

Abstract
The lipid-sensing transcription factor PPARγ is the target of antidiabetic thiazolidinediones (TZD). At two sites within its ligand binding domain, it also binds oxidized vitamin E metabolites and the vitamin E mimetic garcinoic acid. While the canonical interaction within the TZD binding site mediates classical PPARγ activation, the effects of the second binding on PPARγ activity remain elusive. Here, we identified an agonist mimicking dual binding of vitamin E metabolites and developed a selective ligand of the second site, unveiling potential noncanonical regulation of PPARγ activities. We found that this alternative binding event can simultaneously occur with orthosteric ligands and it exerted different effects on PPARγ-cofactor interactions compared to both orthosteric PPARγ agonists and antagonists, indicating the diverse roles of the two binding sites. Alternative site binding lacked the pro-adipogenic effect of TZD and mediated no classical PPAR signaling in differential gene expression analysis but markedly diminished FOXO signaling, suggesting potential therapeutic applications.
Introduction
The peroxisome proliferator-activated receptor γ (PPARγ) is a fatty acid-activated transcription factor acting as a pivotal metabolic regulator.1,2 It is mainly found in adipose tissue and immune cells with a regulatory role in lipid and glucose metabolism as well as adipogenesis.1,2 PPARγ has been an important drug target for type 2 diabetes treatment, but adverse effects of the thiazolidinediones (TZD) including weight gain, bone fractures, and possibly higher cardiovascular risk have restricted the therapeutic use of PPARγ agonists.3 Nonetheless, small molecules that modulate PPARγ activity still hold a great therapeutic potential not only for type 2 diabetes but, e.g., also for nonalcoholic steatohepatitis and neurodegenerative diseases.2,4,5
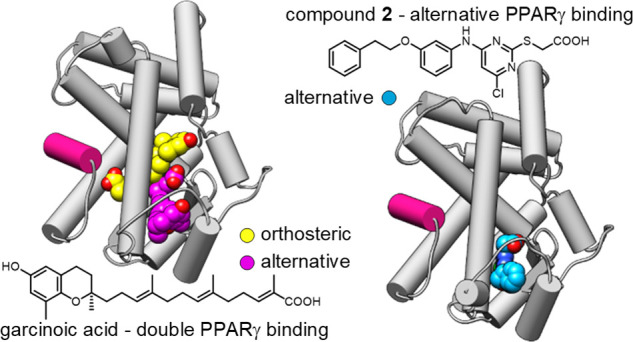
Several chemical scaffolds have been developed as PPARγ agonists, nearly all of which act via orthosteric binding for classical transcription factor activation. Allosteric modulation of nuclear receptors emerges as a concept to achieve more selective and more subtle control over the activity of proteins in this class potentially offering unprecedented therapeutic opportunities.6 Allosteric binding to PPARγ has also been observed for some ligand chemotypes which exhibit double binding to the orthosteric and a second, alternative binding site or span both binding regions.7−11 Previously, we discovered oxidized vitamin E metabolites and garcinoic acid (GA, Scheme 1) as another class of PPARγ ligands that modulate the activity of this transcription factor through both binding regions supporting biological relevance of the alternative binding site.12 Clinical use of vitamin E for the treatment of nonalcoholic fatty liver disease13 as well as promising neuroprotective effects of GA in an Alzheimer’s disease model14 suggest that alternative PPARγ modulation might present another interesting avenue toward novel PPARγ targeting drugs. However, while orthosteric PPARγ activation is well-studied, the mechanisms and effects of selective binding to the alternative site remained elusive. Here, we identified a synthetic PPARγ ligand (1) mimicking the dual binding mode of vitamin E metabolites and developed a selective ligand (2) of the second site by structure-guided design. This novel ligand bound solely to the alternative site without extending to the orthosteric region that is targeted by traditional PPARγ agonists like pioglitazone which acts as an anti-diabetic, insulin-sensitizing agent and is the most widely used PPARγ activating drug. In functional studies with 2 as a chemical tool, we observed remarkably different mechanistic, phenotypic, and gene expression effects of PPARγ modulation through the alternative site compared to traditional agonists.
Scheme 1. PPARγ Ligands Investigated and Developed (1, 2) in this Study.

Results and Discussion
Our recent discovery of PPARγ modulation by double binding natural vitamin E metabolites12 intriguingly suggested new modes of targeting PPARγ with potential advantages in therapeutic efficacy and safety. However, binding to both sites of PPARγ, the complex biological effects and poor synthetic accessibility limited the use of these metabolites as a chemical tool to interrogate PPARγ modulation through the noncanonical site. Therefore, synthetic ligands mimicking selectively the alternative site binding of vitamin E metabolites with improved properties are needed to capture the effects of the second binding site individually.
Identification of nuclear receptor ligands binding at alternative epitopes can be facilitated by occluding the orthosteric site.6,15 The irreversible PPARγ antagonist GW9662 serves this purpose by permanently blocking the orthosteric site via a covalent adduct with Cys28516 which thus enabled screening for compounds capable of modulating PPARγ through other epitopes.17 In a search for a ligand binding to the alternative site, we tested our in-house collection of PPARγ modulators in a Gal4 hybrid reporter gene assay against both ligand-free and GW9662-bound PPARγ. We identified 1(18) (Scheme 1) showing an ability to activate both forms of PPARγ (w/o GW9662: EC50 = 2.1 ± 0.2 μM; w/10 μM GW9662: EC50 = 4 ± 1 μM; Figure 1a). These results indicated that GW9662 blocked access to the orthosteric site without preventing PPARγ activation (Figure S1) and that 1 binding to a different region than the site occupied by the irreversible antagonist can stabilize an activated state of PPARγ, implying likely an allosteric mode-of-action.
Figure 1.
