

Abstract
Alphaviruses are emerging and reemerging viruses that cause disease syndromes ranging from incapacitating arthritis to potentially fatal encephalitis. While infection by arthritogenic and encephalitic alphaviruses results in distinct clinical manifestations, both virus groups induce robust innate and adaptive immune responses. However, differences in cellular tropism, type I interferon induction, immune cell recruitment, and B and T cell responses result in differential disease progression and outcome. In this review, we discuss aspects of immune responses that contribute to protective or pathogenic outcomes after alphavirus infection.
Keywords: alphavirus, arbovirus, immunity, mouse models, tropism, pathogenesis
INTRODUCTION
Alphaviruses are small, enveloped, positive-sense RNA viruses in the Togaviridae family and continue to be a public health concern. Alphaviruses have reemerged in recent decades, causing numerous epidemics and outbreaks worldwide (1). The alphavirus genome is approximately 12 kilobases and encodes four nonstructural proteins (nsP1–4) and five structural proteins (capsid, E3, E2, 6K, and E1) in two open reading frames. The nonstructural polyproteins are translated from the genomic RNA in the host cell cytoplasm and regulate viral replication and protein processing, including synthesis of minus-strand RNA replicative intermediates. The structural proteins are translated from the subgenomic RNA and required for virion formation and budding (2, 3). Alphaviruses are transmitted mainly by hematophagous arthropods between vertebrate reservoir hosts (e.g., nonhuman primates, birds, rodents, and marsupials) (1).
The Alphavirus genus comprises seven complexes classified based on antigenic characteristics and includes over 30 viral species (4) (Figure 1). Historically, alphaviruses were categorized as Old World or New World based upon their area of endemicity and human disease characteristics. Old World alphaviruses, include chikungunya virus (CHIKV), Sindbis virus (SINV), o’nyong-nyong virus (ONNV), Semliki Forest virus (SFV), and Ross River virus (RRV), were first isolated in Africa and Australia and mainly cause rheumatic disease affecting joints and musculoskeletal tissues (5). Though part of the Old World alphavirus classification, Mayaro virus (MAYV) was isolated in the West Indies and is responsible for outbreaks of arthritogenic disease in South and Central America (6). New World alphaviruses, including Venezuelan (VEEV), eastern (EEEV), and western (WEEV) equine encephalitis viruses, are found in North, Central, and South America, and infection can progress to neurological disease (7, 8). This classification generally aligns with symptomatic manifestation and geography but has become less useful with recent geographic patterns and reports of atypical disease presentation. For example, CHIKV is now endemic to several continents, reemerging in Southeast Asia and islands in the Indian Ocean and spreading to the Caribbean Islands and Americas (9). Additionally, SFV, SINV, and CHIKV can cause central nervous system (CNS) infection and neurological disease instead of, or in addition to, rheumatic manifestations (10). In this review, we characterize the alphaviruses based on their primary clinical manifestations, arthritogenic or encephalitic.
Figure 1.
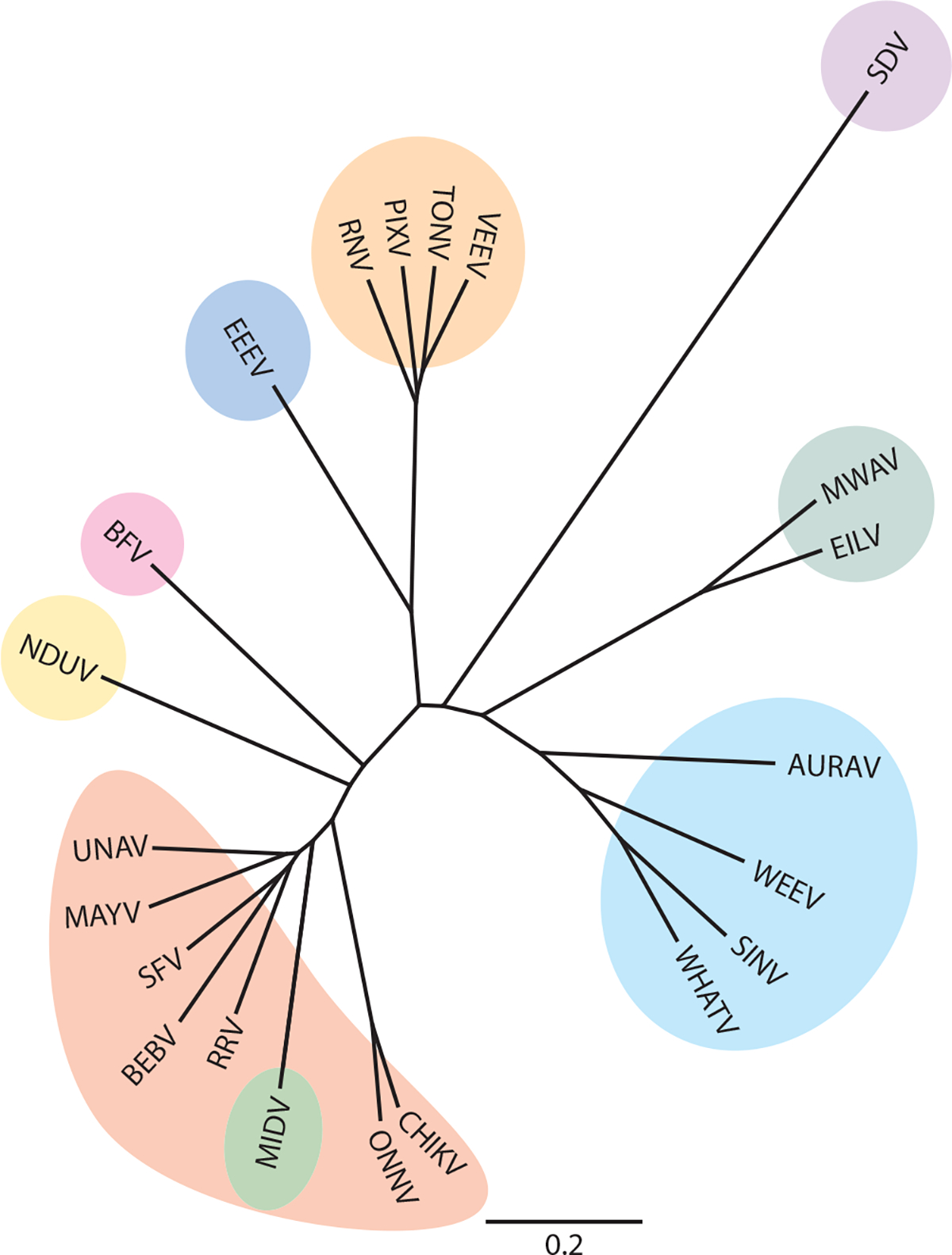
Genetic relationships of alphaviruses. Aquatic alphavirus (purple): sleeping disease virus (SDV) (accession number Q8QL52.1). Insect-only alphaviruses (teal): Mwinilunga virus (MWAV) (BBC45635) and Eilat virus (EILV) (QBG67157). Western equine encephalitis virus complex (cyan): Aura virus (AURAV) (Q86925), western equine encephalitis virus (WEEV) (QEX51909.1), Sindbis virus (SINV) (AAM10630.1), and Whataroa virus (WHATV) (AAO33329). Semliki Forest virus complex (red): chikungunya virus (CHIKV) (ABO38821), o’nyong-nyong virus (ONNV) (AAC97205), Ross River virus (RRV) (AYI50356.1), bebaru virus (BEBV) (AEJ36225), Semliki Forest virus (SFV) (QQZ00842), Mayaro virus (MAYV) (QDL88200.1), and Una virus (UNAV) (YP_009665989). Middelburg virus complex (green): Middelburg virus (MIDV) (AAO33343). Ndumu virus complex (yellow): Ndumu virus (NDUV) (AAO33345). Barmah Forest virus complex (pink): Barmah Forest virus (BFV) (AAO33347). Eastern equine encephalitis virus complex (dark blue): eastern equine encephalitis virus (EEEV) (AMT80322.1). Venezuelan equine encephalitis virus complex (orange): Rio Negro virus (RNV) (YP_009507803), Pixuna virus (PIXV) (YP_009507801), Tonate virus (TONV) (AAD14557), and Venezuelan equine encephalitis virus (VEEV) (AAC19322.1). The C-E3-E2–6K-E1 amino acid sequences were aligned using Geneious Aligner, and a phylogenic tree was built using Geneious Tree Builder.
Acute alphavirus infections are characterized by fever, rash, and myalgia that can progress to severe arthritis or encephalitis based on the tropism of the virus. For both the arthritogenic and encephalitic alphaviruses, viral RNA has been shown to persist past the acute stage of disease and result in long-term neurological sequelae or chronic joint pain and inflammation (11–13). Although most alphaviruses typically cause non-life-threatening acute disease in humans, the economic and social costs incurred during outbreaks are significant (14, 15). The severity and chronicity of disease often depend on the initial immune response to infection. Thus, an enhanced understanding of the pathogenesis of acute alphavirus infection may be critical for the development of counter-measures that limit morbidity.
Cell culture systems and animal models, including experimental infections of mice and nonhuman primates, have been used to identify the cell targets of viral tropism and the factors that contribute to a protective or pathogenic immune response. While recognition of shared viral features and interferon induction may be similar between the arthritogenic and encephalitic alphaviruses, cell tissue tropism, immunoevasion mechanisms, and immune cell recruitment differ between the viruses. For the encephalitic alphaviruses, a key component leading to clinical disease is entry into the CNS and infection of neuronal or glial cells (16). Based on the virus, invasion of the nervous system occurs through hematogenous spread or retrograde and anterograde transport along axons of neurons. Once in the brain, encephalitic alphaviruses infect and replicate within neuronal cells and induce proinflammatory cytokines that compromise the blood-brain barrier (BBB). Chemokine expression recruits innate immune cells, including monocytes and natural killer (NK) cells, followed by adaptive immune cells into the brain (17). Infiltrating T cells induce most of the pathological effects during alphavirus encephalitis but also are required for clearance of virus (17) (Figure 2). Indeed, encephalitic alphavirus infection can result in lethal encephalitis. Mouse models of encephalitic alphavirus disease closely mimic the biphasic nature of disease characterized by febrile illness and neurologic sequelae that is seen in equines and some humans (18). In comparison, the arthritogenic alphaviruses infect and replicate in the joint-associated and musculoskeletal tissues (19). Proinflammatory cytokine and chemokine expression induced by infection recruits immune cells, including monocytes, NK cells, neutrophils, and T cells, which limit infection but also cause tissue damage, edema, and inflammation (13). Additionally, infection of osteoblasts by some arthritogenic alphaviruses promotes osteoclastogenesis and bone erosion (13) (Figure 3). Mouse models of the arthritogenic alphaviruses induce foot swelling, myositis, and/or bone loss, similar to disease observed in humans (20). In this review, we identify shared and distinct factors within the innate and adaptive immune response to acute infection between the encephalitic and arthritogenic alphaviruses that lead to infection, immunity, and disease resolution and pathogenesis.
Figure 2.
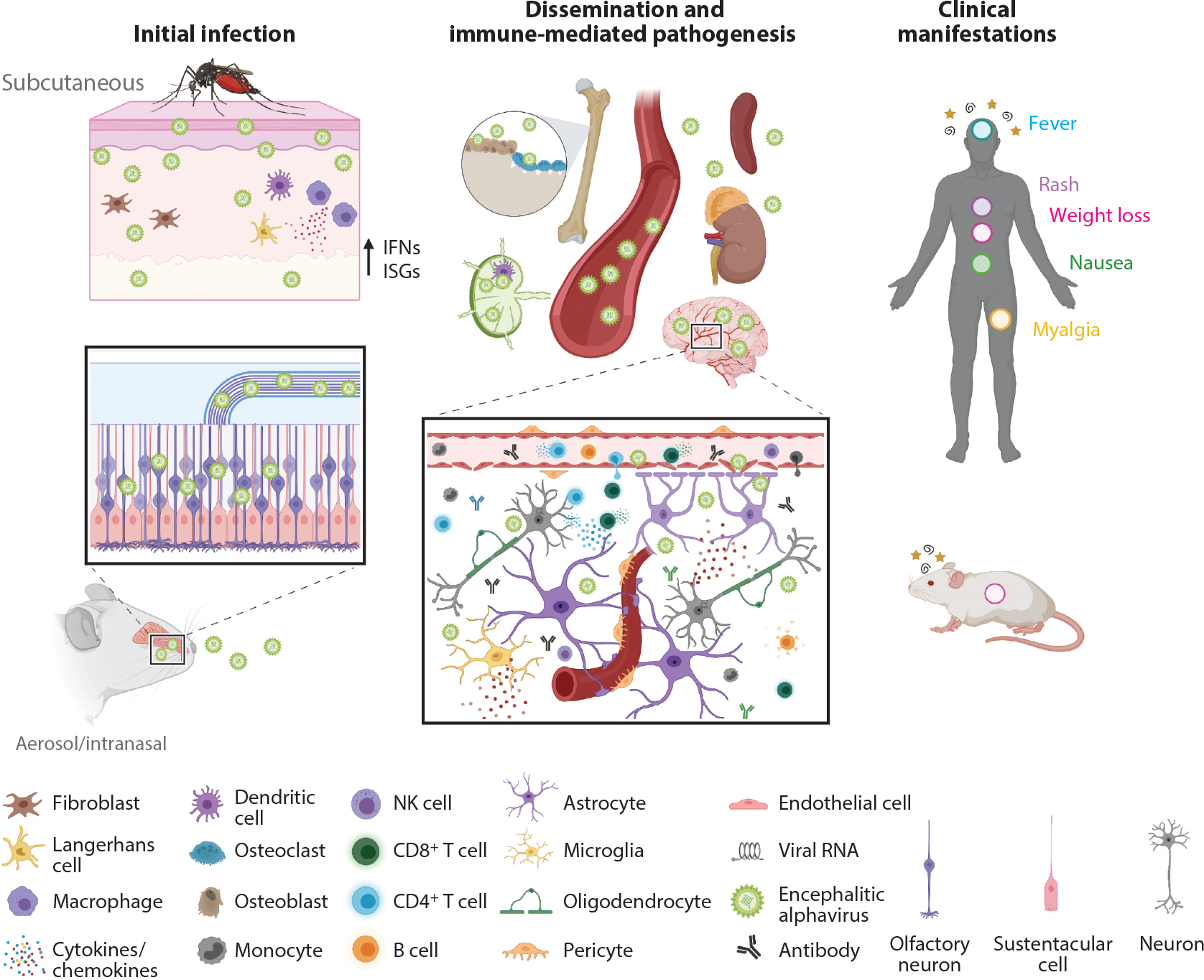
Infection and pathogenesis of encephalitic alphaviruses. Inoculation by encephalitic alphaviruses can occur subcutaneously through the bite of a mosquito or through an intranasal or aerosolized exposure. In the skin, some encephalitic alphaviruses (e.g., VEEV) will replicate locally in dendritic cells and macrophages, prompting release of type I interferons and expression of ISGs. Virus then travels to the draining lymph node or through the bloodstream to disseminate to peripheral tissues. Other encephalitic alphaviruses, like EEEV, replicate in fibroblasts and osteoblasts but not in lymphoid tissues and do not induce a measurable peripheral inflammatory response. Virus spreads rapidly to the CNS via several routes, including retrograde transport in neurons of the olfactory bulb and trigeminal nerves, hematogenous routes, and across the BBB. Release of proinflammatory cytokines and chemokines alters and compromises the BBB, contributing to further CNS infection. Immune cells are recruited into the brain parenchyma, and resident glia become activated, both of which are essential for control of infection. Nonetheless, activated immune cells can also damage neurons and cause demyelination, which drives the neurological syndromes observed in humans (stars and spirals). Depending on the virus strain, neurons and glia can survive infection and harbor viral RNA after acute infection. Other signs and symptoms of systemic disease include fever (blue circle), rash (purple circle), weight loss (pink circle), nausea (green circle), and myalgia (yellow circle). Abbreviations: BBB, blood-brain barrier; CNS, central nervous system; EEEV, eastern equine encephalitis virus; IFN, interferon; ISG, interferon-stimulated gene; NK, natural killer; VEEV, Venezuelan equine encephalitis virus. Figure adapted from images created with BioRender.com.
Figure 3.
