

ABSTRACT
Eukaryotic cells rely upon dynamic, multifaceted regulation at each step of RNA biogenesis to maintain mRNA pools and ensure normal protein synthesis. Studies in budding yeast indicate a buffering phenomenon that preserves global mRNA levels through the reciprocal balancing of RNA synthesis rates and mRNA decay. In short, changes in transcription impact the efficiency of mRNA degradation and defects in either nuclear or cytoplasmic mRNA degradation are somehow sensed and relayed to control a compensatory change in mRNA transcription rates. Here, we review current views on molecular mechanisms that might explain this apparent bidirectional sensing process that ensures homeostasis of the stable mRNA pool.
Keywords: Transcription, mRNA buffering, RNA exosome, Xrn1
Introduction
The central dogma of molecular biology implies a linear flow of information; DNA is converted to RNA, and RNA is translated to protein. This linear view is reinforced by the compartmentalization of eukaryotic transcription in the nucleus and the majority of mRNA degradation and its translation in the cytoplasm. However, studies in budding yeast have provided strong evidence for mutual feedback regulation between nuclear transcript synthesis and mRNA degradation, leading to a more circular view of gene expression control (Figure 1). This model implies that there may be sensor molecules that detect both nascent transcript levels and mRNA abundance in the cytoplasm. Such sensors would provide a means of communication between these processes and their cellular compartments, stabilizing the stable mRNA pool, and ensuring that protein production stays relatively constant. Here, we review evidence for molecular mechanisms that may control this mRNA buffering process, and we discuss how they might be impacted by cellular stress.
Figure 1.
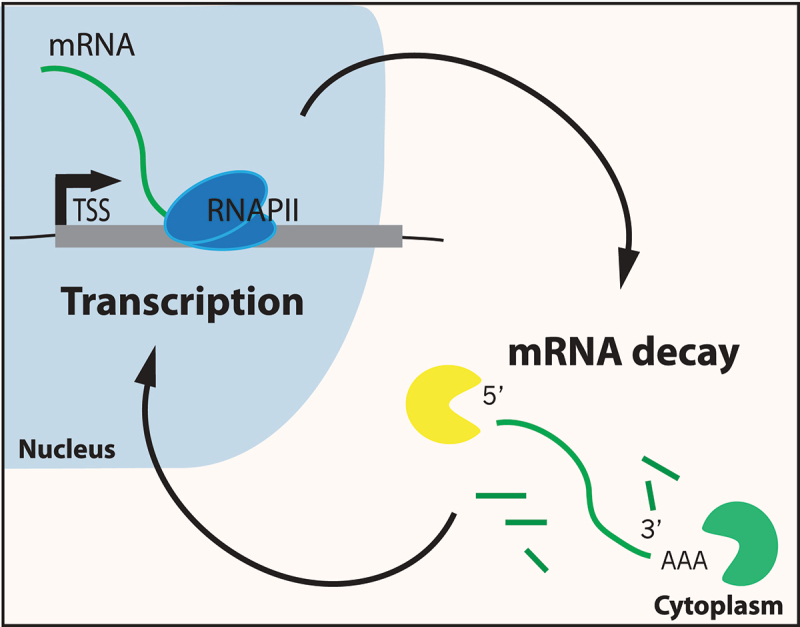
A circular model for gene expression. Arrows indicate communication between the transcription and mRNA decay machineries that buffer the stable mRNA pool.
Information transfer: signaling nascent transcript synthesis to mRNA degradation
The steady state level of an mRNA reflects a balance between the rate of transcript synthesis and the rate of mRNA degradation. Whereas a simple view might posit that these processes occur independently, there are a host of studies over the past 10 or more years demonstrating a remarkable degree of coupling between mRNA synthesis and its decay within both the nuclear and cytoplasmic compartments. One of the first examples in mammalian cells demonstrated that non-transcribed, upstream gene regulatory sequences can impact the post-transcriptional decay of an mRNA. This study investigated the synthesis and decay of a β-globin mRNA that contained a premature stop codon [1]. When linked to the normal β-globin promoter sequences, the stop codon de-stabilized the mRNA; the post-transcriptional process that is now known as the nonsense decay pathway [2]. However, simply swapping the non-transcribed, β-globin promoter sequences with the promoter from the thymidine kinase gene eliminated the impact of the stop codon on mRNA decay. A similar study in budding yeast demonstrated that the cytoplasmic degradation of several mRNAs was dependent on specific promoter elements, as swapping promoters imparted the mRNA stability associated with the normal gene target of the promoter, rather than the actual sequence of the transcription unit [3,4]. Finally, the decay rates of galactose-inducible mRNAs in budding yeast are regulated by carbon source, but this regulation requires non-transcribed, GAL gene promoter elements [5]. Notably, in each of these cases, changes in the mRNA decay rate were not linked to increased or decreased synthesis rate but rather seemed coupled to a distinct set of promoter binding factors.
