10.1021/acs.jmedchem.5b01187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Penthala N.R., Balasubramaniam M., Dachavaram S.S., Morris E.J., Bhat-Nakshatri P., Ponder J., Jordan C.T., Nakshatri H., Crooks P.A. Antitumor properties of novel sesquiterpene lactone analogs as NFκB inhibitors that bind to the IKKβ ubiquitin-like domain (ULD) Eur. J. Med. Chem. 2021;224:113675. doi: 10.1016/j.ejmech.2021.113675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mischenko N.P., Fedoreyev S.A., Pokhilo N.D., Anufriev V.P., Denisenko V.A., Glazunov V.P. Echinamines A and B, first aminated hydroxynaphthazarins from the sea urchin Scaphechinus mirabilis. J. Nat. Prod. 2005;68:1390–1393. doi: 10.1021/np049585r. [DOI] [PubMed] [Google Scholar]
- 80.Mishchenko N.P., Krylova N.V., Iunikhina O.V., Vasileva E.A., Likhatskaya G.N., Pislyagin E.A., Tarbeeva D.V., Dmitrenok P.S., Fedoreyev S.A. Antiviral potential of sea urchin aminated spinochromes against herpes simplex virus type 1. Mar. Drugs. 2020;18:550. doi: 10.3390/md18110550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.You H., Vegi S.R., Lagishetti C., Chen S., Reddy R.S., Yang X., Guo J., Wang C., He Y. Synthesis of bioactive 3,4-dihydro-2H-naphtho[2,3-b][1,4]oxazine-5,10-dione and 2,3,4,5-tetrahydro-1H-naphtho[2,3-b]azepine-6,11-dione derivatives via the copper-catalyzed intramolecular coupling reaction. J. Org. Chem. 2018;83:4119–4130. doi: 10.1021/acs.joc.8b00020. [DOI] [PubMed] [Google Scholar]
- 82.Kirkpatrick D.L., Powis G. Inhibitors of Redox Signaling for Restoration of Apoptosis and Inhibition of Abnormal Cell Proliferation. WO2000006088 A2. 2000 October 2;
- 83.Liu L., Richard J., Kim S., Wojcik E.J. Small molecule screen for candidate antimalarials targeting plasmodium kinesin-5. J. Biol. Chem. 2014;289:16601–16614. doi: 10.1074/jbc.M114.551408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tomek P., Palmer B.D., Flanagan J.U., Sun C., Raven E.L., Ching L.-M. Discovery and evaluation of inhibitors to the immunosuppressive enzyme indoleamine 2,3-dioxygenase 1 (IDO1): Probing the active site-inhibitor interactions. Eur. J. Med. Chem. 2017;126:983–996. doi: 10.1016/j.ejmech.2016.12.029. [DOI] [PubMed] [Google Scholar]
- 85.Kulkarni A., Soni I., Kelkar D.S., Dharmaraja A.T., Sankar R.K., Beniwal G., Rajendran A., Tamhankar S., Chopra S., Kamat S.S., et al. Chemoproteomics of an indole-based quinone epoxide identifies druggable vulnerabilities in vancomycin-resistant Staphylococcus aureus. J. Med. Chem. 2019;62:6785–6795. doi: 10.1021/acs.jmedchem.9b00774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bergamasco R., Porter Q.N., Yap C. Vinylindenes and some heteroanalogues in the Diels-Alder reaction. V. 3-Vinylindoles and quinones. Aust. J. Chem. 1978;31:1841–1844. doi: 10.1071/CH9781841. [DOI] [Google Scholar]
- 87.Kuo C.-W., Konala A., Lin L., Chiang T.-T., Huang C.-Y., Yang T.-H., Kavala V., Yao C.-F. Synthesis of benzo[a]carbazole derivatives from 3-ethylindoles by exploiting the dual character of benzoquinone as an oxidizing agent and dienophile. Chem. Commun. 2016;52:7870–7873. doi: 10.1039/C6CC03124B. [DOI] [PubMed] [Google Scholar]
