

Abstract
Kratom is a botanical natural product belonging to the coffee family, with stimulant effects at low doses and opioid-like effects at higher doses. During the last two decades, kratom has been purported as a safer alternative to pharmaceutical and illicit drugs to self-manage pain and opioid withdrawal symptoms. Kratom alkaloids, typically mitragynine, have been detected in biologic samples from overdose deaths. These deaths are often observed in combination with other drugs and are suspected to result from polyintoxications. This review focuses on the potential for kratom to precipitate pharmacokinetic interactions with object drugs involved in these reported polyintoxications. The legal status, chemistry, pharmacology, and toxicology are also summarized. The aggregate in vitro and clinical data identified kratom and select kratom alkaloids as modulators of cytochrome P450 (P450) enzyme activity, notably as inhibitors of CYP2D6 and CYP3A, as well as P-glycoprotein–mediated efflux activity. These inhibitory effects could increase the systemic exposure to co-consumed object drugs, which may lead to adverse effects. Collectively, the evidence to date warrants further evaluation of potential kratom-drug interactions using an iterative approach involving additional mechanistic in vitro studies, well designed clinical studies, and physiologically based pharmacokinetic modeling and simulation. This critical information is needed to fill knowledge gaps regarding the safe and effective use of kratom, thereby addressing ongoing public health concerns.
SIGNIFICANCE STATEMENT
The botanical kratom is increasingly used to self-manage pain and opioid withdrawal symptoms due to having opioid-like effects. The legal status, chemistry, pharmacology, toxicology, and drug interaction potential of kratom are reviewed. Kratom-associated polyintoxications and in vitro-in vivo extrapolations suggest that kratom can precipitate pharmacokinetic drug interactions by inhibiting CYP2D6, CYP3A, and P-glycoprotein. An iterative approach that includes clinical studies and physiologically based pharmacokinetic modeling and simulation is recommended for further evaluation of potential unwanted kratom-drug interactions.
Introduction
The kratom plant (Mitragyna speciosa Korth.) is one of ten species from the Mitragyna genus belonging to the coffee family (Rubiaceae) (Brown et al., 2017; Ramanathan et al., 2021). Kratom has been valued for multiple purported medicinal properties since at least the 19th century (Field, 1921). Native to Southeast Asia, this tropical tree is prevalent in Thailand (ithang, kratom, thom), Indonesia (keton, kadamba), Malaysia (biak-biak, ketum), Myanmar (beinsa), Philippines (mambog, lugub, polapoput), and Vietnam (giam) (Grundmann et al., 2023). As a long-standing cultural tradition, manual laborers, particularly farmers and fishermen, chew fresh kratom leaves for the stimulant properties to prevent fatigue (Singh et al., 2015). The leaves also are chewed, smoked, and consumed as a tea during religious ceremonies (Singh et al., 2017). Higher quantities of the leaves are used for pain-relieving and relaxing properties. These properties, along with the wide availability and relatively low cost, led to kratom becoming a substitute for opium in some countries (Brown et al., 2017). Other traditional uses of kratom include appetite suppression and treatment of stomach cramps, diarrhea, and diabetes (Warner et al., 2016).
The effects of kratom have been associated with the plant strain, which is visually characterized based on vein color, including white, green, or red (Suwanlert, 1975; Hartley et al., 2022). Vein color changes as the plant ages, from white at the young stage to green and finally to red after maturation (Ngernsaengsaruay et al., 2022). Vein color has been purported to be associated with different pharmacological effects. For example, the white vein variety is used for mild stimulant effects, the green vein variety is used to manage mild to moderate pain, and the red vein variety is used for stronger pain-relieving and sedating effects. Substrains are based on the geographical location of the plant (Sengnon et al., 2023). However, the validity of these claims has not been scientifically evaluated.
Relative to other species, only M. speciosa has been extensively cultivated and used for medicinal benefits. The United States market is dominated by plant products imported from Southeast Asia (Cinosi et al., 2015). At low doses (1–5 g of dried leaf powder), kratom is said to have stimulant properties, whereas at higher doses (>10 g of dried leaf powder), opioid-like effects ensue. Terms such as “legal highs” or “herbal highs” give the perception that kratom is a safe alternative to opioids (Anand and Hosanagar, 2022). Sales of these products at affordable prices via internet vendors or at gas stations and smoke/head shops are widespread (Prozialeck et al., 2012; Williams and Nikitin, 2020).
Dried kratom leaf powder is consumed in western countries as a capsule, pill, or tea (Cinosi et al., 2015). The powder is also swallowed with a drink, otherwise known as the “toss and wash method.” Kratom powder is added to smoothies, cocktails, caffeinated beverages, and even cough syrup. Powders artificially fortified with the kratom alkaloids mitragynine and 7-hydroxymitragynine, termed “enhanced kratom,” are also commonly available. As detailed later, both alkaloids are believed to drive the opioid-like effects of kratom, with ∼10-fold lower and ∼10-fold higher potency than morphine, respectively. Some kratom products available in the United States have been suspected to be artificially enriched with 7-hydroxymitragynine by up to 5-fold of the typical content, which may drive abuse potential (Lydecker et al., 2016). Finally, kratom extracts, tinctures, and resins are available that are more concentrated than the regular dried leaf powders and are sold as low-volume products. To mask the unpleasant taste, kratom is infused in a variety of edible goods, akin to cannabis, that are gaining in popularity.
According to a cross-sectional survey, approximately 3.3 million (1.3%) people in the United States report lifetime kratom use. Most kratom users are male (61%), white (82%), and 18–34 years old (55%) (Schimmel et al., 2021). The rampant use of this once-known-to-be-safe traditional medicine, especially in conjunction with pharmaceutical or illicit drugs, may have exacerbated the risks associated with kratom use in western countries. The number of calls to US poison control centers associated with kratom increased more than 50-fold from 2011 to 2017 (Post et al., 2019).
This review begins with the current legal status and regulation of kratom and active phytoconstituents and the chemistry of these phytoconstituents in kratom leaves. Consideration of the preclinical and clinical pharmacology, toxicology, pharmacokinetics, and drug interaction potential of kratom follows. Our objective was to integrate the aggregate kratom-related knowledge to highlight potential risks associated with consuming kratom with drugs. An iterative approach encompassing clinical evaluations and physiologically based pharmacokinetic modeling and simulation is recommended, with the goal of accurately predicting and simulating real-world kratom-drug interactions. Such a thorough evaluation will facilitate the safe and effective use of this increasingly popular natural product.
Policy and Legal Status
Kratom is either prohibited or heavily regulated in several countries, including Australia, Bhutan, Canada, Denmark, Finland, Ireland, Italy, Japan, Latvia, Lithuania, Malaysia, Myanmar, New Zealand, Poland, Romania, Sweden, and the United Kingdom (Shah et al., 2021). Until 2021, kratom was prohibited in Thailand, where it was regulated under the Kratom Act of 1943 (Kerrigan and Basiliere, 2022). The US Food and Drug Administration (FDA) has not approved kratom as a safe and effective drug for any medical use. As is common with botanical products, kratom products are regulated differently than FDA-approved drugs. Although not recognized as a dietary supplement according to the Dietary Supplement Health and Education Act, the regulation of kratom is not unlike that of dietary supplements (Paine and Roe, 2018; Paine, 2020). That is, kratom products are not required to undergo extensive safety testing prior to marketing. Accordingly, the onus is on the manufacturers and distributors to ensure that their products are safe and appropriately labeled. However, if a public health concern via adulteration, fraudulence, or other violation of the law is suspected, the FDA can inspect manufacturing facilities or monitor already marketed products.
Because kratom engages the same opioid receptors as morphine, the FDA is concerned that it may pose a risk for addiction, abuse, and dependence (Prozialeck et al., 2019). Accordingly, the FDA imposed an import ban on kratom-containing products, first in 2012 and again in 2014. In 2016, citing safety concerns and to prevent imminent hazards to public safety, the US Drug Enforcement Administration (DEA) announced its intention to place mitragynine and 7-hydroxymitragyine into Schedule I of the Controlled Substance Act of 1970. In response, more than 20,000 kratom advocates, which included users, vendors, and congresspersons, filed comments in the Federal Register (Docket ID: DEA-2016-0015) supporting the usefulness of kratom for the self-treatment of pain and opioid withdrawal symptoms with low potential for abuse. This public outcry contributed to the DEA subsequently withdrawing their decision and listing kratom as a “Drug of Concern.” However, there are several county and statewide (Alabama, Arkansas, Indiana, Rhode Island, Vermont, Wisconsin, and Washington, DC) regulations and/or bans against the possession and use of kratom.
In 2017, the FDA recommended “more research to better understand kratom’s safety profile, including the use of kratom combined with other drugs” to make an informed decision about the regulation of kratom (https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-agencys-scientific-evidence-presence-opioid-compounds). The legal status of kratom within and outside the United States is constantly changing, with increasing kratom-related adverse events and scientific literature reports highlighting the benefit-to-risk ratio associated with kratom use. Kratom has been under Expert Committee on Drug Dependence surveillance since 2020 in response to reports suggesting the potential for abuse, dependence, harm to public health, and fatalities associated with kratom use. However, due to a lack of sufficient evidence about the abuse or dependence potential in humans, the committee recommended against a critical review of kratom and that it instead be kept under surveillance by the World Health Organization secretariat (https://apps.who.int/iris/handle/10665/352462). Continued, fast-paced research on the safety and efficacy of kratom is needed to enable informed decisions about the regulation of kratom.
Chemistry
Kratom has been investigated extensively for its phytochemical constitution due to having antinociceptive and psychoactive properties. The chemical composition of kratom encompasses alkaloids, terpenoids, flavonoids, tannins, saponins, and phenols. Alkaloids (i.e., basic nitrogen-containing organic phytochemicals) in the kratom plant have been studied predominantly for their pharmacological effects. Total alkaloid content in leaves ranges from 0.5% to 1.5% of dried plant material (Hassan et al., 2013). The alkaloid content and composition in kratom can vary based on the plant’s age, location, or other environmental factors, as well as processing methods. The biosynthesis of alkaloids occurs via a typical indole alkaloid synthesis process, beginning from the shikimic acid pathway combined with the methyl-erythritol phosphate pathway to form corynanthe-type alkaloids (Karunakaran et al., 2022). The kratom plant produces at least 54 alkaloids (Flores-Bocanegra et al., 2020). These alkaloids are largely classified based on the presence of either a tetracyclic indole or pentacyclic oxindole nucleus (Fig. 1). Rings A and B of the tetracyclic indole alkaloids are aromatic and therefore planar in structure, but conformational flexibility is possible in rings C and D (Kerrigan and Basiliere, 2022). The spatial arrangement of these alkaloids has been reported to be influenced by the position of the lone pair of electrons on the nitrogen shared by rings C and D in relation to the exocyclic ethyl/vinyl group on the tetracycle.
Fig. 1.