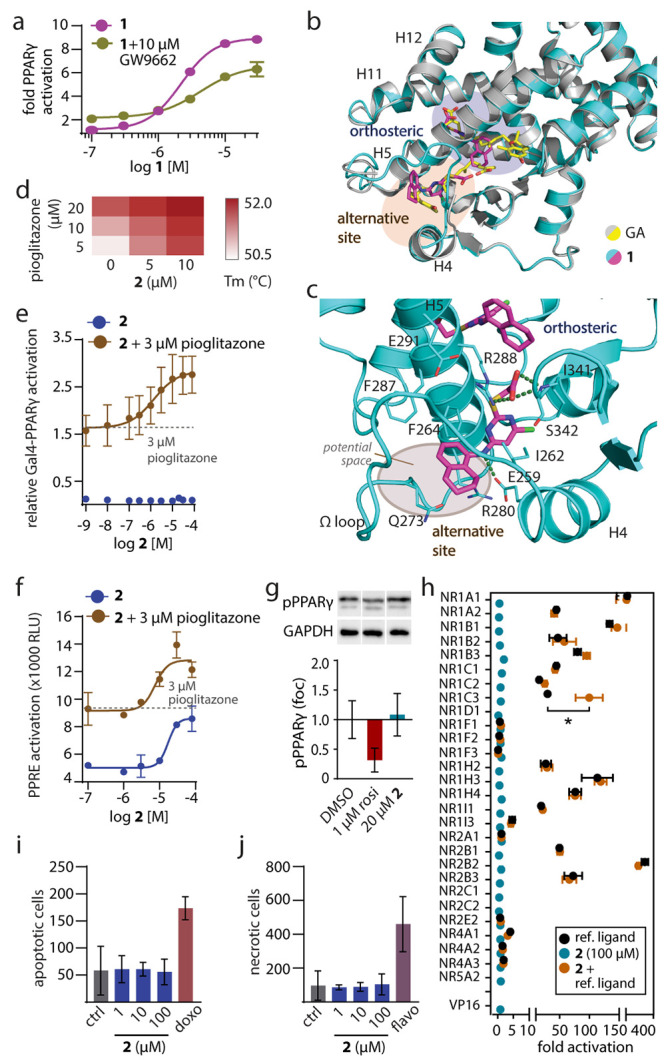
Characterization of 1 mimicking the dual PPARγ binding of GA and of the selective alternative site PPARγ ligand 2. (a) Dose–response curves of 1 in a Gal4-PPARγ reporter gene assay in the absence and presence of the irreversible orthosteric antagonist GW9662 (10 μM). Data are the mean ± S.E.M., n = 3. (b) The X-ray structure of the PPARγ LBD in complex with 1 (pdb ID: 8aty) revealed two molecules binding to the LBD and highly aligned with the PPARγ-GA complex (pdb ID: 7awd).12 Electron density map for 1 in Figure S2. (c) Second site binding of 1 and GA revealed an interaction with the side chain of Arg288 and potential space for an extension of 1 to achieve selective binding to this site. (d) Thermal stability of the PPARγ LBD in the presence of different concentrations of 2 and pioglitazone. The heat map shows the mean Tm, n = 3. (e, f) Effects of 2 on Gal4-PPARγ (e) and PPRE (f) activity in the absence and presence of pioglitazone. Data are the mean ± S.E.M., n ≥ 3. (g) Effects of 2 on PPARγ Ser273 phosphorylation. Data are the mean ± S.E.M., n = 3–6. Rosiglitazone as positive control. Western blots in Supporting Information. (h) Selectivity profiling of 2 on nuclear receptors. Data are the mean ± S.E.M., n ≥ 3. * p < 0.05 (t-test 2 vs 2 + ref. ligand). (i, j) Effects of 2 on apoptosis (i) and necrosis (j) in COS7 cells after 24 h. Doxorubicin (doxo, 100 μM) and flavopiridol (flavo, 100 μM) as positive controls. Data are the mean ± S.E.M., n = 3.
The co-crystal structure of the PPARγ LBD in complex with 1 (pdb ID: 8aty; Figures 1b, S2) indeed confirmed orthosteric and alternative site binding with a 1:2 stoichiometry (PPARγ:1) as also observed for the vitamin E analogue GA.12 Both 1- and GA-bound structures shared a highly similar conformation of the PPARγ LBD. At the canonical site, 1 formed polar contacts with Ser289, His449, and Tyr473, resembling the patterns observed for TZD19 and GA.12 In the second site, 1 was accommodated within the cavity located between helix 4 and helix 5 and made an ionic interaction with Arg288 via its carboxylate group that was also observed for the binding of GA12 (Figure 1c). Additional contacts included hydrogen bonds between the carboxylate of 1 and the backbone of Ser342 as well as between the secondary amine and the Glu259 side chain.
In contrast to the canonical site, which is buried within the PPARγ LBD, the second site is accessible, and the 5- and 6-positions of the tetrahydronaphthalene of 1 in the second site oriented toward the solvent-exposed region. The PPARγ-1 structure thus indicated an avenue to obtain a PPARγ ligand selectively addressing the alternative binding pocket of GA and 1 without canonical orthosteric binding, which could be achieved by an extension from the tetrahydronaphthalene motif of 1. Structure-based design and molecular modeling (Figure S3) suggested substitution of the solvent-exposed tetrahydronaphthalene to a more elongated moiety that could prevent orthosteric binding and provide a better fit to the rather shallow surface in the β-sheet region. Compound 2 was designed based on these considerations as a candidate selective ligand of the alternative site and was synthesized over seven convergent steps with 12% overall yield (Supporting Information).
