

Abstract
Essential Tremor (ET) is a prevalent neurological disease characterized by an 8–10 Hz action tremor. Molecular mechanisms of ET remain poorly understood. Clinical data suggest the importance of the cerebellum in disease pathophysiology, and pathological studies indicate Purkinje Cells (PCs) incur damage. Our recent cerebellar cortex and PC-specific transcriptome studies identified alterations in calcium (Ca2+) signaling pathways that included ryanodine receptor type 1 (RyR1) in ET. RyR1 is an intracellular Ca2+ release channel located on the Endoplasmic Reticulum (ER), and in cerebellum is predominantly expressed in PCs. Under stress conditions, RyR1 undergoes several post-translational modifications (protein kinase A [PKA] phosphorylation, oxidation, nitrosylation), coupled with depletion of the channel-stabilizing binding partner calstabin1, which collectively characterize a “leaky channel” biochemical signature. In this study, we found markedly increased PKA phosphorylation at the RyR1-S2844 site, increased RyR1 oxidation and nitrosylation, and calstabin1 depletion from the RyR1 complex in postmortem ET cerebellum. Decreased calstabin1-RyR1-binding affinity correlated with loss of PCs and climbing fiber-PC synapses in ET. This ‘leaky’ RyR1 signature was not seen in control or Parkinson’s disease cerebellum. Microsomes from postmortem cerebellum demonstrated excessive ER Ca2+ leak in ET vs. controls, attenuated by channel stabilization. We further studied the role of RyR1 in tremor using a mouse model harboring a RyR1 point mutation that mimics constitutive site-specific PKA phosphorylation (RyR1-S2844D). RyR1-S2844D homozygous mice develop a 10 Hz action tremor and robust abnormal oscillatory activity in cerebellar physiological recordings. Intra-cerebellar microinfusion of RyR1 agonist or antagonist, respectively, increased or decreased tremor amplitude in RyR1-S2844D mice, supporting a direct role of cerebellar RyR1 leakiness for tremor generation. Treating RyR1-S2844D mice with a novel RyR1 channel-stabilizing compound, Rycal, effectively dampened cerebellar oscillatory activity, suppressed tremor, and normalized cerebellar RyR1-calstabin1 binding. These data collectively support that stress-associated ER Ca2+ leak via RyR1 may contribute to tremor pathophysiology.
Keywords: Calcium, Cellular stress, Cerebellum, Essential tremor, Mouse model, RyR1
Introduction
Essential Tremor (ET) is a highly prevalent neurological disorder characterized by an 8–10 Hz kinetic tremor, primarily of the arms and hands [45]. ET affects approximately 2.2% of the entire US population, and is chronic and progressive in nature, with an estimated excess of 20% of adults over 90 affected [58, 59]. Despite this high prevalence, the pathophysiology of ET is not completely understood, and there are limited therapeutic options. To shed light on the pathophysiology of ET, our lab has collected and cataloged the largest ET brain repository in the world, which has led to identification of a series of structural and molecular changes in the ET cerebellum [54].
Evidence that the cerebellum is involved in disease pathophysiology comes from extensive clinical [6], physiological [21], neuroimaging [10] and neuropathologic studies [46, 57]. With respect to the latter, neuropathological studies have identified numerous degenerative changes centered in and around Purkinje Cells (PCs), including the cell body (PC loss [11, 52], heterotopic PCs [34, 55], empty PC basket plexus [40]), dendrites (dendritic swellings [82], spine loss [56]), axons (axonal swellings [torpedoes] [51, 60], axon branching [4], recurrent axon collaterals [4], thickened axon profiles [4]), and synaptic connections with adjacent neuronal populations including basket cells (“hairy” baskets [18]) and climbing fibers (decreased climbing fiber synaptic density [39], increased climbing fiber extension into parallel fiber territory [42]). Most recently, we performed RNA-sequencing study of whole cerebellar cortex in ET vs. controls [62], followed by the first-ever laser capture microdissection isolation of postmortem PCs for PC enriched RNA-sequencing in ET [63]. Among other differences, we identified dysregulation of biological processes related to Endoplasmic Reticulum (ER) transport, as well as calcium (Ca2+) signaling and synapse organization. Principal component analysis of normalized transcript reads showed a high degree of heterogeneity among ET samples in our transcriptome studies, but significant enrichment of ET samples in positive principal component 2 space (PC2+). Expression of the gene coding for ryanodine receptor type 1 (RyR1) was also found to be highly enriched in PC2+, correlating with an ET diagnosis, and was an overlapping gene implicated in both transcriptome studies. The potential mechanism of RyR1 protein as it pertains to the pathophysiology of ET remains to be explored, as RyR1 is a novel target in ET with no published studies currently available.
Ryanodine receptors are intracellular Ca2+ release channels localized to the endo/sarcoplasmic reticula of most cell types, where they release Ca2+ from intracellular stores required for critical functions such as neurotransmitter release and muscle contraction [1, 36]. There are three isoforms of ryanodine receptors—RyR1, RyR2 and RyR3—first identified in skeletal muscle, cardiac muscle, and brain, respectively, although all RyR subtypes are present in human brain [36]. The RyR1 macromolecular complex is a homotetramer that includes four RyR1 protomers, with each protomer of the channel having a mass of ~565 kDa. Each RyR1 protomer is comprised of a large N-terminal cytoplasmic domain, which serves as a scaffold for multiple channel regulators (Supplemental Fig. 1). These regulators include kinases (PKA, Ca2+/calmodulin kinase [CaMKII]), a phosphatase (PP1), calmodulin, and a phosphodiesterase (PDE4D3) [64, 65]. Additionally, the RyR1 channel-stabilizing subunit—calstabin1 (FKBP12)—is critical for maintaining a channel ‘closed state’ and averting a pathological leak of Ca2+ via the channel [83]. The RyR1 macromolecular complex is expressed in multiple cell types throughout the body. RyR1 in the brain is most highly expressed in cerebellar cortex compared to other regions, such as hippocampus and striatum, and is highly expressed in PCs [22, 78]. This is physiologically important as PCs have two unique features: very high basal firing rates [13] and dependence on Ca2+ signaling to control the precision of their firing rate and rhythmicity [70]. Among neurons, PCs can efficiently handle large intracellular Ca2+ loads due to a very high affinity for Ca2+ binding [19], and the Sarco-Endoplasmic Reticulum Calcium Pump (SERCA) contributes greatly to Ca2+ clearance from cytoplasm into the ER [20]. Therefore, alterations in regulatory ER Ca2+ channels, such as RyR1, could have substantial effects on PC neuronal firing, conceivably contributing to tremor.
Under conditions of cellular stress, RyR1 ER-Ca2+ channel can undergo site-specific phosphorylation at serine 2844 (S2844) by protein kinase A (PKA) [74], accompanied by stress-induced oxidation and nitrosylation of the channel, leading to the displacement of the channel-stabilizing subunit calstabin1 [3, 5, 83]. The combination of S2844 phosphorylation, increased oxidation and nitrosylation, coupled with depletion of calstabin1 renders RyR1 channels ‘leaky’, enhancing the efflux of ER-Ca2+ into the cytosol. The high metabolic demand of PCs may incur higher levels of oxidative stress, particularly in disease states, and as PCs are likely to be central to the pathogenesis of ET, alterations in RyR1-mediated ER-Ca2+ handling could result in aberrant firing patterns, leading to tremor.
The present study investigates stress-induced post-translational modifications in the RyR1 macromolecular complex from postmortem human ET cerebellum. We identified a ‘leaky’ channel phenotype present only in ET patients compared to age-matched controls and patients with another tremor disorder, Parkinson’s Disease (PD). We additionally studied this RyR1 ‘leaky’ phenotype in mice that harbor a genetically engineered phosphomimetic RyR1-S2844D mutation that induces a constitutively ‘leaky’ RyR1 channel. We demonstrate that homozygous RyR1-S2844D+/+ mice develop a 10 Hz action tremor and abnormal oscillatory activity in cerebellar physiological recordings, and these features are bidirectionally modulated by RyR1 pharmacologic ligands and attenuated by treating mice with a novel drug, Rycal, that fixes RyR1 leakiness [3]. In sum, our studies identify RyR1 as being post-translationally dysregulated in cerebellum of the ET patients we studied and that the RyR1-S2844D+/+ mouse may provide a model to further our mechanistic insights into the pathophysiology of action tremor.
Methods
Human sample selection and clinical criteria
All study subjects signed informed consent forms approved by their respective university or institutional ethics boards.
Clinical and demographic information for all human samples is presented in Supplemental Table 1. Samples are indicated as being used in RyR1 or RyR2 biochemistry and RyR1 binding assay, and samples were selected based on their age for either RyR1 or RyR2 investigation.
Eighteen ET brains were obtained from the Essential Tremor Centralized Brain Repository (ETCBR), a longstanding collaboration between investigators at UT Southwestern and Columbia University [54]. These samples were chosen from a larger inventory of frozen samples used in recent sequencing studies and were selected so that for each decade of life, from 7 to 10th, there would be several analyzed samples. ET diagnosis was confirmed by a senior movement disorder neurologist (EDL) using three sequential methods, as detailed previously [54]. No ET patients had a history of heavy ethanol use, traumatic brain injury or exposure to any medication known to cause cerebellar damage. In addition to their initial videotaped neurological assessment, all patients were prospectively followed with structured telephone assessments and as-needed additional in-person videotaped neurological assessments. Assessment of Parkinsonian features and other movement disorders, such as dystonic postures, were also evaluated during this assessment. None of the chosen ET cases in this current study exhibited Parkinsonism or dystonia.
Ten PD brains were obtained from the New York Brain Bank at Columbia University and were chosen from an inventory sampling of aged-available frozen PD tissue, selected based on decade of age, as noted above for ET samples.
There were 24 total control brains. Fifteen control brains were obtained from the New York Brain Bank at Columbia University. Individuals were prospectively followed at the Alzheimer’s Disease Research Center or the Washington Heights Inwood Columbia Aging Project at Columbia University. During serial neurological examinations, these individuals were clinically free of ET and other neurodegenerative disorders, including PD, Alzheimer’s disease, or progressive supranuclear palsy. Nine control brains were obtained from the National Institutes of Health NeuroBioBank (six from University of Miami, Miami FL; three from University of Maryland, Baltimore MD). Control samples were also chosen from an inventory sampling of aged-available frozen tissue, selected based on decade of age, as noted above for ET samples.
The final sample numbers for RyR1 biochemistry (11 ET, 10 PD and 20 controls) reflect an approximate 1:1 for disease state comparison (ET:PD) and 1:1 for combined disease to control comparison (ET + PD:control). For a calstabin1-RyR1-binding assay, 11 ET and 11 control age-matched samples were used for a 1:1 disease state comparison (ET:control).
The final sample numbers for RyR2 biochemistry (12 ET and 12 controls) reflects a 1:1 matching of disease state to controls. The control samples selected expanded the age range of analysis to include 6 ‘young’ controls (age 36–59 years) and 6 ‘aged’ controls (age 60–99 years).
ET and control samples were chosen based on a lack of widespread marked hypoxic-ischemic damage, concurrent Alzheimer’s-type changes that would meet criteria for high likelihood of AD [67], or evidence of other neurodegenerative disease pathology (e.g., corticobasal degeneration, progressive supranuclear palsy, traumatic encephalopathy). A senior neuropathologist (PLF) examined stained cerebellar sections to determine adequate tissue preservation, including absence of autolytic changes in the granule cell layer. Postmortem interval (PMI) was assessed for all samples, which is the time between patient death and brain samples freezing. Brains had Braak AD staging for neurofibrillary tangles [8, 9] as well as Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) ratings for neuritic plaques [66].
Quantification of cerebellar pathological metrics
Quantification of PCs and torpedoes were performed as previously described [54]. Briefly, a standard 3 × 20 × 25-mm formalin-fixed tissue block from each brain was obtained from a parasagittal slice located 1–1.5 cm from the cerebellar midline, containing anterior and posterior quadrangulate lobules overlying the dentate nucleus. Paraffin embedded Sections (7 μm thick) were stained with Luxol fast blueHematoxylin&Eosin (LH&E). PC body linear density was counted in at least fifteen 100 × microscopic fields across the section and expressed as PC per mm. Torpedoes were counted in the entire tissue section and normalized to the PC layer length, expressed as torpedoes per mm.
Climbing fiber (CF)-PC synaptic changes to evaluate the distribution of CF-PC synapses on PC dendrites was performed by immunohistochemistry to vesicular glutamate transporter type 2 (VGlut2) as previously described [42]. This includes measurement of the density of CF-PC synaptic puncta (CF synapses/mm) and the re-distribution of these synapses to the parallel fiber synaptic territory in the outer 20% of the cerebellar molecular layer (ML) (CFs in outer 20% of ML).
