

Abstract
We previously reported 1H-imidazo[4,5-c]quinolin-4-amines as A3 adenosine receptor (A3AR) positive allosteric modulators (PAMs). A3AR agonists, but not PAMs, are in clinical trials for inflammatory diseases and liver conditions. We synthesized new analogues to distinguish 2-cyclopropyl antagonist 17 (orthosteric interaction demonstrated by binding and predicted computationally) from PAMs (derivatives with large 2-alkyl/cycloalkyl/bicycloalkyl groups). We predicted PAM binding at a hydrophobic site on the A3AR cytosolic interface. Although having low Caco-2 permeability and high plasma protein binding, hydrophobic 2-cyclohept-4-enyl-N-3,4-dichlorophenyl, MRS7788 18, and 2-heptan-4-yl-N-4-iodophenyl, MRS8054 39, derivatives were orally bioavailable in rat. 2-Heptan-4-yl-N-3,4-dichlorophenyl 14 and 2-cyclononyl-N-3,4-dichlorophenyl 20 derivatives and 39 greatly enhanced Cl-IB-MECA-stimulated [35S]GTPγS binding Emax, with only 12b trending toward decreasing the agonist EC50. A feasible route for radio-iodination at the p-position of a 4-phenylamino substituent suggests a potential radioligand for allosteric site binding. Herein, we advanced an allosteric approach to developing A3AR-activating drugs that are potentially event- and site-specific in action.
Graphical Abstract
INTRODUCTION
Agonists of the four subtypes of adenosine receptors (ARs) are an appealing ligand class for drug development due to the many salutary actions of adenosine, such as tissue repair and protection against ischemia and other organ stress.1 Activation of the Gi-coupled A3AR subtype is associated with attenuating chronic neuropathic pain, heart and brain ischemic preconditioning, and anti-inflammatory effects, without causing cardiovascular side effects.2,3 The A3AR is overexpressed in immune and cancer cells, adding to its potential as a possible therapeutic target.3 At present, A3AR agonists are in Phase 2/3 clinical trials for psoriasis, due to their anti-inflammatory properties, and liver diseases, i.e., hepatocellular carcinoma and non-alcoholic steatohepatitis (NASH).4
Allosteric modulators bind to sites that are topographically distinct from the orthosteric binding site for native agonists and can exert their effects through receptor conformation changes beyond those induced by orthosteric ligands.5 Various types of allosteric modulators differ in their pharmacological effects. Positive allosteric modulators (PAMs) may improve agonist affinity, potency, and/or efficacy, while negative allosteric modulators (NAMs) do the opposite. Some allosteric modulators can induce a characteristic functional response in the absence of an agonist, i.e., ago-PAMs.6
The hallmark advantage of PAMs over orthosteric agonists is that they can be event- and site-specific in action.7 Because adenosine is endogenously elevated in response to localized distress signals within the body, a pure PAM will enhance the protective function of adenosine only when and where it is elevated, thereby reducing the risk of side effects.8 A second advantage of developing AR PAMs over orthosteric ligands is the possibility of achieving a high selectivity for a single AR subtype, since PAMs typically bind to regions that are more variable between receptor subtypes. Third, AR agonists have multiple potential pharmaceutical applications in the central nervous system (CNS),9 but current A3AR orthosteric agonists, mainly nucleosides, tend to have low blood–brain barrier (BBB) permeability, with typically only a few percent or less available in the CNS.10–13 Therefore, BBB-penetrating PAMs might be preferable over AR nucleoside agonists for CNS applications. Lastly, it might be possible to develop biased PAMs that selectively enhance certain A3AR-induced signaling pathways.14,15
At least four heterocyclic classes have been reported to act as A3AR allosteric modulators (Chart 1): amilorides,16 represented by 5-(N,N-hexamethylene)amiloride (HMA, 1); PAMs shown are 2 (3-(2-pyridinyl) isoquinolines;17 2,4-disubstituted quinolines, represented by 3; and, 1H-imidazo[4,5-c]quinolin-4-amines (4–12),18–20 including N-(3,4-dichloro-phenyl)-2-cyclohexyl-1H-imidazo[4,5-c]quinolin-4-amine (7, LUF6000).21
Chart 1.

(A–D) Four Classes of Heterocyclic A3AR Allosteric Modulators [Amiloride Derivative 1 (A), 3-(2-Pyridinyl)isoquinoline 2 (B), 2,4-Disubstituted Quinoline 3 (C), and 1H-Imidazo[4,5-c]quinolin-4-amines with 2-Alkyl and 2-Cycloalkyl Substitutions (4–12) (D), Including Alternative 4-Substituted Phenylamino Derivatives and the Bulky 2-Bicycloalkyl (exo-Norbornanyl, 12a) and 2-Tricycloalkyl (Adamantan-1-yl, 12b) Derivatives (E)19,20
We previously explored the structure–activity relationship (SAR) of the 1H-imidazo[4,5-c]quinolin-4-amine family of A3AR PAMs.19,20,22 Prototypical A3AR PAM 7, which is now being considered for clinical testing for erectile dysfunction,23 slowed A3AR agonist dissociation.19,20 It enhanced maximal efficacy (Emax) at the human (h) A3AR (but not the mouse homologue)24 without influencing agonist potency, possibly due to simultaneous competitive interactions at the orthosteric binding site.17,18 The A3AR PAM activity of imidazoquinolinamine derivatives is often accompanied by variable degrees of apparent receptor antagonist activity.19,20 Based on findings with chimeric receptors composed of an A3AR from a responding (human) and a non-responding (mouse) species, we predict that PAM activity results from binding to an allosteric site located within inner/cytosolic A3AR regions, exclusive of the orthosteric binding site.24 The chimeric receptors indicated that a 2-cyclopropyl derivative interacts primarily with the orthosteric site as a competitive antagonist, and the corresponding 2-cyclohexyl derivative 7 can interact with both sites. The possibility that 7 might negatively modulate A3AR agonists by binding to the same intracellular allosteric site, i.e., NAM activity, has been ruled out.24
In this study, we have extended the recent expansion and contraction of the 2-cyclohexyl ring of 7 by Fisher et al.24 with additional 1H-imidazo[4,5-c]quinoline-4-amine derivatives as hA3AR PAMs through modifications at the 4-arylamino and/or the 2 position of the imidazoquinoline scaffold. We functionalized the rings with polar groups to try to reduce lipophilicity and improve aqueous solubility, as well as introduced bridging groups to determine if there is an A3AR-preferred conformation of 2 position substituent. We also explored the effect of various 4-arylamino substitutions on positive allosteric enhancement. Here we describe the synthetic methods for the newly prepared analogues, including those reported by Fisher et al.24
The objectives of this study were: to expand the SAR of 1H-imidazo[4,5-c]quinolin-4-amine derivatives related to 7 to identify high efficacy A3AR PAMs, including those bearing hydrophilic and conformationally constrained groups; to devise a shorter synthetic route to derivatize 1H-imidazo[4,5-c]quinolin-4-amines than the ones previously reported;18–20 and to obtain a baseline example of the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of potential lead compounds from this series.
RESULTS AND DISCUSSION
Description of Synthetic Compound List.
Four groups of 1H-imidazo[4,5-c]quinolin-4-amine derivatives were synthesized for A3AR pharmacological characterization, as listed in Table 1 (5–8, 13–39), including six compounds reported earlier (5–8 and 12).19 The final step reaction yields are shown in Table S1. The first group of derivatives (5–8, 13–23) has hydrophobic alkyl and cycloalkyl substitutions at the 2 position of the 1H-imidazo[4,5-c]quinolin-4-amine scaffold with the 3,4-dichlorophenyl group at the 4-amino position, which was shown in previous SAR studies to improve A3AR PAM activity and subtype selectivity.19,20 The second group of derivatives (24–29) includes bridged 2-bicyclic substitutions, based on the favorable PAM activity of compounds 8 and 19, as well as the exo-norbornanyl and adamantan-1-yl derivatives 12a and 12b (Chart 1E). The third group (30–34) has hydrophilic oxygen-containing functionality introduced on a 2-cycloheptyl ring and a 3,4-dichlorophenyl group at the 4-amino position. Lastly, the fourth group (35–39) has a cyclohexyl ring or a heptan-4-yl moiety at the 2 position combined with various p-substituted 4-phenylamino groups. Prior studies showed tolerance of 4-methoxy and 4-chloro substitutions at the 4-phenylamino position (10 and 11, Chart 1D), resulting in promising A3AR allosteric enhancement based on a slower dissociation rate and increased Emax of agonists.19 Compounds 29 and 32–34 were racemic mixtures. For most of the analogues, we incorporated the 4-(3,4-dichlorophenylamino) substitution based on previous studies in which this group, compared to other haloaryl groups (3,5- and 2,4-dichloroaniline derivatives), had favorable PAM activity as indicated by effects on agonist dissociation kinetics and efficacy.20
Table 1.
1H-Imidazo[4,5-c]quinolin-4-amine Derivatives Synthesized and Effects on Agonist-Induced A3AR Activation
| |||
|---|---|---|---|
| Compound | R2 | R1 | Emaxa(%) |
| 2-alkyl and 2-cycloalkyl derivatives | |||
| 13 | 3,4-Cl2 |
|
153 ± 12* |
| 14 | 3,4-Cl2 |
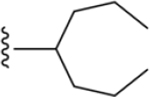
|
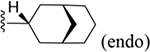
216 ± 12* |
| 15 | 3,4-Cl2 |
|
159 ±13* |
| 16 | 3,4-Cl2 |
|
150 ± 5* |
| 17 | 3,4-Cl2 |
|
111 ± 16 |
| 5 | 3,4-Cl2 |
|
109 ± 3 |
| 6 | 3,4-Cl2 |
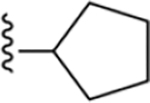
|
142 ± 5* |
| 7 | 3,4-Cl2 |
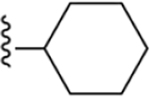
|
225 ± 10* |
| 8 | 3,4-Cl2 |
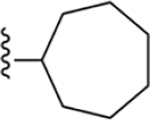
|
175 ± 3* |
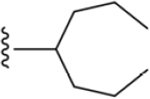
| 18 | 3,4-Cl2 |
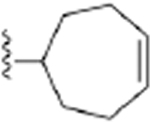
|
241 ± 9* |
| 19 | 3,4-Cl2 |
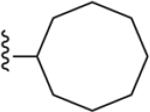
|
146 ± 2* |
| 20 | 3,4-Cl2 |
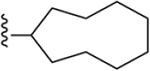
|
241 ± 12* |
| 21 | 3,4-Cl2 |
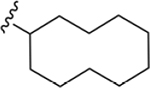
|
135 ± 7* |
| 22 | 3,4-Cl2 |
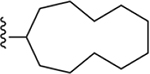
|
122 ± 6* |
| 23 | 3,4-Cl2 |
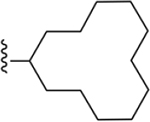
|
101 ± 3 |
| 2-bicycloalkyl derivatives | |||
| 24 | 3,4-Cl2 |
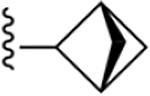
|
187 ± 14* |
| 25 | 3,4-Cl2 |
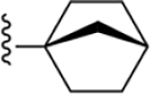
|
170 ± 6* |
| 26 | 3,4-Cl2 |
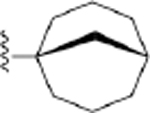
|
154 ± 7* |
| 27 | 3,4-Cl2 |
|
237 ± 12* |
| 28 | 3,4-Cl2 |
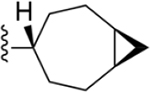
|
219 ± 16* |
| 29 | 3,4-Cl2 |
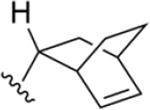
|
187 ±8* |
| 2-cycloalkyl derivatives with hydrophilic substitution | |||
| 30 | 3,4-Cl2 |
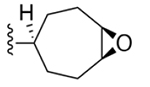
|
136 ± 11 |
| 31 | 3,4-Cl2 |
|
173 ± 14* |
| 32 | 3,4-Cl2 |
|
160 ± 16* |
| 33 | 3,4-Cl2 |
|
118 ± 14 |
| 34 | 3,4-Cl2 |
|
156 ± 10* |
| 2-alkyl and 2-cycloalkyl derivatives with modified 2-arylamino groups | |||
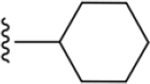
| 35 | 4-I |
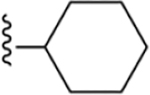
|
184 ± 9* |
| 36 | 4-Br |
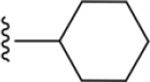
|
207 ± 17* |
| 37 |
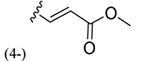
|
|
164 ± 10* |
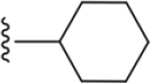
| 38 |
|
|
170 ± 12* |
| 39 | 4-I |
|
223 ± 10* |
| 48 | 4-Sn(CH3)3 |
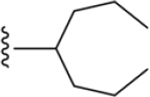
|
ND |
| 49 | 4-Sn((CH2)3CH3)3 |
|
ND |
Effect of PAM derivative (1.0 μM) on [35S]GTPγS binding induced by 52 using WT hA3ARs (n = 3).
P ≤ 0.05 (one-way ANOVA with Bonferroni-adjusted t test for multiple comparisons) with respect to control in the absence of a PAM. Effects of 0.1 and 10 μM are shown in Table S4. ND, not determined.
Chemical Synthesis.
3,4-Diaminoquinoline intermediate 53 and final compounds 13, 14, and 16 were prepared by literature procedures (Scheme S1).19 A shorter synthesis than the previously reported 9-step route,19 requiring only 6 steps, was developed to create a new series of 1H-imidazo[4,5-c]quinolin-4-amine derivatives (Scheme 1). The first four steps closely followed a reported synthetic route used to synthesize a non-adenosine-related series of 2-(p-substituted-phenyl)-4-phenyl-1H-imidazo[4,5-c]quinoline derivatives, which differ from the present compounds in having a 4-phenyl instead of a 4-aminophenyl substitution.25
Scheme 1. Six-Step Synthetic Route for 1H-Imidazo[4,5-c]quinolin-4-amine Derivatives with 2 Position and 4-Arylamino Substitutions (B), Including the Synthesis of a Common Diamino Intermediate 44 (A)a.

aReagents and conditions: (i) HNO3, 75 °C, 95%; (ii) PhPOCl2, 135 °C, 87%; (iii) 28% aq. ammonia, CH3CN, 50 °C, 97%; (iv) Fe powder, HCl, CH3CH2OH/H2O, 75 °C, 70%; (v) PPA, 120 °C; (vi) TCFH, NMI, ACN, 60 °C; (vii) NaOH, H2O:MeOH (1:1), 90 °C; (viii) Pd2(dba)3, tBuXPhos, t-BuONa, 1,4-dioxane, 100 °C, 5–51%, (ix) Pd(OAc)2, tBuXPhos, t-BuONa, H2O, 1,4-dioxane, 100 °C, 2–33%; (x) EtOH, microwave, 130 °C, 10–61%; (xi) 18, Et2Zn, CH2I2, 0 °C, 13%; (xii) 18, m-CPBA, CHCl3, 6–12%, 30 and 31 separated chromatographically; (xiii) 18, (CH3)2S·BH3, THF, NaOH, H2O2, 0 °C, 8–12%, 33 and 34 separated chromatographically; (xiv) 33/34, DMP, CHCl3, 23%; (xv) 36, Pd(OAc)2, CH2═CHCOOCH3, Et3N, 140 °C, 18%; (xvi) 35, Pd(Ph3P)2Cl2, 5-Cl-thien-2-yl-acetylene, CuI, Et3N, 80 °C, 10%.
In the first step of the 6-step route (Scheme 1), quinoline-2,4-diol 40 was nitrated with nitric acid to produce 3-nitroquinoline-2,4-diol 41. 41 was then chlorinated with phenylphosphonic dichloride to afford 2,4-dichloro-3-nitroquinoline 42. The step-2 product was then aminated in step-3 with 28% aq. ammonia to give regioselectively 2-chloro-3-nitroquinolin-4-amine 43. Subsequently, in step-4 Fe powder and hydrochloric acid reduced the 3-nitro group to an amine to provide the vicinal diamine, 2-chloroquinoline-3,4-diamine 44.25
Two alternative step-5 protocols condensed the vicinal diamine with a carboxylic acid followed by cyclization. The first reaction protocol (see Scheme 1, general procedure A (v)), utilized polyphosphoric acid (PPA) for the condensation between 2-chloroquinoline-3,4-diamine and an appropriate carboxylic acid 45, followed by cyclization to the imidazole 46.19 The second reaction protocol (see general procedure B (vi)), required two steps without isolation of the intermediate. Initially, an acyl imidazolium adduct formed between the coupling agent tetramethylchloroformamidinium hexafluorophosphate (TCFH) and N-methylimidazole (NMI), which provided an electrophile to activate the appropriate carboxylic acid 45.26 The vicinal diamine 44 was acylated to produce an amide intermediate (not shown). A similar published vicinal diamine reaction uses room temperature,26 but we found that heating at 60 °C brought the reaction to completion, increasing the yield. The crude amide was subjected to a subsequent base-catalyzed cyclization reaction to form the imidazole ring in 46 and incorporate a 2 position substitution on the imidazoquinoline scaffold.
The last step, a C–N cross-coupling reaction, was performed using three different reaction protocols. The first protocol used the palladium catalyst tris(dibenzylideneacetone) dipalladium(0) (Pd2(dba)3) (general procedure C (viii) in Scheme 1),27–29 while the second used a water-activated palladium acetate (Pd(OAc)2) catalyst (general procedure D (ix) in Scheme 1).28 We attempted to improve the cross-coupling reaction yield using Pd(OAc)2 rather than the Pd2(dba)3 catalyst, because the removal of the sizable dba (dibenzylideneacetone) ligand during the palladium catalyst activation might sterically hinder the oxidative addition of the aryl halide during the catalytic cycle.27 However, higher yields were obtained using the step-6 general procedure C than with the general procedure D. The third reaction protocol was a microwave-assisted reaction in ethanol at 130 °C to achieve the final 1H-imidazo[4,5-c]quinolin-4-amine derivative (general procedure E (x) in Scheme 1).19
Compounds 13–15 have 2-alkyl substitutions, with compound 15 included to investigate the effect of terminal hexafluoro substitution on the pharmacological activity of compound 14.30 The carboxylic acid precursor of compound 15 was prepared by a procedure reported in a patent (Scheme S2).31,32 Compound 16 introduced a trifluoromethyl group at the 4 position of the cyclohexyl ring of a reference PAM, 7. Final compounds 13–22 were prepared with the step-6 general procedure C, having yields ranging from 5% to 51%. Compound 23 was synthesized using the step-6 general procedure E with the C–N cross-coupling of the 4-chloro in step-5 product to produce 3,4-dichloroaniline in 25% yield. Most carboxylic acids used for the construction of the cycloaliphatic rings in compounds 7 and 17–23 (starting from compounds 46e–o using the synthetic procedures in Scheme 1) were commercially available, except for the cyclononane- and cyclodecanecarboxylic acids, which were synthesized from a Favorskii ring contraction of their respective α-bromo-cycloketones (Scheme S3).
