

Abstract
The diagnosis of nephrotic syndrome relies on clinical presentation and descriptive patterns of injury on kidney biopsies, but not specific to underlying pathobiology. Consequently, there are variable rates of progression and response to therapy within diagnoses. Here, an unbiased transcriptomic-driven approach was used to identify molecular pathways which are shared by subgroups of patients with either minimal change disease (MCD) or focal segmental glomerulosclerosis (FSGS). Kidney tissue transcriptomic profile-based clustering identified three patient subgroups with shared molecular signatures across independent, North American, European, and African cohorts. One subgroup had significantly greater disease progression (Hazard Ratio 5.2) which persisted after adjusting for diagnosis and clinical measures (Hazard Ratio 3.8). Inclusion in this subgroup was retained even when clustering was limited to those with less than 25% interstitial fibrosis. The molecular profile of this subgroup was largely consistent with tumor necrosis factor (TNF) pathway activation. Two TNF pathway urine markers were identified, tissue inhibitor of metalloproteinases-1 (TIMP-1) and monocyte chemoattractant protein-1 (MCP-1), that could be used to predict an individual’s TNF pathway activation score. Kidney organoids and single nucleus RNA-sequencing of participant kidney biopsies, validated TNF-dependent increases in pathway activation score, transcript and protein levels of TIMP-1 and MCP-1, in resident kidney cells. Thus, molecular profiling identified a subgroup of patients with either MCD or FSGS who shared kidney TNF pathway activation and poor outcomes. A clinical trial testing targeted therapies in patients selected using urinary markers of TNF pathway activation is ongoing.
Keywords: Nephrotic Syndrome, Transcriptomics, TNF, Data Integration
Introduction
Nephrotic syndrome is characterized by proteinuria, hypoalbuminemia, hyperlipidemia and edema. Two nephrotic diseases, minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS), currently diagnosed based on histopathologic features, have broadly overlapping clinical presentations and treatment approaches1. Within each diagnosis, however, some patients respond well to current therapy, while others either do not respond or relapse upon stopping therapy with variable risk of loss of kidney function. These clinical observations suggest a heterogenous biology underlying current disease classification.2–4 Due to limited understanding of pathobiology, interpretation of the clinical outcome variability in observational studies and clinical trials is challenging5, 6, and molecularly-driven7, 8, personalized6 treatments for MCD and FSGS are unavailable.
Nevertheless, nephrotic syndrome is well-positioned for implementing precision medicine. Clinically procured kidney biopsy tissue allows for unbiased identification of molecular signatures that can be linked to histopathology, non-invasive biomarkers and evaluated against clinical outcomes. With the goal of identifying molecular pathways which are shared by subgroups of MCD and FSGS patients, this study implemented a multi-dimensional data integration approach (Figure 1) in the prospective North American Nephrotic Syndrome Study Network (NEPTUNE)9, then replicated in the European Renal cDNA Bank (ERCB)10, 11 and the Human Heredity and Health in Africa Kidney Disease Research Network cohort (H3Africa)12, 13.
Figure 1: Analysis strategy.
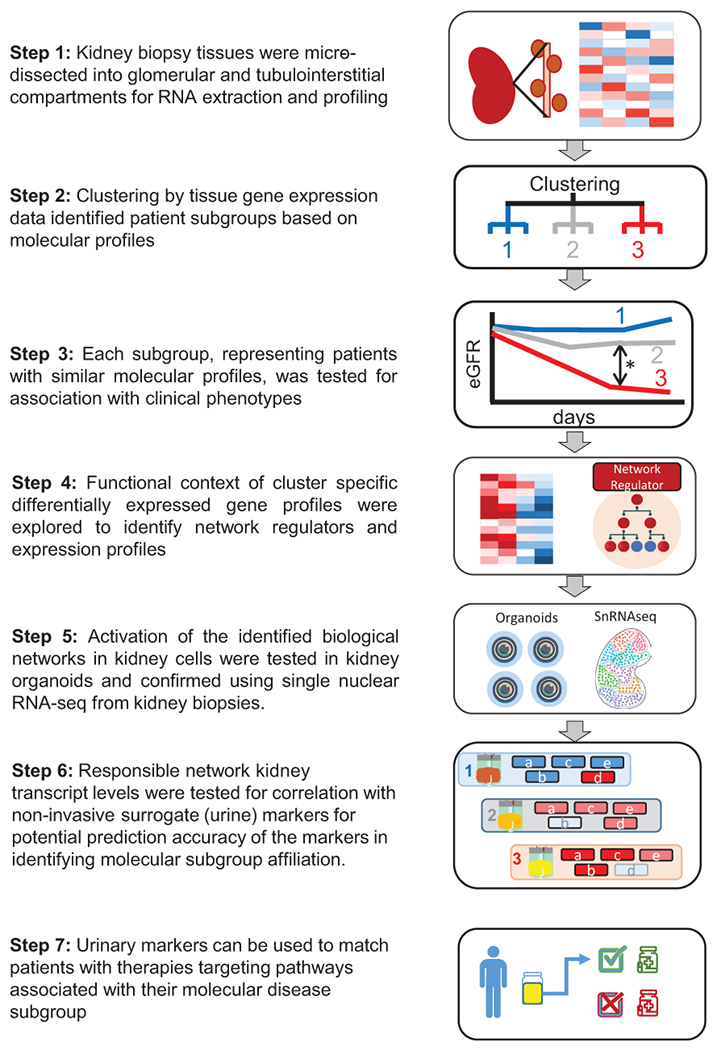
Flowchart of tubulointerstitial compartment gene expression to identify molecular subgroups and associated non-invasive urinary markers.
The study aims to agnostically identify groups of patients with shared molecular signatures, to identify the relevant pathways from that signature that could then be evaluated in individual patients using potential non-invasive markers. Such markers of disease mechanism may enable targeted therapeutic interventions.
Methods
Study Participants
The study included 220 NEPTUNE9, 35 H3Africa 12, and 30 ERCB10, 11 participants with biopsy-proven MCD or FSGS and compartment-enriched genome-wide kidney mRNA expression profiles.
NEPTUNE (NCT01209000) is a prospective study of children and adults with proteinuria, recruited from 21 sites at the time of their first clinically indicated kidney biopsy9, 14. ERCB is a European study of adults recruited at 28 sites, with biopsy tissue for gene expression profiling and cross-sectional clinical information, at the time of a clinically indicated kidney biopsy10, 11. H3 Africa12 is a prospective study of participants aged 15 years and above, eligible for a kidney biopsy, recruited from 13 clinical centers in Nigeria and Ghana with estimated glomerular filtration rate (eGFR) ≥15 mL/min/1.73m2 and proteinuria (albuminuria >500 mg/day) 12.
For all cohorts, informed consent was obtained from individual patients or parents/guardians, on approval by Institutional Review Boards or local ethics committees of participating institutions.
Clinical Data
NEPTUNE participants were followed 2-3 times per year, for up to 5 years. Medical history, medication use, laboratory results, blood and urine samples for measurement of serum creatinine and urine protein/creatinine ratio (UPCR) were collected at each study visit. eGFR (mL/min/1.73m2) was calculated using the CKD-Epi formula for participants ≥18 years old and the modified CKiD-Schwartz formula for participants <18 years old, with an average taken for young adults age 18-26 years 15–17. The endpoint of kidney functional loss was defined by 40% reduction in eGFR or onset of ESKD (initiation of dialysis, receipt of kidney transplant or eGFR <15 mL/min/1.73m2 measured at two sequential visits)18. Complete remission was defined as UPCR <0.3 mg/mg on a single void urine or a 24-hour urine collection. In ERCB and H3 Africa, clinical information, including demographics and clinical laboratory results, were obtained at time of biopsy.
Kidney Pathology:
Diagnosis in NEPTUNE was assigned by study pathologists based on biopsy reports and/or digital images. The degree of interstitial fibrosis (IF) was visually assessed and scoredby 2-5 pathologists using biopsy whole slide images of trichrome, PAS, or silver-stained sections, recorded as the percent of cortex involved, and averaged across the pathologists’ measures19, 20.
Transcriptome profiling
In NEPTUNE, RNA-sequencing (RNAseq) was performed on manually micro-dissected kidney biopsy tissue that separated tubulointerstitial and glomerular compartments of the research core. For H3 Africa, a 5 mm cortical segment (not needed for clinical diagnosis) was manually micro-dissected, RNA isolated, and sequenced to generate RNAseq profiles (see Supplementary Methods).
In ERCB, compartment specific transcriptomic profiles of the research core were generated using the Affymetrix microarray platform (Santa Clara, CA). NEPTUNE RNAseq and ERCB microarray data are available at Nephroseq.org and through Gene Expression Omnibus21 (GSE219185, GSE197307, GSE104954, GSE104948).
Cluster analysis, differential expression and functional enrichment analysis
R Software (R Core Team (2013), Vienna) was used for Kmeans, PAM and hierarchical HCL clustering analyses22, 23. Optimal clustering was determined using delta-K and the Consensus Cluster Plus Package22. Linear models for microarray data (limma) package24 was used for differential expression analysis. Differentially expressed genes (absolute fold change >1.5 and q-value<0.05) between clusters were analyzed for enrichment of canonical pathways using the Ingenuity Pathway Analysis Software Suite (IPA, Qiagen, Hilden, Germany)25. Previously published26 single cell RNA-seq (scRNAseq) cell-type selective expression clusters from adult reference kidney tissue can be accessed at http://nephrocell.miktmc.org/ and GEO (GSE14098).