Immunoprecipitation assays and blotting
Human or mouse cerebellar samples were isotonically lysed in buffer (5 × volume/weight) containing 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 20 mM NaF, and protease inhibitors. For immunoprecipitation of RyR1, an anti-RyR1-specific antibody (RyR1–1327, an affinity-purified rabbit polyclonal antibody raised against a KLH-conjugated peptide with the amino acid sequence CAEPDTDYENLRRS, corresponding to residues 1327–1339 of mouse skeletal RyR1) was used to immunoprecipitate RyR1 from 0.15 mg of brain homogenates. For immunoprecipitation of RyR2, an RyR2-specific antibody was used (affinity-purified polyclonal rabbit antibody raised against KLH-conjugated peptide with the amino acid sequence CKPEFNNHKDYAQEK, corresponding to amino acids 1367–1380 of mouse RyR2 with a cysteine residue added to the amino terminus). The samples were incubated with either RyR1 or RyR2 antibody in 0.5 ml of a modified RIPA buffer (50 mM Tris–HCl pH 7.4, 0.9% NaCl, 5.0 mM NaF, 1% Triton-X100 and protease inhibitors) for 1 h at 4 °C. The immune complexes were incubated with protein A–Sepharose beads (Sigma) overnight at 4 °C and the beads were washed twice with modified RIPA buffer. Proteins were separated on 4–20% Tris–glycine gels (Life Technologies) and transferred to nitrocellulose for 1 h at 100 V. Immunoblots were developed with anti-RyR (5029 antibody [28], 1:2500 dilution), Cys-NO (Alpha Diagnostic International, 1:1000), or an anti-calstabin antibody (Santa Cruz Biotechnology, cat. sc-6173, Santa Cruz, CA; 1:2,500). To determine RyR oxidation, the immunoprecipitates were treated with 2,4-dinitrophenyl hydrazine, and the derivatized carbonyls were detected using the Oxyblot detection kit (Chemicon). Immunoblots were developed and quantified using the Odyssey Infrared Imaging System (LICOR Biosystems, Lincoln, NE) and infrared-labeled secondary antibodies.
Microsomal calcium release assay
Cerebellar samples were isotonically lysed in 0.25 mL of lysis buffer (50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 20 mM NaF, and protease inhibitors). Microsomes were prepared by centrifuging the lysates at 45,000×g for 30 min. Pellets were resuspended in lysis buffer containing 300 mM sucrose. Microsomes (5 μg/ml) were diluted into a 20 mM HEPES buffer (pH 7.2) containing 7 mM NaCl, 1.5 mM MgCl2, 120 mM K-gluconate, 5 mM K-phosphate, 8 mM K-phosphocreatine, 1 μM EGTA and 2 μM CaCl2 mixed with 3 μM Fluo-4 and added to multiple wells of a 96-well plate. Ca2+ loading of the microsomes was initiated by adding 1 mM ATP. After Ca2+ uptake, 3 μM Thapsigargin was added to inhibit further Ca2+ uptake by the calcium pump (SERCA). The subsequent ‘leak’ of Ca2+ out of the ER was measured by the increase in intensity of the Fluo-4 signal (measured in a Tecan infinite F500 fluorescence plate reader). The Ca2+ leak was quantified as the increase in Fluo-4 signal after addition of thapsigargin.
Calstabin1—RyR1 channel-binding assay
RyR1 was immunoprecipitated from 0.1 mg of cerebellum lysate using an anti-RyR1 specific antibody [5] (2.0 μg) in 1.0 ml of a modified RIPA buffer overnight at 4 °C. The immune complexes were isolated by incubation with protein A-Sepharose beads (Sigma) at 4 °C for 1 h, and the beads were then washed three times with RIPA buffer, followed by two washes with binding buffer (10 mM Tris–HCl, pH 6.8, 150 mM NaCl, and protease inhibitors). Immunoprecipitates were incubated in 0.2 ml of binding buffer containing 5, 10, 50, 100, 500, or 1000 nM [35S]-calstabin1 for 1 h at RT. Samples were diluted with 1 ml of ice-cold washing buffer (25 mm Hepes, pH 7.1, 0.25 m KCl) and filtered through Whatman GF/B membrane filters pre-soaked with 0.015% polyethyleneimine in washing buffer. Filters were washed three times with 5 ml of washing buffer. The radioactivity remaining on the filters was determined by liquid scintillation counting to obtain bound [35S]-calstabin1. Nonspecific binding was determined in the presence of 100-fold excess of non-labeled calstabin1. 3H Ryanodine binding was used to quantify RyR1 in immunoprecipitate.
RyR1-S2844D mice
RyR1-S2844D mice were engineered and fully characterized as previously described [3]. Mice with successfully germline-introduced RyR1-S2844D mutation, as confirmed by sequencing, were subsequently backcrossed into C57Bl/6J strain for 6 generations, to minimize the chance of any offtarget mutations. Homozygote and heterozygote mice of both sexes were studied with food and water ad libitum. All animal studies were carried out under an approved IACUC animal protocol according to the institutional guidelines at Columbia University Irving Medical Center.
Tremor measurement and assessment
Tremor measurement and assessment followed our recently published protocols [71]. Briefly, we applied a platform (Convuls-1, Columbus Instruments) for weight-based vibration detection with linear weight-voltage transformation (141 mV per 32 g of mass). Mice were placed on the platform for tremor measurement, which was co-registered with a video-based motion detection system (NeuroMotive, BlackRock microsystem) to separate action vs. rest tremor. The vibratory signals were filtered (low pass filter: 250 Hz) and digitized at 1000 Hz with DAQ device (Cerebus, BlackRock microsystem). Digitized data were processed offline using in-house MATLAB scripts developed for frequency analysis. Behavioral data were transformed into frequency domains via the power spectrum density function (by Welch’s method with Hanning windowing, sampling rate at 1024 Hz in data block of 1 s, given the frequency resolution of 1 Hz and half of the data overlap in each step). Each power spectrum density data point was constructed from a 20-s window and one-second shift for the next data point until the end of data stream. The spectrum data were further normalized to the overall power between 5 and 40 Hz to eliminate the effect of 60 Hz noises and the variability of absolute powers between mice. Data analyses of all mice were performed by these preset programs automatically. We tested the tremor responsiveness to drugs in the mouse model by intraperitoneal (IP) administration of caffeine (20 mg/kg), dantrolene (20 mg/kg) or Rycal (25 mg/kg).
Stereotaxic surgery for implantation of cannulas and electrode
Briefly, RyR1-S2844D homozygous mice underwent IP injection with 2 mg/kg dantrolene prior to the surgery, to prevent anesthesia related complications and subsequent death. Mice were then anesthetized by isoflurane prior to head-fixation on the stereotaxic frame (David Kopf Instruments). Subcutaneous bupivacaine was administered for local anesthesia. The scalp was incised above the cerebellar region after fur removal. Adequate sterilization was applied, and the mouse skull was drilled, and a cannula was implanted into the mouse cerebellum at the following coordinates (to Bregma AP: − 6.4 mm, ML: 0 mm, DV: − 0.15 mm). To record cerebellar local field potential, mice had four small pilot holes placed in the skull at the following stereotactic coordinates relative to Bregma: Ground/Reference: AP: + 4.2; ML: ± 1.5; Ch8/Ch3: AP: − 7.0; ML ± 1.5. Four stainless steel Electroencephalogram (EEG) screws were then inserted through openings in the headmount and manually rotated into the pilot holes. The scalp was filled with radiopaque l-powder and mice received standard postoperative care. In the cerebellar microinfusion experiments, 20 μg caffeine and 4 μg dantrolene were delivered, at a rate of 2 μl every 5 min.
Rycal rescue for biochemical analysis
Assessment of the RyR1 ‘leaky’ biochemical signature in the RyR1-S2844D mice was performed via oral administration of Rycal (S107). S107 derivative of Rycal was utilized as it crosses the blood–brain barrier. Mice were randomized into two groups. The first group received Rycal (50 mg/kg/day) freely in the drinking water while the second group received water only. Drug administration continued daily (weekends included) for 2 months. These animals were sacrificed, and cerebellar tissue was utilized in biochemical analysis of the ‘leaky’ RyR1 channel.
Statistical analysis
All data were expressed as mean ± SEM. We tested for normality in continuous variables using the Kolmogorov–Smirnov test. For comparison of mean values between 2 experimental groups, the Student’s t test was used or, if not normally distributed, a non-parametric test (i.e., Mann–Whitney) was used. The one-way ANOVA test was used to compare mean values between three or more experimental groups with a Tukey’s or Dunn’s post hoc experimental comparison for parametric and non-parametric analysis, respectively. Correlations between metrics were performed using Pearson’s method for normally distributed data or by Spearman’s method if not normally distributed. A value of P < 0.05 was considered statistically significant.
Results
RyR1 channels are ‘leaky’ in ET cerebellar cortex
We first examined whether ET cerebellum has post-translational modifications of RyR1 channels consistent with the ‘leaky’ RyR1 biochemical signature by immunoprecipitation of the RyR1 macromolecular complex. A ‘leaky’ RyR1 channel biochemical signature is characterized by PKA phosphorylation at RyR1-S2844, increased levels of oxidation (DNP) and nitrosylation (Cys-NO), coupled with depletion of calstabin1 [37]. Compared to control cerebellar cortex (n = 20), ET cerebellar cortex had extensive remodeling of RyR1 channels in ET cerebellar cortex (n = 11), with significant increases in PKA phosphorylation, oxidation, and nitrosylation of RyR1, as well as significant depletion of calstabin1 (Fig. 1a–e). As RyR1 phosphorylation is regulated by dynamic levels of interactions between kinases and phosphatases present on the large cytoplasmic domain of RyR1, we further investigated the levels of other RyR1 macromolecular complex scaffold proteins (Supplemental Fig. 1), including the PKA catalytic subunit, protein phosphatase 1 (PP1) and its targeting protein spinophilin, as well as a phosphodiesterase (PDE4D3) (Fig. 1a). There was a significant decrease in PP1 in the RyR1 macromolecular complex in ET compared to control (Fig. 1f), with a trending decrease of its targeting protein spinophilin (Supplemental Fig. 2a; p = 0.0626) in ET, but no difference in PDE4 or PKA (Supplemental Fig. 2b, c), suggesting a protein-level disturbance in the phosphatase axis associated with RyR1. In the present study, age did not affect calstabin1 binding to cerebellar RyR1 across a wide age range in controls (r = 0.0340, p = 0.8868; Fig. 1g).
Fig. 1.
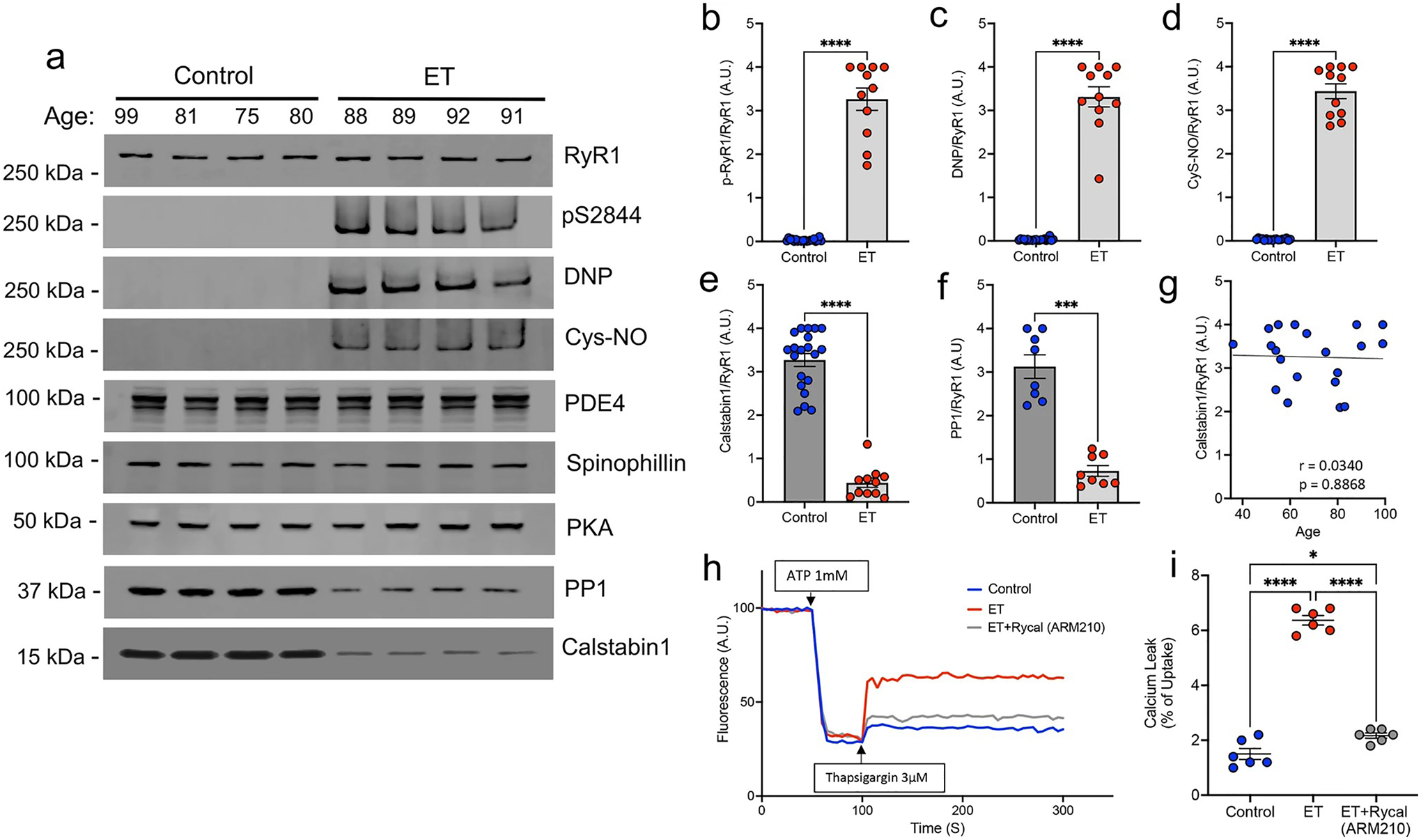
RyR1 channels are ‘leaky’ in ET cerebellar cortex. a Representative western blot of immunoprecipitated RyR1 macromolecular complex probing for total RyR1, RyR1-pS2844, oxidation (DNP), nitrosylation (Cys-NO), and other RyR1 interactors, PDE4, Spinophillin, PKA, PP1 and Calstabin1. Quantification of protein levels normalized to total RyR1 in n = 20 controls and n = 11 ETs for b p-RyR1/RyR1, c DNP/RyR1, d Cys-NO/RyR1, e Calstabin1/RyR1, and f PP1/RyR1 [Mann–Whitney t test ****p < 0.0001]. g Calstabin1/RyR1 vs. age in control cerebellar samples only, aged 36–99 (n = 20), demonstrating that normal aging does not significantly increase calstabin1 depletion from RyR1. h Representative graph of ER calcium (Ca2+) leak measured in microsomes isolated from postmortem control and ET cerebellum, demonstrating increased Ca2+ leak in ET versus control. Pharmacologically inhibiting ER Ca2+ leak by in vitro incubation of ET microsomes with RyCal (ARM210), an RyR1 channel stabilizer, prior to the assay attenuates the Ca2+ leak. i. Quantification of microsomal calcium release assays. Bar graph represents the leak (after thapsigargin) as a percentage of the calcium update after ATP (n = 6). [One Way ANOVA w/Tukey’s multiple comparisons ****p < 0.0001, *p < 0.05]
Consistent with the results of post-translational modifications of RyR1, microsomes isolated from postmortem ET cerebellar cortex exhibited increased ER Ca2+ leak compared to those isolated from controls (Fig. 1h, i). When ET microsomes were separately incubated with the RyR-targeted small molecule Rycal (ARM210), which stabilizes binding of calstabin1 to PKA phosphorylated and oxidized/nitrosylated RyR1 channels, the ER Ca2+ leak was attenuated (Fig. 1h, i). Taken together, these data indicate that ‘leaky’ RyR1 channels are present in the cerebellum of the ET cases we studied, with a significantly increased cerebellar ER Ca2+ leak in ET cases that was attenuated by treating the samples with an RyR1 channel-stabilizing drug.