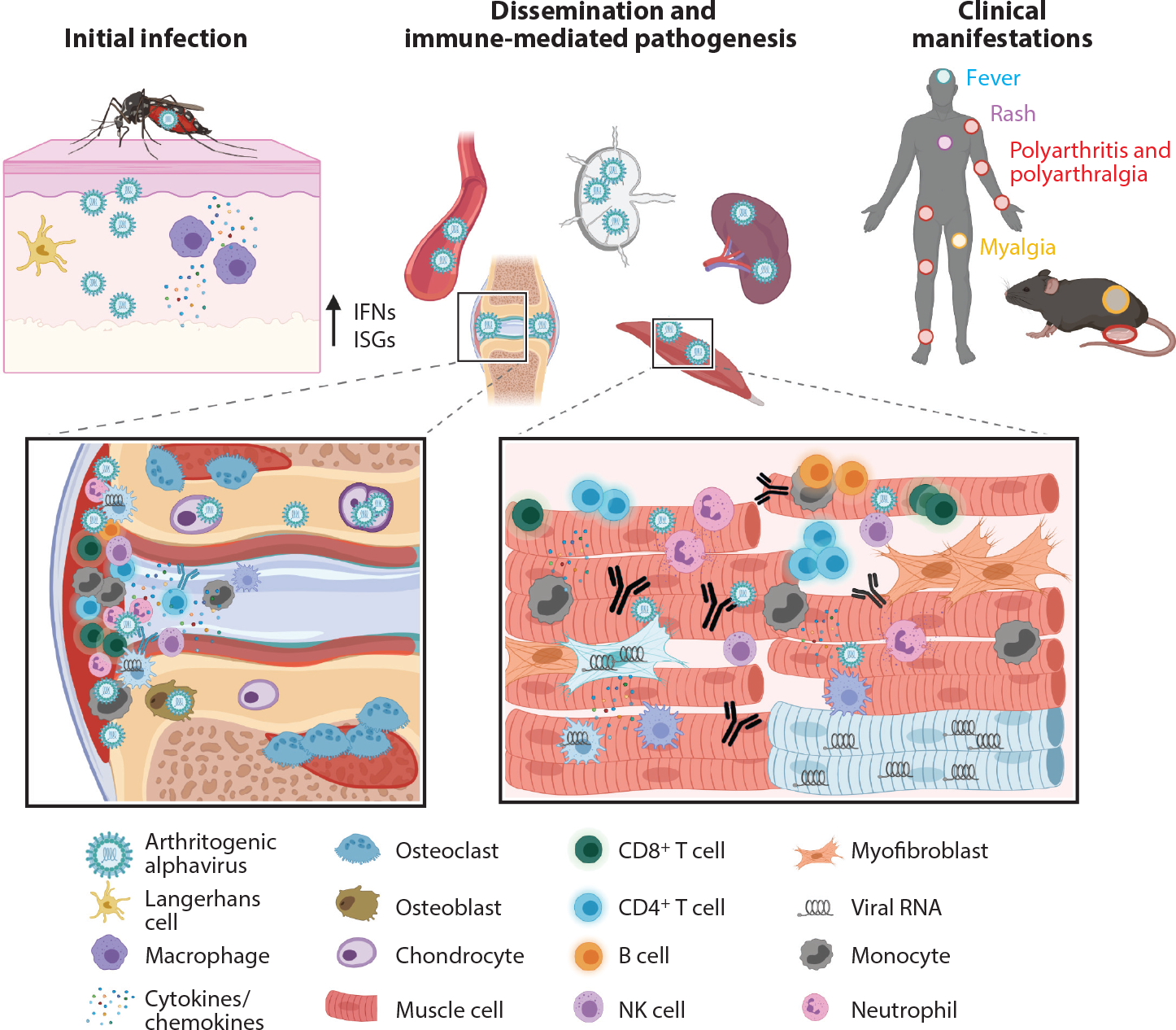
Infection and pathogenesis of arthritogenic alphaviruses. Following a bite from an infected mosquito, arthritogenic alphaviruses replicate locally in fibroblasts, keratinocytes, epithelial and endothelial cells, and/or macrophages. Cellular infection results in induction of type I interferons and expression of ISGs. Virus travels to the lymph node or through the bloodstream to disseminate to peripheral organs, where it replicates to high titers in the joint-associated and musculoskeletal tissues. Infection of osteoblasts promotes osteoclastogenesis and bone erosion. Proinflammatory cytokines and chemokines are released and recruit cells that are essential to control infection but also mediate damage including synovitis, bone reabsorption, and muscle fiber destruction. Macrophages, myofibroblasts, and muscle cells can survive infection and harbor viral RNA many weeks after acute infection. These processes drive the clinical syndromes observed in humans, including fever (blue circle), rash (purple circle), polyarthritis and polyarthralgia (red circles), and myalgia (yellow circle). Abbreviations: IFN, interferon; ISG, interferon-stimulated gene; NK, natural killer. Created with BioRender.com.
CELLULAR TROPISM
The distinction between disease syndromes caused by encephalitic and arthritogenic alphaviruses can be attributed in part to their differences in cellular tropism, which is influenced by receptor usage, a permissive environment for replication, and immunological changes to the target cell. Arthritogenic alphaviruses replicate at or near the site of infection and then disseminate to target organs. CHIKV, a prototype arthritogenic alphavirus, replicates in fibroblasts, keratinocytes, melanocytes, endothelial cells, epithelial cells, and possibly tissue-resident and -infiltrating macrophages (21, 22). Infected cells or free virus seed the draining lymph node with virus, which leads to viremia and dissemination to target organs and tissues, including the spleen, joint-associated tissues, and muscle (23). In the muscle, CHIKV infects satellite cells, myoblasts, and fibroblasts, and in the joint, it targets chondrocytes, osteoblasts, monocytes, and synovial fibroblasts (24, 25), ultimately leading to polyarthralgia and polyarthritis (23). Using a CHIKV reporter virus strain that creates a fate map for infected cells, one study showed skeletal muscle myofibers and dermal and muscle fibroblasts could survive CHIKV infection and harbor viral RNA for weeks (26). Although CHIKV infects skeletal muscle tissue, it is not the principal source of infectious virus in vivo. Preventing skeletal muscle infection by using a CHIKV strain encoding a skeletal muscle microRNA (miRNA) resulted in similar viral titers as control virus but caused less joint swelling, CD4+ T cell recruitment, and proinflammatory cytokine expression, implicating skeletal muscle infection as a driver of clinical disease (27).
Encephalitic alphaviruses vary in their cellular tropism and spread, which impact their clinical manifestations. VEEV initially infects Langerhans cells in the skin and then transits to the draining lymph node, where it targets dendritic cells (DCs) and to a lesser extent lymphocytes and macrophages (28). In cell culture, VEEV replicates efficiently in DCs, macrophages, and mesenchymal cells, including fibroblasts and osteoblasts (29). EEEV also infects mesenchymal cells (29, 30) but replicates poorly in DCs and macrophages, leading to limited replication in the draining lymph node and a delay in dissemination (29). However, the lack of EEEV infection in the draining lymph node is not related to type I interferon signaling, as replication blocks in the draining lymph node persist in IFN-α/β receptor (IFNAR1)−/− mice, suggesting an additional mechanism of restriction. A landmark study identified a hematopoietic-specific miRNA (miR-142–3p) that targets the 3′ untranslated region of the EEEV genome and suppresses its replication in myeloid cells (31). The lack of replication in myeloid cells limited induction of type I interferons and enhanced spread to the CNS (31, 32). The WEEV genome also contains a miR-142–3p seed sequence. Similar to the case of EEEV, WEEV miRNA seed sites limited replication in macrophages or peripheral blood leukocytes (32, 33). The high levels of expression of type I interferons during VEEV infection coupled with the lack of type I interferons during EEEV infection suggest that myeloid cell infection and production of type I interferons during encephalitic alphavirus infection can dictate the pathogenesis sequence (29, 31).
Expression of attachment and entry receptors also determines the cellular tropism of alphaviruses. Distinct cell surface receptors have been identified for the arthritogenic and encephalitic alphaviruses, which may contribute to the differential clinical manifestations. Matrix-remodeling associated 8 (MXRA8) is an adhesion molecule and entry receptor for multiple arthritogenic alphaviruses, including CHIKV, MAYV, RRV, and ONNV, but not for the encephalitic alphaviruses (34). MXRA8 is expressed in tissues that are relevant for arthritogenic alphavirus infection, including those in ankle, muscle, spleen, and lymph node (35). Blockade of MXRA8 in cell culture reduced CHIKV infection in human primary dermal and synovial fibroblasts, osteoblasts, chondrocytes, and skeletal muscle cells (34). Mxra8−/− mice infected with CHIKV, MAYV, RRV, or ONNV showed reduced viral burden and diminished clinical disease with decreased neutrophil recruitment and proinflammatory cytokine and chemokine expression in the foot (35). Recently, low-density lipoprotein receptor class A domain–containing 3 (LDLRAD3) was identified as a key entry receptor for VEEV (but not EEEV or WEEV) in mouse and human neuroblastoma cells and primary human dermal epithelial and fibroblast cells (36). LDLRAD3 is highly expressed in VEEV-tropic tissues, including brain tissue (37–39). Mice with deletions in Ldlrad3 were completely resistant to lethal challenge with VEEV (36). Administration of the recombinant domain 1 of LDLRAD3 as an Fc fusion protein before VEEV infection reduced viremia and viral burden in the CNS to undetectable levels (36). Analogously, very low-density lipoprotein receptor (VLDLR) mediated entry of SFV into human and mouse neuronal cells. Blockade of VLDLR in vivo using an Fc-fusion protein of the ligand-binding domain of VLDLR prolonged time to death in neonate mice. Expression of VLDLR and the closely related apolipoprotein E receptor 2 (ApoER2) also was sufficient for binding and internalization of EEEV and SINV (39a).
Arthritogenic and encephalitic alphaviruses can utilize glycosaminoglycans as attachment factors (40). While the use of glycosaminoglycans, particularly heparan sulfate, typically arises from passage in cell culture and results in attenuation in vivo, North American isolates of EEEV naturally use heparan sulfate, which has been attributed to enhanced neurovirulence (41–44). Compared to a mutated EEEV strain with reduced heparan sulfate binding, wild-type EEEV strains caused decreased viral replication in peripheral tissues but increased viral replication in the brain (41).
Other potential receptors for alphaviruses have been identified for specific cell types, including prohibitin 1 on microglial cells for CHIKV, α1β1 integrin on fibroblasts for RRV, and natural resistance–associated macrophage protein 2 (NRAMP2) on fibroblasts for SINV (45–47). CD147 also was recently identified as a potential entry factor for CHIKV and displays structural similarities to MXRA8, suggesting a preferred structure for entry (48). However, these studies have not been validated extensively or repeated in vivo, so their impact on tropism and the proinflammatory cytokine and cellular response remains unclear. The future identification of additional attachment and entry receptors for different alphaviruses may refine our understanding of cellular tropism, virus-induced inflammation, and mechanisms of pathogenesis and possibly enable the design of targeted therapeutics that block virus entry and infection.
INNATE IMMUNE RESPONSES TO ALPHAVIRUSES
Pattern Recognition Receptors
Host pattern recognition receptors (PRRs) can sense the RNA genome of alphaviruses and trigger antiviral responses. Endosomal Toll-like receptors (TLRs)—including TLR3, which detects double-stranded RNA (dsRNA), and TLR7 and TLR8, which recognize single-stranded RNA (ssRNA)—and their downstream signaling molecules can be triggered during entry of virus or uptake of cellular debris (49). Once in the cell cytoplasm, the RIG-I-like receptors RIG-I and MDA5, which recognize dsRNA, and cyclic GMP-AMP (cGAMP) synthase (cGAS), which senses endogenous or foreign DNA, are stimulated. This leads to the activation of downstream adaptor and signaling molecules [TRIF, MyD88, mitochondrial antiviral-signaling protein (MAVS), and stimulator of interferon genes (STING)], resulting in translocation of interferon regulatory factors (IRFs) into the nucleus and induction of type I interferons and proinflammatory cytokines and chemokines (49–51). While the majority of studies have been performed with arthritogenic alphaviruses, given that they share RNA architectural features with encephalitic alphaviruses, both likely use common pathways of viral recognition (Figure 4).
Figure 4.
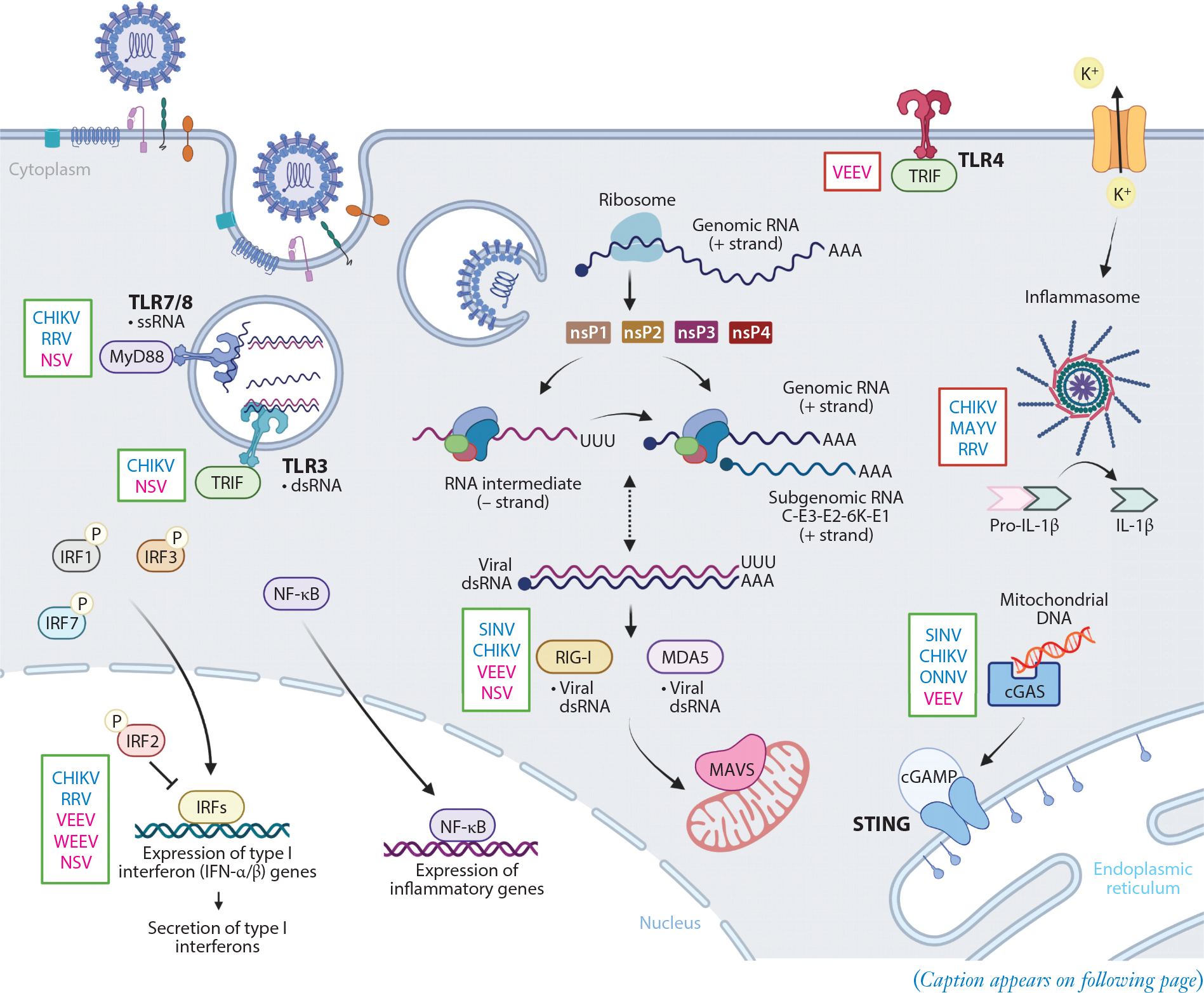
Overview of innate immune signaling pathways impacting alphavirus infection. Alphavirus entry is mediated by attachment factors and receptors at the host cell surface. Once internalized in an endosome, alphaviruses undergo fusion of the viral envelope with the endosomal membrane to release nucleocapsid and positive-strand viral genomic RNA into the cytoplasm. This RNA is translated to generate nonstructural proteins (nsP1–4) that form an initial viral replication complex to synthesize a negative-strand RNA intermediate. The viral replication complex will then switch to generate positive-strand genomic RNA and (positive-strand) subgenomic RNA from these negative-strand RNA intermediates. Subgenomic RNA encodes the structural polyproteins (C-E3-E2–6K-E1), and these are translated and processed in the endoplasmic reticulum before delivery to the cell surface to encapsulate genomic RNA during virion budding. PRRs at the cell surface (TLR4) and within the endosome (TLR3, TLR7, and TLR8) will recognize PAMPs and signal through distinct adaptor proteins (e.g., MyD88 and TRIF). TLR activation, along with secondary signaling through potassium efflux, can activate the inflammasome. Transcription of viral RNA in the cytoplasm leads to formation of dsRNA, which can activate cytoplasmic PRRs, RIG-I and MDA5, to interact with MAVS on the mitochondrial membrane. cGAS can recognize endogenous or foreign DNA released during infection, leading to activation of STING. These recognition events initiate translocation of IRFs and NF-κB into the nucleus and regulation of type I interferons and proinflammatory gene expression. Innate immune pathways that confer presumed protective (green box) or pathological (red box) responses to arthritogenic (blue font) or encephalitic (pink font) alphavirus infection are indicated to the left of each represented signaling molecule. Abbreviations: cGAMP, cyclic GMP-AMP; cGAS, cGAMP synthase; CHIKV, chikungunya virus; dsRNA, double-stranded RNA; IRF, interferon regulatory factor; MAVS, mitochondrial antiviral-signaling protein; MAYV, Mayaro virus; NF-κB, nuclear factor kappa B; NSV, neuroadapted SINV; ONNV, o’nyong-nyong virus; PAMP, pathogen-associated molecular pattern; PRR, pattern recognition receptor; RRV, Ross River virus; SINV, Sindbis virus; ssRNA, single-stranded RNA; STING, stimulator of interferon genes; TLR, Toll-like receptor, VEEV, Venezuelan equine encephalitis virus; WEEV, western equine encephalitis virus. Adapted from images created with BioRender.com.
TLRs.
Most TLRs signal through MyD88, but TLR4 and TLR3 signal through TRIF instead. Single-nucleotide polymorphism analysis of CHIKV-infected patients revealed that TLR3 (rs6552950), TLR7 (rs179010, rs5741880, rs3853839), and TLR8 (rs3764879) polymorphisms were associated with disease severity, implicating these TLRs in alphavirus pathogenesis (52, 53). A deficiency in TLR3 in the context of CHIKV infection resulted in increased viremia and dissemination, reduced disease, and increased macrophage and neutrophil recruitment to joint-associated tissue, which was corroborated by experiments with mice lacking the TLR3 adaptor molecule, TRIF (52, 54). Bone marrow chimera studies showed that TLR3 signaling on hematopoietic cells controls viremia, whereas TLR3 signaling on nonhematopoietic cells is needed to limit disease (52). Nevertheless, more research may be warranted. A conflicting study suggested no role for TLR3 in control of CHIKV replication (22). Loss of TLR7 or signaling through its adaptor molecule, MyD88, during CHIKV infections increased viremia at early time points (55). A lack of MyD88 signaling resulted in increased CHIKV infection in Ly6Chi monocytes, but this translated to only a mild increase in foot swelling (54, 55). In comparison, RRV infection of Tlr7−/− or Myd88−/− mice resulted in enhanced clinical disease and muscle damage; however, loss of these adaptor molecules did not impact early viral load but rather delayed virus clearance in blood and muscle (56).