How might a cis-acting promoter element impact the post-transcriptional fate of an mRNA? A simple view is that promoter-bound factors influence the recruitment of either mRNA decay factors or decay regulators to the nascent RNA during synthesis, imparting a gene-specific and promoter-specific post-transcriptional fate. One candidate factor is the Rpb4/7 subcomplex of RNA polymerase II (RNAPII). The Rpb4/7 module can dissociate from the core RNAPII, bind nascent transcripts, and regulate mRNA export and subsequent mRNA decay [6,7]. It has been proposed that the binding of Rpb4/7 to mRNAs may provide an “imprinting” mark that controls cytoplasmic decay [8,9]. In this model, regulatory factors that bind to upstream promoter elements may influence the amount of Rpb4/7 that is associated with RNAPII and that is available for association with the nascent transcript. Interestingly, Choder and colleagues identified a set of temperature-sensitive alleles of yeast RPB7 that impact RNA decay rates but do not decrease transcription, further reinforcing the view that this RNAPII subunit functions directly in both pathways [10]. Unfortunately, this interesting model has been controversial. Cramer and colleagues created a yeast strain where Rpb4 was fused to the core RNAPII subunit, Rpb2, blocking the dissociation of Rpb4 from RNAPII [11]. Surprisingly, this fusion protein appears to perform most functions of Rpb4, suggesting that its cytoplasmic roles are less important. Notably, these results do not rule out a key imprinting role for Rpb7, independent of Rpb4. However, work from the Choder group found that this Rpb4-Rpb2 fusion protein is prone to a proteolysis event that generates significant amounts of free, functional Rpb4. Furthermore, the Rpb4-Rpb2 fusion protein appears to bind to mRNAs in the cytoplasm [12]. Consequently, the Rpb4/7 imprinting model appears to be viable once again.
In a distinct model, Choder and colleagues have suggested that major components of the cytoplasmic mRNA decay machinery are themselves recruited to gene proximal promoter elements [13]. Specifically, they reported that the Xrn1 exonuclease, which is responsible for the majority of cytoplasmic mRNA decay, can be detected at many 5’gene regulatory regions by chromatin immunoprecipitation (ChIP) analyses. Furthermore, Xrn1 appears to co-localize by ChIP with a host of other mRNA decay factors. Choder and colleagues found that the decay factors shuttle between the cytoplasm and the nucleus, and they proposed the idea of a decaysome that links transcript synthesis and decay in a circular pathway [13]. More recently, the Choder group have identified the nuclear localization signals within Xrn1, as well as the Kap120 importin that directs its nuclear entry [14]. Surprisingly, these authors find that nuclear import of Xrn1 is not only key to maintain proper transcription levels but also for mRNA decay in the cytoplasm. These data suggest a compelling model in which Xrn1 transits with the decaying mRNA from the nucleus to the cytoplasm [14]. To date, it is not clear if differential recruitment of this decaysome is responsible for previous observations regarding promoter-specific impacts on mRNA decay.
In addition to gene-specific impacts on mRNA decay rates, global changes in transcript synthesis rates also appear to be sensed by the mRNA decay machinery. For instance, some of the earliest indicators of a connection between transcription rates and mRNA stability arose from discrepancies between measurements of steady state mRNA levels and nascent transcript levels. For instance, Helenius and colleagues found that deletion of the gene encoding the Mat1 subunit of the mammalian TFIIH general transcription factor had little impact on the transcriptome of mouse embryonic stem cells, when assayed by gene tiling arrays [15]. This was quite surprising, as TFIIH activity is expected to be essential for all mRNA transcription, as this factor promotes both formation of the transcription bubble via its helicase activity and phosphorylation of RNAPII that governs promoter escape. Indeed, mat1−/− cells showed a global defect in RNAPII phosphorylation, which should have been indicative of a global transcription defect. These surprising results were reconciled by analysis of nascent transcript synthesis by pulse labeling with nucleotide analogs, uncovering the expected, global defect in transcription. The authors proposed that the global defect in transcript synthesis must have been masked by a stabilization of mRNAs [15].