Reduced binding affinity of calstabin1 for RyR1 channel in ET
The RyR1 channel is a tetramer comprised of four RyR1 monomers, each binding to a single calstabin1 molecule that stabilizes the closed state of the channel. Post-translational modifications of the RyR1 channel cause dissociation of calstabin1, resulting in the ‘leaky’ phenotype. We next directly tested the affinity of calstabin1 binding for the RyR1 channel in 11 ET and 11 age-matched control cerebellar samples using an in vitro binding assay (Supplemental Table 1). The calstabin1-RyR1 dissociation constants (Kd values) were significantly higher for ET cases (235.0 ± 11.86 nM) compared to controls (79.09 ± 2.81 nM) (p < 0.0001; Fig. 2a), indicating that the binding affinity of calstabin1 for the RyR1 channel is much lower in ET (i.e., higher Kd = lower binding affinity). We then chose four metrics of cerebellar pathology that provide the greatest distinction between ET and controls [57] to correlate with the calstabin1-RyR1 Kd values in this initial set of ET cases (Supplemental Table 1). We identified a strong negative correlation between the calstabin1-RyR1 Kd values and the linear density of PCs in ET cerebellar cortex (PC/mm) (r = − 0.8760, p = 0.0004; Fig. 2b), indicating that as PC loss increases the affinity of calstabin1 binding for the RyR1 channel decreases in ET. This finding is of particular interest as RyR1 is highly expressed within PCs in the cerebellar cortex and PC loss is a feature of ET pathology. In addition, there was a significant negative correlation between calstabin1-RyR1 Kd values and the climbing fiber (CF) synaptic density in ET (r = − 0.6865, p = 0.0196; Fig. 2c), suggesting that cerebellar RyR1 dysfunction may also lead to loss of PC synaptic inputs. There was no correlation between calstabin1-RyR1 Kd values and CFs in the outer 20% of the molecular layer (r = 0.0235, p = 0.9454) or torpedoes/mm (r = − 0.1052, p = 0.7583) in ET. These pathological metrics were not significantly correlated with calstabin1-RyR1 Kd values in control samples (for PCs, r = − 0.0523, p = 0.8787; for CF synaptic density, r = 0.0569, p = 0.8679; for CFs in outer 20% of molecular layer, r = − 0.0337, p = 0.9216; for torpedoes/mm, r = − 0.1043, p = 0.7602), as expected given the lack of RyR1 biochemical modifications in controls.
Fig. 2.
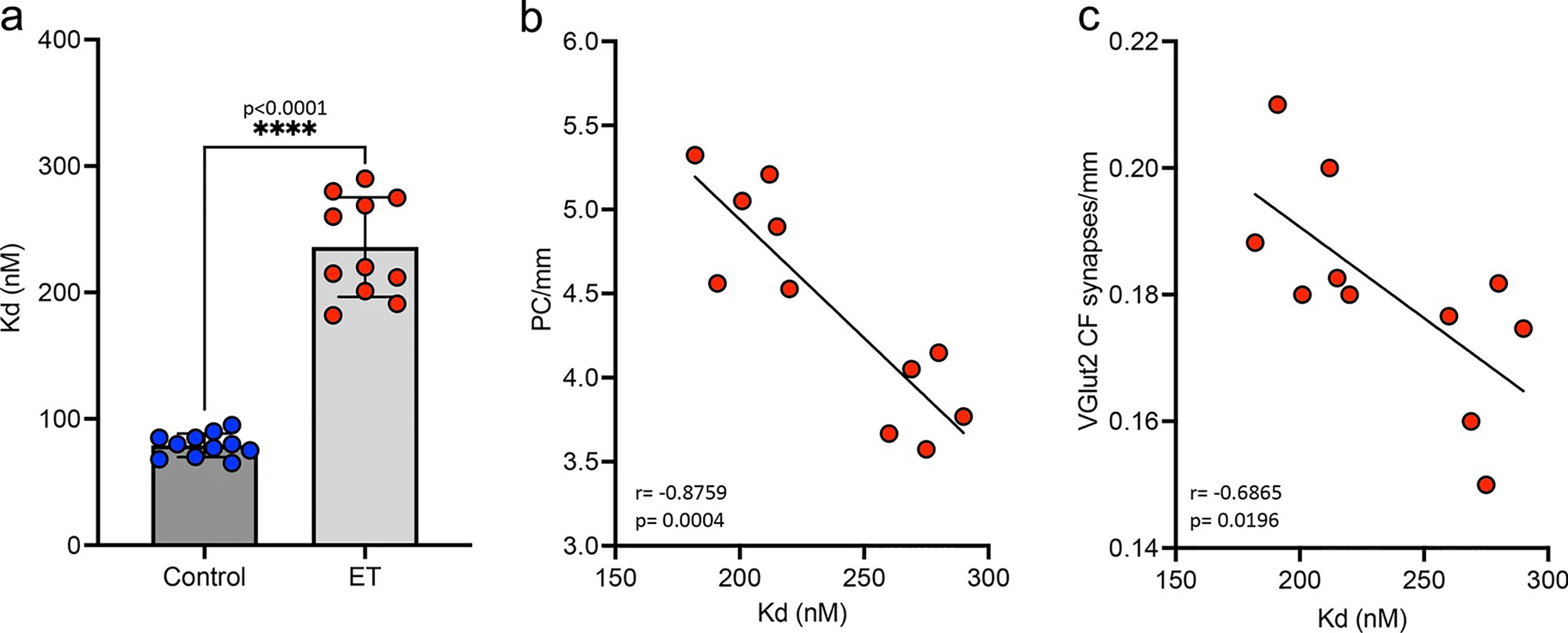
Binding affinity of calstabin1 for RyR1 channel. a An in vitro assay was used to measure the dissociation constants (Kd values) for calstabin1 binding to the RyR1 channel in ET and control cerebellar samples. As Kd values increase, the binding affinity of calstabin1 for the RyR1 channel decreases, reflecting greater dissociation of calstabin1 from RyR1. [Mann–Whitney t test **** p < 0.0001] b Correlation of the linear density of PCs (PC/mm) vs. calstabin1-RyR1 Kd values demonstrates that ET cases with greater PC loss (lower PCs/mm) have lower binding affinity of calstabin1 for RyR1. [Pearson correlation ***p = 0.0004]. c Correlation of the synaptic density of VGlut2 labeled climbing fibers (CFs) on PCs in ET cases demonstrates that greater loss of CF synapses on PC dendrites associates with lower binding affinity of calstabin1 for RyR1. [Pearson correlation ***p = 0.0196]. VGlut2 vesicular glutamate transporter type 2
‘Leaky’ RyR1 channels are not found in the PD cerebellum
To begin to examine the specificity of ‘leaky’ RyR1 channels for ET, we next investigated the post-translational remodeling of the RyR1 macromolecular complex in the second most common tremor disorder: PD. Immunoprecipitation of the RyR1 complex from ET (n = 11), PD (n = 10), and control (n = 20) demonstrated that ‘leaky’ RyR1 channels, with excessive phosphorylation at RyR1-S2844, increased oxidation and nitrosylation, coupled with depletion of calstabin1, was only seen in ET and not in control or PD cerebellar cortex (Fig. 3a–e). These results demonstrate a first step in understanding the disease specificity for the presence of excessively ‘leaky’ RyR1 channels in the ET cerebellum.
Fig. 3.
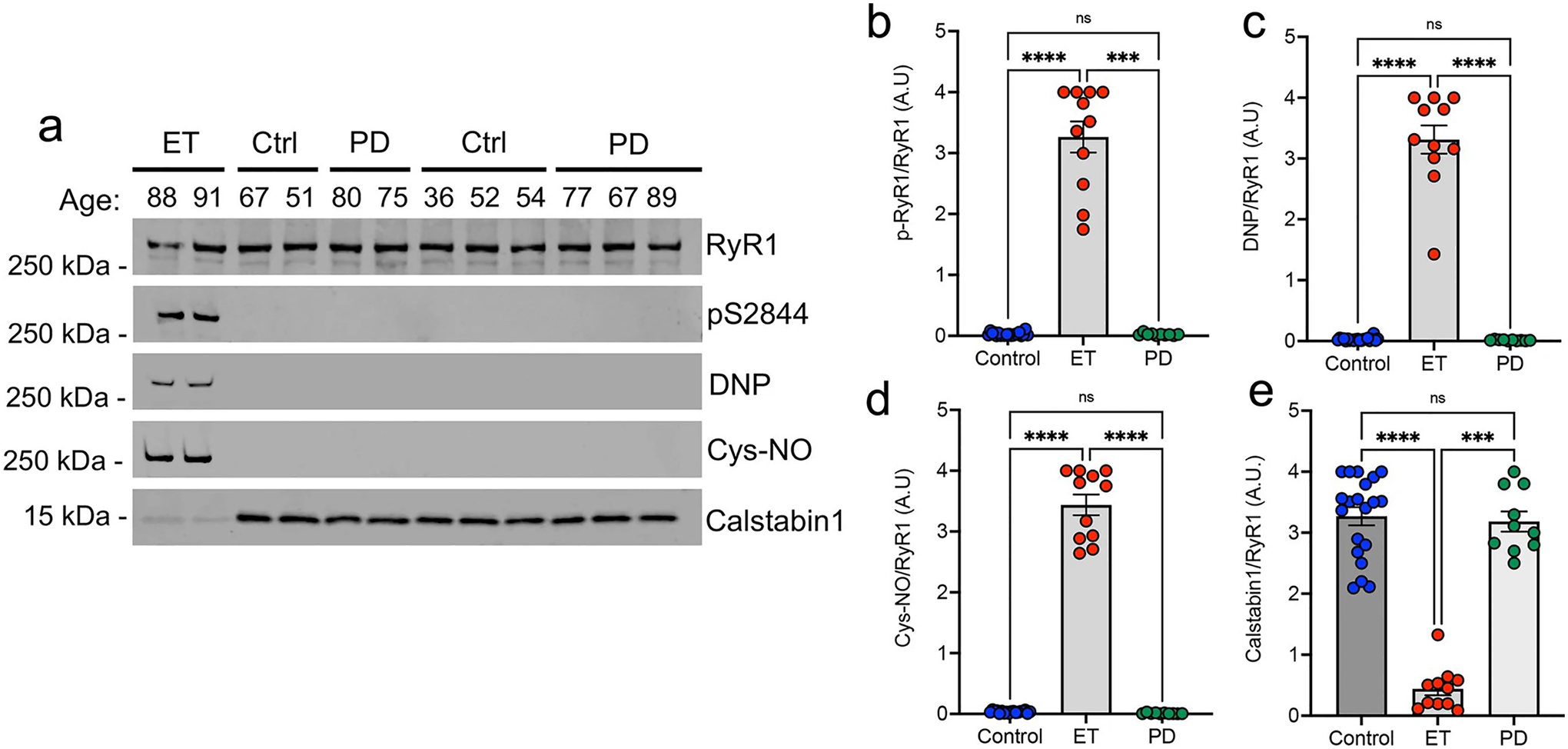
‘Leaky’ RyR1 channels are not found in the Parkinson’s disease cerebellum. a Immunoprecipitation of RyR1 complex from postmortem control, ET and Parkinson’s Disease (PD) human cerebellum probing using antibodies against RyR1, RyR1-pS2844, oxidation (DNP), nitrosylation (Cys-NO) and Calstabin1. Quantification of blots for b p-RyR1/RyR1, c DNP/RyR1, d Cys-NO/RyR1, e Calstabin1/RyR1 (n = 20 control, n = 10 PD, n = 11 ET) [One Way ANOVA w/Dunns multiple comparisons ***p < 0.001, ****p < 0.0001]
RyR2 post-translational modifications are age-dependent in cerebellum
To confirm whether this ‘leaky’ channel phenotype is specific to RyR1 in ET cerebellar cortex, we also examined the RyR2 macromolecular complex in ET (n = 12) and controls (n = 12). ‘Leaky’ RyR2 channels have been associated with other neurodegenerative diseases, including in the hippocampus of Alzheimer’s disease [37] and in several brain regions in Huntington’s disease [15]. In brain, RyR2 is predominantly expressed in hippocampus and cerebral cortex, but is also present in granule cells in the cerebellar cortex, and not detected in PCs [22, 78]. Post-translational modifications of the RyR2 complex were present to a similar degree in both ET and control cerebellum in individuals over 60 years of age (Fig. 4a) and were not detected in younger controls (ages 36–59) (Fig. 4b–e). The youngest available ET sample for analysis in our cohort was 67 years of age. These data demonstrate a strong correlation between aging and remodeling of the RyR2 complex in the human cerebellum (r = 0.7656, p < 0.0001; Fig. 4f). In contrast to the strong association of RyR1 channel leak with the ET clinical phenotype (Fig. 1), the similar extent of RyR2 modifications in age-matched ET and controls suggest that RyR2 channel leak is likely not driving the ET disease phenotype.