Initial assessment on binding of 2 by differential scanning fluorimetry (DSF, Figure 1d) revealed additive thermal stabilization of the PPARγ LBD by 2 and pioglitazone, providing an indication of simultaneous binding of both ligands with 2 occupying the alternative site. In cellular setting, Gal4-PPARγ activation by 2 alone was weak (Figure 1e); however the ligand markedly enhanced the activity of pioglitazone in a dose-dependent manner (EC50 1.4 ± 0.5 μM in the presence of 3 μM pioglitazone). 2 also induced transcriptional activity via the human PPAR response element (PPRE, Figure 1f) and enhanced pioglitazone-mediated PPRE activation. Unlike TZD, 2 did not prevent PPARγ phosphorylation at Ser273, an effect that has been linked to anti-diabetic activity20 (Figure 1g). Selectivity of 2 (100 μM) on nuclear receptors was assessed in Gal4-hybrid reporter gene assays (Figure 1h) which neither revealed agonism nor antagonistic effects on the respective reference ligands. We also detected no other ago-positive activity of 2 than the potentiating effect on PPARγ activation by pioglitazone, but it should be noted that this technique cannot reveal ″silent″ binding to alternative sites. Moreover, toxicity profiling of 2 showed no cytotoxic effects up to 100 μM (Figure 1i,j).
The co-crystal structure of PPARγ in complex with 2 (pdb ID: 8atz, Figures 2a, S2 and S4) confirmed the selective engagement of the ligand within the expected alternative binding site. The carboxylic acid of 2 formed a water-mediated hydrogen bond to Arg288 and a direct contact to the backbone of Ser342, while the extended phenethyl group was situated in a solvent-exposed region and had its ether linker forming a contact to Glu259. Despite a lack of contacts between the ligand and the activation function 2 (AF-2) in helix 12, 2 nonetheless induced an active conformation of the PPARγ LBD with AF-2 stabilized and bound to the core of the LBD, similarly to that observed in the PPARγ-1 structure. Importantly, the PPARγ-2 complex was crystallized without a co-activator peptide that might induce this active state.21 Thus, the observed conformation of the PPARγ LBD was likely achieved through the ligand binding at the alternative site. This might be explained by the ordered Ω-loop connecting H4 and H5, which had a slightly different conformation compared to the 1-bound structure and likely contributed to the overall stabilization of the PPARγ LBD and AF-2.11
Figure 2.

Binding of 2 to PPARγ (pdb ID: 8atz). (a) The co-crystal structure of the PPARγ LBD in complex with 2 (pdb ID: 8atz) revealed selective binding of the ligand to the alternative site and a stabilized active conformation of helix 12. Binding of 2 to the PPARγ LBD was mediated by H-bonds to Arg288 and Ser342. Electron density map for 2 in Figure S2. (b) PPARγ ligands 1, 2, BVT.13 (pdb ID: 2q6s11), and WY14643 (pdb ID: 8cph, 8cpi) induce different conformations of helix 12. (c) Compared to BVT.13, binding of 2 is shifted outward from the orthosteric region. (d) Comparison of PPARγ LBD structures in complex with various alternative sites and double binding ligands reveals differences in ligand binding sites and conformations. Apo (pdb ID: 8cpj) and pioglitazone-complexed (pdb ID: 5y2o27) PPARγ structures are shown as representatives of inactive and active forms, respectively. (e) Superposition of the bound ligands GA, 2, BVT.13, MRL-871, and WY14643 demonstrates different binding modes within the orthosteric and alternative binding regions. (f) Isothermal titration calorimetry (ITC) for the binding of 1 and 2 to the ligand-free and the GW9662-bound PPARγ LBD. The fitting of the heat of binding is shown with the isotherms as insets. (g, h) LC–MS-based binding experiments demonstrated dose-dependent specific binding of 2 (0.2–4 μM) to the PPARγ LBD (1 μM) in the absence and presence of 1 μM pioglitazone (g) and specific binding of 2 and pioglitazone (5 μM each) in the absence or presence of the respective other ligand (h); data are the mean ± S.E.M., n = 3.
Alternative site binding in PPAR is not unprecedented as demonstrated by MRL-871 and BVT.13.7,8,11,22 However, in addition to their diverse binding modes, these ligands induce different protein conformations and thus different activation of the receptor which critically depends on the dynamic position of the C-terminal helix 12.23−25 We next aimed to compare the structural consequences of binding of 2 and these ligands addressing similar epitopes. For direct comparison, we further solved an inactive apo PPARγ structure and two complexes with WY-14643 that interestingly revealed two distinct binding modes: one in an active form with ligand binding to both the orthosteric and alternative sites like the analogue 1 and a second binding mode inducing an inactive form with an unusual single ligand location occupying partially both orthosteric and alternative sites similar to MRL-871 (Figure S5). These two complexes with WY-14643 were consistent with recent findings that typical PPARγ agonist binding follows a two-step mechanism.26 Structural comparison revealed that 1, 2, WY-14643, MRL-871, and BVT.13 induced different structural alterations noted essentially by three distinct conformations of helix 12 (Figure 2b–d). MRL-871 and single WY-14643 binding between the orthosteric and alternative pockets failed to stabilize an active conformation of helix 12 in the crystals, which was either disordered or extended outward from the LBD core, similar to the conformation observed in the inactive apo form due to crystal contacts.24,26 This was in contrast to the other alternative site ligands, whose binding triggered an inward swing and thus stabilization of helix 12. However, only simultaneous binding of GA, 1, or WY-14643 at both orthosteric and alternative sites as well as binding of 2 to the alternative site alone but not BVT.13 induced and stabilized the active conformation of the PPARγ LBD characterized by the fully “in” conformation of helix 12 and reminiscent to that observed for pioglitazone (pdb ID: 5y2o27). These distinct effects might correlate with different ligand binding modes evident from examining the superposition of their bound states (Figure 2e), revealing that 2 spared the orthosteric region while MRL-871 and BVT.13 occupied parts of the pocket typically bound by orthosteric ligands such as GA.