- 88.Brimble M.A., Duncalf L.J., Nairn M.R. Pyranonaphthoquinone antibiotics—Isolation, structure and biological activity. Nat. Prod. Rep. 1999;16:267–281. doi: 10.1039/a804287j. [DOI] [PubMed] [Google Scholar]
- 89.Naysmith B.J., Hume P.A., Sperry J., Brimble M.A. Pyranonaphthoquinones—Isolation, biology and synthesis: An update. Nat. Prod. Rep. 2017;34:25–61. doi: 10.1039/C6NP00080K. [DOI] [PubMed] [Google Scholar]
- 90.Bergy M.E. Kalafungin, a new broad spectrum antibiotic: Isolation and characterization. J. Antibiot. 1968;21:454–457. doi: 10.7164/antibiotics.21.454. [DOI] [PubMed] [Google Scholar]
- 91.Kakinuma S., Ikeda H., Omura S., Hopwood D.A. Biosynthesis of kalafungin in Streptomyces tanashiensis. J. Antibiot. 1990;43:391–396. doi: 10.7164/antibiotics.43.391. [DOI] [PubMed] [Google Scholar]
- 92.Omura S., Tanaka H., Okada Y., Marumo H. Isolation and structure of nanaomycin D, an enantiomer of the antibiotic kalafungin. J. Chem. Soc. Chem. Commun. 1976;1976:320–321. doi: 10.1039/C39760000320. [DOI] [Google Scholar]
- 93.Heapy A.M., Patterson A.V., Smaill J.B., Jamieson S.M.F., Guise C.P., Sperry J., Hume P.A., Rathwell K., Brimble M.A. Synthesis and cytotoxicity of pyranonaphthoquinone natural product analogues under bioreductive conditions. Bioorganic Med. Chem. 2013;21:7971–7980. doi: 10.1016/j.bmc.2013.09.052. [DOI] [PubMed] [Google Scholar]
- 94.Tanaka H., Minami-Kakinuma S., Omura S. Biosynthesis of nanaomycin. III. Nanaomycin A formation from nanaomycin D by nanaomycin D reductase via a hydroquinone. J. Antibiot. 1982;35:1565–1570. doi: 10.7164/antibiotics.35.1565. [DOI] [PubMed] [Google Scholar]
- 95.Salaski E.J., Krishnamurthy G., Ding W.-D., Yu K., Insaf S.S., Eid C., Shim J., Levin J.I., Tabei K., Toral-Barza L., et al. Pyranonaphthoquinone lactones: A new class of AKT selective kinase inhibitors alkylate a regulatory loop cysteine. J. Med. Chem. 2009;52:2181–2184. doi: 10.1021/jm900075g. [DOI] [PubMed] [Google Scholar]
- 96.Kuck D., Caulfield T., Lyko F., Medina-Franco J.L. Nanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cells. Mol. Cancer Ther. 2010;9:3015–3023. doi: 10.1158/1535-7163.MCT-10-0609. [DOI] [PubMed] [Google Scholar]
- 97.Tsuji N., Kobayashi M., Wakisaka Y., Kawamura Y., Mayama M., Matsumoto K. New antibiotics, griseusins A and B. Isolation and characterization. J. Antibiot. 1976;29:7–9. doi: 10.7164/antibiotics.29.7. [DOI] [PubMed] [Google Scholar]
- 98.Masamoto K., Takeuchi Y., Takeda K., Yoshii E. Synthesis of pentacyclic structure of griseusin A. Heterocycles. 1981;16:1659–1664. [Google Scholar]
- 99.Brimble M.A., Nairn M.R. Synthesis of the Griseusin A ring system. J. Chem. Soc. Perkin Trans. 1. 1990;1990:169–171. doi: 10.1039/p19900000169. [DOI] [Google Scholar]
- 100.Brimble M.A., Nairn M.R. Synthesis of a pyranonaphthoquinone-spiroacetal. J. Chem. Soc. Perkin Trans. 1. 1992;1992:579–583. doi: 10.1039/p19920000579. [DOI] [Google Scholar]
- 101.Brimble M.A., Nairn M.R., Park J. Synthesis of Analogues of Griseusin, A. Org. Lett. 1999;1:1459–1462. doi: 10.1021/ol991020c. [DOI] [Google Scholar]