TLR signaling impacts encephalitic alphavirus survival through modulation of cellular immunity or BBB permeability rather than direct control of viral replication. During infection of neuroadapted SINV (NSV), the lack of TLR3, TLR7, and TLR9 did not impact CNS infection but increased time to death and reduced the number of CD11b+ myeloid cells within the brain and spinal cord (57), suggesting TLR signaling modulated chemokine expression or survival of CD11b+ cells in the CNS. In contrast, TLR4 signaling was detrimental to infection with an attenuated VEEV strain. While TLR4 signaling did not alter the viral load in the CNS, it did increase BBB permeability through increased expression of proteins associated with endothelial cell integrity and leukocyte trafficking, including MMP-9, MMP-2, ICAM-1, CCL2, and IFN-γ (58). Lack of TLR4 signaling also increased the number of B cells in the brain, suggesting an enhanced antibody response, although this was not evaluated (58).
Inflammasome.
The NLRP3 inflammasome is a multiprotein complex that results in the secretion of IL-1β and IL-18. Stimulation of various receptors, including TLRs, induces expression of the inactive forms of NLRP3, procaspase-1, pro-IL-1β, and pro-IL-18 (59). Secondary signaling through potassium efflux or reactive oxygen species initiates the cascade to oligomerize NLRP3 with the adaptor protein, ASC, and effector, caspase-1, which activates the inflammasome (59, 60). Alphavirus infection can activate the NLRP3 inflammasome, resulting in production of IL-1β and IL-18 (60, 61). In vitro studies identified human dermal fibroblasts and murine bone marrow–derived macrophages as agents of inflammasome activity and IL-1β secretion following CHIKV or MAYV infection (60, 62, 63). The lack of NLRP3 or caspase-1/11 during MAYV infection resulted in a slight increase in clinical disease with fewer monocytes and neutrophils but more NK cells infiltrating joint-associated tissues compared to wild-type mice (60). Since NLRP3 or caspase-1/11 activity did not alter viral burden in this model, it can be inferred that the difference in clinical disease is associated with changes in immune cell recruitment (60). In CHIKV-infected patients, NLRP3, IL-18, ASC, and caspase-1 mRNA expression in peripheral blood mononuclear cells and IL-1β levels in serum were increased and correlated with the high viremia seen during acute infection (61). In mice, CHIKV infection induced expression of NLRP3 and caspase-1 in ankle tissues, and treatment with an NLRP3 or caspase-1 inhibitor reduced foot swelling and osteoclastogenesis without altering viral burden in musculoskeletal tissues (61). As similar reductions in clinical disease and bone loss were observed with the NLRP3 inhibitor in the context of RRV infection, NLRP3 inflammasome activation and signaling likely contribute to pathogenesis (61).
RIG-I-like receptors.
In the cytoplasm, replication of the alphavirus genome produces dsRNA intermediates that can bind to and activate RIG-I and MDA5, leading to an interaction with MAVS on the mitochondrial membrane. These events trigger a signaling cascade resulting in IRF3 nuclear translocation and induction of type I interferons, proinflammatory cytokines, and chemokines. It is less clear whether alphavirus RNAs signal primarily through RIG-I or MDA5 (64–67). An in vitro ectopic expression screen of interferon-stimulated genes (ISGs) identified RIG-I and MDA5 as inhibitors of alphavirus replication (68). Both RIG-I and MDA5 induced expression of IFN-β following infection of immortalized mouse fibroblast cells with mutants of VEEV or SINV that do not inhibit host translation following infection (65). However, another study identified MDA5 as the key inducer of type I interferon following SINV infection in mouse fibroblasts, but not in bone marrow–derived macrophages (66). For CHIKV, ectopic expression of RIG-I reduced viral replication and infection (67). Analysis of the CHIKV genome binding to RIG-I identified the 3′ untranslated region as the targeted region (67). In contrast, CHIKV infection in Rig-I−/− or Mda5−/− mice showed no difference in viral burden compared to wild-type mice, whereas Mavs−/− mice had increased viremia and foot swelling (22, 54). Similarly, NSV caused increased mortality and viral burden in the brain and spinal cord of Mavs−/− mice (69). These in vivo studies suggest a redundant role for RIG-I and MDA5 in restricting alphavirus infection.
cGAS-STING.
The cGAS-STING signaling pathway, which detects endogenous or nonself DNA in the cytoplasm, also has been shown to reduce alphavirus infection (51, 70). DNA binding to cGAS produces cGAMP, which binds to and activates STING, resulting in IRF3 nuclear translocation and type I interferon production (50). Ectopic expression of cGAS in STAT1−/− fibroblasts inhibited infection with SINV, ONNV, VEEV, and CHIKV (70). A genetic deficiency of cGAS or STING in Raw 264.7 myeloid cells enhanced replication of CHIKV, and Stinggt/gt loss-of-function mouse embryonic fibroblasts sustained a large increase in viral load (51). Loss of STING signaling during CHIKV infection resulted in increased viral burden; foot swelling; and mRNA expression of cytokines and chemokines, including that encoded by Cxcl10 (71). Consistent with increased levels of Cxcl10, more F4/80+ or Ly6C+ cells were present in the feet of Stinggt/gt mice (71). Although alphavirus RNA is not believed to engage cGAS directly, CHIKV infection and the resulting cellular injury can lead to translocation of nuclear or mitochondrial DNA into the cytoplasm. This DNA can then bind and stimulate cGAS (51).
Type I Interferon Induction and Response
Engagement of PRRs triggers the nuclear translocation of IRFs and NF-κB transcription factors, resulting in expression of proinflammatory cytokines and chemokines and type I interferons. Signaling of type I interferons in an autocrine and paracrine manner induces ISG expression that primes the antiviral state, which can block multiple stages of the alphavirus life cycle, including entry, translation, replication, and virion morphogenesis.
Interferon regulatory factors.
Activation of IRFs (IRF1, IRF3, IRF5, and IRF7) through PRR signaling and recruitment of adaptor molecules initiates IRF homo- or heterodimerization and translocation into the nucleus. In the nucleus, the IRF dimers bind to DNA and regulate the expression of type I interferons and ISGs (72). Additionally, IRF1 and IRF7 are activated by IFN-β signaling, resulting in a positive-feedback loop for ISG expression and IFN-α production (73, 74). Signaling through IRF1, IRF3, or IRF7 is implicated in the control of tropism and disease caused by alphaviruses. CHIKV infection of mice deficient in IRF1 resulted in increased infection of muscle cells and expression of the neutrophil and granulocyte chemoattractants CXCL1, CXCL2, CCL3, and CCL4, which correlated with recruitment of neutrophils and eosinophils and foot swelling (75). Similar results with RRV infection in Irf1−/− mice corroborated findings that muscle cell infection modulates inflammation (75). Although in-depth studies are lacking for the encephalitic alphaviruses, IRF1 expression reduced mortality during VEEV infections, suggesting a protective role (76).
IRF3 and IRF7 provide protective functions during alphavirus infection. In the absence of IRF3 and IRF7, CHIKV infection led to a lethal shock syndrome associated with a massive proinflammatory cytokine response (54). Bone marrow chimera experiments showed that expression of IRF3 and IRF7 in either hematopoietic or nonhematopoietic cells was sufficient to prevent mortality, suggesting that PRR signaling and type I interferon production could occur in both cell types (77). While a deficiency of either IRF3 or IRF7 enhanced CHIKV-mediated foot swelling and VEEV mortality, IRF7 expression was critical to the control of viral replication and production of type I interferons in vivo (54, 77, 78). In analogous studies, NSV-infected Irf7−/− mice had increased viral burden in the CNS and an accelerated time to death, with reduced type I interferons compared to wild-type mice (57, 78, 79). Consistent with in vivo results, macrophages and DCs required IRF7 to produce type I interferons following VEEV infection, further validating the concept that myeloid cells are the main producers of type I interferons (78). In contrast, WEEV infection of human neuroblastoma cells expressing a dominant negative IRF3 mutant showed that this transcription factor is critical for induction of type I interferon and control of viral replication (80). Overall, these results suggested a cell type–specific requirement of IRF3 and IRF7.
IRF2 negatively regulates type I interferon signaling through suppression of IRF1 and limits mortality after encephalitic alphavirus infection (81). Irf2−/− mice infected with NSV or an attenuated VEEV strain showed increased lethality compared to wild-type mice (81, 82). For NSV infection, lethality is associated with reduced B cells and virus-specific IgG in the brain, which resulted in higher viral burden in the CNS (81). Additionally, IRF2 expression increased inducible nitric oxide synthase (iNOS) expression in the brain during VEEV infection (82). Based on in vitro studies, microglia cells likely produce iNOS following VEEV infection, and thus a lack of iNOS in infected Irf2−/− mice would result in reduced nitric oxide and increased viral burden (82).
Type I interferon signaling is critical for protection against arthritogenic and encephalitic alphaviruses. Administration of IFN-α during VEEV or CHIKV infection reduced disease by limiting viral replication (83, 84). Likewise, alphavirus infection in mice lacking the type I interferon receptor IFNAR uniformly resulted in enhanced viral replication and dissemination and rapid death (22, 76, 85, 86). For the arthritogenic alphaviruses, specifically CHIKV, type I interferon signaling through IFNAR on nonhematopoietic cells was required to prevent lethality (22). When the individual contributions of IFN-α and IFN-β were dissected during CHIKV infection, both IFN-α and IFN-β reduced foot swelling (87). However, lack of IFN-α-enhanced virus dissemination and IFN-β deficiency increased neutrophil recruitment to the musculoskeletal tissue (87). These results are similar to those of earlier studies using lymphocytic choriomeningitis virus. IFN-β expression did not impact early virus replication, but blockade of IFN-β enhanced the host response, specifically the virus-specific T cell responses (88). Thus, these studies further validated distinct immunomodulatory and antiviral roles of specific type I interferons.
Interferon-stimulated genes and control of alphaviruses.
Interferon binding to the IF-NAR1/IFNAR2 heterodimeric receptor activates the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway to induce the transcription of ISGs that control alphavirus infection (89). Alternatively, some ISGs can be induced directly following PRR activation (89). While hundreds of ISGs have been identified as differentially expressed or regulated during alphavirus infection, a limited number have demonstrated inhibitory activity against the arthritogenic and encephalitic alphaviruses (Table 1). Protein kinase R (PKR) and 2′,5′-oligoadenylate synthetase (OAS) proteins are expressed constitutively at low levels in cells and then increased following alphavirus infection (90, 91). PKR recognizes dsRNA and then phosphorylates eIF2α, leading to inhibition of translation (90). In cell culture, PKR induction in fibroblasts reduced replication of SFV, SINV, and CHIKV and delayed protein translation during SFV infection (92–94). Nonetheless, there are discrepancies in findings about the role of PKR for translation inhibition during CHIKV infection. One group suggested that CHIKV-mediated translation inhibition was PKR-independent, whereas another reported the opposite (93, 95). This disparity could be related to differences in the experimental method of altering PKR expression and the method of analysis. The first group used small interfering RNAs to silence PKR and analyzed puromycin incorporation into de novo polypeptide chains by immunoblotting. While there was substantial knockdown, it was not complete (93). The other group used genetically deficient PKR−/− mouse embryonic fibroblasts and evaluated puromycin labeling of de novo proteins via confocal microscopy (95). Ostensibly in agreement with both studies, SINV infection uses both PKR-dependent and PKR-independent mechanisms of translation inhibition (94). PKR activation upregulates GADD34 (growth arrest and DNA damage–inducible protein 34), which is a phosphatase that negatively regulates eukaryotic initiation factor 2α (eIF2α) (96). Gadd34−/− neonatal mice quickly succumbed to CHIKV infection and showed increased viral load in noncanonical target tissues, such as the heart, which correlated with reduced levels of type I interferons (95). Similarly, PKR−/− mice had prolonged SFV infection in the brain (92). OAS also recognizes dsRNA, which activates RNase L and leads to mRNA or viral RNA degradation (90). The large OAS isoform OAS3 restricted replication of CHIKV, SFV, and SINV (91, 97). Unlike in Ifnar1−/− mice, where SINV infection resulted in 100% mortality, Rnasel−/− or PKR−/− mice were protected from lethal SINV challenge, indicating other factors contribute to type I interferon–mediated protection (98). ISG20 is an interferon-induced endonuclease that preferentially targets ssRNA for degradation (99). However, during alphavirus infection, ISG20 inhibits translation, and not degradation, of the viral RNA genome through upregulation of other ISGs through IRF3 (100). In cell culture, ectopic expression of ISG20 reduced replication of SINV, CHIKV, and VEEV (100, 101). However, the absence of ISG20 during CHIKV or virulent VEEV infection did not impact clinical disease or lethality, but there was increased lethality and clinical disease with an attenuated strain of VEEV (100).
Table 1.
Function of interferon-stimulated genes against alphaviruses
| ISG | Viruses | In vitro studies | In vivo studies | References |
|---|---|---|---|---|
| PKR | SFV SINV CHIKV |
Human PKR siRNA KD in foreskin fibroblasts: ■ No change in cellular translation or type I IFN levels (CHIKV) |
PKR−/− mouse SFV: ■ No death ■ Delayed viral clearance in brain SINV: ■ 100% survival |
92–95, 98 |
|
Mouse PKR−/− MEFs or PKR siRNA KD in NIH-3T3 cells: ■ ↑ Infection/viral titer (SFV, CHIKV, SINV) ■ ↓ Type I IFNs (SFV, CHIKV) ■ ↓ Cellular translation (CHIKV) ■ Prolonged infection (SINV) PKR−/− BMDCs: ■ ↑ Viral titer (SINV) ■ ↑ Replication or translation (SINV) Dominant-negative PKR mutant in NIH-3T3 cells: ■ Partial ↓ cellular translation (SINV) ■ No change in viral titers (SINV) | ||||
| GADD34 | CHIKV |
Mouse Gadd34−/− MEFs: ■ ↑ Viral titer (CHIKV) ■ ↓ IFN-β (CHIKV) |
Gadd34−/− neonate mouse CHIKV: ■ ↑ Lethality ■ ↑ Viral load ■ ↓ Type I IFNs |
95 |
| OAS | SFV SINV CHIKV |
Human Oas3−/− or Rnasel−/− A549 cells: ■ No total RNA degradation (SINV) ■ ↑ Viral titers (SINV) Ectopic expression of OAS3 in HeLa cells: ■ ↓ Viral titers (CHIKV, SINV, SFV) ■ ↓ Viral RNA accumulation (CHIKV) Mouse Rnasel−/− BMDCs: ■ No change in viral titer (SINV) ■ ↓ Replication or translation (SINV) |
Rnasel−/− mouse SINV: ■ 100% survival |
91, 97 |
| ISG20 | SINV CHIKV VEEV |
Mouse Ectopic expression in MEFs: ■ ↓ Replication (SINV) ■ ↓ Viral titers (CHIKV, VEEV) ■ ↓ Translation (CHIKV) |
Mouse SINV encoding ISG20 (neonate): ■ ↑ Survival Isg20−/− mouse CHIKV (neonate): ■ No change in survival VEEV: ■ Virulent strain—no change in survival ■ Attenuated strain—↑ lethality and disease |
100, 101 |
| Bst2/Tetherin | SFV CHIKV VEEV |
Human Ectopic expression in 293T and HeLa cells: ■ ↓ Viral titer (SFV, CHIKV) ■ ↓ Virus release (SFV, CHIKV) Rodent Bst2−/− MEFs and macrophages: ■ ↑ Viral titers in the presence of type I IFNs (CHIKV) NIH-3T3 or BHK-21 cells infected with VEEV expressing Bst2: ■ ↓ Virus release (VEEV) |
Bst2−/− mouse CHIKV: ■ ↑ Viral burden ■ ↓ Type I IFNs, IFN-γ, and CD40 expression |
102–105 |
| RSAD2/Viperin | SINV CHIKV |
Mouse Ectopic expression in MEFs: ■ ↓ replication (SINV) Rsad2−/− tail fibroblasts: ■ ↑ Viral antigen and load (CHIKV) ■ ↑ Type I IFN expression (CHIKV) |
Mouse SINV encoding Viperin (neonate): ■ ↑ Survival Rsad2−/− mouse CHIKV (young): ■ ↑ Foot titer early ■ ↑ Viremia ■ ↑ Cytokines, chemokines, and inflammation ■ ↑ F4/80+ cells and monocyte recruitment ■ CD4+ T cell–mediated disease with ↑ IFN-γ |
101, 106, 107 |
| IFITM | CHIKV VEEV SFV ONNV MAYV |
Human Loss of IFITM3 activity in HeLa cells: ■ ↑ Infection (CHIKV, MAYV) ■ ↑ Virus spread (CHIKV) Ectopic expression of IFITM1 or IFITM2 in HeLa cells: ■ ↓ Infection (CHIKV, MAYV) Mouse Ifitm3−/− MEFs: ■ ↓ Infection (CHIKV, VEEV, SFV, ONNV) ■ ↓ Viral fusion (CHIKV) |
Ifitm3−/− mouse CHIKV (young): ■ ↑ Foot swelling ■ ↑ Tissue titers and macrophage infection ■ ↑ Inflammatory cytokines VEEV: ■ ↑ Mortality ■ ↑ Early CNS titers |
108, 109 |
| PLZF | SFV |
Mouse Zbtb16−/− MEFs: ■ ↓ Infection Zbtb16−/− BMMs: ■ ↓ PLZF-dependent ISGs: Oas1g, Viperin, Ifit2, Cxcl10 Zbtb16−/− NK cells: ■ ↓ Effector function |
Zbtb16−/− mouse ■ ↑ Tissue titers ■ No change in serum IFN levels ↓ PLZF-dependent ISGs: ■ Oas1g, Viperin, Ifit2, Cxcl10 ■ ↓ NK cell effector function ■ ↓ Granzyme B |
110 |
| ZC3HAV1/ZAP | SINV SFV RRV VEEV |
Human ZC3HAV1−/− 293T cells: ■ Cofactor TR1M25 silenced ■ ↑ Viral replication (SINV) siRNA KD in Huh-7 cells: ■ ↑ Viral replication (SINV) Rodent ZAP-transduced fibroblasts: ■ ↓ Viral replication (SINV, SFV, RRV, VEEV) ■ ↓ Viral RNA translation (SINV) BHK-21 cells with ZAP overexpression: ■ + KD of 69 ISGs: ↑↑ viral replication (SINV) Mouse Overexpression in MEFs: ■ ↓ Virus release (SINV) ■ ↓↓ Virus release with IFN treatment (SINV) siRNA KD in MEFs: ■ ↑ Viral replication (SINV) |
Mouse Inoculation with SINV encoding ZAP (neonate): ■ ↑ Survival |
101, 111–114 |
| ISG15 | SINV CHIKV |
Mouse Ectopic expression in MEFs: ■ ↓ Virus release (SINV) siRNA KD in MEFs: ■ ↑ Viral replication (SINV) |
Mouse Inoculation with SINV encoding ISG15 (neonates): ■ No change in survival (SINV) IFN-α/βR−/− mouse inoculated with SINV encoding ISG15: ■ ↑ Survival (SINV) ■ ↓ Viral titers (SINV) ■ ↓ Immunopathology (SINV) ISG15−/− mouse ■ ↑ Lethality (SINV, CHIKV) ■ No change in tissue titers and type I IFN (CHIKV) ■ ↑ TNF-α, IL-1β, IL-6, CCL2, CCL3, CCL5 (CHIKV) |
101, 115–117 |
Abbreviations: BMDC, bone marrow–derived dendritic cell; BMM, bone marrow–derived macrophage; CHIKV chikungunya virus; CNS, central nervous system; GADD34, growth arrest and DNA damage–inducible protein 34; IFN, interferon; ISG, interferon-stimulated gene; ITIFM, interferon-induced transmembrane; KD, knockdown; MAYV, Mayaro virus; MEF, mouse embryonic fibroblast; OAS, 2′,5′-oligoadenylate synthetase; ONNV, o’nyong-nyong virus; PKR, protein kinase R; PLZF, promyelocytic zinc finger; RRV Ross River virus; SFV, Semliki Forest virus; SINV, Sindbis virus; siRNA, small interfering RNA; VEEV Venezuelan equine virus; VLP, virus-like particle; ZAP, zinc finger antiviral protein.