To validate this unique alternative site binding mode of 2 which unlike other ligands did not partially or fully occupy the orthosteric site (Figure 2e), we conducted binding studies both in the presence and absence of an orthosteric ligand using isothermal titration calorimetry (ITC) and mass spectrometry (MS). The ITC results demonstrated a 1:2 protein to ligand binding stoichiometry of 1 while 2 exhibited 1:1 interaction with the PPARγ LBD (Figure 2f). When using GW9662-bound PPARγ LBD in which the orthosteric site was occluded by the covalent antagonist, we observed a binding stoichiometry of 1:1 for both 1 and 2 without changes in affinities. Consistent with this, MS-based binding assays showed that 2 interacted with the protein in a dose-dependent manner regardless of the presence of the orthosteric agonist pioglitazone (Figure 2g). These results suggested therefore the selective binding of 2 to the alternative site in solution since the presence of GW9662 or pioglitazone did not affect binding of 2, demonstrating that both orthosteric and alternative site ligands can bind to PPARγ simultaneously without competition. Vice versa, binding of pioglitazone was invariant to the presence of 2 (Figure 2h), confirming that 2 allowed binding of the orthosteric agonist and molecular modeling supported simultaneous binding of 2 and pioglitazone to the PPARγ LBD (Figure S6).
Our results obtained from cellular, structural, and biophysical characterization thus presented 2 as a chemical tool to investigate selective modulation of PPARγ through the alternative ligand binding site. To capture the molecular effects of PPARγ modulation by 2, we compared how 2, pioglitazone, and GW9662 affected co-regulator recruitment to the PPARγ LBD using homogenous time-resolved fluorescence resonance energy transfer (HTRF)-based assays (Figures 3a, S7). From a diverse panel of 29 canonical nuclear receptor co-regulators, pioglitazone mainly induced the binding of CBP-1, PGC-1α, NCoA6, and DRIP2 but displaced SMRT and NCOR. The antagonist GW9662 enhanced binding of NCOR and SMRT to PPARγ but displaced CBP-1 and fully reversed the effects of pioglitazone upon co-incubation. Interestingly, slightly enhanced binding of the co-activators PGC-1α, NCoA6, and DRIP indicated also a weak activating stabilization effect of GW9662, which was consistent with its weak partial agonistic activity. The alternative site ligand 2 alone mediated weaker recruitment of PGC-1α and NCoA6, and, in contrast to pioglitazone, displaced CBP-1 and DRIP2, but enhanced binding of SMRT and NCOR. In comparison to the agonist pioglitazone and the antagonist GW9662, this highlighted a unique profile of PPARγ modulation by 2 with activating (PGC-1α and NCoA6) and inactivating (CBP-1, DRIP2, SMRT, and NCOR) contributions.
Figure 3.

Mechanistic and biological effects of the alternative site PPARγ ligand 2. (a) 2 caused distinguished effects on co-regulator recruitment by PPARγ with activating and inactivating contributions. Pioglitazone (3 μM), 2 (30 μM), GW9662 (10 μM). Data are the mean ± SD ΔHTRF vs DMSO; n = 4. (b) Pioglitazone (10 μM) and the RXR agonist SR11237 (10 μM) increased and 2 (50 μM) diminished heterodimerization of PPARγ with RXR. 2 also blocked the dimer-stabilizing effects of pioglitazone and SR11237. Data are the mean ± SD HTRF vs DMSO; n = 3. (c) In contrast to pioglitazone, 2 induced no differentiation of human adipocyte-derived stem cells (ASC). Data are the mean ± SD relative Oil Red O (ORO) deposition compared to pioglitazone; n = 3. (d) Representative images of ASC differentiation experiments stained with ORO. (e) Differential gene expression in HepG2 cells treated with 2 (20 μM) versus DMSO (0.1%). Volcano plot shows log2(fold change) in the gene expression level (x-axis) versus the statistical significance level (−log10(p-value); y-axis). (f) Compared effects of 2 and GA on gene expression. Heat map shows induced (blue), downregulated (red), and nonregulated (black) genes. (g–i) Selected genes regulated by treatment with 2 (20 μM) associated with FOXO signaling (g), adipo–/lipogenesis (h), TOR signaling (i), cell cycle (i), apoptosis (i), and ATP generation (i). Heat maps show log2(fold change) in the gene expression of significantly (p-value <0.05) regulated genes. (j) Treatment with 2 (20 μM, 16 h) enhanced the inactivating phosphorylation of FOXO3a at Ser253. Data are the mean ± S.E.M.; n = 6. Western blots in the Supporting Information. (k) Pioglitazone enhanced, 2 decreased FOXO activity in HepG2 cells over time. Data are the mean ± S.E.M. FHRE activity; n ≥ 3.
Since the binding of 2 could occur concomitantly with pioglitazone, we questioned whether such a co-interaction scenario might exert different effects. Indeed, we found an interesting co-factor recruitment profile that mixed parts of the individual signatures of both ligands. The alternative site ligand 2 dominated the effects on corepressor binding (SMRT, NCOR) but did not fully reverse the pioglitazone-induced coactivator recruitment (CBP-1 and DRIP2) which was also evident from cross-titration experiments (Figure S7).
