

Abstract
Double-stranded breaks (DSBs) are toxic DNA damage and a serious threat to genomic integrity. Thus, all living organisms have evolved multiple mechanisms of DNA DSB repair, the two principal ones being classical-non homologous end joining (C-NHEJ), and homology dependent recombination (HDR). In mammals, C-NHEJ is the predominate DSB repair pathway, but how a cell chooses to repair a particular DSB by a certain pathway is still not mechanistically clear. To uncover novel regulators of DSB repair pathway choice, we performed a kinome-wide screen in a human cell line engineered to express a dominant-negative C-NHEJ factor. The intellectual basis for such a screen was our hypothesis that a C-NHEJ-crippled cell line might need to upregulate other DSB repair pathways, including HDR, in order to survive. This screen identified Bromodomain-containing Protein 3 (BRD3) as a protein whose expression was almost completely ablated specifically in a C-NHEJ-defective cell line. Subsequent experimentation demonstrated that BRD3 is a negative regulator of HDR as BRD3-null cell lines proved to be hyper-recombinogenic for gene conversion, sister chromatid exchanges and gene targeting. Mechanistically, BRD3 appears to be working at the level of Radiation Sensitive 51 (RAD51) recruitment. Overall, our results demonstrate that BRD3 is a novel regulator of human DSB repair pathway choice.
Keywords: BRD3, Kinome, HDR, RAD51, PRKDC
1. Introduction
Most living organisms have evolved at least two discrete mechanisms to repair DNA double-stranded breaks (DSBs): homology dependent recombination (HDR) and classical non-homologous end joining (C-NHEJ). In HDR, a DSB is repaired using an undamaged homologue or sister chromatid in a process that generally requires extensive regions of homology between the damaged chromosome and the undamaged donor [1,2]. Most mitotic and meiotic recombination and the repair of DSBs in late S and G2 phases of the cell cycle [e.g., stalled replication forks; [3]] are carried out by HDR. Thus, HDR accounts for an important portion of the DNA DSB activity in a wild-type human cell. Not surprisingly therefore, mutations of many HDR genes are associated with cancer predisposition in humans [4,5]. One of the most critical HDR genes is Radiation Sensitive 51 (RAD51), which is responsible for the homology searches and strand exchanges required during HDR [6].
The bulk of DSB repair in higher eukaryotes proceeds, however, more frequently by a process that does not require large regions of homology. Specifically, mammalian cells have evolved an efficient ability to join nonhomologous DNA molecules together [7] using the C-NHEJ pathway [8]. C-NHEJ is critically required for the proper development of the immune system and it is especially clinically relevant because it is the preferred pathway that cells utilize to repair ionizing radiation (IR)-induced DSBs [9,10]. Three important C-NHEJ genes are the protein kinase, DNA-activated, catalytic subunit (PRKDC or DNA-PKcs), and its DNA binding heterodimeric subunit, Ku70:Ku86 [11,12]. Relevantly, mutations in PRKDC have been associated with IRs (IR sensitivity), immune deficiency and/or cancer predisposition in humans [13–15].
Although much work has been carried out and great progress has been made over the last decade [16,17], a complete mechanistic understanding of how a cell decides to repair a DSB via HDR or C-NHEJ is still lacking. The mammalian DNA damage response (DDR) is regulated by PRKDC and the related kinases ataxia telangiectasia mutated (ATM), and ataxia telangiectasia and RAD3 related (ATR) [18,19]. Some of the relevant substrates for these kinases have been identified, but their pathways are also still not completely defined. In the past, this problem has been addressed by knocking out or knocking down the expression of ATM, ATR, and/or PRKDC in a favorite model system and then identifying those proteins phosphorylated in the control, but not the treated, cells following IR exposure [20]. Alternatively, mutations were made in one of these kinases and then mRNA was quantitated to identify genes that were either up or down regulated plus or minus IR exposure [21–23]. We have taken a unique approach to this problem. First, we generated human cell models for PRKDC including a knockout [24] and a kinase-dead knock-in [25]. We then used the kinase-dead and the parental cell line to perform “kinome” analyses [26] following IR exposure. In this approach, cellular extracts are incubated with beads onto which a dozen ATP-analog inhibitors have been immobilized. Since every kinase has an ATP-binding domain, these beads bind mostly kinases and other proteins containing ATP-binding domains (e.g., ATPases, chromatin remodelers, etc.). The beads are then centrifuged down, washed and subjected to quantitative proteomics. In this fashion, ~70 % of all human kinases and a multitude of additional ATP-binding proteins can be queried [26]. These analyses identified BRD3 (bromodomain containing protein 3) as a protein whose expression was almost completely downregulated in the PRKDC kinase-dead cell line. Importantly, although BRD3 is not a kinase, it is likely an ATP-dependent chromatin remodeler [27] and we believe that either its putative ATP-interaction domain [28] or its interaction with a bona fide ATP-binding protein, allowed it to be identified by this screen.
There are 46 bromodomain-containing proteins (BRDs) encoded in the human genome. A BRD subfamily, called the bromodomain (BD) and extra terminal (ET) domain (BET) family, consists of 4 members [BRD2, BRD3, BRD4, and BRDT (T = testis specific)] each of which contains two N-terminal BD domains and an “extra” C-terminal domain [29,30]. BDs are modules that facilitate a protein’s ability to bind to acetylated lysines [31]. Since acetylation is often associated with histone modification it is not surprising that the majority of BRDs (including the BET family members) are involved in chromatin regulation and transcription [27, 32]. The most intensely researched BET family member is BRD4, an important transcription factor [33] and a protein that has been implicated in the DDR [34,35] potentially by preventing the accumulation of R-loops [36]. Most recently, BRD4 has also been directly implicated in HDR-mediated DSB repair using an in vitro Xenopus extract system [37].
In stark contrast, there is little known about BRD3 and there are no reports of BRD3′s involvement in HDR. To date, the best function for BRD3 was inferred from an isolation of proteins on nascent DNA analysis where it was determined that BRD3 can bind to and regulate a protein called ATPase Family AAA domain containing 5 [ATAD5; [38]]. ATAD5, in turn, is the main component of a complex that unloads proliferating nuclear cell antigen (PCNA) from replicating DNA. Thus, BRD3, through its interaction with ATAD5, negatively regulates the amount of PCNA on a replication fork via ATAD5′s PCNA unloading activity [38–41]. Other than these few reports, however, there is little biochemical information available about BRD3.
Here we demonstrate that BRD3-null human cells are hyper-recombinogenic. Thus, human cells that have been genetically engineered to lack BRD3 expression show elevated levels of gene conversion, sister chromatid exchanges and gene targeting. Biochemically, this increase in HDR activity appears to be related to the altered kinetics of RAD51 recruitment at DSBs. Thus, we have identified BRD3 as a potent negative regulator of human HDR and this function may explain why BRD3 expression was specifically ablated in PRKDC/DNA-PKcs kinase-dead cells, which are incapable of performing C-NHEJ.
2. Materials and methods
2.1. Cell lines and cell culture
Human hTERT-RPE1 (ATCC® CRL-4000™) cells were cultured in Dulbecco’s modified Eagle’s medium F12 (DMEM-F12) supplemented with 10 % fetal bovine serum and 100 U/ml penicillin and 100 μg/ml streptomycin. HCT116 (ATCC® CCL-247™) cells and U-2 OS (ATCC® HTB-96™) cells were cultured in McCoy’s 5 A medium supplemented with 10 % fetal bovine serum and 100 U/ml penicillin and 100 μg/ml streptomycin.
BRD3-null cell lines were constructed using Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR):CRISPR-associated 9 (Cas9)-mediated gene editing technology [42–44]. Human hTERT-RPE1 cells were electroporated with a pSpCas9(BB)–2A-GFP (px458) plasmid containing a single-guide RNA (AGCAATTCGAGAACATCAGC) targeting exon 7 of BRD3. These cells were then flow sorted to select for green fluorescent protein (GFP)-positive cells, which were then subsequently subjected to single cell subcloning. Candidates were screened by PCR and confirmed by DNA sequencing to obtain clones containing indels resulting in frameshift mutations.
To complement BRD3−/− cell lines, wild type (WT) and mutant BRD3 cDNAs were cloned into a PiggyBac transposon vector that co-expresses a neomycin resistance selection cassette [45]. Candidate clones were selected by resistance to G418 and then the expression of WT and/or mutant BRD3 proteins were identified/quantitated by western blot analysis.
2.2. Antibodies
The following antibodies were used in this study: anti-BRD3 (A302–367A, A302–368A, Bethyl Laboratories), anti-53bp1 (Ab133534, Abcam), anti-RAD51 (PC-130, EMD Millipore Corp), anti-ATAD5 (Ab72111, Abcam), anti-actin (NB600–501, NOVUS Bio), anti-gamma H2A.X (phosphoS139; Ab81299, Abcam), anti-Flag (F31165–1MG, Sigma), anti-mouse IgG:HRP (170–6516, Biorad), anti-rabbit IgG:HRP (5213–2504, Biorad).