Since this early case, there have been numerous examples where a global defect in transcription is masked by changes in mRNA degradation rates (for an excellent review, see [16]). In the majority of instances, investigators have employed a powerful approach called comparative dynamic transcriptome analysis (cDTA), pioneered by the Cramer group [17]. In this analysis, budding yeast cells are pulse-labeled with a thymidine analog, 4-thiouridine (4tsU), and the total and labeled RNAs are analyzed by DNA microarrays. Each sample also contains a ‘spike-in’ of 4tsU-labeled RNA from fission yeast, S. pombe, which allows direct comparison between budding yeast samples. The rates of both transcript synthesis and mRNA decay are extracted from the microarray data using a kinetic model. cDTA is now widely used by many groups to investigate the rates of transcript synthesis and mRNA degradation. In one of the first tests of the cDTA method, the Cramer group found that an elongation defective version of RNAPII (rpb1-N488D) caused a global decrease in transcript synthesis rates, as expected, but mRNA decay rates were also decreased [17]. Consequently, mRNA levels remained virtually unchanged. Since this first demonstration, cDTA has been employed by many groups to demonstrate similar, compensatory changes in mRNA decay rates in response to global changes in transcription. For example, inactivation of components of the general transcription machinery, such as TFIID, TFIIH, or the Mediator complex, all lead to global decreases in transcription rates, but compensatory increases in mRNA decay rates buffer to maintain the mRNA pool [16].
Recently, we reported that a global transcriptional impact of a histone modification, histone H3-K56 acetylation (H3-K56Ac) also appears to be masked by changes in mRNA decay rates [18,19]. H3-K56 is not found on the exposed N-terminal “tail” of histone H3, but rather it is localized underneath the final turn of nucleosomal DNA on the entry and exit positions where it makes a water-mediated, histone-DNA contact. Acetylation of H3-K56 eliminates this histone-DNA contact, enhancing the unwrapping frequency of DNA from the nucleosome edge, predicting a decrease in nucleosome stability [20]. H3-K56Ac is catalyzed by a single histone acetyltransferase in yeast, Rtt109, but inactivation of Rtt109 has almost no impact on the stable mRNA pool [19,21]. However, analysis of nascent transcription by Native-elongating transcript sequencing (Net-Seq) or Transient-transcriptome sequencing (TT-Seq) demonstrated a ~ 2-fold reduction in global yeast transcription rates [19]. To date, this is the sole histone acetylation event that promotes global transcript synthesis. Interestingly, inactivation of the deacetylases that remove H3-K56Ac, Hst3, and Hst4 also has little impact on the yeast transcriptome when analyzed by RNA-seq, whereas, Net-seq revealed a global ~2-fold increase in transcription [18]. In both cases, inactivation of the nuclear RNA exosome, a major player in mRNA quality control and mRNA decay, revealed the expected increase or decrease in stable mRNA levels that was predicted by the nascent transcript analyses. Thus, these studies suggest that global changes in transcription due to alterations in H3-K56Ac was buffered by regulating the levels or activity of the nuclear RNA exosome.
How might global changes in nascent transcription rates be sensed by the mRNA degradation machinery so that mRNA levels are properly buffered? The simplest model posits that a global decrease in transcription results in decreased protein levels for components of the decay machinery, such as the RNA exosome or the Xrn1 exonuclease. Decreased or increased levels of decay enzymes would lead to compensatory changes in mRNA decay rates. This model was initially proposed by Cramer and colleagues to explain how levels of the Xrn1 exonuclease might impact global decay rates [22]. This model also predicts that mRNA levels should change transiently as transcription rates are altered, and that the compensatory changes in mRNA decay rates should occur after a lag phase that reflects changes in decay factor synthesis. Indeed, this is exactly what was found following rapid depletion of a general transcription factor, the Spt7 subunit of the SAGA complex. In this study, the authors found that mRNA buffering was characterized by a lag phase of ~30 minutes, consistent with this simple model [23].
More complex models to explain the sensing of transcription rates have also been proposed. In general, these models require that factors involved in mRNA decay are also bound to, or regulate, nascent transcripts, providing a sensor for subsequent decay in the cytoplasm. Examples would include the Xrn1 decaysome, Rpb4/7 (discussed above), or the CCR-NOT deadenylase complex (Figure 2). Each of these factors is important for global transcription rates, as well as decay, and each of these factors appears to shuttle between the nucleus and cytoplasm. As noted above, the Choder group found that the Xrn1 exonuclease is bound upstream of many RNAPII genes, and genome-wide studies of RNAPII occupancy and transcription rates by three different groups (including the Choder group) demonstrated that loss of Xrn1 leads to a global decrease in transcription [13,14,24–26]. Notably, however, transcriptional defects due to loss of Rpb4 or CCR-NOT subunits are still buffered by slower rates of mRNA decay, ruling out simple versions of these models [16].
Figure 2.