Tumor Necrosis Factor (TNF) Activation Score:
A TNF activation network was generated from expert curated interactions from NETPro annotations in the Genomatix Genome Analyzer database (Precigen Bioinformatics, Germany). From the database, 272 causally downstream genes or proteins that increased expression from TNF exposure were used to generate a TNF activation score. Individual gene expression values were first Z-transformed. The TNF activation score for each participant was the average Z-score of the 272 genes in their kidney RNAseq profile.
Urine Marker Profiling:
Urine proteins were identified and measured using the multiplex Luminex platform (Eve Technologies, Alberta, Canada) composed of a panel of 54 urinary cytokines, matrix metalloproteinases and tissue inhibitor of metalloproteinases (see Supplementary Methods). A candidate protein had to satisfy the following criteria to be considered a potential non-invasive marker of TNF activation: 1) protein must be a product of a gene expressed downstream of TNF; 2) urine protein expression must correlate with the corresponding intra-renal tissue gene expression (mRNA levels) and 3) gene expression must correlate with the TNF activation score.
Putative urine markers were assayed in duplicate using Quantikine ELISA kit Human chemokine (C-C motif) ligand 2 (CCL2) / monocyte chemoattractant protein-1 (MCP-1) (DCP00) and tissue inhibitor of metalloproteinases 1(TIMP-1, DTM100, R&D Systems, Minneapolis, MN). Absorbance was measured with a VersaMax ELISA plate reader, and results were calculated with SoftMax Pro (Molecular Devices). Markers were normalized to urine creatinine concentration and log2 transformed.
Statistical Analysis of the Association with Clinical data and Urine Markers:
Descriptive statistics were used to characterize baseline (time of biopsy) participant characteristics by molecular cluster. Differences in Kaplan-Meier curves, by molecular cluster, were tested by the log rank test. Univariate Cox proportional hazard models were fit separately for time from biopsy to complete remission and time to the composite of ESKD and 40% decline in eGFR to assess association of molecular cluster and TNF score with clinical outcomes. Models were adjusted for diagnosis (MCD, FSGS), eGFR, and UPCR. Interstitial fibrosis and glomerular sclerosis were assumed to be on the causal pathway and therefore not included in the models. Pearson’s correlation was used to assess the relationship between TNF score, marker tissue mRNA expression and urinary concentration. Linear regression models were fit to assess the association of urinary markers and clinical features with TNF score and to calculate a predicted score. Pearson’s correlation was used to correlate the predicted and observed TNF activation scores. Analyses were performed using STATA, v12.1 (College Station, TX).
Single nuclear RNA-seq
Nuclei were prepared from biopsy tissue of 10 NEPTUNE participants (5 from cluster 3, with high TNF activity scores (defined as TNF High) and 5 from clusters 1 and 2, with lower TNF activity scores (defined as TNF Low)) stored in RNAlater using protocols from the Kidney Precision Medicine Project27, 28 (Supplementary Methods, GSE213030). Nuclear cluster annotation was determined by defining enriched genes in each cell cluster and comparing cluster selective gene profiles with previously identified human kidney cell marker gene sets26–28.
Kidney organoid culture, treatment and analysis
Kidney organoids were generated from UM77-2 human embryonic stem cells (hESC) as previously described29. Organoids were treated with TNF (R&D Systems, Cat# 10291-TA) resuspended in phosphate-buffered saline (PBS) on day 23 at the indicated concentrations. Organoid supernatants were removed at specified times, RNA extracted and sequenced (Supplementary Methods). Organoid culture supernatants and cell lysates were diluted 150-fold to measure MCP-1 and 10-fold to measure TIMP-1 using ELISA as described above. Quantitative real-time PCR analysis were performed in triplicate using TaqMan Fast Universal PCR Master Mix (2X) for CCL2, TIMP1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Results
Unbiased Consensus Clustering of Gene Expression Profiles Identifies Shared Molecular Signatures
Transcriptomic profiles of micro-dissected kidney biopsy compartments were used to group participants into distinct subclusters. Due to the known association of tubulointerstitial changes with risk of loss of eGFR, tubulointerstitial transcriptional data were analyzed first. Transcriptomes from NEPTUNE participants clustered into three groups (n=85, 76 and 59, respectively), with one cluster (T3) demonstrating the highest cluster stability (Figure 2A). The delta-K revealed the 3-cluster solution was optimal across clustering approaches (Supplementary figure S1A). To validate the molecular profiles identified in NEPTUNE, Kmeans, PAM and hierarchical consensus clustering22, 23 were also applied to the tubulointerstitial transcriptome data from two independent FSGS/MCD cohorts, ERCB (N=30) and H3 Africa (N=35) resulting in three distinct clusters (Figure 2B and C) with high cluster stability. Glomerular compartment clustering also identified three clusters (Supplementary Figure S2A, B and C) with a transcriptional signature largely shared with the tubulointerstitium (Figure 2D). Differential expression analysis of the tubulointerstitial transcripts between T3 and the other two clusters in each cohort showed a robust, directionally conserved molecular signal across cohorts (correlation of fold change 0.94, p<0.001 for NEPTUNE vs. ERCB and 0.93, p<0.001 for NEPTUNE vs. H3, Figure 2E and F). Finally, of the 179 NEPTUNE participants with measured interstitial fibrosis, clustering was repeated in only those participants with interstitial fibrosis <25% (n=148). All 26 participants originally in T3 from this group, again clustered together.
Figure 2: Kidney transcriptomic cluster membership and unadjusted Kaplan Meier curves.
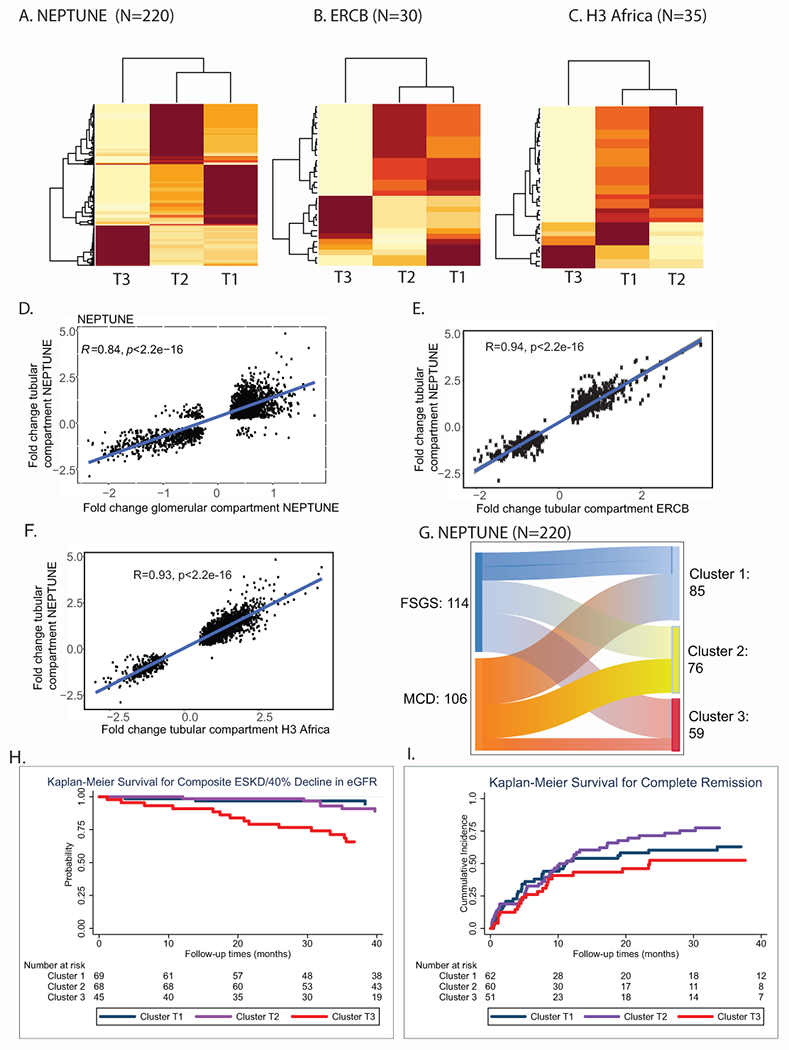
Consensus clustering using kmeans identified optimal cluster membership from tubulointerstitial transcriptomic profiles with 3 clusters (Clusters were designated by tissue compartment and cluster number, T1, T2, T3 for tubulointerstitial clusters) in a cluster matrix from (A) NEPTUNE, (B) ERCB, and (C) H3 Africa cohorts. The values ranged from 0 (pale yellow, samples do not cluster together) to 1 (brown, samples demonstrate high affinity and cluster together). Scatter plots showed strong correlation of significant fold change differences of genes differentially expressed in (D) tubulointerstitial and glomerular compartments in NEPTUNE; (C) Tubulointerstitium Cluster 3 (T3) compared to T2 and T1 from NEPTUNE (y-axis) and cluster T3 compared to T1 and T2 from ERCB (x-axis); and, similarly, for (F) H3 Africa (x-axis). (G) Alluvial plot of correspondence between diagnosis and cluster membership of participants in the NEPTUNE cohort. Unadjusted Kaplan Meier survival curves by NEPTUNE tubulointerstitial cluster for (H) composite endpoint of 40% loss of eGFR or ESKD and (I) complete remission.