When testing the combination of 2 and GW9662, additive displacement of CBP-1 was observed, while NCOR and SMRT corepressor recruitment was abrogated, suggesting cooperative effects on the PPARγ LBD. These alterations in co-factor recruitment patterns correlated with different ligand-induced PPARγ LBD conformations. The ability of 2 to induce an active state similar to the orthosteric agonist pioglitazone indicated that the binding of this ligand can mediate sufficient stabilization to induce partial activation. In addition, simultaneous binding of 2 and the antagonist GW9662 observed in ITC was in agreement with the ability of 1 and 2 to activate GW9662-bound PPARγ, suggesting cooperative stabilization by orthosteric and alternative site ligands.28,29
Since heterodimerization with the retinoid X receptor (RXR) is a well-described consequence of PPARγ activation, we next assessed an influence of 2 on this mechanism (Figure 3b). We interestingly observed that while pioglitazone and the RXR agonist SR11237 enhanced the PPARγ:RXR interaction,302 not only decreased heterodimer formation but also abrogated dimer stabilization by pioglitazone. A similar effect was also evident for the combination of 2 and SR11237, suggesting that destabilization of the heterodimer was not due to competition with pioglitazone. Overall, distinct co-regulator binding and dimerization profiles thus differentiated 2 from orthosteric PPARγ agonists and demonstrated different consequences of orthosteric and alternative site PPARγ modulation with a complex crosstalk between ligands of both binding regions.
As a selective alternative site ligand, 2 emerged as a tool to determine the biological effects of PPARγ modulation through this epitope. We thus studied whether 2 would promote differentiation of human adipocyte-derived stem cells (ASC; ASC52telo, hTERT) into adipocytes, a process that is regulated by PPARγ31 (Figure 3c,d). In contrast to pioglitazone, 2 did not cause adipogenesis even at a high 50 μM concentration but actually diminished pioglitazone-induced adipogenesis, illustrating that alternative site binding did not activate classical, pro-adipogenic PPARγ signaling. To elucidate the effects of this noncanonical PPARγ modulation, we next studied how 2 affected gene expression in hepatocytes (HepG2) in an unbiased fashion by mRNAseq. Treatment with 2 significantly altered the expression of 1750 protein coding genes (p-value <0.05, |log2(fold change)| > 1; Figure 3e, Table S1), yet no induction of canonical PPAR signaling was observable (Figures S8–S10). In fact, 2 merely decreased expression of several PPARγ regulated genes involved in adipogenesis, lipid metabolism, and transport (e.g., CPT-1, perilipin, FABP1, SCD-1),32 which was consistent with the lack of pro-adipogenic effects in ASC differentiation. Nevertheless, 230 of the 1750 genes regulated by 2 comprise experimentally confirmed or predicted PPAR response elements (Table S2; pioglitazone: 62),33 supporting PPAR-mediated effects of 2.
Gene expression changes by 2 and GA (Figure 3f) shared common effects but with a more specific activity of the selective alternative site binder 2. Differential gene expression analysis indicated downregulation of forkhead box O (FOXO) signaling, anti-apoptotic and anti-proliferative activity, as well as reduced lipogenesis as prominent effects of 2 (Figures 3g–i, S11–S13).32 Diminished FOXO signaling promotes expression of antioxidant and cytoprotective genes and decreases expression of negative cell cycle regulators, while activated forms of FOXO stimulate proapoptotic gene expression (Bim, TRAIL, FasL).34 FOXO also plays a role in insulin signaling and glucose homeostasis (G6Pase, PEPCK) and its downregulation can reverse hyperglycemia and insulin resistance.35,36 FOXO activity is regulated by acetylation and phosphorylation.37 Activating deacetylation mediated, e.g., by SIRT1 and activating phosphorylation, triggered by oxidative (JNK, MST1) and nutrient stress (AMPK) or external stimuli (STAT3) cause FOXO to (re-)enter the nucleus.37−39 On the contrary, inactivating acetylation (e.g., by CBP) and phosphorylation stimulated by growth factors or insulin-induced cell growth lead to nuclear export and subsequent degradation of FOXO.37,39,40
Detailed gene expression analysis revealed that 2 caused downregulation of FOXO3 and activators of FOXO signaling (e.g., SIRT1, MAPK8, TNFSF10), while inhibitory regulators (e.g., SGK1) were upregulated (Figures 3g, S11). In contrast, no effect on FOXO signaling-related gene expression was detected for pioglitazone (Figure S12).12 Inhibition of FOXO signaling by 2 was accompanied by downregulation of genes associated with lipogenesis (e.g., LDLRAP1, SERPINA3, ATP10A, Figure 3h), TOR signaling (e.g., LAMTOR1, 2, mTORC1, PIP4K, Figure 3i), cell cycle progression (e.g., ANAPC15, CDC16), apoptosis (e.g., DDIT3, BID), and ATP generation (e.g., SLC25A17, NDUFA9),32 all of which indicated reduced metabolic activity and cells entering a resting state in response to diminished FOXO activity. Suppressive effects of 2 on FOXO signaling were also evident in orthogonal cellular experiments. Treatment with 2 promoted phosphorylation of FOXO3a at Ser253 (Figure 3j), which has been shown to enhance its export from the nucleus.41 Additionally, 2 diminished transcriptional activity of FOXO in a time-dependent manner, supporting a genomic mechanism causing FOXO suppression (Figure 3k).41
Binding of endogenous vitamin E metabolites12 and other natural ligands10,17 suggests considerable relevance of the alternative PPARγ binding site and may even support the hypothesis that this region constitutes a second orthosteric site for natural ligand binding. Here, we sought to elucidate the impact of this site on PPARγ regulation by developing a ligand (2) that selectively mimicked the binding of vitamin E metabolites to the alternative site.