Compounds 24–27 were produced using the step-6 general procedure D, having yields ranging from 2% to 33%. In the cyclopropanation reaction of alkene derivative 18 to form compound 28,33 only the cis stereoisomer was recovered (Scheme 1, the imidazole group is trans with respect to the cyclopropane), which was confirmed by 1D-NOE NMR.
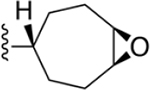
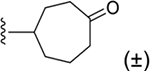
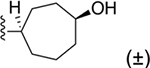
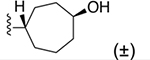
Bicyclo[2.2.2]oct-2-ene derivative 29 was the only two-carbon bridged derivative with a double bond to provide extra rigidity to the ring system. The precursor bicyclic carboxylic acids were obtained upon saponification of the methyl ester products formed from a Diels–Alder reaction between 1,3-cyclohexadiene and methyl acrylate (Scheme S4).34,35 Two pairs of enantiomers that are diastereoisomers to one another were isolated from the Diels–Alder reaction, the majority being the endo enantiomeric pair, likely due to the lower transition state energy favoring the formation of the endo over exo stereoisomers. Thus, compound 29 is a racemic mixture of endo enantiomers, which was confirmed by 1D-NOE. The double bond of 2-cycloheptenyl derivative 18 allowed hydrophilic oxygen-containing functional groups to be installed (Scheme 1). Two diastereomeric oxirane compounds, 30 and 31, were produced and resolved from the same epoxidation reaction of compound 18 using m-CPBA.36 The distinguishing characteristic between these two diastereomers is the chemical shift of the ring proton (CH) on the carbon atom connected to the 2 position of the 1H-imidazo[4,5-c]quinolin-4-amine, which was δ2.71 and δ3.07 ppm for 30 and 31, respectively, in the 1H NMR spectrum. The structures of 27, 29 and 30 were confirmed by 1D and 2D NMR (Supporting Information). Compounds 33 and 34 were made in the same hydroboration–oxidation reaction37 and isolated as racemic mixtures, being enantiomeric pairs that are diastereomers to one another. The assignment of the configuration of compound 33 was similarly confirmed by 1D-NOE (Supporting Information). Compound 32 was made from treating a mixture of compounds 33 and 34 with the oxidizing agent Dess–Martin periodinane,38 producing the racemic carbonyl enantiomers.
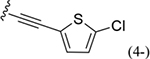
Compounds 35 and 36 have 4-iodophenyl and 4-bromophenyl substitutions, respectively. Both were produced using the step-6 general procedure E microwave-assisted reaction between a 4-chloro-2-cyclohexyl-imidazo[4,5-c]-quinolinamine 46, the step-5 product, with the corresponding p-iodo- or p-bromo-aniline in ethanol (Scheme 1). Compound 37, having a methyl acrylate 4 position substitution on the phenylamino moiety, was generated from a Heck reaction between compound 36 and methyl acrylate using a Pd(OAc)2 catalyst (Scheme 1)39 as the E-isomer. Compound 38 has a p-(5-chlorothiophen-2-yl)ethynyl substitution, and was prepared by a Sonogashira reaction between compound 35 and 2-chloro-5-ethynylthiophene using a bis(triphenylphosphine) palladium II dichloride catalyst (Scheme 1).40,41 A reaction using p-bromo derivative 36 was unsuccessful, but using p-iodo derivative 35 provided the desired product 38 (Scheme 1). Compound 39 was produced to study the effect of an I-label and to provide a potential allosteric radioligand. Two trialkylstannyl derivatives, 48 and 49, were synthesized from compound 39 using a bistriphenylphosphine palladium dichloride catalyst with hexamethylditin or hexabutylditin, respectively (Scheme S5).42 They are potential precursors for a radio-iodination reaction to produce a 125I radioligand (Scheme S6).43
Pharmacological Characterization.
Two assays using fixed PAM concentrations characterized the effects of the new derivatives on hA3AR agonist radioligand ([125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide 50, [125I]I-AB-MECA) binding. Pharmacological results for a subset of these compounds (5–8, 14 and 17–23) were reported by Fisher et al. and are included here for comparison.24 This first assay (Figure 1) consisted of measuring the degree of radioligand dissociation after a standard time delay (3 h) following the addition of an excess of an unlabeled AR agonist (adenosine-5′-N-ethylcarboxamide 51, NECA), reflecting the effect on dissociation rate (as a PAM would slow the dissociation). The second assay (Figure 2) consisted of measuring the influence of each modulator on the relative % binding of the radioligand at the hA3AR after a long duration of incubation (18 h), at which point equilibrium conditions were achieved. A third assay, a functional assay, consisted of a [35S]GTPγS binding assay to determine the potency of agonist (2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine 52, Cl-IB-MECA) as an EC50 value (concentration of half-maximal activation) in hA3AR activation and its Emax in the presence of the indicated compound. All three assays used membranes prepared from human embryonic kidney (HEK) 293 cells overexpressing the hA3AR.
Figure 1.

Effect of 1H-imidazo[4,5-c]quinolin-4-amine derivatives on the dissociation of [125I]50 using hA3ARs. HEK-293 membranes stably overexpressing the hA3AR were incubated with ~0.3 nM [125I]50 and 10 μM of the indicated modulator for 3 h. The addition of 100 μM 51 initiated dissociation. The amount of radioligand left remaining after 60 min was measured. Statistical significance was calculated by a two-tailed Student’s t test (n = 3; * denotes P < 0.05). Data are presented as mean ± SEM.
Figure 2.

Effect of PAM derivatives on equilibrium binding of [125I]50 at the hA3AR. HEK-293 cell membranes stably overexpressing the hA3AR were incubated with ~0.3 [125I]50 and 10 μM of the indicated modulator for ~18 h to reach equilibrium. The amount of specific binding was determined, and % change from the vehicle was calculated. Statistical significance was determined by two-tailed paired Student’s t test of raw counts per min values for modulator compared to vehicle (n = 3; * denotes P < 0.05). Data are presented as mean ± SEM.
In Figure 1, single-point dissociation assays were conducted to measure the relative influence of each derivative on dissociation of 50 from the hA3AR 60 min after initiation of dissociation by the addition of a competitive AR agonist 51 at a saturating concentration of 100 μM (Table S2). The horizontal dotted line represents the control amount of agonist radioligand that remained bound in the absence of a 1H-imidazo[4,5-c]quinolin-4-amine derivative, i.e., 26% of the amount bound at time 0. Thus, bars significantly above the line represent compounds that had a net effect to increase the amount of radioligand remaining bound, presumably by allosterically slowing the agonist dissociation rate, i.e., PAM activity.
Compounds 13–16 showed varying results, with 2-(heptan-4-yl) derivative 14 slowing agonist dissociation the most, with 71% of the radioligand remaining bound. Despite being the hexa-fluoro equivalent of compound 14, compound 15 did not slow agonist dissociation compared to the control (P-value = 0.958).
Compounds 17 (cyclopropyl), 5 (cyclobutyl) through 8 (cycloheptyl), and 18 (cyclooctyl) through23 (cyclododecyl) represent the 2-cycloalkyl-substituted analogues, ranging from 3 to 12 carbons. As the hydrophobic ring system increased from cyclopropyl 17 to cyclo-octyl 19, there was a steady increase in percent radioligand remaining hA3AR-bound compared to control. The following compounds displayed the greatest reduction in dissociation (P-values < 0.05, Student’s t test), ranging from 45% to 54% left remaining: 2-cyclohexyl 7, 2-cycloheptyl 8, 2-cyclohept-4-enyl 18, and 2-cyclooctyl 19. With larger rings, radioligand dissociation was not significantly slowed compared to control (P-values = 0.188, 0.786, 0.808, and 0.735, for cyclononyl 20 to cyclododecyl 23, respectively).
exo-Norbornanyl 12a, adamantan-1-yl 12b, and 24–29 represent bi- and tricyclic ring systems of varying sizes. Except for compounds 12a and 29, all compounds slowed the agonist dissociation compared to the control. The percent of radioligand remaining bound in the presence of modulators 12b and 24–28 ranged from 45% to 52%. The compounds with hydrophilic substitutions on the 2-cycloheptyl ring, 30–34, showed no net slowing of radioligand dissociation compared to the control.
Among the p-substituted 4-phenylamino derivatives 35–38, compounds 35 (4-iodophenyl) and 36 (4-bromophenyl) slowed the radioligand dissociation rate the most, with 57% and 56% radioligand remaining, respectively. However, compounds 37 ((E)-methyl-3-(4-aminophenyl acrylate) and 38 (5-chlorothiophen-2-yl)ethynyl)phenyl)) did not significantly slow the agonist dissociation rate compared to the control (P-values = 0.052 and 0.450, respectively). Compound 39, with combined features of compounds 14 and 35, i.e., 2-(heptan-4-yl) and 4-iodophenylamino substitutions, considerably slowed radioligand dissociation with 65% remaining A3AR-bound.
Figure 2 shows single-point equilibrium binding assay results: the positive, negative, or neutral percent change from the vehicle of the radioligand in the presence of a fixed concentration (10 μM) of each modulator under equilibrium conditions (Table S3). The radioligand bound was measured at 18 h, which ensures equilibrium (kon = koff) conditions were achieved.24,44 Thus, bars above the 0 line represent compounds that positively modulate radioligand binding as PAMs, possibly from increased agonist affinity. Bars significantly below the 0 line represent a net reduction in agonist radioligand binding, presumable due to competitive antagonism (5, 12a, 13, 17, 18 and 28–36). Only three (28, 35 and 36) of these fourteen derivatives slowed agonist radioligand dissociation (Figure 1).24
Comparing open-chain alkyl and fluorinated compounds 13–16, the n-propyl derivative 13 decreased radioligand binding the most with a −45% change from the vehicle. Compound 14 increased radioligand binding the most with a 70% change from the vehicle.
Among the 2-cycloalkyl-substituted derivatives, compound 17 (2-cyclopropyl) decreased radioligand binding by 71% compared to the control, and compound 19 (2-cyclooctyl) increased radioligand binding by 41% compared to the control. Compounds 6–8 and 21–23 did not produce a net change in radioligand equilibrium binding compared to the control.
Among the bridged compounds, i.e., 12a, 12b and 24–29, compounds 12b (2-adamantan-1-yl), 26 (2-bicyclo[3.3.1]-nonan-1-yl), and 27 (2-((1R,3s,5S)-bicyclo[3.3.1]nonan-3-yl)) significantly increased radioligand binding with a 19%, 38% and 41% change from vehicle, respectively. Compounds 24 and 25 had no effect on radioligand binding, while compounds 12a, 28 and 29 decreased radioligand binding with −19%, −37% and −21% change from vehicle, respectively.
All the derivatives with hydrophilic substitutions, 30–34, decreased A3AR radioligand binding at equilibrium, with compound 33 having the most significant decrease in binding with a 36% change from vehicle when compared to the control (P-value = 0.023).
Of the p-substituted phenylamino derivatives, compound 37 increased agonist equilibrium binding the most with a 15% change from the vehicle. Compounds 35 and 36 decreased agonist equilibrium binding with −31% and −49% change from the vehicle, respectively. Compounds 38 and 39 did not affect radioligand binding at equilibrium compared to control.
In addition to the kinetic binding studies, the ability of each compound to allosterically modulate hA3AR-dependent G protein activation by agonist 52 was measured in the [35S]GTPγS functional assay (Figure 3 and Table S4). Concentration–response curves were generated for each compound, including a curve for the control (agonist 52 with DMSO) and curves for the agonist 52.24 Compounds 12a and 12b were reported in Kim et al.20 and re-assayed here with a new protocol.
Figure 3.

Effect of 2-alkyl- and 2-cycloalkyl-substituted PAM derivatives (5–8, 13–23), bridged PAM derivatives (12a, 12b, 24–29), PAM derivatives bearing hydrophilic substituents (30–34), and p-phenylamino-substituted PAM derivatives (35–39) on hA3AR activation by 52 determined using [35S]GTPγS binding (n = 3). HEK-293 cell membranes expressing hA3AR were pretreated with 52 and modulators for 1 h, followed the addition of radiolabeled [35S]GTPγS to initiate the reaction, and incubated for an additional 2 h at room temperature. AR antagonists 8-[4-[4-(4-chlorophenzyl)piperazide-1-sulfonyl)phenyl]]-1-propylxanthine (62, PSB-603) and 4-[2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a]-[1,3,5]triazin-5-yl-amino]ethyl]phenol (63, ZM241385) were added (0.3 μM each final concentration) to block any endogenously expressed A2BARs. 1 unit/mL of adenosine deaminase (ADA) was added to degrade endogenous adenosine. The incubation was terminated by rapid filtration through a Whatman GF/B filter and membrane-bound radioactivity was measured by liquid scintillation spectrometry. Non-specific binding of [35S]GTPγS was measured in the presence of 10 μM of unlabeled GTPγS. 52 potency and maximal efficacy was measured using DMSO, 0.1 μM, 1.0 μM, and 10 μM of the modulator. Results were normalized to concentration–response curve Emax values obtained by 52 with DMSO and expressed as % activation.
The effect on [35S]GTPγS binding of each modulator (5–8 and 12–39) at concentrations of 0.1, 1.0, and 10 μM was determined (Tables 1 and S4, Figure 3). Compounds 7, 14, 18 and 20 were particularly efficacious and at least doubled the agonist Emax compared to control. Comparing the progression of effects of N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine derivatives at 1.0 μM, there was a trend to increase the Emax from the 2-cyclopropyl derivative 17 to the 2-cyclononyl derivative 20, except for 2-cyclo-octyl 19. While not significant, there was a 2-fold increase in agonist potency in the presence of 1 μM of 2-cyclononyl derivative 20 compared to the agonist alone (P-value = 0.3712, one-way ANOVA with multiple comparisons). As the ring size increased from 2-cyclodecyl 21 to 2-cyclododecyl 23, there was no change in agonist potency but a steady decrease in its Emax. The fluorinated compounds, 15 and 16, at 1 μM moderately improved agonist Emax but did not influence agonist potency.
Of the bridged derivatives, 12b and 26 at 1 μM enhanced the efficacy of agonist 52 to the greatest extent, having an Emax of 237 ± 12 and 219 ± 16 (P-value = 0.0002 and 0.0005, respectively). While neither 12b nor 26 significantly improved agonist potency, 12b at 1.0 μM tended to improve potency almost 4-fold from 46 nM to 10 nM (P-value = 0.058). Compounds 12a, 24, 27 and 28 moderately enhanced the [35S]GTPγS binding in the presence of agonist 52 compared to control. 2-(Bicyclo[2.2.1]heptan-1-yl) derivative 25 was the only bicyclic compound to induce a decrease in agonist potency at 1 μM (4-fold compared to the control, P-value = 0.0257). Compound 29 did not influence agonist potency and led to a moderate increase in agonist Emax (167 ± 12). All bicyclic compounds tended toward inverse agonist activity, and compound 29 at 10 μM decreased the basal efficacy the most, by ~100%, at the lowest agonist concentration.
The derivatives with hydrophilic substitutions, compounds 30–34, moderately allosterically enhanced [35S]GTPγS binding (Figure 3 and Table S4). However, these modulators at concentrations of 0.1 μM and 1.0 μM had no significant effect on agonist potency compared to the control. At 1.0 μM, only compounds 31, 32, and 34 increased agonist Emax compared to the control, ranging from 167% to 186% maximal efficacy of the agonist alone. Like compound 29, compound 34 behaved as an inverse agonist at 10 μM, decreasing basal efficacy at the lowest agonist concentration by ~50%. The decreased lipophilicity of 34 relative to 2-cyclononyl derivative 20 was predicted using Stardrop software45 with the following parameters for 34: Log D = 5.53, topological polar surface area (TPSA) = 73.8 Å2, and blood–brain barrier (BBB) log([brain]:[blood]) = −0.442, i.e., not BBB penetrant. The same parameters for 20 were Log D = 7.08, TPSA = 53.6 Å2, and BBB log([brain]:[blood]) ratio = −0.0326, i.e., BBB penetrant. Efficacious PAMs 14, 18 and 39 were also predicted to penetrate the BBB. The calculated parameters for all compounds tested pharmacologically are provided in Table S5.
Of the p-substituted phenylamino derivatives, compounds 35, 37, and 38 at 1.0 μM moderately increased agonist efficacy compared to control (Figure 3 and Table S4). At 1 μM, compound 36 doubled the agonist efficacy compared to control. None of the p-substituted phenylamino derivatives influenced agonist potency compared to control. Compound 39 combined the favorable features of compounds 14 and 35, based on their favorable modulation of dissociation kinetics and [35S]GTPγS binding. Compound 39 contains both the 2-heptan-4-yl substitution of 14 and the 4-iodophenyl substitution of 35. Compared to the control, compound 39 at 1 μM did not improve agonist potency but doubled agonist Emax.
The off-target screening was performed by the NIMH Psychoactive Drug Screening Program (PDSP) (Table 2).46 A total of ten derivatives were tested for off-target binding activity against 45 other receptors, transporters, and channels. Only a few weak off-target interactions were observed, the lowest Ki being for compound 39 at the translocator protein (TSPO) with Ki = 0.123 μM. The two 4-iodophenyl derivatives tested, compounds 35 and 39, were the only derivatives interacting with TSPO. Most derivatives interacted with one or two sigma receptors, specifically σ1 and σ2. Compound 39 had the lowest Ki observed, 0.891 μM, among the derivatives interacting with a sigma receptor. Compound 27 interacted with the most off-target sites: κ (KOR) and μ (MOR) opioid receptors, σ1, σ2, dopamine transporter (DAT), and serotonin 5HT2B receptors, interacting most strongly with DAT at Ki = 0.467 μM.
Table 2.
Off-Target Analysis of Selected PAM Derivatives at 45 Other Receptors, Transporters, and Channels, As Determined in Radioligand Binding Assays by the PDSPa
| derivative | protein | Ki (μM) |
|---|---|---|
| 14 | 5HT2B | 1.36 |
| σ 1 | 1.48 | |
| σ 2 | 1.36 | |
| 18 | σ 1 | 4.64 |
| σ 2 | 0.575 | |
| 20 | σ 1 | 5.15 |
| σ 2 | 0.907 | |
| 21 | σ 2 | 3.62 |
| 22 | D3 | 5.28 |
| 23 | D3 | 7.63 |
| σ 1 | 3.02 | |
| σ 2 | 2.84 | |
| 26 | σ 1 | 2.34 |
| σ 2 | 1.34 | |
| 27 | MOR | 7.22 |
| σ 1 | 2.42 | |
| σ 2 | 3.43 | |
| KOR | 5.50 | |
| DAT | 0.467 | |
| 5HT2B | 3.25 | |
| 35 | D3 | 0.846 |
| TSPO | 0.427 | |
| σ 2 | 3.01 | |
| 39 | 5HT2B | 2.60 |
| TSPO | 0.123 | |
| σ 2 | 0.891 |
These are the only hits with >50% inhibition in the primary screen at 10 μM.