2.3. Multiplexed inhibitor bead (MIBs) affinity chromatography
MIBs chromatography was performed essentially as described [26]. Briefly, cells were lysed on ice for 20 min in buffer containing 50 mM HEPES (pH 7.5), 0.5 % Triton X-100, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10 mM sodium fluoride, 2.5 mM sodium orthovanadate, 1X protease inhibitor cocktail from Roche, and 1 % each of phosphatase inhibitor cocktails 2 and 3 from Sigma. The cell lysate was sonicated (3×10 s) on ice, centrifuged for 15 min (13,000 rpm) at 4 °C and then the supernatant was collected and filtered through a 0.2 μM membrane. The filtered lysate was brought to 1 M NaCl and pre-cleared by flowing over 500 μl of blocked and washed NHS-activated Sepharose 4 Fast Flow beads. The flow-through was collected and passed through a column of layered inhibitor-conjugated beads [Bisindoylmaleimide-X (50 μl), SB203580 (50 μl), Lapatinib (100 μl), Dasatinib (100 μl), Purvalanol B (100 μl), VI16832 (100 μl), PP58 (100 μl)] to isolate protein kinases from the lysates. Kinase-bound inhibitor beads were washed with 20 ml of high-salt buffer and 10 ml of low-salt buffer, each containing 50 mM HEPES (pH 7.5), 0.5 % Triton X-100, 1 mM EDTA, 1 mM EGTA, and 10 mM sodium fluoride, and 1 M NaCl or 150 mM NaCl, respectively. A final wash of 1 ml 0.1 % SDS was applied to the columns before elution in 1 ml of a 0.5% SDS solution in high heat. Elutions from all columns were combined and cysteines were alkylated by sequential incubations with DTT for 20 min at 60 °C and iodoacetamide for 30 min at room temperature in the dark. The elution was spin-concentrated to 100 μl and detergents were removed by a chloroform/methanol extraction. After a final mixing, the sample was centrifuged for 5 min to pellet the protein at the interface and the upper phase was removed with care to leave the protein pellet intact. The protein pellet and lower phase were resuspended in 300 μl of methanol, and the sample was again vortexed and centrifuged for 5 min to pellet the protein at the bottom of the tube. The supernatant was removed and one or more methanol washes were performed to ensure the removal of detergents.
2.4. Immunoblotting
For whole cell lysates, cells were collected and resuspended in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitors. The cells were incubated in RIPA for 30 min at 4 °C and then centrifuged at 4 °C for 15 min
For chromatin fraction lysates, cells were collected and resuspended in Buffer A (10 mM Hepes, pH = 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10 % glycerol, 0.1 % Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1 μg/ml leupeptin). The cells were incubated in Buffer A for 5 min at 4 °C and then centrifuged for 5 min at 4 °C. The pellets were then resuspended in TSE 500 buffer (20 mM Tris, pH = 8.0, 2 mM EDTA, 500 mM NaCl, 0.1 % SDS, 1 % Triton X-100, 1 mM PMSF, and 1 μg/ml leupeptin), followed by sonication and centrifugation at 4 °C. The resulting protein concentrations were determined using a Bradford protein assay and appropriate samples were then subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to nitrocellulose membranes. The membranes were blocked with 5 % milk in Tris-buffered saline:0.1 % Tween (TBST) for 1 hr at room temperature followed by overnight incubation with the primary antibody (diluted in 5 % milk in TBST). Proteins were detected by incubating membranes with horseradish peroxidase-conjugated secondary antibodies for 1 hr at room temperature and an enhanced chemiluminescence kit.
2.5. Immunofluorescence staining
Cells were grown in chamber slides to 70 % confluency. Cells were washed with phosphate-buffed saline (PBS) and then fixed with 4 % formaldehyde at room temperature for 10 min. After fixing, cells were treated with 0.5 % NP-40 at room temperature for 10 min. Cells were blocked with 1 % bovine serum albumin (BSA) in PBS for 30 min. Slides were incubated with primary antibodies overnight at 4 °C followed by application of fluorescent secondary antibodies. Slides were mounted with ProLong™ Gold and Diamond Antifade Mountant with DAPI. Images were obtained with a Nikon inverted TI-E deconvolution microscope and analyzed with Image J.
2.6. Flow cytometry
Cells were collected in PBS and fixed in 4 % formaldehyde. After fixing, the cells were treated with FLAER (Alexa 488 proaerolysin variant) in a liquid format for in vitro detection. Cells were then processed on a Becton Dickson Fortessa X-20 machine and profiles were analyzed with Flowjo.
2.7. Sister-chromatid exchange (SCE) assay
Cells were seeded in 6-well plates and treated with thymidine for 18 hr. Cells were then washed with PBS and grown in fresh media for 9 hr. After first-round release, the cells were treated with thymidine and 5-ethynyl-2′-deoxyuridine (EdU) for 15 hr. Then, cells were washed with PBS and grown in fresh media with EdU for 9 hr. After the second-round release, cells were washed with PBS and grown in fresh media for 33 hr. At this juncture, many cells were in metaphase.
The cells were then treated with 0.25 μg/ml colcemid for 1.5 hr. Cells were collected in PBS followed by 0.075 M KCl treatment at 37 °C. After KCl treatment, cells were slowly fixed by the addition of fixative (1:3 acetic acid to methanol) while vortexing. The cells were then dropped onto pre-warmed slides in a 37 °C water bath. Slides were then incubated in a 37 °C water bath for 5 min and air-dried overnight.
The slides were rehydrated in PBS for 5 min and fixed with fresh 3.7 % formaldehyde/PBS. After washing with PBS, slides were incubated with click reaction buffer (10 μM biotin-azide, 10 mM sodium ascorbate, and 2 mM CuSO4 in PBS) for 1 hr. After washing with PBS, slides were blocked in ABDIL buffer (20 mM Tris, pH 7.5, 2 % BSA, 0.2 % fish gelatin, 150 mM NaCl, 0.1 % sodium azide) for 1 hr at room temperature followed by anti-streptavidin Alexa Flour 488 conjugate incubation for 1 hr at room temperature. After washing with PBS, slides were mounted with ProLong™ Gold and Diamond Antifade Mountant with DAPI.
2.8. Gene targeting assay
The gene-targeting assay was performed essentially as described [46] by first knocking out the expression of the Phosphatidylinositol Glycan Anchor Biosynthesis Class A (PIGA) gene and then restoring its expression via gene targeting using a donor plasmid. To first inactivate PIGA expression in RPE1-hTERT WT, BRD3−/−, and BRD3−/−:+BRD3cDNA 1–8 cell lines, a TGATCGGGTATCAGTGGAAG sgRNA was used with CRISPR/Cas9 methodology. To reconstitute the expression of PIGA and minimize the effect of the NHEJ pathway, we used a sgRNA (TGGGTGAAAGTGCTCACACT) targeting a distal intronic region. To perform the gene targeting assay, the following nucleic acids were transfected into RPE1-hTERT cells (1 × 106) using a Neon Transfection System: 1 μg CleanCap® Cas9 mRNA; 1 μg donor plasmid; 1 μg mCherry expression plasmid (from Clontech); 50 pmol PIGA intron 5 sgRNA from Synthego Corporation. The cultures were grown for a subsequent ~14 days. All cells were then fixed with 4 % formaldehyde for 15 min. The fixed cells were then resuspended in phosphate buffered saline (PBS) at a low density and stained with 5 × 10−9 M fluorescent Alexa aerolysin (FLAER) 488 (Pinewood Scientific Services) for 15 min. Eventually, the percentage of FLAER-negative and FLAER-positive cells was quantitated by flow cytometry (FACSCanto II, BD Biosciences).
2.9. DNA repair assays
The pEGFP-Pem1-Ad2 plasmid based NHEJ assay was performed essentially as described [47]. Prior to transfection, the pEGFP-Pem1-Ad2 plasmid was digested by either the I-SceI or HindIII restriction enzymes (NEB) for 1 hr. After gel purification, 0.4 μg of digested plasmids and 0.1 μg of an mCherry transfection control expression plasmid (Clontech) were co-transfected into 24-well plates using Lipofectamine 3000 Reagent. Cells were collected 48 hr post transfection and analyzed by flow cytometry for GFP expression as described [47].
For the DR-GFP assay, 0.2 μg DR-GFP plasmid, 0.2 μg I-SceI expression plasmid (from Clontech), and 0.1 μg mCherry expression plasmid (from Clontech) were co-transfected into 24-well plates. Cells were collected 48 hr post transfection and analyzed by flow cytometry for GFP expression as described [47].
2.10. Imaging and analysis
Images were captured on a Nikon inverted TI-E deconvolution microscope and analyzed with Image J.
2.11. Clonogenic survival assay
Cells were seeded in 6-well plates (300–500 cells/well based upon the doubling time of a specific cell line) and incubated at 37 °C with 5 % CO2 overnight. The cells were then treated with different doses of IR or etoposide and incubated at 37 °C with 5 % CO2. After approximately one week of incubation, the media was removed from each well by aspiration. The cells were then rinsed with PBS, fixed with 10 % methanol:10 % acetic acid at room temperature for 15 min. The cells were then stained with 0.5 % crystal violet staining buffer at room temperature for 30 min. After rinsing thoroughly with PBS, colonies (defined as > 50 cells) were ready to be counted.