NEPTUNE participants in T3 were older, and had a lower eGFR, greater interstitial fibrosis and higher UPCR at biopsy (Table 1, Supplementary Figure S3). In ERCB and H3Africa, participants in T3 also had lower eGFR and were older. Although T3 had a greater proportion of FSGS in all three cohorts, it also included participants with MCD (Figure 2G, Supplementary Figure S1D and E). In an unadjusted survival model, NEPTUNE T3 participants were more likely to reach the composite of ESKD or 40% decline in eGFR [unadjusted HR 5.23 (95% CI 1.9, 14.5), p<0.001 for overall differences in curves, Figure 2H] and fewer complete proteinuria remission events were observed [unadjusted HR 0.73 (95% CI 0.43, 1.26), p=0.068 for overall difference in curves, Figure 2I] compared to T1.
Table 1.
Clinical characteristics of participants summarized by cluster identity and cohort
| NEPTUNE | ALL (N = 220) |
Cluster1 (N = 85) |
Cluster 2 (N = 76) |
Cluster 3 (N = 59) |
P-value |
|---|---|---|---|---|---|
| AGE mean (sd) | 28.2 (21.7) | 22.3 (19.5) | 26.8 (22.6) | 38.5 (20.0) | < 0.0001 |
| Female (%) | 89 (40) | 33 (39) | 30 (39) | 26 (44) | 0.82 |
| Adult (%) | 112(59) | 31(36) | 35(46) | 46(78) | < 0.0001 |
| eGFR mean (sd) | 89.5 (46.8) | 102.1 (40.7) | 104.2 (48.9) | 52.3 (29.2) | < 0.0001 |
| Albumin mean (sd) | 3.62 (0.89) | 3.68(0.86) | 3.62(0.89) | 3.54(0.95) | 0.3916 |
| FSGS (%) | 114 (52) | 32 (38) | 37 (49) | 45 (76) | < 0.0001 |
| UPCR median (IQR) | 2.76 (1.0, 7.04) | 1.9 (0.6, 7.1) | 2.56 (1.02, 5.5) | 5.35 (2.0, 10.6) | <0.0001 |
| % IF median (IQR) | 4 (0, 16) | 1.5 (0,5) | 3 (0, 8) | 20 (10, 55) | <0.0001 |
| Disease duration prior to biopsy (months) mean (sd) | 29.47 (69.46) | 26.78 (46.56) | 18 (50.6) | 49 (108) | 0.30 |
| On RAAS Blockade (%) | 86 (39) | 34 (40) | 26 (34) | 26 (44) | 0.0005 |
| On IST (%) | 101 (46) | 49 (58) | 37 (49) | 15 (25) | 0.0005 |
| ERCB | Cluster 1 (N = 18) |
Cluster 2 (N = 4) |
Cluster 3 (N = 8) |
P-value | |
|
| |||||
| Age mean (sd) | 42.2 (19.0) | 29.4 (6.6) | 49.2 (18.3) | 0.63 | |
| Female (%) | 10 (56) | 1 (25) | 3 (38) | 0.59 | |
| eGFR mean (sd) | 92.2 (34.5) | 119.0 (4.0) | 43.2 (31.0) | 0.02 | |
| FSGS (%) | 8 (44) | 2 (50) | 7 (87) | 0.135 | |
| H3 Africa | Cluster1 (N = 14) |
Cluster 2 (N = 16) |
Cluster3 (N = 5) |
P-value | |
|
| |||||
| Age mean (sd) | 24.1 (10.2) | 30.0 (14.1) | 30.8 (10.7) | 0.19 | |
| Female (%) | 3 (21) | 4 (25) | 0 (0) | 0.90 | |
| eGFR mean (sd) | 89.8 (42.6) | 109.8 (14.7) | 25.8 (11.7) | 0.03 | |
| FSGS (%) | 7 (50) | 4 (25) | 4 (80) | 0.07 | |
| UPCR median (IQR) | 1.4 (1.1, 2.2) | 1.1 (0.3, 2.8) | 3.6 (1.8, 7.5) | 0.05 | |
| % IF median (IQR) | 0 (0, 9) | 0 (0, 1) | 40 (0, 40) | 0.01 | |
| Disease duration less than 6 months (%) | 2 (14) | 4 (25) | 4 (80) | 0.02 | |
| IST in past 6 months (%) | 5 (36) | 4 (25) | 1 (20) | 0.71 | |
Abbreviations: FSGS, Focal segmental glomerulosclerosis; eGFR, estimated glomerular filtration rate (mL/min/1.73m2); UPCR, Urine protein to creatinine ratio (mg/mg); IF – interstitial fibrosis; IST – immunosuppressive therapy; RAAS-renin angiotensin aldosterone system.
Biological and molecular relevance of cluster 3
Differential mRNA expression profiles were used to elucidate the molecular functions associated with cluster T3. In NEPTUNE, there were 2721 transcripts in T3 with an absolute 1.5-fold-change and q<0.05 (2199 up-regulated, 522 down-regulated), compared to T1 and T2 (Supplementary Table S1a). This gene set was analyzed to identify enriched canonical pathways, predicted upstream regulators30, and gene interaction networks. These analyses converged on TNF pathway activation.
In signal transduction pathway over-representation (enrichment), the granulocyte adhesion and diapedesis signal transduction pathway had the highest enrichment score (−log(p)=21.4). Of the 180 genes in this pathway, 70 (38.9%) were found differentially regulated in T3, including TNF (2.4-fold up-regulated in T3, q<0.001, Figure 3A). Causal analysis of predicted upstream regulators predicted TNF as the top biological mediator activated in T3 (IPA network Z-score=13.2, enrichment p=2.09E-120). An expanded causal mechanistic network centered on downstream effects of predicted TNF activation (Figure 3B) explained 48% (1299/2721) of the differentially expressed genes between T3 and T1 and 2. Regulated transcripts included multiple transcription factors previously implicated in chronic kidney disease progression, such as nuclear factor kappa B [including NFκB 1 (p105/p50), RELA (p65) subunits]11, 31, 32, signal transducer and activator of transcription (STAT1 and 3)33. In the gene interaction network analysis, TNF was identified as the hub gene connecting the T3 regulated gene set (Figure 3C). Mapping the upstream regulators using differential expression profiles from each cohort (Supplementary Table S1a) recapitulated the NEPTUNE signal in the ERCB and H3 Africa cohorts, with TNF identified as the top upstream regulator (Supplementary Table S1b).
Figure 3. Molecular and functional context of cluster T3 expression profiles.
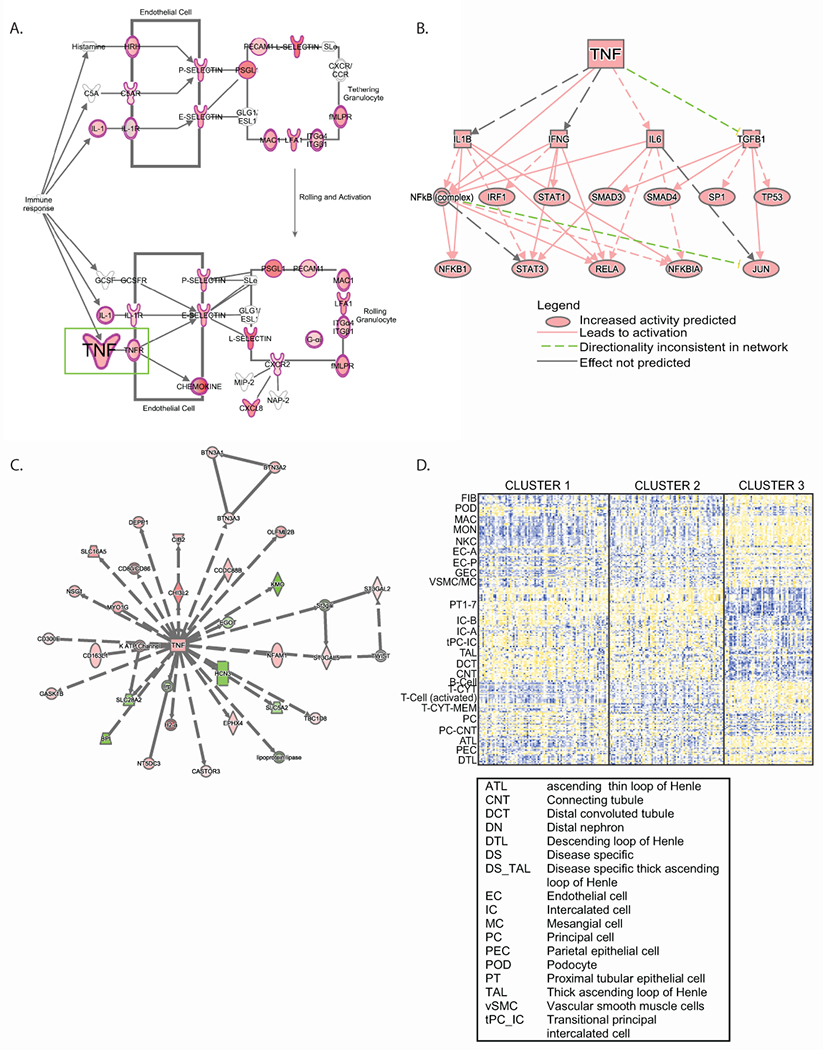
Differential expression profiles from T3 compared to T1 and T2 in the NEPTUNE cohort were generated, and enrichment analysis was performed using Ingenuity Pathways Analysis. (A) Granulocyte adhesion and diapedesis was the top enriched canonical pathway; a subset of the pathway is shown highlighting TNF as an input to the pathway. Genes highlighted in red were up-regulated in the differential expression profile. (B) A mechanistic network of predicted upstream regulators from the differential expression profile indicating TNF as an input. (C) TNF was identified in a gene interaction network (red indicates the gene was up-regulated in the differential expression profile, while green indicates down-regulation). (D) Cell selective gene expression markers were previously identified26 and were intersected with voom-transformed gene (row) normalized expression data (yellow indicates higher expression, blue indicates lower expression) to elucidate probable cell contribution to differential expression profiles.