As demonstrated by the 2-bound PPARγ LBD structure and binding studies, 2 exhibited a unique binding mode that still allowed simultaneous binding of orthosteric ligands. Using 2 as a chemical tool for biological studies, we demonstrated that selective interaction with the alternative site led to non-canonical PPARγ modulation on the molecular and cellular level. Mechanistically, 2 induced a PPARγ-cofactor interaction pattern distinct from orthosteric reference agonist and antagonist and prevented PPARγ:RXR dimerization. A lack of pro-adipogenic activity, no inhibition of PPARγ phosphorylation at Ser273, as well as the absence of classical PPAR signaling by the selective alternative site ligand illustrated biological effects different from the activity of insulin-sensitizing, orthosteric TZD.2,19,20 These molecular and cellular consequences accentuated different modulation effects of alternative site binding, suggesting another PPARγ regulatory mechanism that may involve an interplay and cooperativity between (endogenous) orthosteric and alternative site PPARγ ligands.6,17,28,29 Differential gene expression analysis further revealed unprecedented cellular effects of 2, highlighted by suppression of FOXO signaling. Such activity may have a potential therapeutic value due to a link between FOXO activation and diverse pathologies. For example, downregulation of this signaling pathway42 may have a beneficial neuroprotective effect as indicated by enhanced neurogenesis upon FOXO inhibition in vivo.43 These findings unveil some non-canonical consequences of PPARγ modulation through the alternative site that may open new therapeutic opportunities.
Associated Content
The crystal structures of the PPARγ LBD associated to this study have been deposited in the PDB with the accession codes 8aty, 8atz, 8cph, 8cpi, and 8cpj. The mRNAseq dataset has been deposited in Array Express with the accession code E-MTAB-12166. All other data generated in this study are available from the corresponding author on request.
Acknowledgments
This research was financially supported by the European Research Council (ERC StG 101040355 to D.M.), the Innovative Medicines Initiative (Grant Agreement No. 875510), and the Aventis Foundation (Life Science Bridge Award to D.M.). E.P. and J.H. thank the Fraunhofer Leistungszentrum innovative Therapeutics (TheraNova) for financial support. FHRE-Luc was a gift from Michael Greenberg (Addgene plasmid # 1789).
Glossary
Abbreviations
- AF-2
activation function 2
- ASC
adipocyte-derived stem cells
- CBP-1
CREB-binding protein
- DRIP
vitamin D receptor interacting protein
- DSF
differential scanning fluorimetry
- FOXO
forkhead box O
- GA
garcinoic acid
- HTRF
homogenous time-resolved fluorescence resonance energy transfer
- ITC
isothermal titration calorimetry
- LBD
ligand binding domain
- NCoA
nuclear receptor co-activator
- NCOR
nuclear receptor co-repressor
- PPAR
peroxisome proliferator-activated receptor
- PGC-1α
PPARγ co-activator 1α
- RXR
retinoid X receptor
- SMRT
silencing mediator for retinoid and thyroid hormone receptor.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c03417.
The authors declare no competing financial interest.
Supplementary Material
References
- Michalik L.; Auwerx J.; Berger J. P.; Chatterjee V. K.; Glass C. K.; Gonzalez F. J.; Grimaldi P. A.; Kadowaki T.; Lazar M. A.; O’Rahilly S.; Palmer C. N. A.; Plutzky J.; Reddy J. K.; Spiegelman B. M.; Staels B.; Wahli W. International Union of Pharmacology. LXI. Peroxisome Proliferator-Activated Receptors. Pharmacol. Rev. 2006, 58, 726–741. 10.1124/pr.58.4.5. [DOI] [PubMed] [Google Scholar]
- Montaigne D.; Butruille L.; Staels B. PPAR Control of Metabolism and Cardiovascular Functions. Nat. Rev. Cardiol. 2021, 18, 809–823. 10.1038/s41569-021-00569-6. [DOI] [PubMed] [Google Scholar]
- Tolman K. G. The Safety of Thiazolidinediones. Expert Opin. Drug Saf. 2011, 10, 419–428. 10.1517/14740338.2011.534982. [DOI] [PubMed] [Google Scholar]
- Francque S.; Szabo G.; Abdelmalek M. F.; Byrne C. D.; Cusi K.; Dufour J. F.; Roden M.; Sacks F.; Tacke F. Nonalcoholic Steatohepatitis: The Role of Peroxisome Proliferator-Activated Receptors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 24–39. 10.1038/s41575-020-00366-5. [DOI] [PubMed] [Google Scholar]
- Willems S.; Zaienne D.; Merk D. Targeting Nuclear Receptors in Neurodegeneration and Neuroinflammation. J. Med. Chem. 2021, 64, 9592–9638. 10.1021/acs.jmedchem.1c00186. [DOI] [PubMed] [Google Scholar]
- Meijer F. A.; Leijten-van de Gevel I. A.; de Vries R. M. J. M.; Brunsveld L. Allosteric Small Molecule Modulators of Nuclear Receptors. Mol. Cell. Endocrinol. 2019, 485, 20–34. 10.1016/j.mce.2019.01.022. [DOI] [PubMed] [Google Scholar]
- Östberg T.; Svensson S.; Selén G.; Uppenberg J.; Thor M.; Sundbom M.; Sydow-Bäckman M.; Gustavsson A. L.; Jendeberg L. A New Class of Peroxisome Proliferator-Activated Receptor Agonists with a Novel Binding Epitope Shows Antidiabetic Effects. J. Biol. Chem. 2004, 279, 41124–41130. 10.1074/jbc.M401552200. [DOI] [PubMed] [Google Scholar]