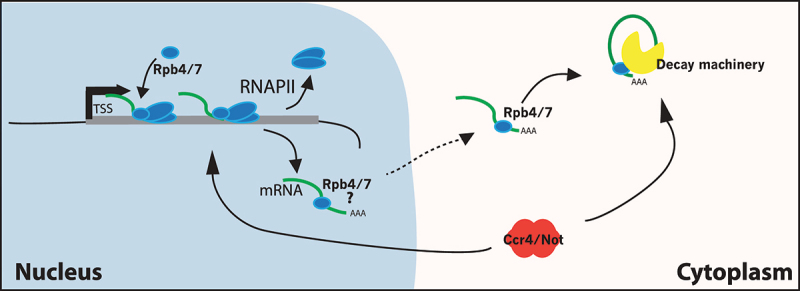
Co-transcriptional imprinting mRNAs with decay mediators. Model proposes that the Rpb4/7 module of RNAPII associates with mRNAs co-transcriptionally, remains associated with mRNAs as they are exported from the nucleus, and their abundance regulates subsequent decay events. Question mark represents the controversial role of Rpb4/7 in mRNA decay and synthesis. Also shown is the CCR/NOT mRNA deadenylase complex that may also coordinate both processes.
Information transfer: signaling mRNA degradation to transcription
Not only are transcription rates coupled to the mRNA decay machinery but changes in mRNA half-lives also influence the rates of transcription. For instance, inactivation of the cytoplasmic Ccr4 nuclease, a key enzyme that controls mRNA de-adenylation, leads to a global increase in mRNA half-lives. However, the increased stability of mRNAs is buffered by a compensatory decrease in transcription rates, leading to little change in mRNA levels [17]. Subsequent studies by both the Cramer and Choder groups extended these analyses to a host of enzymes that impact mRNA processing and decay [13,22]. The nearly universal conclusion is that inactivation of factors that control mRNA decay rates leads to compensatory decreases in global transcription rates.
How does the nuclear, transcription machinery “know” the stability of cytoplasmic mRNAs? One model was discussed above – that the decay machinery, or a decaysome, is also a direct, positive regulator of transcription (Figure 3). In this case, inactivation of the decaysome leads to longer half-lives for mRNAs but also decreased transcription rates. This model is a bit controversial, as cDTA results from the Cramer group suggested that one component of the decaysome, Xrn1, is a global repressor of transcription, not an activator [22]. These differing results are hard to reconcile, as the work from Choder and colleagues was quite extensive, involving RNAPII ChIP-seq, Gro-seq analysis of nascent transcription, and visualization of nascent transcripts by in situ RNA FISH [13]. More recently, the Choder group has also confirmed their findings with Net-seq analysis [27], and the Friedman lab used a modified version of the cDTA method and reported a global decrease in transcription as well [25].
Figure 3.
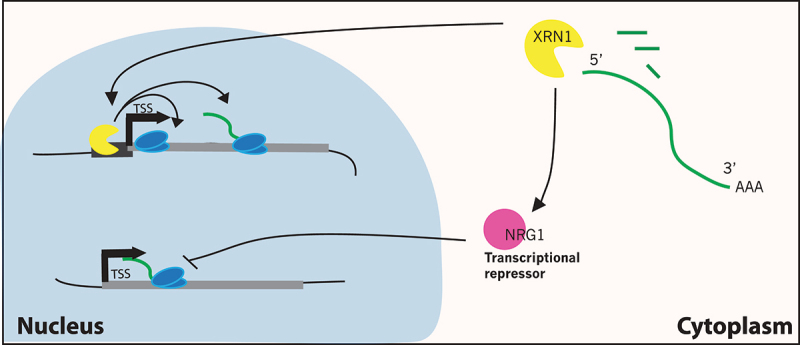
Global regulation of mRNA transcription by the decay machinery. Two models that explain how the Xrn1 nuclease might regulate transcription. Top, Xrn1 is a subunit of a decaysome complex that directly associates with gene promoter sequences and activates both transcription initiation and elongation by RNAPII. Bottom, Xrn1 promotes expression of a global negative repressor of transcription, Nrg1.