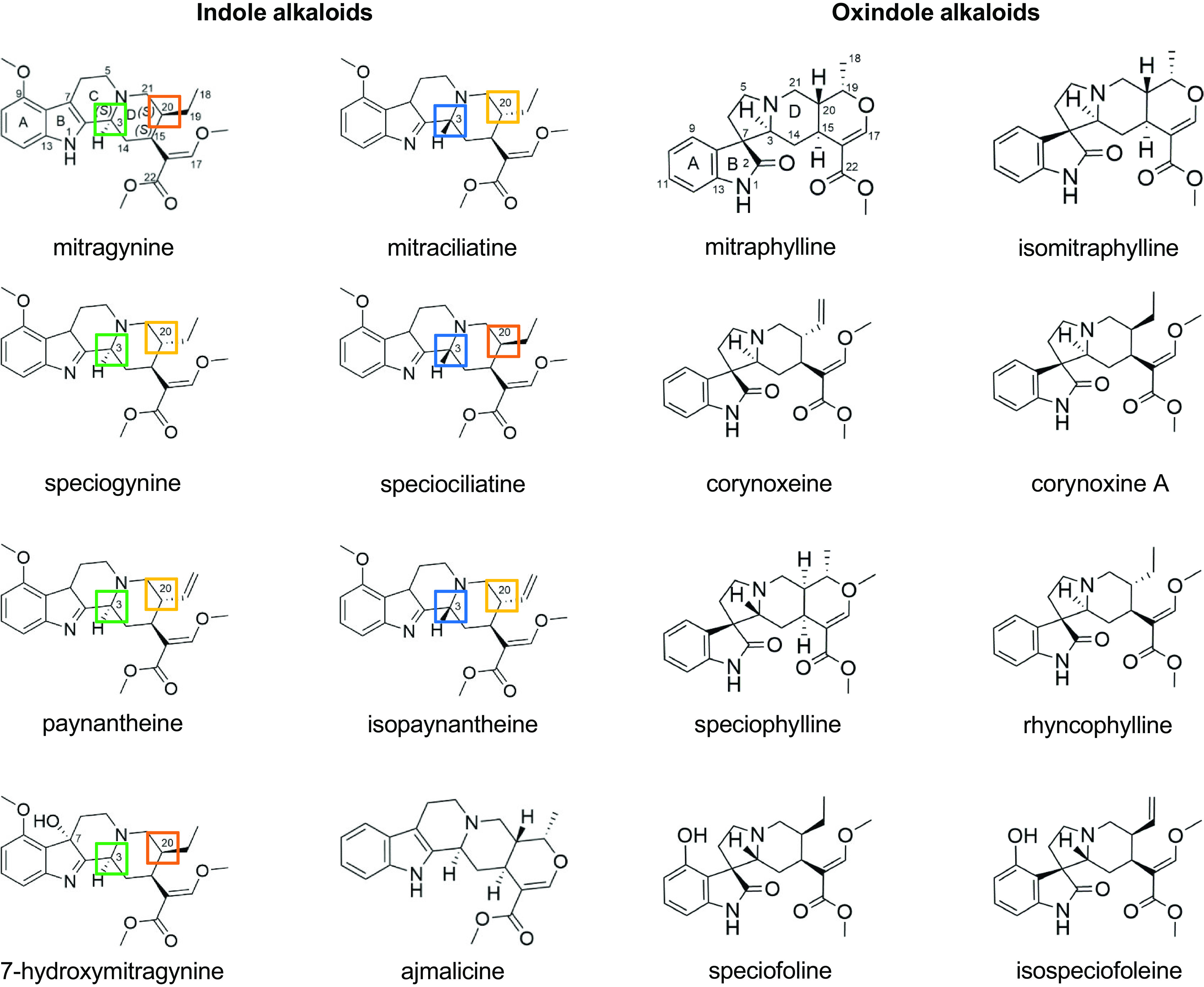
Chemical structures of key indole and oxindole kratom alkaloids. Structures of mitragynine and mitraphylline are numbered to serve as references for the indole and oxindole alkaloids, respectively. Stereochemical differences at C3 and C20 of the indole alkaloids are indicated by the colored boxes. These differences may explain the pharmacokinetic differences observed between those with the 3S configuration (mitragynine, speciogynine, paynantheine) and the 3R configuration (mitraciliatine, speciociliatine, isopaynantheine) in human adult participants.
Mitragynine (Fig. 1; Table 1) is the major alkaloid in some of the kratom cultivars that are most widely used medicinally (Takayama, 2004). The pharmacological activity of kratom has been attributed largely to mitragynine, along with 7-hydroxymitragynine, which is also formed in vivo via P450-mediated metabolism (Kruegel and Grundmann, 2018; Kruegel et al., 2019; Tanna et al., 2022). Previous studies failed to detect 7-hydroxymitragynine in fresh kratom leaves, leading to the hypothesis that its presence in marketed kratom products may be due to chemical oxidation (autoxidation) of mitragynine during the postharvest phase (Ponglux et al., 1994; Chear et al., 2021).
TABLE 1.
Typical alkaloidal content in kratom products and dried leaves
| Alkaloid | Range of Alkaloid Contenta (mg/g of Product) |
|---|---|
| mitragynine | 0.5–270 |
| speciogynine | 3.18–33.4 |
| mitraciliatine | 0.647–4.75 |
| speciociliatine | 0.185–41.7 |
| paynantheine | 1.46–70.4 |
| isopaynantheine | 0.269–3.80 |
| 7-hydroxymitragynine | 0.124–1.10 |
| speciofoline | 0.122–5.90 |
| corynoxeine | 0.290–1.22 |
| corynoxine A | 0.205–11.4 |
| ajmalicine | 0.192–0.648 |
| rhynchophylline | 0.239 |
aBased on two powdered plant products, a loose-leaf product, a liquid product, and an encapsulated powder (Manwill et al., 2022).
Researchers have investigated the pharmacological potential of other kratom alkaloids. Indole alkaloids contain three chiral centers (C3, C15, C20), resulting in multiple prominent mitragynine diastereoisomers that include speciogynine, mitraciliatine, and speciociliatine (Takayama, 2004) (Fig. 1; Table 1). This stereochemistry is responsible for the spatial arrangement of alkaloids, which has been postulated to account for differences in the physicochemical and pharmacological properties among the alkaloids. Related alkaloids include the mitragynine analogs paynantheine and isopaynantheine, which contain a vinyl group instead of an ethyl group at C20. Mitragynine, speciogynine, paynantheine, and 7-hydroxymitragynine possess the 3S configuration, whereas speciociliatine, mitraciliatine, and isopaynantheine possess the 3R configuration.
Kratom has multiple chemotypes reflective of its geographical origin (Manwill et al., 2022). Metabolomic analysis of more than 50 commercial kratom products revealed at least two different chemotypes predominantly containing either indole or oxindole alkaloids (Fig. 1). These differences led to a wide range of individual alkaloidal content (Todd et al., 2020) (Table 1). Given this reported variability, thorough characterization and appropriate labeling of marketed products are needed to inform the pharmacology, toxicology, or drug interaction potential of kratom. Methods that can be used to characterize botanical and other natural products are described elsewhere (Kellogg et al., 2019).
Pharmacology
The pharmacology of kratom that elicits the stimulant effects at low doses and relaxing/sedative effects at higher doses remains equivocal. Kratom alkaloids, along with their metabolites, including 7-hydroxymitragynine, bind and act differentially at the opioid, adrenergic, and serotonergic pathway receptors (Table 2). In vitro assays characterizing the binding affinities and functional activities of the alkaloids, followed by the in vivo functional evaluations of the individual alkaloids and leaf extracts, are described below.
TABLE 2.
Binding affinities (Ki) of kratom alkaloids toward opioid (OR), adrenergic (α), and serotonergic (5-HT) receptors
| Receptor | Mitragynine | 7-Hydroxy-Mitragynine | Speciogynine | Paynantheine | Speciociliatine | Corynantheidine | Corynoxeine | Corynoxine B | Speciofoline |
|---|---|---|---|---|---|---|---|---|---|
| Ki ± S.E.M. (nM) | |||||||||
| μOR | 238 ± 28 233 ± 48 161 ± 10 |
16 ± 1 47 ± 18 7.16 ± 0.94 |
728 ± 61 | 410 ± 152 | 54.5 ± 4.4 560 ± 168 |
118 ± 12 | 16.3 ± 1.4 >10,000 |
109.8 ± 8.1 1600 |
>10,000 |
| κOR | 482 ± 29 772 ± 207 198 ± 30 |
113 ± 37 188 ± 38 74.1 ± 7.8 |
3200 ± 360 116 ± 36 |
2560 ± 370 | 116 ± 36 329 ± 112 |
1910 ± 50 | >10,000 | >10,000 | >10,000 |
| δOR | >10,000 | 137 ± 21 219 ± 41 |
>10,000 | >10,000 | >10,000 | >10,000 | >10,000 | 7600 | >10,000 |
| 5-HT1A | 5880 ± 828 | — | 95.5 ± 34.7 | 71.8 ± 13.2 | — | — | — | — | — |
| 5-HT7A | >10,000 | — | 1600 ± 82 | 870 ± 72 | — | — | — | — | — |
| 5-HT2A | 5010 ± 1150 | — | 1320 ± 365 | 815 ± 192 | — | — | — | — | — |
| 5-HT2B | 1260 ± 138 | — | 23.0 ± 5.7 | 20.0 ± 2.8 | — | — | — | — | — |
| 5-HT2C | >10,000 | — | 5430 ± 922 | 1770 ± 417 | — | — | — | — | — |
| α-1A | 1340 ± 100 | — | — | — | — | >10,000 | — | — | — |
| α-1B | 4770 ± 120 | — | — | — | — | >10,000 | — | — | — |
| α-1D | 5480 ± 540 | — | — | — | — | 41.7 ± 4.7 | — | — | — |
| α-2A | 4720 ± 120 | >10,000 | — | — | — | >10,000 | — | — | — |
| α-2B | 9290 ± 30 | >10,000 | — | — | — | >10,000 | — | — | — |
| α-2C | 2320 ± 140 | >10,000 | — | — | — | >10,000 | — | — | — |
—, information not available.
In Vitro
Both mitragynine and 7-hydroxymitragynine have been shown to be partial agonists at the μ-opioid receptor based on inhibition of forskolin-stimulated cyclic AMP (cAMP) accumulation (Todd et al., 2020) and antagonists at the δ and κ opioid receptors using the G-protein bioluminescence resonance energy transfer functional assay (Kruegel et al., 2016). In contrast to other opioids like morphine, mitragynine and 7-hydroxymitragynine selectively activate the G-protein second messenger upon binding to the G-protein–coupled receptor rather than the β-arrestin-2 signaling pathway, which is responsible for many of the adverse effects associated with typical opioids. Based on results from the guanosine 5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) stimulation assay, mitragynine was reported to be a competitive antagonist at μ-opioid receptors (Obeng et al., 2021), suggesting that other alkaloids may contribute to the opioid-like effects. Other kratom alkaloids, such as speciociliatine and mitraciliatine, have been reported to be partial agonists at the μ-opioid receptor (Hiranita et al., 2022). Mitragynine has also been shown to engage several nonopioid receptors, including α2 adrenergic, adenosine A2a, dopamine D2, and the serotonin receptors 5-HT2C and 5-HT7 (Anand and Hosanagar, 2022). Mitragynine was shown to be a partial agonist at α1A,D receptors and a competitive antagonist at α1A,B,D,2C receptors (Obeng et al., 2020). Corynanthidine, a 9-demethoxy analog of mitragynine, was ∼130-fold more potent than mitragynine at α1A,D receptors (Table 2). Mitragynine exhibited low binding affinity to serotonergic receptors, whereas speciogynine and paynantheine demonstrate high binding affinity to the 5-HT1A and 5-HT2B serotonergic receptors (León et al., 2021).
In Vivo
Kratom extract, tea, and purified alkaloids including mitragynine, 7-hydroxymitragynine, speciociliatine, mitraciliatine, isopaynantheine, corynanthidine, and corynoxeine have shown robust antinociceptive effects in rodents using the hot plate and/or tail-flick assays (Matsumoto et al., 1996a,b; Chin and Mark-Lee, 2018). Upon oral or parenteral administration of mitragynine, a partial antinociceptive effect was observed in rats and mice compared with the full opioid agonist morphine. In contrast, 7-hydroxymitragynine produced a ∼3- to 10-fold more potent antinociceptive effect than morphine (Chakraborty et al., 2021). The observed antinociceptive effects decreased when the μ-opioid receptor antagonist naltrexone was administered intraperitoneally, suggesting that these effects are primarily μ-opioid receptor mediated. Although 7-hydroxymitragynine is more potent than mitragynine, its role in the overall antinociceptive effect after mitragynine administration is inconclusive, largely due to the lower brain concentration of 7-hydroxymitragynine (Berthold et al., 2022). Mitragynine was shown to have more potent effects in rodent models when administered orally compared with intraperitoneal or subcutaneous routes, suggesting potential first-pass bioactivation of this alkaloid to more potent metabolites (Kruegel and Grundmann, 2018). Overall, the in vivo efficacy of kratom appears to be mediated by mitragynine, other alkaloids, and their metabolites. However, definitive roles of several other alkaloids isolated from the kratom leaves remain uncertain.
One study objectively assessed the effects of kratom on pain tolerance in humans (Vicknasingam et al., 2020). This randomized, placebo-controlled, double-blind study involved 26 healthy male long-term kratom users. A significant increase in pain tolerance in the cold pressor task (∼11 to ∼25 seconds) was reported 1 hour after kratom ingestion. Clinical evaluation of the stimulant effects of kratom at lower doses remains lacking.