Next, previously published cell-selective transcripts from human kidney scRNAseq data sets26 were used to interrogate the cluster-specific tubulointerstitial expression profiles in NEPTUNE. The top 10 genes selectively enriched in kidney cell types26 served as cell type specific markers. Bulk RNAseq expression data were filtered for markers from each cell type to see which cell types contributed most to the transcriptional signal in T3. Increased contributions in T3 were observed from kidney cells (fibroblasts, endothelial, parietal epithelial, ascending thin loop of Henle and descending loop of Henle) and immune cell lineages while genes specific for proximal tubules, intercalated cells, thick ascending loop of Henle, distal convoluted tubule, connecting tubule, principal and transitioning cells were expressed at lower levels in T3 (Figure 3D). Taken together, these findings demonstrate TNF activation in T3 represents a molecular signature derived from both resident kidney and immune cells.
Patient-level TNF activation score and relationship to cluster information
Multiple lines of evidence converged on TNF; therefore, individual-level kidney TNF activation was assessed. Using a rich knowledge base34–36, in silico analysis extracted a set of 272 genes causally downstream of TNF activation (Supplementary Table S2). Expression of these 272 genes formed the readout of TNF activation in kidney biopsies. A TNF activation score was calculated for each participant 19, 37, 38 and evaluated across the three cohorts (Figure 4A). Consistent with TNF activation accounting for the clustering, the range of TNF activation scores were similar, with the highest scores in cluster T3 (Figure 4A). The TNF activation score was also calculated from glomerular samples and found to be strongly correlated with the tubulointerstitial TNF activation score in the two cohorts where matched gene expression samples were available (Figure 4B). A TNF z-score was generated using the PROGENy TNF-pathway signature gene set39. This 98 gene signature shared 41 genes with our in silico TNF signature. Both signatures were strongly correlated with one another (R2>0.94, p<0.0001) in the tubulointerstitial NEPTUNE transcriptomic data.
Figure 4:
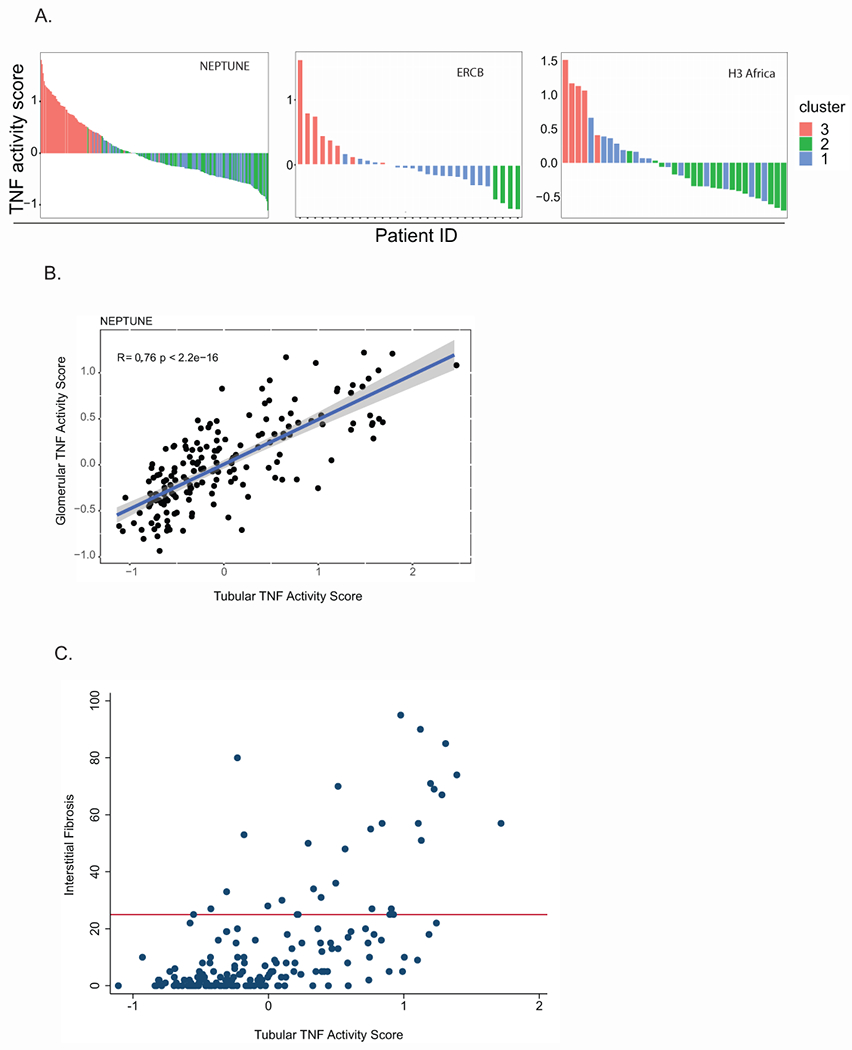
TNF activity scores across all profiled participants (A) from the indicated cohorts colored by cluster membership in tubular transcriptomes (B) Pearson’s correlation of TNF activity scores from the glomeruli (y-axis) and tubular (x-axis) transcriptomes in the same NEPTUNE participants. (C) Correlation of the TNF activity score from the tubular transcriptome with interstitial fibrosis. The horizontal bar (red) indicates 25% interstitial fibrosis.
Association of cluster 3 and TNF activation with interstitial fibrosis and clinical outcomes
The Spearman correlation of TNF activation scores with severity of interstitial fibrosis in NEPTUNE was significant (n=179, rho = 0.59, p<0.001, Figure 4C). However, among the 148 participants with minimal interstitial fibrosis (<25% of the kidney cortex), elevated TNF activation scores (TNF activation score >0) were observed in 47 (32%), indicating that the TNF activation score may be more sensitive to early signs of kidney damage that are not yet visible by histopathological examination.
To evaluate the extent to which the molecular information from the kidney tissue captured the variability in loss of eGFR over time observed in T3 versus T1 and 2, a survival model was fit separately with cluster membership (Model 1, Table 2) and TNF activation score (Model 2, Table 2) as primary predictors of interest in NEPTUNE . After adjustments for diagnosis (MCD vs. FSGS), baseline eGFR and UPCR, cluster 3 was associated with a higher hazard of reaching the composite outcome, HR 3.8, p=0.035. T2 was not significantly different from T1. Similarly, an increase in TNF activation score was associated with higher hazard of the composite outcome (unadjusted HR 2.6, p<0.001). After adjusting for diagnosis, the HR remained elevated (2.3, p=0.003). The association was attenuated after further adjustment for eGFR and UPCR (HR 1.7, p=0.12), suggesting that these factors may be on the causal pathway of GFR decline.
Table 2:
Unadjusted and adjusted Cox proportional hazards models for composite of ESKD and 40% decline in eGFR from baseline in the NEPTUNE study.
| Unadjusted Model | Adjusted for MCD/FSGS | Adjusted for MCD/FSGS, eGFR and UPCR | |||||
|---|---|---|---|---|---|---|---|
| Predictor | HR (95% CI) | p-value | HR (95% CI) | p-value | HR (95% CI) | p-value | |
| Model 1: Cluster Membership | Cluster 1 | Ref | Ref. | Ref. | |||
| Cluster 2 | 1.7 (0.6, 5.1) | 0.34 | 1.6 (0.5, 4.8) | 0.40 | 2.3 (0.69, 7.57) | 0.18 | |
| Cluster 3 | 5.2 (1.9, 14.5) | 0.001 | 4.5 (1.6, 12.9) | 0.005 | 3.80 (1.1, 13.1) | 0.035 | |
| Model 2: TNF activation score * | TNF Activation Score | 2.6 (1.5, 4.4) | 0.001 | 2.3 (1.3, 4.1) | 0.003 | 1.7 (0.9, 3.5) | 0.12 |
HR for increase in z-score by 1.
MCD, Minimal Change disease; FSGS, Focal segmental glomerulosclerosis; eGFR, estimated glomerular filtration rate (mL/min/1.73m2); UPCR, Urine protein to creatinine ratio (mg/mg)
Identification of non-invasive surrogates of TNF activation
Based on prior work40, an intra-renal pathway activation signal might be reflected in participants’ urine profiles. In NEPTUNE, genes in the TNF activation signature were cross-referenced with the urine proteomic profile. Fourteen genes in the TNF activation network had corresponding urinary proteins (Figure 5A). Of these, intra-renal gene expression of CCL2 and TIMP1 correlated with urine protein levels (r=0.58, p<0.0001 and r=0.50, p<0.0001, respectively). Urinary MCP-1 (the protein encoded by CCL2) and TIMP-1 were also correlated with the TNF activation score (p<0.0001, r≥0.50 for both biomarkers, Figure 5B and 5C, respectively). Mean levels of both markers were higher in MCD and FSGS participants in cluster T3 (Supplementary Figure S4). Thus, these two urine proteins were identified as potential non-invasive surrogates reflective of intra-renal TNF activation.
Figure 5: Non-invasive surrogate selection for TNF activation.