Bone marrow stromal cell antigen 2 (BST-2), also known as tetherin, prevents extracellular release of CHIKV and SFV, resulting in retention of virus particles on the surface of the cell (102, 103). Bst2−/− mice infected with CHIKV showed increased viral burden and reduced expression of IFN-γ and CD40L in the foot and draining lymph node; this result suggested that BST-2 may also impact other aspects of immunity (e.g., T cell activation or DC maturation) to control CHIKV spread (104). VEEV infection upregulated BST-2 in fibroblasts, and ectopic expression of BST-2 reduced viral loads (105). Similarly, ectopic expression of poly(ADP-ribose) polymerases 7, 10, or 12 (PARP7, 10, or 12) in BHK cells and fibroblasts reduced VEEV viral loads (105). Viperin, which is encoded by RSAD2, was highly expressed in peripheral blood mononuclear cells isolated from CHIKV-infected individuals and controlled SINV infection in vitro (101, 106). Mice lacking viperin showed increased viremia and joint inflammation during CHIKV infection and increased mortality during SINV infection (106, 107). Interferon-induced transmembrane 3 (IFITM3) is recruited to the late endosome and reduces infection of SFV, SINV, ONNV, VEEV, MAYV, and CHIKV (108, 109). Ifitm3−/− mice infected with CHIKV showed increased foot swelling and viral infection (108). A lack of IFITM3 expression enhanced infection in neutrophils and macrophages, thus altering the tropism of CHIKV (108). In analogous studies with VEEV, infected Ifitm3−/− mice developed higher viral load in the CNS at early time points and had increased mortality (108). Multiple interferon-induced zinc finger proteins also suppress alphavirus infection. Neonatal mice lacking promyelocytic leukemia zinc finger (PLZF), encoded by Zbtb16, and infected with SFV sustained markedly higher viral burdens with diminished NK cell activation and ISG responses (110). PLZF binds to the promoter regions and regulates expression of some ISGs, including IFIT2 and viperin, suggesting a potential protective mechanism in vivo (110). Additionally, zinc finger antiviral protein (ZAP) impaired translation of the SINV genome through interactions with tripartite motif-containing 25 (TRIM25) and controlled infection in neonatal mice (101, 111–114). ISG15 is induced rapidly by type I interferons following infection with CHIKV or SINV and reduces viral replication (101, 115, 116). The lack of ISG15 during SINV or CHIKV infection resulted in a shorter survival time (115, 117). Reconstitution of ISG15 protected mice from SINV replication and lethality (116, 117). Interestingly, CHIKV lethality in ISG15-deficient mice was not mediated through increased viral load or changes in type I interferon expression. This was likely due to the high levels of proinflammatory cytokines (115).
Cellular Immunity
As part of the innate immune response to alphavirus infection, hematopoietic cells are mobilized to sites of infection. These immune cells can be critical to control viral replication, promote clearance of infected cells, and stimulate a protective adaptive immune response. However, infiltrating and tissue-resident cells produce proinflammatory cytokines and chemokines that also can contribute to immunopathogenesis and disease progression. Here we discuss the protective and pathogenic cellular immune responses that occur during alphavirus infection.
Myeloid cells.
DCs, macrophages, monocytes, and neutrophils are rapidly recruited to the site of infection and contribute to control or pathogenesis of alphavirus infections. The activity of these cells in the context of immunity has been studied extensively during arthritogenic alphavirus infection. While there are limited studies directly evaluating myeloid cells during encephalitic alphavirus infection, chemokine and cytokine expression and analysis of cellular infiltrates suggest that myeloid cells also may contribute to protection or pathogenesis. Some alphaviruses, including SFV, VEEV, and SINV, replicate in myeloid DCs (mDCs), also referred to as conventional DCs, and/or Langerhans cells, which can traffic the virus to DLNs to promote virus dissemination (28, 29, 118, 119). Infection of myeloid DCs by RRV and Barmah Forest virus (BFV) is dictated by N-linked glycans present on the E2 protein (120). Unprocessed viruses displaying high-mannose glycans derived from mosquito cells infected mDCs more efficiently than viruses with complex glycans acquired from mammalian cells (120). Additionally, VEEV, SINV, CHIKV, MAYV, and RRV replicate in macrophages (21, 29, 78, 84, 121). While alphavirus RNA persistence is not extensively covered in this review, macrophages are proposed reservoirs for CHIKV RNA in joint-associated tissues (122). Beyond viral replication, subsets of recruited or tissue-resident myeloid cells produce type I interferons and cytokines that assist in viral clearance yet also promote inflammatory tissue damage.
Plasmacytoid dendritic cells (pDCs) specialize in type I interferon production predominantly following stimulation of the endosomal TLR7, sensing single-stranded RNA, or TLR9, sensing unmethylated CpG DNA, and signaling through IRF7 (123). The importance of pDCs during alphavirus infection has been evaluated only for arthritogenic alphaviruses. One study used mice with pDC-specific forced expression of IRF7 on an Irf3/Irf7−/− background, ensuring pDCs were the sole producers of type I interferons. This model established that production of type I interferons by pDCs was sufficient to protect Irf3/Irf7−/− mice from lethal CHIKV challenge and control viral replication (124). The type I interferons produced by pDCs during CHIKV or MAYV infection can limit infection of monocytes (55). Perturbation of pDC production of type I interferons through changes in microbiome composition resulted in increased CHIKV or MAYV infection of Ly6Chi monocytes (55). pDCs protected mice infected with an interferon-sensitive mutant of RRV from clinical disease and viral load in a MAVS-independent manner (125). These results were supported by studies showing that direct interaction of pDCs and infected epithelial cells or fibroblasts stimulated TLR7 signaling to induce type I interferon secretion (124). Since pDCs control arthritogenic alphavirus infections through IRF7-dependent production of type I interferons, and IRF7 signaling is important for the control of the encephalitic alphavirus VEEV (78), pDCs likely also produce type I interferons during encephalitic alphavirus infection. To date, there are no data showing that alphaviruses replicate in pDCs, which, in theory, could alter type I interferon expression and immune protection.
Shortly after alphavirus infection, chemoattractants such as CCL2, CXCL10, and MIF, presumably secreted by fibroblasts, endothelial and epithelial cells, and macrophages, recruit monocytes to the site of infection, where they differentiate based on the cytokine milieu (126–134). The presence of monocytes and macrophages in tissues is associated with increased expression of IL-6, TNF-α, IL-1β, and type I interferons and correlates with cytokine release following alphavirus infection of monocytes or macrophages in vitro (84, 135–137). During arthritogenic alphavirus infection, monocytes and macrophages accumulate in musculoskeletal tissues and serve both protective and pathogenic functions (137, 138). Clodronate liposome depletion of macrophages reduced skeletal muscle damage and clinical disease during RRV and CHIKV infection but did not impact viral burden in tissues (84, 135). Limiting monocyte and/or macrophage recruitment through a deficiency of MIF or CXCL10 expression also reduced clinical disease and immune cell infiltration during RRV, CHIKV, or ONNV infection (126, 131). However, Ccr2−/− mice, which lack the receptor for CCL2, infected with CHIKV developed severe foot swelling due to a compensatory influx of neutrophils and eosinophils in joint-associated tissue (139). Whereas complete abrogation of monocyte recruitment through CCR2 deletion leads to increased pathogenesis (presumably due to neutrophils), administration of bindarit, a small molecule that inhibits p50/p65 activation of the CCL2 promoter, during CHIKV or RRV infection reduced expression of proinflammatory cytokines. This decreased recruitment of CD11b+ and F4/80+ cells and alleviated clinical disease and muscle damage (127, 138, 140). These studies show that recruitment of monocytes and macrophages drives a proinflammatory response resulting in tissue damage and disease.
The resolution of acute musculoskeletal disease caused by alphaviruses is associated with an increase in alternative M2 macrophages expressing arginase 1 (Arg1) and chitinase-like protein 3 (also called Ym1) and in CX3CR1+ macrophages, which could support healing of the damaged tissue (130, 141). Indeed, CX3CR1-deficient mice showed increased disease and muscle damage following RRV infection, consistent with a protective role of these cells (130). However, specific depletion of Arg1 in monocytes, macrophages, and neutrophils did not alter acute infection but rather enhanced clearance of RRV at late time points (e.g., 14 and 21 days after inoculation) and increased tissue recovery, indicating that Arg1+ macrophages and neutrophils prolong infection and tissue damage (141).
The impact of myeloid cells on encephalic alphavirus infection has been less extensively characterized. However, some studies do provide insight into their possible roles during infection. VEEV infection induces expression of CCL2, MIF, and CXCL10 in the brain, suggesting that monocytes and/or macrophages are recruited into the brain (128, 129, 142). Indeed, CX3CR1+CCR2+ monocytes enter the brain at early time points following intranasal VEEV infection, and this correlated with increased levels of TNF-α and IL-1β (16). These cytokines can destabilize the BBB, and indeed, monocytes were localized near areas of BBB permeability (16, 143, 144). Since VEEV-infected myeloid cells are one of the main producers of type I interferons in cell culture, the recruitment of these cells to the brain may be protective (78). Contrasting protective and pathological roles have been proposed for neutrophils during encephalitic alphavirus infection. Infection of cynomolgus macaques with VEEV resulted in neutropenia, whereas EEEV infection produced neutrophilia (145). Future studies are needed to determine whether these differences in neutrophil mobilization might be caused by differential type I interferon production in VEEV- or EEEV-infected myeloid cells.
NK cells.
NK cells are rapidly activated and recruited to the site of viral infections. NK cells mediate clearance of infected cells through release of cytolytic perforin and granzyme molecules, secretion of IFN-γ, and interaction of death receptors on cell targets with Fas ligand (146, 147). Activated NK cells are detectable in the blood and musculoskeletal tissues following CHIKV or RRV infection in humans and mice and in the brains of SFV-, SINV-, or VEEV-infected mice (148–153). A pathogenic role of NK cells has been proposed during arthritogenic alphavirus infection, though this finding may be CHIKV genotype dependent (154). NK cell depletion during a lethal challenge with an attenuated VEEV strain resulted in protection of mice, and adoptive transfer of NK cells into depleted mice resulted in increased lethality (155). NK cell–depleted mice had reduced levels of IL-12p40, granulocyte colony-stimulating factor (G-CSF), and RANTES; higher levels of TNF-α; and equivalent IFN-γ expression in the brain compared to control mice (155). Since the levels of IFN-γ were similar between the groups, this suggests there was increased T cell activity in the NK cell–depleted mice, which may have contributed to the protection observed (155). Though the absence of NK cells during SFV infection prolonged time to death, the mice succumbed to infection at a higher rate than controls (152). The clinical disease associated with NK cell depletion after SFV infection was not typical (e.g., paralysis), suggesting that NK cells are pathogenic in the brain but may have protective roles at other infection sites (152). These studies suggest that NK cells may be pathogenic during alphavirus infection, but their relative contribution to disease can vary between the different viruses.
γδ T cells.
The most abundant skin-resident T lymphocytes, γδ T cells, constitute a first line of defense in the immune response at the local site of alphavirus infection (156). These cells lack MHC restriction and do not require conventional processing to react to antigens. They have a critical role in cytotoxicity, cytokine secretion, and promoting DC maturation (157, 158). In CHIKV infection, γδ T cells play a protective role. CHIKV infection in mouse footpads prompted an increase in γδ T cells in the foot and adjacent popliteal lymph nodes (159). An absence of γδ T cells yields increased monocyte infiltration into joints and higher levels of the proinflammatory mediators IFN-γ, CCL2, and CXCL9. This leads to increased tissue damage and disease severity (160). The role of γδ T cells in controlling CHIKV infection in humans remains unclear (161). Indeed, many functions of γδ T cells are unknown, particularly in the CNS (162). The early antiviral response to encephalitic alphavirus infection in the brain may be independent of γδ T cells, since γδ T cell–deficient mice showed no change in survival compared to wild-type mice following VEEV and SINV challenge (163). VEEV persisted in the brains of γδ T cell–deficient mice for up to four weeks in the absence of clinical disease, suggesting these cells function in clearance (163).
ADAPTIVE IMMUNE RESPONSES TO ACUTE ALPHAVIRUS INFECTION
Compared to the extensive work on innate immunity to alphaviruses, studies exploring the roles of adaptive immunity in modulating alphavirus infection, pathogenesis, and disease severity are more limited. A hallmark of acute alphavirus infection is rapid viral dissemination from a local site of replication to distant target tissues (11). During this initial phase, CD8+ T cells, CD4+ T cells, and B cells become activated in the context of antigen-presenting cells and cytokine stimulation. Antigen-experienced cells migrate from lymphoid tissues to sites of infection to contribute to local inflammation, control of replication, and clearance for some, but not all, alphaviruses (84, 164–166). In this section, we review differences in lymphocyte contributions to the acute immune response against arthritogenic and encephalitic alphaviruses.
CD8+ T Cell Responses
Studies examining the functions of CD8+ T cells in alphavirus infection have identified limited tissue-specific roles in controlling replication of some alphavirus species. For others, the contribution of CD8+ T cells to protective immunity is uncertain. During acute alphavirus infection, CD8+ T cells become activated and express markers such as CD69, CD107a, granzyme B, and perforin (167, 168). Infiltrating activated CD8+ T cell numbers increase in the circulation and in tissues targeted by arthritogenic (169, 170) and encephalitic (163) alphaviruses. One primary function of CD8+ T cells is direct cytolysis of virus-infected cells and production of antiviral and immunomodulatory proteins (171). Studies in mice suggest that CD8+ T cells mediate damage to neuronal cells in the CNS in the context of SFV infection. SFV infects oligodendrocytes, which upregulate MHC-I antigens, making them targets for cytotoxic killing (172). Depletion of CD8+ T cells reduced inflammation and eliminated demyelinating lesions in mice infected with SFV, confirming their pathogenic role (173, 174). For neurotropic alphaviruses like NSV and VEEV, the latter of which primarily infects neurons, CD8+ T cells do not target neurons directly, possibly because of the paucity of MHC-I expressed in these cells. Instead, CD8+ T cells may protect the host via noncytolytic mechanisms of viral clearance (175, 176). CD8+ T cells are not absolutely required for clearance of NSV and VEEV, since they were eventually cleared, though more slowly, in CD8+ T cell–deficient mice (163, 176). However, recovery from NSV and VEEV infections in B cell–deficient mice required CD8+ and CD4+ T cells (175, 177), and adoptive transfer of primed T cells prevented lethal VEEV-induced encephalitis in TCRαβ−/− mice (163, 178). Studies suggest CD8+ T cells may act in concert with other lymphocytes (e.g., CD4+ T cells and γδ T cells) to secrete cytokines (TNF-α and IFN-γ) that aid in clearance of intracellular viral RNA, though the antiviral proteins responsible for this control remain poorly characterized (176, 179–183). Studies with NSV showed the CD8+ T cell–dependent clearance from neurons is localized in the brain stem and spinal cord, but not in the cerebral cortex (180, 184). This phenotype was attributed to differences in MHC-I surface expression and differential susceptibility of neuronal subpopulations to IFN-γ-mediated clearance (176, 184). In comparison, CD8+ T cells seem to have less antiviral effect in joint-associated tissues in the context of infection by the arthritogenic alphaviruses CHIKV and MAYV (168, 185–188). The mechanisms by which CHIKV and other arthritogenic alphaviruses evade the CD8+ T cell response could be due to impaired antigen presentation by infected dermal cells, lack of migration or accessibility of CD8+ T cells to infected cells, or an immunosuppressive microenvironment within specific target tissues (26, 168). This may explain why CHIKV infection persists in joint-associated tissues (24, 26, 122, 150, 189, 190). Analogous studies with RRV suggested that CD8+ T cells control RRV infection in muscle but not in joint tissues (191, 192). Different immune environments among target tissues may have different influences on the immune control of arthritogenic alphaviruses by CD8+ T cells.
CD4+ T Cell Responses
Naive CD4+ T cells are activated after MHC-II-restricted antigen presentation and, depending on the cytokine milieu, differentiate into one of several effector T helper (Th) subsets—Th1, Th2, or Th17 cells—or regulatory T cells (Tregs) (193). Each CD4+ T cell lineage is distinguished by its cytokine production profile, which can modulate the acute response to infection via activation, enhancement, and/or suppression of innate and adaptive immune components. Though our understanding of CD4+ T cells during alphavirus infection remains limited, studies have shown they can function in protection and/or pathogenesis depending on the alphavirus.