Fig. 4.
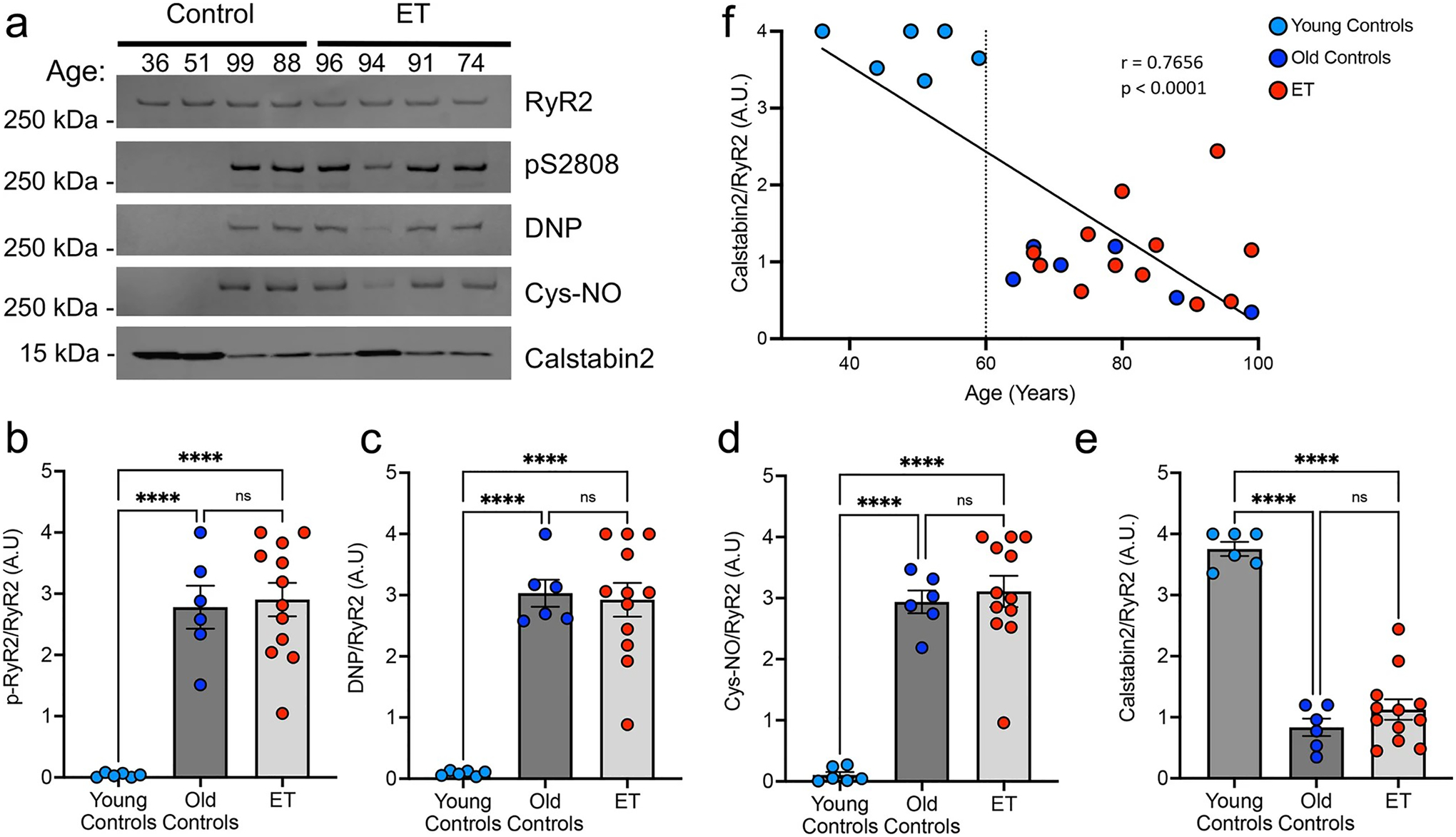
RyR2 post-translational modifications are aging associated in human cerebellum. a Representative western blot of immunoprecipitated RyR2 macromolecular complex in the postmortem human ET and control cerebellum. Quantification of blots for b p-RyR2/pS2808, c DNP/RyR2, d Cys-NO/RyR2 and e Calstabin2/RyR2 (n = 6 young controls, n = 6 old controls, n = 12 ET). The RyR2 macromolecular complex is post-translationally modified in old controls (> 60 years) and ET patients (youngest ET 67 years) but not in young controls (< 59 years) [One-way ANOVA with Tukey’s multiple comparisons ****p < 0.0001]. f Linear regression (r = 0.7747) and Spearman correlation (p < 0.0001) of calstabin2 depletion from RyR2 (Calstabin2/RyR2) with age, indicating a significant correlation between RyR2 remodeling and age, which occurs after age 60 years
RyR1-S2844D+/+ mice develop action tremor
To further study the relationship between ‘leaky’ RyR1 and tremor, we utilized a mouse with phosphomimetic mutation of RyR1 at S2844, which mimics constitutive site-specific phosphorylation leading to RyR1 leak [3]. We examined whether RyR1-S2844D mice have tremor using a sensitive force plate, coupled with camera for motion detection, in a freely moving setting (Fig. 5a). This measures the distribution of the power spectral density (PSD, power of the vibrations per Hz) across frequency ranges, which allows an unbiased evaluation whether the mice have a consistent PSD peak at a given frequency (i.e., tremor is a frequency dependent involuntary movement). The Homozygous RyR1-S2844D+/+ mice were found to have a robust 10 Hz action tremor that was not observed in wild-type (WT) or heterozygous RyR1-S2844D± mice (Fig. 5b–d). Since ET patients predominantly have action tremor, we further studied the tremor characteristics of homozygous RyR1-S2844D+/+ mice and found they predominantly have a 10 Hz action tremor (Fig. 5e–g).
Fig. 5.
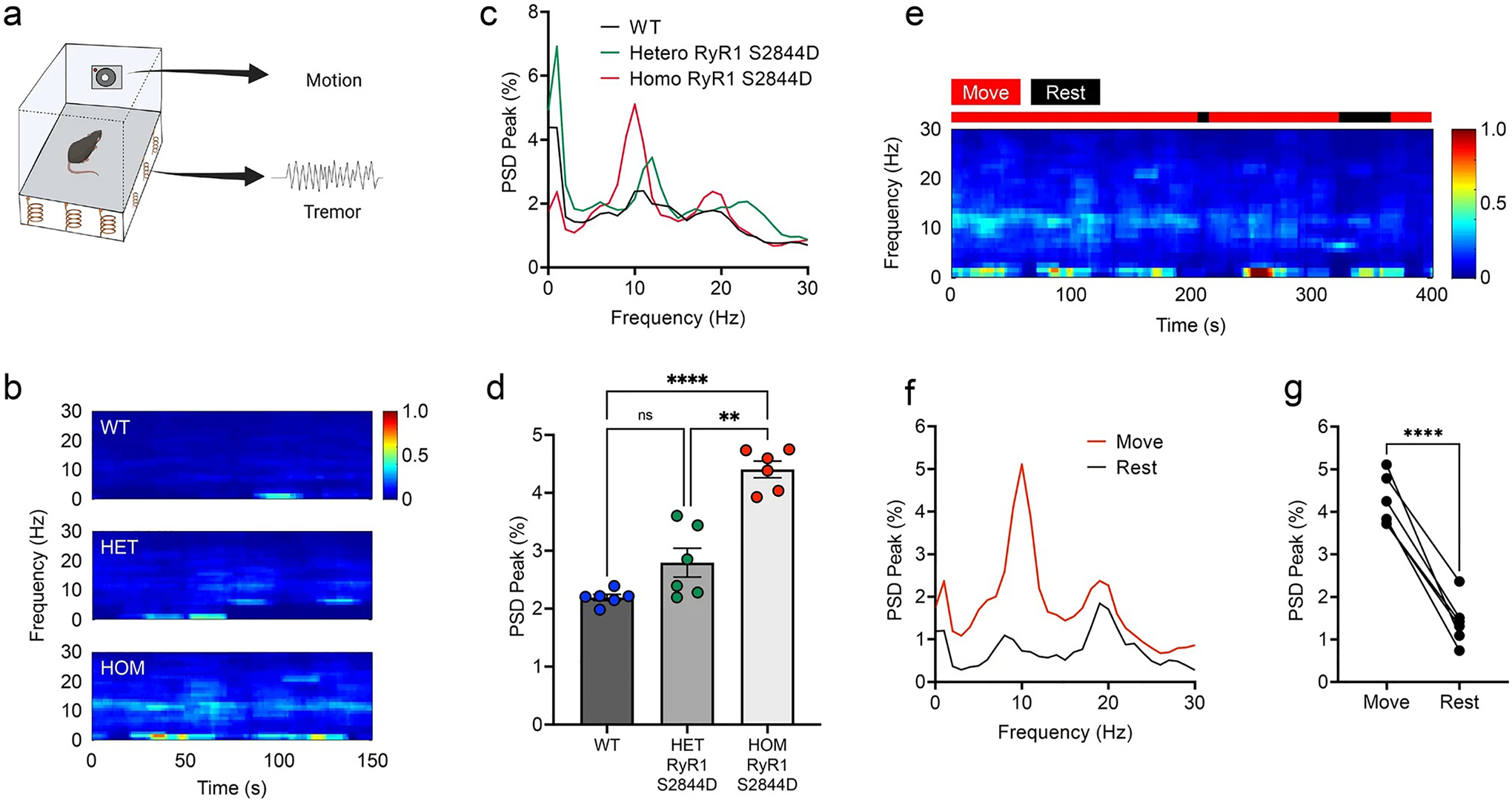
Mice with RyR1-S2844D+/+ mutation have an action tremor. a A scheme showing tremor recording with simultaneous motion monitoring in a freely moving mouse. b Representative time–frequency plots and c, d Quantified spectral diagrams (n = 6 mice per group) of tremor in 6-month-old wild-type (WT), RyR1-S2844D± (HET) and RyR1-S2844D+/+ (HOM) mice. d RyR1-S2844D+/+ mice developed robust tremor at 10 Hz. e A representative time–frequency plot in RyR1-S2844D+/+ mouse [** p = 0.0015, **** p < 0.0001 by Brown-Forsythe and Welch ANOVA] and f, g corresponding spectral diagram quantification, co-registered by video-based motion detection, showed kinetic-predominant tremor (action tremor; n = 6 mice). PSD power spectral density. ****p < 0.0001 by two-tailed paired t test
Bi-directional manipulation of RyR1 modulates tremor in RyR1-S2844D+/+ mice
We next examined the effect of bi-directional pharmacological manipulations on tremor in the RyR1-S2844D+/+ mice. We found that systemic intraperitoneal administration of the RyR1 agonist caffeine significantly enhanced tremor in RyR1-S2844D+/+ mice (Fig. 6a–c), whereas intraperitoneal administration of the RyR1 blocker, dantrolene, suppressed tremor in real time (Fig. 6d–f). To further test the contribution of cerebellar RyR1 in tremor, we performed cannula microinfusion of caffeine and dantrolene directly onto the cerebellum of RyR1-S2844D+/+ mice. We found that microinfusion of caffeine significantly increased action tremor (Fig. 6g–i), whereas microinfusion of dantrolene significantly inhibited action tremor (Fig. 6j–l). These results support a role for RyR1 in action tremor and a direct role of cerebellum for tremor generation in RyR1-S2844D+/+ mice.
Fig. 6.