Experimental ADMET Properties.
We chose 2-cyclohept-4-enyl 18 and 2-heptan-4-yl-N-4-iodophenyl 39 derivatives as relatively potent (estimated) and efficacious A3AR PAMs having only weak off-target interactions, to assess the pharmacological and ADMET properties in vivo and in vitro, as determined by reported methods.47 In fasted Wistar rats, the plasma concentration of 18 and 39 was determined following three different doses administered by oral gavage (p.o.) or one dose administered intravenously (i.v.). Multiple in vitro assays assessed drug metabolism and pharmacokinetics (DMPK): plasma stability, HepG2 cytotoxicity, human ether-à-go-go-related gene (hERG) potassium channel inhibition, cytochrome P450 (CYP) inhibition, microsomal stability, pION solubility, plasma protein binding, and chemical stability in simulated gastric and intestinal fluids.
In vitro and in vivo pharmacokinetic parameters of compounds 18 and 39 are shown in Table 3 and Tables S6 and S7. When dissolved in simulated intestinal and gastric fluids, 88.1% and 69.1% of compound 18 remained after 2 h, respectively, and 39 was more stable in gastric fluid. Out of the five CYP enzymes tested, the inhibition with CYP1A2 was the most concerning, with IC50 values of 6.99 (18) and 1.31 (39) μM. Both compounds had IC50 values above 30 μM for the other four CYP enzymes measured. The plasma stability was satisfactory, with compound 18 having 88.7% remaining in rat plasma after 120 min. The highest half-life of compound 18 in plasma was achieved in the mouse plasma at 695 min. Both compounds were relatively stable in liver microsomal assays, having the most degradation with rat microsomes, i.e., 52% of 18 remaining after 120 min. In the microsomal stability assays, compound 18 had a greater % remaining in all species than the reference compound, testosterone. In the hERG potassium ion channel inhibition assay, compounds 18 and 39 displayed IC50 values of 6.06 and >30 μM, respectively. Both compounds were not toxic to HepG2 liver cells, evidenced by an IC50 greater than 30 μM. In all three species, human, rat, and mouse, compound 18 was strongly bound to plasma protein, having % bound values of ~100, 99.1, and ~100, respectively. 39 was less water-soluble but had a much higher free fraction (93.6% bound) than 18 in human plasma. The measured concentration of compound 18 in the pION solution buffer (pH 7.4) was 0.39 μg/mL (0.92 μM), i.e., low aqueous solubility.
Table 3.
In Vitro and In Vivo Experimental Pharmacokinetic Parameters of Compounds 18 and 39
| test | reference compound(s) | compound 18 | compound 39 |
|---|---|---|---|
| simul. intestinal fluid % remaining at 120 min (t1/2, min) | Verapamil 100 (>240) | 100 (573) | 86.2 (533) |
| simul. gastric fluid % remaining at 120 min t1/2, min) | Omeprazole 0.0 (15.3) | 69.1 (204) | 99.7 (>240) |
| plasma stability % remaining at 120 min t1/2, min, 3 species) | 0.0 (0.11, h, procaine); 83.9 (414, r, diltiazem); 0.0 (0.11, m, procaine) |
69.7 (159, h); 88.7 (580, r); 83.5 (695, m) |
53.2 (140, h); ~ 100 (>240, r); ~ 100 (>240, m); |
| CYP1A2, IC50 (μM) | Miconazole, 4.55 | 6.99 | 1.31 |
| CYP2C9, IC50 (μM) | Miconazole, 0.38 | >30 | 32.3 |
| CYP2C19, IC50 (μM) | Miconazole, 0.00002 | >30 | 83.6 |
| CYP2D6, IC50 (μM) | Miconazole, 1.64 | >30 | 60.8 |
| CYP3A4 (IC50, μM) | Miconazole (0.0010) | >30 | 61.6 |
| microsomal stability % remaining at 120 min (t1/2, min, 3 species) | Testosterone 5.44 (15.6, h); 0 (1.43, r); 0 (4.33, m) |
81.2 (200, h); 52.1 (70, r); 76.4 (194, m) | 92.1 (>120, h); 86.3 (>120, r); 75.2 (>120, m); |
| plasma protein binding % bound (3 species) | Warfarin, 96.3 (h); 100 (r); 83.6 (m) | ~100 (h);99.1 (r);~100 (m) | 93.6 (h);100 (r); 100 (m) |
| hERG, IC50 (μM)a | n/a | 6.06 | >30b |
| HepG2 cell toxicity, IC50 (μM) | Verapamil, 65.2 | >30 | 35.4 |
| aqueous solubility pH 7.4 (μg/mL) | n/a | 0.39 | <0.01 |
| oral bioavailability, t1/2 (h) (3 mg/kg, p.o.) | n/a | 28.7%F, 1.29 | 64.0%F, 3.44 |
Fluorescence polarization hERG assay.
27% inhibition at 30 μM.
Caco-2 permeability assay assessed intestinal permeability of compounds 18 and 39 (Table S7). Compound 18 had no measurable permeability (Papp) for either apical to basolateral (A to B) and basolateral to apical (B to A) and was therefore classified as having low permeability. An efflux ratio was not calculated due to low Papp in A to B and B to A. B to A had a relatively large % recovery of 78.0%, which was substantially more than the 48.1% seen for the A to B % recovery.
A comparison of the in vivo pharmacokinetics of 18 and 39 (Figure 4, Table 3, Table S8), indicated that 39 had considerably longer in vivo half-life and improved oral bioavailability (3.44 h, 64.0%F at 3 mg/kg; 3.84 h, 61.5%F at 10 mg/kg) than 18, indicating substantial oral bioavailability. The longest half-life of 2.60 h of 18 was with an oral dose of 10 mg/kg, similar to the half-life of 2.38 h seen with the i.v. dose of 0.5 mg/kg 18. 28.7%F and 47.5%F were determined for compound 18. The source of the discrepancy between the low passive drug absorption predicted using Caco-2 cells48 and the moderate oral bioavailability remains unexplored.
Figure 4.

Mean plasma concentrations of compound 18 (A) and compound 39 (B) in Wistar rats vs multiple time points. The dose and administration route are specified above for each of the four groups (three rats in each group).
An oral dose of 10 mg/kg of compound 18 produced higher plasma concentrations (Cmax at 2 h of 1780 ng/mL, equal to 4.2 μM) compared to the other groups during each time point measured. All groups of rats administered compound 18 orally, i.e., at 1, 3, and 10 mg/kg, achieved the maximum plasma concentration at ~2 h. However, other parameters were similar for 18 and 39. Rats given 3 mg/kg had almost double the clearance rate (1580 mL/(h·kg)) of compound 18 from the plasma compared to 10 mg/kg (975 mL/(h·kg)) and roughly three times the clearance rate compared to 1 mg/kg (447 mL/(h·kg)) (Table S8). A 3 mg/kg dose had double the elimination rate constant, kel, compared to other p.o. doses, having ~50% of the remaining compound in the body excreted every h. A 3 mg/kg dose produced the longest mean residence time (MRT, length of time a compound remains in the system before elimination) of 3.98 h.
Molecular Modeling.
In general a mixture of positive and negative modulatory hA3AR effects was observed for this class of PAMs, with the negative modulation likely due to competitive antagonism at the orthosteric site.24
As reported in Fisher et al.,24 the 2-cyclopropyl analogue 17 was proposed to function predominantly as a competitive antagonist of the orthosteric site of the hA3AR, because it retained the ability to right-shift the concentration–response curve of agonist 52 in [35S]GTPγS binding assays with a chimeric mousein/humanout A3AR that is non-responsive to the efficacy-enhancing effect of the imidazoquinolinamine PAMs. Chimeric receptor studies proved that compound 7 interacts with both the allosteric and the orthosteric site. The binding affinity (KB) of 17 for the orthosteric site of the chimeric receptor was estimated by Schild analysis to be 140 nM (Figure S1B). The affinity of 7, which also reduces potency of Cl-IB-MECA in [35S]GTPγS binding assays at the mousein/humanout A3AR, was estimated to be 799 nM at this chimeric receptor (Figure S1A). This conclusion was further supported in equilibrium binding studies with the antagonist radio-iodinated ligand [125I]2-(4-(3-(4-amino-3-iodobenzyl)-2,6-dioxo-1-propyl-2,3,6,7-tetrahydro-1H-purin-8-yl)phenoxy)-acetic acid ([125I]I-ABOPX), where 7 and 17 were found to displace specific binding to the WT hA3AR (Figure S2). Compounds 7 and 17 were roughly equipotent in inhibiting orthosteric radioligand binding (Ki ≈ 2.0 μM), whereas compound 14 was slightly less potent (~5.40 μM). Compound 17 did not inhibit orthosteric antagonist binding to the hA1, hA2A, or hA2B ARs at concentrations as high as 10 μM (Supporting Information).
Molecular modeling studies to rationalize the PAM activity and predict the allosteric binding site(s) are still ongoing and not part of this study, because further pharmacological characterization is needed. A PAM-bound hA1AR experimental structure (PDB ID: 7LD3) is available,6 with 2-amino-4-(3,5-bis(trifluoromethyl)phenyl)thiophen-3-yl)(4-chlorophenyl)-methanone (MIPS521, an hA1AR PAM) bound to an intramembrane region at the interface among TM1, TM6, and TM7. Homology modeling of hA3AR using structure 7LD36 as template (46% hA1AR and hA3AR sequence identity, computed with GPCRdb),66 followed by docking of compound 7 to a binding pocket equivalent to the site of MIPS521 in the A1AR, did not provide any reasonable poses (data not reported). This is possibly due to a more hindered pocket in hA3AR, where the bulkier A2737.44 and M2767.47 replace the non-conserved G2797.44 and A2827.47 of hA1AR (alignment reported in Figure S3). This, together with ongoing pharmacological experiments,24 suggests that hA1AR and hA3AR allosteric sites are distinct, at least for MIPS521 at hA1AR and the series of compounds of this manuscript at hA3AR.
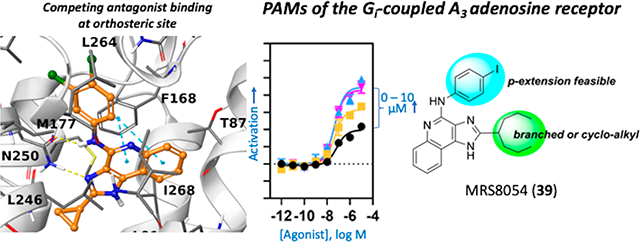
Conversely, we could model the ability of 17 to be accommodated in hA3AR orthosteric site, which is well known for ARs. Given the lack of an experimental hA3AR structure, we employed a previously reported homology model built on the experimental structure of an antagonist-bound hA1AR X-ray structure.49 The compound was docked to the orthosteric site through an induced fit docking (IFD) procedure50 and refined by minimization through molecular mechanics with a generalized Born and surface area solvation (MM-GBSA).51 The selected hA3AR pose of compound 17 (Figure 5) was compared to other known A3AR ligands. In the suggested binding mode, the compound’s 1H-imidazo[4,5-c]pyridin-4-amine moiety mimics the adenine scaffold of adenosine-like agonists47 and the [1,2,4]triazolo[1,5-c]-pyrimidin-5-amine scaffold of the known antagonist N-(9-chloro-2-(furan-2-yl)-[1,2,4]triazolo[1,5-c]quinazolin-5-yl)-2-phenylacetamide (64, MRS1220) in its previously predicted pose.49 A conserved π–π stacking interaction is observed between the aromatic scaffold of 17, similar to 64, and F168EL2, while nitrogen N3 and the exocyclic amino group at position 4 are engaged in a bidentate H-bond with N2506.55. The 2-cyclopropyl moiety is located deep into the orthosteric binding pocket, surrounded by T943.36, M1775.38, S1815.42, I1865.47, W2436.48, L2466.51, and N2506.55 (residues located within 3 Å), while the 3,4-dichlorophenyl group points toward the extracellular portion of the receptor, surrounded by the tip of TM6, TM7, EL2, and EL3, and specifically by residues Q167EL2, F168 EL2, I2496.54, N2506.55, I2536.58, V259EL3, L2647.35, and I2687.39.
Figure 5.

Predicted favored pose of compound 17 (orange) at hA3AR orthosteric binding site (gray). The pose was generated though IFD followed by MM-GBSA minimization.
Considering that compound 7 shares with 17 the ability to inhibit orthosteric antagonist radioligand binding, we assessed its ability to fit the orthosteric binding pocket. We applied induced fit docking and MM-GBSA minimization and selected by visual inspection a possible orthosteric binding mode. Poses with the dichlorophenyl group anti respect to atom C5 (Figure S4B) were discarded as they are less favored and require overcoming a high energy barrier, as suggested by a dihedral scan performed with the semiempirical quantum mechanical methods GFNn-xTB (Figure S5). Compound 7 was thus predicted to bind the hA3AR orthosteric binding pocket similarly to compound 17 (Figure S4A), which is in agreement with the experimentally demonstrated antagonist effect of the two compounds. This suggest that the two compounds’ differential ability to enhance agonist activity (present for 7 and absent for 17) is not due to a major antagonist character of 17 as compared to 7, since their antagonist behavior is similar, but to a major PAM effect of compound 7.
This study extended previous SAR studies of hA3AR PAMs having an imidazoquinolinamine scaffold.14,52 A ligand structure-guided design approach was pursued given that structural information for the A3AR is not yet available to enable a target-guided structural approach for PAMs. Although modeling predictions can be made for ligand recognition at the hA3AR orthosteric binding site by close similarity to the hA1AR and hA2AAR,53 prediction of the precise allosteric ligand binding site is uncertain.
This class of PAMs is selective for the A3AR with respect to allosteric modulation at other AR subtypes.18–20,22 However, a mixture of positive and negative modulatory A3AR effects is generally observed in this family. The relatively low competitive binding of 4 and 719,20 and the lack of allosteric enhancement by 4 and 717,54 at other AR subtypes were reported. Based on chimeric receptors, we now know that the site of PAM action is on the cytosolic side of the receptor, while the negative effects in this series appear to arise from the canonical orthosteric site binding.24 The A3AR orthosteric antagonism by 2-cyclopropyl derivative 17 and prototypical A3AR PAM 7 was quantified here using functional data from the mousein/humanout chimeric A3AR and radioligand binding at the WT receptor,22 and their orthosteric binding was modeled. The ability of these derivatives and compound 14 to bind to varying degrees to the orthosteric site is also consistent with the fact that this 1H-imidazo[4,5-c]quinolin-4-amine scaffold was initially characterized as potent A1AR antagonists.55 Curiously, the topical anti-tumor agent imiquimod, which contains the same scaffold, was reported to antagonize ARs with μM affinity.56
Like past studies, this series of derivatives showed variable allosteric modulatory effects, with positive effects on [125I]50 dissociation, potency and/or maximal efficacy of 52 in functional assays. Although PAM effects were demonstrated here with synthetic agonists [125I]50 and 52, the enhancing effects apply similarly to endogenous adenosine, as 7 increased the maximal efficacy of adenosine in [35S]GTPγS binding.24
Prior 2-(n-pentyl) substitution (9) of the 1H-imidazo[4,5-c]quinolin-4-amine scaffold with the 3,4-dichlorophenylamino group at the 4 position did not significantly enhance A3AR activity.19 However, another acyclic analogue, 2-heptan-4-yl derivative 14, originating from the synthetic precursor valproic acid, at 1 μM, enhanced agonist Emax compared to control (Emax = 216 ± 12%, P-value < 0.0001). This 2-fold increase in Emax of 52 at the receptor was comparable to the enhancement by 2-cyclohexyl derivative 7. Yet, it did not increase agonist potency compared to the control (P-value = 0.2041). Thus, for the first time we demonstrated that acyclic alkyl substitutions at the 2 position provides pharmacological advantages.
Other PAMs that significantly enhanced agonist efficacy at 0.1 μM were 7, 8, 12b, 16, 18, 20, 27, 37 and 39 (Table S4). When evaluating other cycloalkyl derivatives compared to compound 7, compounds 18 (2-cyclohept-4-enyl), and 20 (2-cyclononyl) stand out. Both potentiated the maximal efficacy of agonist 52 similarly to compound 7 (225 ± 10%) at 1 μM, having Emax values of 241 ± 9% and 242 ± 9%, respectively. Compounds 18 and 20 also showed PAM characteristics by slowing the rate of agonist radioligand dissociation from the receptor.
Use of bridged alicyclic rings at the 2 position for steric constraint failed to identify a hypothetical receptor-preferred conformation. All bridged modulators at 1 μM increased agonist efficacy, with an approximately 2-fold increase by compounds 12b, 24 and 28. The tricyclic 2-(adamantan-1-yl) substitution enhanced agonist Emax and trended toward increasing potency (P-value = 0.058). Thus, none of the bicyclic derivatives improved agonist potency compared to control in the [35S]GTPγS assay, but uniformly acted as PAMs by slowing the radioligand dissociation and improving the maximal efficacy of the agonist. The binding of bridged derivatives at both the orthosteric and allosteric binding sites is the most likely explanation for lack of substantial improvement in agonist potency. Another potential explanation is that the activation and binding cooperativities of these compounds are separate parameters controlling both agonist Emax and potency changes. Interestingly, the effect on agonist EC50 of bicyclic alkene 29 was similar to competitive antagonist 17, significantly decreasing the potency in a concentration-dependent manner.