2.12. Immunofluorescence staining
Cells were grown in chamber slides to 70 % confluency, washed with PBS and then fixed with 4 % formaldehyde (FA) at room temperature for 10 min. After fixing, cells were treated with 0.5 % NP-40 at room temperature for 10 min and then blocked with 1 % bovine serum albumin (BSA) in PBS for 30 min. Slides were incubated with primary antibodies overnight at 4 °C followed by incubation with fluorescent secondary antibodies. Slides were mounted with ProLong™ Diamond Antifade Mountant with DAPI. Images were taken on a Nikon inverted TI-E deconvolution microscope and analyzed with Image J. At least 50 images were scored for each experimental condition.
2.13. siRNA transfections
SMARTPool ATAD5 siRNA were ordered from Horizon Discovery Ltd., (Catalog ID: L-004738–00–0005) and transfected into RPE1-hTERT cell line using the DharmaFECT™ Transfection Reagents - siRNA transfection protocol.
In brief, siRNA was resuspended into DNase/RNase-free water at a final concentration or 5 μM. To transfect siRNA into cells growing on 24-well plates, two tubes were prepared to dilute the siRNA and DharmaFECT 1 transfection reagent separately: tube 1- a 50 μl volume of the siRNA in serum-free medium was made by adding 2.5 μl of the siRNA to 47.5 μl of serum-free medium; tube 2- a 50 μl volume of diluted DharmaFECT 1 transfection reagent in serum-free medium was generated by adding 1.25 μl of DharmaFECT 1 transfection reagent to 48.75 μl of serum free-medium. The contents of tube 1 and tube 2 were then mixed by carefully pipetting them together and then incubating them for 20 min at room temperature. 400 μl of antibiotic-free complete medium was then added for a total volume of 500 μl transfection medium. This transfection medium was then added into each well of a 24-well plate.
2.14. Structure/function analyses
To complement BRD3−/− cell line, wild type BRD3 and structure mutant BRD3 cDNA were cloned into a PiggyBac transposase vector with neomycin selection cassette. The expression of WT and mutant BRD3 were identified by western blot.
2.15. Statistics and reproducibility
For the statistics of all experiments, p-values were calculated using the student test (t-distribution).
For reproducibility: 1) the DR-GFP assay was repeated up to five times except for the ATAD5-knock down samples, which were repeated only twice). 2) The SCE assay was repeated three times except for the samples using a mutant BRD3, which were repeated only twice. A double-blind method was used for quantification. 3) The gene targeting assay was repeated three times. 4) The focus formation assay was repeated twice.
2.16. Nucleic acid sequences
All nucleic acid sequences can be found in Supplemental Table S1.
3. Results
3.1. Identification of BRD3 as a gene differentially regulated in PRKDC-KD cells
PRKDC is essential for C-NHEJ [11, 20, 48]. In the course of investigating the roles of PRKDC in C-NHEJ, we utilized gene editing methodologies to generate two PRKDC deficient cell lines: [24] and [KD = kinase dead; hereafter PRKDC-KD cells; [25]] in the human colorectal carcinoma HCT116 cell background. In particular, the dominant negative activity of the KD version of PRKDC makes these cells exquisitely sensitive to IR and essentially ablates C-NHEJ in comparison to wild-type cells [25]. Despite this severe phenotype, PRKDC-KD cells are nonetheless viable [25]. We hypothesized that one explanation for this viability might be due to a compensatory upregulation of other DSB repair pathways, including HDR. On a simplistic level, this compensatory upregulation could be accomplished by up-regulating activators or inactivating inhibitors of other DSB pathways. Thus, we were interested in identifying proteins whose expression was either enhanced (potential activators) or decreased (potential inhibitors) in a PRKDC-KD cell line.
To identify such putative novel DSB repair factors, we performed a kinome-wide screen using multiplexed inhibitor beads (MIBs) and mass spectrometry (MS) [26]. MIBs are composed of a combination of sepharose beads with covalently immobilized kinase inhibitors of selected kinases or pan-kinases. Using this methodology, approximately ~70 % of all human kinases can be queried [26]. Thus, this technology reveals the impact of a mutation NOT on downstream substrates per se, but on the other kinase signaling pathways in the cell. The rationale for utilizing this approach was the hope for identifying regulatory kinases/factors (activators or inhibitors) whose activity might be altered in the mutant cell lines.
Accordingly, proteins were pulled-down by MIBs and then processed by MS using extracts prepared from HCT116 wild-type (WT) and PRKDC-KD cells. WT cells were used as a baseline and proteins whose expression level changed in the PRKDC-KD cells were identified (Fig. 1A). As a control and as expected, the level of PRKDC was only slightly reduced in the PRKDC-KD cells compared to the WT parental cell line (Fig. 1A). In total, 3,088 proteins were identified whose relative abundance was either unchanged, enhanced or reduced (Supplemental Table S2). Of these, 51 proteins in the PRKDC-KD cells exhibited relative abundance changes (reduced) greater than five-fold (Table S3). A Gene Ontology (GO) analysis (data not shown) was not particularly informative, but one protein, the BET family member, BRD3, whose relative abundance was reduced the most (Table S3) in the PRKDC-KD cell line (Fig. 1A), caught our attention. This protein, which is not a kinase, is a putative chromatin remodeling protein and moreover the BET family of proteins have been implicated in DNA DSB repair [37]. A Western blot analysis of the parental WT HCT116 and the PRKDC-KD cells confirmed that BRD3 abundance was reduced in the latter cell line (Fig. 1B).
Fig. 1.

Kinome analysis identifies BRD3 as a protein whose expression is greatly reduced in PRKDC-KD cells. (A) The number of peptides that could be unequivocally identified as belonging to a particular protein are shown along the X and Y axes for the parental HCT116 and the PRKDC-KD cell lines, respectively. The red dashed line corresponds to the theoretical equal expression in both cell lines for any given protein and the points above and below the line correspond to proteins whose expression was enhanced or decreased, respectively, in the PRKDC-KD cells. The data points corresponding to PRKDC and BRD3 are shown. (B) Western blot analysis confirms that BRD3 expression is greatly reduced in PRKDC-KD cells. Whole cell extracts were prepared from the indicated cell lines and subjected to a immunoblot analysis using either an antibody to BRD3 or, as a loading control β-actin. (C) Schematic of gene targeting approach for generation of BRD3 KO cell lines. Exon 7, which encodes one of the bromodomains of BRD3 was targeted using a CRISPR sgRNA. The sequence of the sgRNA is shown and the scissors indicate the predicted site of cleavage. The adjacent PAM site is shown in red. (D) Western blot analysis of RPE1 BRD3-KO cell lines. Whole cell extracts were prepared from the indicated cell lines (negative = PRKDC-KD and positive = HCT116 WT) and subjected to an immunoblot analysis using either an antibody to BRD3 or, as a loading control, β-actin.
3.2. Derivation of BRD3 knockout (KO) cell lines
To pursue BRD3 in more detail, we next generated human BRD3-KO cell lines. For these experiments, the parental cell line was the retinal pigment epithelial 1 cell line immortalized by the expression of human telomerase (hTERT-RPE1 or simply RPE1 cells). We chose this cell line, because it has a stable, diploid karyotype and although it is immortalized (allowing for easy propagation in tissue culture), it is not transformed (unlike the HCT116 cell line) and thus conforms to a more “normal” human cell model. The KO cell lines were generated using CRISPR/Cas9 via the strategy of allowing a CRISPR-induced DSB to be mutagenically repaired by the endogenous C-NHEJ pathway. A sgRNA was designed that targeted exon 7, which encodes one of the BDs of BRD3 (Fig. 1C). Single cell clones were isolated, expanded and subjected to DNA sequence analysis. Three independent BRD3 KO (BRD3−/−) clones were obtained containing out-of-frame indels of − 1 and − 2 (#22), + 1 and + 1 (#43) and − 2 and + 1 (#126). These cell lines were subsequently subjected to a Western blot analysis to confirm the absence of BRD3 protein expression (Fig. 1D). Since the KO clones showed no differences in growth properties (Fig. S1A) in comparison to the parental cell line we used them interchangeably in subsequent experiments.