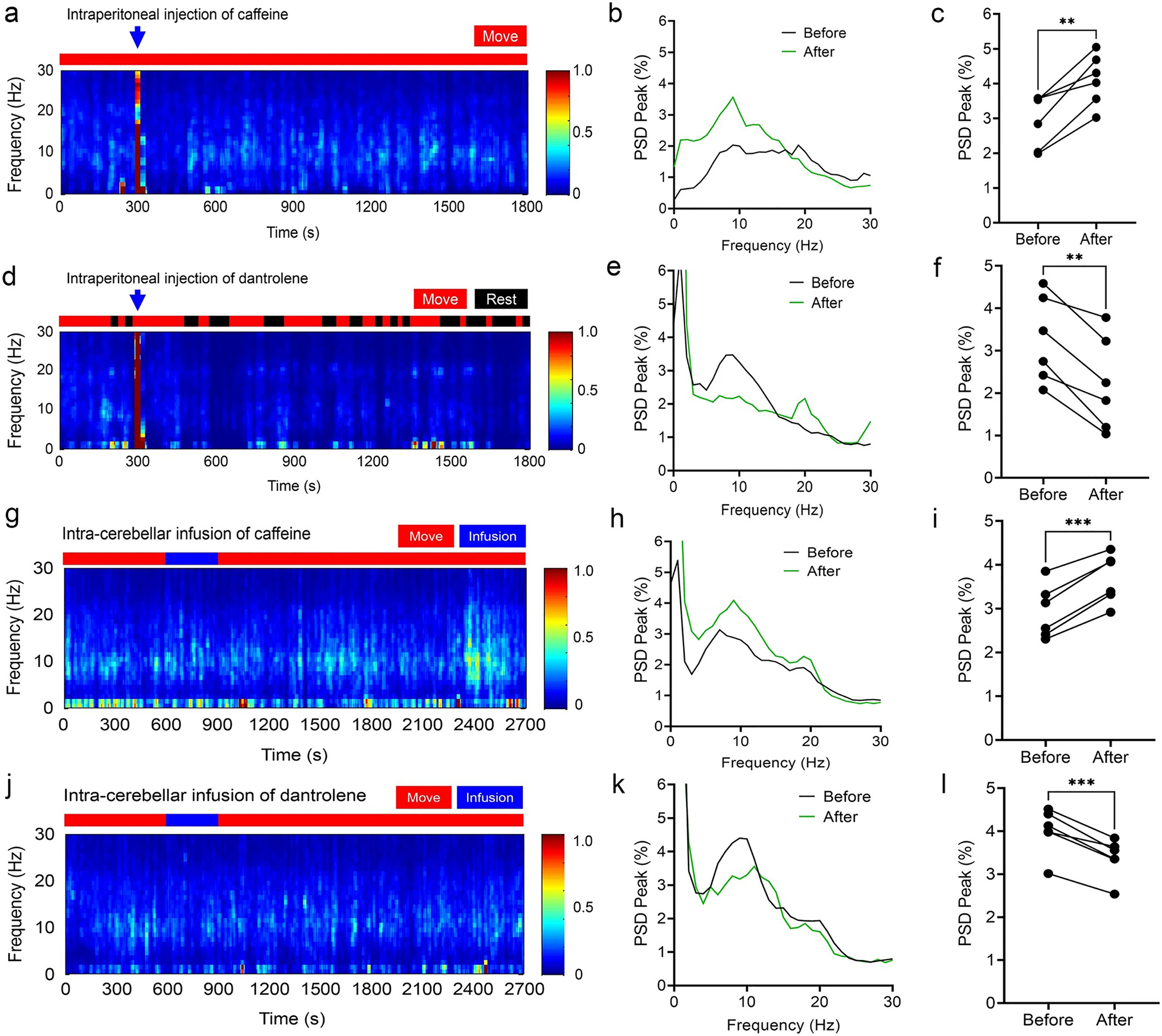
Bi-directional pharmacological manipulation of RyR1 modulates tremor in RyR1 S2844D +/+ mice by agonist caffeine and antagonist dantrolene. a Representative time–frequency plot following intraperitoneal (IP) injections of caffeine in RyR1-S2844D+/+ mice, with b corresponding spectral diagram and c tremor quantification showing increased tremor by IP caffeine injection. d Representative time–frequency plot following IP injections of dantrolene in RyR1-S2844D+/+ mice, with e corresponding spectral diagram and f tremor quantification showing decreased tremor by IP dantrolene injection. (n = 6 mice at 6–12 months old for each ligand. [**p < 0.01 by two-tailed paired t test] g A representative time–frequency plot following cerebellar microinfusion of caffeine in RyR1-S2844D+/+ mice, with h corresponding spectral diagram and i tremor quantification showing increased tremor by intracerebellar caffeine infusion. j A representative time–frequency plot following cerebellar microinfusion of dantrolene in RyR1-S2844D+/+ mice, with k corresponding spectral diagram and l tremor quantification showing decreased tremor by intracerebellar dantrolene infusion. (n = 3 mice at 6–12 months old for each ligand, two independent runs for each mouse). PSD power spectral density. ***p < 0.001, ****p < 0.0001 by two-tailed paired t test
Abnormal cerebellar physiology related to RyR1-S2844D+/+ tremor
Through our decades of long research into the pathophysiology of ET, we have cataloged numerous pathological changes in the cerebellar cortex [54]. As oscillatory activity in the cerebellum has been linked to tremor in one study [71], we recorded cerebellar physiology by examining Local Field Potentials (LFPs) in the mouse cerebellum utilizing a surface electrode in a freely moving setting (Fig. 7a). We observed robust cerebellar oscillatory activity at 10 Hz in RyR1-S2844D+/+ mice, similar to the tremor frequency of ET (Fig. 7b–f). Together, these data suggest that ‘leaky’ RyR1 in the cerebellum can alter cerebellar physiology that could drive a tremor phenotype.
Fig. 7.
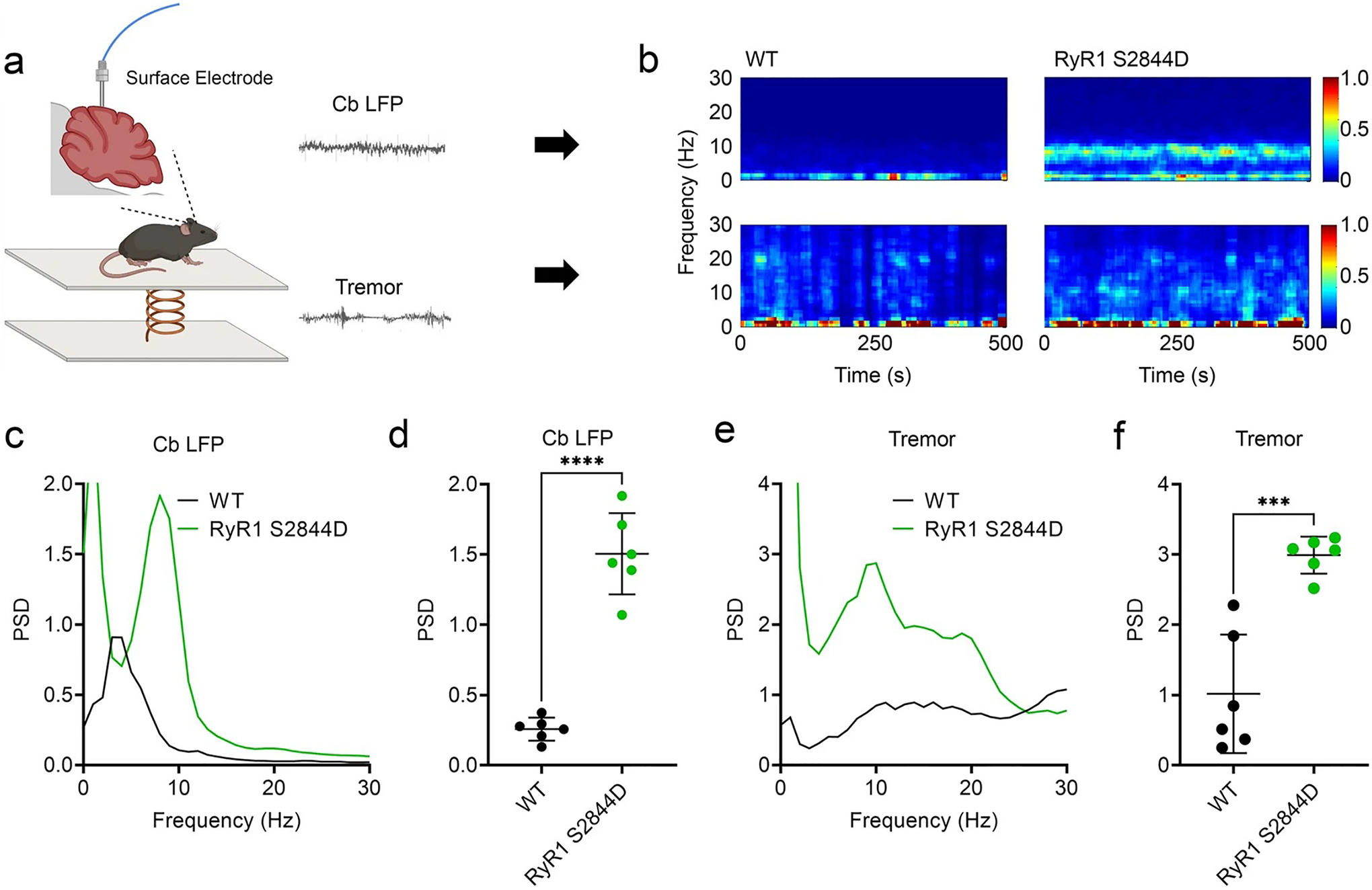
Altered cerebellar physiology in RyR1-S2844D+/+ mice. a Scheme showing simultaneous recordings of tremor and cerebellar local field potentials (LFPs) of lobules V and VI in a freely moving mouse. b Representative time–frequency plots and c, d quantified corresponding spectral diagrams of cerebellar LFP in wild-type (WT) and RyR1-S2844D+/+ mice, with a significant cerebellar LFP spike at 10 Hz in the RyR1-S2844D+/+ mice compared to WT. e A quantified spectral diagram of tremor in a WT mouse and a RyR1-S2844D+/+ mouse, f with significant tremor at 10 Hz in a RyR1-S2844D+/+ mice compared to WT. (n = 3 mice at 6–12 months old for each genotype, two independent runs for each mouse). **p = 0.0016, ****p < 0.0001 by Welch’s paired t test
Stabilizing RyR1 complex reduces tremor
A small molecule compound, Rycal, can stabilize RyR1 channels and prevent RyR1-mediated ER Ca2+ leak [3]. We sought to determine whether tremor and abnormal cerebellar oscillations could be attenuated by systemic treatment with Rycal S107, as this compound can cross the blood–brain barrier [44]. We administered S107 (25 mg/kg) IP to RyR1-S2844D+/+ mice and found that S107 significantly attenuated cerebellar oscillatory activity (Fig. 8a–c) and induced significant tremor suppression (Fig. 8d–f). Altogether, these data show that RyR1-S2844D+/+ mice display a 10 Hz action tremor that can be attenuated via pharmacological intervention by RyR1 channel stabilization.
Fig. 8.
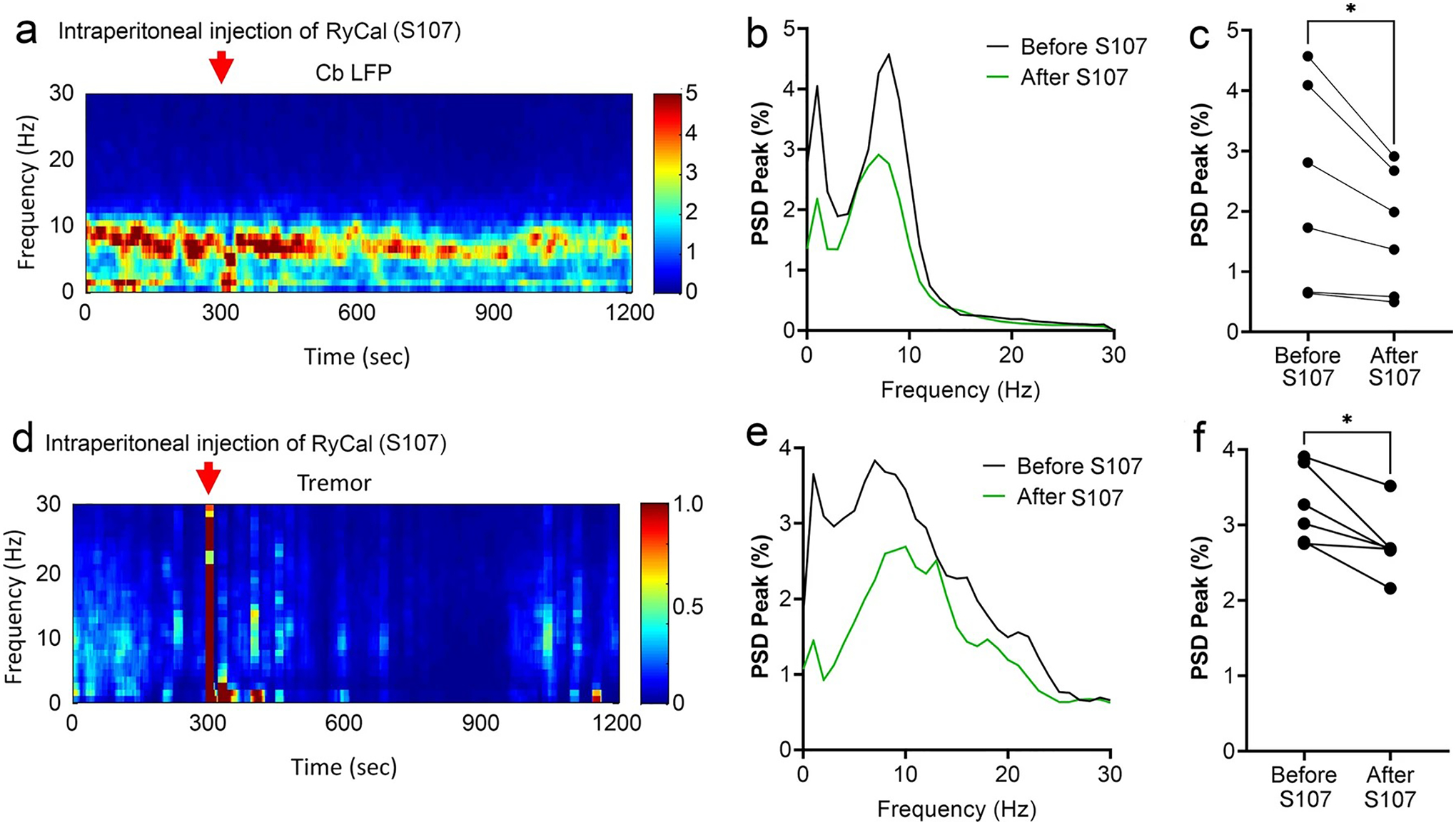
RyCal administration reduces cerebellar oscillatory activity and action tremor in RyR1-S2844D+/+ mice. a A representative time–frequency plot showing cerebellar oscillatory activity in local field potential (LFP) recording following intraperitoneal injection of RyCal, an RyR1 complex stabilizing agent, and b a corresponding spectral diagram demonstrating cerebellar oscillatory activity before and after RyCal administration in RyR1-S2844D+/+ mice, c Quantification of cerebellar LFP shows significantly suppressed cerebellar oscillatory activity in RyR1-S2844D+/+ mice following RyCal treatment. d A representative time–frequency plot showing tremor following intraperitoneal injection of RyCal in a RyR1-S2844D+/+ mouse, and e a corresponding spectral diagram demonstrating tremor before and after RyCal administration. f Tremor quantification shows significantly suppressed tremor in RyR1-S2844D+/+ mice following RyCal treatment. (n = 3 mice at 6–12 months old for each measurement, two independent runs for each mouse); *p < 0.05 by Two-tailed paired t test
RyR1-S2844D+/+ mice recapitulate the human biochemical phenotype of ‘leaky’ RyR1
We next examined the biochemical signature of the RyR1 channel in cerebellum of RyR1-S2844D+/+ versus WT mice and the effect on cerebellar RyR1 after treating RyR1-S2844D+/+ mice with Rycal S107. RyR1-S2844D+/+ mice have the characteristic ‘leaky’ RyR1 channel biochemical signature in cerebellum, with significantly increased oxidation (DNP/RyR1), nitrosylation (Cys-NO/RyR1) and depletion of calstabin1 from RyR1 compared to WT mice (Fig. 9a–d). After 2 months of oral Rycal S107 treatment (50 mg/kg/day), cerebellar RyR1 oxidation was unchanged (Fig. 9a, b), but there was a strong reduction in RyR1 nitrosylation (Fig. 9a, c) and the depletion of calstabin1 from the RyR1 channel complex was rescued (Fig. 9a, d). To further assess RyR1 leak, microsomes were prepared from cerebellum of WT and RyR1-S2844D+/+ mice and separately treated in vitro with the Rycal ARM210 (same mechanism of action as S107 but does not cross the blood–brain barrier) (Fig. 9e). The RyR1-S2844D+/+ mice had significantly more ER Ca2+ leak compared to WT mice, and this leak was again rescued in vitro with Rycal ARM210 treatment (Fig. 9f). Together, these data demonstrate that the RyR1-S2844D+/+ mice recapitulate biochemical aspects observed in our human ET samples, exhibiting ‘leaky’ RyR1 channels in vivo in the cerebellum, and that fixing the RyR1-mediated ER Ca2+ leak with a Rycal compound can reverse the cerebellar RyR1 biochemical remodeling and reduce tremors in mice.