We installed different hydrophilic groups on compound 18 to potentially improve PAM water solubility while maintaining enhancement of the agonist dissociation and functional effects at the A3AR. These derivatives with various oxygen substitutions (i.e., oxirane, alcohol, and carbonyl groups) did not influence agonist potency, but had similar improvements in agonist efficacy compared to the bicyclic library of derivatives. Thus, they act as PAMs,57 but with limited cooperativity due to their mediocre slowing of radioligand dissociation, and they reduced radioligand binding at equilibrium, suggestive of orthosteric site binding.58 Although efficacy enhancement compared to control was moderate, these derivatives were the first example of A3AR PAM activity with any hydrophilic substitution, in light of the failure of previously tested polar heterocyclic derivatives.20
In prior studies, certain halogenated and 4-substituted phenylamino derivatives were tolerated with respect to A3AR PAM effects.20 Overall, all p-phenylamino substitutions were tolerated here, but none having exceptional binding or functional effects compared to compound 7. At 1 μM concentration, potency remained unchanged and Emax doubled. Compounds 35 and 36, with the 4-iodo- and 4-bromophenylamino substitutions, respectively, had a similar non-optimal influence on potency and efficacy as compounds 37 and 38; though, they both considerably slowed the radioligand dissociation (58% and 56% remaining, respectively) compared to control (P-values = 0.010 and 0.015, respectively). Although compounds 35 and 36 slowed radioligand dissociation, they both considerably decreased the radioligand binding at equilibrium by 31% and 49% compared to the vehicle (P-values = 0.040 and 0.036, respectively). Thus, 35 and 36 bound to both orthosteric and allosteric sites, with greater inhibition of radioligand binding compared to other PAMs. Important to note is the substantial increase in agonist Emax at 1 μM between compounds 35 and 39, (184 ± 9% and 223 ± 10%, P-value = 0.044), correlating with the 2-heptan-4-yl substitution of 39, which was also evident in improved equilibrium radioligand binding.
Interestingly, compound 37, having a methyl acrylate p-phenylamino substitution, slowed radioligand dissociation and had favorable equilibrium binding results, with low % competitive inhibition of radioligand binding at the orthosteric binding site. These are promising results because 37 has more hydrophilic character (predicted Log D = 5.12, TPSA = 79.7 Å2, and BBB log([brain]:[blood]) = −0.450, i.e., not BBB penetrant) than the rest of the library due to the 4-aminophenyl substitution. Of the compounds with hydrophilic substitutions, compound 37 most represents a PAM. A hydrophilic p-phenylamino substitution might achieve a hydrophilic PAM, suggested by the differences in SAR between 37 and the derivatives with hydrophilic substitutions at the 2 position. Altogether, it would be worth investigating the effects of other polar p-phenylamino substitutions.
Compounds 18 and 39 proved to be orally bioavailable, despite having low A-B and B-A permeability in Caco-2 cells. Neither was strongly depleted by simulated digestive fluid, while 39 and to a lesser extent 18 inhibited CYP1A2. 18 displayed extremely high plasma protein binding in all three species tested, attributed to its hydrophobic nature. It is conceivable that the high plasma protein binding could provide a reservoir of the PAM in the body for prolonged activity, as it would release gradually from a bound to unbound state. On the other hand, this could limit the amount of compound available to traverse to the cellular environment for event- and site-specific action.
CONCLUSIONS
This study aimed to further explore the SAR of A3AR PAMs and to determine the suitability of the new analogues for translational development. We achieved our objectives of creating a shorter 6-step synthetic route for 1H-imidazo[4,5-c]quinolin-4-amine PAM derivatives, preparing a new series of 1H-imidazo-[4,5-c]quinolin-4-amine PAM derivatives, determining the SAR of the series of derivatives, and obtaining the first reported baseline ADMET of this structural family.
Although we did not discover a PAM derivative that induced a greater agonist efficacy-enhancement than compound 7, we did reveal a trend toward higher efficacy with certain 2- and 4-amino substitutions, including 12, 14, 18, 20, 30, and 39. The heptan-4-yl 2 position substitution slowed agonist dissociation from the A3AR the most, with both 3,4-dichlorophenyl and 4-iodophenyl substitutions at the 4-amino position. In addition to the 2-heptan-4-yl substitution, the 2-cyclohept-4-enyl and 2-cyclononyl substitutions improved agonist efficacy comparable to the reference 2-cyclohexyl. Other 2 position substitutions such as bicyclic and hydrophilic groups did not significantly enhance PAM effects, although these hydrophilic substitutions were more successful than previous attempts to introduce polar groups.
The human/mouse chimeric A3AR data showed the dependence on the 2 position substitution of orthosteric site affinity, which is spatially distinct from the allosteric binding site located on the cytosolic half of the receptor. Thus, fine-tuning of this family of PAMs is achievable, by separating distinct orthosteric (undesired) and allosteric binding effects. These separate activities could be further probed using the chimeric A3ARs as screening tools.
Developing a radioligand specific for the 1H-imidazo[4,5-c]quinolin-4-amine PAM binding site would significantly aid the pharmacological characterization of this PAM family. We prepared a precursor for introducing a 125I label and will perform future labeling studies.
Thus, we have developed a series of 1H-imidazo[4,5-c]quinolin-4-amine modulators with promising characteristics. Selected compounds will be utilized in future preclinical studies once an animal disease model is identified, furthering the allosteric approach to developing drugs to activate the A3AR that are event- and site-specific in action.
EXPERIMENTAL METHODS
Reagents and Instrumentation.
All glassware and stir bars were oven-dried before use in a reaction. All reactions were conducted in a ventilated hood. Pyrophoric reagents were handled and administered under a nitrogen gas atmosphere to reactions in an evacuated oven-dried glass round-bottom flask. Room temperature (rt) refers to 25 ± 5 °C. All final compounds were stored at 4 °C in a parafilm-sealed vial. Unless otherwise stated, all the reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). Unless noted, the reagents and solvents were of reagent grade and used without further purification. The following suppliers supplied the following commercial compounds: TCFH was purchased from TCI (Portland, OR), bicyclo[3.3.1]nonane-1-carboxylic acid was purchased from Chemspace US (Monmouth Junction, NJ); bicyclo[3.3.1]nonane-3-carboxylic acid was purchased from AstaTech (Bristol, PA); cyclohept-4-ene carboxylic acid was purchased from Ambeed (Arlington Heights, IL); bicyclo[2.2.1]heptane-1-carboxylic acid was purchased from Enamine (Kyiv, Ukraine). All reagents used were of commercial grade.
NMR spectra were recorded on a Bruker 400, 500, or 600 MHz spectrometer at 25 °C under an optimized parameter setting for each sample (Supporting Information). For compounds 14, 17, 18–22, 24–39, 48, and 49 1H NMR chemical shifts were measured relative to the residual solvent peak of 7.26 ppm for CDCl3 or CDCl3/CD3OD. For compounds 15 and 23, 1H NMR chemical shifts were measured to the residual solvent peak of 2.5 ppm for DMSO-d6. For compound 13, 1H NMR chemical shifts were measured to the residual solvent peak of 3.34 ppm for CD3OD. For compounds 16, 1H NMR chemical shifts were measured to the residual solvent peak of 1.94 for CD3CN. 1H NMR chemical shifts were measured relative to tetramethylsilane at 0.00 ppm in CDCl3 and the residual water peak at 3.30 ppm in CD3OD. 19F NMR spectra were recorded for derivatives 15, 16, and 20–23.
Analytical TLC was performed on 0.2 mm silica-coated sheets with an F254 indicator (Sigma-Aldrich). TLC visualization of the products was aided using UV light or by staining with a solution of potassium permanganate (1.5 g of KMnO4, 10 g K2CO3, and 1.25 mL 10% NaOH in 200 mL water). Column chromatography was performed on 230–400 mesh silica gel (pore size of 60 Å, Sigma-Aldrich).
Accurate mass data were obtained using a Xevo G2-XS QTof mass spectrometer (Supporting Information). The instrument was operated in positive-ion ESI mode at a resolution of 25,000. The ESI capillary voltage was 2.8 kV and the desolvation temperature was 280 °C. Accurate masses were determined using trifluoroacetic acid (TFA) sodium salt as an internal standard. An Acquity I-class Ultra Performance Liquid Chromatography (Waters, Milford, MA) was the liquid chromatography system. Solvent A was 100% water and solvent B was an 80:20 mixture of ACN:MeOH with 0.1% TFA and 0.2% formic acid added. The column was a ProSwift RP-4H 1 × 50 mm monolithic (ThermoFisher, Waltham, MA). The LC gradient was 0% B to 100% B in 10 min at a flow rate of 0.250 mL per min. Accurate mass high-resolution LC/ESI/MS at the Mass Spectrometry Facility, NIDDK, NIH.
HPLC analysis was carried out with Agilent 1100 Series HPLC equipped with Agilent Eclipse 5 μm XDB-C18 analytical column (250 × 4.6 mm; Agilent Technologies Inc., Palo Alto, CA, USA). Mobile phase: linear-gradient solvent system, 10 mM TEAA (triethylammonium acetate): CH3CN from 50:50 to 0:100 in 20 min, and then 100% CH3CN for 5 min; the flow rate was 1.0 mL min, unless noted. The RP-HPLC was performed using a Phenomenex Luna 5 μm C18(2)100A, AXIA, 21.2 mm × 250 mm column. The peaks were detected by UV absorption with a diode array detector at 210, 230, 254, and 280 nm. All derivatives tested for biological activity showed ≥95% purity in the HPLC systems, unless noted.
General Procedure for the Synthesis of Compounds 13–22.
Procedure C.
An oven-dried 10 mL round-bottom flask (cooled to rt under nitrogen) equipped with a stir bar was charged with the appropriate 2-substituted 4-chloro-1H-imidazo[4,5-c]quinoline derivative (46a–n) (1.0 equiv), 3,4-dichloroaniline (1.5 equiv), Pd2(dba)3 (5–10 mol%), tBuXPhos (20 mol%), and sodium butoxide (1.5–2.0 equiv). The flask was evacuated for 2 min and backfilled with N2 (gas). Dioxane (~1 mL) was added via syringe, and the reaction mixture was flushed/purged with nitrogen for 5 min while stirring. The reaction mixture was stirred at preheated oil bath (95–100 °C) under nitrogen for 16–24 h. Subsequently, the reaction mixture was cooled to rt and diluted with EtOAc (5 mL). The solution was then filtered through a silica plug. The filtrate was concentrated by rotary evaporation to obtain the crude product, purified by silica column with 10–25% ethyl acetate in hexane as the eluent system to obtain the product. Few products were further purified using reverse-phase high-pressure liquid chromatography.
2-Propyl-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (13).
Compound 13 was synthesized following the general procedure C described above using 46a (15.0 mg, 0.061 mmol), Pd2(dba)3 (5 mol%) and sodium t-butoxide (1.5 equiv). The crude was purified by silica gel column (10–20% ethyl acetate/hexane) to afford 6.0 mg (23%) of a white solid (HPLC tR = 14.9 min): 1H NMR (400 MHz, methanol-d4) δ 8.60–8.55 (m, 1H), 8.00 (d, J = 8.1 Hz, 1H), 7.84 (dd, J = 19.6, 8.8 Hz, 2H), 7.55 (t, J = 7.9 Hz, 1H), 7.48–7.30 (m, 2H), 2.99 (t, J = 7.5 Hz, 2H), 1.96 (h, J = 7.4 Hz, 2H), 1.09 (t, J = 7.4 Hz, 3H); HRMS calcd for C19H17N4Cl2 (M+H)+ 371.0830, found 371.0827.
2-(Heptan-4-yl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (14).
Compound 14 was synthesized following the general procedure C described above using 46b (26.0 mg, 0.086 mmol), Pd2(dba)3 (5 mol%) and sodium t-butoxide (1.5 equiv). The crude was purified by silica gel column (10–20% ethyl acetate/hexane) to afford 4.2 mg (11%) of a white solid (HPLC tR = 19.7 min): 1H NMR (400 MHz, chloroform-d) δ 8.50 (d, J = 2.5 Hz, 1H), 8.01 (d, J = 8.4 Hz, 1H), 7.87–7.77 (m, 3H), 7.56 (t, J = 7.8 Hz, 1H), 7.44–7.30 (m, 2H), 3.03 (p, J = 7.3 Hz, 1H), 1.81 (td, J = 8.9, 8.3, 4.4 Hz, 4H), 1.43–1.26 (m, 4H), 0.92 (t, J = 7.3 Hz, 6H); HRMS calcd for C13H25N4Cl2 (M+H)+ 427.1456, found 427.1462.
2-(1,1,1,7,7,7-Hexafluoroheptan-4-yl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (15).
Compound 15 was synthesized following the general procedure C described above using 46c (4.8 mg, 0.012 mmol), Pd2(dba)3 (10 mol%) and sodium t-butoxide (2.0 equiv) gave 15 (3.2 mg, 50%) as a white solid. The crude was purified by silica gel column (10–20% ethyl acetate/hexane) followed by semipreparative HPLC (RP-HPLC linear gradient solvent system: ACN:H2O from 80:20 to 100:0 in 40 min, at a rate of 5 mL/min. tR = 29 min) to afford 3.2 mg (50%) of a white solid (HPLC purity: 91%; tR = 18.1 min): 19F NMR (DMSO-d6) δ −64.75; 1H NMR (400 MHz, DMSO-d6) δ 8.27–8.09 (m, 1H), 8.09–7.94 (m, 1H), 7.85 (s, 1H) 7.62 (d, J = 11.5 Hz, 2H), 7.49 (s, 2H), 2.34 (d, J = 14.0, 2H), 2.10 (d, J = 7.5 Hz, 6H); HRMS calcd for C23H19N4F6Cl2 (M+H)+ 535.0891, found 535.0901.
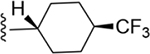
2-(4-(Trifluoromethyl)cyclohexyl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (16).
Compound 16 was synthesized following the general procedure C described above using 46d (14.0 mg, 0.040 mmol), Pd2(dba)3 (5 mol%) and sodium t-butoxide (2.0 equiv). The crude was purified by silica gel column (10–20% ethyl acetate/hexane) to afford 4.0 mg (21%) of a white solid (HPLC tR = 18.2 min): 19F NMR (acetonitrile-d3) δ −74.30; 1H NMR (400 MHz, acetonitrile-d3) δ 8.65 (d, J = 2.5 Hz, 1H), 8.14 (s, 1H), 8.01 (dd, J = 8.8, 2.6 Hz, 1H), 7.88 (d, J = 8.3 Hz, 1H), 7.55 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H), 7.50–7.38 (m, 2H), 3.05–2.94 (m, 1H), 2.31 (d, J = 12.9 Hz, 2H), 2.13 (d, J = 22.1 Hz, 4H), 1.80–1.71 (m, 1H), 1.61–1.49 (m, 2H); HRMS calcd for C23H20N4F3Cl2 (M +H)+ 479.1017, found 479.1019.
2-(Cyclopropyl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (17).
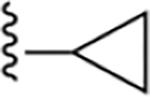
Compound 17 was synthesized following the general procedure C described above using 46e (11.0 mg, 0.045 mmol), Pd2(dba)3 (10 mol%) and sodium t-butoxide (2.0 equiv). The crude was purified by silica gel column (10–20% ethyl acetate/hexane) to afford 3.0 mg (18%) of a white solid (HPLC tR = 14.4 min): 1H NMR (400 MHz, chloroform-d) δ 8.48 (s, 1H), 7.91 (d, J = 8.3 Hz, 1H), 7.86–7.71 (m, 2H), 7.48 (t, J = 7.6 Hz, 1H), 7.39–7.27 (m, 2H), 2.12 (p, J = 6.7 Hz, 1H), 1.14 (d, J = 6.5 Hz, 4H); HRMS calcd for C19H15N4Cl2 (M+H)+ 369.0674, found 369.0676.
2-(Cyclohept-4-en-1-yl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (18).
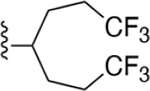
Compound 18 was synthesized following the general procedure C described above using 46j (9.0 mg, 0.030 mmol), Pd2(dba)3 (10 mol%) and sodium t-butoxide (2.0 equiv). The crude was purified by silica gel column (10–20% ethyl acetate/hexane) to afford 3.0 mg (23%) of a white solid (HPLC tR = 18.4 min): 1H NMR (400 MHz, chloroform-d) δ 8.49 (s, 1H), 8.00 (d, J = 8.3 Hz, 1H), 7.79 (d, J = 8.4 Hz, 2H), 7.60–7.52 (m, 1H), 7.43–7.34 (m, 2H), 5.91 (dd, J = 4.9, 2.4 Hz, 2H), 3.27 (dq, J = 6.8, 3.3 Hz, 1H), 2.43 (d, J = 15.8 Hz, 2H), 2.37–2.20 (m, 4H), 1.85 (dt, J = 13.5, 10.9 Hz, 2H); HRMS calcd for C23H21N4Cl2 (M +H)+ 423.1143, found 423.1137.
2-(Cyclooctyl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (19).
Compound 19 was synthesized following the general procedure C described above using 46k (40.0 mg, 0.13 mmol), Pd2(dba)3 (10 mol%) and sodium t-butoxide (2.0 equiv). The crude was purified by silica gel column (10–20% ethyl acetate/hexane) to afford 15 mg (27%) of a white solid (HPLC tR = 20.5 min): 1H NMR (400 MHz, chloroform-d) δ 8.43 (s, 1H), 8.04–7.65 (m, 3H), 7.61–7.47 (m, 1H), 7.46–7.29 (m, 2H), 3.27 (t, J = 9.6 Hz, 1H), 2.21–2.10 (m, 2H), 2.09–1.95 (m, 2H), 1.84 (s, 2H), 1.67 (s, 8H); HRMS calcd for C24H25N4Cl2 (M+H)+ 439.1456, found 439.1452.
2-(Cyclononyl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (20).
Compound 20 was synthesized following the general procedure C described above using 46l (6.0 mg, 0.018 mmol), Pd2(dba)3 (10 mol%) and sodium t-butoxide (2.0 equiv). The product was obtained as a white solid (TFA salt) after lyophilization (2.0 mg, 5%; RP-HPLC, linear gradient solvent system: ACN:0.1% aq. TFA from 70:30 to 100:0 in 40 min, tR = 21.7 min): 1H NMR (400 MHz, chloroform-d) δ 8.01 (s, 1H), 7.68 (d, J = 8.3 Hz, 1H), 7.60–7.54 (m, 1H), 7.48 (s, 1H), 7.36 (s, 1H), 7.29 (dd, J = 8.5, 2.5 Hz, 2H), 3.29 (s, 1H), 1.97 (d, J = 4.6 Hz, 5H), 1.77–1.53 (m, 9H), 1.26 (s, 2H); HRMS calcd for C25H27N4Cl2 (M+H)+ 453.1613, found 453.1616.
2-(Cyclodecyl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (21).
Compound 21 was synthesized following the general procedure C described above using 46m (13.0 mg, 0.038 mmol), Pd2(dba)3 (10 mol%) and sodium t-butoxide (2.0 equiv). The product was obtained as a white solid (TFA salt) after lyophilization (2.8 mg, 16%; RP-HPLC, linear gradient solvent system: ACN:0.1% aq. TFA from 70:30 to 100:0 in 40 min, at a rate of 5 mL/min; HPLC purity: 77%; tR = 23.8 min)): 1H NMR (400 MHz, chloroform-d) δ 8.44 (s, 1H), 7.89 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 8.3 Hz, 1H), 7.47 (t, J = 7.8 Hz, 1H), 7.37–7.27 (m, 2H), 3.53 (s, 1H), 2.05 (t, J = 10.1 Hz, 1H), 1.94 (s, 2H), 1.59 (s, 8H), 0.81 (s, 4H), 0.71–0.57 (m, 3H); HRMS calcd for C26H29N4Cl2 (M+H)+ 467.1769, found 467.1770.