3.3. BRD3-KO cell lines have aberrant DNA damage responses
We treated RPE1 WT and BRD3−/− cells with IR. BRD3−/− cells were more sensitive to IR (Fig. S1B) but the sensitivity was mild (D37 for WT = 1.2 Gy; D37 for BRD3-KO cells = 0.8 Gy). Consistent with this observation, the disappearance of γ-H2AX foci (Fig. S2A) following IR exposure (2 Gy) was slightly, but significantly, retarded in BRD3-KO cells (Fig. S2B). To assess if there was a frank defect in DSB repair, we utilized the pEGFP-Pem1-Ad2 extrachromosomal reporter assay that has been vetted extensively [47,49,50]. This reporter permits the analysis of the relative C-NHEJ repair frequency. The plasmid contains an enhanced GFP (EGFP) gene interrupted by a 2.4 kb intron derived from the rat phosphatidylethanolamine methyltransferase 1 (PEM1) gene that is itself interrupted by an adenoviral (Ad2) exon. The Ad2 exon, in turn, is flanked on both sides with HindIII and I-SceI restriction enzyme recognition sites (Fig. S3A). In its unmodified form, the presence of an adenoviral (Ad2) exon within the PEM1 intron prevents the expression of EGFP due to the Ad2 exon being incorporated into the GFP mRNA (Fig. S3C). Upon digestion with either a HindIII or I-SceI restriction enzyme, linearized plasmids lacking the Ad2 exon with either compatible cohesive ends (HindIII) or incompatible ends (I-SceI) are generated. While the digestion of the HindIII recognition sites results in cohesive 4-bp overlapping ends, the fact that the I-SceI sites have been arranged in an inverted orientation requires the processing of the DNA ends before they can be rejoined (Fig. S3B). Importantly, both undigested and partially digested plasmids will not contribute to the GFP positive readout for this assay as any inclusion of the Ad2 exon will create mRNAs with an interrupted EGFP coding region (Fig. S3C). In addition, the introns spanning the Ad2 exon provide a substrate that can undergo long-range resection events before losing the capability of expressing EGFP. Therefore, the impact of the loss of BRD3 expression on end joining could be assessed by FACS (fluorescent activated cell sorting) analysis. In order to determine the repair efficiency, cells are co-transfected with a pCherry expression vector that acts as a transfection control. The repair efficiency is reported as a percentage of cherry-positive cells that are also green (i.e., repaired). Using this assay, BRD3 KO cells appeared to have no quantifiable defect in C-NHEJ; neither with HindIII- nor with I-SceI-generated ends (Fig. S3D).
In summary, human BRD3 KO cells were mildly IR-sensitive and appeared to have difficultly completing repair in a timely fashion as indicated by the slower disappearance of γ-H2AX foci, but they did not appear to be defective in C-NHEJ.
3.4. BRD3-KO cell lines are hyper-recombinogenic
To interrogate the phenotype of BRD3 KO cells in more detail we next assessed their ability to perform HDR using a plasmid reporter. The DR-GFP reporter [51] (Fig. 2A) contains two inactive GFP genes along with a recognition site for the I-SceI meganuclease. Expression of I-SceI, can cleave one of the inactive GFP genes and if HDR occurs it can restore GFP expression resulting in easily-quantitated green fluorescing cells. This reporter measures non-crossover products generated by the HDR sub-pathway of gene conversion [2]. Two independent BRD3 KO clones showed ~2-fold and 2.5-fold, respectively, greater HDR activity relative to the parental cell line (Fig. 2B). A HCT116 parental cell line and a RAD52 KO cloned derived from it, the latter of which is known to be reduced for HDR activity [46], were used as controls and the RAD52−/− cell line showed an expected ~2-fold reduction in HDR activity. Thus, BRD3 KO cell lines appeared to be hyper-recombinogenic.
Fig. 2.

BRD3-null cells are hyper-recombinogenic for gene conversion. (A) A schematic of the DR-GFP extrachromosomal reporter is shown. The reporter contains two inactive copies of the GFP open reading frame (ORF). One of the copies, however, contains the recognition site for the I-SceI restriction enzyme. Following cleavage by I-SceI, a non-crossover (NCO) HDR event will restore one of GFP copies into a functional ORF, which can be quantitated by scoring for green fluorescence using FACS. (B). BRD3-KO cells are hyper-recombinogenic. The amount of green fluorescence was quantitated in the respective cell lines. The data shown represent 2 independent experiments, performed in duplicate.
To confirm the hyper-recombination phenotype of BRD3−/− cells using endogenous chromosomes, we next conducted a sister chromatid exchange (SCE) assay. In this assay, chromosomes are labeled for 1 cell cycle with the nucleoside analog, EdU. After a 2nd round of replication in the absence of EdU, only one strand of one sister chromatid will contain the EdU label (Fig. 3A). If exchanges between the sister chromatids subsequently occur before mitosis, these exchanges can be visualized with a fluorescent dye using click chemistry (Fig. 3A). Thus, SCEs were quantitated in both the parental RPE1 +hTERT cell line as well as in the BRD3 KO clone #126 (Fig. 3B). The BRD3-null cells had a highly significant ~2-fold greater incidence of SCEs than the parental cells (Fig. 3C).
Fig. 3.

BRD3-null cells have elevated levels of SCEs. (A) A schematic of the EdU labeling protocol used for measuring SCEs. (B) A representative metaphase image for the parental RPE1 +hTERT (left) or the BRD3-null #126 (right) cell lines are shown including an enlargement of metaphase chromosomes that have undergone SCEs. (C) Quantitation of the SCEs for the parental (WT) and the BRD3-null clone #126. The data represent three independent experiments and a double-blind method was used for quantification.
Finally, we assessed HDR activity by measuring the frequency of gene targeting. To accomplish this assessment, CRISPR was first utilized to disrupt the phosphatidylinositol glycan anchor biosynthesis class A (PIGA) gene in both WT and BRD3−/− cells by inserting a single nucleotide into exon 6 to disrupt its reading frame (Fig. 4A). PIGA can serve as a screening marker because it is required for the biosynthesis of glycophosphatidylinositol (GPI) anchors and the inactivation of PIGA leads to the loss of GPI anchors. The absence of GPI anchors, in turn, can be quantitated using a specific stain (an Alexa-488 conjugated proaerolysin variant called FLAER) that normally binds to the anchors [46,52]. To correct the PIGA mutation, we induced a second CRISPR-mediated DSB into the intronic region 47 bp away from the insertion site during gene-editing and provided a plasmid-based donor DNA (Fig. 4A) that should facilitate the conversion of PIGA-null (PIGAo/−) cells back to PIGA-proficient (PIGAo/+). Thus, the gene targeting efficiency can be calculated as the ratio of FLAER-positive (PIGAo/+) cells to FLAER-negative (PIGAo/−) [46,52]. In the parental cells, correction could be achieved about 1 % of the time. In two independent BRD3 KO cell lines, however, PIGA correction occurred at frequencies of ~3 % and 4 %, respectively (Fig. 4B).
Fig. 4.

BRD3-null cells are hyper-recombinogenic for gene targeting. (A) A schematic of the PIGA locus that was utilized for the gene editing experiments. In the first step, CRISPR/Cas9 was used to introduce an out-of-frame mutation into exon 6 (scissors). In the second step, a second CRISPR/Cas9-mediated DSB was introduced 47 nt downstream of the mutation and then (step 3) a donor plasmid containing WT BRD3 exon 6 sequences was used to restore PIGA expression. (B) The percentage of PIGA-null cells that were converted to PIGA-positive for WT, and two independent BRD3-null clones (#43 and #126) are shown. The data shown are derived from three independent experiments.
In summary, by 3 completely independent methodologies: 1) exogenous reporter assays, 2) SCEs and 3) gene targeting, BRD3-null cells were reproducibly hyper-recombinogenic.
3.5. Reconstitution of BRD3 expression rescues the hyper-recombination phenotype of BRD3−/− cells
Two endogenous BRD3 isoforms have been reported for human cells in the NCBI database (Fig. 5A). Isoform 1 conforms to the expected wild type BRD3 protein. Isoform 2 is identical to isoform 1 except that it is missing a single amino acid (alanine 647) in its C-terminus. Although isoform 2 is likely just a cloning artefact, we nonetheless isolated subclones of BRD3-null cells (clone #43) stably expressing either isoform 1 or isoform 2. Three subclones were selected for analysis. Subclones 1–2 and 2–6 (expressing isoforms 1 and 2, respectively) expressed supra levels of BRD3 whereas subclone 1–8 (expressing isoform 1) expressed approximately wild-type levels (Fig. 5B). These cell lines were then tested for their ability to carry out HDR gene conversion (Fig. 5C), the frequency at which they generated SCEs (Fig. 5D) and the frequency at which they underwent gene targeting at the PIGA locus (Fig. 5E). In all cases, regardless of whether isoform 1 or isoform 2 was expressed and regardless of the level of that expression, the restoration of BRD3 expression complemented the hyper-recombination phenotypes of the BRD3-null cells. From these experiments we concluded that one of the normal functions of endogenous BRD3 is to suppress hyper-recombination.
Fig. 5.

Expression of a wild type BRD3 cDNA rescues all of the hyper-recombination phenotypes of a BRD3-null cell. (A) Schematics of two BRD3 wild-type cDNAs listed in the National Center for Biotechnology Information (NCBI) database. The cDNAs are identical except that isoform 2 lacks a single amino acid (647 A) in the C-terminus. (B) BRD3-null subclones stably expressing either isoform 1 or isoform 2 were subjected to Western blot analysis to demonstrate the re-expression of BRD3. A higher non-specific band is seen in all the lanes probed with the anti-BRD3 antibody whereas the red arrow indicates the position of BRD3. Actin was used as a loading control. Three subclones (1–2, 1–6 and 1–8) expressing isoform 1 and two subclones (2–2 and 2–6) expressing isoform 2 are shown. (C) Complementation of the elevated HDR gene conversion activity in BRD3-null clone #43 by the re-expression of either isoform 1–2 or 2–6. (D) Complementation of the elevated SCE activity in BRD3-null clone #43 by the re-expression of either isoform 1–2 or 2–6. (E) Complementation of the elevated PIGA gene targeting activity in BRD3-null clone #43 by the re-expression of isoform 1–8. The data shown are derived from two or three independent experiments.