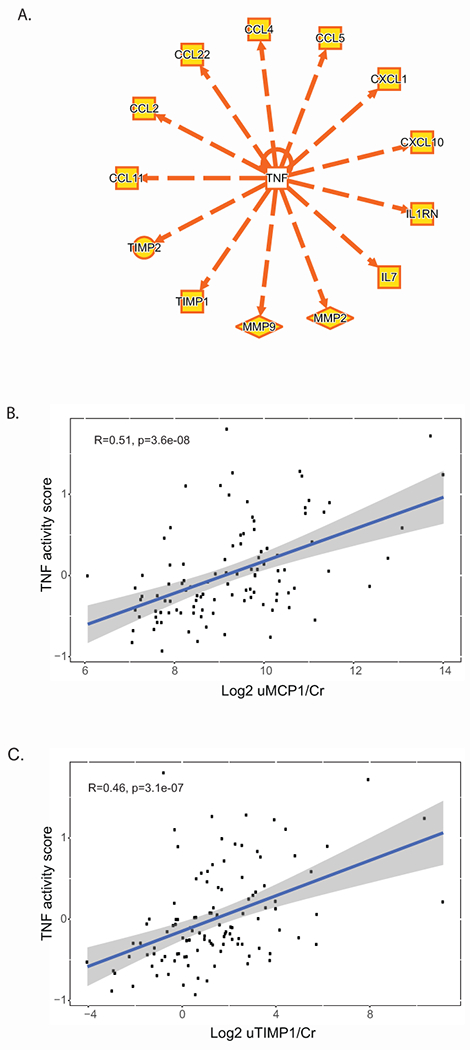
(A) Fourteen genes up-regulated in cluster T3, were downstream of TNF activation, as characterized by curated cause and effect relationships, and were present on the Luminex panel used to profile urine profile from NEPTUNE participants. TNF activation score plotted against (B) Log2 uMCP1/Cr and (C) Log2 uTIMP1/Cr.
TNF effect on kidney organoids
Human organoids were used to further support the relationship of the non-invasive surrogate markers to intra-renal TNF activation. TNF treatment of organoids resulted in an early, dose-dependent increase in TNF activation scores (3h), that was slightly dampened but sustained for 20 hours in culture (Figure 6A). TNF activation was reflected in the up-regulation of CCL2 and TIMP1 mRNA expression (Figure 6B) followed by increased detection of the encoded proteins MCP-1 and TIMP-1 in the organoid culture medium (Figure 6C).
Figure 6: TNF effects on kidney organoids and surrogate markers.
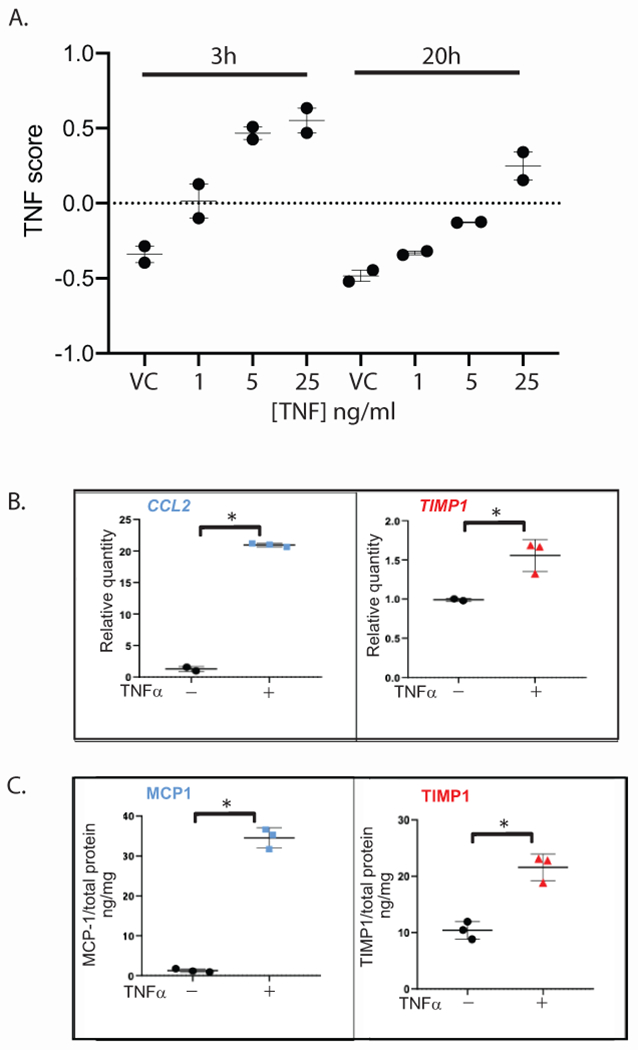
TNF directly stimulates TNF activation and expression of the selected surrogate markers in human pluripotent stem cell derived-kidney organoids. (A) TNF activation scores were calculated from bulk RNAseq data obtained from kidney organoids treated with vehicle control (VC) or 1, 5, 25ng/ml TNF for 3 or 20h. Quantification of (B) CCL2 (left) and TIMP1 (right) transcript levels in kidney organoid cell lysates by qRT-PCR relative to control, and of (C) MCP-1 (left) and TIMP-1 (right) protein levels in kidney organoid culture supernatant by ELISA normalized to total protein, generated from the same samples following treatment with 5 ng/ml TNF or vehicle control for 24h. Each data point was generated from a unique sample and represents the average of analysis in triplicate. Long bar, mean; short bar, 1 S.D. ; *p-value < 0.05 by Student’s t-test. Representative experiment (1 of 4 independent) shown.
TNF activation and expression of surrogate markers in kidney cells
To test the cellular source of the non-invasive surrogate markers in human biopsies, snRNAseq analysis of 10 NEPTUNE biopsies, five with high and five with moderate to low TNF activation scores in the tubulointerstitial profiles (Supplementary Table S3). The analysis identified 22 unique nuclear clusters representing the major cell types of the kidney (Figure 7A). In the TNF High samples, cell type specific gene expression for both CCL2 and TIMP1 were higher across both immune and intrinsic kidney cell types (Figure 7B), consistent with findings from kidney organoids. Thus, the TNF-responsive surrogate markers reflect alterations in inflammatory and resident kidney cells in participants with TNF pathway activation.
Figure 7: TNF effects on kidney and immune cell types by single nucleus RNAseq from glomerular-depleted biopsies.
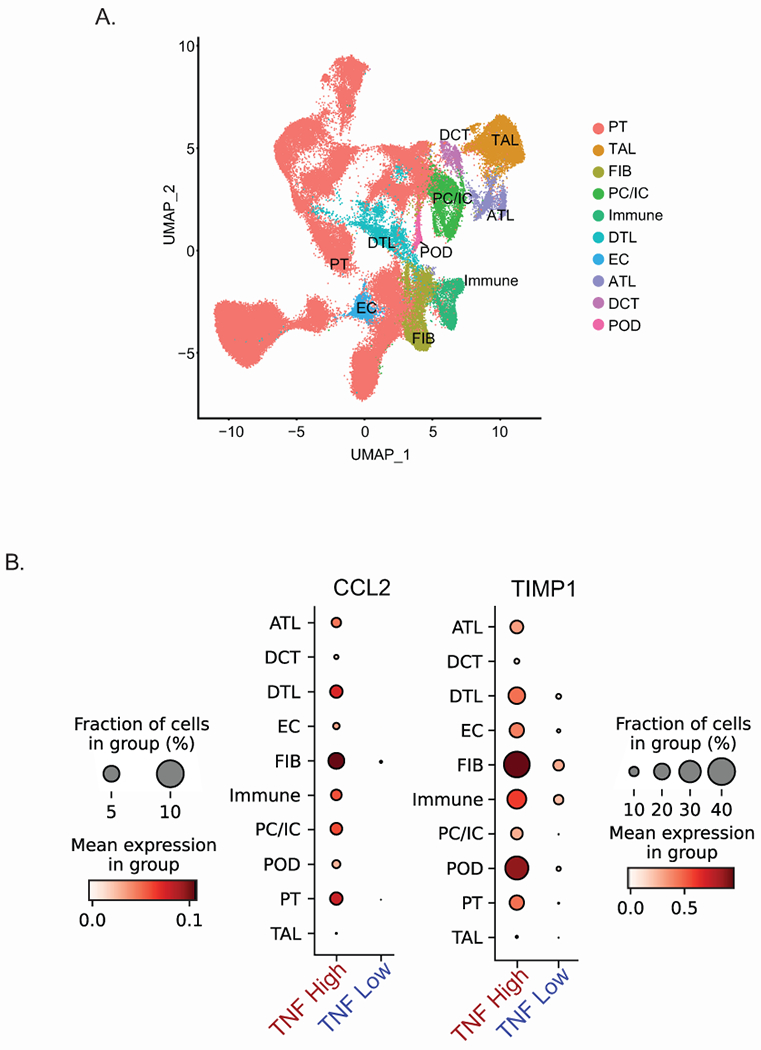
(A) UMAP plot of snRNAseq profiles from TI of selected NEPTUNE participants found to have elevated TNF activation scores (TNF high) and low to moderate TNF activation scores (TNF low) and (B) Single nuclear cluster expression of CCL2 and TIMP1 by TNF activation status. Cell type specific markers were based on Lake et al, 202128.
Abbreviations: ATL = ascending thin limb cells; DCT = distal convoluted tubule cells; DTL = descending thin limb cells; EC-GC = glomerular endothelial cells; FIB = fibroblast cells; Immune = several types of immune cells; PC/IC = principal cells / intercalated cells; POD = podocytes; PT = proximal tubule cells; TAL = thick ascending limb cells.