2-(Cycloundecyl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (22).
Compound 22 was synthesized following the general procedure C described above using 46n (14.0 mg, 0.039 mmol), Pd2(dba)3 (10 mol%) and sodium t-butoxide (2.0 equiv). The product was obtained as a white solid (TFA salt) after lyophilization (2.82 mg, 15%; RP-HPLC, linear gradient solvent system: ACN:0.1% aq. TFA from 70:30 to 100:0 in 40 min, at a rate of 5 mL/min; HPLC purity: 80%; tR = 22.7 min): 1H NMR (400 MHz, chloroform-d) δ 8.10 (s, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 8.4 Hz, 1H), 7.60–7.42 (m, 2H), 7.36 (d, J = 9.0 Hz, 2H), 3.19 (s, 1H), 1.88 (td, J = 13.1, 12.7, 6.8 Hz, 2H), 1.41 (d, J = 11.0 Hz, 6H), 1.27–1.14 (m, 10H), 0.78 (d, J = 7.0 Hz, 2H); HRMS calcd for C27H31N4Cl2 (M+H)+ 481.1926, found 481.1921.
General Procedure for the Synthesis of Compounds 23, 29, 35, 36, and 39.
Procedure E.
The appropriate 4-chloro-2-substituted 1H-imidazo[4,5-c]quinoline (46b,h,o,t) starting material (0.05 mmol, 1 equiv) and the appropriate halogenated aniline compound (47a–c) (0.15–0.25 mmol, 3–5 equiv) were added to 1 mL of ethanol in a 2 to 5 mL microwaveable vial. The reaction contents were degassed with N2(g) for 15 min, and the reaction was set up in an Initiator microwave reactor (Biotage, Charlotte, NC) at 130 °C for 6 h. The reaction mixture was filtered through a silica plug. The filtrate was evaporated in vacuo, and the product was purified by flash chromatography with a 15% ethyl acetate in hexane eluent system.
2-(Cyclododecyl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (23).
Compound 23 was synthesized following the general procedure E described above using 46o (6.0 mg, 0.016 mmol) and 47a (13.0 mg, 0.08 mmol, 5.0 equiv). The product was obtained as a white solid (TFA salt) after lyophilization (2.0 mg, 25%; RP-HPLC acetonitrile/0.1% aq. TFA, 70/30 in 40 min at a rate of 5 mL/min, HPLC tR = 21.6 min): 1H NMR (400 MHz, DMSO-d6) δ 8.22 (d, J = 7.4 Hz, 1H), 7.87 (s, 1H), 7.61 (s, 2H), 7.50 (s, 2H), 2.97 (s, 1H), 1.98 (s, 3H), 1.83 (s, 3H), 1.43 (d, J = 32.5 Hz, 17H), 1.27 (s, 3H); HRMS calcd for C28H33N4Cl2 (M+H)+ 495.2082, found 495.2085.
General Procedure for the Synthesis of Compounds 24–27.
Procedure D.
To a 10 mL round-bottom flask (cooled to rt under N2(g)) equipped with a stir bar were added Pd(OAc)2 (1 mol%), tBuXPhos (3 mol%), and H2O (40 mol %) and dissolved in 1.0 mL of 1,4-dioxane. The solution was degassed with nitrogen for 15 min and allowed to stir at 80 °C for 5 min. The appropriate 2-substituted 4-chloro-1H-imidazo-[4,5-c]quinoline derivative (46p–s) (1.0 equiv), 3,4-dichloroaniline (1.2 equiv), and sodium butoxide (2.0 equiv) were dissolved in 3 mL of dry 1,4-dioxane in a separate 25 mL round-bottom flask. The reaction mixture in the 25 mL round-bottom flask was degassed at rt with nitrogen for 15 min. The activated catalyst from the 10 mL flask was cooled and then transferred to the 25 mL reaction mixture using a cannula. The 25 mL flask was slightly immersed in a 100 °C oil bath, and the reaction continued for 16–20 h. The reaction mixture was diluted with ethyl acetate (5 mL) and filtered through a short silica plug. A rotary evaporator concentrated the filtrate to obtain a residue, purified by silica column chromatography with 10% ethyl acetate in hexane as the eluent system to afford the product.
2-(Bicyclo[1.1.1]heptan-1-yl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (24).
Compound 24 was synthesized following the general procedure D described above using 46p (47.0 mg, 0.17 mmol). The crude was purified by silica gel column (10–20% ethyl acetate/hexane) to afford 9.0 mg (13%) of a white solid (HPLC tR = 16.1 min): 1H NMR (400 MHz, chloroform-d) δ 8.47 (s, 1H), 8.00 (s, 1H), 7.80 (d, J = 24.6 Hz, 2H), 7.57 (s, 1H), 7.41 (d, J = 9.6 Hz, 2H), 2.68 (s, 1H), 2.40 (s, 6H); HRMS calcd for C21H17N4Cl2 (M+H)+ 395.0830, found 395.0835.
2-(Bicyclo[2.2.1]heptan-1-yl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (25).
Compound 25 was synthesized following the general procedure D described above using 46q (383 mg, 1.29 mmol). The crude was purified by silica gel column (10–20% ethyl acetate/hexane) to afford 179 mg (33%) of a white solid (HPLC tR = 18.3 min): 1H NMR (400 MHz, chloroform-d) δ 8.49 (d, J = 2.6 Hz, 1H), 8.01 (d, J = 8.4 Hz, 1H), 7.82 (dd, J = 8.7, 2.7 Hz, 2H), 7.56 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H), 7.43–7.34 (m, 2H), 2.53 (d, J = 4.8 Hz, 1H), 1.95–1.84 (m, 6H), 1.57 (d, J = 11.2 Hz, 2H), 1.26 (t, J = 7.1 Hz, 2H); HRMS calcd for C23H21N4Cl2 (M +H)+ 423.1143, found 423.1140.
2-(Bicyclo[3.3.1]nonan-1-yl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (26).
Compound 26 was synthesized following the general procedure D described above using 46r (333 mg, 1.02 mmol). The crude was purified by silica gel column (10–20% ethyl acetate/hexane) to afford 101 mg (22%) of a white solid (HPLC tR = 21.0 min): 1H NMR (400 MHz, chloroform-d) δ 8.49 (d, J = 2.6 Hz, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.82 (dd, J = 11.5, 8.5 Hz, 2H), 7.55 (t, J = 7.7 Hz, 1H), 7.44–7.34 (m, 2H), 2.27–2.08 (m, 6H), 1.79 (q, J = 8.4, 6.6 Hz, 6H), 1.26 (t, J = 7.1 Hz, 3H); HRMS calcd for C25H25N4Cl2 (M+H)+ 451.1456, found 451.1452.
2-((1R,3s,5S)-Bicyclo[3.3.1]nonan-3-yl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (27).
Compound 27 was synthesized following the general procedure D described above using 46s (255 mg, 0.78 mmol). The crude was purified by silica gel column (10–20% ethyl acetate/hexane) to afford 5.7 mg (2%) of a white solid (HPLC tR = 19.4 min): 1H NMR (400 MHz, chloroform-d) δ 8.47 (s, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.85–7.75 (m, 3H), 7.60–7.51 (m, 1H), 7.37 (t, J = 8.3 Hz, 2H), 3.23 (ddt, J = 19.1, 12.9, 5.7 Hz, 1H), 2.37 (td, J = 12.7, 5.7 Hz, 2H), 2.23 (d, J = 11.4 Hz, 2H), 1.93–1.81 (m, 2H), 1.72–1.62 (m, 3H), 1.51 (dd, J = 8.3, 4.8 Hz, 5H); HRMS calcd for C25H25N4Cl2 (M+H)+ 451.1456, found 451.1460.
Synthesis of 28 by the Cyclopropanation of 18.
2-((1R,4r,7S)-bicyclo[5.1.0]octan-4-yl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (28).
2-(Cyclohept-4-en-1-yl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (18, 30.0 mg, 0.071 mmol) was added to 1 mL of dichloromethane in a 10 mL round-bottom flask. The reaction mixture was cooled to 0 °C and degassed with N2(g) for 15 min. Diethylzinc (249 μL, 0.25 mmol) was slowly added to the reaction vessel, followed by the slow addition of diiodomethane (29 μL, 0.35 mmol). The reaction mixture was stirred at 0 °C for 30 min and then the silicone stopper was replaced with a plastic stopper and wrapped with parafilm. The reaction mixture was stirred at rt overnight. Saturated ammonium chloride (0.5 mL) was added to the reaction vessel, and the reaction continued to stir for 30 min. The product was extracted with ethyl acetate, and the organic layer was washed with water (1 × 5 mL) and brine (1 × 5 mL). The organic layer was dried over magnesium sulfate, filtered, and concentrated by a rotary evaporator to obtain a residue purified by flash chromatography using 15% ethyl acetate in hexane eluent system to provide 4.0 mg (13%) of product as a white solid (HPLC tR = 19.0 min): 1H NMR (400 MHz, chloroform-d) δ 8.47 (s, 1H), 7.92 (d, J = 8.4 Hz, 1H), 7.86 (s, 1H), 7.79–7.72 (m, 1H), 7.50 (t, J = 7.7 Hz, 1H), 7.38 (d, J = 8.7 Hz, 1H), 7.31 (t, J = 7.5 Hz, 1H), 2.95–2.84 (m, 1H), 2.33 (dt, J = 13.3, 5.7 Hz, 2H), 2.17 (dt, J = 9.0, 5.3 Hz, 2H), 1.88 (q, J = 12.5 Hz, 2H), 1.13–1.00 (m, 2H), 0.95 (q, J = 7.3, 6.3 Hz, 2H), 0.86 (d, J = 7.7 Hz, 1H), 0.77 (td, J = 7.8, 4.5 Hz, 1H). HRMS calcd for C24H23N4Cl2 (M+H)+ 437.1300, found 437.1304.
2-((1R,2R,4R)- and (1S,2S,4S))-Bicyclo[2.2.2]oct-5-en-2-yl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (29).
Compound 29 was synthesized following the general procedure E described above using 46t (8.0 mg, 0.026 mmol) and 47a (13 mg, 0.077 mmol). The crude was purified by silica gel column (10–20% ethyl acetate/hexane) to afford 4.0 mg (36%) of a white solid (HPLC tR = 17.9 min): 1H NMR (400 MHz, chloroform-d) δ 8.50 (d, J = 2.6 Hz, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.75 (d, J = 8.1 Hz, 3H), 7.55 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H), 7.44–7.33 (m, 2H), 6.70 (t, J = 7.5 Hz, 1H), 6.40 (t, J = 7.4 Hz, 1H), 3.47 (ddd, J = 10.3, 5.4, 2.1 Hz, 1H), 2.97–2.91 (m, 1H), 2.87–2.78 (m, 1H), 2.25 (ddd, J = 13.1, 10.2, 2.7 Hz, 1H), 1.86–1.76 (m, 1H), 1.71–1.62 (m, 2H), 1.49–1.34 (m, 2H); HRMS calcd for C24H21N4Cl2 (M+H)+ 435.1143, found 435.1136.
Synthesis of 30 and 31 by the Epoxidation of 18.
m-Chloroperoxybenzoic acid (17.0 mg, 0.10 mmol, 2.0 equiv) was added to a 10 mL round-bottom flask containing a solution of 2-(cyclohept-4-en-1-yl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine 18 (21.0 mg, 0.05 mmol, 1.0 equiv) in 2 mL of dichloromethane. The reaction mixture was stirred at rt for 3 h. Once the starting material disappeared via TLC, 1 mL of acetone and 1 mL of 10% aq. sodium bicarbonate were added, and the mixture was allowed to stir for 30 min. The product was extracted with ethyl acetate (5 mL), and the organic layer was washed with water (10 mL) and brine (10 mL). The organic layer was dried using magnesium sulfate and then filtered. A rotary evaporator evaporated the solvent to obtain a crude residue, first purified by flash chromatography using a 30% acetone in hexane eluent system to obtain a mixture of 30 and 31. A second flash chromatography column used a 0.75–1.0% methanol in dichloromethane eluent system to separate and isolate 30 (2.0 mg, 6%, red solid, analytical HPLC tR = 14.1 min) and 31 (4.0 mg, 12%, red solid, analytical HPLC tR = 14.3 min).
2-((1R,4s,7S)-8-Oxabicyclo[5.1.0]octan-4-yl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (30).
1H NMR (400 MHz, chloroform-d) δ 8.47 (s, 1H), 7.93 (t, J = 8.7 Hz, 2H), 7.76 (dd, J = 8.7, 2.6 Hz, 1H), 7.56–7.49 (m, 1H), 7.41–7.31 (m, 2H), 3.27 (dd, J = 4.4, 1.8 Hz, 2H), 2.71 (s, 1H), 2.46 (d, J = 15.4 Hz, 2H), 2.09–1.80 (m, 6H); HRMS calcd for C23H21N4OCl2 (M+H)+ 439.1092, found 439.1093.
2-((1R,4r,7S)-8-Oxabicyclo[5.1.0]octan-4-yl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (31).
1H NMR (400 MHz, chloroform-d) δ 8.50 (s, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.79 (dd, J = 8.9, 2.6 Hz, 1H), 7.56–7.48 (m, 1H), 7.35 (dd, J = 15.7, 8.2 Hz, 2H), 3.15 (q, J = 4.6 Hz, 2H), 3.11–3.02 (m, 1H), 2.41 (dt, J = 13.8, 6.4 Hz, 2H), 2.14–2.05 (m, 3H), 2.01–1.88 (m, 3H); HRMS calcd for C23H21N4OCl2 (M+H)+ 439.1092, found 439.1090.
Synthesis of 32 by the Oxidation of a Mixture of Compounds 33 and 34.
(R)- and (S)-4-(4-((3,4-dichlorophenyl)amino)-1H-imidazo[4,5-c]quinolin-2-yl)cycloheptan-1-one (32). A 10 mL round-bottom flask containing a solution of (1R,4S)-, (1S,4R)-, (1R,4R)-, and (1S,4S)-4-(4-((3,4-dichlorophenyl)amino)-1H-imidazo[4,5-c]quinolin-2-yl)cycloheptan-1-ol (17.0 mg, 0.039 mmol) 33, and 34 in 1.5 mL dichloromethane, Dess–Martin periodinane (25.0 mg, 0.058 mmol) was added in one portion. The reaction mixture was stirred at rt until the starting material disappeared (monitored by TLC). The reaction mixture was quenched with saturated aq. NaHCO3 (2 mL). The product was extracted with dichloromethane (5 mL) and the organic layer washed with water (10 mL) and brine (10 mL). The organic layer was dried over magnesium sulfate, filtered, and concentrated by rotary evaporator to obtain a crude residue, which was purified by flash chromatography using a 0.75 to 1.0% methanol in dichloromethane eluent system to provide 3.9 mg (23%) of compound 32 as a red solid (HPLC purity: 95%; tR = 13.2 min): 1H NMR (400 MHz, chloroform-d) δ 8.46 (s, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.79 (s, 2H), 7.60–7.53 (m, 1H), 7.42–7.34 (m, 2H), 3.27–3.18 (m, 1H), 2.77 (t, J = 6.1 Hz, 2H), 2.67 (q, J = 5.1 Hz, 2H), 2.38 (d, J = 17.5 Hz, 2H), 2.15 (td, J = 12.9, 10.8, 7.3 Hz, 2H), 2.04–1.88 (m, 2H); HRMS calcd for C23H21N4OCl2 (M+H)+ 439.1092, found 439.1100.
Synthesis of Hydroxy Derivatives 33 and 34 by the Hydroboration–Oxidation of 18.
2-(Cyclohept-4-en-1-yl)-N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine 18 (20 mg, 0.05 mmol, 1.0 equiv) was added to 0.5 mL of dry tetrahydrofuran (THF) in a 25 mL round-bottom flask and flushed with N2(g). The reaction mixture was cooled to 0 °C, and borane dimethyl sulfide complex solution in 2.0 M THF (47 μL, 0.1 mmol) was added to the reaction vessel. The reaction mixture was stirred for 30 min at 0 °C and then overnight at rt. The reaction flask was placed in a 0 °C ice–water bath when all the starting material was consumed. 10% aq. NaOH (0.5 mL, 0.15 mmol) and H2O2 (1 mL, 0.26 mmol) were added successively to the reaction vessel. The reaction mixture reacted at 0 °C for 3 h. The product was extracted with ethyl acetate (5 mL), and the organic layer was washed with water (10 mL) and brine (10 mL). The organic layer was dried over magnesium sulfate, filtered, and then evaporated by a rotary evaporator. Compounds 33 (1.6 mg, 8%, red solid, analytical HPLC tR = 12.2 min) and 34 (2.6 mg, 12%, red solid (analytical HPLC tR = 12.4 min) were isolated and purified from the crude residue by flash chromatography using a 0.75–1.0% methanol in dichloromethane eluent system.
(1R,4S)- and (1S,4R)-4-(4-((3,4-Dichlorophenyl)amino)-1H-imidazo[4,5-c]quinolin-2-yl)cycloheptan-1-ol (33).
1H NMR (400 MHz, chloroform-d) δ 8.47 (d, J = 2.6 Hz, 1H), 7.93 (t, J = 7.7 Hz, 2H), 7.77 (dd, J = 8.8, 2.6 Hz, 1H), 7.50 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.38–7.28 (m, 2H), 4.06 (dq, J = 9.3, 4.5 Hz, 1H), 3.15 (tt, J = 9.6, 4.4 Hz, 1H), 2.22–2.03 (m, 3H), 2.00–1.79 (m, 5H), 1.63 (dddd, J = 13.7, 10.3, 8.0, 2.1 Hz, 1H), 1.52–1.39 (m, 1H); HRMS calcd for C23H23N4OCl2 (M+H)+ 441.1249, found 441.1247.
(1R,4S)- and (1S,4R)-4-(4-((3,4-Dichlorophenyl)amino)-1H-imidazo[4,5-c]quinolin-2-yl)cycloheptan-1-ol (34).
1H NMR (400 MHz, chloroform-d) δ 8.48 (d, J = 2.7 Hz, 1H), 7.93 (d, J = 8.3 Hz, 1H), 7.87 (d, J = 7.9 Hz, 1H), 7.78 (dd, J = 8.8, 2.6 Hz, 1H), 7.50 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.40–7.28 (m, 2H), 3.97 (h, J = 4.3 Hz, 1H), 3.18–3.09 (m, 1H), 2.13 (pd, J = 8.0, 7.5, 4.5 Hz, 3H), 2.03–1.62 (m, 7H); HRMS calcd for C23H23N4OCl2 (M+H)+ 441.1249, found 441.1252.