- 102.Perry N.B., Blunt J.W., McCombs J.D., Munro M.H.G. Discorhabdin C, a highly cytotoxic pigment from a sponge of the genus Latrunculia. J. Org. Chem. 1986;51:5476–5478. doi: 10.1021/jo00376a096. [DOI] [Google Scholar]
- 103.Hu J.-F., Fan H., Xiong J., Wu S.-B. Discorhabdins and pyrroloiminoquinone-related alkaloids. Chem. Rev. 2011;111:5465–5491. doi: 10.1021/cr100435g. [DOI] [PubMed] [Google Scholar]
- 104.Copp B.R., Fulton K.F., Perry N.B., Blunt J.W., Munro M.H.G. Natural and synthetic derivatives of discorhabdin C, a cytotoxic pigment from the New Zealand sponge Latrunculia cf. bocagei. J. Org. Chem. 1994;59:8233–8238. doi: 10.1021/jo00105a047. [DOI] [Google Scholar]
- 105.Na M.K., Ding Y., Wang B., Tekwani B.L., Schinazi R.F., Franzblau S., Kelly M., Stone R., Li X.-C., Ferreira D., et al. Anti-infective discorhabdins from a deep-water Alaskan sponge of the genus Latrunculia. J. Nat. Prod. 2010;73:383–387. doi: 10.1021/np900281r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Goey A.K.L., Chau C.H., Sissung T.M., Cook K.M., Venzon D.J., Castro A., Ransom T.R., Henrich C.J., McKee T.C., McMahon J.B., et al. Screening and biological effects of marine pyrroloiminoquinone alkaloids: Potential inhibitors of the HIF-1α/p300 interaction. J. Nat. Prod. 2016;79:1267–1275. doi: 10.1021/acs.jnatprod.5b00846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mąkosza M., Stalewski J., Maslennikova O.S. Synthesis of 7,8-dimethoxy-4-oxo-1,3,4,5-tetrahydropyrrolo[4,3,2-de]quinoline. A key intermediate en route to makaluvamines, discorhabdin C and other marine alkaloids of this group via vicarious nucleophilic substitution of hydrogen. Synthesis. 1997;1997:1131–1133. doi: 10.1055/s-1997-1325. [DOI] [Google Scholar]
- 108.Roberts D., Joule J.A., Bros M.A., Alvarez M. Synthesis of pyrrolo[4,3,2-de]quinolines from 6,7-dimethoxy-4-methylquinoline. Formal total syntheses of damirones A and B, batzelline C, isobatzelline C, discorhabdin C, and makaluvamines A–D. J. Org. Chem. 1997;62:568–577. doi: 10.1021/jo961743z. [DOI] [PubMed] [Google Scholar]
- 109.Kita Y., Tohma H., Inagaki M., Hatanaka K., Yakura T. Total synthesis of discorhabdin C: A general aza spiro dienone formation from O-silylated phenol derivatives using a hypervalent iodine reagent. J. Am. Chem. Soc. 1992;114:2175–2180. doi: 10.1021/ja00032a036. [DOI] [Google Scholar]
- 110.Sadanandan E.V., Pillai S.K., Lakshmikantham M.V., Billimoria A.D., Culpepper J.S., Cava M.P. Efficient syntheses of the marine alkaloids makaluvamine D and discorhabdin C: The 4,6,7-trimethoxyindole approach. J. Org. Chem. 1995;60:1800–1805. doi: 10.1021/jo00111a043. [DOI] [Google Scholar]
- 111.Aubart K.M., Heathcock C.H. A Biomimetic approach to the discorhabdin alkaloids: Total syntheses of discorhabdins C and E and dethiadiscorhabdin D. J. Org. Chem. 1999;64:16–22. doi: 10.1021/jo9815397. [DOI] [PubMed] [Google Scholar]
- 112.Kublak G.G., Confalone P.N. The preparation of the aza-spirobicyclic system of discorhabdin C via an intramolecular phenolate alkylation. Tetrahedron Lett. 1990;31:3845–3848. doi: 10.1016/S0040-4039(00)97484-7. [DOI] [Google Scholar]