Predictive ability of surrogate markers
NEPTUNE participants with TNF activation scores, and urinary cytokine measurements obtained within 45 days following biopsy (N=90), were included in predictive models. Using a combination of urine biomarkers (MCP-1 and TIMP-1), eGFR and UPCR, a predicted TNF score was calculated and highly correlated with the transcriptionally derived intra-renal TNF activation score (r=0.61, p<0.001), Figure 8. Thus, a non-invasive biomarker signature of MCP-1 and TIMP-1, coupled with routinely obtained clinical information, predicts intra-renal TNF activation profiles in participants with FSGS and MCD.
Figure 8: Correlation of observed TNF activation score with a predicted score based on urinary biomarkers and clinical features.
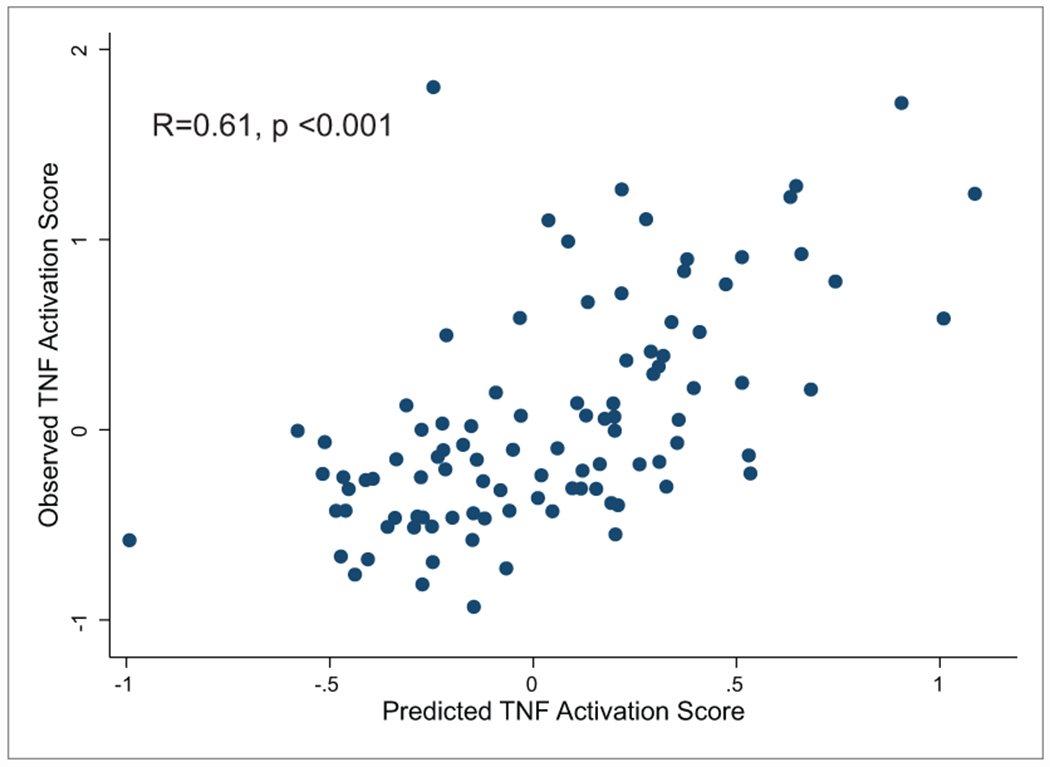
Linear regression models were used to generate predicted tissue TNF activation scores based on eGFR, UPCR, urinary TIMP1 and urinary MCP1. Correlation was 0.61, p-value <0.001.
Discussion
This study used tissue transcriptomics to address the heterogeneity in disease progression under routine clinical care, in children and adults with biopsied MCD and FSGS, to agnostically identify patient subgroups with a shared molecular signature. In the subgroup with highest risk of eGFR loss, activation in the kidney of a targetable pathway was identified that could be quantified at an individual level, and predicted non-invasively, through urine surrogate markers. Such approaches for identifying molecular markers provide a strategy for precision medicine in nephrology, where patients can be matched to mechanistically relevant therapies2.
For the first time, from a cluster of participants with shared molecular profiles in three geographically diverse cohorts, a subgroup of high-risk participants, comprising both MCD and FSGS diagnoses, was identified. Clustering remained consistent when limiting to those with low levels of interstitial fibrosis and, after adjusting for diagnosis, eGFR and UPCR, this subgroup had increased risk of GFR loss. This suggests that transcriptomic data captures prognostic information not currently captured by clinical-pathologic evaluation, and potential disease pathways not targeted by current treatments. The pathways within the subgroup’s molecular profile could represent a variety of biological process, including both disease initiating and progression mechanisms, both of which could be valuable targets for therapy.
The kidney tissue molecular profile of the poor outcome subgroup centered on TNF activation, a cytokine linked to a range of diseases41–44. From several lines of evidence, including human and animal studies in kidneys 45, 46, TNF is produced in immune and resident kidney cells47, 48, has been implicated early in disease causation49, 50 and progression44, 47, 49, 51, but has not been implemented in clinical care. Although TNF activation was associated with the degree of fibrosis in this study, many participants without significant scarring had elevated TNF activation scores, demonstrating pathway activation may occur early in the disease course. Two candidate urine markers of TNF activation were identified; MCP-1, a marker of active inflammation,52 and TIMP-1, associated with tissue remodeling and scarring53. These markers were elevated in the poor outcome cluster and along with existing clinical measures, accurately predicted intra-renal TNF activation. Cell-specific transcriptional signals characterizing the poor outcome cluster were derived from both infiltrating immune cells and resident kidney cells, including endothelial cells. This was confirmed in the snRNAseq data where participants with high TNF scores showed increased expression of CCL2 and TIMP1 across multiple cell types. Additionally, TNF-treated organoids had increased TNF pathway activation scores, transcript and protein levels of TIMP-1 and MCP-1.
Case reports and small studies suggest anti-TNF therapy may be effective in a subset of patients with nephrotic syndrome, but intra-renal TNF activation was not assessed 54–56. The FONT trial (Novel Therapies for Resistant FSGS) tested the TNF inhibitor adalimumab in patients with multi-drug resistant FSGS 57, 58. Of the 17 patients treated in the combined phase I and phase II studies, 4 patients had ≥50% reduction in proteinuria; 2 patients achieved dramatic improvements, from UPCR of 17 to 0.6 mg/mg in one and from 3.6 to 0.6 mg/mg in the other. Although the study was considered unsuccessful in demonstrating efficacy of anti-TNF therapy for FSGS patients as a group, a response in any patient with this severe phenotype is notable and consistent with underlying biologic heterogeneity within the diagnosis. Other clinical trials in MCD and FSGS have also observed variable responses to other interventions 59, 60.
These examples highlight the need for precision medicine and for clinical trials to incorporate markers (serum, urine, or genetic) indicating activation of the targeted injury pathway to ensure alignment of the molecular profile of participants with that of the intervention. Molecular categorization could be combined with consensus clustering based on clinical and laboratory data, as outlined here for nephrotic syndrome, and recently applied to the Chronic Renal Insufficiency Cohort 61, for precise delineation of patient prognosis and optimization of therapy.
The subgroup of highest clinical need, those with greatest risk of GFR loss, was the focus for this initial study. Tubulointerstitial damage and fibrosis have been shown to strongly associate with eGFR decline, and treatment response across diagnoses19, 62–64. Therefore, although MCD and FSGS are glomerular diseases, the premise for this study was that pathways associated with disease progression were more likely to be detected in the tubulointerstitial profiles. Nevertheless, the TNF activation signal in the glomerular and tubular compartments were strongly correlated, capturing similar activation in both compartments. Further, even when only the participants with low fibrosis were clustered, their high-risk subgroup membership persisted. Therefore, the tubulointerstitial TNF pathway merits investigation and may represent both disease initiating as well as chronic progression mechanisms in glomerular diseases.
Several limitations of the present study are acknowledged. This study included only biopsy proven MCD and FSGS. The TNF pathway and associated urine markers are likely relevant to other kidney diseases that could be addressed in future studies using this pipeline. Relatedly, this study included all MCD and FSGS participants to be broadly inclusive of the spectrum of presentation at the time of a clinical biopsy. However, MCD patients, particularly children, who respond well to therapy and may not receive a clinical biopsy, are not represented in this cohort. Similar analyses could identify pathways relevant within clinical subgroups of these diagnoses (e.g., only those with high degrees of proteinuria). Future work using non-tissue based -omic technology could identify molecular subclusters in non-biopsied patients, not included here. Genetic information is expanding our understanding of nephrotic syndrome. However, in NEPTUNE, a low frequency of mutations in 21 monogenic nephrotic syndrome-associated genes were detected65, and likely prevented us from finding an association of TNF with monogenic glomerular diseases. Similar data are unavailable for ERCB and H3 Africa. In many patients, a single pathway is unlikely to drive disease progression and additional or combination therapies may be appropriate. Future work using this pipeline and data from this study can uncover additional pathways and surrogate markers relevant to each of the tubular and glomerular transcriptomic clusters.
As next steps, clinical studies are needed to validate the use of predicted TNF activation scores as a target engagement biomarker during the treatment of FSGS and other glomerular diseases. Reprising the FONT trial design by limiting trial eligibility to patients with high predicted TNF pathway activation would enable assessment of response to TNF inhibition, stratified by TNF activation levels. A Phase IIa proof of concept trial with this design has been initiated for children and adults with biopsy proven FSGS and MCD (clinicaltrials.gov/NCT04009668).