Th1 cells.
In animal models of alphavirus pathogenesis, virus-specific CD4+ T cells migrate to infected sites in peripheral organs and the CNS (25, 126, 164, 194). These cells express the transcription factor T-bet and produce high levels of IFN-γ, consistent with a Th1 subset of CD4+ T cells (84, 164, 185, 195). Th1 cells mediate control of intracellular pathogens by recruiting and activating myeloid cells to sites of infection (196–199). During acute CHIKV infection, Th1 cells contribute to the pathogenesis of arthritis (150, 166, 185). Mice with genetic or acquired deficiencies of MHC-II or CD4+ T cells developed minimal or no joint pathology when challenged with CHIKV, with little impact on tissue viral RNA levels (84, 185, 200). Adoptive transfer of CD4+ T cells from CHIKV-infected donors into infected TCRαβ−/− mice restored joint swelling (185). Therapies targeting CD4+ T cells, such as fingolimod, which blocks lymphocyte exit from lymphoid organs (201), and anti-CTLA-4 blocking antibodies (202), reduced joint pathology in CHIKV-infected mice. Though IFN-γ expression levels are elevated in Th1 cells during CHIKV infection, this cytokine does not seem to contribute to CD4+ T cell–mediated joint swelling (150), possibly because CHIKV interferes with downstream JAK/STAT signaling to promote its replication (203). Instead, Th1 cells might mediate arthritis through production of granzyme A, a serine protease involved in T cell–mediated cytolysis (204). Accordingly, mice deficient in granzyme A and infected with CHIKV did not develop joint swelling (205).
The few studies of Th1 cells in neurotropic alphavirus infection have reported conflicting results regarding their role in disease pathogenesis. This may be due to differences in animal models used, including variations in host genetic background, route of virus inoculation, and inherent pathogenicity of challenge strains. In studies using VEEV, IFN-γ-expressing CD4+ T cells promoted disease resolution in the CNS. Adoptive transfer of primed CD4+ T cells into TCRαβ−/− mice conferred protection against lethal intranasal VEEV challenge (163, 178). One study evaluated a nonlethal strain of VEEV in B cell–deficient mice and showed recovery from infection, except when CD4+ and CD8+ T cells were depleted. The CD4+ compartment contributed to the majority of the T cell–associated antiviral activity and produced more IFN-γ and had higher levels of degranulation activity than CD8+ T cells (177). Possible mechanisms of Th1-mediated VEEV clearance from the CNS include direct cytotoxic effects and IFN-γ-mediated activation of protective monocytes and microglia (177). In NSV infections of mice, CD4+ T cells have both pathogenic and protective effects (175, 195, 206) that may be potentiated by their expression of IFN-γ (180). Understanding the functional roles of CD4+ T cells and the cytokines they produce in alphavirus encephalitis remains a complex problem that likely will require studies in mice with conditional cytokine deletions.
Th2 cells.
The Th2 subset of CD4+ T cells contributes to the response against extracellular pathogens by producing cytokines (e.g., IL-4, IL-5, IL-10, and IL-13) that modulate humoral immunity (207). In general, few studies have reported on the effects of Th2 cells and their cytokines on alphavirus pathogenesis, and those that have are focused mostly on CHIKV (208). Th2 cytokines can be measured in the joint tissues of CHIKV-infected humans, and higher levels of some Th2 cytokines (e.g., IL-13) are associated with delayed disease resolution and prolonged musculoskeletal symptoms (209–212). In mice, the T cell response is shifted from Th2 skewing early in infection and toward a Th1 bias later in the response (154, 212), which appears to contrast with that seen in humans (209). In human CHIKV infections, it is speculated that the enhanced Th2 response is an attempt to curtail damaging proinflammatory Th1 cell activity (209, 213).
Th17 cells and Tregs.
Differentiation of naive CD4+ cells into Th17 or Tregs is promoted by tumor growth factor β (TGF-β) in the context of additional proinflammatory signals. In the presence of IL-6 or IL-12, CD4+ T cells differentiate into Th17 cells, which have a pathogenic effect in arthritogenic and encephalitic alphavirus infections. The cytokines secreted by Th17 cells, including IL-17, IL-22, IL-23, and granulocyte-macrophage colony-stimulating factor (GM-CSF), are implicated in neutrophil recruitment to infected musculoskeletal tissues (214, 215) and linked to bone erosion during CHIKV infection (216). In NSV infection, Th17 cells have been associated with fatal encephalomyelitis in the absence of IFN-γ expression (217), although the effector functions causing disease remain unclear. Plausible explanations include Th17 cell production of IL-17, which contributes to autoimmune encephalitis (218), and GM-CSF and IL-22, which can cause neuronal damage through activation of microglial cells, recruitment of myeloid cells to the CNS, and BBB disruption (219, 220). Th17 cells can also directly target neurons, which express the IL-17 receptor under physiological stress (221).
In the absence of proinflammatory signals, TGF-β promotes differentiation of naive CD4+ T cells into Tregs (222, 223). Tregs produce the anti-inflammatory cytokines IL-10 and TGF-β, which suppress activity of immune cells and confer a protective role in arthritogenic and encephalitic alphavirus infection (222, 224). Selective expansion of Tregs can downregulate costimulation by antigen-presenting DCs and limit pathogenic CD4+ T cell–mediated joint swelling (224). Interestingly, MAYV infection induces a higher level of Tregs than CHIKV does during acute infection. This may be one reason why there are more asymptomatic cases of MAYV infection than CHIKV infection (225–227). Most studies describing the function of Tregs in alphavirus encephalitis have focused on NSV. Tregs are implicated as a source of IL-10 in the regulatory immune response to NSV infection in the brain (228), and deficiencies in IL-10 accelerate fatal disease (206). In comparative studies of BALB/c mice that are resistant to fatal NSV and susceptible C57BL/6 mice, there are more Tregs producing IL-10 in the former (206). The exact mechanism by which Tregs and IL-10 protect against NSV-induced CNS disease remains unclear.
B Cell Responses
During alphavirus infection, B cells produce virus-specific antibodies that clear virus from the bloodstream. B cells and antibodies also have important roles in the clearance of arthritogenic and encephalitic alphaviruses from target tissues and modulate disease progression (229, 230). Infection of B cell–deficient mice with CHIKV (230) or VEEV (177) resulted in persistent infection in the joint or brain, respectively. Resolution of infection and disease depended on both antibody neutralizing activity and effector functions, including antibody-mediated cellular cytotoxicity, complement activation, and virus opsonization (231). Indeed, passive transfer of monoclonal antibodies or immune serum protected against arthritogenic and encephalitic alphavirus infection and disease (232–240). Protective antibodies interfered with virus attachment and entry, endosomal fusion, virion assembly, virus egress, and cell-to-cell spread (231, 238, 241–243).
Neutralizing IgM responses early in infection are associated with reduced viral load and recovery in models of CHIKV and SINV infection (244). However, antiviral IgM persistence is associated with chronic disease and a failure to clear alphavirus infection (245–247). Natural IgM antibodies in the sera of mice can recognize CHIKV and also contribute to protection, although this has not been confirmed in humans (208, 248). In humans, high levels of CHIKV replication early during infection triggered production of anti-CHIKV IgG3 antibodies that promoted disease resolution (249). In C57BL/6 mice, CHIKV infection largely induced IgG2c antibodies, which efficiently engaged complement components and activating Fcγ receptors. During alphavirus infection, CD4+ T cell help is important for antibody isotype switching in B cells. When MHC-II−/− mice, deficient in CD4+ T cells, were infected with SFV, infectious virus was initially cleared at the same rate as in wild-type mice. However, early levels of IgM and IgG2b were lower, levels of SFV RNA in the brain were higher, and infectious virus was detected one month after infection (250). Interestingly, the antibody-mediated control of CHIKV infection can occur independently of CD4+ T cell help. Cd4−/− mice infected with CHIKV still controlled infection despite lower levels of neutralizing antibodies. Similarly, in MHC-II−/− mice infected with CHIKV, class switching from IgM to IgG still occurred in the absence of T cell help (200). Thus, CD4+ T cell–independent antibody responses can be protective in arthritogenic alphavirus infection.
Long-term control of virus replication in the CNS is associated with infiltration and retention of B cells that secrete virus-specific antibodies (251). High levels of neutralizing antibodies, however, were insufficient for eliminating NSV and SFV from the brain during acute infection (250). Initial clearance of infectious NSV from the brains of mice with severe combined immune deficiency by hyperimmune serum was followed by reemergence of infectious virus (252). This was also seen in arthritogenic alphavirus infection. Treatment of CHIKV-infected Rag1−/− mice with exogenous neutralizing monoclonal antibodies eliminated infectious virus from circulation, but as antibody levels waned, infectious virus reemerged (200, 230). During infection, pathogenic strains of CHIKV can subvert or delay induction of humoral immune responses by triggering a rapid influx of neutrophils and monocytes into the draining lymph node that disrupts follicle organization and impairs B cell responses to CHIKV (253).
CONCLUSIONS
Alphaviruses remain a global concern as climate change, international travel, and urbanization expand the mosquito vector distribution, vector exposure, and potential virus adaptation to mosquito species. This pattern has already been observed with CHIKV, which now causes explosive outbreaks worldwide. A single–amino acid mutation in the E1 structural protein of CHIKV enhanced its replication in Aedes albopictus mosquitoes and enabled epidemic spread into new regions of the world (254). VEEV, EEEV, and WEEV are emerging viruses and classified as biodefense threats owing to their high lethality following aerosol transmission. While there are no available counter-measures against alphavirus infection, multiple promising vaccine approaches are being evaluated in clinical trials, including virus-like particle, live-attenuated strains, or viral-vectored platforms against CHIKV, VEEV, EEEV, or WEEV (NCT03879603, NCT00584805, NCT02466750, and NCT03051386) (255–258). Furthermore, numerous studies have documented in vivo protection against arthritogenic and encephalitic alphaviruses using monoclonal antibodies and reduced infection in vitro with repurposed drugs and small-molecule inhibitors (232, 237, 239, 242, 259, 260). As our understanding of the similarities and differences in pathogenesis of alphaviruses grows (Figure 5), new approaches and broadly effective immune-targeted therapies may become possible in the not-so-distant future.
Figure 5.
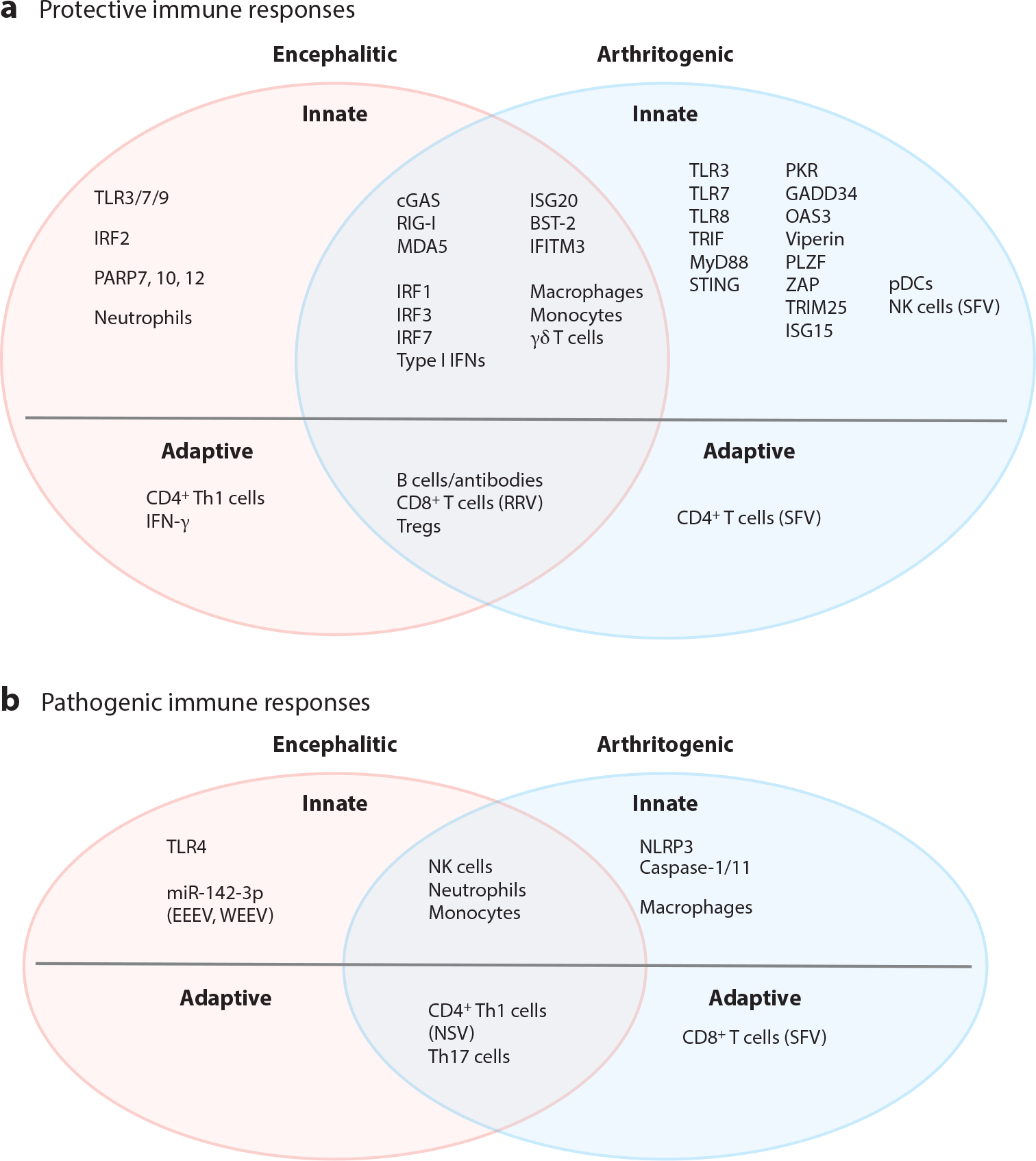
Protective and pathogenic components of the immune response to alphavirus infections. Pattern recognition receptors, type I interferons, interferon-stimulated genes, and immune cells involved in the innate and adaptive immune responses to alphaviruses that are encephalitic (pink ovals), arthritogenic (light blue ovals), or both encephalitic and arthritogenic (mauve overlap) are categorized by their known or presumed (a) protective and/or (b) pathogenic contributions. Specific viruses are indicated when the type of immune response differs compared to other viruses in the respective group. Abbreviations: BST-2, bone marrow stromal cell antigen 2; cGAS, cyclic GMP-AMP synthase; EEEV, eastern equine encephalitis virus; GADD34, growth arrest and DNA damage–inducible protein 34; IFITM3, interferon-induced transmembrane 3; IRF1, interferon regulatory factor 1; ISG20, interferon-stimulated gene 20; NK, natural killer; NSV, neuroadapted Sindbis virus; OAS3, 2′,5′-oligoadenylate synthetase 3; PARP7, poly(ADP-ribose) polymerase 7; pDC, plasmacytoid dendritic cell; PKR, protein kinase R; PLZF, promyelocytic leukemia zinc finger; RRV, Ross River virus; SFV, Semliki Forest virus; STING, stimulator of interferon genes; Th1, T helper type 1; TLR3, Toll-like receptor 3; Treg, regulatory T cell; TRIM25, tripartite motif-containing 25; WEEV, western equine encephalitis virus; ZAP, zinc finger antiviral protein.
ACKNOWLEDGMENTS
Our research was supported in part by the Intramural Research Program of the NIH and by contracts and grants from the NIH (T32 AI007172, R01 AI143673, U19 AI142790, R01AI164653, R01AI152484, R01AI141436, R01AI127513) and the Defense Threat Reduction Agency (HDTRA1–15-1–0013).
Footnotes
DISCLOSURE STATEMENT
M.S.D. is a consultant for Inbios, Vir Biotechnology, Senda Biosiences, and Carnival Corporation and is on the Scientific Advisory Boards of Moderna and Immunome. The Diamond laboratory has received funding support in sponsored research agreements from Moderna, Vir Biotechnology, and Emergent BioSolutions.