Fig. 9.
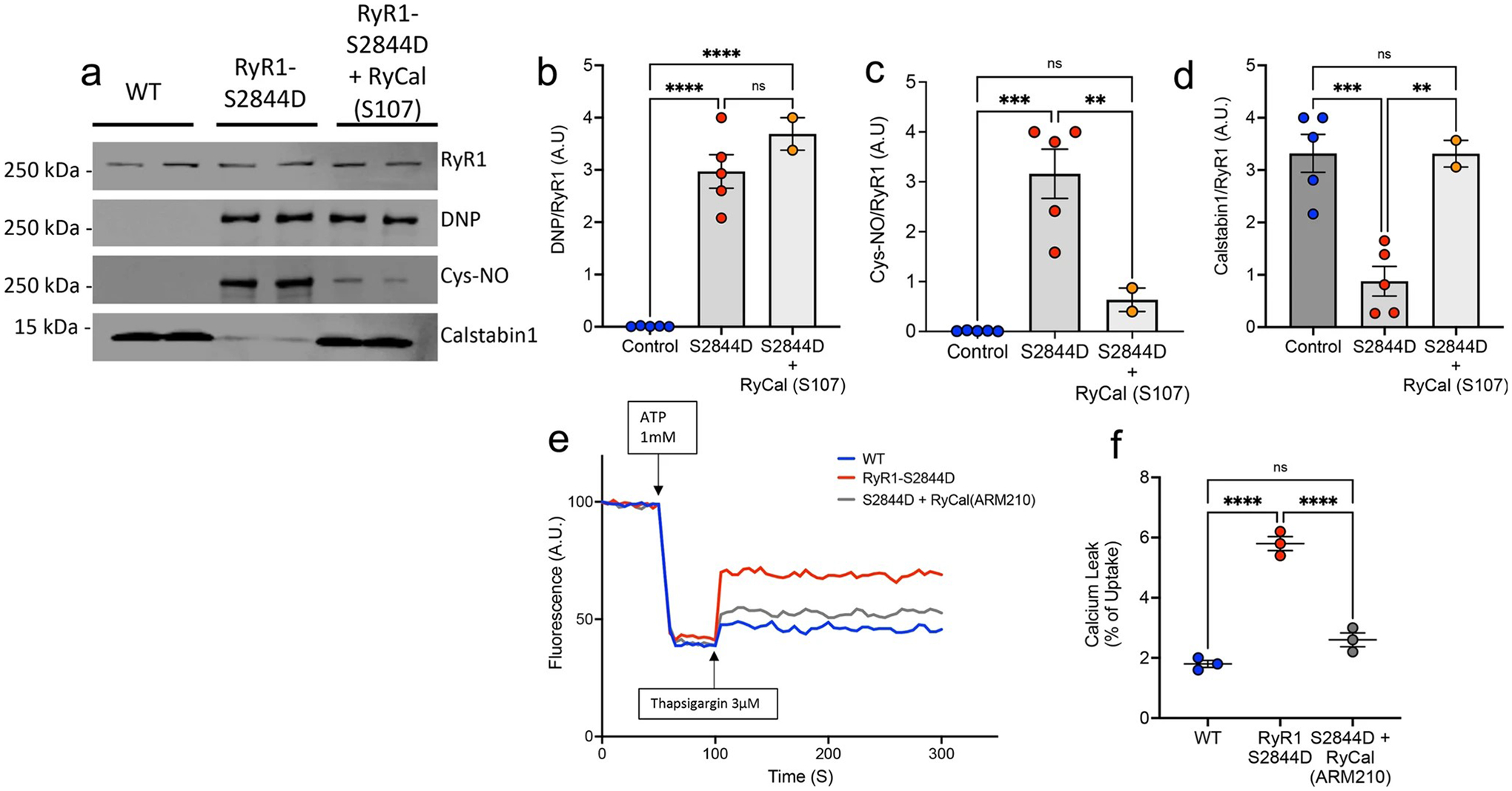
RyR1-S2844D+/+ mice have ‘leaky’ cerebellar RyR1 that is stabilized by RyCal treatment. a Immunoprecipitated RyR1 macromolecular complex probing for total RyR1, oxidation (DNP), nitrosylation (Cys-NO) and calstabin1 in wild-type (WT) and RyR1-S2844D+/+ mouse cerebellum showing characteristic ‘leaky’ RyR1 biochemical signature in mutant mice, and effect of treatment of RyR1-S2844D+/+ mice with RyCal (S107), a RyR1 channel stabilizer that crosses the blood–brain barrier. Quantification of b DNP/RyR1, c Cys-NO/RyR1, d calstabin1/RyR1 in immunoblot (n = 5 for WT and RyR1-S2844D; n = 2 for RyR1-S2844D + RyCal) demonstrates the ‘leaky’ RyR1 channel in untreated RyR1-S2844D+/+ mice, and that RyCal treatment of RyR1-S2844D+/+ mice d normalized calstabin1 binding to RyR1 and c strongly reduced RyR1 nitrosylation, whereas b RyR1 oxidation (DNP) was unaltered. e Representative graph of ER calcium (Ca2+) leak measured in microsomes isolated from WT and RyR1-S2844D+/+ mice cerebellum, demonstrating increased Ca2+ leak in RyR1-S2844D+/+ mice versus WT. Pharmacologically inhibiting ER Ca2+ leak by incubation of RyR1-S2844D+/+ mouse microsomes in vitro with RyCal (ARM210), an RyR1 channel stabilizer, prior to the assay attenuates the Ca2+ leak f Quantification of microsomal Ca2+ release assays. Bar graph represents the leak (after thapsigargin) as a percentage of the Ca2+ uptake after ATP (n = 2). One-way ANOVA with Tukey’s multiple comparisons **p = 0.006, ***p < 0.001, ****p < 0.0001
Discussion
While research into the pathogenesis of ET has identified diverse findings, dysfunction of PCs and Ca2+ signaling abnormalities have both been implicated [50, 62, 63]. PCs are among the principal neurons of the cerebellum and are the sole efferent neuron in the cerebellar cortex, making them immensely important for cerebellar function [24]. While ER Ca2+ handling is a general biological process that occurs in cells throughout the body, the cerebellum and in particular the PC, seem to be highly vulnerable to ER Ca2+ mishandling, and this could lead to neurodegeneration [43]. Notably, PCs are heavily dependent on ER Ca2+ channels for their intrinsic pacemaking activity [23, 33, 81]. As RyR1 is expressed in PCs, our findings of excessively ‘leaky’ RyR1 channels in these samples of human ET cerebellum and from RyR1-S2844D+/+ mice provide what may be a novel molecular link between dysfunction of PC ER Ca2+ handling and certain phenotypes of tremor.
Dysregulation of Ca2+ signaling is strongly implicated in aging and neurologic disease, where elevated intracellular Ca2+ levels can lead to cellular damage, impairment of synaptic neuronal function, and eventually neuronal death [44, 61, 72, 76, 77]. Thus, long-term cerebellar ER Ca2+ leak and resulting cellular excitotoxic damage could be a risk factor for neuronal death, leading to PC loss. PC loss has been observed in several, although not all [50], studies of ET pathology [11, 30], and alterations in CF synapses on PCs are also a pathological feature of ET [42, 57]. In this study, we directly demonstrated that the Kd values for calstabin1-RyR1 channel binding were significantly increased in ET vs. controls (Fig. 2a), consistent with increased dissociation of calstabin1 from RyR1 that would lead to increased ER Ca2+ leak in ET. In addition, this reduced calstabin1-binding affinity for the RyR1 channel strongly correlated with both the extent of PC loss and reduced density of CF synapses on PC dendrites in the ET cerebellum (Fig. 2b, c). The strength of these correlations further implicates that chronically ‘leaky’ cerebellar RyR1 channels may contribute to the loss of synaptic inputs onto PC dendrites and ultimately cell death of PCs. While torpedoes/mm in ET did not significantly correlate with calstabin1-RyR1 Kd values, this is not unexpected as this pathological change has been viewed as partially regenerative and reparative rather than solely degenerative, in contrast to PC loss which is degenerative [50].
The cellular stressors that lead to ‘leaky’ RyR1 channels in ET have not been specifically identified in this study. ET has increasingly been conceptualized as a neurodegenerative disease [49], which in and of itself is a highly stressed cellular state. Furthermore, the intrinsically high firing rate of PCs requires a high cellular metabolic demand that predisposes them to cellular stress [27]. Investigations into familial ET have found isolated mutations in plasmalemmal T-type Ca2+ channels [69], which may contribute to aberrant Ca2+ influx in PCs and result in increased RyR1-mediated ER Ca2+ release through a Ca2+-induced Ca2+-release process [76]. While our recent RNA transcriptome studies highlighted defects in ER function and Ca2+ signaling pathways in ET [62, 63], genetic mutations in RyR1 have not been linked to ET in several GWAS analyses and genome studies in ET families [29]. Instead, the findings in this study implicate cellular stress in the cerebellum as a potential contributor to the post-translational remodeling of RyR1 channels that may then contribute to ET pathogenesis. It is noteworthy that ‘leaky’ RyR1 channels in this study are only seen in the ET cerebellar samples studied herein, and not in those from controls or another tremor disorder, PD. As RyR1 is highly expressed in cerebellar PCs, and current studies of ET pathology center around PC degeneration and dysfunction, this study provides a further convergence of findings suggesting that the RyR1 biochemical remodeling we observe could reflect a distinct biological stressor in ET PCs. However, significantly more samples from ET as well as samples from other cerebellar neurodegenerative disorders need to be examined to fully test this hypothesis.
Genetic mutations in RyR1 are known to cause malignant hyperthermia and several types of myopathies, with both autosomal dominant and autosomal recessive inheritance patterns [38]. ‘Leaky’ RyR1 channels have been identified in skeletal muscle biopsies from 17 individuals with RyR1-related myopathies, with mutations spanning domains across the large RyR1 molecule [35]. It is probable that individuals with RyR1-related myopathies or malignant hyperthermia would also have ‘leaky’ RyR1 channels in cerebellar PCs, but these genetic disorders are not known to be clinically associated with tremor, a finding also observed in a clinical trial of Rycal for treatment of RyR1-related myopathy (NCT04141670; Dr. P. Mohassel, personal communication). Thus, there may be additional factors that determine whether an individual with leaky RyR1 channels exhibits tremor and the mechanism whereby ‘leaky’ cerebellar RyR1 channels leads to tremor remains to be determined.