A recent study from the Chavez group appears to at least partially rectify the conflicting results regarding the impact of Xrn1 loss and transcriptional changes [24]. This group monitored nascent transcription by crosslinking analysis of cDNAs (CRAC) which provides strand-specific, nucleotide level localization of engaged RNAPII, similar to Net-Seq. Similar to previous work from the Choder group, CRAC analysis demonstrated that Xrn1 is a global, positive regulator of transcription, as indicated by a large negative impact of an xrn1∆ on transcription rates. Furthermore, rapid depletion of Xrn1 with an auxin-inducible degron allele also yielded a global decrease in transcription. The authors hypothesized that metabolic labeling methods that use 4tsU, like cDTA, may be misleading if 4tsU incorporation impacts mRNA decay rates. To test this idea, Chavez and colleagues monitored the half-lives of GAL gene mRNAs following transcriptional repression by addition of glucose. They made the remarkable finding that a short pulse label with 4tsU stabilized the GAL mRNAs, but only in the absence of Xrn1. Furthermore, 4tsU labeling increased the abundance of noncoding RNAs that are targeted by the nuclear RNA exosome, even in wild-type cells. These data indicate that 4tsU incorporation inhibits the activity of the RNA exosome, which becomes the major mRNA decay machinery in the absence of Xrn1. Thus, the authors suggest that the cDTA data may not yield an accurate measurement of transcription in mutant backgrounds where decay kinetics relay on the exosome. Notably, these data do not provide an explanation for why a recent cDTA approach also concluded that Xrn1 is a positive regulator of transcription [25]. Perhaps, strain differences are a contributing factor. Together, however, it now seems clear that Xrn1 does indeed function as a global positive regulator of transcription, potentially as a subunit of a larger decaysome.
A simple alternative to the decaysome model posits that the decay machinery regulates the abundance of a global transcriptional repressor. For instance, the mRNA for the putative repressor may be a sensitive target for the decay machineries, such that inactivation of decay factors leads to increased production of repressor protein. As predicted from this model, the global repression of transcription that follows stabilization of mRNAs occurs after a significant lag period [25]. Alternatively, loss of the decay machinery may lead, directly or indirectly, to increased transcription of the repressor mRNA. In the latter case, the repressor mRNA must “escape” the global down-regulation of transcription that buffers the crippling of mRNA decay pathways. The Cramer group reported just such an example, where they found that inactivation of several decay factors leads to increased transcription of the NRG1 gene, which encodes a known transcriptional repressor [22]. How these decay factors regulate NRG1 transcription is not clear, but loss of Nrg1 does lead to a small, global increase in transcription, as assayed by cDTA, consistent with this simple model.
Recently, we reported that the nuclear version of the RNA exosome regulates the expression of the Hst3 sirtuin deacetylase, which has the hallmarks of a global transcriptional repressor [28]. The RNA exosome is a 3’ to 5’ exonuclease that functions within the nucleus or cytoplasm depending on which catalytic subunit is incorporated; yeast Rrp6 functions within the nuclear RNA exosome, while Dis3 is the nuclease for the cytoplasmic enzyme [29]. The RNA exosome is highly conserved across eukaryotes and is considered a key RNA quality control pathway that regulates cryptic unstable transcripts (CUTs), non-coding RNAs, and abnormal coding transcripts [30–34]. The RNA exosome is also a key decay factor for 3’-5’ degradation of mRNAs following the shortening of the polyA tail and prior to 5’ mRNA de-capping. Initially, Cramer and colleagues used cDTA to show that deletion of the RRP6 gene caused a nearly 2-fold stabilization of global mRNAs, which was buffered by a similar decrease in transcript synthesis rates [22,28]. Likewise, we used the anchor-away method to rapidly deplete Rrp6 from the yeast nucleus, and found that this resulted in a global decrease of RNAPII transcription, as assayed by either TT-seq or Net-seq [28] .
To test the simple model that the RNA exosome might regulate a global transcriptional repressor, mRNA levels were evaluated for several well-characterized repressors that impact either preinitiation complex (PIC) assembly or histone modifications. Remarkably, the nuclear depletion of Rrp6 led to an increase in both mRNA and protein for the Hst3 sirtuin histone deacetylase. The HST3 transcript contains several consensus binding sites for the Nrd1-Nab3-Sen1 (NNS) complex, and previous work found that the Nrd1 and Nab3 components of the NNS complex bind to the Hst3 transcript in vivo [35]. The NNS complex promotes early termination of RNAPII transcripts in the absence of a polyA signal, and thus a simple model predicts that the RNA exosome regulates HST3 transcript levels by promoting termination by the NNS complex (Figure 4).
Figure 4.