Toxicology
Nonhuman
After oral administration of a methanolic kratom extract to rats (100–1000 mg/kg) for 14 days, mild nephrotoxicity and moderate to severe hepatotoxicity were observed, with elevations in alanine aminotransferase, aspartate aminotransferase, albumin, triglycerides, and cholesterol (Harizal et al., 2010). After oral administration of a different kratom extract to mice, the LD50 (lethal dose, 50%) ranged from ∼170 to 590 mg/kg (Reanmongkol et al., 2007; Sabetghadam et al., 2013a). After oral administration of mitragynine to rats (1–10 mg/kg) for 28 days, no adverse effects were observed (Sabetghadam et al., 2013b). At a higher dose of mitragynine (100 mg/kg), biochemical, hematologic, and histopathological abnormalities involving the liver, brain, and kidney surfaced, but there were no deaths. A single oral dose of mitragynine administered to dogs (80 mg/kg) produced no adverse effects, including respiratory depression (Kruegel and Grundmann, 2018). A subchronic low oral dose of mitragynine administered to dogs (5–20 mg/kg per day) for 21 days also produced no adverse effects; however, after oral administration of a higher dose (40 mg/kg per day) to these dogs for the subsequent 21 days, changes in blood chemistry, liver cell morphology, and lymphatic hyperplasia were observed. Overall, based on the evidence from preclinical species, kratom as an extract and mitragynine as the purified alkaloid may be toxic only with chronic use at high doses.
Human
Both acute and chronic kratom toxicology have been investigated. Kratom-associated risks and adverse events have been reported based on cross-sectional surveys from active kratom users, national poison data systems, and clinical case reports (Kerrigan and Basiliere, 2022). Common adverse effects include agitation, tachycardia, drowsiness, vomiting, and confusion (Eggleston et al., 2019). Severe adverse effects include hepatotoxicity, cardiotoxicity, respiratory depression, seizure, neonatal abstinence syndrome, hypothyroidism, overdose toxidrome, and fatalities (Alsarraf et al., 2019). Kratom-induced intrahepatic cholestasis has been reported after chronic (∼1–4 weeks) use of large kratom doses (>10 g per day) (Schimmel and Dart, 2020). In some cases, kratom-induced hepatotoxicity was completely reversed after kratom cessation. However, due to polysubstance use, typical with kratom use, the toxicology of kratom remains unclear. According to the FDA Adverse Event Reporting System (FAERS) database, from October 2012 through September 2021, 489 adverse events associated with kratom (i.e., mitragynine) have been reported, including 244 deaths (Li et al., 2023). Most of these deaths were believed to result from co-consumption with other drugs.
Depending on the symptoms, kratom-related overdose has been managed with the μ-opioid receptor antagonist naloxone to reverse the opioid-like effects (either from kratom or coingested opioids), whereas the dual α and β adrenergic antagonist labetalol has been used to manage sympathetic overactivation (Peran et al., 2023). Kratom-related withdrawal symptoms have been managed with a combination of naloxone and the partial μ-opioid agonist buprenorphine (Suboxone) (Weiss and Douglas, 2021).
Pharmacokinetics
The pharmacokinetics of key kratom alkaloids have been characterized in both preclinical species and human participants. Intravenous and oral administration of kratom extracts and individual alkaloids, including mitragynine, have been extensively investigated in rodents and dogs, as reviewed elsewhere (Hiranita et al., 2022). In contrast, only two studies to date have focused on the pharmacokinetics of mitragynine and other kratom alkaloids in humans (Trakulsrichai et al., 2015; Tanna et al., 2022) (Table 3). Regarding the earlier study (Trakulsrichai et al., 2015), blood was collected from 0 to 24 hours, yet an average terminal half-life of ∼24 hours was reported, raising concerns about the accuracy of this and other fundamental pharmacokinetic parameters reported, including oral clearance (CL/F) and apparent volume of distribution (Vd/F) (Tanna et al., 2022). Accordingly, results from the latter study, where blood was collected from 0 to 120 hours, are presented (Table 3). These latter pharmacokinetic observations in humans are further supported by pertinent in vitro studies (discussed below). The following section summarizes the physicochemical and pharmacokinetic properties of these alkaloids as they may relate to the risk of kratom-drug interactions in humans.
TABLE 3.
Clinical pharmacokinetics of key kratom alkaloids after oral administration of kratom tea to healthy adult participants
| Study | Trakulsrichai et al. (2015) | Tanna et al. (2022) | ||||||
|---|---|---|---|---|---|---|---|---|
| Participants (n) | 10 | 5 | ||||||
| Mean ± S.D. | Median (Range) | |||||||
| Alkaloid | Mitragynine | Mitragynine | Speciogynine | Paynantheine | Speciociliatine | Mitraciliatine | Isopaynantheine | 7-Hydroxy-Mitragynine |
| Dose (mg) | Variablea | 39 | 6.4 | 12 | 10 | 1.3 | 1.0 | — |
| Cmax (nM) | — | 81.9 (50.1–177) | 51.4 (34.2–121) | 61.1 (56.4–157) | 308 (154–380) | 73.5 (34.9–98.6) | 48.8 (26.2–68.2) | 16.1 (11.9–22.2) |
| tmax (h) | 0.83 ± 0.35 | 1 (0.75–1.5) | 2 (1–3.5) | 1 (0.75–2.5) | 2.5 (1–3.5) | 4.5 (3.5–6.5) | 4.5 (2.5–6.5) | 1 (0.75–2.5) |
| t1/2 (h) | 23 ± 16 | 45.3 (31.9–50.2) | 23.5 (16.1–28.3) | 27.0 (17.7–30.8) | 12.3 (10.4–21.1) | 17.8 (11.2–24.7) | 14.4 (11.8–20.9) | 5.67 (5.03–6.52) |
| AUCinf (nMh) | — | 420 (324–1360) | 477 (379–1120) | 438 (389–956) | 5120 (3200–7560) | 1160 (1040–3520) | 794 (667–2130) | 106 (60.8–126) |
| Vz/F (L) | 2900 ± 1850b | 12,700 (5190–19,700) | 962 (584–1235) | 1940 (1370–2620) | 130 (60.1–159) | 46.0 (26.2–74.0) | 55.5 (36.6–76.0) | — |
| CL/F (l/h) | 7550 ± 3900b | 233 (71.7–302) | 33.5 (14.3–42.1) | 67.4 (30.9–76.0) | 5.01 (3.40–8.04) | 2.78 (0.92–3.11) | 3.25 (1.21–3.87) | — |
—, information not available.
AUCinf, area under the plasma concentration-time profiles from time zero to infinity; CL/F, oral clearance; t1/2, terminal elimination half-life; Tmax, time to reach Cmax; Vz/F, apparent volume of distribution during the terminal phase.
aA range of doses (6.25–23 mg) of mitragynine contained in a kratom tea were administered, precluding direct comparison of Cmax and AUCinf between studies.
bObtained by multiplying the Vd/F in l/kg with the mean weight (77 kg) of the participants; blood was collected from 0 to 24 hours, yet an average terminal half-life of ∼24 hours was reported, raising concerns about the accuracy of the reported pharmacokinetic outcomes, including t1/2, CL/F, and Vz/F.
Physicochemical Properties
Mitragynine is categorized as a Biopharmaceutics Classification System class 2 xenobiotic, meaning it has high permeability and low aqueous solubility. Mitragynine and other kratom alkaloids, including 7-hydroxymitragynine, speciogynine, and paynantheine, were shown to have variable stability across a wide range of temperatures and pH (Basiliere and Kerrigan, 2020). In general, the extent of degradation was higher at elevated temperatures (>40°C) and under more acidic (pH 2–4) or alkaline (pH 8–10) conditions. Both mitragynine and 7-hydroxymitragynine degraded by <10% in simulated gastric (pH 1.2) and intestinal (pH 6.8) fluids after 30 minutes and 3 hours, respectively. However, near the physiologic conditions of temperature and pH of the stomach, intestine, tissues, and systemic circulation, the extent of degradation is expected to be more limited. These observations suggest that upon oral administration, the entire doses of the kratom alkaloids administered may be available at the site of absorption. Mitragynine was shown to have an octanol:water partition coefficient (Log P) of 1.7 (Ramanathan et al., 2015) but a higher in silico predicted Log P of 3.4–4.2 using ADMET Predictor (v10.4; Simulations Plus, Inc., Lancaster, CA). The lipophilicity of kratom alkaloids has been reported to be dependent on the stereochemistry at the chiral centers (Trager et al., 1967; Beckett and Dwuma‐Badu, 1969). Alkaloids with the 3S configuration (e.g., mitragynine, speciogynine, and paynantheine) were shown to have a planar conformation and to be more lipophilic compared with alkaloids with the 3R configuration (e.g., mitraciliatine, speciociliatine, and isopaynantheine) (Fig. 1), which were shown to have a bent conformation. The log ionization constant (pKa) of these alkaloids was also dependent on the stereochemistry but within a narrow range (7.06–7.95).
Absorption
After oral administration of a kratom tea to six human participants, the key kratom alkaloids were rapidly absorbed and measurable in the systemic circulation (Tanna et al., 2022). The rate of kratom alkaloid absorption was variable, with the maximum plasma concentration (Cmax) achieved within 1–4.5 hours. Kratom indole alkaloids with the 3S configuration generally exhibited a shorter time to reach Cmax (tmax) than those with the 3R configuration (1 to 2 vs. 2.5–4.5 hours). This difference has been attributed to differences in the partition coefficients of the alkaloids (Beckett and Dwuma‐Badu, 1969). The absolute oral bioavailability (Foral) of mitragynine in humans is unknown, and preclinical data suggest species-specific differences. For example, the Foral of mitragynine (3%–30%) after administration of 20–50 mg/kg to rats was lower than that in beagle dogs, which exhibited an Foral of ∼70% after a single dose of 5 mg/kg (Parthasarathy et al., 2010; Avery et al., 2019; Maxwell et al., 2020). Based on an allometric method involving the intravenous clearance of mitragynine in beagle dogs and the oral clearance of mitragynine (administered as kratom) in humans (Tanna et al., 2022), human Foral of mitragynine was estimated at ∼30%.
Using the parallel artificial membrane permeability assay (PAMPA), mitragynine flux through the phospholipid bilayer at pH 4 and 7.4 was 0.23 × 10−6 and 11 × 10−6 cm/s, respectively (Kong et al., 2017). Higher permeability at pH 7.4 is consistent with mitragynine permeating as the unionized form. Using Caco-2 cell monolayers (mimicking the intestinal barrier), absorptive [apical (A)→basolateral (B)] and exsorptive (B→A) flux for mitragynine at 10 μM was 2.5 × 10−5 and 2.9 × 10−5 cm/s, respectively, suggesting moderate to high permeability (Manda et al., 2014b). The flux ratio [(B→A)/A→B)] was approximately unity, suggesting that mitragynine is not a substrate for the efflux transporter P-glycoprotein (P-gp). Likewise, mitragynine flux through Madin-Darby canine kidney cell monolayers overexpressing multidrug resistance 1 protein (MDR-MDCK) at 10 μM was 16 × 10−6 and 18 × 10−6 cm/s, respectively, with a flux ratio of 1.1. In contrast, the minor alkaloid mitraphylline was shown to be a substrate for P-gp (flux ratio ∼6 to 7) (Manda et al., 2014b).
Distribution
After administration of kratom tea to healthy human adult participants, concentration-time profiles for the key kratom alkaloids were best described by a two-compartment model (Tanna et al., 2022). In general, the extent of tissue distribution was higher for the indole alkaloids with the 3S configuration compared with those with the 3R configuration, as indicated by a larger apparent volume of distribution, specifically a larger apparent peripheral volume of distribution. Alkaloids with the 3S configuration were also shown to be rapidly distributed into the peripheral tissues and to redistribute slowly out of the peripheral tissues relative to the alkaloids with the 3R configuration. Preclinical pharmacokinetic studies of mitragynine and speciociliatine in rodents reported similar trends in distribution (Berthold et al., 2021).