Environmental exposures likely play a role in the pathogenesis of ET, including heavy metals such as lead and the tremorgenic β-carboline alkaloid harmane, both of which are elevated in the blood of ET patients compared to controls [14, 48, 53]. The neurotoxicity of lead [2] and harmane [75] may be related to increased generation of reactive oxygen species, as well as an imbalanced metabolism and cellular dysregulation of redox resulting in excessive free radicals [80]. RyR1 is particularly redox-sensitive, as the channel has approximately 100 cysteine residues per protomer, and almost half of these thiols are maintained at a reduced state under resting conditions [32]. This makes the RyR1 channel susceptible to nitrosylative and oxidative post-translational modifications [5]. Additionally, numerous genes and pathways associated with oxidative stress were found in our RNA-sequencing study of enriched PCs in ET [63]. These data support the notion that ET could be a disease of genetic predispositions with environmental influences, converging at RyR1 and the ER Ca2+ handling pathway.
Another key finding in this study is that expression of a single phospho-mimetic point mutation in RyR1 (S2844D) in a homozygous state is sufficient to induce neuronal intracellular ER Ca2+ leak in cerebellum associated with a 10 Hz action tremor in mice, a similar tremor frequency as seen in ET. Furthermore, we found that this tremor is not only linked to abnormal cerebellar activity, but additionally that the tremor, cerebellar oscillations and RyR1 biochemical remodeling can be modulated by RyR1 pharmacological treatments (caffeine, dantrolene, Rycals) in RyR1-S2844D+/+ mice. The power spectral density (PSD) peak at the tremor frequency is a key parameter used to objectively measure the severity of tremor in animal models and is the same method to quantify tremor severity as recorded by accelerometers in ET patients [26, 79]. Therefore, the methods and results in this study are directly applicable to clinical measurements used in humans. In addition, burst stimulation of parallel fibers in mouse cerebellum generates a localized increase in nitric oxide (NO) concentration in PC dendrites, and this NO then directly activates ER Ca2+ release by reversible S-nitrosylation of RyR1, a process termed NO-induced Ca2+ release [31, 32]. It is of interest that Rycal treatment of RyR1-S2844D+/+ mice also significantly reduced the Cys-nitrosylation of cerebellar RyR1 on biochemical analysis (Fig. 8), raising the possibility that excessive NO may contribute to the abnormal cerebellar oscillations observed in RyR1-S2844D+/+ mice. Notably, excessive cerebellar oscillations that correlate with tremor severity have been observed in one study in individuals with ET [71]. Thus, the RyR1-S2844D+/+ mice have a form of action tremor and ‘leaky’ cerebellar RyR1, similar to human ET, and could provide a novel preclinical model to test the efficacy of Rycals and other drugs for tremor therapy.
Both RyR1 (expressed in PCs) and RyR2 (expressed in granule cells and other cerebellar interneurons) are present in cerebellar cortex, but the current study suggests that it is specifically RyR1 remodeling that is associated with ET. Biochemical remodeling of the RyR2 isoform was only seen in cerebellum of individuals over age 60 years. We postulate that this could indicate a cell-type and disease-dependent vulnerability of RyR channels that are distinct to different cerebellar neuronal populations. Age-dependent ER Ca2+ leak in RyR channels occurs due to progressive oxidative stress [3] and may lead to sustained intracellular Ca2+ elevations and excitotoxicity in aging neurons [12, 77]. As normal age-dependent loss of PCs in humans is accelerated after age 60 [25], this raises the possibility that defective RyR2-mediated ER Ca2+ handling in granule cells and/or other cerebellar interneurons may be a contributing factor to age-dependent PC death. It has also been noted that features of clinical disease progression in ET often worsen after ~ age 60 years, raising the possibility that age-dependent onset of RyR2-mediated defective ER Ca2+ handling in cerebellar interneurons may further accentuate PC dysfunction and/or loss in the setting of excessive RyR1 Ca2+ leak as seen in ET PCs [47]. In contrast to RyR2, increased RyR1 biochemical remodeling was not seen in human control cerebellar samples across a wide age range. However, we are not able to determine when RyR1 remodeling might begin in ET as younger individuals with ET are not available to us for study.
In spinocerebellar ataxias (SCAs), it is proposed that multiple cellular pathways in PCs are disrupted by inherited gene mutations, but SCA pathogenesis does not begin until Ca2+ signaling pathways are disturbed [16, 73]. Recently, we have highlighted clinical and pathological similarities between ET and SCAs [54, 57] and propose that ET is a mild form of cerebellar degeneration [49]. In addition to RyR1 channels, PCs prominently express the 1,4,5-inositol trisphosphate receptor type 1 (IP3R1), an intracellular Ca2+ release channel closely related to RyR channels that is activated by inositol-1,4,5-trisphosphate generated after upstream activation of G protein-coupled receptors, including the plasmalemmal metabotropic glutamate receptor type 1. Both overlapping and distinct roles for RyR1 versus IP3R1 ER Ca2+ channels have been demonstrated in PCs [7, 17, 68]. Future studies are needed to determine if there are defects in IP3R1 or other molecules involved in ER Ca2+ handling in ET and RyR1-S2844D+/+ mice.
The RyR1-S2844D+/+ mice used in this study have been extensively studied [3], however, this is the first detailed description of action tremor in these mice. It should be noted that these mice are not presented here as an animal model of ET, as the similarities to the human disease are currently limited to the ‘leaky’ RyR1 biochemical signature in cerebellum and a similar frequency of action tremor. Indeed, several other human tremors may have a 10 Hz frequency, such as enhanced physiologic tremor and possibly dystonic tremor [41]. Other clinical features in ET, such as its chronic progressive nature are not evident in these mice, and the extent to which the array of pathologic findings described in human ET may be observed in the RyR1-S2844D+/+ mice requires future studies. Nonetheless, this is the first study in humans demonstrating that a ‘leaky’ RyR1 biochemical signature is associated with ET, a common human central nervous system disorder. Therefore, these mice, that also have ‘leaky’ cerebellar RyR1 channels, may provide some insight into general mechanisms underlying action tremor.
In conclusion, we show that the ‘leaky’ RyR1 channel biochemical signature was present in human ET cerebellar samples we studied. This initial study provides preliminary evidence for a novel molecular link between dysfunction of PC ER Ca2+ handling and certain phenotypes of tremor. Further research is warranted to examine the extent of ‘leaky’ RyR1 channels in more ET patients, more controls and other tremor-related disorders or cerebellar degenerations, in addition to the prospective efficacy of RyR1 channel-stabilizing drugs for the treatment/prevention of tremor disorders.
Supplementary Material
Acknowledgements
Human brain tissue was derived from the New York Brain Bank at Columbia University and the NIH NeuroBioBank (University of Miami, Miami FL; University of Maryland, Baltimore MD).
We would like to thank all the patients and families that contributed to brain donation and the staff at the ETCBR at the New York Brain Bank and the NIH NeuroBioBank. We thank Dr. Marco C. Miotti for creating the illustration of the RyR1 macromolecular complex of kinases, phosphatases, and accessory scaffold proteins.
Funding
Funding for this project was provided from the National Institutes of Health (NIH)/NINDS (R01 NS124854 [SHK and PLF], R01 NS118179 [SHK], R01 NS117745 [PLF and EDL], RF1 NS114570 [ARM], R01 NS104423 [SHK], R01 NS088257 [EDL], R01 NS086736 [EDL]; from the NIH/NHLBI (R25 HL156002 [ARM], R01 HL145473 [ARM], R01 HL140934 [ARM], R01 HL142903 [ARM], T32 HL120826 [ARM]) and from the NIH/NIDDK (R01 DK118240 [ARM]).
Footnotes
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s00401-023-02602-z.
Conflict of interest ARM and Columbia University own shares in ARMGO Pharma, Inc. a biotech company developing RyR targeted therapeutics. All other authors declare no competing interests or conflicts.
Data availability
Basic, anonymized demographic, clinical and tissue data are available as Supplemental Online material.
References
- 1.Abu-Omar N, Das J, Szeto V, Feng ZP (2018) Neuronal ryanodine receptors in development and aging. Mol Neurobiol 55(2):1183–1192. 10.1007/s12035-016-0375-4 [DOI] [PubMed] [Google Scholar]
- 2.Almeida Lopes ACB, Peixe TS, Mesas AE, Paoliello MMB (2016) Lead exposure and oxidative stress: a systematic review. Rev Environ Contam Toxicol 236:193–238. 10.1007/978-3-319-20013-2_3 [DOI] [PubMed] [Google Scholar]
- 3.Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W et al. (2011) Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab 14(2):196–207. 10.1016/j.cmet.2011.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Babij R, Lee M, Cortes E, Vonsattel JP, Faust PL, Louis ED (2013) Purkinje cell axonal anatomy: quantifying morphometric changes in essential tremor versus control brains. Brain 136(Pt 10):3051–3061. 10.1093/brain/awt238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L et al. (2009) Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med 15(3):325–330. 10.1038/nm.1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benito-Leon J, Labiano-Fontcuberta A (2016) Linking essential tremor to the cerebellum: clinical evidence. Cerebellum 15(3):253–262. 10.1007/s12311-015-0741-1 [DOI] [PubMed] [Google Scholar]
- 7.Bezprozvanny I, Watras J, Ehrlich BE (1991) Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351:751–754. 10.1038/351751a0 [DOI] [PubMed] [Google Scholar]
- 8.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112(4):389–404. 10.1007/s00401-006-0127-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braak H, Braak E (1997) Diagnostic criteria for neuropathologic assessment of Alzheimer’s disease. Neurobiol Aging 18:S85–S88. 10.1016/s0197-4580(97)00062-6 [DOI] [PubMed] [Google Scholar]
- 10.Cerasa A, Quattrone A (2016) Linking essential tremor to the cerebellum-neuroimaging evidence. Cerebellum 15(3):263–275. 10.1007/s12311-015-0739-8 [DOI] [PubMed] [Google Scholar]
- 11.Choe M, Cortes E, Vonsattel JP, Kuo SH, Faust PL, Louis ED (2016) Purkinje cell loss in essential tremor: random sampling quantification and nearest neighbor analysis. Mov Disord 31(3):393–401. 10.1002/mds.26490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clodfelter GV, Porter NM, Landfield PW, Thibault O (2002) Sustained Ca2+ −induced Ca2+ −release underlies the postglutamate lethal Ca2+ plateau in older cultured hippocampal neurons. Eur J Pharmacol 447:189–200. 10.1016/s0014-2999(02)01843-5 [DOI] [PubMed] [Google Scholar]
- 13.De Zeeuw CI (2021) Bidirectional learning in upbound and downbound microzones of the cerebellum. Nat Rev Neurosci 22(2):92–110. 10.1038/s41583-020-00392-x [DOI] [PubMed] [Google Scholar]
- 14.Dogu O, Louis ED, Tamer LT, Unal O, Yilmaz A, Kaleagasi H (2007) elevated blood lead concentrations in essential tremor: a case-control study in Mersin, Turkey. Environ Health Perspect 115(11):1564–1568. 10.1289/ehp.10352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dridi H, Liu X, Yuan Q, Reiken S, Yehia M, Sittenfeld L et al. (2020) Role of defective calcium regulation in cardiorespiratory dysfunction in Huntington’s disease. JCI Insight. 10.1172/jci.insight.140614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duenas AM, Goold R, Giunti P (2006) Molecular pathogenesis of spinocerebellar ataxias. Brain 129(Pt 6):1357–1370. 10.1093/brain/awl081 [DOI] [PubMed] [Google Scholar]
- 17.Egorova PA, Bezprozvanny IB (2022) Electrophysiological studies support utility of positive modulators of SK channels for the treatment of spinocerebellar ataxia type 2. Cerebellum 21(5):742–749. 10.1007/s12311-021-01349-1 [DOI] [PubMed] [Google Scholar]
- 18.Erickson-Davis CR, Faust PL, Vonsattel J-PG, Gupta S, Honig LS, Louis ED (2010) ‘“Hairy Baskets”‘ associated with degenerative Purkinje cell changes in essential tremor. Neuropathol Exp Neurol 69(3):262–271. 10.1097/NEN.0b013e3181d1ad04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fierro L, Llano I (1996) High endogenous calcium buffering in Purkinje cells from rat cerebellar slices. J Physiol 496(3):617–625. 10.1113/jphysiol.1996.sp021713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fierro L, DiPolo R, Llano I (1998) Intracellular calcium clearance in Purkinje cell somata from rat cerebellar slices. J Physiol 510:499–512. 10.1111/j.1469-7793.1998.499bk.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Filip P, Lungu OV, Manto MU, Bares M (2016) Linking essential tremor to the cerebellum: physiological evidence. Cerebellum 15(6):774–780. 10.1007/s12311-015-0740-2 [DOI] [PubMed] [Google Scholar]
- 22.Giannini G, Conti A, Mammarella S, Scrobogna M, Sorrentino V (1995) The ryanodine receptor: calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. JCB 128(5):893–904. 10.1083/jcb.128.5.893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomez LC, Kawaguchi S-Y, Collin T, Jalil A, Del Pilar M, Gomez EN et al. (2020) Influence of spatially segregated IP 3-producing pathways on spike generation and transmitter release in Purkinje cell axons. Proc Natl Acad Sci 117(20):11097–11108. 10.1073/pnas.2000148117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haines DE, Dietrichs E (2012) The cerebellum—structure and connections. Handb Clin Neurol 103:3–36. 10.1016/B978-0-444-51892-7.00001-2 [DOI] [PubMed] [Google Scholar]