- Leijten-van de Gevel I. A.; van Herk K. H. N.; de Vries R. M. J. M.; Ottenheym N. J.; Ottmann C.; Brunsveld L. Indazole MRL-871 Interacts with PPARγ via a Binding Mode That Induces Partial Agonism. Bioorg. Med. Chem. 2022, 68, 116877 10.1016/j.bmc.2022.116877. [DOI] [PubMed] [Google Scholar]
- Hughes T. S.; Shang J.; Brust R.; De Vera I. M. S.; Fuhrmann J.; Ruiz C.; Cameron M. D.; Kamenecka T. M.; Kojetin D. J. Probing the Complex Binding Modes of the PPARγ Partial Agonist 2-Chloro-N-(3-Chloro-4-((5-Chlorobenzo[d]Thiazol-2-Yl)Thio)Phenyl)-4-(Trifluoromethyl)Benzenesulfonamide (T2384) to Orthosteric and Allosteric Sites with NMR Spectroscopy. J. Med. Chem. 2016, 59, 10335–10341. 10.1021/acs.jmedchem.6b01340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T.; Fairall L.; Amin K.; Inaba Y.; Szanto A.; Balint B. L.; Nagy L.; Yamamoto K.; Schwabe J. W. R. Structural Basis for the Activation of PPARγ by Oxidized Fatty Acids. Nat. Struct. Mol. Biol. 2008, 15, 924–931. 10.1038/nsmb.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning J. B.; Chalmers M. J.; Prasad S.; Busby S. A.; Kamenecka T. M.; He Y.; Nettles K. W.; Griffin P. R. Partial Agonists Activate PPARγ Using a Helix 12 Independent Mechanism. Structure 2007, 15, 1258–1271. 10.1016/j.str.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Willems S.; Gellrich L.; Chaikuad A.; Kluge S.; Werz O.; Heering J.; Knapp S.; Lorkowski S.; Schubert-Zsilavecz M.; Merk D. Endogenous Vitamin E Metabolites Mediate Allosteric PPARγ Activation with Unprecedented Co-Regulatory Interactions. Cell Chem. Biol. 2021, 28, 1489–1500.e8. 10.1016/j.chembiol.2021.04.019. [DOI] [PubMed] [Google Scholar]
- Rotman Y.; Sanyal A. J. Current and Upcoming Pharmacotherapy for Non-Alcoholic Fatty Liver Disease. Gut 2017, 66, 180–190. 10.1136/gutjnl-2016-312431. [DOI] [PubMed] [Google Scholar]
- Marinelli R.; Torquato P.; Bartolini D.; Mas-Bargues C.; Bellezza G.; Gioiello A.; Borras C.; De Luca A.; Fallarino F.; Sebastiani B.; Mani S.; Sidoni A.; Vina J.; Leri M.; Bucciantini M.; Nardiello P.; Casamenti F.; Galli F. Garcinoic Acid Prevents β-Amyloid (Aβ) Deposition in the Mouse Brain. J. Biol. Chem. 2020, 295, 11866–11876. 10.1074/jbc.RA120.013303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer F. A.; Van Den Oetelaar M. C. M.; Doveston R. G.; Sampers E. N. R.; Brunsveld L. Covalent Occlusion of the RORγt Ligand Binding Pocket Allows Unambiguous Targeting of an Allosteric Site. ACS Med. Chem. Lett. 2021, 12, 631–639. 10.1021/acsmedchemlett.1c00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leesnitzer L. M.; Parks D. J.; Bledsoe R. K.; Cobb J. E.; Collins J. L.; Consler T. G.; Davis R. G.; Hull-Ryde E. A.; Lenhard J. M.; Patel L.; Plunket K. D.; Shenk J. L.; Stimmel J. B.; Therapontos C.; Willson T. M.; Blanchard S. G. Functional Consequences of Cysteine Modification in the Ligand Binding Sites of Peroxisome Proliferator Activated Receptors by GW9662. Biochemistry 2002, 41, 6640–6650. 10.1021/bi0159581. [DOI] [PubMed] [Google Scholar]
- Shang J.; Brust R.; Mosure S. A.; Bass J.; Munoz-Tello P.; Lin H.; Hughes T. S.; Tang M.; Ge Q.; Kamenekca T. M.; Kojetin D. J. Cooperative Cobinding of Synthetic and Natural Ligands to the Nuclear Receptor PPARγ. Elife 2018, 7, e43320 10.7554/eLife.43320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollinger J.; Gellrich L.; Schierle S.; Kilu W.; Schmidt J.; Kalinowsky L.; Ohrndorf J.; Kaiser A.; Heering J.; Proschak E.; Merk D. Tuning Nuclear Receptor Selectivity of Wy14,643 towards Selective Retinoid X Receptor Modulation. J. Med. Chem. 2019, 62, 2112–2126. 10.1021/acs.jmedchem.8b01848. [DOI] [PubMed] [Google Scholar]
- Jang J. Y.; Bae H.; Lee Y. J.; Il Choi Y.; Kim H.-J.; Park S. B.; Suh S. W.; Kim S. W.; Han B. W. Structural Basis for the Enhanced Anti-Diabetic Efficacy of Lobeglitazone on PPARγ. Sci. Rep. 2018, 8, 31. 10.1038/s41598-017-18274-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. H.; Banks A. S.; Estall J. L.; Kajimura S.; Boström P.; Laznik D.; Ruas J. L.; Chalmers M. J.; Kamenecka T. M.; Blüher M.; Griffin P. R.; Spiegelman B. M. Anti-Diabetic Drugs Inhibit Obesity-Linked Phosphorylation of PPARγ by Cdk5. Nature 2010, 466, 451–456. 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vink P. J.; Koops A. A.; D’Arrigo G.; Cruciani G.; Spyrakis F.; Brunsveld L. Cooperativity as Quantification and Optimization Paradigm for Nuclear Receptor Modulators. Chem. Sci. 2022, 13, 2744–2752. 10.1039/D1SC06426F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardes A.; Souza P. C. T.; Muniz J. R. C.; Ricci C. G.; Ayers S. D.; Parekh N. M.; Godoy A. S.; Trivella D. B. B.; Reinach P.; Webb P.; Skaf M. S.; Polikarpov I. Molecular Mechanism of Peroxisome Proliferator-Activated Receptor α Activation by WY14643: A New Mode of Ligand Recognition and Receptor Stabilization. J. Mol. Biol. 2013, 425, 2878–2893. 10.1016/j.jmb.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Shang J.; Mosure S. A.; Zheng J.; Brust R.; Bass J.; Nichols A.; Solt L. A.; Griffin P. R.; Kojetin D. J. A Molecular Switch Regulating Transcriptional Repression and Activation of PPARγ. Nat. Commun. 2020, 11, 956. 