These differences in alkaloid distribution were again attributed to differences in the lipophilicity of the 3S and the 3R configured indole alkaloids (Tanna et al., 2022) but could also be due to lysosomal trapping of the alkaloids in various cells, as reported for other cationic lipophilic drugs like chloroquine (Macintyre and Cutler, 1988). Kratom alkaloids are highly bound to plasma proteins, with a fraction unbound of <0.06 at 1 μM (Tanna et al., 2022). Human blood-to-plasma ratios for kratom alkaloids ranged from 0.65 to 1.05 at 1 μM, indicating limited partitioning into erythrocytes. Tissue distribution of mitragynine in mice was perfusion rate limited, wherein the ratio of AUCorgan (area under the organ concentration vs. time curve) to AUCplasma was directly proportional to the blood flow of the respective organ (AUCorgan/AUCplasma in liver, 28.6 > kidney, 17.1 > lung, 15.9 > spleen, 5.1) except the brain due the blood-brain barrier (Yusof et al., 2019; Berthold et al., 2022). The AUCbrain/AUCplasma for the more polar 7-hydroxymitragynine was <1.
Elimination
In general, kratom alkaloids demonstrated a long elimination half-life upon administration of kratom tea to healthy adult participants (Tanna et al., 2022). Following the trend observed for absorption and distribution, the terminal half-lives for the indole alkaloids with the 3S configuration (24–45 hours) were longer than analogs with the 3R configuration (∼12–18 hours). In addition, the fraction of the dose excreted unchanged in the urine (fe) was higher for the 3R compared with the 3S configured alkaloids. However, for all alkaloids, fe was <0.2%, indicating that urinary excretion is a minor route of systemic elimination. Consistent with that interpretation, renal clearance (CLR) for all alkaloids was much lower than effective renal plasma flow (<0.5 vs. 36 l/h).
Kratom alkaloids, including mitragynine, primarily undergo oxidative metabolism, which can subsequently be either glucuronidated or sulfated based on the species (Philipp et al., 2009; Basiliere et al., 2018) (Fig. 2). These alkaloids were extensively metabolized in an NADPH-dependent manner in both enteric (human intestinal microsomes) and hepatic (human liver microsomes) tissue fractions (Tanna et al., 2022). The extent of metabolism of kratom alkaloids is stereoselective (i.e., the indole alkaloids with the 3S configuration are more rapidly metabolized than those with the 3R configuration). Cytochrome P450 (P450) 3A4 is the major enzyme that metabolizes mitragynine to 7-hydroxymitragynine (Kamble et al., 2019; Kruegel et al., 2019). 7-Hydroxymitragynine was further reported to be metabolized by an unknown human plasma enzyme to a 31-fold more potent activator of the μ-opioid receptor, mitragynine pseudoindoxyl (Kamble et al., 2020a). CYP2C9, CYP2C19, and CYP2D6 have minor roles in the metabolism of mitragynine (Kamble et al., 2019). Mitragynine acid formation from mitragynine was catalyzed by human carboxylesterase (hCES) 1c (Km = 87 μM; Vmax = 0.7 nmol/min per mg) but not by hCES2 (Meyer et al., 2015). However, based on the low intrinsic clearance, the clinical relevance of the hCES1c pathway is unlikely.
Fig. 2.

Proposed metabolic scheme for mitragynine based on reported in vitro and in vivo evaluations. Boxes suggest the tentative site of metabolism.
Kratom-Drug Interactions
In Vitro Evidence
Kratom has been extensively evaluated in vitro as a precipitant of pharmacokinetic drug interactions. Methanolic extracts of kratom and purified kratom alkaloids were shown to alter the activity of key drug metabolizing enzymes, including P450s and UDP-glucuronosyltransferases (UGTs) responsible for the elimination of xenobiotics (Table 4). Kratom extracts were initially shown to inhibit CYP1A2, CYP2C19, CYP2D6, and CYP3A using high-throughput fluorometric assays, with IC50 ranging from ∼0.6 (CYP2D6) to 140 (CYP3A) μg/ml (Kong et al., 2011). Methanolic extracts prepared from well characterized kratom products were later shown to inhibit CYP2C9 (∼65% at 20 μg/ml), CYP2D6 (∼90% at 20 μg/ml), and CYP3A (∼50% at 20 μg/ml) activities using selective probe drug substrates and a liquid chromatography–mass spectrometry (LCMS)-based assay (Todd et al., 2020). The extent of inhibition remained consistent among three kratom extracts despite differences in alkaloidal content and composition.
TABLE 4.
IC50 values for mitragynine and kratom extracts against P450 and UGT activities using various test systems
| Enzyme | Substrate | Reaction | Enzyme System | Monitoring Method | Mitragynine (μM) | Extract (μg/ml) |
|---|---|---|---|---|---|---|
| CYP1A2 | phenacetin | O-deethylation | HLMs | LCMS | >45 | — |
| CEC | O-deethylation | recombinant | fluorometric | — | 39 | |
| CYP2C8 | amodiaquine | N-deethylation | HLMs | LCMS | 33.5 | — |
| CYP2C9 | diclofenac | 4’-hydroxylation | HLMs | LCMS | >45 | — |
| luciferin H | 9.7 | — | ||||
| diclofenac | 4’-hydroxylation | HLMs | LCMS | 39.7 | — | |
| CYP2C19 | S-mephenytoin | 4’-hydroxylation | HLMs | LCMS | 10.5 | — |
| CEC | O-deethylation | recombinant | fluorometric | — | 85 | |
| CYP2D6 | dextromethorphan | O-demethylation | HLMs | LCMS | 2.2 | — |
| AMMC | O-demethylation | recombinant | fluorometric | — | 0.64 | |
| luciferin ME-EGE | luminometric | 0.45 | — | |||
| luciferin ME-EGE | luminometric | — | 3.6 | |||
| dextromethorphan | O-demethylation | HLMs | LCMS | 0.67 | — | |
| CYP3A4/5 | Midazolam | 1’-hydroxylation | HLMs | LCMS | 11.4 | — |
| testosterone | 6β-hydroxylation | HLMs | LCMS | >45 | — | |
| BFC | O-debenzylation | recombinant | fluorometric | — | 0.78 | |
| midazolam | 1’-hydroxylation | recombinant | HPLC-UV | 17.31 | — | |
| testosterone | 6β-hydroxylation | recombinant | HPLC-UV | 3.98 | — | |
| luciferin-BE | luminometric | 41.32 | — | |||
| luciferin-BE | luminometric | — | 142.8 | |||
| midazolam | 1’-hydroxylation | HLMs | LCMS | 18.9 | — | |
| midazolam | 1’-hydroxylation | HIMs | LCMS | 21.9 | — | |
| UGT1A1 | 4-MU | glucuronidation | recombinant | HPLC-UV | >100 | — |
| UGT2B7 | zidovudine | glucuronidation | HLMs | HPLC-UV | 8.11 | — |
| 4-MU | glucuronidation | recombinant | HPLC-UV | >100 | — |
—, information not available.
AMMC, 3-[2-(N,N-diethyl-N-methylammonium)ethyl]-7-methoxy-4-methylcoumarin; BFC, 7-benzyloxy-4-(trifluoromethyl)-coumarin; CEC, 3-cyano-7-ethoxycoumarin; HPLC-UV, high pressure liquid chromatography coupled with UV detector; luciferin-BE, luciferin 6’ benzyl ether; luciferin H, 6’deoxyluciferin; luciferin ME-EGE, ethylene glycol ester of luciferin 6’ methyl ether; 4-MU, 4-methylumbelliferone.
Mitragynine inhibited the activity of several P450 enzymes in human liver microsomes (HLMs), including CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A, with IC50 values ranging from 0.45 μM (CYP2D6) to ∼40 μM (CYP2C9) (Hanapi et al., 2013; Lim et al., 2013; Kamble et al., 2020b) (Table 4). Inhibition kinetics alone do not necessarily indicate the risk of a pharmacokinetic kratom-drug interaction. The likelihood of an interaction depends on several factors, including route of administration, dose of the precipitant, enzyme expression, extent of plasma protein binding, and disposition of the precipitant. Using basic static models recommended by the FDA, a 40-mg oral dose of mitragynine (contained in a 2-g oral dose of kratom) was predicted to precipitate a presystemic intestinal interaction for CYP3A, a presystemic hepatic interaction for CYP2D6, but no systemic interaction (Table 5). No interaction was predicted for any of the other tested P450s. These predictions warrant further investigation of the drug interaction potential of kratom when consumed with drugs metabolized by CYP3A and CYP2D6.
TABLE 5.
Prediction of the pharmacokinetic drug interaction potential of kratom using mitragynine as the precipitant.
A basic static model for reversible inhibition was used to predict the drug interaction potential of mitragynine administered orally via inhibition of cytochromes P450. R values indicate the predicted AUC ratio of the object drug in the presence to absence of inhibitor for intestine (R1,gut) or using hepatic (R1,hep) and systemic (R1,sys) concentration.
| Enzyme | IC50 (μM) | Kia (μM) | R1,gut | R1,hep | R1,sys | Reference |
|---|---|---|---|---|---|---|
| CYP1A2 | >45 | >22.5 | NA | 1.01 | 1.00 | (Kamble et al., 2020b) |
| CYP2C8 | 33.5 | 16.8 | NA | 1.01 | 1.00 | (Kamble et al., 2020b) |
| CYP2C9 | 39.7 | 19.9 | NA | 1.01 | 1.00 | (Tanna et al., 2021) |
| CYP2C19 | 10.5 | 5.25 | NA | 1.04* | 1.00 | (Kamble et al., 2020b) |
| CYP2D6 | 0.67 | 0.34 | NA | 1.57* | 1.01 | (Tanna et al., 2021) |
| CYP3A | 11.4 | 5.7 | 71* | 1.03* | 1.00 | (Tanna et al., 2021) |
| P-gpb | 18.2 | — | 22* | — | — | (Manda et al., 2014a) |
—, information not available.
aKi values are estimated as IC50/2 using the Cheng-Prusoff equation assuming competitive inhibition.
bR1,gut = 1 + (Igut/Ki) or (Igut/IC50) for P-gp, where intestinal luminal concentration (Igut) was calculated as dose/250 ml (∼400 μM).
*R1,gut ≥11 and R1,hep or R1,sys ≥1.02 indicates a potential interaction. R1,hep = 1 + (Ihep,u/Ki), where Ihep,u was calculated as fu,p × (Cmax + Fa × ka × dose/Qh/RB) (∼0.19 μM); R1,sys = 1 + (Isys,u/Ki), where Isys,u is fu,p × Cmax.
Mitragynine caused an ∼7-fold leftward shift in IC50 toward CYP3A activity upon a 30-minute preincubation with NADPH (Tanna et al., 2021) and was confirmed to be a time-dependent inhibitor of CYP3A activity in both HLMs [time-dependent inhibition constant (KI) = 4.1 ± 0.9 μM; maximum rate of inactivation (kinact) = 0.068 ± 0.01 min−1] and human intestinal microsomes (HIMs) (4.2 ± 2.5 μM; kinact = 0.079 ± 0.02 min−1) (Table 6). The mechanism of the observed time-dependent inhibition was not ascertained. Such NADPH-dependent inhibition over time is most commonly attributed to P450-mediated bioactivation of the compound, generating a reactive species that irreversibly binds to the enzyme, rendering the enzyme inactive. Although no direct structural alerts are present in mitragynine, based on literature evidence gathered for compounds with similar structural features as mitragynine, it was speculated that bioactivation of the quinolizidine moiety to an imine, 3-methylindolenine, p-quinone, or o-quinoneimine intermediate may result in covalent binding to enzyme nucleophilic residues (Tanna et al., 2021). Other compounds, including evodiamine, rutaecarpine, and zafirlukast, have been shown to inactivate CYP3A4 via a 3-methylindolenine–like intermediate, rendering this mechanism the most probable. Further in vitro evaluation is needed to confirm the exact mechanism. No IC50 shift was observed for the inhibition of CYP2C9 and CYP2D6 activities. However, strong inhibition of CYP2D6 activity was observed and of a reversible competitive nature (Ki ∼1.2 μM) (Table 6).
TABLE 6.