3.6. The depletion of BRD3 facilitates the formation of RAD51 filaments and impairs 53BP1 recruitment to DSB sites
To gain some mechanistic insight into the underlying role of BRD3 in hyper-recombination/DSB repair, we queried the status of RAD51, 53BP1 and γ-H2AX as indicators of HDR, C-NHEJ and DSBs, respectively. Immunofluorescence staining was performed with both RAD51 and 53BP1 antibodies on IR-treated (2 Gy) or untreated WT, BRD3−/−, and BRD3−/−:+BRD3cDNA #1–8 cells at different time points (Fig. 6A). There was no significant difference in the number of γ-H2AX foci introduced by IR exposure into the cell lines nor in their temporal resolution (Fig. 6B). These data suggested that the levels of initial DSB damage in the 3 cell lines was comparable and that in the short term (i.e., within 16 hr) there was no significant difference in the number of DSBs resolved. BRD3-null cells did show a reduction in the formation of 53BP1 foci 2 hr after IR exposure (Fig. 6C). However, since this phenotype was not rescued by the re-expression of BRD3 and there was no difference at any other time points the potential significance of this reduction at this single time point was ambiguous (Fig. 6C). In contrast, the kinetics of RAD51 focus formation were clearly altered (Fig. 6D). In BRD3-null cells, RAD51 foci accumulated at the earliest time point examined (2 hr) and then were rapidly resolved. The parental WT cells and the complemented cell line, on the other hand, showed an expected slower accumulation of RAD51 foci (peaking at 4 hr) as well as a slower resolution (Fig. 6D). This alteration in RAD51 focus formation was observed in an independent experiment that was carried out for 18 hr, with the exception that the distinction between the nulls and the parental/complemented cell lines was even more extreme, principally because the parental/complemented cell lines did not show a peak of RAD51 accumulation until 8 hr (Fig. S4). The slow [4 hr (Fig. 6D) to 8 hr (Fig. S4)] accumulation of RAD51 foci in the parental and complemented cell lines was consistent with the HDR repair kinetics observed in normal human cells [9] and contrasted sharply with the kinetics observed in BRD3-null cells (Fig. 6D and Fig. S4).
Fig. 6.

The kinetics of RAD51 foci formation are altered in BRD3-null cells. (A) Representative images for immunofluorescent analyses of the parental cell line (WT), BRD3-null cells (clone #43) and BRD3-null cells complemented clone #1–8 at the indicated times following exposure to 2 Gy of X-irradiation. The images are arranged in sets of 3 with a DAPI image, the corresponding antibody-stained image (RAD51, 53BP1 or γ-H2AX as indicated) and a merged image. Panels (B) γ-H2AX, (C) 53BP1 and (D) RAD51 show the quantitation of the number of fluorescent foci per nucleus for at least 50 cells for each indicated antibody, respectively. The data shown are derived from two independent experiments. n.s. = not significant; * = p < 0.05; ** = p < 0.01; *** = p < 0.0001.
3.7. The BD and ET domains are important for BRD3 activity
BRD3 is a presumed chromatin remodeler. One of the reasons for believing that BRD3 is a remodeler is that BRD3 contains two BD domains, which facilitate binding to acetylated lysines, a moiety often found on histones [31]. To investigate if these domains played a role in BRD3′s ability to suppress hyper-recombination, we generated stable cell lines containing cDNAs deleted for BD1, BD2 or the ET domain (Fig. 7A). To facilitate identification of the BRD3 variants a FLAG epitope moiety was appended onto their C-termini. These variants were then stably introduced in BRD3-null cells and individual clones expressing the deletion variants were isolated (Fig. 7B) although it should be noted that for the BD1Δ isoform our best clone contained only a low level of expression. Each of the cell lines was then assessed for the frequency of SCEs. The BRD3-null cells once again showed an increase in SCEs compared to the parental WT cell line (Fig. 7C). Importantly, while the expression of a WT BRD3 cDNA completely rescued this hyper-SCE formation phenotype, BD1Δ-, BD2Δ- and ETΔ-expressing clones were just as hyper-recombinogenic as the null cells (Fig. 7C). From these experiments we concluded that the 3 known functional domains of BRD3 were all required for BRD3′s ability to suppress hyper-recombination.
Fig. 7.

The BD and ET domains of BRD3 are required for significant complementation. (A) Representative cartoons of the 3 BRD3 cDNA constructs lacking either BD1, BD2 or the ET domain that were used in the complementation experiments. (B) Immunoblot analysis using an anti-FLAG antibody to demonstrate the expression of the indicated BRD3 derivative proteins in BRD3-null cells. β-actin was used as a loading control. (C) Inability of the BRD3 deletion constructs to rescue the SCE hyper-recombination phenotype of BRD3-null cells. SCEs were scored as described in the Material and Methods and in the legend to Fig. 2. As a positive control, a clone expressing a full-length, wild-type BRD3 cDNA (#1–8) was assessed, and this clone showed full complementation. The data are derived from two independent experiments. Each of the BD domain deletions reduces the wild-type protein size by ~10 kDa (95 AAs) whereas the ET deletion reduces the wild-type protein size by ~7 kDa (62 AAs). The actin band migrates around 40 kDa, whereas the wild-type BRD3 protein with a predicted MW of around 80 kDa and its deletion derivatives generally migrated higher in the gels around 110 kDa.
3.8. ATAD5 is epistatic to BRD3
The inability of BD1Δ and BD2Δ BRD3 cDNAs to rescue the hyper-recombination activity of BRD3-null cells was somewhat expected as BD1 and BD2 are the domains through which BRD3 interacts with chromatin. We were, however, surprised that the ETΔ expressing clone was as equally inactive. Little is known about the biochemical activity of ET domains, other than that they appear to be protein:protein interaction modules [29,30]. One protein that the BRD3 ET domain is known to interact with is ATAD5. ATAD5, in turn, has been implicated in the chromatin unloading of PCNA [38–41] and the loading of RAD51 [53], the latter protein being one whose recruitment kinetics are clearly altered in BRD3-null cells (Fig. 6D). Thus, we inquired whether BRD3′s anti-recombination activity required ATAD5 expression. To answer this inquiry, we utilized siRNA to knockdown the expression of ATAD5 (Fig. 8 A). Importantly, the reduction in ATAD5 expression completely suppressed the hyper-recombination activity of BRD3-null cells using the DR-GFP assay (Fig. 8B). As importantly, a reduction in ATAD5 expression also completely suppressed the aberrant RAD51 recruitment kinetics to DSBs in BRD3-null cells (Fig. 8C). From these experiments, we conclude that ATAD5 is epistatic to BRD3 and this, in turn, is consistent with the requirement for the presence of the ET domain (i.e., the ATAD5 interaction domain) in BRD3 (Fig. 7).
Fig. 8.

ATAD5 is epistatic to BRD3. (A) An immunoblot analysis demonstrates significant knockdown of ATAD5 expression in BRD3-null cells. An irrelevant band is marked by the asterisk (*) and ATAD5 expression by the arrow. Ku86 was used as a loading control. (B) The knockdown of ATAD5 expression suppresses the hyper-recombination of BRD3-null cells. Either wild type (WT) or BRD3-null (clone #43) cells are shown treated either with a control siRNA or one specific for ATAD5. The HDR activity was assessed by a DR-GFP assay as described in the Material and Methods and in the legend to Fig. 2. (C) The knockdown of ATAD5 expression suppresses the aberrant RAD51 recruitment kinetics in BRD3-null cells. Either wild type (WT) or BRD3-null (clone #43) cells exposed to 2 Gy of IR are shown treated either with a control siRNA or one specific for ATAD5. n.s. = not significant; * = p < 0.05; *** = p < 0.0001.
4. Discussion
4.1. A kinome analysis approach to identifying novel regulators of DNA DSB repair
Although the regulation of DSB repair is complicated, this complexity has not detracted a veritable bevy of researchers from trying to identify the genes involved in, and the mechanism(s) required for, this process [1,2]. Because much of the mammalian DDR is regulated by kinases and kinase cascades [16, 18, 19] a significant amount of effort (which has yielded a corresponding significant amount of success) has gone into identifying the immediate downstream substrates for the regulatory kinases [20]. Alternatively, in a more indirect manner, differences in gene expression (generally measured at the mRNA level) have been quantitated in cells that were either genetically modified and/or exposed to DNA damaging agents [21–23]. To augment these strategies, we have taken what we believe is a unique approach to identifying novel regulators of DSB repair. In a kinome analysis, the difference in ATP-binding protein expression between two cell lines is determined. While not all cellular signaling occurs via kinases and phosphorylation events, much of it does occur this way. This is especially true of DSB repair, which appears to be regulated predominately by 3 PI-like kinases: ATM, ATR and PRKDC. Consequently, we hypothesized that if we were to inactivate one of these kinases (specifically PRKDC), we might be able to observe compensatory or rescuing alterations in other kinase cascades or kinase substrates and in so doing identified BRD3. This result confirms that the kinome approach can be added to the toolbox of approaches currently used to dissect signaling pathways. In addition, a kinome analysis offers the additional advantage of identifying proteins that are neither kinases nor kinase substrates, but which simply bind ATP. With all that said, it is nonetheless unclear why BRD3 was identified in this screen since 1) BRD3 is not a kinase, 2) BRD3 has never been reported to be a substrate for a kinase, 3) nor does BRD3 contain canonical ATP-binding motifs, such as Walker A/B boxes [54]. It should be noted, however, that BRD3 does contain a cluster of residues (AA69 to 78; NlpDYhKliK) that have been reported to be (at least theoretically) ATP-interacting [28]. Alternatively, if BRD3 does not directly interact with ATP, then it is also possible that it may have been identified because it was interacting with another protein that was binding to the ATP analogs on the MIBs [e.g., NuRD, BAF, ATAD5 or INO80 complexes; [55]]. Regardless, and importantly, the identification of BRD3 — a gene that has never been directly implicated in regulating DSB repair — validates the utility of the kinome methodology.