10.1038/s41467-020-14750-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrisman I. M.; Nemetchek M. D.; De Vera I. M. S.; Shang J.; Heidari Z.; Long Y.; Reyes-Caballero H.; Galindo-Murillo R.; Cheatham T. E.; Blayo A. L.; Shin Y.; Fuhrmann J.; Griffin P. R.; Kamenecka T. M.; Kojetin D. J.; Hughes T. S. Defining a Conformational Ensemble That Directs Activation of PPARγ. Nat. Commun. 2018, 9, 1794. 10.1038/s41467-018-04176-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J.; Corzo C.; Chang M. R.; Shang J.; Lam V. Q.; Brust R.; Blayo A. L.; Bruning J. B.; Kamenecka T. M.; Kojetin D. J.; Griffin P. R. Chemical Crosslinking Mass Spectrometry Reveals the Conformational Landscape of the Activation Helix of PPARγ; a Model for Ligand-Dependent Antagonism. Structure 2018, 26, 1431–1439.e6. 10.1016/j.str.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J.; Kojetin D. J. Structural Mechanism Underlying Ligand Binding and Activation of PPARγ. Structure 2021, 29, 940–950.e4. 10.1016/j.str.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. A.; Tan L.; Yang H.; Im Y. G.; Im Y. J. Structures of PPARγ Complexed with Lobeglitazone and Pioglitazone Reveal Key Determinants for the Recognition of Antidiabetic Drugs. Sci. Rep. 2017, 7, 16837. 10.1038/s41598-017-17082-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vink P. J.; Andrei S. A.; Higuchi Y.; Ottmann C.; Milroy L. G.; Brunsveld L. Cooperativity Basis for Small-Molecule Stabilization of Protein-Protein Interactions. Chem. Sci. 2019, 10, 2869–2874. 10.1039/C8SC05242E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries R. M. J. M.; Meijer F. A.; Doveston R. G.; Leijten-Van de Gevel I. A.; Brunsveld L. Cooperativity between the Orthosteric and Allosteric Ligand Binding Sites of RORγt. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2021287118 10.1073/pnas.2021287118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilu W.; Merk D.; Steinhilber D.; Proschak E.; Heering J. Heterodimer Formation with Retinoic Acid Receptor RXRα Modulates Coactivator Recruitment by Peroxisome Proliferator-Activated Receptor PPARγ. J. Biol. Chem. 2021, 297, 100814 10.1016/j.jbc.2021.100814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolbank S.; Peterbauer A.; Fahrner M.; Hennerbichler S.; Van Griensven M.; Stadler G.; Redl H.; Gabriel C. Dose-Dependent Immunomodulatory Effect of Human Stem Cells from Amniotic Membrane: A Comparison with Human Mesenchymal Stem Cells from Adipose Tissue. Tissue Eng. 2007, 13, 1173–1183. 10.1089/ten.2006.0313. [DOI] [PubMed] [Google Scholar]
- Carlson M.Org.Hs.Eg.Db: Genome Wide Annotation for Human. 2020, p R package version 3.12.0. 10.18129/B9.bioc.org.Hs.eg.db [DOI]
- Fang L.; Zhang M.; Li Y.; Liu Y.; Cui Q.; Wang N. PPARgene: A Database of Experimentally Verified and Computationally Predicted PPAR Target Genes. PPAR Res. 2016, 2016, 6042162 10.1155/2016/6042162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Tang N.; Hadden T. J.; Rishi A. K. Akt, FoxO and Regulation of Apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. 10.1016/j.bbamcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- Lee S.; Dong H. H. FoxO Integration of Insulin Signaling with Glucose and Lipid Metabolism. J. Endocrinol. 2017, 233, R67–R79. 10.1530/JOE-17-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accili D.; Arden K. C. FoxOs at the Crossroads of Cellular Metabolism, Differentiation, and Transformation. Cell 2004, 117, 421–426. 10.1016/S0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- Eijkelenboom A.; Burgering B. M. T. FOXOs: Signalling Integrators for Homeostasis Maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- Farhan M.; Wang H.; Gaur U.; Little P. J.; Xu J.; Zheng W. FOXO Signaling Pathways as Therapeutic Targets in Cancer. Int. J. Biol. Sci. 2017, 13, 815–827. 10.7150/ijbs.20052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt P. K.; Jiang H.; Aoki M. Triple Layer Control: Phosphorylation, Acetylation and Ubiquitination of FOXO Proteins. Cell Cycle 2005, 4, 908–913. 10.4161/cc.4.7.1796. [DOI] [PubMed] [Google Scholar]
- Van Der Horst A.; Tertoolen L. G. J.; De Vries-Smits L. M. M.; Frye R. A.; Medema R. H.; Burgering B. M. T. FOXO4 Is Acetylated upon Peroxide Stress and Deacetylated by the Longevity Protein HSir2SIRT1. J. Biol. Chem. 2004, 279, 28873–28879. 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- Brunet A.; Bonni A.; Zigmond M. J.; Lin M. Z.; Juo P.; Hu L. S.; Anderson M. J.; Arden K. C.; Blenis J.; Greenberg M. E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. 10.1016/S0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Wang X.; Wang Z.; Chen Y.; Huang X.; Hu Y.; Zhang R.; Ho M. S.; Xue L. FoxO Mediates APP-Induced AICD-Dependent Cell Death. Cell Death Dis. 2014, 5, e1233 10.1038/cddis.2014.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegrist S. E.; Haque N. S.; Chen C. H.; Hay B. A.; Hariharan I. K. Inactivation of Both Foxo and Reaper Promotes Long-Term Adult Neurogenesis in Drosophila. Curr. Biol. 2010, 20, 643–648. 10.1016/j.cub.2010.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.