Inhibition kinetics for mitragynine against P450 activities
| Enzyme | Substrate | Reaction | Enzyme System | Monitoring Method | Mode of Inhibition | Inhibition Kinetics |
|---|---|---|---|---|---|---|
| CYP2C9 | luciferin H | luminometric | noncompetitive | Ki = 155 μM | ||
| CYP2D6 | dextromethorphan | O-demethylation | HLMs | LCMS | competitive | Ki = 1.17 μM |
| dextromethorphan | O-demethylation | HLMs | LCMS | Ki = 1.1 μM | ||
| luciferin ME-EGE | luminometric | noncompetitive | Ki = 12.86 μM | |||
| CYP3A4/5 | midazolam | 1’-hydroxylation | HLMs | LCMS | time dependent | KI = 4.1 μM; kinact = 0.068 min−1 |
| midazolam | 1’-hydroxylation | HIMs | LCMS | time dependent | KI = 4.2 μM; kinact = 0.079 min−1 | |
| luciferin-BE | luminometric | competitive | Ki = 380 μM |
luciferin-BE, luciferin 6’ benzyl ether; luciferin H, 6’deoxyluciferin; luciferin ME-EGE, ethylene glycol ester of luciferin 6’ methyl ether.
An in vitro to in vivo extrapolation (IVIVE) method was used to predict the likelihood of P450-mediated pharmacokinetic drug interactions upon oral consumption of a low dose (2 g) of kratom. Using an established mechanistic static model for time-dependent inhibition, a high interaction risk was predicted with the CYP3A probe substrate midazolam, as indicated by an AUCR (area under the plasma concentration vs. time curve in the presence to absence of inhibitor) of 5.7. Such a high magnitude of interaction can be attributed to the abundant expression of CYP3A in enterocytes coupled with a high mitragynine concentration in the intestinal lumen relative to the liver (Thelen and Dressman, 2009). In contrast, the mechanistic static model using reversible inhibition predicted a low interaction risk with the CYP2D6 probe substrate dextromethorphan (AUCR = 1.1). Although the likelihood of an interaction with CYP2D6 substrates was predicted to be low, clinically relevant interactions may still result at higher kratom doses.
Other alkaloids, including paynantheine, speciogynine, speciociliatine, corynantheidine, and 7-hydroxymitraynine, have been shown to differentially inhibit P450 activities (Kamble et al., 2020b). In general, like mitragynine, these other alkaloids reversibly inhibited CYP2D6 (IC50 ∼4–13 μM), CYP3A (IC50 ∼7–26 μM), and CYP2C19 (IC50 ∼8–38 μM). The extent of inhibition of CYP2C9, CYP2D6, and CYP3A activities by kratom extracts (20 μg/ml) containing 1 μM mitragynine was higher compared with an equimolar concentration of purified mitragynine (1 μM) tested alone (Todd et al., 2020). Mitragynine accounted for most of the inhibitory effects, which may be due to its relatively high abundance in the extracts and strong inhibitory potential compared with other alkaloids. Collectively, these observations suggest that more than one alkaloid is responsible for the total P450 inhibitory effects. The total inhibitory effect in vivo likely depends on alkaloid abundance in the product used, concentration at the enzyme site, and the inhibition kinetics of individual phytoconstituents.
Mitragynine has also been shown to inhibit UGT activity. UGT2B7 was inhibited in HLMs (IC50 ∼8 μM), whereas UGT1A1 was unaffected (IC50 >100 μM) (Abdullah and Ismail, 2018) (Table 4). However, the effects of kratom and its alkaloids on other major intestinal and hepatic UGTs, including UGT1A4, UGT1A6, UGT1A9, UGT1A10, UGT2B4, UGTB15, and UGT2B17, remain to be evaluated.
Mitragynine and 7-hydroxymitragynine were reported to inhibit the efflux transporter P-glycoprotein (P-gp). Both alkaloids inhibited P-gp–mediated efflux in MDR-MDCK cells and using the calcein-AM uptake assay, with an IC50 of ∼18 and 32 μM, respectively (Manda et al., 2014a). Additionally, mitragynine (10 μM) inhibited digoxin transport in Caco-2 cell monolayers to the same extent as the clinically relevant P-gp inhibitor quinidine (Rusli et al., 2019). High concentrations of mitragynine and other alkaloids attained in the intestinal lumen (∼400 μM based on a 2-g kratom dose containing ∼40 mg mitragynine in 250 ml) could inhibit intestinal P-gp, attenuating the efflux of P-gp substrates to potentially increase oral bioavailability. Applying a basic static model, mitragynine was predicted to precipitate an interaction with P-gp (Table 5). Inhibition of another intestinal efflux transporter, breast cancer resistance protein (BCRP), has also been tested, but the IC50 (∼360 μM) may be inconsequential (Wagmann et al., 2018). Effects of kratom on other drug transporters recognized to be of clinical importance by the FDA, including organic anion transporting polypeptides (OATPs), organic anion transporters (OATs), organic cation transporters (OCTs), multidrug resistance proteins (MRPs), and bile salt export pump (BSEP), are lacking (https://www.fda.gov/media/134582/download).
Kratom extracts and alkaloids, including mitragynine, have been reported to activate the pregnane X receptor in HepG2 cells (EC50 ∼5 μg/ml and 1–5 μM, respectively) (Manda et al., 2017; Hartley et al., 2022), which may induce the expression of certain drug metabolizing enzymes and transporters, including CYP3A and P-gp (Tolson and Wang, 2010). Although mitragynine did not increase CYP3A4 mRNA expression nor enzyme activity, the oxindole alkaloids isorotundifoline, isospeciofoline, corynoxine B, and corynoxine increased both markers by ∼2- to 3-fold relative to vehicle. However, given the typically lower abundance of these alkaloids compared with other well studied alkaloids in kratom products, the induction potential of kratom remains uncertain. Additional in vitro (e.g., human hepatocytes) and clinical studies, combined with physiologically based pharmacokinetic modeling and simulation, are warranted to determine the net effect of kratom-mediated time-dependent inhibition and induction of drug metabolism and transport upon chronic use.
CYP3A and CYP2D6 metabolize >50% of marketed drugs (that are cleaved by oxidation) belonging to a broad range of therapeutic areas. Inhibition of these P450 enzymes could have major implications for pharmacotherapy. Specifically, both metabolize and eliminate several centrally acting drugs, including opioids and benzodiazepines (Mercadante, 2015). Thus, kratom users co-consuming such drugs may be at high risk of experiencing a pharmacokinetic/pharmacodynamic drug interaction (Fig. 3). Regarding conjugative enzymes, UGT2B7 is widely recognized as the major isoform responsible for metabolizing opioids, including morphine, codeine, some codeine metabolites, and other related compounds (Mercadante, 2015). These UGT2B7 drug substrates could also be involved in adverse drug interactions with kratom.
Fig. 3.
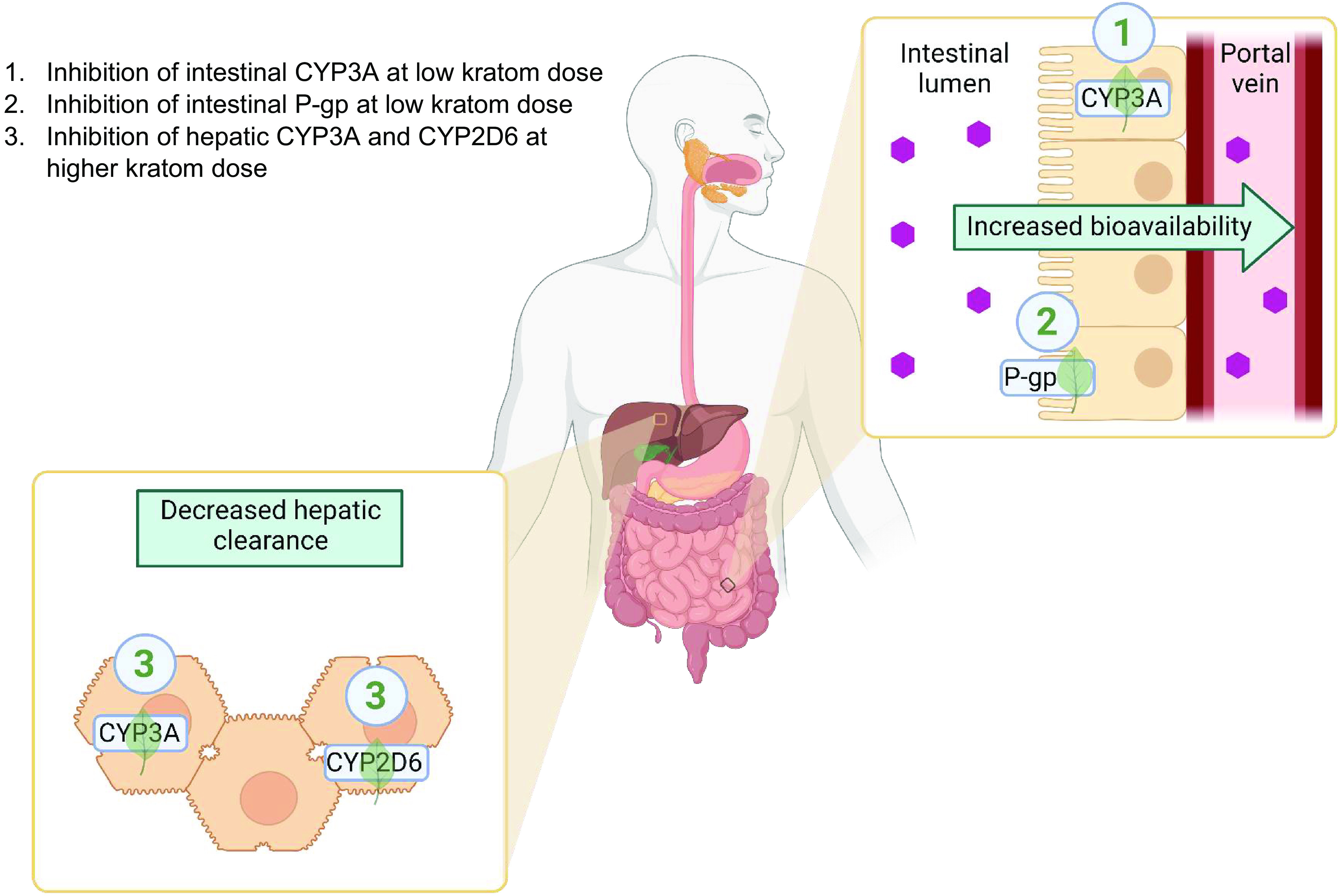
Identified targets and relevant mechanisms of potential pharmacokinetic kratom-drug interactions after oral administration of kratom (created with https://www.biorender.com/).
Suspected Cases of Kratom-Drug Interactions in Humans
A recent report that compiled a total of 156 deaths associated with kratom use in the United States and Europe highlighted that 87% of the cases involved polysubstance use (Corkery et al., 2019). These cases were predominant in white (100%) males (80%) with a mean age of ∼32 years and a history of drug abuse (95%). Three cases that exemplify a suspected pharmacokinetic kratom-drug interaction are described.
Case 1. In Sweden, nine deaths within <1 year were linked to krypton (Kronstrand et al., 2011), which consists of kratom and O-desmethyltramadol, a metabolite of tramadol that is a more potent opioid than the parent drug. Postmortem blood samples from all nine decedents contained mitragynine and O-desmethyltramadol along with at least two or more other centrally acting drugs, including anxiolytics, antipsychotics, antidepressants, psychedelics, and sedatives. Pulmonary edema and bladder distension were the most commonly observed features postmortem, both of which are associated with opioid-like toxicity. These toxicities may be due to kratom/mitragynine, but the blood concentrations of mitragynine varied considerably (0.02–0.18 μg/g). Because the observed toxicities were not directly correlated with mitragynine concentration, these effects may have been due to a toxic build-up of O-desmethyltramadol and/or other unknown opioids as part of the multidrug exposure. Kratom may have increased the systemic concentration of O-desmethyltramadol via inhibition of CYP3A4 or UGT2B7, contributing to overdose and death.