LITERATURE CITED
- 1.Weaver SC, Barrett AD. 2004. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat. Rev. Microbiol. 2:789–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leung JY, Ng MM, Chu JJ. 2011. Replication of alphaviruses: a review on the entry process of alphaviruses into cells. Adv. Virol. 2011:249640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li L, Jose J, Xiang Y, Kuhn RJ, Rossmann MG. 2010. Structural changes of envelope proteins during alphavirus fusion. Nature 468:705–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weaver SC, Winegar R, Manger ID, Forrester NL. 2012. Alphaviruses: population genetics and determinants of emergence. Antivir. Res. 94:242–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levi LI, Vignuzzi M. 2019. Arthritogenic alphaviruses: a worldwide emerging threat? Microorganisms 7:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Endy TP. 2020. Viral febrile illnesses and emerging pathogens. In Hunter’s Tropical Medicine and Emerging Infectious Diseases, ed. Ryan ET, Hill DR, Solomon T, Aronson NE, Endy TP, pp. 325–50. London: Elsevier. 10th ed. [Google Scholar]
- 7.Zacks MA, Paessler S. 2010. Encephalitic alphaviruses. Vet. Microbiol. 140:281–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guzmán-Terán C, Calderón-Rangel A, Rodriguez-Morales A, Mattar S. 2020. Venezuelan equine encephalitis virus: the problem is not over for tropical America. Ann. Clin. Microbiol. Antimicrob. 19:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yactayo S, Staples JE, Millot V, Cibrelus L, Ramon-Pardo P. 2016. Epidemiology of chikungunya in the Americas. J. Infect. Dis. 214:S441–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mehta R, Gerardin P, de Brito CAA, Soares CN, Ferreira MLB, Solomon T. 2018. The neurological complications of chikungunya virus: a systematic review. Rev. Med. Virol. 28:e1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atkins GJ. 2013. The pathogenesis of alphaviruses. ISRN Virol. 2013:861912 [Google Scholar]
- 12.Ronca SE, Dineley KT, Paessler S. 2016. Neurological sequelae resulting from encephalitic alphavirus infection. Front. Microbiol. 7:959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zaid A, Burt FJ, Liu X, Poo YS, Zandi K, et al. 2021. Arthritogenic alphaviruses: epidemiological and clinical perspective on emerging arboviruses. Lancet Infect. Dis. 21:e123–33 [DOI] [PubMed] [Google Scholar]
- 14.Seyler T, Hutin Y, Ramanchandran V, Ramakrishnan R, Manickam P, Murhekar M. 2010. Estimating the burden of disease and the economic cost attributable to chikungunya, Andhra Pradesh, India, 2005–2006. Trans. R. Soc. Trop. Med. Hyg. 104:133–38 [DOI] [PubMed] [Google Scholar]
- 15.Forrester NL, Wertheim JO, Dugan VG, Auguste AJ, Lin D, et al. 2017. Evolution and spread of Venezuelan equine encephalitis complex alphavirus in the Americas. PLOS Negl. Trop. Dis. 11:e0005693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cain MD, Salimi H, Gong Y, Yang L, Hamilton SL, et al. 2017. Virus entry and replication in the brain precedes blood-brain barrier disruption during intranasal alphavirus infection. J. Neuroimmunol. 308:118–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein RS, Garber C, Funk KE, Salimi H, Soung A, et al. 2019. Neuroinflammation during RNA viral infections. Annu. Rev. Immunol. 37:73–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ronca SE, Smith J, Koma T, Miller MM, Yun N, et al. 2017. Mouse model of neurological complications resulting from encephalitic alphavirus infection. Front. Microbiol. 8:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suhrbier A, Jaffar-Bandjee M-C, Gasque P. 2012. Arthritogenic alphaviruses—an overview. Nat. Rev. Rheumatol. 8:420–29 [DOI] [PubMed] [Google Scholar]
- 20.Haese NN, Broeckel RM, Hawman DW, Heise MT, Morrison TE, Streblow DN. 2016. Animal models of chikungunya virus infection and disease. J. Infect. Dis. 214:S482–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sourisseau M, Schilte C, Casartelli N, Trouillet C, Guivel-Benhassine F, et al. 2007. Characterization of reemerging chikungunya virus. PLOS Pathog. 3:e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schilte C, Couderc T, Chretien F, Sourisseau M, Gangneux N, et al. 2010. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J. Exp. Med. 207:429–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matusali G, Colavita F, Bordi L, Lalle E, Ippolito G, et al. 2019. Tropism of the chikungunya virus. Viruses 11:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ozden S, Huerre M, Riviere J-P, Coffey LL, Afonso PV, et al. 2007. Human muscle satellite cells as targets of chikungunya virus infection. PLOS ONE 2:e527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noret M, Herrero L, Rulli N, Rolph M, Smith PN, et al. 2012. Interleukin 6, RANKL, and osteoprotegerin expression by chikungunya virus–infected human osteoblasts. J. Infect. Dis. 206:455–57 [DOI] [PubMed] [Google Scholar]
- 26.Young AR, Locke MC, Cook LE, Hiller BE, Zhang R, et al. 2019. Dermal and muscle fibroblasts and skeletal myofibers survive chikungunya virus infection and harbor persistent RNA. PLOS Pathog. 15:e1007993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lentscher AJ, McCarthy MK, May NA, Davenport BJ, Montgomery SA, et al. 2020. Chikungunya virus replication in skeletal muscle cells is required for disease development. J. Clin. Investig. 130:1466–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacDonald GH, Johnston RE. 2000. Role of dendritic cell targeting in Venezuelan equine encephalitis virus pathogenesis. J. Virol. 74:914–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gardner CL, Burke CW, Tesfay MZ, Glass PJ, Klimstra WB, Ryman KD. 2008. Eastern and Venezuelan equine encephalitis viruses differ in their ability to infect dendritic cells and macrophages: impact of altered cell tropism on pathogenesis. J. Virol. 82:10634–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vogel P, Kell WM, Fritz DL, Parker MD, Schoepp RJ. 2005. Early events in the pathogenesis of eastern equine encephalitis virus in mice. Am. J. Pathol. 166:159–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trobaugh DW, Gardner CL, Sun C, Haddow AD, Wang E, et al. 2014. RNA viruses can hijack vertebrate microRNAs to suppress innate immunity. Nature 506:245–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trobaugh DW, Sun C, Bhalla N, Gardner CL, Dunn MD, Klimstra WB. 2019. Cooperativity between the 3′ untranslated region microRNA binding sites is critical for the virulence of eastern equine encephalitis virus. PLOS Pathog. 15:e1007867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levitt NH, Miller HV, Edelman R. 1979. Interaction of alphaviruses with human peripheral leukocytes: in vitro replication of Venezuelan equine encephalomyelitis virus in monocyte cultures. Infect. Immun. 24:642–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang R, Kim AS, Fox JM, Nair S, Basore K, et al. 2018. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 557:570–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang R, Earnest JT, Kim AS, Winkler ES, Desai P, et al. 2019. Expression of the Mxra8 receptor promotes alphavirus infection and pathogenesis in mice and Drosophila. Cell Rep. 28:2647–58.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma H, Kim AS, Kafai NM, Earnest JT, Shah AP, et al. 2020. LDLRAD3 is a receptor for Venezuelan equine encephalitis virus. Nature 588:308–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, et al. 2015. Proteomics: tissue-based map of the human proteome. Science 347:1260419. [DOI] [PubMed] [Google Scholar]
- 38.Sjöstedt E, Zhong W, Fagerberg L, Karlsson M, Mitsios N, et al. 2020. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367(6482):eaay5947. [DOI] [PubMed] [Google Scholar]
- 39.Hum. Protein. Atlas. 2021. LDLRAD3. Human Protein Atlas, updated November 18. https://www.proteinatlas.org [Google Scholar]; 39a. Clark LE, Clark SA, Lin C, Liu J, Coscia A, et al. 2022. VLDLR and ApoER2 are receptors for multiple alphaviruses. Nature 602:475–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holmes AC, Basore K, Fremont DH, Diamond MS. 2020. A molecular understanding of alphavirus entry. PLOS Pathog. 16:e1008876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gardner CL, Ebel GD, Ryman KD, Klimstra WB. 2011. Heparan sulfate binding by natural eastern equine encephalitis viruses promotes neurovirulence. PNAS 108:16026–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ashbrook AW, Burrack KS, Silva LA, Montgomery SA, Heise MT, et al. 2014. Residue 82 of the Chikungunya virus E2 attachment protein modulates viral dissemination and arthritis in mice. J. Virol. 88:12180–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silva LA, Khomandiak S, Ashbrook AW, Weller R, Heise MT, et al. 2014. A single-amino-acid polymorphism in Chikungunya virus E2 glycoprotein influences glycosaminoglycan utilization. J. Virol. 88:2385–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Byrnes AP, Griffin DE. 2000. Large-plaque mutants of Sindbis virus show reduced binding to heparan sulfate, heightened viremia, and slower clearance from the circulation. J. Virol. 74:644–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wintachai P, Wikan N, Kuadkitkan A, Jaimipuk T, Ubol S, et al. 2012. Identification of prohibitin as a Chikungunya virus receptor protein. J. Med. Virol. 84:1757–70 [DOI] [PubMed] [Google Scholar]
- 46.Linn ML, Eble JA, Lübken C, Slade RW, Heino J, et al. 2005. An arthritogenic alphavirus uses the α1β1 integrin collagen receptor. Virology 336:229–39 [DOI] [PubMed] [Google Scholar]
- 47.Rose PP, Hanna SL, Spiridigliozzi A, Wannissorn N, Beiting DP, et al. 2011. Natural resistance-associated macrophage protein is a cellular receptor for Sindbis virus in both insect and mammalian hosts. Cell Host Microbe 10:97–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Caluwé L, Coppens S, Vereecken K, Daled S, Dhaenens M, et al. 2021. The CD147 protein complex is involved in entry of chikungunya virus and related alphaviruses in human cells. Front. Microbiol. 12:615165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kawasaki T, Kawai T. 2014. Toll-like receptor signaling pathways. Front. Immunol. 5:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hopfner K-P, Hornung V 2020. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 21:501–21 [DOI] [PubMed] [Google Scholar]
- 51.Webb LG, Veloz J, Pintado-Silva J, Zhu T, Rangel MV, et al. 2020. Chikungunya virus antagonizes cGAS-STING mediated type-I interferon responses by degrading cGAS. PLOS Pathog. 16:e1008999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Her Z, Teng TS, Tan JJ, Teo TH, Kam YW, et al. 2015. Loss of TLR3 aggravates CHIKV replication and pathology due to an altered virus-specific neutralizing antibody response. EMBO Mol. Med. 7:24–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dutta SK, Tripathi A. 2017. Association of Toll-like receptor polymorphisms with susceptibility to chikungunya virus infection. Virology 511:207–13 [DOI] [PubMed] [Google Scholar]
- 54.Rudd PA, Wilson J, Gardner J, Larcher T, Babarit C, et al. 2012. Interferon response factors 3 and 7 protect against chikungunya virus hemorrhagic fever and shock. J. Virol. 86:9888–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Winkler ES, Shrihari S, Hykes BL Jr., Handley SA, Andhey PS, et al. 2020. The intestinal microbiome restricts alphavirus infection and dissemination through a bile acid-type I IFN signaling axis. Cell 182:901–18.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Neighbours LM, Long K, Whitmore AC, Heise MT. 2012. Myd88-dependent Toll-like receptor 7 signaling mediates protection from severe Ross River virus-induced disease in mice. J. Virol. 86:10675–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Esen N, Blakely PK, Rainey-Barger EK, Irani DN. 2012. Complexity of the microglial activation pathways that drive innate host responses during lethal alphavirus encephalitis in mice. ASN Neuro 4:207–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hollidge BS, Cohen CA, Akuoku Frimpong J, Badger CV, Dye JM, Schmaljohn CS. 2021. Toll-like receptor 4 mediates blood-brain barrier permeability and disease in C3H mice during Venezuelan equine encephalitis virus infection. Virulence 12:430–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Swanson KV, Deng M, Ting JP. 2019. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19:477–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Castro-Jorge LA, de Carvalho RVH, Klein TM, Hiroki CH, Lopes AH, et al. 2019. The NLRP3 inflammasome is involved with the pathogenesis of Mayaro virus. PLOS Pathog. 15:e1007934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen W, Foo SS, Zaid A, Teng TS, Herrero LJ, et al. 2017. Specific inhibition of NLRP3 in chikungunya disease reveals a role for inflammasomes in alphavirus-induced inflammation. Nat. Microbiol. 2:1435–45 [DOI] [PubMed] [Google Scholar]
- 62.Ekchariyawat P, Hamel R, Bernard E, Wichit S, Surasombatpattana P, et al. 2015. Inflammasome signaling pathways exert antiviral effect against Chikungunya virus in human dermal fibroblasts. Infect. Genet. Evol. 32:401–8 [DOI] [PubMed] [Google Scholar]
- 63.Soares-Schanoski A, Baptista Cruz N, de Castro-Jorge LA, de Carvalho RVH, Santos CA, et al. 2019. Systems analysis of subjects acutely infected with the Chikungunya virus. PLOS Pathog. 15:e1007880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rehwinkel J, Gack MU. 2020. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat. Rev. Immunol. 20:537–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Akhrymuk I, Frolov I, Frolova EI. 2016. Both RIG-I and MDA5 detect alphavirus replication in concentration-dependent mode. Virology 487:230–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burke CW, Gardner CL, Steffan JJ, Ryman KD, Klimstra WB. 2009. Characteristics of alpha/beta interferon induction after infection of murine fibroblasts with wild-type and mutant alphaviruses. Virology 395:121–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sanchez David RY, Combredet C, Sismeiro O, Dillies MA, Jagla B, et al. 2016. Comparative analysis of viral RNA signatures on different RIG-I-like receptors. eLife 5:e11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, et al. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wollish AC, Ferris MT, Blevins LK, Loo YM, Gale M Jr., Heise MT. 2013. An attenuating mutation in a neurovirulent Sindbis virus strain interacts with the IPS-1 signaling pathway in vivo. Virology 435:269–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, et al. 2014. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505:691–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Geng T, Lin T, Yang D, Harrison AG, Vella AT, et al. 2021. A critical role for STING signaling in limiting pathogenesis of chikungunya virus. J. Infect. Dis. 223:2186–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thompson CD, Matta B, Barnes BJ. 2018. Therapeutic targeting of IRFs: pathway-dependence or structure-based? Front. Immunol. 9:2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. 1998. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 441:106–10 [DOI] [PubMed] [Google Scholar]
- 74.Michalska A, Blaszczyk K, Wesoly J, Bluyssen HAR. 2018. A positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls type I and type II IFN responses. Front. Immunol. 9:1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nair S, Poddar S, Shimak RM, Diamond MS. 2017. Interferon regulatory factor 1 protects against chikungunya virus-induced immunopathology by restricting infection in muscle cells. J. Virol. 91:e01419–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grieder FB, Vogel SN. 1999. Role of interferon and interferon regulatory factors in early protection against Venezuelan equine encephalitis virus infection. Virology 257:106–18 [DOI] [PubMed] [Google Scholar]
- 77.Schilte C, Buckwalter MR, Laird ME, Diamond MS, Schwartz O, Albert ML. 2012. Cutting edge: independent roles for IRF-3 and IRF-7 in hematopoietic and nonhematopoietic cells during host response to chikungunya infection. J. Immunol. 188:2967. [DOI] [PubMed] [Google Scholar]
- 78.Bhalla N, Gardner CL, Downs SN, Dunn M, Sun C, Klimstra WB. 2019. Macromolecular synthesis shutoff resistance by myeloid cells is critical to IRF7-dependent systemic interferon alpha/beta induction after alphavirus infection. J. Virol. 93:e00872–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Esen N, Rainey-Barger EK, Huber AK, Blakely PK, Irani DN. 2014. Type-I interferons suppress microglial production of the lymphoid chemokine, CXCL13. Glia 62:1452–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Peltier DC, Lazear HM, Farmer JR, Diamond MS, Miller DJ. 2013. Neurotropic arboviruses induce interferon regulatory factor 3-mediated neuronal responses that are cytoprotective, interferon independent, and inhibited by Western equine encephalitis virus capsid. J. Virol. 87:1821–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li MM, Bozzacco L, Hoffmann HH, Breton G, Loschko J, et al. 2016. Interferon regulatory factor 2 protects mice from lethal viral neuroinvasion. J. Exp. Med. 213:2931–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schoneboom BA, Lee JS, Grieder FB. 2000. Early expression of IFN-alpha/beta and iNOS in the brains of Venezuelan equine encephalitis virus-infected mice. J. Interferon Cytokine Res. 20:205–15 [DOI] [PubMed] [Google Scholar]
- 83.Lukaszewski RA, Brooks TJ. 2000. Pegylated alpha interferon is an effective treatment for virulent Venezuelan equine encephalitis virus and has profound effects on the host immune response to infection. J. Virol. 74:5006–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gardner J, Anraku I, Le TT, Larcher T, Major L, et al. 2010. Chikungunya virus arthritis in adult wild-type mice. J. Virol. 84:8021–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE. 2000. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J. Virol. 74:3366–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Couderc T, Chrétien F, Schilte C, Disson O, Brigitte M, et al. 2008. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLOS Pathog. 4:e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cook LE, Locke MC, Young AR, Monte K, Hedberg ML, et al. 2019. Distinct roles of interferon alpha and beta in controlling chikungunya virus replication and modulating neutrophil-mediated inflammation. J. Virol. 94:e00841–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ng CT, Sullivan BM, Teijaro JR, Lee AM, Welch M, et al. 2015. Blockade of interferon beta, but not interferon alpha, signaling controls persistent viral infection. Cell Host Microbe 17:653–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schoggins JW. 2019. Interferon-stimulated genes: What do they all do? Annu. Rev. Virol. 6:567–84 [DOI] [PubMed] [Google Scholar]