2-Cyclohexyl-N-(4-iodophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (35).
Compound 35 was synthesized following the general procedure E described above using 46h (10 mg, 0.035 mmol) and 47b (38.0 mg, 0.17 mmol). The crude was purified by silica gel column (10–20% ethyl acetate/hexane) to afford 10.0 mg (61%) of a white solid (HPLC tR = 16.7 min): 1H NMR (400 MHz, chloroform-d) δ 7.97 (d, J = 8.3 Hz, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.80–7.62 (m, 3H), 7.54 (t, J = 7.8 Hz, 1H), 7.36 (t, J = 7.6 Hz, 1H), 3.06–2.94 (m, 1H), 2.22 (d, J = 12.8 Hz, 2H), 1.95 (d, J = 12.8 Hz, 2H), 1.82 (d, J = 13.0 Hz, 1H), 1.69 (q, J = 12.2 Hz, 2H), 1.56–1.31 (m, 3H); HRMS calcd for C22H22N4I (M+H)+ 469.0889, found 469.0898.
2-Cyclohexyl-N-(4-bromophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (36).
Compound 36 was synthesized following the general procedure E described above using 46h (15.0 mg, 0.052 mmol) and 47c (49.0 mg, 0.28 mmol). The crude was purified by silica gel column (10–20% ethyl acetate/hexane) to afford 2.0 mg (10%) of a white solid (HPLC tR = 15.7 min): 1H NMR (400 MHz, chloroform-d) δ 7.98 (t, J = 9.4 Hz, 2H), 7.81–7.68 (m, 2H), 7.58–7.51 (m, 1H), 7.46 (d, J = 2.0 Hz, 1H), 7.36 (t, J = 7.5 Hz, 1H), 3.00 (tt, J = 11.9, 3.6 Hz, 1H), 2.26–2.17 (m, 2H), 1.94 (dt, J = 12.3, 3.8 Hz, 2H), 1.82 (d, J = 13.1 Hz, 1H), 1.69 (qd, J = 12.3, 3.4 Hz, 2H), 1.55–1.31 (m, 4H); HRMS calcd for C22H22N4Br (M+H)+ 421.1028, found 421.1029.
Synthesis of 37 from a Heck Reaction Using 36.
Methyl (E)-3-(4-((2-cyclohexyl-1H-imidazo[4,5-c]quinolin-4-yl)amino)phenyl)acrylate (37).
2-Cyclohexyl-N-(4-bromophenyl)-1H-imidazo[4,5-c]quinolin-4-amine 36 (15.0 mg, 0.036 mmol), methyl acrylate (9 μL, 0.10 mmol, 1 equiv), palladium acetate (1.0 mg, 4.4 μmol, 2.8 equiv), and triethylamine (15 μL, 0.11 mmol, 3 equiv) were added successively to 2 mL of DMF in a 50 mL sealed tube. The reaction mixture was purged with N2(g) at rt for 30 min and then stirred at 140 °C for 24 h. The product was diluted with ethyl acetate (5 mL) and filtered through a short silica plug. The filtrate was concentrated by rotary evaporator and then co-evaporated with toluene (2 × 2 mL) to obtain a crude residue, which was purified by flash chromatography using a 0.75–1.0% methanol in dichloromethane solvent system to afford 2.8 mg (18%) of compound 37 as a white solid (E-isomer, HPLC purity: 98%; tR = 13.3 min): 1H NMR (400 MHz, CDCl3) δ 9.74 (s, 1H), 8.12 (s, 2H), 8.00 (d, J = 8.4 Hz, 1H), 7.82 (s, 1H), 7.70 (d, J = 15.9 Hz, 1H), 7.55 (dd, J = 8.0, 5.9 Hz, 3H), 7.37 (t, J = 7.5 Hz, 1H), 6.37 (d, J = 15.9 Hz, 1H), 3.81 (s, 3H), 2.99 (d, J = 17.2 Hz, 1H), 2.22 (d, J = 12.8 Hz, 2H), 1.94 (d, J = 12.9 Hz, 3H), 1.88–1.74 (m, 1H), 1.69 (td, J = 12.4, 3.3 Hz, 2H), 1.48 (q, J = 12.6 Hz, 2H). HRMS calcd for C26H27N4O2 (M+H)+ 427.2134, found 427.2133.
Synthesis of 38 by a Sonogashira Reaction of 35.
2-Cyclohexyl-N-(4-((5-chlorothiophen-2-yl)ethynyl)phenyl)-1H-imidazo[4,5-c]quinolin-4-amine (38).
2-Chloro-5-ethynylthiophene (141 mg, 1.06 mmol, 5.0 equiv), 2-cyclohexyl-N-(4-iodophenyl)-1H-imidazo[4,5-c]quinolin-4-amine 35 (100 mg, 0.214 mmol, 1.0 equiv), bis(triphenylphosphine) palladium(II) dichloride (30.0 mg, 42.8 μmol, 5 mol%), copper(I) iodide (4.0 mg, 21.4 μmol, 2.5 mol%), and triethylamine (298 μL, 2.14 mmol, 10 equiv) were added successively to 4 mL of dry DMF in a round-bottom flask. The reaction mixture was purged with N2(g) at rt for 30 min and then stirred at 80 °C for 4 h under N2(g). The reaction mixture was cooled to the rt, diluted with ethyl acetate (10 mL), and filtered through a silica plug. The filtrate was concentrated by rotary evaporator and then co-evaporated with toluene (2 × 3 mL) to obtain a crude residue, which was purified by flash chromatography using a 15% ethyl acetate in hexane eluent system to provide 10 mg (10%) of compound 38 as a white solid (HPLC purity: 96%; tR = 16.3 min): 1H NMR (400 MHz, chloroform-d) δ 8.07 (d, J = 7.5 Hz, 2H), 8.00 (d, J = 8.3 Hz, 1H), 7.79 (d, J = 7.1 Hz, 1H), 7.57–7.45 (m, 3H), 7.35 (s, 1H), 7.02 (d, J = 3.9 Hz, 1H), 6.81 (d, J = 3.9 Hz, 1H), 2.97 (tt, J = 11.8, 3.6 Hz, 1H), 2.19 (d, J = 11.8 Hz, 2H), 1.92 (d, J = 13.0 Hz, 2H), 1.80 (d, J = 12.7 Hz, 1H), 1.68 (d, J = 12.2 Hz, 2H), 1.45 (d, J = 12.7 Hz, 4H); HRMS calcd for C28H24N4SCl (M+H)+ 483.1410, found 483.1412.
2-(Heptan-4-yl)-N-(4-iodophenyl)-1H-imidazo[4,5-c]quinolin-4-amine (39).
Compound 39 was synthesized following the general procedure E described above using 46h (195 mg, 0.64 mmol) and 47b (424 mg, 1.94 mmol). The crude was purified by silica gel column (10–20% ethyl acetate/hexane) to afford 33 mg (11%) of a white solid (HPLC tR = 18.6 min): 1H NMR (400 MHz, chloroform-d) δ 8.14–7.98 (m, 1H), 7.88 (d, J = 8.4 Hz, 1H), 7.67 (t, J = 7.4 Hz, 2H), 7.59–7.46 (m, 3H), 7.32 (t, J = 7.5 Hz, 1H), 3.05 (tt, J = 9.0, 5.7 Hz, 1H), 1.90–1.67 (m, 4H), 1.34–1.15 (m, 4H), 0.86 (t, J = 7.3 Hz, 6H). HRMS calcd for C23H26N4I (M+H)+ 485.1202, found 485.1200.
6-Step Synthesis Protocol for 1H-Imidazo-[4,5-c]quinolin-4-amine Derivatives.
3-Nitroquinoline-2,4-diol (41).
According to the literature procedure,19 quinoline-2,4-diol (2.0 g, 12.41 mmol) was added to 12 mL of concentrated nitric acid in a 50 mL round-bottom flask. The reaction mixture was stirred for 10 min at rt and heated in an oil bath at 75 °C for 15 min. The reaction mixture was cooled to rt and poured on a crushed ice–water mixture to obtain a precipitate. The suspension was filtered, and the solid was washed with cold water and dried to obtain 2.43 g (95%) product as a nitrate salt (yellow solid).
2,4-Dichloro-3-nitroquinoline (42).19
The nitrate salt of 3-nitroquinoline-2,4-diol (41, 2.0 g, 9.7 mmol) was added to 17 mL of phenylphosphonic dichloride in a 50 mL round-bottom flask. The flask was fitted with a condenser and stirred the reaction mixture in an oil bath at 135 °C for 3 h. The reaction mixture was cooled to rt, poured slowly on a crushed ice–water mixture, and stirred vigorously to obtain the light brown clay-like precipitate. The mixture was filtered using a fritted funnel, and the precipitate was washed with cold water and dried under air to afford 2.05 g (87%) of crude product as an orange solid.
2-Chloro-3-nitroquinolin-4-amine (43).19
2,4-Dichloro-3-nitroquinoline (42, 2.0 g, 8.0 mmol) and 25% aq. ammonia (6 mL, 80 mmol, 10 equiv) were added to 20 mL of acetonitrile in a 100 mL glass pressure vessel. The mixture was stirred at 50 °C for 6 to 7 h. The reaction mixture was diluted with water (20 mL) and extracted with a mixture of ethyl acetate and methanol (95:5, 2 × 50 mL). The organic layer was separated, dried over MgSO4, filtered, and concentrated by rotary evaporation to obtain 1.7 g (97%) of crude product as a yellow solid.
2-Chloroquinoline-3,4-diamine (44).19
To a 100 mL sealed vessel, 2-chloro-3-nitroquinolin-4-amine (43, 2.0 g, 9.0 mmol, 1.0 equiv), 30 mL of EtOH: H2O (4:1), 4 N hydrochloric acid (~10 mL) and Fe powder (2.5 g, 45 mmol, 5 equiv) were added, and the reaction mixture was stirred at 75 °C for 3 h. The reaction mixture was cooled to rt and filtered through a short silica plug, and the plug was washed with EtOH:H2O (95:5, 20 mL). The combined filtrate was neutralized with aq. NaOH/KOH until pH ~7.0. The product was extracted with a mixture of ethyl acetate and methanol (95:5, 2 × 100 mL). The organic layer was separated, dried over MgSO4, filtered, and concentrated by rotary evaporation, leaving a residue. The residue was purified by silica chromatography with 2% methanol in dichloromethane as an eluent to obtain 1.2 g (70%) product as a brown solid.
Synthesis of Commercially Unavailable Carboxylic Acids (45c,l,m,t).
5,5,5-Trifluoro-2-(3,3,3-trifluoropropyl)-pentanoic Acid (45c).
32 In a 50 mL round-bottom flask equipped with a stir bar were added ethyl 2-cyanoacetate (0.91 mL, 8.54 mmol, 1.0 equiv, Scheme S2), 3-bromo-1,1,1-trifluoropropane (2.0 mL, 18.8 mmol, 2.2 equiv), potassium carbonate (2.48 g, 17.9 mmol, 2.1 equiv) and 21 mL of DMF (~0.4 M). The reaction mixture was flushed with nitrogen and stirred at 60 °C for 48 h. The reaction mixture was cooled to rt, and the solvent was removed under reduced pressure by rotary evaporation to obtain a crude residue as a mixture of ethyl 2-cyano-5,5,5-trifluoropentanoate and ethyl 2-cyano-5,5,5-trifluoro-2-(3,3,3-trifluoropropyl)pentanoate 57. The residue (1.37 g, 4.49 mmol, 1.0 equiv) was dissolved in aq. NaOH (21 mL, ~9.0 g, 225 mmol, 50 equiv), and tetrabutylammonium bromide (303 mg, ~21 mol%) was added to the flask. The reaction mixture was stirred under reflux at 90 °C for 36 h. 1 M aq. HCl was added to the reaction mixture until pH ~7. The mixture was extracted with ethyl acetate, and the organic layer was washed with water. The organic layer was dried over MgSO4, filtered, and solvent evaporated in vacuo to provide the product as a black-brown oil, which was distilled under high vacuum at 170–180 °C to afford 300 mg (16%) crude product, assuming purity of ~50% (by 19F and 1H NMR): 19F NMR (chloroform-d) δ −66.65; 1H NMR (400 MHz, chloroform-d) δ 10.91–10.86 (m, 1H), 3.06–2.98 (m, 1H), 2.46 (ddt, J = 14.1, 9.6, 5.4 Hz, 1H), 2.27–2.01 (m, 3H), 1.97–1.83 (m, 2H), 1.75 (ddt, J = 13.7, 10.8, 5.5 Hz, 2H), 1.68–1.55 (m, 1H), 1.45–1.29 (m, 1H), 0.94 (td, J = 7.4, 1.2 Hz, 2H).
General Method for Favorskii Rearrangement59,60 to Prepare 45l,m.
The appropriate cyclic ketone (1.25 mmol, 1.0 equiv, Scheme S3), N-bromosuccinimide (1.40 mmol, 1.0–1.1 equiv) and p-toluenesulfonic acid (0.13 mmol, 10 mol%)61 were added to 5 mL of dichloromethane in a 15 mL round-bottom flask. The reaction mixture was stirred at rt for 16 h. The solvent was removed under reduced pressure by rotary evaporation to obtain a residue. The residue was dissolved in 10% ethyl acetate in hexane and then passed through a short silica plug. The filtrate was concentrated by rotary evaporation to obtain the α-brominated cyclic ketone as an oily residue, which was used for the next step without further purification.
In a 25 mL round-bottom flask, the crude α-brominated cyclic ketone 59a,b (0.43 mmol) and sodium methoxide (232 mg, 4.3 mmol) were dissolved in 8 mL of methanol. The reaction mixture was stirred overnight and then refluxed for 30 min. 1 M aq. HCl was added to the reaction mixture until pH ~7. The reaction mixture was diluted with water (10 mL) and extracted with ethyl acetate (2 × 20 mL). The organic layer was separated, dried over MgSO4, filtered, and concentrated by rotary evaporation to get a crude product. Purification by silica gel chromatography using a 20% ethyl acetate in hexane eluent system to provide white solid products.
Cyclononanecarboxylic acid (45l).
Compound 45l was synthesized following the Favorskii rearrangement procedure described above using 58a (100 mg, 0.65 mmol). The crude was purified by silica gel column (20% ethyl acetate in hexane) to afford 79 mg (27%) of a white solid: 1H NMR (400 MHz, DMSO-d6) δ 11.97 (s, 1H), 2.38 (ddq, J = 12.7, 8.6, 4.5 Hz, 1H), 1.74 (d, J = 4.0 Hz, 2H), 1.44 (d, J = 7.0 Hz, 14H). Alternatively, a solution of crude α-brominated cyclic ketone (59a, 145 mg, 0.622 mmol, 1.0 eq.) in 3 mL of 10% aq. KOH (10 eq.) was refluxed for 3 h. The reaction mixture was cooled to the room temperature and was acidified with dil. HCl until the pH ~ 5–6. The reaction mixture was diluted with EtOAc (20 mL), and separated the layers. The aqueous phase was washed with EtOAc (2×10 mL), and the combined EtOAc layer was dried over Na2SO4 and filtered. The filtrate was concentrated by rotary evaporation to obtain the crude material, which was purified by silica column to obtain a colorless oil. Yield: 410 mg (76%).
Cyclodecanecarboxylic acid (45m).
Compound 45m was synthesized following the Favorskii rearrangement procedure described above using 58b (50 mg, 0.30 mmol). The crude was purified by silica gel column (20% ethyl acetate in hexane) to afford 89 mg (29%) of a white solid: 1H NMR (400 MHz, DMSO-d6) δ 12.01 (s, 1H), 2.57 (d, J = 6.4 Hz, 1H), 1.82–1.45 (m, 18H).
Synthesis of 45t by a Diels–Alder Reaction and a Subsequent Hydrolysis Reaction.
((1R,2R,4R)- and (1S,2S,4S)-bicyclo[2.2.2]oct-5-ene carboxylic acid (45t). Toluene (2 mL) was added to a 15 mL glass pressure tube containing 1,3-cyclohexadiene 60 (0.50 mL, 5.2 mmol, 1.0 equiv, Scheme S4) and methyl acrylate (0.52 mL, 5.78 mmol, 1.1 equiv). The solution was purged with N2(g) and then sealed. The reaction mixture was stirred at 180 °C for 20 h. The reaction mixture was cooled to rt, and the solvent evaporated by rotary evaporation to afford the mixture of endo and exo isomers as a clear oil. The racemic endo product (carboxylate methyl ester) was isolated as a clear oil (402 mg, 49% yield) by silica gel chromatography using 5% ethyl acetate in hexane as the eluent system.
The endo carboxylate methyl ester (80.0 mg, 0.48 mmol) was dissolved in 5 mL of methanol in a 25 mL round-bottom flask, and an aq. NaOH solution (1.5 M, 5 mL) was added. The reaction mixture was stirred at rt for 1 h. 1 M aq. HCl was added to the reaction mixture until pH ~ 7. The reaction mixture was diluted with water (10 mL) and extracted with ethyl acetate (2 × 20 mL). The organic layer was separated, dried over MgSO4, filtered, and concentrated by rotary evaporation to obtain the desired 400 mg (54%) of the carboxylic acid 45t: 1H NMR (400 MHz, DMSO-d6) δ 11.91 (s, 1H), 6.25 (s, 1H), 6.10 (s, 1H), 2.85 (d, J = 3.4 Hz, 1H), 2.53 (d, J = 7.4 Hz, 2H), 1.65 (s, 1H), 1.60–1.48 (m, 2H), 1.48–1.39 (m, 1H), 1.16 (ddd, J = 13.4, 8.9, 3.5 Hz, 2H).
General Procedures for 2-Substituted-4-chloro-1H-imidazo[4,5-c]quinolines (46b,c,e,h,j–t).
Procedure A.
2.0 g PPA (per 50 mg of 2-chloroquinoline-3,4-diamine) was weighed in a 5 mL round-bottom flask. 2-Chloroquinoline-3,4-diamine (1.0 equiv) and the appropriate carboxylic acid 45c,e,h,k–n (1.2 equiv) were added to the flask. The reaction mixture was stirred at 120 °C for 5 h. The reaction mixture was quenched by pouring on a crushed ice–water mixture. The ice–water mixture was neutralized with aq. K2CO3 (2 M) until pH 8–9 under stirring. The reaction mixture was extracted with ethyl acetate and then washed with water and brine several times. The organic layer was dried over MgSO4, filtered, and removed the solvent by rotary evaporation to obtain the crude product, purified by silica column to obtain the 1H-imidazo[4,5-c]quinoline derivative with 15–25% ethyl acetate in hexane as the eluent system.