Case 2. A probable pharmacokinetic interaction between ingested kratom and the antipsychotic quetiapine was described for a 27-year-old male with a history of bipolar disorder, Asperger’s syndrome, and substance abuse (Hughes, 2019). He was found deceased with a toxic blood concentration of quetiapine (12 μg/ml), which was ∼100-fold higher than therapeutic concentrations, along with valproic acid (8.8 μg/ml) and qualitative detection of mitragynine. Based on pill count, the decedent was believed not to have ingested an excessive amount of quetiapine to attain the observed supratherapeutic blood concentration. Quetiapine is a substrate for CYP3A4 and P-gp, leading to suspicions of a pharmacokinetic kratom-drug interaction. Mitragynine-mediated time-dependent inhibition of intestinal and hepatic CYP3A4 activity could have diminished the first-pass metabolism of quetiapine via CYP3A4 (fraction metabolized ∼0.85), at least in the liver (fraction escaping gut metabolism ∼0.99) (Gjestad et al., 2017). Alternatively, mitragynine-mediated inhibition of intestinal P-gp could have increased the oral bioavailability of quetiapine, resulting in toxic systemic concentrations (Boulton et al., 2002).
Case 3. A 36-year-old male presented to the emergency department with serotonin syndrome and electrocardiogram abnormalities, which were suspected to result from adverse effects of the antidepressant venlafaxine and/or quetiapine, which were taken concurrently with an ultrahigh amount of kratom (∼90 g per day) (Brogdon et al., 2022). Kratom was speculated to inhibit the metabolism of either or both drugs, which is mediated by CYP2D6 and CYP3A, leading to drug build-up in the systemic circulation. Upon discontinuation of venlafaxine and quetiapine (but not kratom), the adverse effects gradually resolved. Reversal of these effects further supports that a kratom-drug interaction occurred.
In addition to pharmacokinetic mechanisms, some of the observed polyintoxications may have been exclusively pharmacodynamic in nature. For example, the presence of other drugs may lower the seizure threshold to increase the likelihood of kratom-mediated seizures. This mechanism may underlie the presumed interaction between kratom and the stimulant modafinil (Boyer et al., 2008), which is not a substrate for CYP2D6 or CYP3A nor does the drug cause seizures. However, modafinil, quetiapine, tricyclic antidepressants such as amitriptyline, and over-the-counter antihistaminic agents such as diphenhydramine and propylhexedrine are known to lower the seizure threshold (Holler et al., 2011; Umaharan et al., 2021). Accordingly, these drugs may increase the risk of seizures caused by kratom depending on the dose of kratom and the susceptibility of the user.
Overall, the information garnered from kratom-associated polyintoxications and the in vitro inhibitory effects of kratom (and its alkaloids) on P450 and P-gp activity suggests a high likelihood of observing additional pharmacokinetic kratom-drug interactions that affect drug pharmacodynamics. Mitragynine concentrations measured in postmortem blood or plasma (up to 12 μM) approached or exceeded the inhibition constants (IC50, Ki, KI) toward CYP2D6 and CYP3A (Papsun et al., 2019). The interaction risk may be higher for drugs that are extensively metabolized by intestinal CYP3A and/or are substrates of P-gp assuming a higher concentration of mitragynine in the intestine compared with the liver (Fig. 3). In addition, the overall risk of a pharmacokinetic kratom-drug interaction will depend on multiple factors, including kratom consumption pattern, coingested drugs, and the user’s genetic make-up.
Perspectives and Path Forward
Increasing kratom use likely will continue due to the common (mis)perception that it is safe. The ready availability of kratom products and low cost make kratom the preferred treatment of pain and opioid withdrawal symptoms among kratom users (Coe et al., 2019). People often use kratom in an attempt to wean themselves from other dangerous drugs on the market, but they may relapse (White, 2018; Japarin et al., 2023). Hence, the likelihood of concomitant use of kratom with other psychoactive substances like opioids, benzodiazepines, or antidepressants is high. Taken together, thorough clinical evaluation of potential pharmacokinetic, as well as pharmacodynamic, kratom-drug interactions is warranted. The in vitro evidence and clinical case reports to date point to CYP3A, CYP2D6, and P-gp as targets of potential clinical pharmacokinetic kratom-drug interactions. An iterative approach involving physiologically based pharmacokinetic (PBPK) modeling and simulation and clinical pharmacokinetic studies can be used to comprehensively evaluate these interactions (Fig. 4).
Fig. 4.

Proposed iterative approach for rigorous assessment of potential pharmacokinetic kratom-drug interactions (created with https://www.biorender.com/).
Clinical Assessment
Like pharmacokinetic drug-drug interactions recommended by regulatory agencies, kratom-drug interactions should be studied in a controlled clinical environment to obtain direct, actionable evidence. As mentioned earlier, clinical kratom-drug interaction studies are warranted for targets identified in this review, particularly CYP2D6, CYP3A, and P-gp. Studies should be conducted using well characterized kratom products and relevant probe drugs as described elsewhere for evaluating potential pharmacokinetic natural product-drug interactions (Kellogg et al., 2019; Cox et al., 2022). Evaluation of a well characterized kratom product representative of the majority of marketed products, administered in a form mimicking typical usage, will ensure generalizability of the results. An advantage of using probe drugs is that the results can be extrapolated to other drugs that either fully or partially rely on the corresponding metabolic or transporter pathways. The probe drugs can be administered at subtherapeutic doses, as the objective of the study is to observe a pharmacokinetic interaction, rather than a change in pharmacodynamics, in heathy participants. Chronic administration of kratom should also be considered because time-dependent inhibition of CYP3A was identified, which may be underestimated with a single dose.
Subsequent to studies involving probe drugs, pharmacokinetic interaction studies using clinically relevant object drugs causing a measurable change in pharmacodynamics can be undertaken. Pharmacodynamic changes can be measured using clinical biomarkers relevant to the object drugs. Clinical evaluation can later be expanded to other target drug metabolizing enzymes and transporters using larger probe cocktails (five to six object drugs), which may reveal other drug interactions that were either not tested or missed in vitro (Nguyen et al., 2021). Alternatively, emerging endogenous biomarkers such as coproporphyrins for OATPs, N-methyl nicotinamide for OCTs, and homovanillic acid/pyridoxic acid for OATs (Li et al., 2021) can be monitored for alteration in transporter activity with kratom administration.
PBPK Modeling and Simulation
As for natural product-drug interactions in general, the complexities involved in assessing kratom-drug interactions, including the compositional variability of marketed kratom products and diverse consumption patterns, can be overcome using in vitro to in vivo extrapolation (IVIVE) approaches (Cox et al., 2021). Dynamic mathematical models, including PBPK models, which consider both human physiology and drug-related parameters, have been extensively used to support the drug discovery and development process. PBPK models can be applied to kratom-drug interactions to simulate various scenarios that are otherwise difficult or unethical to test in humans. Such model predictions are increasingly accepted by regulatory agencies in lieu of results generated from formal clinical pharmacokinetic interaction studies. The available mechanistic in vitro information about the P450 and P-gp inhibitory effects of kratom and the clinical pharmacokinetics of key kratom alkaloids using a well characterized product can be used to develop robust PBPK models. The next steps in this iterative approach (Fig. 4) would be to clinically assess potential pharmacokinetic kratom-drug interactions, the data from which can be used to refine the PBPK models, potentially prompting follow-up clinical studies. Unforeseen observations from clinical studies can also prompt follow-up clinical studies. The end goal would be to continue refining the PBPK model with these additional clinical, as well as mechanistic in vitro, data to accurately simulate various real-world scenarios (e.g., different object drugs, kratom doses, kratom consumption patterns, special populations).
Summary
The research community is beginning to fill critical knowledge gaps regarding the epidemiology, chemistry, pharmacology (pharmacokinetics and pharmacodynamics), and toxicology of kratom. The increasing number of kratom-related adverse events involving polyintoxication warrants a thorough investigation of potential kratom-drug interactions, which can be accomplished using an iterative approach involving additional mechanistic in vitro, PBPK modeling and simulation, and clinical assessments. The widespread availability and use of kratom products underscore the urgency of this research. Multidisciplinary collaborations among the natural products industry, academia, and government agencies are essential to reduce the time lag in providing this essential information. National Institutes of Health National Center for Complementary and Integrative Health, National Institute on Drug Abuse, and other agencies are funding several research projects to create awareness among kratom users, healthcare providers, and federal regulators. Results from these projects will help various authorities develop scientifically informed policies to promote the safe use of kratom and mitigate further public health hazards.
Acknowledgments
M.F.P. dedicates this article to David P. Paine.
Data Availability
The authors declare that all of the data included in this review are available within the paper and/or are openly available in the Natural Product-Drug Interaction Research (NaPDI) Center Database (https://repo.napdi.org/).
Abbreviations
- AUC
area under the concentration versus time curve
- DEA
US Drug Enforcement Administration
- FDA
US Food and Drug Administration
- Foral
absolute oral bioavailability
- hCES
human carboxylesterase
- HIM
human intestinal microsome
- HLM
human liver microsome
- KI
time-dependent inhibition constant
- Ki
reversible inhibition constant
- kinact
maximum rate of inactivation
- LCMS
liquid chromatography–mass spectrometry
- P450
cytochrome P450
- PBPK
physiologically based pharmacokinetic
- P-gp
P-glycoprotein
- UGT
UDP-glucuronosyltransferase
Authorship Contributions
Performed data analysis: Tanna.
Wrote or contributed to the writing of the manuscript: Tanna, Cech, Oberlies, Rettie, Thummel, Paine.
Footnotes
This work was supported by National Institutes of Health (NIH) National Center for Complimentary and Integrative Health and Office of Dietary Supplements [Grant U54 AT008909] (to M.F.P.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
The authors have declared a conflict of interest. N.H.O. serves on the Scientific Advisory Boards of Clue Genetics, Inc., Mycosynthetix, Inc., and Ionic Pharmaceuticals, LLC.