- 113.Kadela-Tomanek M., Bebenek E., Chrobak E., Boryczka S. 5,8-Quinolinedione scaffold as a promising moiety of bioactive agents. Molecules. 2019;24:4115. doi: 10.3390/molecules24224115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bolzán A.D., Bianchi M.S. Genotoxicity of streptonigrin: A review. Mutat. Res. 2001;488:25–37. doi: 10.1016/S1383-5742(00)00062-4. [DOI] [PubMed] [Google Scholar]
- 115.Rao K.V., Cullen W.P. Streptonigrin, an antitumor substance. In: Welch H., Martilhanez F., editors. Antibiotics Annual: 1959–1960. Medical Encyclopedia, Inc.; New York, NY, USA: 1960. pp. 950–953. [PubMed] [Google Scholar]
- 116.Doyle T.W., Balitz D.M., Grulich R.E., Nettleton D.E., Gould S.J., Tann C.-H., Moews A.E. Structure determination of lavendamycin- a new antitumor antibiotic from Streptomyces lavendulae. Tetrahedron Lett. 1981;22:4595–4598. doi: 10.1016/S0040-4039(01)82990-7. [DOI] [Google Scholar]
- 117.Behforouz M., Cai W., Stocksdale M.G., Lucas J.S., Jung J., Briere D., Wang A., Katen K.S., Behforouz N.C. Novel lavendamycin analogues as potent HIV-reverse transcriptase inhibitors: Synthesis and evaluation of anti-reverse transcriptase activity of amide and ester analogues of lavendamycin. J. Med. Chem. 2003;46:5773–5780. doi: 10.1021/jm0304414. [DOI] [PubMed] [Google Scholar]
- 118.Hassani M., Cai W., Koelsch K.H., Holley D.C., Rose A.S., Olang F., Lineswala J.P., Holloway W.G., Gerdes J.M., Behforouz M., et al. Lavendamycin antitumor agents: Structure-based design, synthesis, and NAD(P)H:quinone oxidoreductase 1 (NQO1) model validation with molecular docking and biological studies. J. Med. Chem. 2008;51:3104–3115. doi: 10.1021/jm701066a. [DOI] [PubMed] [Google Scholar]
- 119.Porter T.H., Skelton F.S., Folkers K. Synthesis of new alkylamino- and alkylaminomethyl-5,8-quinolinequinones as inhibitors of Coenzyme Q and as antimalarials. J. Med. Chem. 1971;14:1029–1033. doi: 10.1021/jm00293a003. [DOI] [PubMed] [Google Scholar]
- 120.Cossy J., Belotti D., Brisson M., Skoko J.J., Wipf P., Lazo J.S. Biological evaluation of newly synthesized quinoline-5,8-quinones as Cdc25B inhibitors. Bioorganic Med. Chem. 2006;14:6283–6287. doi: 10.1016/j.bmc.2006.05.053. [DOI] [PubMed] [Google Scholar]
- 121.Santoso K.T., Menorca A., Cheung C.-Y., Cook G.M., Stocker B.L., Timmer M.S.M. The synthesis and evaluation of quinolinequinones as anti-mycobacterial agents. Bioorganic Med. Chem. 2019;27:3532–3545. doi: 10.1016/j.bmc.2019.06.002. [DOI] [PubMed] [Google Scholar]
- 122.Egu S.A., Ibezim A., Onoabedje E.A., Okoro U.C. Biological and in silico evaluation of quinolinedione and naphthoquinone derivatives as potent antibacterial agents. ChemistrySelect. 2017;2:9222–9226. doi: 10.1002/slct.201700692. [DOI] [Google Scholar]
- 123.Ciftci H.I., Bayrak N., Yıldız M., Yıldırım H., Sever B., Tateishi H., Otsuka M., Fujita M., Tuyun A.F. Design, synthesis and investigation of the mechanism of action underlying anti-leukemic effects of the quinolinequinones as LY83583 analogs. Bioorganic Chem. 2021;114:105160. doi: 10.1016/j.bioorg.2021.105160. [DOI] [PubMed] [Google Scholar]