Procedure B.
The appropriate carboxylic acid 45b,j,o–t (1.4 equiv) and NMI (3.5 equiv), were added to 4 mL acetonitrile in a 15 mL round-bottom flask with stirring, followed by 2-chloroquinoline-3,4-diamine (1 equiv) and TCFH (1.5 equiv). The reaction mixture was stirred at 60 °C for 5 h. The cooled reaction mixture was diluted with ethyl acetate, and the organic layer was washed with brine (3×). The organic layer was dried over MgSO4 and filtered, transferred to a 50 mL round-bottom flask, and the solvent evaporated by a rotary evaporator. A 1:1 solution of aq. NaOH (15 equiv of NaOH in 10 mL of H2O):methanol (10 mL) was added with stirring. The reaction mixture was refluxed at 90 °C for 3 h. The cooled reaction mixture was diluted with ethyl acetate, and the organic layer was washed with brine (3 × 10 mL) followed by water (3 × 10 mL). The organic layer was dried over MgSO4, evaporated in vacuo, and the resulting residue was used in the final reaction step without further purification.
General Stannylation Procedure to Prepare 48 and 49 from 39.
2-(Heptan-4-yl)-N-(4-iodophenyl)-1H-imidazo-[4,5-c]quinolin-4-amine 39 (0.10 mmol, 1.0 equiv), bis(triphenylphosphine)palladium(II) dichloride (20 μmol, 20 mol%), and hexamethylditin or hexabutylditin (0.5 mmol, 5.0 equiv) were added to 2 mL of 1,4-dioxane in a 10 mL round-bottom flask. The reaction mixture was purged with N2(g) at rt for 30 min and stirred at 70 °C for 2.5 h or until the starting material disappeared (monitored by TLC). The reaction mixture was cooled to the rt, diluted with ethyl acetate (10 mL), and filtered through a short silica plug. The filtrate was concentrated by rotary evaporator and then co-evaporated with toluene (2 × 3 mL) to obtain a crude residue, purified by flash chromatography using a 10% ethyl acetate in hexane eluent system.
2-(Heptan-4-yl)-N-(4-(trimethylstannyl)phenyl)-1H-imidazo[4,5-c]quinolin-4-amine (48).
Compound 48 was synthesized following the stannylation procedure described above using 39 (49.0 mg, 0.10 mmol) and hexamethylditin (105 μL, 0.50 mmol). The crude was purified by silica gel column (20% ethyl acetate in hexane) to afford 5.0 mg (10%) of a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.04 (s, 2H), 7.83 (s, 1H), 7.57–7.47 (m, 3H), 7.40–7.28 (m, 2H), 3.11–2.95 (m, 1H), 1.87–1.70 (m, 4H), 1.40–1.28 (m, 4H), 0.89 (d, J = 7.3 Hz, 6H), 0.39–0.17 (m, 9H); HRMS calcd for C26H35N4Sn (M+H)+ 523.1884, found 523.1882.
2-(Heptan-4-yl)-N-(4-(tributylstannyl)phenyl)-1H-imidazo[4,5-c]quinolin-4-amine (49).
Compound 49 was synthesized following the stannylation procedure described above using 39 (50.0 mg, 0.10 mmol) and hexabutylditin (300 μL, 0.50 mmol). The crude was purified by silica gel column (20% ethyl acetate in hexane) to afford 9.0 mg (13%) of a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.05 (d, J = 8.8 Hz, 1H), 7.97 (s, 1H), 7.79 (d, J = 21.8 Hz, 2H), 7.57–7.44 (m, 2H), 7.38 (t, J = 7.7 Hz, 1H), 7.32 (s, 1H), 3.04 (s, 1H), 1.80 (dq, J = 12.3, 7.1, 6.3 Hz, 4H), 1.65 (tt, J = 8.2, 6.5 Hz, 6H), 1.56 (ddt, J = 10.4, 8.0, 3.7 Hz, 4H), 1.38–1.28 (m, 19H), 0.91 (dd, J = 8.9, 7.3 Hz, 18H); HRMS calcd for C35H53N4Sn (M+H)+ 649.3292, found 649.3293.
Pharmacological Methods.
Cell Culture.
HEK-293 cell lines expressing wild-type human, mouse, or chimeric A3AR were cultured and maintained in Dulbecco’s modified eagle medium (DMEM) along with 10% fetal bovine serum, 1% penicillin/streptomycin, and 0.6 mg/mL G418.
Membrane Preparation.
HEK-293 cells stably expressing wild-type human, mouse, or chimeric A3ARs were washed in phosphate-buffered saline followed by homogenization in hypotonic lysis buffer containing 10 mM Na+-HEPES, 10 mM EDTA, and 1 mM benzamidine (pH 7.4) and then centrifuged at 27000g for 30 min at 4 °C. Cell pellets were subsequently resuspended in He buffer containing 10 mM Na+-HEPES, 1 mM EDTA, and 1 mM benzamidine (pH 7.4) and re-centrifuged. Supernatant was discarded and remaining pellets were resuspended in He buffer containing 10% sucrose and stored at −20 °C.
Single-Point Dissociation Study.
~0.3 nM of [125I]50 was incubated with 50 μg of HEK-293 cell membranes expressing A3ARs for 2 h at rt in 100 μL of binding buffer (50 mM Tris-HCl [pH 7.4], 10 mM MgCl2, 1 mM EDTA, and 1 unit/mL adenosine deaminase). The assay was started by adding 100 μM of the nonselective agonist 51, along with 10 μM of the PAM or vehicle (DMSO). After 60 min, the bound [125I]50 was measured following rapid filtration using GF/C filters. Data are expressed as the amount of radioligand left remaining after 60 min as a percent of the vehicle. A gamma counter measured [125I]50-bound receptors in the filters. Nonspecific binding was determined for all assays by incubation of membranes in the presence of 100 μM 51. All experiments were performed in technical and biological triplicate. Raw CPM values for agonist and modulator tubes and control tubes were used to calculate statistical significance using a two-tailed paired Student’s t test.
Single-Point Equilibrium Binding Study.
HEK-293 membranes (50 μg) were incubated at rt with ~0.3 nM [125I]50 and 10 μM modulator of vehicle (DMSO) for 18 h in 100 μL of binding buffer (50 mM Tris-HCl [pH 7.4], 10 mM MgCl2, 1 mM EDTA and 1 unit/mL adenosine deaminase). At that point, the amount of radioligand was measured by rapid filtration using GF/C filters. Data are expressed as the amount of specific binding as a % change from vehicle. Nonspecific binding was determined for all assays by incubation in the presence of 100 μM 51. All experiments were performed in technical and biological triplicate. Binding data of agonists influenced by modulators were statistically compared to the control using a two-tailed unpaired Student’s t test.
GTPγS Binding Method for Measuring Receptor Activation.
[35S]GTPγS binding assays were conducted to assess the direct effects of modulators on receptor activation. Five μg membranes overexpressing the indicated A3AR were pretreated with modulators for 1 h in 100 μL GTPγS binding buffer (50 mM Tris-HCl [pH 7.4], 10 mM MgCl2, 1 mM EGTA, 100 mM NaCl, 0.004% CHAPS, an 0.5% BSA). To block endogenously expressed A2BARs found in HEK-293 cells, AR antagonists 62 and 63 were added, with each at a final concentration of each of 300 nM. 1 Unit/mL of ADA was added to break down any endogenous adenosine that might have been present. Reactions were initiated by the addition of ~0.2 nM [35S]GTPγS and agonist in 100 μL GTPγS binding buffer and incubated for 2 h. Reactions were stopped with membranes being harvested by rapid filtration through Whatman GF/B filters presoaked for 2 h in GTPγS binding buffer containing an additional 0.02% CHAPS using a cell harvester (Brandel, Gaithersburg, MD). Radioactivity trapped in the filters was measured by liquid scintillation counting. Nonspecific binding of [35S]GTPγS was measured in the presence of 10 μM unlabeled GTPγS. Agonist potency and maximal efficacy at 0.1 μM, 1.0 μM, and 10 μM were statistically compared to the control using one-way ANOVA with multiple comparisons and Bonferroni post hoc test.
ADMET Methods.
Both in vitro and in vivo ADMET properties were determined by JRF India of JRF Global (Gujarat, India).
In Vivo Assays.47
The animal breeding facility, Jai Research Foundation, provided healthy adult male rats (Rattus norvegicus) of Wistar strain (RccHan:WIST). Rats were 6 to 10 weeks old at the start of acclimation. The weight variation of rats was ±20% of mean body weight (b.wt.) at the start of acclimation. Table S6 shows the experimental outline for the in vivo study of pharmacokinetics using rats.
Two routes of administration, intravenous and oral, were used to determine A3AR PAM bioavailability. The dose level for the G1 intravenous route was 0.5 mg/kg b.wt., and the dose levels for the G2–4 oral route were 1, 3, and 10 mg/kg b.wt. The A3AR PAM-DMSO solution was diluted with an aq. solution of 20% 2-hydroxypropyl-β-cyclodextrin (HPBCD) for the intravenous administration, creating a final volume ratio of DMSO to HPBCD, 10:90. An equal volume of Kolliphor EL (polyoxyl castor oil) was added to the A3AR PAM-DMSO solution for oral administration, followed by phosphate buffer saline (PBS). The final volume ratio of DMSO:Kolliphor:PBS for the oral administration was 15:15:70.
200 μL blood was collected from the jugular vein from each rat group on the day of dosing at 0.083, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 h post dosing to assess the A3AR PAM PK profile through intravenous administration. Blood samples were collected in prelabeled microcentrifuge tubes with the anticoagulant heparin (20 IU/mL of blood). After collection, samples were inverted 4 to 5 times, placed on ice, and centrifuged at 9000 rpm for 10 min. Plasma was frozen at −70 °C.
Pharmacokinetic analysis of the frozen plasma concentration–time data was performed using the non-compartmental model of the WinNonlin software. Estimated parameters were the maximum plasma concentration (Cmax), the time to achieve peak plasma concentration (Tmax), the area under the plasma concentration–time curve until the last measured time point (AUC0-last), the area under the plasma concentration–time curve extrapolated to infinity (AUC0−∞), the terminal elimination half-life (T1/2), the mean residence time (MRT), volume of distribution (Vd), the elimination rate constant (kel), the bioavailability (%F), and the clearance (Cl).
In Vitro Assays.
Plasma stability was measured in three species: human, rat, and mouse at 0 to 120 min by liquid chromatography mass spectroscopy–mass spectroscopy (LCMS-MS). % A3AR PAM remaining was provided at 120 min and t1/2 (min). The methods were as described.47
HepG2 cytotoxicity was measured using the CellTiter-Glo Luminescent Cell Viability assay. This assay determined the number of live cells in culture by measuring the ATP metabolism of active cells. 10 to 40 K cells were placed in a 96-well plate. The A3AR PAMs were incubated with live cells for 48 h. Eight dilutions were made with varying concentrations of A3AR PAM ranging from 30 μM to 0.2 μM and incubated. Verapamil/Rifampicin were used as a reference compound. Analysis was done via luminometry. Data was reported as an IC50 value, the concentration of A3AR PAM that reduces cell viability by 50% (ATP measurement).
Human Ether-à-go-go Related Gene (hERG) potassium channels are involved in cardiac action potential re-polarization. hERG assay measures the % inhibition of the potassium channels by the A3AR PAM. The assay used HEK-293 cells stably transfected with the hERG potassium channel. A3AR PAM concentrations were prepared using serial half-log dilutions, starting at 30 μM. A3AR PAMs were incubated with the hERG-expressing cells for 2 h. A3AR PAMs bound to the hERG ion channel were identified by their ability to displace the tracer (Predictor hERG Tracer Red), which resulted in a lower fluorescence polarization using a TAMRA fluorescent polarization filter. E-4031 is a reference compound. % inhibition was reported as the ratio of half-maximum inhibitory concentration of the hERG channel (hERG IC50, μM) to the peak serum concentration of unbound drug (Cmax).
CYP inhibition was determined with a cocktail approach. A3AR PAMsat six concentrations in duplicate (30 μM to 0.12 μM)were incubated with liver microsomes (human, rat, mice) containing the CYP panel with known substrates (1A2, phenacetin (10 μM); 2C9, diclofenac (5 μM); 2C19, omeprazole (5 μM); 2D6, dextromethorphan (5 μM); 3A4, midazolam (2 μM)), 1 mM of NADPH cofactor, 1 μM miconazole (pan inhibitor) in plasma. Analysis of substrates was done by LC-MS/MS. This approach reported A3AR PAM concentrations which produced 50% inhibition of CYPs (IC50 value, μM). LCMS-MS quantified metabolites by area ratio: acetaminophen (1A2), 4-OH-diclofenac (2C9), 5-OH-omeprazole (2C19), dextrorphan (2D6), and 1-OH-midazolam (3A4).
Microsomal stability assays were done using human, rat, and mouse liver microsomes containing the following enzymes: CYPs, flavin monooxygenases, carboxylesterases, and epoxide hydrolase. A3AR PAMs were in 3 μM concentrations duplicates and mixed with 1 mM NADPH and 0.5 g/mL of human, rat, or mouse liver microsomes. Samples were taken from five time periods (0.5, 15, 30, 60, 90, 120 min) and analyzed by LC-MS/MS. The average half-life (min) and % remaining at 120 min was reported.
Caco-2 cell permeability assay measured the rate of transport of a compound across the Caco-2 cell line (21-day process consisting of bidirectional monitoring of absorptive (A-B) and secretory (B-A) fluxes). The Caco-2 cell line originated from a human colon carcinoma, having a polarized monolayer, an apical surface, and intercellular junctions. The cells were exposed to the A3AR PAM (10 μM) in an HBSS buffer with 2% BSA. Samples were gathered at 0- and 120 min. Samples were analyzed by LCMS-MS. Positive controls were atenolol, digoxin, and propranolol. Apparent permeability (Papp – 106 cm/sec), efflux ratio, and % recovery were reported.
pION solubility of each A3AR PAM at pH 7.4 was measured using the pION buffer method. Each A3AR PAM was added to the pION buffer in sufficient amount to reach 500 μM (hypothetically). Sampling was done at 18 h. Positive controls were albendazole and flurbiprofen. Solubility was reported for each A3AR PAM as mean solubility (μg/mL).
Plasma protein binding was determined and expressed as the % unbound A3AR PAM in the plasma of three species (rat, mouse, and human). Cellulose membranes in potassium phosphate buffer (100 mM, pH 7.4) were exposed to 10 μM of A3AR PAM and 500 μL of plasma. Samples were loaded into dialysis cells and maintained in an incubator at 37 °C at 100 rpm. Samples were collected into prelabeled microfuge tubes at 0 an 5 h. The samples were vortexed and centrifuged. Supernatants were analyzed by LC-MS/MS. % binding reported: 1–40% low bound, 41–70% medium bound, 71–100% highly bound.
Simulated gastrointestinal fluids were used to determine whether A3AR PAMs are chemically stable in the stomach at low pH 1–2 or in the intestine pH 6–8. Five μM of each A3AR PAM was added to simulated gastric fluid (SGF) and simulated intestinal fluid (SIF). Sampling was done at 0- and 120 min time points. Method of analysis was LC-MS/MS by area ratio. % A3AR PAM at 120 min was reported.
Prediction of Log D, TPSA, and BBB log([brain]:[blood]) was performed using the StarDrop software (v. 7.2), https://www.optibrium.com/stardrop-installers/.45
Molecular Docking.
Maestro (Maestro 2021–2) of the Schrödinger (New York, NY, USA) suite was used for basic molecular modeling operations. Ligands were drawn and minimized with OPLS462 force field with water solution model. The model of hA3AR was generated in a previous work,49 using an antagonist-bound hA1AR X-ray structure as a template and a refinement with Induced Fit Docking of the antagonist 64. The following tautomeric states were employed for histidines: protonation at Nδ nitrogen for H2727.43 (HSD according to the CHARMM nomenclature) and at the Nε nitrogen for H793.21, H953.37, H1244.39, H158EL2, and H3048.65. The Ballesteros–Weinstein sequence-based numbering scheme63 is indicated as superscript for all residues throughout the text.
Compounds 17 and 7 were docked to the receptor structure using the IFD tool of the Schrödinger suite.50,65 An inner box of 10 Å box and outer box of 30 Å, centered on the barycenter of residues F168EL2 and N2506.55, were employed. Residues within 3 Å of the ligand (with the addition of Q167EL2 and V169EL2) were optimized, and the XP scoring function67 was used for the refinement stage. A maximum of 20 poses was generated for each ligand, and the 5 top scoring ones (according to the IFDScore) were minimized with Prime MM-GBSA tool, using implicit membrane, VSGB solvation model, OPLS4 force field, and minimizing residues within 3 Å from the ligand.
The Semiempirical Extended Tight-Binding Program Package, XTB,64 was used for the dihedral scan. The geometry of the common N-(3,4-dichlorophenyl)-1H-imidazo[4,5-c]quinolin-4-amine scaffold was optimized with the GFN2-xTB method, and then a relaxed dihedral scan of the torsional angle defined by atoms N5–C4–N(amino)–Cp(phenyl) was performed from 0° to 360° in 72 steps (freezing the scanned dihedral with a force constant of 50 Eh/a02).
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge support from the NIH Intramural Research Program (NIDDK, ZIADK031117), the NIH Extramural Research Program (NHLBI, R01 grant HL133589), and the MCW Therapeutic Accelerator Program. We thank John Lloyd (NIDDK) for mass spectral determinations and Robert O’Connor (NIDDK) for NMR spectra and Joel Linden (Univ. of Virginia) for providing ABOPX for iodination. We thank Dilip K. Tosh, Young-Hwan Jung, Zhiwei Wen, and Kiran S. Toti (NIDDK) for chemical advice. We thank Dr. Bryan L. Roth (Univ. North Carolina at Chapel Hill) and National Institute of Mental Health’s Psychoactive Drug Screening Program (Contract No. HHSN-271-2008-00025-C) for screening data.
ABBREVIATIONS USED
- ADA
adenosine deaminase
- AR
adenosine receptor
- DAT
dopamine transporter
- DIPEA
diisopropylethylamine
- DPFGSE
Double Pulsed Field Gradient Selective Echo
- HBSS
Hanks’s balanced salt solution
- HEK-293
human embryonic kidney 293
- HMA
hexamethylene amiloride
- HPBCD
2-hydroxypropyl-β-cyclodextrin
- IFD
induced fit docking
- KOR
κ opioid receptor
- MOR
μ opioid receptor
- NAM
negative allosteric modulator
- NMI
N-methylimidazole
- PAM
positive allosteric modulator
- PDSP
NIMH Psychoactive Drug Screening Program
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- PPA
polyphosphoric acid
- RMSF
root-mean-square-fluctuation
- TCFH
tetramethylchloroformamidinium hexafluorophosphate
- TPSA
topological polar surface area
- TSPO
translocator protein
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01170.