In conclusion, this study provides a road map for implementing precision medicine in primary podocytopathies. It identified a molecularly defined subset of patients with nephrotic syndrome who have poor clinical outcomes and increased activation of a targetable pathway, TNF, as a key driver of disease progression. Non-invasive markers, validated in an organoid model system, are available to identify the participants with TNF activation, an approach currently being tested in an interventional trial. The concepts developed here for FSGS/MCD, and the TNF pathway represents a first step towards a comprehensive map of targetable pathways for glomerular diseases and a move towards precision medicine where the right medicine is administered to the right patient with glomerular disease.
Supplementary Material
Supplementary Figures:
Supplementary Figure S1. Cluster dendrogram and assignment of MCD and FSGS participants based on kidney biopsy tubulointerstitial gene expression data in NEPTUNE, ERCB and H3 Africa
Supplementary Figure S2. Comparison of cluster assignment and clinical measures in NEPTUNE and ERCB using glomerular compartment expression data
Supplementary Figure S3. Comparison of clinical factors and cluster assignment across cohorts
Supplementary Figure S4. Urine marker levels by diagnosis and cluster
Supplementary Tables:
Supplementary Table S1a. List of differentially expressed genes (absolute FC>1.5, q<0.05) in Cluster 3 versus Clusters1 and 2, in each cohort, used for enrichment analyses.
Supplementary Table S1b. Predicted Upstream Regulators based on DEG profiles of patients in cluster 3 relative to clusters 1 and 2.
Supplementary Table S2. Genes and gene products activated by TNF used to generate the TNF activation score.
Supplementary Table S3. Clinical characteristics of NEPTUNE participants with biopsies used for snRNAseq
Acknowledgements
We thank Dr. Lalita Subramanian for help with writing, editing and formatting this manuscript. The Nephrotic Syndrome Study Network Consortium (NEPTUNE), U54-DK-083912, is a part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through collaboration between the Office of Rare Diseases Research, National Center for Advancing Translational Sciences and the National Institute of Diabetes, Digestive, and Kidney Diseases. Additional funding and/or programmatic support for this project has also been provided by the Else Kröner-Fresenius Foundation (ERCB), University of Michigan, the NephCure Kidney International and the Halpin Foundation, and the Applied Systems Biology Core at the University of Michigan George M. O’Brien Kidney Translational Core Center (2P30-DK-08194). Dr Mariani is supported through funding from NIH/NIDDK, K08 DK115891-01. We acknowledge the role of the H3Africa Consortium in making this research possible though the sharing of data and knowledge. The National Institutes of Health (USA) and Wellcome Trust (UK) have provided the core funding for the H3Africa Consortium and more information is available at https://h3africa.org/. This research was supported by the following grants from NIH/NHGRI/NIDDK: H3Africa Kidney Disease Study (U54 HG006939), H3Africa Kidney Disease Cohort Study (U01 DK107131), H3Africa Kidney Disease Collaborative Centers (9U54 DK116913). The views expressed in this paper do not represent the views of the H3Africa Consortium or their funders. ERCB, NEPTUNE and H3 Africa contributing members are listed in supplementary acknowledgement.
Disclosures
Dr. M. Kretzler reports grants from NIH, Chan Zuckerberg Initiative, JDRF, AstraZeneca, NovoNordisk, Eli Lilly, Gilead, Goldfinch Bio, Janssen, Boehringer-Ingelheim, Moderna, European Union Innovative Medicine Initiative, Certa, Chinook, amfAR, Angion, RenalytixAI, Travere, Regeneron, IONIS , outside the submitted work. In addition, Dr. Kretzler has a patent PCT/EP2014/073413 “Biomarkers and methods for progression prediction for chronic kidney disease” licensed. Also, outside of submitted work, Dr. L. Mariani has served on the advisory board of Reata Pharmaceuticals, Calliditas Therapeutics and Travere Therapeutics; Dr. Eddy reports grant support from AstraZeneca, NovoNordisk, Eli Lilly, Gilead, Janssen, Moderna, Certa, Chinook, amfAR, Angion, IONIS outside the submitted work. Dr. H. Trachman consults for Travere Therapeutics, Goldfinch Bio, Akebia, Natera, Otsuka and Chemocentryx; Dr. K.L. Gibson serves on Reata CKD Advisory Board and the Travere Inc. FSGS & IgA Advisory WorkGroup; Dr. V.K. Derebail reports funding from Novartis, honoraria from UpToDate and has served on the advisory board of Retrophin and Bayer. Dr. Lieske reports grants from Dicerna, Allena, Orfan-Bridgebio, Synlogic, Novobiome, OxThera, Oxidien, Alnylam, Federation Bio outside the submitted work. Dr. L.A.Greenbaum reports grants from Reata Pharmaceuticals and Vertex Pharmaceuticals. He has served as a paid consultant for Roche Pharmaceuticals. He is a paid member of a data safety monitoring board for Travere Pharmaceuticals. Dr. D.S. Gipson reports grants from Travere Therapeutics, Reata, Goldfinch Bio, Novartis, Boehringer Ingelheim and serves on advisory or consultancy through University of Michigan with Roche, Genentech, AstraZeneca, and Vertex. All other authors have no disclosures to report.
Funding:
National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN) grant U54-DK-083912; additional funding and/or programmatic support by the Else Kröner-Fresenius Foundation (ERCB), University of Michigan, the NephCure Kidney International and the Halpin Foundation; NIH grant 2P30-DK-08194; grants from NIH/NHGRI/NIDDK: H3Africa Kidney Disease Study (U54 HG006939), H3Africa Kidney Disease Cohort Study (U01 DK107131), H3Africa Kidney Disease Collaborative Centers (9U54 DK116913). Dr. Mariani is supported through funding from NIH/NIDDK, K08 DK115891-01.
References:
- 1.Rovin BH, Adler SG, Barratt J, et al. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int 2021; 100: S1–S276. [DOI] [PubMed] [Google Scholar]
- 2.D’Agati VD, Alster JM, Jennette JC, et al. Association of histologic variants in FSGS clinical trial with presenting features and outcomes. Clin J Am Soc Nephrol 2013; 8: 399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg AZ, Kopp JB. Focal Segmental Glomerulosclerosis. Clin J Am Soc Nephrol 2017; 12: 502–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gipson DS, Troost JP, Lafayette RA, et al. Complete Remission in the Nephrotic Syndrome Study Network. Clin J Am Soc Nephrol 2016; 11: 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trachtman H, Nelson P, Adler S, et al. DUET: A Phase 2 Study Evaluating the Efficacy and Safety of Sparsentan in Patients with FSGS. J Am Soc Nephrol 2018; 29: 2745–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janiaud P, Serghiou S, Ioannidis JPA. New clinical trial designs in the era of precision medicine: An overview of definitions, strengths, weaknesses, and current use in oncology. Cancer Treat Rev 2019; 73: 20–30. [DOI] [PubMed] [Google Scholar]
- 7.Collins FS. Reengineering translational science: the time is right. Sci Transl Med 2011; 3: 90cm17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eddy S, Mariani LH, Kretzler M. Integrated multi-omics approaches to improve classification of chronic kidney disease. Nat Rev Nephrol 2020. [DOI] [PubMed] [Google Scholar]
- 9.Barisoni L, Nast CC, Jennette JC, et al. Digital pathology evaluation in the multicenter Nephrotic Syndrome Study Network (NEPTUNE). Clin J Am Soc Nephrol 2013; 8: 1449–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yasuda Y, Cohen CD, Henger A, et al. Gene expression profiling analysis in nephrology: towards molecular definition of renal disease. Clin Exp Nephrol 2006; 10: 91–98. [DOI] [PubMed] [Google Scholar]
- 11.Schmid H, Boucherot A, Yasuda Y, et al. Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes 2006; 55: 2993–3003. [DOI] [PubMed] [Google Scholar]
- 12.Osafo C, Raji YR, Burke D, et al. Human Heredity and Health (H3) in Africa Kidney Disease Research Network: A Focus on Methods in Sub-Saharan Africa. Clin J Am Soc Nephrol 2015; 10: 2279–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osafo C, Raji YR, Olanrewaju T, et al. Genomic approaches to the burden of kidney disease in Sub-Saharan Africa: the Human Heredity and Health in Africa (H3Africa) Kidney Disease Research Network. Kidney Int 2016; 90: 2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gadegbeku CA, Gipson DS, Holzman LB, et al. Design of the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate primary glomerular nephropathy by a multidisciplinary approach. Kidney Int 2013; 83: 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levey AS, Bosch JP, Lewis JB, et al. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med 1999; 130: 461–470. [DOI] [PubMed] [Google Scholar]
- 16.Schwartz GJ, Work DF. Measurement and estimation of GFR in children and adolescents. Clin J Am Soc Nephrol 2009; 4: 1832–1843. [DOI] [PubMed] [Google Scholar]
- 17.Ng DK, Schwartz GJ, Schneider MF, et al. Combination of pediatric and adult formulas yield valid glomerular filtration rate estimates in young adults with a history of pediatric chronic kidney disease. Kidney Int 2018; 94: 170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zee J, Mansfield S, Mariani LH, et al. Using All Longitudinal Data to Define Time to Specified Percentages of Estimated GFR Decline: A Simulation Study. Am J Kidney Dis 2019; 73: 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grayson PC, Eddy S, Taroni JN, et al. Metabolic pathways and immunometabolism in rare kidney diseases. Ann Rheum Dis 2018; 77: 1226–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barisoni L, Troost JP, Nast C, et al. Reproducibility of the NEPTUNE descriptor-based scoring system on whole-slide images and histologic and ultrastructural digital images. Mod Pathol 2016; 29: 671–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002; 30: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics (Oxford, England) 2010; 26: 1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monti S, Tamayo P, Mesirov JP, et al. Consensus Clustering: A Resampling-Based Method for Class Discovery and Visualization of Gene Expression Microarray Data. Machine Learning 2004; 52: 91–118. [Google Scholar]