- 90.Sadler AJ, Williams BR. 2008. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8:559–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li Y, Banerjee S, Wang Y, Goldstein SA, Dong B, et al. 2016. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. PNAS 113:2241–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barry G, Breakwell L, Fragkoudis R, Attarzadeh-Yazdi G, Rodriguez-Andres J, et al. 2009. PKR acts early in infection to suppress Semliki Forest virus production and strongly enhances the type I interferon response. J. Gen. Virol. 90:1382–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.White LK, Sali T, Alvarado D, Gatti E, Pierre P, et al. 2011. Chikungunya virus induces IPS-1-dependent innate immune activation and protein kinase R-independent translational shutoff. J. Virol. 85:606–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gorchakov R, Frolova E, Williams BR, Rice CM, Frolov I. 2004. PKR-dependent and -independent mechanisms are involved in translational shutoff during Sindbis virus infection. J. Virol. 78:8455–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Clavarino G, Cláudio N, Couderc T, Dalet A, Judith D, et al. 2012. Induction of GADD34 is necessary for dsRNA-dependent interferon-β production and participates in the control of Chikungunya virus infection. PLOS Pathog. 8:e1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brush MH, Weiser DC, Shenolikar S. 2003. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol. Cell. Biol. 23:1292–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bréhin AC, Casadémont I, Frenkiel MP, Julier C, Sakuntabhai A, Desprès P. 2009. The large form of human 2′,5′-oligoadenylate synthetase (OAS3) exerts antiviral effect against Chikungunya virus. Virology 384:216–22 [DOI] [PubMed] [Google Scholar]
- 98.Ryman KD, White LJ, Johnston RE, Klimstra WB. 2002. Effects of PKR/RNase L-dependent and alternative antiviral pathways on alphavirus replication and pathogenesis. Viral Immunol. 15:53–76 [DOI] [PubMed] [Google Scholar]
- 99.Nguyen LH, Espert L, Mechti N, Wilson DM 3rd. 2001. The human interferon- and estrogen-regulated ISG20/HEM45 gene product degrades single-stranded RNA and DNA in vitro. Biochemistry 40:7174–79 [DOI] [PubMed] [Google Scholar]
- 100.Weiss CM, Trobaugh DW, Sun C, Lucas TM, Diamond MS, et al. 2018. The interferon-induced exonuclease ISG20 exerts antiviral activity through upregulation of type I interferon response proteins. mSphere 3:e00209–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang Y, Burke CW, Ryman KD, Klimstra WB. 2007. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J. Virol. 81:11246–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jones PH, Maric M, Madison MN, Maury W, Roller RJ, Okeoma CM. 2013. BST-2/tetherin-mediated restriction of chikungunya (CHIKV) VLP budding is counteracted by CHIKV non-structural protein 1 (nsP1). Virology 438:37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ooi YS, Dubé M, Kielian M. 2015. BST2/tetherin inhibition of alphavirus exit. Viruses 7:2147–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mahauad-Fernandez WD, Jones PH, Okeoma CM. 2014. Critical role for bone marrow stromal antigen 2 in acute Chikungunya virus infection. J. Gen. Virol. 95:2450–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Atasheva S, Akhrymuk M, Frolova EI, Frolov I. 2012. New PARP gene with an anti-alphavirus function. J. Virol. 86:8147–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Teng TS, Foo SS, Simamarta D, Lum FM, Teo TH, et al. 2012. Viperin restricts chikungunya virus replication and pathology. J. Clin. Investig. 122:4447–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Carissimo G, Teo TH, Chan YH, Lee CY, Lee B, et al. 2019. Viperin controls chikungunya virus-specific pathogenic T cell IFNγ Th1 stimulation in mice. Life Sci. Alliance 2:e201900298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Poddar S, Hyde JL, Gorman MJ, Farzan M, Diamond MS. 2016. The interferon-stimulated gene IFITM3 restricts infection and pathogenesis of arthritogenic and encephalitic alphaviruses. J. Virol. 90:8780–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Franz S, Pott F, Zillinger T, Schüler C, Dapa S, et al. 2021. Human IFITM3 restricts chikungunya virus and Mayaro virus infection and is susceptible to virus-mediated counteraction. Life Sci. Alliance 4:e202000909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xu D, Holko M, Sadler AJ, Scott B, Higashiyama S, et al. 2009. Promyelocytic leukemia zinc finger protein regulates interferon-mediated innate immunity. Immunity 30:802–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li MMH, Lau Z, Cheung P, Aguilar EG, Schneider WM, et al. 2017. TRIM25 enhances the antiviral action of zinc-finger antiviral protein (ZAP). PLOS Pathog. 13:e1006145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bick MJ, Carroll JW, Gao G, Goff SP, Rice CM, MacDonald MR. 2003. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J. Virol. 77:11555–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Karki S, Li MM, Schoggins JW, Tian S, Rice CM, MacDonald MR. 2012. Multiple interferon stimulated genes synergize with the zinc finger antiviral protein to mediate anti-alphavirus activity. PLOS ONE 7:e37398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.MacDonald MR, Machlin ES, Albin OR, Levy DE. 2007. The zinc finger antiviral protein acts synergistically with an interferon-induced factor for maximal activity against alphaviruses. J. Virol. 81:13509–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Werneke SW, Schilte C, Rohatgi A, Monte KJ, Michault A, et al. 2011. ISG15 is critical in the control of Chikungunya virus infection independent of UbE1L mediated conjugation. PLOS Pathog. 7:e1002322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lenschow DJ, Giannakopoulos NV, Gunn LJ, Johnston C, O’Guin AK, et al. 2005. Identification of interferon-stimulated gene 15 as an antiviral molecule during Sindbis virus infection in vivo. J. Virol. 79:13974–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos NV, Lutz A, et al. 2007. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. PNAS 104:1371–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hidmark AS, McInerney GM, Nordström EK, Douagi I, Werner KM, et al. 2005. Early alpha/beta interferon production by myeloid dendritic cells in response to UV-inactivated virus requires viral entry and interferon regulatory factor 3 but not MyD88. J. Virol. 79:10376–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gardner JP, Frolov I, Perri S, Ji Y, MacKichan ML, et al. 2000. Infection of human dendritic cells by a Sindbis virus replicon vector is determined by a single amino acid substitution in the E2 glycoprotein. J. Virol. 74:11849–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shabman RS, Morrison TE, Moore C, White L, Suthar MS, et al. 2007. Differential induction of type I interferon responses in myeloid dendritic cells by mosquito and mammalian-cell-derived alphaviruses. J. Virol. 81:237–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cavalheiro MG, Costa LS, Campos HS, Alves LS, Assunção-Miranda I, Poian AT. 2016. Macrophages as target cells for Mayaro virus infection: involvement of reactive oxygen species in the inflammatory response during virus replication. An. Acad. Bras Cienc. 88:1485–99 [DOI] [PubMed] [Google Scholar]
- 122.Labadie K, Larcher T, Joubert C, Mannioui A, Delache B, et al. 2010. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J. Clin. Investig. 120:894–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Swiecki M, Colonna M. 2015. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 15:471–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Webster B, Werneke SW, Zafirova B, This S, Coléon S, et al. 2018. Plasmacytoid dendritic cells control dengue and Chikungunya virus infections via IRF7-regulated interferon responses. eLife 7:e34273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Haist KC, Carpentier KS, Davenport BJ, Morrison TE. 2021. Plasmacytoid dendritic cells mediate control of Ross River virus infection via a type I interferon-dependent, MAVS-independent mechanism. J. Virol. 95:e01538–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Herrero LJ, Sheng KC, Jian P, Taylor A, Her Z, et al. 2013. Macrophage migration inhibitory factor receptor CD74 mediates alphavirus-induced arthritis and myositis in murine models of alphavirus infection. Arthritis Rheum. 65:2724–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rulli NE, Rolph MS, Srikiatkhachorn A, Anantapreecha S, Guglielmotti A, Mahalingam S. 2011. Protection from arthritis and myositis in a mouse model of acute chikungunya virus disease by bindarit, an inhibitor of monocyte chemotactic protein-1 synthesis. J. Infect. Dis. 204:1026–30 [DOI] [PubMed] [Google Scholar]
- 128.Sharma A, Bhattacharya B, Puri RK, Maheshwari RK. 2008. Venezuelan equine encephalitis virus infection causes modulation of inflammatory and immune response genes in mouse brain. BMC Genom. 9:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sharma A, Maheshwari RK. 2009. Oligonucleotide array analysis of Toll-like receptors and associated signalling genes in Venezuelan equine encephalitis virus-infected mouse brain. J. Gen. Virol. 90:1836–47 [DOI] [PubMed] [Google Scholar]
- 130.Zaid A, Tharmarajah K, Mostafavi H, Freitas JR, Sheng KC, et al. 2020. Modulation of monocyte-driven myositis in alphavirus infection reveals a role for CX3CR1+ macrophages in tissue repair. mBio 11:e03353–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lin T, Geng T, Harrison AG, Yang D, Vella AT, et al. 2020. CXCL10 signaling contributes to the pathogenesis of arthritogenic alphaviruses. Viruses 12:1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Deshmane SL, Kremlev S, Amini S, Sawaya BE. 2009. Monocyte chemoattractant protein-1 (MCP-1): an overview. J. Interferon Cytokine Res. 29:313–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gregory JL, Morand EF, McKeown SJ, Ralph JA, Hall P, et al. 2006. Macrophage migration inhibitory factor induces macrophage recruitment via CC chemokine ligand 2. J. Immunol. 177:8072–79 [DOI] [PubMed] [Google Scholar]
- 134.Selvi E, Tripodi SA, Catenaccio M, Lorenzini S, Chindamo D, et al. 2003. Expression of macrophage migration inhibitory factor in diffuse systemic sclerosis. Ann. Rheum. Dis. 62:460–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lidbury BA, Rulli NE, Suhrbier A, Smith PN, McColl SR, et al. 2008. Macrophage-derived proinflammatory factors contribute to the development of arthritis and myositis after infection with an arthrogenic alphavirus. J. Infect. Dis. 197:1585–93 [DOI] [PubMed] [Google Scholar]
- 136.Assunção-Miranda I, Bozza MT, Da Poian AT. 2010. Pro-inflammatory response resulting from Sindbis virus infection of human macrophages: implications for the pathogenesis of viral arthritis. J. Med. Virol. 82:164–74 [DOI] [PubMed] [Google Scholar]
- 137.Haist KC, Burrack KS, Davenport BJ, Morrison TE. 2017. Inflammatory monocytes mediate control of acute alphavirus infection in mice. PLOS Pathog. 13:e1006748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rulli NE, Guglielmotti A, Mangano G, Rolph MS, Apicella C, et al. 2009. Amelioration of alphavirus induced arthritis and myositis in a mouse model by treatment with bindarit, an inhibitor of monocyte chemotactic proteins. Arthritis Rheum. 60:2513–23 [DOI] [PubMed] [Google Scholar]
- 139.Poo YS, Nakaya H, Gardner J, Larcher T, Schroder WA, et al. 2014. CCR2 deficiency promotes exacerbated chronic erosive neutrophil-dominated chikungunya virus arthritis. J. Virol. 88:6862–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen W, Foo SS, Taylor A, Lulla A, Merits A, et al. 2015. Bindarit, an inhibitor of monocyte chemotactic protein synthesis, protects against bone loss induced by chikungunya virus infection. J. Virol. 89:581–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stoermer KA, Burrack A, Oko L, Montgomery SA, Borst LB, et al. 2012. Genetic ablation of arginase 1 in macrophages and neutrophils enhances clearance of an arthritogenic alphavirus. J. Immunol. 189:4047–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Salimi H, Klein RS. 2019. Disruption of the blood-brain barrier during neuroinflammatory and neuroinfectious diseases. In Neuroimmune Diseases: From Cells to the Living Brain, ed. Mitoma H, Manto M, pp. 195–234. Cham, Switz.: Springer Int. [Google Scholar]
- 143.Pan W, Stone KP, Hsuchou H, Manda VK, Zhang Y, Kastin AJ. 2011. Cytokine signaling modulates blood-brain barrier function. Curr. Pharm. Des. 17:3729–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang Y, Jin S, Sonobe Y, Cheng Y, Horiuchi H, et al. 2014. Interleukin-1β induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLOS ONE 9:e110024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Smith DR, Schmaljohn CS, Badger C, Ostrowski K, Zeng X, et al. 2020. Comparative pathology study of Venezuelan, eastern, and western equine encephalitis viruses in non-human primates. Antivir. Res. 182:104875. [DOI] [PubMed] [Google Scholar]
- 146.Smyth MJ, Cretney E, Kelly JM, Westwood JA, Street SE, et al. 2005. Activation of NK cell cytotoxicity. Mol. Immunol. 42:501–10 [DOI] [PubMed] [Google Scholar]
- 147.Brandstadter JD, Yang Y. 2011. Natural killer cell responses to viral infection. J. Innate Immun. 3:274–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hazelton RA, Hughes C, Aaskov JG. 1985. The inflammatory response in the synovium of a patient with Ross River arbovirus infection. Aust. N. Z. J. Med. 15:336–39 [DOI] [PubMed] [Google Scholar]
- 149.Petitdemange C, Becquart P, Wauquier N, Béziat V, Debré P, et al. 2011. Unconventional repertoire profile is imprinted during acute chikungunya infection for natural killer cells polarization toward cytotoxicity. PLOS Pathog. 7:e1002268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hoarau J-J, Jaffar Bandjee M-C, Krejbich Trotot P, Das T, Li-Pat-Yuen G, et al. 2010. Persistent chronic inflammation and infection by chikungunya arthritogenic alphavirus in spite of a robust host immune response. J. Immunol. 184:5914. [DOI] [PubMed] [Google Scholar]
- 151.Schanoski AS, Le TT, Kaiserman D, Rowe C, Prow NA, et al. 2019. Granzyme A in chikungunya and other arboviral infections. Front. Immunol. 10:3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Alsharifi M, Lobigs M, Simon MM, Kersten A, Müller K, et al. 2006. NK cell-mediated immunopathology during an acute viral infection of the CNS. Eur. J. Immunol. 36:887–96 [DOI] [PubMed] [Google Scholar]
- 153.Griffin DE, Hess JL. 1986. Cells with natural killer activity in the cerebrospinal fluid of normal mice and athymic nude mice with acute Sindbis virus encephalitis. J. Immunol. 136:1841–45 [PubMed] [Google Scholar]
- 154.Teo TH, Her Z, Tan JJ, Lum FM, Lee WW, et al. 2015. Caribbean and La Réunion chikungunya virus isolates differ in their capacity to induce proinflammatory Th1 and NK cell responses and acute joint pathology. J. Virol. 89:7955–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Taylor K, Kolokoltsova O, Patterson M, Poussard A, Smith J, et al. 2012. Natural killer cell mediated pathogenesis determines outcome of central nervous system infection with Venezuelan equine encephalitis virus in C3H/HeN mice. Vaccine 30:4095–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hayday AC. 2000. γδ cells: a right time and a right place for a conserved third way of protection. Annu. Rev. Immunol. 18:975–1026 [DOI] [PubMed] [Google Scholar]
- 157.Born W, Cady C, Jones-Carson J, Mukasa A, Lahn M, O’Brien R. 1999. Immunoregulatory functions of gamma delta T cells. Adv. Immunol. 71:77–144 [PubMed] [Google Scholar]
- 158.Carding SR, Egan PJ. 2002. γδ T cells: functional plasticity and heterogeneity. Nat. Rev. Immunol. 2:336–45 [DOI] [PubMed] [Google Scholar]
- 159.Long KM, Ferris MT, Whitmore AC, Montgomery SA, Thurlow LR, et al. 2016. Gamma delta T cells play a protective role in chikungunya virus-induced disease. J. Virol. 90:433–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Fu YX, Roark CE, Kelly K, Drevets D, Campbell P, et al. 1994. Immune protection and control of inflammatory tissue necrosis by gamma delta T cells. J. Immunol. 153:3101. [PubMed] [Google Scholar]
- 161.Poh CM, Chan Y-H, Ng LFP. 2020. Role of T cells in chikungunya virus infection and utilizing their potential in anti-viral immunity. Front. Immunol. 11:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Born WK, Reardon CL, O’Brien RL. 2006. The function of γδ T cells in innate immunity. Curr. Opin. Immunol. 18:31–38 [DOI] [PubMed] [Google Scholar]
- 163.Paessler S, Yun NE, Judy BM, Dziuba N, Zacks MA, et al. 2007. Alpha-beta T cells provide protection against lethal encephalitis in the murine model of VEEV infection. Virology 367:307–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Morrison TE, Oko L, Montgomery SA, Whitmore AC, Lotstein AR, et al. 2011. A mouse model of chikungunya virus-induced musculoskeletal inflammatory disease: evidence of arthritis, tenosynovitis, myositis, and persistence. Am. J. Pathol. 178:32–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Torres-Ruesta A, Teo T-H, Chan Y-H, Rénia L, Ng LFP. 2020. Pathogenic Th1 responses in CHIKV-induced inflammation and their modulation upon Plasmodium parasites co-infection. Immunol. Rev. 294:80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wauquier N, Becquart P, Nkoghe D, Padilla C, Ndjoyi-Mbiguino A, Leroy EM. 2011. The acute phase of Chikungunya virus infection in humans is associated with strong innate immunity and T CD8 cell activation. J. Infect. Dis. 204:115–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dias CNS, Gois BM, Lima VS, Guerra-Gomes IC, Araújo JMG, et al. 2018. Human CD8 T-cell activation in acute and chronic chikungunya infection. Immunology 155:499–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Davenport BJ, Bullock C, McCarthy MK, Hawman DW, Murphy KM, et al. 2020. Chikungunya virus evades antiviral CD8+ T cell responses to establish persistent infection in joint-associated tissues. J. Virol. 94:e02036–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Miner JJ, Aw-Yeang HX, Fox JM, Taffner S, Malkova ON, et al. 2015. Chikungunya viral arthritis in the United States: a mimic of seronegative rheumatoid arthritis. Arthritis Rheumatol. 67:1214–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hoarau J-J, Gay F, Pellé O, Samri A, Jaffar-Bandjee M-C, et al. 2013. Identical strength of the T cell responses against E2, nsP1 and capsid CHIKV proteins in recovered and chronic patients after the epidemics of 2005–2006 in La Reunion Island. PLOS ONE 8:e84695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kägi D, Hengartner H. 1996. Different roles for cytotoxic T cells in the control of infections with cytopathic versus noncytopathic viruses. Curr. Opin. Immunol. 8:472–77 [DOI] [PubMed] [Google Scholar]
- 172.Fazakerley JK, Pathak S, Scallan M, Amor S, Dyson H. 1993. Replication of the A7(74) strain of Semliki Forest virus is restricted in neurons. Virology 195:627–37 [DOI] [PubMed] [Google Scholar]
- 173.Subak-Sharpe I, Dyson H, Fazakerley J. 1993. In vivo depletion of CD8+ T cells prevents lesions of demyelination in Semliki Forest virus infection. J. Virol. 67:7629–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fazakerley JK, Webb HE. 1987. Semliki Forest virus-induced, immune-mediated demyelination: adoptive transfer studies and viral persistence in nude mice. J. Gen. Virol. 68 (Part 2):377–85 [DOI] [PubMed] [Google Scholar]