Additional synthetic routes, spectral and HPLC data, off-target binding inhibition data, pharmacological data, ADMET procedures, and data for compounds 18 and 39, including Schemes S1–S6, Tables S1–S8, and Figures S1–S5 (PDF)
Molecular formula strings file (CSV)
Heavy atoms coordinates of the predicted poses of compounds 7 and 17 at the orthosteric binding site (ZIP)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.2c01170
Contributor Information
Lucas B. Fallot, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States; Department of Biochemistry and Molecular Biology, Uniformed Services University of the Health Sciences, Bethesda, Maryland 20814, United States; Department of Chemistry and Life Science, United States Military Academy, West Point, New York 10996, United States.
R. Rama Suresh, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States.
Courtney L. Fisher, Department of Pharmacology and Toxicology, Medical College of Wisconsin, Milwaukee, Wisconsin 53226, United States
Veronica Salmaso, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States.
Robert D. O’Connor, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States
Noy Kaufman, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States.
Zhan-Guo Gao, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States.
John A. Auchampach, Department of Pharmacology and Toxicology and Cardiovascular Center, Medical College of Wisconsin, Milwaukee, Wisconsin 53226, United States
Kenneth A. Jacobson, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States
REFERENCES
- (1).Gessi S; Merighi S; Varani K Adenosine receptors: The status of the art. In The Adenosine Receptors; Borea PA., Varani K., Gessi S., Merighi S., Vincenzi F., Eds.; Springer International Publishing, 2018; pp 1–11. [Google Scholar]
- (2).Linden J Adenosine in tissue protection and tissue regeneration. Mol. Pharmacol. 2005, 67 (5), 1385. [DOI] [PubMed] [Google Scholar]
- (3).Borea PA; Varani K; Vincenzi F; Baraldi PG; Tabrizi MA; Merighi S; Gessi S The A3 adenosine receptor: History and perspectives. Pharmacol. Rev. 2015, 67 (1), 74. [DOI] [PubMed] [Google Scholar]
- (4).Jacobson KA; Tosh DK; Jain S; Gao Z-G Historical and current adenosine receptor agonists in preclinical and clinical development. Front. Cell. Neurosci. 2019, 13, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Changeux J-P; Edelstein SJ Allosteric mechanisms of signal transduction. Science 2005, 308 (5727), 1424–1428. [DOI] [PubMed] [Google Scholar]
- (6).Draper-Joyce CJ; Bhola R; Wang J; Bhattarai A; Nguyen ATN; Cowie-Kent I; O’Sullivan K; Chia LY; Venugopal H; Valant C; Thal DM; Wootten D; Panel N; Carlsson J; Christie MJ; White PJ; Scammells P; May LT; Sexton PM; Danev R; Miao Y; Glukhova A; Imlach WL; Christopoulos A Positive allosteric mechanisms of adenosine A1 receptor-mediated analgesia. Nature 2021, 597 (7877), 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Conn PJ; Christopoulos A; Lindsley CW Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug. Discovery 2009, 8 (1), 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Merighi S; Simioni C; Lane R; IJzerman AP Regulation of second messenger systems and intracellular pathways. In A3 Adenosine Receptors from Cell Biology to Pharmacology and Therapeutics; Borea PA., Ed.; Springer Netherlands, 2010; pp 61–73. [Google Scholar]
- (9).Foster DJ; Conn PJ Allosteric modulation of GPCRs: New insights and potential utility for treatment of schizophrenia and other CNS disorders. Neuron 2017, 94 (3), 431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Schaddelee MP; Read KD; Cleypool CG; IJzerman AP; Danhof M; de Boer AG Brain penetration of synthetic adenosine A1 receptor agonists in situ: role of the rENT1 nucleoside transporter and binding to blood constituents. Eur. J. Pharm. Sci. 2005, 24 (1), 59–66. [DOI] [PubMed] [Google Scholar]
- (11).Liston TE; Hinz S; Müller CE.; Holstein DM.; Wendling J; Melton RJ; Campbell M; Korinek WS; Suresh RR; Sethre-Hofstad DA; Gao ZG; Tosh DK; Jacobson KA; Lechleiter JD Nucleotide P2Y1 receptor agonists are in vitro and in vivo prodrugs of A1/A3 adenosine receptor agonists: implications for roles of P2Y1 and A1/A3 receptors in physiology and pathology. Purinergic Signal 2020, 16 (4), 543–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Fredholm BB; IJzerman AP; Jacobson KA; Linden J; Müller, C. E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors–an update. Pharmacol. Rev. 2011, 63 (1), 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liston TE; Hama A; Boltze J; Poe RB; Natsume T; Hayashi I; Takamatsu H; Korinek WS; Lechleiter JD Adenosine A1R/A3R (adenosine A1 and A3 receptor) Agonist AST-004 reduces brain infarction in a nonhuman primate model of stroke. Stroke 2022, 53 (1), 238–248. [DOI] [PubMed] [Google Scholar]
- (14).Jacobson KA; Gao Z-G Allosteric modulators of adenosine, P2Y and P2X receptors. Allosterism in Drug Discovery; The Royal Society of Chemistry, 2017; Chapter 11, pp 247–270. [Google Scholar]
- (15).Slosky LM; Caron MG; Barak LS Biased allosteric modulators: New frontiers in GPCR drug discovery. Trends Pharmacol. Sci. 2021, 42 (4), 283–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Gao Z-G; Melman N; Erdmann A; Kim SG; Müller CE.; IJzerman AP.; Jacobson KA. Differential allosteric modulation by amiloride analogues of agonist and antagonist binding at A1 and A3 adenosine receptors. Biochem. Pharmacol. 2003, 65 (4), 525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gao Z-G; Van Muijlwijk-Koezen JE; Chen A; Müller CE.; IJzerman AP.; Jacobson KA. Allosteric modulation of A3 adenosine receptors by a series of 3-(2-pyridinyl)isoquinoline derivatives. Mol. Pharmacol. 2001, 60 (5), 1057. [PMC free article] [PubMed] [Google Scholar]
- (18).Gao Z-G; Kim SG; Soltysiak KA; Melman N; IJzerman AP; Jacobson KA Selective allosteric enhancement of agonist binding and function at human A3 adenosine receptors by a series of imidazoquinoline derivatives. Mol. Pharmacol. 2002, 62 (1), 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Göblyös A; Gao Z-G; Brussee J; Connestari R; Santiago SN; Ye K; IJzerman AP; Jacobson KA Structure–activity relationships of new 1H-imidazo[4,5-c]quinolin-4-amine derivatives as allosteric enhancers of the A3 adenosine receptor. J. Med. Chem. 2006, 49 (11), 3354–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kim Y; de Castro S; Gao Z-G; IJzerman AP; Jacobson KA Novel 2- and 4-substituted 1H-imidazo[4,5-c]quinolin-4-amine derivatives as allosteric modulators of the A3 adenosine receptor. J. Med. Chem. 2009, 52 (7), 2098–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Heitman LH; Göblyös A; Zweemer AM; Bakker R; Mulder-Krieger T; van Veldhoven JPD; de Vries H; Brussee J; IJzerman AP A series of 2,4-disubstituted quinolines as a new class of allosteric enhancers of the adenosine A3 receptor. J. Med. Chem. 2009, 52 (4), 926–931. [DOI] [PubMed] [Google Scholar]
- (22).Gao Z-G; Verzijl D; Zweemer A; Ye K; Göblyös A; IJzerman AP; Jacobson KA Functionally biased modulation of A3 adenosine receptor agonist efficacy and potency by imidazoquinolinamine allosteric enhancers. Biochem. Pharmacol. 2011, 82 (6), 658–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Itzhak I; Cohen S; Fishman S; Fishman P A3 adenosine receptor allosteric modulator CF602 reverses erectile dysfunction in a diabetic rat model. Andrologia 2022, 54, No. e14498. [DOI] [PubMed] [Google Scholar]
- (24).Fisher CL; Fallot LB; Wan TC; Keyes RF; Suresh RR; Rothwell AC; Gao Z-G; McCorvy JD; Smith BC; Jacobson KA; Auchampach JA Structure activity relationship of dual action purine nucleoside allosteric modulators of the A3 adenosine receptor. ACS Pharmacol. Transl. Sci. 2022, 5 (8), 625–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Gao D; Xiao Q; Zhang M; Li Y Design, synthesis and biological evaluation of benzyloxyphenyl-methylaminophenol derivatives as STAT3 signaling pathway inhibitors. Bioorg. Med. Chem. 2016, 24 (11), 2549–2558. [DOI] [PubMed] [Google Scholar]
- (26).Beutner GL; Young IS; Davies ML; Hickey MR; Park H; Stevens JM; Ye Q TCFH–NMI: Direct access to N-acyl imidazoliums for challenging amide bond formations. Org. Lett. 2018, 20 (14), 4218–4222. [DOI] [PubMed] [Google Scholar]
- (27).Ruiz-Castillo P; Buchwald SL Applications of palladium-catalyzed C–N cross-coupling reactions. Chem. Rev. 2016, 116 (19), 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ghodke PP; Pradeepkumar PI Nucleoside modification using Buchwald-Hartwig amination reactions. In Palladium-Catalyzed Modification of Nucleosides, Nucleotides and Oligonucleotides; Kapdi AR., Maiti D., Sanghvi YS., Eds.; Elsevier, 2018; Chapter 10, pp 295–333. [Google Scholar]
- (29).Fors BP; Krattiger P; Strieter E; Buchwald SL Water-mediated catalyst preactivation: An efficient protocol for C–N cross-coupling reactions. Org. Lett. 2008, 10 (16), 3505–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Meanwell NA Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 2018, 61 (14), 5822–5880. [DOI] [PubMed] [Google Scholar]
- (31).Miller MA; Sletten EM A general approach to biocompatible branched fluorous tags for increased solubility in perfluorocarbon solvents. Org. Lett. 2018, 20 (21), 6850–6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kouji Y; Mikiro Y Novel fluorine-containing compound, its preparation and antiepileptic agent. Patent JPH0421652A, Jan 24, 1992. [Google Scholar]
- (33).Denmark SE; Edwards JP A comparison of (chloromethyl)- and (iodomethyl)zinc cyclopropanation reagents. J. Org. Chem. 1991, 56 (25), 6974–6981. [Google Scholar]
- (34).Singh V; Tosh DK; Mobin SM Synthesis of embellished bicyclo[2.2.2]octenones and a sigmatropic 1,2-acyl shift in an excited state: a novel and stereoselective route to (±)-hirsutic acid C and complicatic acid. Tetrahedron Lett. 2004, 45 (8), 1729–1732. [Google Scholar]
- (35).Singh V; Pal S; Tosh DK; Mobin SM Molecular complexity from aromatics. Cycloaddition of cyclohexa-2,4-dienones, sigmatropic 1,2-acyl shift and ring-closing metathesis: a new, efficient, and stereoselective synthesis of (±)-hirsutic acid C and medium ring carbocycles. Tetrahedron 2007, 63 (11), 2446–2454. [Google Scholar]
- (36).Appendino G; Spagliardi P; Sterner O; Milligan S Structure–activity relationships of the estrogenic sesquiterpene ester ferutinin. modification of the terpenoid core. J. Nat. Prod. 2004, 67 (9), 1557–1564. [DOI] [PubMed] [Google Scholar]
- (37).Dikošová L; Laceková J; Záborský O; Fischer R Synthesis of 3-substituted isoxazolidin-4-ols using hydroboration–oxidation reactions of 4,5-unsubstituted 2,3-dihydroisoxazoles. Beilstein J. Org. Chem. 2020, 16, 1313–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Beghetto V; Scrivanti A; Bertoldini M; Aversa M; Zancanaro A; Matteoli U A Practical, enantioselective synthesis of the fragrances canthoxal and silvial®, and evaluation of their olfactory activity. Synthesis 2015, 47 (02), 272–288. [Google Scholar]
- (39).Qadir M; Möchel T; Hii KK Examination of ligand effects in the Heck arylation reaction. Tetrahedron 2000, 56 (40), 7975–7979. [Google Scholar]
- (40).Thorand S; Krause N Improved procedures for the palladium-catalyzed coupling of terminal alkynes with aryl bromides (Sonogashira coupling). J. Org. Chem. 1998, 63 (23), 8551–8553. [Google Scholar]
- (41).Sonogashira K Development of Pd–Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides. J. Organomet. Chem. 2002, 653 (1), 46–49. [Google Scholar]
- (42).Sloan NL; Luthra SK; McRobbie G; Pimlott SL; Sutherland A A one-pot radioiodination of aryl amines via stable diazonium salts: preparation of 125I-imaging agents. Chem. Commun. 2017, 53 (80), 11008–11011. [DOI] [PubMed] [Google Scholar]
- (43).Auchampach JA; Gizewski E; Wan TC; de Castro S; Brown GG; Jacobson KA Synthesis and pharmacological characterization of [125I]MRS5127, a high affinity, selective agonist radioligand for the A3 adenosine receptor. Biochem. Pharmacol. 2010, 79, 967–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Du L; Gao Z-G; Paoletta S; Wan TC; Gizewski ET; Barbour S; van Veldhoven JPD; IJzerman AP; Jacobson KA; Auchampach JA Species differences and mechanism of action of A3 adenosine receptor allosteric modulators. Purinergic Signalling 2018, 14, 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Segall MD Multi-parameter optimization: Identifying high quality compounds with a balance of properties. Curr. Pharm. Des. 2012, 18, 1292–1310. [DOI] [PubMed] [Google Scholar]
- (46).Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang X-P; Norval S; Sassano MF; Shin AI; Webster LA; Simeons FR; Stojanovski L; Prat A; Seidah NG; Constam DB; Bickerton GR; Read KD; Wetsel WC; Gilbert IH; Roth BL; Hopkins AL Automated design of ligands to polypharmacological profiles. Nature 2012, 492 (7428), 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Tosh DK; Salmaso V; Campbell RG; Rao H; Bitant A; Pottie E; Stove CP; Liu N; Gavrilova O; Gao ZG; Auchampach JA; Jacobson KA A3 adenosine receptor agonists containing dopamine moieties for enhanced interspecies affinity. Eur. J. Med. Chem. 2022, 228, 113983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Larregieu CA; Benet LZ Drug discovery and regulatory considerations for improving in silico and in vitro predictions that use Caco-2 as a surrogate for human intestinal permeability measurements. AAPS J. 2013, 15 (2), 483–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Suresh RR; Gao ZG; Salmaso V; Chen E; Campbell RG; Poe RB; Liston TE; Jacobson KA Selective A3 adenosine receptor antagonist radioligand for human and rodent species. ACS Med. Chem. Lett. 2022, 13 (4), 623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sherman W; Day T; Jacobson MP; Friesner RA; Farid R Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [DOI] [PubMed] [Google Scholar]
- (51).Genheden S; Ryde U The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discovery 2015, 10, 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Jacobson KA; Gao ZG; Göblyös A; IJzerman AP Allosteric modulators of purine and pyrimidine receptors. Adv. Pharmacol. 2011, 61, 187–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Ciancetta A; Jacobson KA Structural probing and molecular modeling of the A3 adenosine receptor: A focus on agonist binding. Molecules 2017, 22 (3), 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Gao ZG; Ye K; Göblyös A; IJzerman AP; Jacobson KA Flexible modulation of agonist efficacy at the human A3 adenosine receptor by an imidazoquinoline allosteric enhancer LUF6000 and its analogues. BMC Pharmacol 2008, 8, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).van Galen PJM; Nissen P; van Wijngaarden I; IJzerman AP; Soudijn W 1H-imidazo[4,5-c]quinolin-4-amines: novel non-xanthine adenosine antagonists. J. Med. Chem. 1991, 34, 1202–1206. [DOI] [PubMed] [Google Scholar]
- (56).Schön MP; Schön M; Klotz K-N The small antitumoral immune response modifier imiquimod interacts with adenosine receptor signaling in a TLR7- and TLR8-independent fashion. J. Invest. Dermatol. 2006, 126 (6), 1338–1347. [DOI] [PubMed] [Google Scholar]
- (57).Christopoulos A; Changeux JP; Catterall WA; Fabbro D; Burris TP; Cidlowski JA; Olsen RW; Peters JA; Neubig RR; Pin JP; Sexton PM; Kenakin TP; Ehlert FJ; Spedding M; Langmead CJ International Union of Basic and Clinical Pharmacology. XC. Multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol. Rev. 2014, 66 (4), 918–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Sato J; Makita N; Iiri T Inverse agonism: the classic concept of GPCRs revisited. Endocr. J. 2016, 63 (6), 507–514. [DOI] [PubMed] [Google Scholar]
- (59).Kende AS The Favorskiĭ rearrangement of haloketones. Org. Reactions 2011, 261–316. [Google Scholar]
- (60).Tsuchida N; Yamazaki S; Yamabe S A new mechanism for the Favorskii rearrangement. Org. Biomol. Chem. 2008, 6 (17), 3109–3117. [DOI] [PubMed] [Google Scholar]
- (61).Sekhar T; Thriveni P; Venkateswarlu A; Daveedu T; Peddanna K; Sainath SB One-pot synthesis of thiazolo[3,2-a]pyrimidine derivatives, their cytotoxic evaluation and molecular docking studies. Spectrochim. Acta Part A: Mol. Biomol. Spectr. 2020, 231, 118056. [DOI] [PubMed] [Google Scholar]
- (62).Lu C; Wu C; Ghoreishi D; Chen W; Wang L; Damm W; Ross GA; Dahlgren MK; Russell E; Von Bargen CD; Abel R; Friesner RA; Harder ED OPLS4: improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [DOI] [PubMed] [Google Scholar]
- (63).Ballesteros JA; Weinstein H [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci 1995, 25, 366–428. [Google Scholar]
- (64).Bannwarth C; Ehlert S; Grimme S GFN2-xTB-an accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [DOI] [PubMed] [Google Scholar]
- (65).Miller EB; Murphy RB; Sindhikara D; Borrelli KW; Grisewood MJ; Ranalli F; Dixon SL; Jerome S; Boyles NA; Day T; Ghanakota P; Mondal S; Rafi SB; Troast DM; Abel R; Friesner RA Reliable and accurate solution to the induced fit docking problem for protein–ligand binding. J. Chem. Theory. Comput. 2021, 17 (4), 2630–2639. [DOI] [PubMed] [Google Scholar]
- (66).Kooistra AJ; Mordalski S; Pándy-Szekeres G; Esguerra M; Mamyrbekov A; Munk C; Keserű, G. M.; Gloriam, D. E. GPCRdb in 2021: integrating GPCR sequence, structure and function. Nucleic Acids Res. 2021, 49, D335–D343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.