- 124.Ryu C.-K., Jeong H.-J., Lee S.K., You H.J., Choi K.U., Shim J.Y., Heo Y.H., Lee C.-O. Effects of 6-arylamino-5,8-quinolinediones and 6-chloro-7-arylamino-5,8-isoquinolinediones on NAD(P)H:quinone oxidoreductase (NQO1) activity and their cytotoxic potential. Arch. Pharm. Res. 2001;24:390–396. doi: 10.1007/BF02975181. [DOI] [PubMed] [Google Scholar]
- 125.Fryatt T., Pettersson H.I., Gardipee W.T., Bray K.C., Green S.J., Slawin A.M.Z., Beall H.D., Moody C.J. Novel quinolinequinone antitumor agents: Structure-metabolism studies with NAD(P)H:quinone oxidoreductase (NQO1) Bioorganic Med. Chem. 2004;12:1667–1687. doi: 10.1016/j.bmc.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 126.Schlager J.J., Powis G. Cytosolic NAD(P)H:(quinone-acceptor)oxidoreductase in human normal and tumor tissue: Effects of cigarette smoking and alcohol. Int. J. Cancer. 1990;45:403–409. doi: 10.1002/ijc.2910450304. [DOI] [PubMed] [Google Scholar]
- 127.Cerecetto H., González M., Lavaggi M.L., Azqueta A., de Cerain A.L., Monge A. Phenazine 5,10-dioxide derivatives as hypoxic selective cytotoxins. J. Med. Chem. 2005;48:21–23. doi: 10.1021/jm0492150. [DOI] [PubMed] [Google Scholar]
- 128.Lavaggi M.L., Cabrera M., Pintos C., Arredondo C., Pachon G., Rodriguez J., Raymondo S., Pacheco J.P., Cascante M., Olea-Azar C., et al. Novel phenazine 5,10-dioxides release •OH in simulated hypoxia and induce reduction of tumour volume in vivo. ISRN Pharmacol. 2011;2011:314209. doi: 10.5402/2011/314209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Reddy S.B., Williamson S.K. Tirapazamine: A novel agent targeting hypoxic tumor cells. Expert Opin. Investig. Drugs. 2009;18:77–87. doi: 10.1517/13543780802567250. [DOI] [PubMed] [Google Scholar]
- 130.Abi-Jaoudeh N., Dayyani F., Chen P.J., Fernando D., Fidelman N., Javan H., Liang P.-C., Hwang J.-I., Imagawa D.K. Phase I trial on arterial embolization with hypoxia activated tirapazamine for unresectable hepatocellular carcinoma. J. Hepatocell. Carcinoma. 2021;8:421–434. doi: 10.2147/JHC.S304275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen E.H., Tanabe K., Saggiomo A.J., Nodiff E.A. Modifications of primaquine as antimalarials. 4. 5-Alkoxy derivatives of primaquine. J. Med. Chem. 1987;30:1193–1199. doi: 10.1021/jm00390a012. [DOI] [PubMed] [Google Scholar]
- 132.Shelke Y.G., Hande P.E., Gharpure S.J. Recent advances in the synthesis of pyrrolo[1,2-a]indoles and their derivatives. Org. Biomol. Chem. 2021;19:7544–7574. doi: 10.1039/D1OB01103K. [DOI] [PubMed] [Google Scholar]
- 133.Bass P.D., Gubler D.A., Judd T.C., Williams R.M. Mitomycinoid alkaloids: Mechanism of action, biosynthesis, total syntheses, and synthetic approaches. Chem. Rev. 2013;113:6816–6863. doi: 10.1021/cr3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Liu L., Wang Y., Zhang L. Formal synthesis of 7-methoxymitosene and synthesis of its analog via a key PtCl2-catalyzed cycloisomerization. Org. Lett. 2012;14:3736–3739. doi: 10.1021/ol301593w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zheng Z., Touve M., Barnes J., Reich N., Zhang L. Synthesis-enabled probing of mitosene structural space leads to improved IC50 over mitomycin C. Angew. Chem. Int. Ed. 2014;53:9302–9305. doi: 10.1002/anie.201402268. [DOI] [PubMed] [Google Scholar]