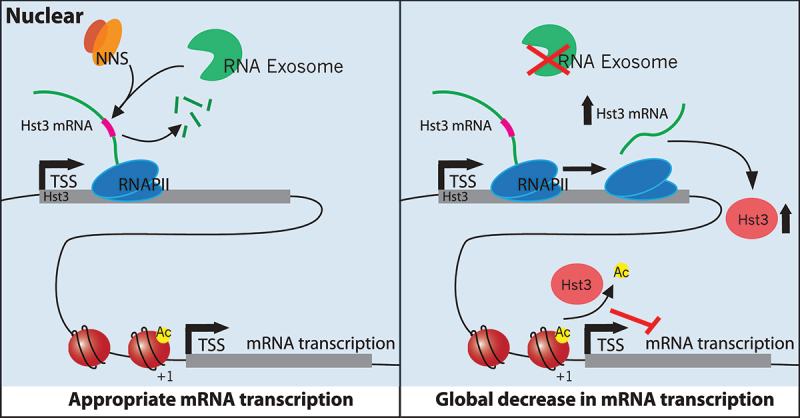
The nuclear RNA exosome inhibits expression of the histone deacetylase, Hst3, a global transcriptional repressor. Left, The RNA exosome functions with the NNS complex to promote premature transcription termination of the HST3 mRNA, down-regulating expression. Right, In the absence of the RNA exosome, the Hst3 transcript is stabilized and leads to increased levels of Hst3, increased deacetylation of gene promoter nucleosomes at histone H3-K56, and globally decreased transcription.
Hst3 and Hst4 are two yeast paralogs of mammalian Sirt6 that target the deacetylation of H3-K56 (Celic et al. 2006). In yeast, H3-K56Ac is a hallmark of nucleosomes located adjacent to promoters of nearly all RNAPII genes, and H3-K56Ac appears to function with the histone variant, H2A.Z, as a global activator of transcription [28]. Previously, we showed that depletion of both Hst3 and Hst4 resulted in hyperacetylation of H3-K56 and a global, ~2x increase in nascent RNA transcription [18]. Furthermore, over-expression of Hst3 is sufficient to repress transcription by ~1.5-fold genome-wide [28]. Thus, a simple model proposes that the RNA exosome regulates expression of the global Hst3 repressor, which provides the buffering mechanism that couples RNA-exosome-mediated mRNA decay to transcription initiation (Figure 4). Whether or how the nuclear RNA exosome communicates with its cytoplasmic counterparts remains an unresolved question.
Regulation of mRNA buffering by stress and the environment
What is the biological role of mRNA buffering? In some cases, communication between transcription and mRNA decay can coordinate steady state levels of proteins that act within multi-subunit complexes or proteins within the same pathway. An example was provided above for genes involved in galactose metabolism, where different GAL mRNAs share transcription induction and mRNA decay rates. Likewise, mRNA encoding subunits of the proteasome are known to be degraded at similar rates in certain conditions [36]. Pipel and colleagues have reported some intriguing results concerning how the mRNA transcription and decay processes work together to regulate the response to different types of environmental stresses [37]. First, when cells are treated with an oxidative stress agent, such as hydrogen peroxide (H2O2), the mRNAs for a large number of genes are rapidly induced, while another set of genes are repressed. However, this response is transient, relaxing back to pre-stress conditions after a time interval. In contrast, exposure of cells to the DNA damaging agent, methyl methanesulfonate (MMS), leads to a sustained response, with a set of genes induced and others repressed.
Analysis of mRNA decay rates following either H2O2 or MMS treatment indicated that novel patterns of mRNA buffering are associated with each type of environmental stress [37]. For oxidative damage, mRNA buffering served to rapidly relax the changes in transcription – induced genes showed increased rates of mRNA decay, while repressed gene mRNAs were stabilized. Importantly, these changes in decay rates cannot be explained by a simple model where increased transcription leads to expression of more decay enzymes, as this model predicts that repressed gene mRNAs should also show shorter mRNA half-lives. When cells were treated with MMS, a different buffering response was observed. Here, induced genes showed a slower decay rate (stabilization) and repressed genes were destabilized. Thus, this cellular response promoted a sustained change in gene expression.
What controls these differential responses to stress-induced transcription? In the case of oxidative damage, Pilpel and colleagues have presented evidence that the Rpb4/7 subunit module of RNAPII may play a key role [9]. In their study, they exploited a mutation in RPB6 that produced an Rpb6 derivative (rpb6Q100R) that cripples binding of Rpb4/7 to the RNAII core enzyme [7]. In this mutant yeast strain, there is a global defect in transcription, as expected for a mutation affecting RNAPII, but also a defect in mRNA decay, leading to buffering of mRNA levels in the absence of stress. When cells were treated with H2O2, the rpb6Q100R derivative nearly eliminated the stress-induced changes in mRNA decay rates. Induced mRNAs were not de-destabilized, and repressed RNAs were not stabilized. The biological consequence was a more sustained change in gene expression following oxidative stress, rather than the normal transient response. To explain this phenomenon, the authors propose a version of the Rpb4/7 imprinting model, where the mRNA for induced genes acquires more Rpb4/7, compared to repressed genes, and the higher levels of Rpb4/7 promote their more efficient degradation in the cytoplasm due to the ability of Rpb4/7 to interact with concentration limiting decay factors.