Finally, we would like to note that the BRD3 downregulation observed in the DNA-PK-KD cell line used in this study [25] is not observed in the DNA-PK-null [24] cell line. Indeed, BRD3 expression is relatively unaltered in DNA-PKcs-null cells (data not shown). We, however, do not view this superficially disparate result as problematic, but actually quite interesting, because while these cell lines might ostensibly seem similar, mechanistically they are not. Thus, in the PRKDC kinase-dead cell line, a defective PRKDC protein is produced that binds onto the broken DNA ends, but is incapable of signaling any downstream steps of repair. Thus, this protein acts in a dominant-negative fashion. As a consequence, the ends are rendered irreparable and the phenotype is severe. In the PRKDC-null cell line, the PRKDC protein is not expressed. This absence of the PRKDC, reduces C-NHEJ activity, but, importantly, it leaves the unrepaired ends available for processing by other subpathways of DSB repair. Consequently, we expected that the effect of these PRKDC mutations on signaling pathways would be different in these two cell lines and they are. To this end, it is not unreasonable to expect to identify different novel regulators by repeating the kinome screen using the PRDKC-null cells, an experiment that is underway.
4.2. The loss of BRD3 results in cells that are IR-sensitive
Cells defective in any of the C-NHEJ pathway components are uniformly IR-sensitive. While the BRD3 cells are not profoundly so, they are nonetheless IR-sensitive (Fig. S1), which was surprising since we could not detect a defect in C-NHEJ activity (Fig. S3). Importantly, there was no difference in DSB induction following IR exposure between the parental and BRD3-null cells and the frank resolution of those DSBs also appeared relatively unaltered (Fig. S3B). Thus, we hypothesize that in a BRD3-null cell, the DSBs are kinetically resolved in a manner similar to wild type, but that a significant fraction of those repair events is faulty such that ultimately they result in the cell lethality that is observed in the clonogenic survival assays (Fig. S1). Importantly, this faulty repair likely extends to other types of DSBs besides IR-induced lesions, as the knockdown of BRD3 also results in hypersensitivity to ATR inhibition as well hydroxyurea exposure [38].
HDR is often referred to as “error-free” repair and superficially one could envision that a hyper-recombination state might be hypomutable. This bias is, however, likely inaccurate. Thus, even when the donor for a recombination reaction is the sister chromatid, aberrant products of recombination such as loss-of-heterozygosity can result. One of the most well-described hyper-recombination situations involves mutations to the Blooms Syndrome (BLM) gene. While BLM-null cells are not particularly IR-sensitive, they have significant genomic instability and the patients are quite cancer-prone [56]. Similarly, when the C-NHEJ factor Ku86 is depleted from human cells, HDR is increased throughout the genome [47] but with unfortunate consequences such that inappropriate HDR occurs at chromosome ends, where it results in lethal telomeric recombination events [57,58]. Thus, in the case of BLM and Ku86 deficiencies, too much HDR is unequivocally a pathological state. We believe that in the absence of BRD3, where a similar hyper-recombination atmosphere reigns, that many DSBs can be slowly (Fig. S2), albeit properly, repaired, but that some fraction of them are also mis-repaired in a manner more deleterious than the indels associated with C-NHEJ processes and that these mutations ultimately result in reduced viability (Fig. S1).
Lastly, it is relevant to point out that other BR-containing proteins have been implicated in DNA repair. For example, Zinc Finger MYND-Type Containing 8 (ZMYND8) has been reported to recognize DNA damage in transcriptionally active regions and to interact with Chromodomain Helicase DNA binding Protein 4 (CHD4) to facilitate HDR [59,60]. Similarly, another BR-containing protein, BRD9, assists in the formation of a RAD51:RAD54 complex to promote HDR [61]. Finally, BRD4 has also been directly implicated in HDR-mediated DSB repair using an in vitro Xenopus extract system [37]. In addition to involvement in HDR, two BR-containing proteins, CREB Binding Protein (CBP) and p300, assist NHEJ by facilitating the localization of the Switch/Sucrose Non-Fermentable) SWI/SNF complex to DSBs [62]. More importantly, a recent BR-containing protein screening has identified more than one third of all BR-containing proteins being recruited to DSB sites [59]. To these studies we now demonstrate that BRD3 appears to be a bona fide HDR inhibitor and its absence results in a hyper-recombination state. Illuminating the role of BR-containing proteins in DSBs repair is critical for understanding of the mechanism(s) of DSB repair and for fulfilling their potential clinical applications.
4.3. The loss of BRD3 results in cells that are hyper-recombinogenic by a mechanism that is ATAD5-dependent
Our data demonstrate that human somatic cells lacking BRD3 are hyper-recombinogenic for extrachromosomal HDR assays (Fig. 2), sister chromatid exchanges (Fig. 3) and chromosomal gene targeting (Fig. 4). An unequivocal conclusion from these observations is the finding that BRD3 is a bona fide inhibitor of HDR. The corollary of this conclusion is that inhibition of BRD3 could potentially be used to enhance HDR. The current commercially available inhibitors (e.g., JQ1) of BRD3 are pan-inhibitors of the whole BET family [63]. This is clearly sub-optimal as, for example, BRD4 is involved in the DDR, DSB repair, in the prevention of R-loops accumulation and its depletion results in accumulated endogenous DNA damage [34, 36, 37]. From that perspective, the development of a BRD3-specific inhibitor to enhance, for example, gene editing experiments seems eminently worthwhile.
Insight into the mechanism of how the loss of BRD3 expression in human cells causes hyper-recombination came from a structure:function analysis of the BRD3 protein. The absence of either BD (BD1 or BD2) resulted in the complete inability to rescue the hyper-recombination phenotypes (Fig. 7). This observation was not unexpected as BD domains are used for interaction with acetylated histones in chromatin [32] and this interaction/activity is per force likely necessary for a chromatin binding protein. In contrast, the inability of BRD3 lacking the ET domain (a protein:protein interaction module) to complement the hyper-recombination phenotype (Fig. 7) was unexpected. One of the few proteins described to interact with BRD3′s ET domain is ATAD5. ATAD5 is a AAA domain ATPase and it has been implicated in regulating the loading [e.g., RAD51; [53]] or unloading [e.g. PCNA; [38–41]] of proteins onto chromatin, although how it precisely regulates these activities is unknown. Importantly, however, subsequent experiments demonstrated that the hyper-recombination phenotype (Fig. 8B) and the aberrant chromatin binding kinetics of RAD51 (Fig. 8C) of BRD3-null cells were completely dependent upon the expression of ATAD5. Thus, our current model is that in BRD3-proficient cells BRD3 is recruited to DSBs via nearby acetylated nucleosomes via its BD modules (Fig. 9A). BRD3, in turn, uses its ET domain to interact with ATAD5 and we postulate that this interaction hinders ATAD5′s ability to load RAD51 onto DSBs (Fig. 9A). This scenario permits predominately C-NHEJ-mediated repair of the DSB (Fig. 9A). In contrast, in BRD3-deficient cells, ATAD5 is now unrestricted and can (hyper)load RAD51 on the DSB and this extra RAD51 facilitates proportionately more HDR, which, as discussed above, is generally associated with ancillary genomic instability (Fig. 9B). In the case where BRD3 and ATAD5 are both deficient, the lack of ATAD5 to bolster RAD51 loading reverses the hyper-recombination phenotype and results in a scenario where C-NHEJ once again predominates (Figs. 8 and 9C). Lastly, this model predicts that BRD3-proficient, ATAD5-null cells should be HDR defective (due to the absence of ATAD5-mediated RAD51 loading; Fig. 9D). We are currently trying to test this prediction directly, but it should be noted that it was recently reported that ATAD5-reduced human A2780 cells were hypersensitive to poly(ADP)ribose polymerase inhibitors, a phenotype often associated with a HDR deficiency [64].
Fig. 9.