- 136.Ayala C.E., Villalpando A., Nguyen A.L., McCandless G.T., Kartika R. Chlorination of aliphatic primary alcohols via triphosgene–triethylamine activation. Org. Lett. 2012;14:3676–3679. doi: 10.1021/ol301520d. [DOI] [PubMed] [Google Scholar]
- 137.Sweeney M. Ph.D. Thesis. National University of Ireland Galway; Galway, Ireland: 2019. Hydrogen Peroxide and Hydrohalic acid Mediated Synthesis of Halogenated Benzimidazolequinones and Anti-cancer Evaluation of Benzotriazinones. [Google Scholar]
- 138.Lee S.-R., Yang K.-S., Kwon J., Lee C., Jeong W., Rhee S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 139.Smith S.L., Pitt A.R., Spickett C.M. Approaches to investigating the protein interactome of PTEN. J. Proteome Res. 2021;20:60–77. doi: 10.1021/acs.jproteome.0c00570. [DOI] [PubMed] [Google Scholar]
- 140.Meuillet E.J., Mahadevan D., Berggren M., Coon A., Powis G. Thioredoxin-1 binds to the C2 domain of PTEN inhibiting PTEN’s lipid phosphatase activity and membrane binding: A mechanism for the functional loss of PTENs tumor suppressor activity. Arch. Biochem. Biophys. 2004;429:123–133. doi: 10.1016/j.abb.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 141.Duan D., Zhang J., Yao J., Liu Y., Fang J. Targeting thioredoxin reductase by parthenolide contributes to inducing apoptosis of HeLa Cells. J. Biol. Chem. 2016;291:10021–10031. doi: 10.1074/jbc.M115.700591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Madkour M.M., Anbar H.S., El-Gamal M.I. Current status and future prospects of p38a/MAPK14 kinase and its inhibitors. Eur. J. Med. Chem. 2021;213:113216. doi: 10.1016/j.ejmech.2021.113216. [DOI] [PubMed] [Google Scholar]
- 143.Paz M.M., Zhang X., Lu J., Holmgren A. A new mechanism of action for the anticancer drug mitomycin C: Mechanism-based inhibition of thioredoxin reductase. Chem. Res. Toxicol. 2012;25:1502–1511. doi: 10.1021/tx3002065. [DOI] [PubMed] [Google Scholar]
- 144.Newsome J.J., Swann E., Hassani M., Bray K.C., Slawin A.M.Z., Beall H.D., Moody C.J. Indolequinone antitumour agents: Correlation between quinone structure and rate of metabolism by recombinant human NAD(P)H:quinone oxidoreductase. Org. Biomol. Chem. 2007;5:1629–1640. doi: 10.1039/b703370b. [DOI] [PubMed] [Google Scholar]
- 145.Yan C., Siegel D., Newsome J.J., Chilloux A., Moody C.J., Ross D. Antitumor indolequinones induced apoptosis in human pancreatic cancer cells via inhibition of thioredoxin reductase and activation of redox signaling. Mol. Pharmacol. 2012;81:401–410. doi: 10.1124/mol.111.076091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nitulescu G.M., Soriga S.G., Socea L.I., Olaru O.T., Plesu V. Structure-activity relationships and chemoinformatic analysis of the anticancer profile of an aminopyrazole derivative. Rev. Chim. 2016;67:162–165. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.