References
- Abdullah NH, Ismail S (2018) Inhibition of UGT2B7 enzyme activity in human and rat liver microsomes by herbal constituents. Molecules 23:2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsarraf E, Myers J, Culbreth S, Fanikos J (2019) Kratom from head to toe—case reviews of adverse events and toxicities. Curr Emerg Hosp Med Rep 7:141–168. [Google Scholar]
- Anand A, Hosanagar A (2022) The addictive potential and challenges with use of the “herbal supplement” kratom: a case report and literature review. Pain Med 23:4–9. [DOI] [PubMed] [Google Scholar]
- Avery BA, Boddu SP, Sharma A, Furr EB, Leon F, Cutler SJ, McCurdy CR (2019) Comparative pharmacokinetics of mitragynine after oral administration of Mitragyna speciosa (kratom) leaf extracts in rats. Planta Med 85:340–346. [DOI] [PubMed] [Google Scholar]
- Basiliere S, Bryand K, Kerrigan S (2018) Identification of five Mitragyna alkaloids in urine using liquid chromatography-quadrupole/time of flight mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 1080:11–19. [DOI] [PubMed] [Google Scholar]
- Basiliere S, Kerrigan S (2020) Temperature and pH-dependent stability of Mitragyna alkaloids. J Anal Toxicol 44:314–324. [DOI] [PubMed] [Google Scholar]
- Beckett AH, Dwuma-Badu D (1969) The influence of stereochemistry on pKa, rate of quaternization and partition coefficients of corynantheidine-type alkaloids. J Pharm Pharmacol 21:162S–168S. [DOI] [PubMed] [Google Scholar]
- Berthold EC, Kamble SH, Raju KS, King TI, Popa R, Sharma A, León F, Avery BA, McMahon LR, McCurdy CR (2021) Preclinical pharmacokinetic study of speciociliatine, a kratom alkaloid, in rats using an UPLC-MS/MS method. J Pharm Biomed Anal 194:113778. [DOI] [PubMed] [Google Scholar]
- Berthold ECKamble SHRaju KSKuntz MASenetra ASMottinelli MLeón FRestrepo LFPatel AHo NP, et al. (2022) The lack of contribution of 7-hydroxymitragynine to the antinociceptive effects of mitragynine in mice: a pharmacokinetic and pharmacodynamic study. Drug Metab Dispos 50:158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton DW, DeVane CL, Liston HL, Markowitz JS (2002) In vitro P-glycoprotein affinity for atypical and conventional antipsychotics. Life Sci 71:163–169. [DOI] [PubMed] [Google Scholar]
- Boyer EW, Babu KM, Adkins JE, McCurdy CR, Halpern JH (2008) Self-treatment of opioid withdrawal using kratom (Mitragyna speciosa korth). Addiction 103:1048–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogdon HD, McPhee MM, Paine MF, Cox EJ, Burns AG (2022) A case of potential pharmacokinetic kratom-drug interactions resulting in toxicity and subsequent treatment of kratom use disorder with buprenorphine/naloxone. J Addict Med 16:606–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PN, Lund JA, Murch SJ (2017) A botanical, phytochemical and ethnomedicinal review of the genus Mitragyna korth: implications for products sold as kratom. J Ethnopharmacol 202:302–325. [DOI] [PubMed] [Google Scholar]
- Chakraborty SUprety RSlocum STIrie TLe Rouzic VLi XWilson LLScouller BAlder AFKruegel AC, et al. (2021) Oxidative metabolism as a modulator of kratom’s biological actions. J Med Chem 64:16553–16572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chear NJ-YLeón FSharma AKanumuri SRRZwolinski GAbboud KASingh DRestrepo LFPatel AHiranita T, et al. (2021) Exploring the chemistry of alkaloids from Malaysian Mitragyna speciosa (kratom) and the role of oxindoles on human opioid receptors. J Nat Prod 84:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin K-Y, Mark-Lee WF (2018) A review on the antinociceptive effects of Mitragyna speciosa and its derivatives on animal model. Curr Drug Targets 19:1359–1365. [DOI] [PubMed] [Google Scholar]
- Cinosi EMartinotti GSimonato PSingh DDemetrovics ZRoman-Urrestarazu ABersani FSVicknasingam BPiazzon GLi J-H, et al. (2015) Following “the roots” of kratom (Mitragyna speciosa): the evolution of an enhancer from a traditional use to increase work and productivity in Southeast Asia to a recreational psychoactive drug in western countries. BioMed Res Int 2015:968786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe MA, Pillitteri JL, Sembower MA, Gerlach KK, Henningfield JE (2019) Kratom as a substitute for opioids: results from an online survey. Drug Alcohol Depend 202:24–32. [DOI] [PubMed] [Google Scholar]
- Corkery JM, Streete P, Claridge H, Goodair C, Papanti D, Orsolini L, Schifano F, Sikka K, Körber S, Hendricks A (2019) Characteristics of deaths associated with kratom use. J Psychopharmacol 33:1102–1123. [DOI] [PubMed] [Google Scholar]
- Cox EJ, Rettie AE, Unadkat JD, Thummel KE, McCune JS, Paine MF (2022) Adapting regulatory drug-drug interaction guidance to design clinical pharmacokinetic natural product-drug interaction studies: a NaPDI Center recommended approach. Clin Transl Sci 15:322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox EJ, Tian D-D, Clarke JD, Rettie AE, Unadkat JD, Thummel KE, McCune JS, Paine MF (2021) Modeling pharmacokinetic natural product-drug interactions for decision-making: a NaPDI Center recommended approach. Pharmacol Rev 73:847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggleston W, Stoppacher R, Suen K, Marraffa JM, Nelson LS (2019) Kratom use and toxicities in the United States. Pharmacotherapy 39:775–777. [DOI] [PubMed] [Google Scholar]
- Field E (1921) XCVIII.—Mitragynine and mitraversine, two new alkaloids from species of mitragyne. J Chem Soc Trans 119:887–891. [Google Scholar]
- Flores-Bocanegra L, Raja HA, Graf TN, Augustinović M, Wallace ED, Hematian S, Kellogg JJ, Todd DA, Cech NB, Oberlies NH (2020) The chemistry of kratom [Mitragyna speciosa]: updated characterization data and methods to elucidate indole and oxindole alkaloids. J Nat Prod 83:2165–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjestad C, Haslemo T, Andreassen OA, Molden E (2017) 4β-Hydroxycholesterol level significantly correlates with steady-state serum concentration of the CYP3A4 substrate quetiapine in psychiatric patients. Br J Clin Pharmacol 83:2398–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundmann O, Hendrickson RG, Greenberg MI (2023) Kratom: history, pharmacology, current user trends, adverse health effects and potential benefits. Dis Mon 69:101442. [DOI] [PubMed] [Google Scholar]
- Hanapi NA, Ismail S, Mansor SM (2013) Inhibitory effect of mitragynine on human cytochrome P450 enzyme activities. Pharmacognosy Res 5:241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harizal SN, Mansor SM, Hasnan J, Tharakan JK, Abdullah J (2010) Acute toxicity study of the standardized methanolic extract of Mitragyna speciosa Korth in rodent. J Ethnopharmacol 131:404–409. [DOI] [PubMed] [Google Scholar]
- Hartley C 2nd, Bulloch M, Penzak SR (2022) Clinical pharmacology of the dietary supplement kratom (Mitragyna speciosa). J Clin Pharmacol 62:577–593. [DOI] [PubMed] [Google Scholar]
- Hassan ZMuzaimi MNavaratnam VYusoff NHSuhaimi FWVadivelu RVicknasingam BKAmato Dvon Hörsten SIsmail NI, et al. (2013) From kratom to mitragynine and its derivatives: physiological and behavioural effects related to use, abuse, and addiction. Neurosci Biobehav Rev 37:138–151. [DOI] [PubMed] [Google Scholar]
- Hiranita T, Obeng S, Sharma A, Wilkerson JL, McCurdy CR, McMahon LR (2022) In vitro and in vivo pharmacology of kratom. Adv Pharmacol 93:35–76. [DOI] [PubMed] [Google Scholar]
- Holler JM, Vorce SP, McDonough-Bender PC, Magluilo J Jr, Solomon CJ, Levine B (2011) A drug toxicity death involving propylhexedrine and mitragynine. J Anal Toxicol 35:54–59. [DOI] [PubMed] [Google Scholar]
- Hughes RL (2019) Fatal combination of mitragynine and quetiapine - a case report with discussion of a potential herb-drug interaction. Forensic Sci Med Pathol 15:110–113. [DOI] [PubMed] [Google Scholar]
- Japarin RA, Harun N, Hassan Z, Shoaib M (2023) Mitragynine, a primary constituent of kratom reinstates morphine-seeking behaviour in rats. Behav Pharmacol 34:123–130. [DOI] [PubMed] [Google Scholar]
- Kamble SHLeón FKing TIBerthold ECLopera-Londoño CSiva Rama Raju KHampson AJSharma AAvery BAMcMahon LR, et al. (2020a) Metabolism of a kratom alkaloid metabolite in human plasma increases its opioid potency and efficacy. ACS Pharmacol Transl Sci 3:1063–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamble SH, Sharma A, King TI, Berthold EC, León F, Meyer PKL, Kanumuri SRR, McMahon LR, McCurdy CR, Avery BA (2020b) Exploration of cytochrome P450 inhibition mediated drug-drug interaction potential of kratom alkaloids. Toxicol Lett 319:148–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamble SH, Sharma A, King TI, León F, McCurdy CR, Avery BA (2019) Metabolite profiling and identification of enzymes responsible for the metabolism of mitragynine, the major alkaloid of Mitragyna speciosa (kratom). Xenobiotica 49:1279–1288. [DOI] [PubMed] [Google Scholar]
- Karunakaran T, Ngew KZ, Zailan AAD, Mian Jong VY, Abu Bakar MH (2022) The chemical and pharmacological properties of mitragynine and its diastereomers: an insight review. Front Pharmacol 13:805986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg JJ, Paine MF, McCune JS, Oberlies NH, Cech NB (2019) Selection and characterization of botanical natural products for research studies: a NaPDI center recommended approach. Nat Prod Rep 36:1196–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerrigan S, Basiliere S (2022) Kratom: a systematic review of toxicological issues. Wiley Interdisciplinary Reviews: Forensic Science 4:e1420. [Google Scholar]
- Kong WM, Chik Z, Mohamed Z, Alshawsh MA (2017) Physicochemical characterization of Mitragyna speciosa alkaloid extract and mitragynine using in vitro high throughput assays. Comb Chem High Throughput Screen 20:796–803. [DOI] [PubMed] [Google Scholar]
- Kong WM, Chik Z, Ramachandra M, Subramaniam U, Aziddin RER, Mohamed Z (2011) Evaluation of the effects of Mitragyna speciosa alkaloid extract on cytochrome P450 enzymes using a high throughput assay. Molecules 16:7344–7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronstrand R, Roman M, Thelander G, Eriksson A (2011) Unintentional fatal intoxications with mitragynine and O-desmethyltramadol from the herbal blend krypton. J Anal Toxicol 35:242–247. [DOI] [PubMed] [Google Scholar]
- Kruegel AC, Gassaway MM, Kapoor A, Váradi A, Majumdar S, Filizola M, Javitch JA, Sames D (2016) Synthetic and receptor signaling explorations of the Mitragyna alkaloids: mitragynine as an atypical molecular framework for opioid receptor modulators. J Am Chem Soc 138:6754–6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruegel AC, Grundmann O (2018) The medicinal chemistry and neuropharmacology of kratom: a preliminary discussion of a promising medicinal plant and analysis of its potential for abuse. Neuropharmacology 134 (Pt A):108–120. [DOI] [PubMed] [Google Scholar]
- Kruegel ACUprety RGrinnell SGLangreck CPekarskaya EALe Rouzic VAnsonoff MGassaway MMPintar JEPasternak GW, et al. (2019) 7-Hydroxymitragynine is an active metabolite of mitragynine and a key mediator of its analgesic effects. ACS Cent Sci 5:992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- León FObeng SMottinelli MChen YKing TIBerthold ECKamble SHRestrepo LFPatel AGamez-Jimenez LR, et al. (2021) Activity of Mitragyna speciosa (“kratom”) alkaloids at serotonin receptors. J Med Chem 64:13510–13523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Ndungu P, Taneja SB, Chapin MR, Egbert SB, Akenapalli K, Paine MF, Kane‐Gill SL, Boyce RD (2023) An evaluation of adverse drug reactions and outcomes attributed to kratom in the US Food and Drug Administration Adverse Event Reporting System from January 2004 through September 2021. Clin Transl Sci. DOI: 10.1111/cts.13505 [published ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Talebi Z, Chen X, Sparreboom A, Hu S (2021) Endogenous biomarkers for SLC transporter-mediated drug-drug interaction evaluation. Molecules 26:5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim EL, Seah TC, Koe XF, Wahab HA, Adenan MI, Jamil MFA, Majid MIA, Tan ML (2013) In vitro evaluation of cytochrome P450 induction and the inhibition potential of mitragynine, a stimulant alkaloid. Toxicol In Vitro 27:812–824. [DOI] [PubMed] [Google Scholar]