- 175.Griffin DE. 2010. Recovery from viral encephalomyelitis: immune-mediated noncytolytic virus clearance from neurons. Immunol. Res. 47:123–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kimura T, Griffin DE. 2000. The role of CD8+ T cells and major histocompatibility complex class I expression in the central nervous system of mice infected with neurovirulent Sindbis virus. J. Virol. 74:6117–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Brooke CB, Deming DJ, Whitmore AC, White LJ, Johnston RE. 2010. T cells facilitate recovery from Venezuelan equine encephalitis virus-induced encephalomyelitis in the absence of antibody. J. Virol. 84:4556–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yun NE, Peng BH, Bertke AS, Borisevich V, Smith JK, et al. 2009. CD4+ T cells provide protection against acute lethal encephalitis caused by Venezuelan equine encephalitis virus. Vaccine 27:4064–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wesselingh SL, Levine B, Fox RJ, Choi S, Griffin DE. 1994. Intracerebral cytokine mRNA expression during fatal and nonfatal alphavirus encephalitis suggests a predominant type 2 T cell response. J. Immunol. 152:1289–97 [PubMed] [Google Scholar]
- 180.Binder GK, Griffin DE. 2001. Interferon-γ-mediated site-specific clearance of alphavirus from CNS neurons. Science 293:303–6 [DOI] [PubMed] [Google Scholar]
- 181.Tau G, Rothman P. 1999. Biologic functions of the IFN-γ receptors. Allergy 54:1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lampson LA. 1995. Interpreting MHC class I expression and class I/class II reciprocity in the CNS: reconciling divergent findings. Microsc. Res. Tech. 32:267–85 [DOI] [PubMed] [Google Scholar]
- 183.Mucke L, Oldstone M. 1992. The expression of major histocompatibility complex (MHC) class I antigens in the brain differs markedly in acute and persistent infections with lymphocytic choriomeningitis virus (LCMV). J. Neuroimmunol. 36:193–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Burdeinick-Kerr R, Wind J, Griffin DE. 2007. Synergistic roles of antibody and interferon in noncytolytic clearance of Sindbis virus from different regions of the central nervous system. J. Virol. 81:5628–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Teo T-H, Lum F-M, Claser C, Lulla V, Lulla A, et al. 2013. A pathogenic role for CD4+ T cells during chikungunya virus infection in mice. J. Immunol. 190:259. [DOI] [PubMed] [Google Scholar]
- 186.Webb EM, Azar SR, Haller SL, Langsjoen RM, Cuthbert CE, et al. 2019. Effects of Chikungunya virus immunity on Mayaro virus disease and epidemic potential. Sci. Rep. 9:20399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Choi H, Kudchodkar SB, Reuschel EL, Asija K, Borole P, et al. 2019. Protective immunity by an engineered DNA vaccine for Mayaro virus. PLOS Negl. Trop. Dis. 13:e0007042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chu H, Das SC, Fuchs JF, Suresh M, Weaver SC, et al. 2013. Deciphering the protective role of adaptive immunity to CHIKV/IRES a novel candidate vaccine against Chikungunya in the A129 mouse model. Vaccine 31:3353–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hawman DW, Stoermer KA, Montgomery SA, Pal P, Oko L, et al. 2013. Chronic joint disease caused by persistent Chikungunya virus infection is controlled by the adaptive immune response. J. Virol. 87:13878–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Messaoudi I, Vomaske J, Totonchy T, Kreklywich CN, Haberthur K, et al. 2013. Chikungunya virus infection results in higher and persistent viral replication in aged rhesus macaques due to defects in anti-viral immunity. PLOS Negl. Trop. Dis. 7:e2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Burrack KS, Montgomery SA, Homann D, Morrison TE. 2015. CD8+ T cells control Ross River virus infection in musculoskeletal tissues of infected mice. J. Immunol. 194:678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Fraser J 1986. Epidemic polyarthritis and Ross River virus disease. Clinics Rheum. Dis. 12:369–88 [PubMed] [Google Scholar]
- 193.Rosendahl Huber S, van Beek J, de Jonge J, Luytjes W, van Baarle D. 2014. T cell responses to viral infections—opportunities for peptide vaccination. Front. Immunol. 5:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Irani DN, Griffin DE. 1991. Isolation of brain parenchymal lymphocytes for flow cytometric analysis: application to acute viral encephalitis. J. Immunol. Methods 139:223–31 [DOI] [PubMed] [Google Scholar]
- 195.Rowell JF, Griffin DE. 2002. Contribution of T cells to mortality in neurovirulent Sindbis virus encephalomyelitis. J. Neuroimmunol. 127:106–14 [DOI] [PubMed] [Google Scholar]
- 196.Boehm U, Klamp T, Groot M, Howard J. 1997. Cellular responses to interferon-γ. Annu. Rev. Immunol. 15:749–95 [DOI] [PubMed] [Google Scholar]
- 197.Abbas AK, Murphy KM, Sher A. 1996. Functional diversity of helper T lymphocytes. Nature 383:787–93 [DOI] [PubMed] [Google Scholar]
- 198.Schroder K, Hertzog PJ, Ravasi T, Hume DA. 2004. Interferon-γ: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 75:163–89 [DOI] [PubMed] [Google Scholar]
- 199.Sallusto F, Lanzavecchia A, Mackay CR. 1998. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol. Today 19:568–74 [DOI] [PubMed] [Google Scholar]
- 200.Poo YS, Rudd PA, Gardner J, Wilson JA, Larcher T, et al. 2014. Multiple immune factors are involved in controlling acute and chronic chikungunya virus infection. PLOS Negl. Trop. Dis. 8:e3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Teo TH, Chan YH, Lee WW, Lum FM, Amrun SN, et al. 2017. Fingolimod treatment abrogates chikungunya virus-induced arthralgia. Sci. Transl. Med. 9:eaal1333. [DOI] [PubMed] [Google Scholar]
- 202.Miner JJ, Cook LE, Hong JP, Smith AM, Richner JM, et al. 2017. Therapy with CTLA4-Ig and an antiviral monoclonal antibody controls chikungunya virus arthritis. Sci. Transl. Med. 9:eaah3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Fros JJ, Liu WJ, Prow NA, Geertsema C, Ligtenberg M, et al. 2010. Chikungunya virus nonstructural protein 2 inhibits type I/II interferon-stimulated JAK-STAT signaling. J. Virol. 84:10877–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Hidalgo L, Einecke G, Allanach K, Halloran P. 2008. The transcriptome of human cytotoxic T cells: similarities and disparities among allostimulated CD4+ CTL, CD8+ CTL and NK cells. Am. J. Transplant. 8:627–36 [DOI] [PubMed] [Google Scholar]
- 205.Wilson JA, Prow NA, Schroder WA, Ellis JJ, Cumming HE, et al. 2017. RNA-seq analysis of chikungunya virus infection and identification of granzyme A as a major promoter of arthritic inflammation. PLOS Pathog. 13:e1006155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kulcsar KA, Baxter VK, Abraham R, Nelson A, Griffin DE, Dermody TS. 2015. Distinct immune responses in resistant and susceptible strains of mice during neurovirulent alphavirus encephalomyelitis. J. Virol. 89:8280–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Spellberg B, Edwards JE Jr. 2001. Type 1/type 2 immunity in infectious diseases. Clin. Infect. Dis. 32:76–102 [DOI] [PubMed] [Google Scholar]
- 208.Tanabe IS, Tanabe EL, Santos EC, Martins WV, Araújo IM, et al. 2018. Cellular and molecular immune response to chikungunya virus infection. Front. Cell. Infect. Microbiol. 8:345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Venugopalan A, Ghorpade RP, Chopra A. 2014. Cytokines in acute chikungunya. PLOS ONE 9:e111305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Chow A, Her Z, Ong EK, Chen J-M, Dimatatac F, et al. 2011. Persistent arthralgia induced by Chikungunya virus infection is associated with interleukin-6 and granulocyte macrophage colony-stimulating factor. J. Infect. Dis. 203:149–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Chaaitanya IK, Muruganandam N, Sundaram SG, Kawalekar O, Sugunan AP, et al. 2011. Role of proinflammatory cytokines and chemokines in chronic arthropathy in CHIKV infection. Viral Immunol. 24:265–71 [DOI] [PubMed] [Google Scholar]
- 212.Patil DR, Hundekar SL, Arankalle VA. 2012. Expression profile of immune response genes during acute myopathy induced by chikungunya virus in a mouse model. Microbes Infect. 14:457–69 [DOI] [PubMed] [Google Scholar]
- 213.Khan M, Dhanwani R, Rao PVL, Parida M. 2012. Subunit vaccine formulations based on recombinant envelope proteins of Chikungunya virus elicit balanced Th1/Th2 response and virus-neutralizing antibodies in mice. Virus Res. 167:236–46 [DOI] [PubMed] [Google Scholar]
- 214.Chen W, Foo SS, Sims NA, Herrero LJ, Walsh NC, Mahalingam S. 2015. Arthritogenic alphaviruses: new insights into arthritis and bone pathology. Trends Microbiol. 23:35–43 [DOI] [PubMed] [Google Scholar]
- 215.Sandquist I, Kolls J. 2018. Update on regulation and effector functions of Th17 cells. F1000Research 7:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Amdekar S, Parashar D, Alagarasu K. 2017. Chikungunya virus-induced arthritis: role of host and viral factors in the pathogenesis. Viral Immunol. 30:691–702 [DOI] [PubMed] [Google Scholar]
- 217.Baxter VK, Griffin DE. 2020. Interferon-gamma modulation of the local T cell response to alphavirus encephalomyelitis. Viruses 12:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, et al. 2006. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J. Immunol. 177:566–73 [DOI] [PubMed] [Google Scholar]
- 219.Wang T, Lee M-H, Choi E, Pardo-Villamizar CA, Lee SB, et al. 2012. Granzyme B-induced neurotoxicity is mediated via activation of PAR-1 receptor and Kv1. 3 channel. PLOS ONE 7(8):e43950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, et al. 2007. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 13:1173–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wang D-D, Zhao Y-F, Wang G-Y, Sun B, Kong Q-F, et al. 2009. IL-17 potentiates neuronal injury induced by oxygen–glucose deprivation and affects neuronal IL-17 receptor expression. J. Neuroimmunol. 212:17–25 [DOI] [PubMed] [Google Scholar]
- 222.Lee GR. 2018. The balance of Th17 versus Treg cells in autoimmunity. Int. J. Mol. Sci. 19:730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, et al. 2006. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441:235–38 [DOI] [PubMed] [Google Scholar]
- 224.Lee WW, Teo T-H, Her Z, Lum F-M, Kam Y-W, et al. 2015. Expanding regulatory T cells alleviates chikungunya virus-induced pathology in mice. J. Virol. 89:7893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Coimbra TLM, Santos CL, Suzuki A, Petrella S, Bisordi I, et al. 2007. Mayaro virus: imported cases of human infection in São Paulo state, Brazil. Rev. Inst. Med. Trop. São Paulo 49:221–24 [DOI] [PubMed] [Google Scholar]
- 226.Danillo Lucas Alves E, Fonseca BAL. 2018. Characterization of the immune response following in vitro mayaro and chikungunya viruses (Alphavirus, Togaviridae) infection of mononuclear cells. Virus Res. 256:166–73 [DOI] [PubMed] [Google Scholar]
- 227.Figueiredo CM, Neris RLS, Gavino-Leopoldino D, Silva MOL, Almeida JS, et al. 2019. Mayaro virus replication restriction and induction of muscular inflammation in mice are dependent on age, type-I interferon response, and adaptive immunity. Front. Microbiol. 10:2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kulcsar KA, Griffin DE. 2016. T cell-derived interleukin-10 is an important regulator of the Th17 response during lethal alphavirus encephalomyelitis. J. Neuroimmunol. 295–296:60–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Metcalf TU, Griffin DE. 2011. Alphavirus-induced encephalomyelitis: antibody-secreting cells and viral clearance from the nervous system. J. Virol. 85:11490–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hawman DW, Fox JM, Ashbrook AW, May NA, Schroeder KMS, et al. 2016. Pathogenic chikungunya virus evades B cell responses to establish persistence. Cell Rep. 16:1326–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Fox JM, Diamond MS. 2016. Immune-mediated protection and pathogenesis of chikungunya virus. J. Immunol. 197:4210–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kim AS, Austin SK, Gardner CL, Zuiani A, Reed DS, et al. 2019. Protective antibodies against Eastern equine encephalitis virus bind to epitopes in domains A and B of the E2 glycoprotein. Nat. Microbiol. 4:187–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Williamson LE, Gilliland T Jr., Yadav PK, Binshtein E, Bombardi R, et al. 2020. Human antibodies protect against aerosolized eastern equine encephalitis virus infection. Cell 183:1884–900.e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Fox JM, Long F, Edeling MA, Lin H, Van Duijl-Richter MKS, et al. 2015. Broadly neutralizing alphavirus antibodies bind an epitope on E2 and inhibit entry and egress. Cell 163:1095–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Earnest JT, Basore K, Roy V, Bailey AL, Wang D, et al. 2019. Neutralizing antibodies against Mayaro virus require Fc effector functions for protective activity. J. Exp. Med. 216:2282–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hunt AR, Bowen RA, Frederickson S, Maruyama T, Roehrig JT, Blair CD. 2011. Treatment of mice with human monoclonal antibody 24 h after lethal aerosol challenge with virulent Venezuelan equine encephalitis virus prevents disease but not infection. Virology 414:146–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Hunt AR, Frederickson S, Hinkel C, Bowdish KS, Roehrig JT. 2006. A humanized murine monoclonal antibody protects mice either before or after challenge with virulent Venezuelan equine encephalomyelitis virus. J. Gen. Virol. 87:2467–76 [DOI] [PubMed] [Google Scholar]
- 238.Pal P, Dowd KA, Brien JD, Edeling MA, Gorlatov S, et al. 2013. Development of a highly protective combination monoclonal antibody therapy against chikungunya virus. PLOS Pathog. 9:e1003312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Powell LA, Fox JM, Kose N, Kim AS, Majedi M, et al. 2020. Human monoclonal antibodies against Ross River virus target epitopes within the E2 protein and protect against disease. PLOS Pathog. 16:e1008517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Stanley J, Cooper SJ, Griffin DE. 1986. Monoclonal antibody cure and prophylaxis of lethal Sindbis virus encephalitis in mice. J. Virol. 58:107–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Lam S, Nyo M, Phuektes P, Yew CW, Tan YJ, Chu JJH. 2015. A potent neutralizing IgM mAb targeting the N218 epitope on E2 protein protects against Chikungunya virus pathogenesis. mAbs 7(6):1178–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Smith SA, Silva LA, Fox JM, Flyak AI, Kose N, et al. 2015. Isolation and characterization of broad and ultrapotent human monoclonal antibodies with therapeutic activity against chikungunya virus. Cell Host Microbe 18:86–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Jin J, Liss NM, Chen D-H, Liao M, Fox JM, et al. 2015. Neutralizing monoclonal antibodies block chikungunya virus entry and release by targeting an epitope critical to viral pathogenesis. Cell Rep. 13:2553–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Petitdemange C, Wauquier N, Vieillard V. 2015. Control of immunopathology during chikungunya virus infection. J. Allergy Clin. Immunol. 135:846–55 [DOI] [PubMed] [Google Scholar]
- 245.Kumar R, Shrivastava T, Samal S, Ahmed S, Parray HA. 2020. Antibody-based therapeutic interventions: possible strategy to counter chikungunya viral infection. Appl. Microbiol. Biotechnol. 104:3209–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Malvy D, Ezzedine K, Mamani-Matsuda M, Autran B, Tolou H, et al. 2009. Destructive arthritis in a patient with chikungunya virus infection with persistent specific IgM antibodies. BMC Infect. Dis. 9:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Schilte C, Staikovsky F, Couderc T, Madec Y, Carpentier F, et al. 2013. Chikungunya virus-associated long-term arthralgia: a 36-month prospective longitudinal study. PLOS Negl. Trop. Dis. 7:e2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Lum FM, Teo TH, Lee WW, Kam YW, Rénia L, Ng LF. 2013. An essential role of antibodies in the control of Chikungunya virus infection. J. Immunol. 190:6295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kam Y-W, Simarmata D, Chow A, Her Z, Teng T-S, et al. 2012. Early appearance of neutralizing immunoglobulin G3 antibodies is associated with chikungunya virus clearance and long-term clinical protection. J. Infect. Dis. 205:1147–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Fragkoudis R, Dixon-Ballany CM, Zagrajek AK, Kedzierski L, Fazakerley JK. 2018. Following acute encephalitis, Semliki Forest virus is undetectable in the brain by infectivity assays but functional virus RNA capable of generating infectious virus persists for life. Viruses 10:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Griffin D, Levine B, Tyor W, Ubol S, Desprès P. 1997. The role of antibody in recovery from alphavirus encephalitis. Immunol. Rev. 159:155–61 [DOI] [PubMed] [Google Scholar]
- 252.Levine B, Griffin DE. 1992. Persistence of viral RNA in mouse brains after recovery from acute alphavirus encephalitis. J. Virol. 66:6429–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.McCarthy MK, Reynoso GV, Winkler ES, Mack M, Diamond MS, et al. 2020. MyD88-dependent influx of monocytes and neutrophils impairs lymph node B cell responses to chikungunya virus infection via Irf5, Nos2 and Nox2. PLOS Pathog. 16:e1008292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Tsetsarkin KA, Vanlandingham DL, McGee CE, Higgs S. 2007. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLOS Pathog. 3:e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Chen GL, Coates EE, Plummer SH, Carter CA, Berkowitz N, et al. 2020. Effect of a chikungunya virus-like particle vaccine on safety and tolerability outcomes: a randomized clinical trial. JAMA 323:1369–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Wressnigg N, Hochreiter R, Zoihsl O, Fritzer A, Bézay N, et al. 2020. Single-shot live-attenuated chikungunya vaccine in healthy adults: a phase 1, randomised controlled trial. Lancet Infect. Dis. 20:1193–203 [DOI] [PubMed] [Google Scholar]
- 257.Ko S-Y, Akahata W, Yang ES, Kong W-P, Burke CW, et al. 2019. A virus-like particle vaccine prevents equine encephalitis virus infection in nonhuman primates. Sci. Transl. Med. 11:eaav3113. [DOI] [PubMed] [Google Scholar]
- 258.Henss L, Yue C, Von Rhein C, Tschismarov R, Lewis-Ximenez LL, et al. 2020. Analysis of humoral immune responses in chikungunya virus (CHIKV)-infected patients and individuals vaccinated with a candidate CHIKV vaccine. J. Infect. Dis. 221:1713–23 [DOI] [PubMed] [Google Scholar]
- 259.Subudhi BB, Chattopadhyay S, Mishra P, Kumar A. 2018. Current strategies for inhibition of chikungunya infection. Viruses 10:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Chung DH, Jonsson CB, Tower NA, Chu YK, Sahin E, et al. 2014. Discovery of a novel compound with anti-Venezuelan equine encephalitis virus activity that targets the nonstructural protein 2. PLOS Pathog. 10:e1004213. [DOI] [PMC free article] [PubMed] [Google Scholar]