Recent work from the Choder group has found that the nuclear-cytoplasmic shuttling of the Xrn1 exonuclease also plays a role in regulating mRNA buffering in response to rapid environmental conditions [14]. Under optimal and constant growth conditions, the shuttling of Xrn1 promotes a “normal” level of both transcript synthesis and decay. Consequently, if the shuttling of Xrn1 is eliminated by removal of its nuclear localization signals or loss of the Kap120 importin, mRNA levels are buffered and proliferation is not affected. However, when cells are starved and re-exposed to nutrients, or if cells are shifted rapidly to new temperatures, cell growth is compromised by loss of Xrn1 shuttling, and mRNA buffering is disrupted. The authors suggest that Xrn1 shuttling, in concert with other decay factors, plays an essential role in re-establishing new steady state levels of mRNAs in response to changing environmental conditions.
Future directions
It now seems clear that cells must utilize a variety of mechanisms to ensure that protein production is buffered from changes in either transcription or mRNA decay. Although some simple mechanisms can explain a subset of buffering scenarios, there is no “one size fits all” explanation. Delineation of molecular mechanisms in both yeast and mammalian systems will clearly involve a substantial number of additional studies. Although the Rpb4/7 subunits of RNAPII are known to play roles in multiple aspects of RNA metabolism, their role in mRNA buffering has been somewhat controversial. Recently, Choder and colleagues reported that Rpb4 is post-translationally modified on a large number of sites, and the patterns of modifications change in response to cellular stress [38]. Rpb4/7 interact with a host of mRNA processing factors, as well as RNAPII, so these modifications may impact numerous post-transcriptional events. Certainly, the idea of a transcript imprinting role for Rpb4/7 becomes quite attractive if modification patterns impact the buffering of mRNA levels.
Funding Statement
This work was supported by the National Institute of General Medical Sciences [R35GM122519].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Enssle J, Kugler W, Hentze MW, et al. Determination of mRNA fate by different RNA polymerase II promoters. Proc Natl Acad Sci. 1993;90(21):10091–10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurosaki T, Popp MW, Maquat LE.. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat Rev Mol Cell Bio. 2019;20(7):406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bregman A, Avraham-Kelbert M, Barkai O, et al. Promoter elements regulate cytoplasmic mRNA decay. Cell. 2011;147(7):1473–1483. [DOI] [PubMed] [Google Scholar]
- 4.Dori-Bachash M, Shalem O, Manor YS, et al. Widespread promoter-mediated coordination of transcription and mRNA degradation. Genome Biol. 2012;13(12):R114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munchel SE, Shultzaberger RK, Takizawa N, et al. Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol Biol Cell. 2011;22(15):2787–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choder M. Rpb4 and Rpb7: subunits of RNA polymerase II and beyond. Trends Biochem Sci. 2004;29(12):674–681. [DOI] [PubMed] [Google Scholar]
- 7.Goler-Baron V, Selitrennik M, Barkai O, et al. Transcription in the nucleus and mRNA decay in the cytoplasm are coupled processes. Gene Dev. 2008;22(15):2022–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choder M. mRNA imprinting. Cell Logist. 2011;1(1):37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shalem O, Groisman B, Choder M, et al. Transcriptome kinetics is governed by a genome-wide coupling of mRNA production and degradation: a role for RNA pol II. PLoS Genet. 2011;7(9):e1002273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lotan R, Goler-Baron V, Duek L, et al. The Rpb7p subunit of yeast RNA polymerase II plays roles in the two major cytoplasmic mRNA decay mechanisms. J Cell Biol. 2007;178(7):1133–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulz D, Pirkl N, Lehmann E, et al. Rpb4 subunit functions mainly in mRNA synthesis by RNA polymerase II ♦. J Biol Chem. 2014;289(25):17446–17452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duek L, Barkai O, Elran R, et al. dissociation of Rpb4 from RNA polymerase II is important for yeast functionality. PLoS One. 2018;13(10):e0206161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haimovich G, Medina DA, Causse SZ, et al. Gene expression is circular: Factors for mRNA degradation also foster mRNA synthesis. Cell. 2013;153(5):1000–1011. [DOI] [PubMed] [Google Scholar]