- Lydecker AG, Sharma A, McCurdy CR, Avery BA, Babu KM, Boyer EW (2016) Suspected adulteration of commercial kratom products with 7-hydroxymitragynine. J Med Toxicol 12:341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIntyre AC, Cutler DJ (1988) The potential role of lysosomes in tissue distribution of weak bases. Biopharm Drug Dispos 9:513–526. [DOI] [PubMed] [Google Scholar]
- Manda V, Avula B, Ali Z, Khan I, Walker L, Khan S (2014a) Determination of ADME properties of mitragynine, 7-hydroxymitragynine, and mitraphylline. Planta Med 80:PU10. [DOI] [PubMed] [Google Scholar]
- Manda VK, Avula B, Ali Z, Khan IA, Walker LA, Khan SI (2014b) Evaluation of in vitro absorption, distribution, metabolism, and excretion (ADME) properties of mitragynine, 7-hydroxymitragynine, and mitraphylline. Planta Med 80:568–576. [DOI] [PubMed] [Google Scholar]
- Manda VK, Avula B, Dale OR, Ali Z, Khan IA, Walker LA, Khan SI (2017) PXR mediated induction of CYP3A4, CYP1A2, and P-gp by Mitragyna speciosa and its alkaloids. Phytother Res 31:1935–1945. [DOI] [PubMed] [Google Scholar]
- Manwill PK, Flores-Bocanegra L, Khin M, Raja HA, Cech NB, Oberlies NH, Todd DA (2022) Kratom (Mitragyna speciosa) validation: quantitative analysis of indole and oxindole alkaloids reveals chemotypes of plants and products. Planta Med 88:838–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K, Mizowaki M, Suchitra T, Murakami Y, Takayama H, Sakai S, Aimi N, Watanabe H (1996a) Central antinociceptive effects of mitragynine in mice: contribution of descending noradrenergic and serotonergic systems. Eur J Pharmacol 317:75–81. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Mizowaki M, Suchitra T, Takayama H, Sakai S, Aimi N, Watanabe H (1996b) Antinociceptive action of mitragynine in mice: evidence for the involvement of supraspinal opioid receptors. Life Sci 59:1149–1155. [DOI] [PubMed] [Google Scholar]
- Maxwell EA, King TI, Kamble SH, Raju KSR, Berthold EC, León F, Avery BA, McMahon LR, McCurdy CR, Sharma A (2020) Pharmacokinetics and safety of mitragynine in beagle dogs. Planta Med 86:1278–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercadante S (2015) Opioid metabolism and clinical aspects. Eur J Pharmacol 769:71–78. [DOI] [PubMed] [Google Scholar]
- Meyer MR, Schütz A, Maurer HH (2015) Contribution of human esterases to the metabolism of selected drugs of abuse. Toxicol Lett 232:159–166. [DOI] [PubMed] [Google Scholar]
- Ngernsaengsaruay C, Leksungnoen N, Boonthasak W, Utharatsamee S, Racharak P, Leetanasakskul K, Pongamorn P, Saengbuapuean A (2022) Additional knowledge on the genus Mitragyna (Rubiaceae) in Thailand. Thai Forest Bulletin (Botany) 50:20–39. [Google Scholar]
- Nguyen JTTian DDTanna RSHadi DLBansal SCalamia JCArian CMShireman LMMolnár BHorváth M, et al. (2021) Assessing transporter‐mediated natural product‐drug interactions via in vitro‐in vivo extrapolation: clinical evaluation with a probe cocktail. Clin Pharmacol Ther 109:1342–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng SKamble SHReeves MERestrepo LFPatel ABehnke MChear NJ-YRamanathan SSharma ALeón F, et al. (2020) Investigation of the adrenergic and opioid binding affinities, metabolic stability, plasma protein binding properties, and functional effects of selected indole-based kratom alkaloids. J Med Chem 63:433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng SWilkerson JLLeón FReeves MERestrepo LFGamez-Jimenez LRPatel APennington AETaylor VAHo NP, et al. (2021) Pharmacological comparison of mitragynine and 7-hydroxymitragynine: in vitro affinity and efficacy for μ-opioid receptor and opioid-like behavioral effects in rats. J Pharmacol Exp Ther 376:410–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine MF (2020) Natural products: experimental approaches to elucidate disposition mechanisms and predict pharmacokinetic drug interactions. Drug Metab Dispos 48:956–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine MF, Roe AL (2018) “Green medicine”: the past, present, and future of botanicals. Clin Pharmacol Ther 104:410–415. [DOI] [PubMed] [Google Scholar]
- Papsun DM, Chan-Hosokawa A, Friederich L, Brower J, Graf K, Logan B (2019) The trouble with kratom: analytical and interpretative issues involving mitragynine. J Anal Toxicol 43:615–629. [DOI] [PubMed] [Google Scholar]
- Parthasarathy S, Ramanathan S, Ismail S, Adenan MI, Mansor SM, Murugaiyah V (2010) Determination of mitragynine in plasma with solid-phase extraction and rapid HPLC-UV analysis, and its application to a pharmacokinetic study in rat. Anal Bioanal Chem 397:2023–2030. [DOI] [PubMed] [Google Scholar]
- Peran D, Stern M, Cernohorsky P, Sykora R, Popela S, Duska F (2023) Mitragyna speciosa (kratom) poisoning: findings from ten cases. Toxicon 225:107054. [DOI] [PubMed] [Google Scholar]
- Philipp AA, Wissenbach DK, Zoerntlein SW, Klein ON, Kanogsunthornrat J, Maurer HH (2009) Studies on the metabolism of mitragynine, the main alkaloid of the herbal drug kratom, in rat and human urine using liquid chromatography-linear ion trap mass spectrometry. J Mass Spectrom 44:1249–1261. [DOI] [PubMed] [Google Scholar]
- Ponglux D, Wongseripipatana S, Takayama H, Kikuchi M, Kurihara M, Kitajima M, Aimi N, Sakai S (1994) A new indole alkaloid, 7 alpha-hydroxy-7H-mitragynine, from Mitragyna speciosa in Thailand. Planta Med 60:580–581. [DOI] [PubMed] [Google Scholar]
- Post S, Spiller HA, Chounthirath T, Smith GA (2019) Kratom exposures reported to United States poison control centers: 2011-2017. Clin Toxicol (Phila) 57:847–854. [DOI] [PubMed] [Google Scholar]
- Prozialeck WCAvery BABoyer EWGrundmann OHenningfield JEKruegel ACMcMahon LRMcCurdy CRSwogger MTVeltri CA, et al. (2019) Kratom policy: the challenge of balancing therapeutic potential with public safety. Int J Drug Policy 70:70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prozialeck WC, Jivan JK, Andurkar SV (2012) Pharmacology of kratom: an emerging botanical agent with stimulant, analgesic and opioid-like effects. J Am Osteopath Assoc 112:792–799. [PubMed] [Google Scholar]
- Ramanathan S, León F, Chear NJ, Yusof SR, Murugaiyah V, McMahon LR, McCurdy CR (2021) Kratom (Mitragyna speciosa Korth.): a description on the ethnobotany, alkaloid chemistry, and neuropharmacology. Stud Nat Prod Chem 69:195–225. [Google Scholar]
- Ramanathan S, Parthasarathy S, Murugaiyah V, Magosso E, Tan SC, Mansor SM (2015) Understanding the physicochemical properties of mitragynine, a principal alkaloid of Mitragyna speciosa, for preclinical evaluation. Molecules 20:4915–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reanmongkol W, Keawpradub N, Sawangjaroen K (2007) Effects of the extracts from Mitragyna speciosa Korth. leaves on analgesic and behavioral activities in experimental animals. Songklanakarin J Sci Technol 29:39–48. [Google Scholar]
- Rusli N, Amanah A, Kaur G, Adenan MI, Sulaiman SF, Wahab HA, Tan ML (2019) The inhibitory effects of mitragynine on P-glycoprotein in vitro. Naunyn Schmiedebergs Arch Pharmacol 392:481–496. [DOI] [PubMed] [Google Scholar]
- Sabetghadam A, Navaratnam V, Mansor SM (2013a) Dose–response relationship, acute toxicity, and therapeutic index between the alkaloid extract of Mitragyna speciosa and its main active compound mitragynine in mice. Drug Dev Res 74:23–30. [Google Scholar]
- Sabetghadam A, Ramanathan S, Sasidharan S, Mansor SM (2013b) Subchronic exposure to mitragynine, the principal alkaloid of Mitragyna speciosa, in rats. J Ethnopharmacol 146:815–823. [DOI] [PubMed] [Google Scholar]
- Schimmel J, Amioka E, Rockhill K, Haynes CM, Black JC, Dart RC, Iwanicki JL (2021) Prevalence and description of kratom (Mitragyna speciosa) use in the United States: a cross-sectional study. Addiction 116:176–181. [DOI] [PubMed] [Google Scholar]
- Schimmel J, Dart RC (2020) Kratom (Mitragyna speciosa) liver injury: a comprehensive review. Drugs 80:263–283. [DOI] [PubMed] [Google Scholar]
- Sengnon N, Vonghirundecha P, Chaichan W, Juengwatanatrakul T, Onthong J, Kitprasong P, Sriwiriyajan S, Chittrakarn S, Limsuwanchote S, Wungsintaweekul J (2023) Seasonal and geographic variation in alkaloid content of kratom (Mitragyna speciosa (Korth.) Havil.) from Thailand. Plants 12:949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, Tankersley W, Mekala H (2021) Kratom: an emerging issue and need for regulations in the United States. Prim Care Companion CNS Disord 23:26032. [DOI] [PubMed] [Google Scholar]
- Singh D, Müller CP, Vicknasingam BK, Mansor SM (2015) Social functioning of kratom (Mitragyna speciosa) users in Malaysia. J Psychoactive Drugs 47:125–131. [DOI] [PubMed] [Google Scholar]
- Singh D, Narayanan S, Vicknasingam B, Corazza O, Santacroce R, Roman-Urrestarazu A (2017) Changing trends in the use of kratom (Mitragyna speciosa) in Southeast Asia. Hum Psychopharmacol 32:e2582. [DOI] [PubMed] [Google Scholar]
- Suwanlert S (1975) A study of kratom eaters in Thailand. Bull Narc 27:21–27. [PubMed] [Google Scholar]
- Takayama H (2004) Chemistry and pharmacology of analgesic indole alkaloids from the rubiaceous plant, Mitragyna speciosa. Chem Pharm Bull (Tokyo) 52:916–928. [DOI] [PubMed] [Google Scholar]
- Tanna RSNguyen JTHadi DLManwill PKFlores-Bocanegra LLayton MEWhite JRCech NBOberlies NHRettie AE, et al. (2022) Clinical pharmacokinetic assessment of kratom (Mitragyna speciosa), a botanical product with opioid-like effects, in healthy adult participants. Pharmaceutics 14:620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanna RS, Tian D-D, Cech NB, Oberlies NH, Rettie AE, Thummel KE, Paine MF (2021) Refined prediction of pharmacokinetic kratom-drug interactions: time-dependent inhibition considerations. J Pharmacol Exp Ther 376:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelen K, Dressman JB (2009) Cytochrome P450-mediated metabolism in the human gut wall. J Pharm Pharmacol 61:541–558. [DOI] [PubMed] [Google Scholar]
- Todd DAKellogg JJWallace EDKhin MFlores-Bocanegra LTanna RSMcIntosh SRaja HAGraf TNHemby SE, et al. (2020) Chemical composition and biological effects of kratom (Mitragyna speciosa): in vitro studies with implications for efficacy and drug interactions. Sci Rep 10:19158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolson AH, Wang H (2010) Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv Drug Deliv Rev 62:1238–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager W, Lee C, Phillipson J, Beckett A (1967) The absolute configuration of paynantheine and hirsutine. Tetrahedron 23:1043–1047. [Google Scholar]
- Trakulsrichai S, Sathirakul K, Auparakkitanon S, Krongvorakul J, Sueajai J, Noumjad N, Sukasem C, Wananukul W (2015) Pharmacokinetics of mitragynine in man. Drug Des Devel Ther 9:2421–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umaharan T, Sivayokan S, Sivansuthan S (2021) Amitriptyline dependence and its associations: a case report and literature review. Case Rep Psychiatry 2021:6647952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicknasingam BChooi WTRahim AARamachandram DSingh DRamanathan SYusof NSMZainal HMurugaiyah VGueorguieva R, et al. (2020) Kratom and pain tolerance: a randomized, placebo-controlled, double-blind study. Yale J Biol Med 93:229–238. [PMC free article] [PubMed] [Google Scholar]
- Wagmann L, Maurer HH, Meyer MR (2018) Inhibition and stimulation of the human breast cancer resistance protein as in vitro predictor of drug-drug interactions of drugs of abuse. Arch Toxicol 92:2875–2884. [DOI] [PubMed] [Google Scholar]
- Warner ML, Kaufman NC, Grundmann O (2016) The pharmacology and toxicology of kratom: from traditional herb to drug of abuse. Int J Legal Med 130:127–138. [DOI] [PubMed] [Google Scholar]
- Weiss ST, Douglas HE (2021) Treatment of kratom withdrawal and dependence with buprenorphine/naloxone: a case series and systematic literature review. J Addict Med 15:167–172. [DOI] [PubMed] [Google Scholar]
- White CM (2018) Pharmacologic and clinical assessment of kratom. Am J Health Syst Pharm 75:261–267. [DOI] [PubMed] [Google Scholar]
- Williams RS, Nikitin D (2020) The internet market for kratom, an opioid alternative and variably legal recreational drug. Int J Drug Policy 78:102715. [DOI] [PubMed] [Google Scholar]
- Yusof SR, Mohd Uzid M, Teh EH, Hanapi NA, Mohideen M, Mohamad Arshad AS, Mordi MN, Loryan I, Hammarlund-Udenaes M (2019) Rate and extent of mitragynine and 7-hydroxymitragynine blood-brain barrier transport and their intra-brain distribution: the missing link in pharmacodynamic studies. Addict Biol 24:935–945. [DOI] [PubMed] [Google Scholar]