- 25.Hall T, Miller A, Corsellis J (1975) Variations in the human Purkinje cell population according to age and sex. Neuropathol Exp Neurol 1(3):267–292. 10.1111/j.1365-2990.1975.tb00652.x [DOI] [Google Scholar]
- 26.Haubenberger D, Abbruzzese G, Bain PG, Bajaj N, Benito-León J, Bhatia KP et al. (2016) Transducer-based evaluation of tremor. Mov Disord 31(9):1327–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang M, Verbeek DS (2019) Why do so many genetic insults lead to Purkinje Cell degeneration and spinocerebellar ataxia? Neurosci Lett 688:49–57. 10.1016/j.neulet.2018.02.004 [DOI] [PubMed] [Google Scholar]
- 28.Jayaraman T, Brillantes AM, Timerman AP, Fleischer S, Erdjument-Bromage H, Tempst P et al. (1992) FK506 binding protein associated with the calcium release channel (ryanodine receptor). J Biol Chem 267(14):9474–9477. 10.1016/S0021-9258(19)50114-4 [DOI] [PubMed] [Google Scholar]
- 29.Jimenez-Jimenez FJ, Alonso-Navarro H, Garcia-Martin E, Alvarez I, Pastor P, Agundez JAG (2021) Genomic markers for essential tremor. Pharmaceuticals (Basel). 10.3390/ph14060516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Axelrad JE, Louis ED, Honig LS, Flores I, Ross W, Pahwa R et al. (2008) Reduced Purkinje cell number in essential tremor: a postmortem study. Arch Neurol 65(1):101–107. 10.1001/archneurol.2007.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kakizawa S, Yamazawa T, Chen Y, Ito A, Murayama T, Oyamada H et al. (2012) Nitric oxide-induced calcium release via ryanodine receptors regulates neuronal function. EMBO J 31(2):417–428. 10.1038/emboj.2011.386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kakizawa S, Yamazawa T, Iino M (2013) Nitric oxide-induced calcium release: activation of type 1 ryanodine receptor by endogenous nitric oxide. Channels (Austin) 7(1):1–5. 10.4161/chan.22555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khodakhah K, Armstrong CM (1997) Inositol trisphosphate and ryanodine receptors share a common functional Ca2+ pool in cerebellar Purkinje neurons. Biophys J 73(6):3349–3357. 10.1016/S0006-3495(97)78359-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuo SH, Erickson-Davis C, Gillman A, Faust PL, Vonsattel JP, Louis ED (2011) Increased number of heterotopic Purkinje cells in essential tremor. JNNP 82(9):1038–1040. 10.1136/jnnp.2010.213330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kushnir A, Todd JJ, Witherspoon JW, Yuan Q, Reiken S, Lin H et al. (2020) Intracellular calcium leak as a therapeutic target for RYR1-related myopathies. Acta Neuropathol 136(6):1089–1104. 10.1007/s00401-020-02150-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kushnir A, Wajsberg B, Marks AR (2018) Ryanodine receptor dysfunction in human disorders. BBA-Mol Cell Res 1865(11 Pt B):1687–1697. 10.1016/j.bbamcr.2018.07.011 [DOI] [PubMed] [Google Scholar]
- 37.Lacampagne A, Liu X, Reiken S, Bussiere R, Meli AC, Lauritzen I et al. (2017) Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol 134(5):749–767. 10.1007/s00401-017-1733-7 [DOI] [PubMed] [Google Scholar]
- 38.Lawal TA, Todd JJ, Witherspoon JW, Bönnemann CG, Dowling JJ, Hamilton SL et al. (2020) Ryanodine receptor 1-related disorders: an historical perspective and proposal for a unified nomenclature. Skelet Muscle. 10.1186/s13395-020-00243-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee D, Gan S, Faust P, Louis E, Kuo S (2018) Climbing fiberPurkinje cell synaptic pathology across essential tremor subtypes. Parkinsonism Relat Disord 51:24–29. 10.1016/j.parkreldis.2018.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee PJ, Kerridge CA, Chatterjee D, Koeppen AH, Faust PL, Louis ED (2018) A quantitative study of empty baskets in essential tremor and other motor neurodegenerative diseases. J Neuropathol Exp Neurol. 10.1093/jnen/nly114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lenka A, Jankovic J (2021) Tremor syndromes: an updated review. Front Neurol. 10.3389/fneur.2021.684835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin C-Y, Louis ED, Faust PL, Koeppen AH, Vonsattel J-PG, Kuo S-H (2014) Abnormal climbing fibre-Purkinje cell synaptic connections in the essential tremor cerebellum. Brain 137:3149–3159. 10.1093/brain/awu281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu J, Tang TS, Tu H, Nelson O, Herndon E, Huynh DP et al. (2009) Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci 29(29):9148–9162. 10.1523/JNEUROSCI.0660-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu X, Betzenhauser MJ, Reiken S, Meli AC, Xie W, Chen B-X et al. (2012) Role of leaky neuronal ryanodine receptors in stress- induced cognitive dysfunction. Cell 150(5):1055–1067. 10.1016/j.cell.2012.06.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Louis ED (2013) The primary type of tremor in essential tremor is kinetic rather than postural: cross-sectional observation of tremor phenomenology in 369 cases. Eur J Neurol 20(4):725–727. 10.1111/j.1468-1331.2012.03855.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Louis ED (2016) Linking essential tremor to the cerebellum: neuropathological evidence. Cerebellum 15(3):235–242. 10.1007/s12311-015-0692-6 [DOI] [PubMed] [Google Scholar]
- 47.Louis ED (2019) The roles of age and aging in essential tremor: an epidemiological perspective. Neuroepidemiology 52(1–2):111–118. 10.1159/000492831 [DOI] [PubMed] [Google Scholar]
- 48.Louis ED, Factor-Litvak P, Gerbin M, Slavkovich V, Graziano JH, Jiang W et al. (2011) Blood harmane, blood lead, and severity of hand tremor: evidence of additive effects. Neurotoxicology 32:227–232. 10.1016/j.neuro.2010.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Louis ED, Faust PL (2020) Essential tremor: the most common form of cerebellar degeneration? Cerebellum Ataxias 7(12):1–10. 10.1186/s40673-020-00121-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Louis ED, Faust PL (2020) Essential tremor pathology: neurodegeneration and reorganization of neuronal connections. Nat Rev Neurol 16(2):69–83. 10.1038/s41582-019-0302-1 [DOI] [PubMed] [Google Scholar]
- 51.Louis ED, Faust PL, Vonsattel JP, Honig LS, Rajput A, Rajput A et al. (2009) Torpedoes in Parkinson’s disease, Alzheimer’s disease, essential tremor, and control brains. Mov Disord 24(11):1600–1605. 10.1002/mds.22567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Louis ED, Faust PL, Vonsattel JP, Honig LS, Rajput A, Robinson CA et al. (2007) Neuropathological changes in essential tremor: 33 cases compared with 21 controls. Brain 130(Pt 12):3297–3307. 10.1093/brain/awm266 [DOI] [PubMed] [Google Scholar]
- 53.Louis ED, Jurewicz EC, Applegate L, Factor-Litvak P, Parides M, Andrews L et al. (2003) Association between essential tremor and blood lead concentration. Environ Health Perspect 111(14):1707–1711. 10.1289/ehp.6404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Louis ED, Kerridge CA, Chatterjee D, Martuscello RT, Diaz DT, Koeppen AH et al. (2019) Contextualizing the pathology in the essential tremor cerebellar cortex: a patholog-omics approach. Acta Neuropathol 138(5):859–876. 10.1007/s00401-019-02043-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Louis ED, Kuo SH, Tate WJ, Kelly GC, Gutierrez J, Cortes EP et al. (2018) Heterotopic Purkinje Cells: a Comparative Postmortem Study of Essential Tremor and Spinocerebellar Ataxias 1, 2, 3, and 6. Cerebellum 17(2):104–110. 10.1007/s12311-017-0876-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Louis ED, Lee M, Babij R, Ma K, Cortes E, Vonsattel JP et al. (2014) Reduced Purkinje cell dendritic arborization and loss of dendritic spines in essential tremor. Brain 137(Pt 12):3142–3148. 10.1093/brain/awu314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Louis ED, Martuscello RT, Gionco JT, Hartstone WG, Musacchio JB, Portenti M et al. (2023) Histopathology of the cerebellar cortex in essential tremor and other neurodegenerative motor disorders: comparative analysis of 320 brains. Acta Neuropathol 145(3):265–283. 10.1007/s00401-022-02535-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Louis ED, McCreary M (2021) How common is essential tremor? Update on the worldwide prevalence of essential tremor. TOHM. 10.5334/tohm.632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Louis ED, Ottman R (2014) How many people in the USA have essential tremor? Deriving a population estimate based on epidemiological data. TOHM. 10.7916/D8TT4P4B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Louis ED, Yi H, Erickson-Davis C, Vonsattel JP, Faust PL (2009) Structural study of Purkinje cell axonal torpedoes in essential tremor. Neurosci Lett 450(3):287–291. 10.1016/j.neulet.2008.11.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marks AR (1997) Intracellular calcium-release channels: regulators of cell life and death. Am J Physiol 2727(2):H597–605. 10.1152/ajpheart.1997.272.2.H597 [DOI] [PubMed] [Google Scholar]
- 62.Martuscello RT, Kerridge CA, Chatterjee D, Hartstone WG, Kuo SH, Sims PA et al. (2020) Gene expression analysis of the cerebellar cortex in essential tremor. Neurosci Lett 721:134540. 10.1016/j.neulet.2019.134540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martuscello RT, Sivprakasam K, Hartstone WG, Kuo S-H, Konopka G, Louis ED et al. (2022) Gene expression analysis of laser captured Purkinje cells in the essential tremor cerebellum. Cerebellum. 10.1007/s12311-022-01483-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marx S, Reiken S, Hisamatsu Y, Gaburjakova M, Gaburjakova J, Yang Y et al. (2001) Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. J Cell Biol 153(4):699–708. 10.1083/jcb.153.4.699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marx S, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N et al. (2000) PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101(4):365–376. 10.1016/s0092-8674(00)80847-8 [DOI] [PubMed] [Google Scholar]
- 66.Mirra SS (1997) The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: a commentary. Neurobiol Aging 18:S91–S94. 10.1016/s0197-4580(97)00058-4 [DOI] [PubMed] [Google Scholar]
- 67.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al. (2011) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123(1):1–11. 10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mouton J, Marty I, Villaz M, Feltz A, Maulet Y (2001) Molecular interaction of dihydropyridine receptors with type-1 ryanodine receptors in rat brain. Biochem J 354:597–603. 10.1042/0264-6021:3540597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Odgerel Z, Hernandez N, Park J, Ottman R, Louis E, Clark L (2019) Whole genome sequencing and rare variant analysis in essential tremor families. PLoS ONE 14(8):e0220512. 10.1371/journal.pone.0220512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Otsu Y, Marcaggi P, Feltz A, Isope P, Kollo M, Nusser Z et al. (2014) Activity-dependent gating of calcium spikes by A-type K+ channels controls climbing fiber signaling in Purkinje cell dendrites. Neuron 84(1):137–151. 10.1016/j.neuron.2014.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pan MK, Li YS, Wong SB, Ni CL, Wang YM, Liu WC et al. (2020) Cerebellar oscillations driven by synaptic pruning deficits of cerebellar climbing fibers contribute to tremor pathophysiology. Sci Transl Med 12(526):eaay1769. 10.1126/scitranslmed.aay1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pchitskaya E, Popugaeva E, Bezprozvanny I (2018) Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70:87–94. 10.1016/j.ceca.2017.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prestori F, Moccia F, D’Angelo E (2019) Disrupted calcium signaling in animal models of human spinocerebellar ataxia (SCA). Int J Mol Sci. 10.3390/ijms21010216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reiken S, Lacampagne A, Zhou H, Kherani A, Lehnart SE, Ward C et al. (2003) PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J Cell Biol 160(7):919–928. 10.1083/jcb.200211012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sammi SR, Agim ZS, Cannon JR (2018) Harmane-induced selective dopaminergic neurotoxicity in Caenorhabditis elegans. Toxicol Sci 161(2):335–348. 10.1093/toxsci/kfx223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Santulli G, Marks AR (2015) Essential roles of intracellular calcium release channels in muscle, brain, metabolism, and aging. Curr Mol Pharmacol 8(2):206–222. 10.2174/1874467208666150507105105 [DOI] [PubMed] [Google Scholar]
- 77.Thibault O, Gant JC, Landfield PW (2007) Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: minding the store. Aging Cell 6(3):307–317. 10.1111/j.1474-9726.2007.00295.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A et al. (2015) Tissue-based map of the human proteome. Science. 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 79.Vial F, Kassavetis P, Merchant S, Haubenberger D, Hallett M (2019) How to do an electrophysiological study of tremor. Clin Neurophysiol Pract 4:134–142. 10.1016/j.cnp.2019.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilkins HM, Kirchhof D, Manning E, Joseph JW, Linseman DA (2013) Mitochondrial glutathione transport is a key determinant of neuronal susceptibility to oxidative and nitrosative stress. J Biol Chem 288(7):5091–5101. 10.1074/jbc.M112.405738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Womack MD, Walker JW, Khodakhah K (2000) Impaired calcium release in cerebellar Purkinje neurons maintained in culture. J Gen Physiol. 10.1085/jgp.115.3.339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu M, Ma K, Faust PL, Honig LS, Cortes E, Vonsattel JP et al. (2012) Increased number of Purkinje cell dendritic swellings in essential tremor. Eur J Neurol 19(4):625–630. 10.1111/j.1468-1331.2011.03598.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zalk R, Lehnart SE, Marks AR (2007) Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem 76:367–85. https://www.annualreviews.org/doi/https://doi.org/10.1146/annurev.biochem.76.053105.094237 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Basic, anonymized demographic, clinical and tissue data are available as Supplemental Online material.