- 24.Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015; 43: e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calvano SE, Xiao W, Richards DR, et al. A network-based analysis of systemic inflammation in humans. Nature 2005; 437: 1032–1037. [DOI] [PubMed] [Google Scholar]
- 26.Menon R, Otto EA, Hoover P, et al. Single cell transcriptomics identifies focal segmental glomerulosclerosis remission endothelial biomarker. JCI Insight 2020; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lake BB, Chen S, Hoshi M, et al. A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nature Communications 2019; 10: 2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lake BB, Menon R, Winfree S, et al. An atlas of healthy and injured cell states and niches in the human kidney. bioRxiv 2021: 2021.2007.2028.454201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harder JL, Menon R, Otto EA, et al. Organoid single cell profiling identifies a transcriptional signature of glomerular disease. JCI Insight 2019; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kramer A, Green J, Pollard J Jr., et al. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014; 30: 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wiggins JE, Patel SR, Shedden KA, et al. NFkappaB promotes inflammation, coagulation, and fibrosis in the aging glomerulus. Journal of the American Society of Nephrology : JASN 2010; 21: 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martini S, Nair V, Keller BJ, et al. Integrative biology identifies shared transcriptional networks in CKD. J Am Soc Nephrol 2014; 25: 2559–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tao J, Mariani L, Eddy S, et al. JAK-STAT signaling is activated in the kidney and peripheral blood cells of patients with focal segmental glomerulosclerosis. Kidney international 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta A, Puri S, Puri V. Bioinformatics Unmasks the Maneuverers of Pain Pathways in Acute Kidney Injury. Sci Rep 2019; 9: 11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schüler-Toprak S, Häring J, Inwald EC, et al. Agonists and knockdown of estrogen receptor β differentially affect invasion of triple-negative breast cancer cells in vitro. BMC Cancer 2016; 16: 951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Supper J, Gugenmus C, Wollnik J, et al. Detecting and visualizing gene fusions. Methods 2013; 59: S24–28. [DOI] [PubMed] [Google Scholar]
- 37.Lee E, Chuang HY, Kim JW, et al. Inferring pathway activity toward precise disease classification. PLoS Comput Biol 2008; 4: e1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tao J, Mariani L, Eddy S, et al. JAK-STAT Activity in Peripheral Blood Cells and Kidney Tissue in IgA Nephropathy. Clin J Am Soc Nephrol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schubert M, Klinger B, Klünemann M, et al. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat Commun 2018; 9: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ju W, Nair V, Smith S, et al. Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Sci Transl Med 2015; 7: 316ra193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ware CF. The TNF Superfamily-2008. Cytokine Growth Factor Rev 2008; 19: 183–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jaattela M Biologic activities and mechanisms of action of tumor necrosis factor-alpha/cachectin. Lab Invest 1991; 64: 724–742. [PubMed] [Google Scholar]
- 43.Ernandez T, Mayadas TN. Immunoregulatory role of TNFalpha in inflammatory kidney diseases. Kidney Int 2009; 76: 262–276. [DOI] [PubMed] [Google Scholar]
- 44.Bakr A, Shokeir M, El-Chenawi F, et al. Tumor necrosis factor-alpha production from mononuclear cells in nephrotic syndrome. Pediatr Nephrol 2003; 18: 516–520. [DOI] [PubMed] [Google Scholar]
- 45.McCarthy ET, Sharma R, Sharma M, et al. TNF-alpha increases albumin permeability of isolated rat glomeruli through the generation of superoxide. J Am Soc Nephrol 1998; 9: 433–438. [DOI] [PubMed] [Google Scholar]
- 46.Le Berre L, Herve C, Buzelin F, et al. Renal macrophage activation and Th2 polarization precedes the development of nephrotic syndrome in Buffalo/Mna rats. Kidney Int 2005; 68: 2079–2090. [DOI] [PubMed] [Google Scholar]
- 47.Pedigo CE, Ducasa GM, Leclercq F, et al. Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J Clin Invest 2016; 126: 3336–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baud L, Fouqueray B, Philippe C, et al. Tumor necrosis factor alpha and mesangial cells. Kidney Int 1992; 41: 600–603. [DOI] [PubMed] [Google Scholar]
- 49.Otalora L, Chavez E, Watford D, et al. Identification of glomerular and podocyte-specific genes and pathways activated by sera of patients with focal segmental glomerulosclerosis. PLoS One 2019; 14: e0222948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee HH, Cho YI, Kim SY, et al. TNF-alpha-induced Inflammation Stimulates Apolipoprotein-A4 via Activation of TNFR2 and NF-kappaB Signaling in Kidney Tubular Cells. Sci Rep 2017; 7: 8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wen Y, Lu X, Ren J, et al. KLF4 in Macrophages Attenuates TNFα-Mediated Kidney Injury and Fibrosis. J Am Soc Nephrol 2019: ASN.2019020111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim MJ, Tam FWK. Urinary monocyte chemoattractant protein-1 in renal disease. Clin Chim Acta 2011; 412: 2022–2030. [DOI] [PubMed] [Google Scholar]
- 53.Ahmed AK, Haylor JL, El Nahas AM, et al. Localization of matrix metalloproteinases and their inhibitors in experimental progressive kidney scarring. Kidney Int 2007; 71: 755–763. [DOI] [PubMed] [Google Scholar]
- 54.Peyser A, Machardy N, Tarapore F, et al. Follow-up of phase I trial of adalimumab and rosiglitazone in FSGS: III. Report of the FONT study group. BMC Nephrol 2010; 11: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raveh D, Shemesh O, Ashkenazi YJ, et al. Tumor necrosis factor-alpha blocking agent as a treatment for nephrotic syndrome. Pediatr Nephrol 2004; 19: 1281–1284. [DOI] [PubMed] [Google Scholar]
- 56.Ito S, Tsutsumi A, Harada T, et al. Long-term remission of nephrotic syndrome with etanercept for concomitant juvenile idiopathic arthritis. Pediatr Nephrol 2010; 25: 2175–2177. [DOI] [PubMed] [Google Scholar]
- 57.Joy MS, Gipson DS, Powell L, et al. Phase 1 trial of adalimumab in Focal Segmental Glomerulosclerosis (FSGS): II. Report of the FONT (Novel Therapies for Resistant FSGS) study group. Am J Kidney Dis 2010; 55: 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trachtman H, Vento S, Herreshoff E, et al. Efficacy of galactose and adalimumab in patients with resistant focal segmental glomerulosclerosis: report of the font clinical trial group. BMC Nephrol 2015; 16: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hogan J, Bomback AS, Mehta K, et al. Treatment of Idiopathic FSGS with Adrenocorticotropic Hormone Gel. Clin J Am Soc Nephrol 2013; 8: 2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu C-C, Fornoni A, Weins A, et al. Abatacept in B7–1–Positive Proteinuric Kidney Disease. N Engl J Med 2013; 369: 2416–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng Z, Waikar SS, Schmidt IM, et al. Subtyping CKD Patients by Consensus Clustering: The Chronic Renal Insufficiency Cohort (CRIC) Study. J Am Soc Nephrol 2021: ASN.2020030239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zee J, Liu Q, Smith AR, et al. Kidney Biopsy Features Most Predictive of Clinical Outcomes in the Spectrum of Minimal Change Disease and Focal Segmental Glomerulosclerosis. J Am Soc Nephrol 2022; 33: 1411–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kolachalama VB, Singh P, Lin CQ, et al. Association of Pathological Fibrosis With Renal Survival Using Deep Neural Networks. Kidney Int Rep 2018; 3: 464–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Srivastava A, Palsson R, Kaze AD, et al. The Prognostic Value of Histopathologic Lesions in Native Kidney Biopsy Specimens: Results from the Boston Kidney Biopsy Cohort Study. J Am Soc Nephrol 2018; 29: 2213–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sampson MG, Gillies CE, Robertson CC, et al. Using Population Genetics to Interrogate the Monogenic Nephrotic Syndrome Diagnosis in a Case Cohort. J Am Soc Nephrol 2016; 27: 1970–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures:
Supplementary Figure S1. Cluster dendrogram and assignment of MCD and FSGS participants based on kidney biopsy tubulointerstitial gene expression data in NEPTUNE, ERCB and H3 Africa
Supplementary Figure S2. Comparison of cluster assignment and clinical measures in NEPTUNE and ERCB using glomerular compartment expression data
Supplementary Figure S3. Comparison of clinical factors and cluster assignment across cohorts
Supplementary Figure S4. Urine marker levels by diagnosis and cluster
Supplementary Tables:
Supplementary Table S1a. List of differentially expressed genes (absolute FC>1.5, q<0.05) in Cluster 3 versus Clusters1 and 2, in each cohort, used for enrichment analyses.
Supplementary Table S1b. Predicted Upstream Regulators based on DEG profiles of patients in cluster 3 relative to clusters 1 and 2.
Supplementary Table S2. Genes and gene products activated by TNF used to generate the TNF activation score.
Supplementary Table S3. Clinical characteristics of NEPTUNE participants with biopsies used for snRNAseq