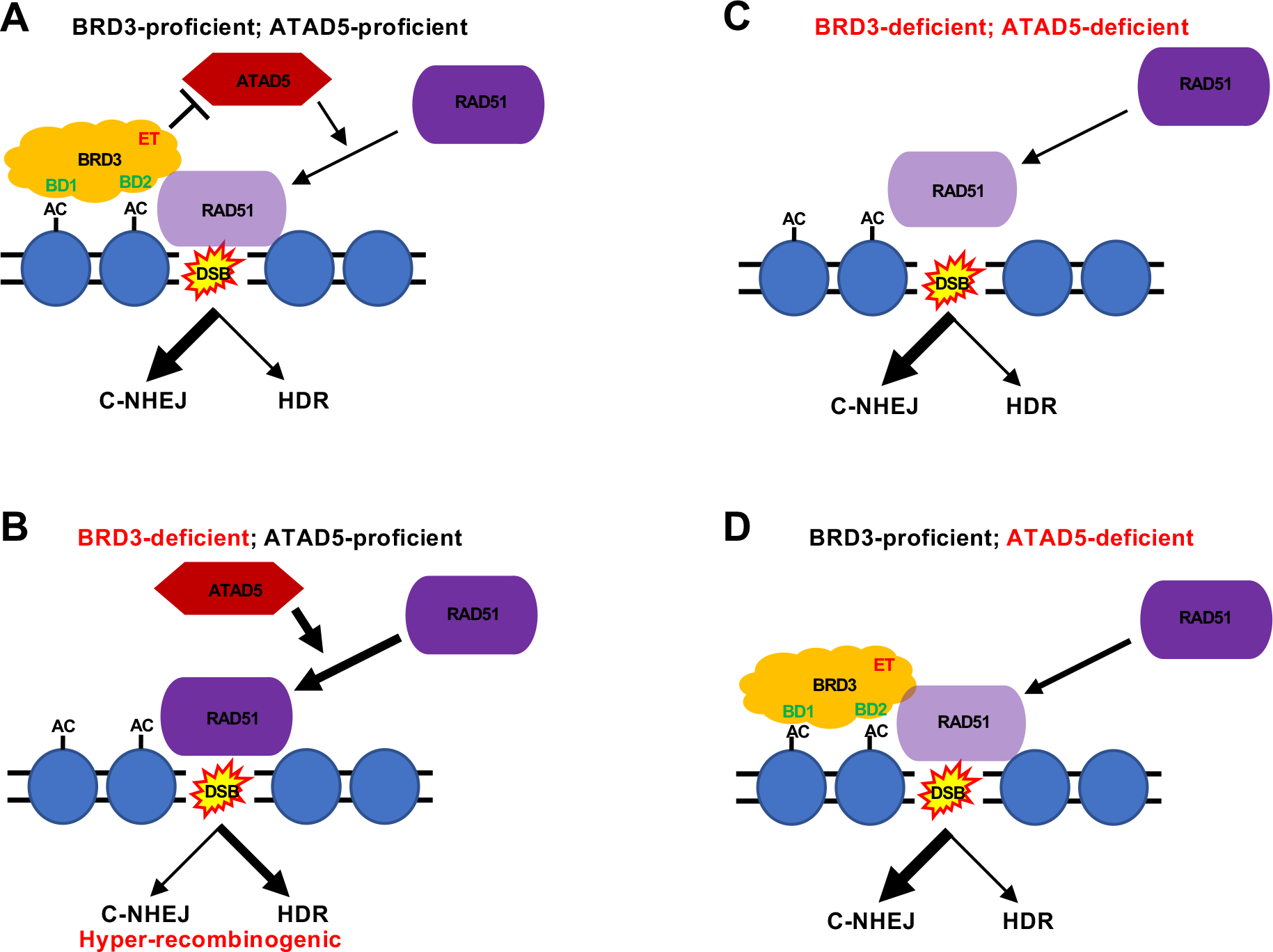
A schematic for the action of BRD3. (A) In BRD3-proficient, ATAD5-proficient cells BRD3 can be localized to chromatin near a DSB via its BD domains (BD1 and BD2) to acetylated (AC) histones in nucleosomes. BRD3, in turn, via its ET domain can interact with ATAD5. This interaction impedes ATAD5′s activity (which is to load RAD51 on the DSB) thus encouraging C-NHEJ and limiting HDR. (B) In BRD3-deficient, ATAD5-proficient cells BRD3 cannot be localized to chromatin near a DSB and hence ATAD5 is unrestricted in its RAD51 loading activity. As a consequence, RAD51 encourages enhanced HDR at the expense of C-NHEJ and results in a hyper-recombinogenic phenotype. (C) In BRD3-deficient, ATAD5-deficient cells the absence of ATAD5 diminishes RAD51′s ability to be loaded at a DSB and this reduction in RAD51 suppresses the hyper-recombination phenotype of a BRD3-deficiency. (D) In BRD3-proficient, ATAD5-deficient cells the absence of ATAD5 again diminishes RAD51′s ability to be loaded at a DSB and this reduction in RAD51 should result in a HDR-deficient phenotype.
Supplementary Material
Funding
Funding for the Hendrickson laboratory was provided in part through grants from the National Cancer Institute (CA154461 and CA266524). This agency had no involvement in the study design, the data collection, the analysis nor the interpretations presented here.
Footnotes
Declaration of Competing Interest
E.A.H. declares that he is a member of the scientific advisory board of Intellia Therapeutics, a company that specializes in applying gene editing technology to basic research and therapeutics.
CRediT authorship contribution statement
D.W.C. was solely responsible for carrying out all of the kinome analyses. C. W. was solely responsible for all other experimental data presented in this manuscript. C. W. wrote the first version of the manuscript. E. A. H. contributed to the experimental design, the data interpretation, the funding and the writing of the manuscript.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.dnarep.2022.103445.
Data Availability
Data will be made available on request.
References
- [1].Verma P, Greenberg RA, Noncanonical views of homology-directed DNA repair, Genes Dev. 30 (2016) 1138–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chen CC, Feng W, Lim PX, Kass EM, Jasin M, Homology-directed repair and the role of BRCA1, BRCA2, and related proteins in genome integrity and cancer, Annu Rev. Cancer Biol. 2 (2018) 313–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rickman K, Smogorzewska A, Advances in understanding DNA processing and protection at stalled replication forks, J. Cell Biol. 218 (2019) 1096–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jackson SP, Bartek J, The DNA-damage response in human biology and disease, Nature 461 (2009) 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Goodarzi AA, Jeggo PA, The repair and signaling responses to DNA double-strand breaks, Adv. Genet 82 (2013) 1–45. [DOI] [PubMed] [Google Scholar]
- [6].Bonilla B, Hengel SR, Grundy MK, Bernstein KA, RAD51 gene family structure and function, Annu Rev. Genet 54 (2020) 25–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Betermier M, Bertrand P, Lopez BS, Is non-homologous end-joining really an inherently error-prone process? PLoS Genet 10 (2014), e1004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Frock RL, Sadeghi C, Meng J, Wang JL, DNA end joining: G0-ing to the core, Biomolecules 11 (2021) 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang H, Zeng ZC, Bui TA, Sonoda E, Takata M, Takeda S, Iliakis G, Efficient rejoining of radiation-induced DNA double-strand breaks in vertebrate cells deficient in genes of the RAD52 epistasis group, Oncogene 20 (2001) 2212–2224. [DOI] [PubMed] [Google Scholar]
- [10].Nickoloff JA, Sharma N, Allen CP, Taylor L, Allen SJ, Jaiswal AS, Hromas R, Roles of homologous recombination in response to ionizing radiation-induced DNA damage, Int J. Radiat. Biol. (2021) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen S, Lees-Miller JP, He Y, Lees-Miller SP, Structural insights into the role of DNA-PK as a master regulator in NHEJ, Genome Instab. Dis. 2 (2021) 195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Matsumoto Y, Asa A, Modak C, Shimada M, DNA-dependent protein kinase catalytic subunit: the sensor for DNA double-strand breaks structurally and functionally related to ataxia telangiectasia mutated, Genes (Basel) 12 (2021) 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Woodbine L, Neal JA, Sasi NK, Shimada M, Deem K, Coleman H, Dobyns WB, Ogi T, Meek K, Davies EG, et al. , PRKDC mutations in a SCID patient with profound neurological abnormalities, J. Clin. Invest 123 (2013) 2969–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Esenboga S, Akal C, Karaatmaca B, Erman B, Dogan S, Orhan D, Boztug K, Ayvaz D, Tezcan I, Two siblings with PRKDC defect who presented with cutaneous granulomas and review of the literature, Clin. Immunol. 197 (2018) 1–5. [DOI] [PubMed] [Google Scholar]
- [15].Chen Y, Li Y, Xiong J, Lan B, Wang X, Liu J, Lin J, Fei Z, Zheng X, Chen C, Role of PRKDC in cancer initiation, progression, and treatment, Cancer Cell Int 21 (2021) 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ackerson SM, Romney C, Schuck PL, Stewart JA, To join or not to join: decision points along the pathway to double-strand break repair vs. chromosome end protection, Front Cell Dev. Biol. 9 (2021), 708763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Becker JR, Clifford G, Bonnet C, Groth A, Wilson MD, Chapman JR, BARD1 reads H2A lysine 15 ubiquitination to direct homologous recombination, Nature 596 (2021) 433–437. [DOI] [PubMed] [Google Scholar]
- [18].Blackford AN, Jackson SP, ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response, Mol. Cell 66 (2017) 801–817. [DOI] [PubMed] [Google Scholar]
- [19].Neal JA, Dunger K, Geith K, Meek K, Deciphering the role of distinct DNA-PK phosphorylations at collapsed replication forks, DNA Repair (Amst. ) 94 (2020), 102925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dylgjeri E, Knudsen KE, DNA-PKcs: a targetable protumorigenic protein kinase, Cancer Res. 82 (2022) 523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. , 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers, Nat. Struct. Mol. Biol. 17 (2010) 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Adamson B, Smogorzewska A, Sigoillot FD, King RW, Elledge SJ, A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response, Nat. Cell Biol. 14 (2012) 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Balmus G, Pilger D, Coates J, Demir M, Sczaniecka-Clift M, Barros AC, Woods M, Fu B, Yang F, Chen E, et al. , ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks, Nat. Commun. 10 (2019) 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ruis BL, Fattah KR, Hendrickson EA, The catalytic subunit of DNA-dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells, Mol. Cell Biol. 28 (2008) 6182–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lu H, Saha J, Beckmann PJ, Hendrickson EA, Davis AJ, DNA-PKcs promotes chromatin decondensation to facilitate initiation of the DNA damage response, Nucleic Acids Res. 47 (2019) 9467–9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al. , Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer, Cell 149 (2012) 307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hsu SC, Blobel GA, The role of bromodomain and extraterminal motif (BET) proteins in chromatin structure, Cold Spring Harb. Symp. Quant. Biol. 82 (2017) 37–43. [DOI] [PubMed] [Google Scholar]