- 14.Chattopadhyay S, Garcia-Martinez J, Haimovich G, et al. RNA-controlled nucleocytoplasmic shuttling of mRNA decay factors regulates mRNA synthesis and a novel mRNA decay pathway. Nat Commun. 2022;13(1):7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helenius K, Yang Y, Tselykh TV, et al. Requirement of TFIIH kinase subunit Mat1 for RNA Pol II C-terminal domain Ser5 phosphorylation, transcription and mRNA turnover. Nucleic Acids Res. 2011;39(12):5025–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Timmers H, Tora L. Transcript buffering: a balancing act between mRNA synthesis and mRNA degradation. Mol Cell. 2018;72(1):10–17. [DOI] [PubMed] [Google Scholar]
- 17.Sun M, Schwalb B, Schulz D, et al. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res. 2012;22(7):1350–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feldman JL, Peterson CL. Yeast sirtuin family members maintain transcription homeostasis to ensure genome stability. Cell Rep. 2019;27(2978–2989.e5):2978–2989.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Topal S, Vasseur P, Radman-Livaja M, et al. Distinct transcriptional roles for histone H3-K56 acetylation during the cell cycle in Yeast. Nat Commun. 2019;10(1):4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neumann H, Hancock SM, Buning R, et al. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol Cell. 2009;36(1):153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsubota T, Berndsen CE, Erkmann JA, et al. Histone H3-K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol Cell. 2007;25(5):703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun M, Schwalb B, Pirkl N, et al. Global Analysis of Eukaryotic mRNA Degradation Reveals Xrn1-Dependent Buffering of Transcript Levels. Mol Cell. 2013;52(1):52–62. [DOI] [PubMed] [Google Scholar]
- 23.Baptista T, Grünberg S, Minoungou N, et al. SAGA Is a general cofactor for RNA polymerase II transcription. Mol Cell. 2017;68(130–143.e5):130–143.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Begley V, Jordán-Pla A, Peñate X, et al. Xrn1 influence on gene transcription results from the combination of general effects on elongating RNA pol II and gene-specific chromatin configuration. RNA Biol. 2021;18(9):1310–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chappleboim A, Joseph-Strauss D, Gershon O, et al. Transcription feedback dynamics in the wake of cytoplasmic mRNA degradation shutdown. Nucleic Acids Res. 2022;50(10):5864–5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fischer J, Song YS, Yosef N, et al. The yeast exoribonuclease Xrn1 and associated factors modulate RNA polymerase II processivity in 5‘ and 3‘ gene regions. J Biol Chem. 2020a;295(33):11435–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischer J, Song YS, Yosef N, et al. The yeast exoribonuclease Xrn1 and associated factors modulate RNA polymerase II processivity in 5‘ and 3‘ gene regions. J Biol Chem. 2020b;295(33):11435–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bryll AR, Peterson CL. Functional interaction between the RNA exosome and the sirtuin deacetylase Hst3 maintains transcriptional homeostasis. Gene Dev. 2021;36(1–2):17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Q, Greimann JC, Lima CD. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell. 2006;127(6):1223–1237. [DOI] [PubMed] [Google Scholar]
- 30.Delan-Forino C, Schneider C, Tollervey D. Transcriptome-wide analysis of alternative routes for RNA substrates into the exosome complex. PLoS Genet. 2017;13(3):e1006699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kiss DL, Andrulis ED. Genome-wide analysis reveals distinct substrate specificities of Rrp6, Dis3, and core exosome subunits. Rna. 2010;16(4):781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitchell P, Petfalski E, Shevchenko A, et al. The Exosome: a conserved eukaryotic RNA processing complex containing multiple 3’→5’ exoribonucleases. Cell. 1997;91(4):457–466. [DOI] [PubMed] [Google Scholar]
- 33.Schneider C, Kudla G, Wlotzka W, et al. Transcriptome-wide analysis of exosome targets. Mol Cell. 2012;48(3):422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Torchet C, Bousquet-Antonelli C, Milligan L, et al. Processing of 3’-extended read-through transcripts by the exosome can generate functional mRNAs. Mol Cell. 2002;9(6):1285–1296. [DOI] [PubMed] [Google Scholar]
- 35.Creamer TJ, Darby MM, Jamonnak N, et al. Transcriptome-wide binding sites for components of the saccharomyces cerevisiae non-poly(a) termination pathway: Nrd1, Nab3, and sen1. PLoS Genet. 2011;7(10):e1002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Liu CL, Storey JD, et al. Precision and functional specificity in mRNA decay. Proc Natl Acad Sci. 2002;99(9):5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shalem O, Dahan O, Levo M, et al. Transient transcriptional responses to stress are generated by opposing effects of mRNA production and degradation. Mol Syst Biol. 2008;4(1):223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richard S, Gross L, Fischer J, et al. Numerous post-translational modifications of RNA polymerase II subunit Rpb4/7 link transcription to post-transcriptional mechanisms. Cell Rep. 2021;34(2):108578. [DOI] [PubMed] [Google Scholar]