- [28].Chauhan JS, Mishra NK, Raghava GP, Identification of ATP binding residues of a protein from its primary sequence, BMC Bioinforma. 10 (2009) 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Taniguchi Y, The bromodomain and extra-terminal domain (BET) family: functional anatomy of BET paralogous proteins, Int J. Mol. Sci. (2016) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Stathis A, Bertoni F, BET proteins as targets for anticancer treatment, Cancer Discov. 8 (2018) 24–36. [DOI] [PubMed] [Google Scholar]
- [31].Gokani S, Bhatt LK, Bromodomains: a novel target for the anticancer therapy, Eur. J. Pharm. 911 (2021), 174523. [DOI] [PubMed] [Google Scholar]
- [32].Chiu LY, Gong F, Miller KM, Bromodomain proteins: repairing DNA damage within chromatin, Philos. Trans. R. Soc. Lond. B Biol. Sci. 372 (2017) 20160286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hajmirza A, Emadali A, Gauthier A, Casasnovas O, Gressin R, Callanan MB, BET family protein BRD4: an emerging actor in NFkappaB signaling in inflammation and cancer, Biomedicines 6 (2018) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Floyd SR, Pacold ME, Huang Q, Clarke SM, Lam FC, Cannell IG, Bryson BD, Rameseder J, Lee MJ, Blake EJ, et al. , The bromodomain protein Brd4 insulates chromatin from DNA damage signalling, Nature 498 (2013) 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kang MS, Kim J, Ryu E, Ha NY, Hwang S, Kim BG, Ra JS, Kim YJ, Hwang JM, Myung K, et al. , PCNA unloading is negatively regulated by BET proteins, Cell Rep. 29 (4632–4645) (2019), e4635. [DOI] [PubMed] [Google Scholar]
- [36].Lam FC, Kong YW, Huang Q, Vu Han TL, Maffa AD, Kasper EM, Yaffe MB, BRD4 prevents the accumulation of R-loops and protects against transcription-replication collision events and DNA damage, Nat. Commun. 11 (2020) 4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Barrows JK, Lin B, Quaas CE, Fullbright G, Wallace EN, Long DT, BRD4 promotes resection and homology-directed repair of DNA double-strand breaks, Nat. Commun. 13 (2022) 3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wessel SR, Mohni KN, Luzwick JW, Dungrawala H, Cortez D, Functional analysis of the replication fork proteome Identifies BET proteins as PCNA regulators, Cell Rep. 28 (3497–3509) (2019), e3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sikdar N, Banerjee S, Lee KY, Wincovitch S, Pak E, Nakanishi K, Jasin M, Dutra A, Myung K, DNA damage responses by human ELG1 in S phase are important to maintain genomic integrity, Cell Cycle 8 (2009) 3199–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lee KY, Fu H, Aladjem MI, Myung K, ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin, J. Cell Biol. 200 (2013) 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kang MS, Ryu E, Lee SW, Park J, Ha NY, Ra JS, Kim YJ, Kim J, Abdel-Rahman M, Park SH, et al. , Regulation of PCNA cycling on replicating DNA by RFC and RFC-like complexes, Nat. Commun. 10 (2019) 2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. , Multiplex genome engineering using CRISPR/Cas systems, Science 339 (2013) 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J, RNA-programmed genome editing in human cells, Elife 2 (2013), e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM, RNA-guided human genome engineering via Cas9, Science 339 (2013) 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Stroik S, Kurtz K, Hendrickson EA, CtIP is essential for telomere replication, Nucleic Acids Res. 47 (2019) 8927–8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kan Y, Batada NN, Hendrickson EA, Human somatic cells deficient for RAD52 are impaired for viral integration and compromised for most aspects of homology-directed repair, DNA Repair (Amst.) 55 (2017) 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Fattah F, Lee EH, Weisensel N, Wang Y, Lichter N, Hendrickson EA, Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells, PLoS Genet. 6 (2010), e1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chaplin AK, Hardwick SW, Stavridi AK, Buehl CJ, Goff NJ, Ropars V, Liang S, De Oliveira TM, Chirgadze DY, Meek K, et al. , Cryo-EM of NHEJ supercomplexes provides insights into DNA repair, Mol. Cell 81 (3400–3409) (2021), e3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Seluanov A, Mittelman D, Pereira-Smith OM, Wilson JH, Gorbunova V, DNA end joining becomes less efficient and more error-prone during cellular senescence, Proc. Natl. Acad. Sci. USA 101 (2004) 7624–7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ruis B, Molan A, Takasugi T, Hendrickson EA, Absence of XRCC4 and its paralogs in human cells reveal differences in outcomes for DNA repair and V(D)J recombination, DNA Repair (Amst.) 85 (2020), 102738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pierce AJ, Johnson RD, Thompson LH, Jasin M, XRCC3 promotes homology-directed repair of DNA damage in mammalian cells, Genes Dev. 13 (1999) 2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Karnan S, Konishi Y, Ota A, Takahashi M, Damdindorj L, Hosokawa Y, Konishi H, Simple monitoring of gene targeting efficiency in human somatic cell lines using the PIGA gene, PLoS One 7 (2012), e47389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Park SH, Kang N, Song E, Wie M, Lee EA, Hwang S, Lee D, Ra JS, Park IB, Park J, et al. , ATAD5 promotes replication restart by regulating RAD51 and PCNA in response to replication stress, Nat. Commun. 10 (2019) 5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Walker JE, Saraste M, Runswick MJ, Gay NJ, Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold, EMBO J. 1 (1982) 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wai DCC, Szyszka TN, Campbell AE, Kwong C, Wilkinson-White LE, Silva APG, Low JKK, Kwan AH, Gamsjaeger R, Chalmers JD, et al. , The BRD3 ET domain recognizes a short peptide motif through a mechanism that is conserved across chromatin remodelers and transcriptional regulators, J. Biol. Chem. 293 (2018) 7160–7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kaur E, Agrawal R, Sengupta S, Functions of BLM helicase in cells: is it acting like a double-edged sword? Front Genet 12 (2021), 634789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Indiviglio SM, Bertuch AA, Ku’s essential role in keeping telomeres intact, Proc. Natl. Acad. Sci. USA 106 (2009) 12217–12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wang Y, Ghosh G, Hendrickson EA, Ku86 represses lethal telomere deletion events in human somatic cells, Proc. Natl. Acad. Sci. USA 106 (2009) 12430–12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gong F, Chiu LY, Cox B, Aymard F, Clouaire T, Leung JW, Cammarata M, Perez M, Agarwal P, Brodbelt JS, et al. , Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination, Genes Dev. 29 (2015) 197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gong F, Clouaire T, Aguirrebengoa M, Legube G, Miller KM, Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair, J. Cell Biol. 216 (2017) 1959–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhou Q, Huang J, Zhang C, Zhao F, Kim W, Tu X, Zhang Y, Nowsheen S, Zhu Q, Deng M, et al. , The bromodomain containing protein BRD-9 orchestrates RAD51-RAD54 complex formation and regulates homologous recombination-mediated repair, Nat. Commun. 11 (2020) 2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ogiwara H, Ui A, Otsuka A, Satoh H, Yokomi I, Nakajima S, Yasui A, Yokota J, Kohno T, Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors, Oncogene 30 (2011) 2135–2146. [DOI] [PubMed] [Google Scholar]
- [63].Schwalm MP, Knapp S, BET bromodomain inhibitors, Curr. Opin. Chem. Biol. 68 (2022), 102148. [DOI] [PubMed] [Google Scholar]
- [64].Giovannini S, Weller MC, Hanzlikova H, Shiota T, Takeda S, Jiricny J, ATAD5 deficiency alters DNA damage metabolism and sensitizes cells to PARP inhibition, Nucleic Acids Res. 48 (2020) 4928–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.