

Abstract
Granulosa cell tumors of the ovary (GCT) are a distinct, hormonally active subset of ovarian cancers. Although it has recently been shown that ∼97 % of all adult GCT harbor a novel somatic missense mutation in the FOXL2 gene, given its almost universal presence, it does not explain differences in tumor stage and/or recurrence. The nuclear factor kappaB (NFκB) transcription factor is constitutively active in two human GCT-derived cell lines, COV434 and KGN, which are useful in vitro models to investigate juvenile and adult GCT, respectively. This study aimed to determine the molecular basis and pathogenetic significance of this aberrant NFκB activity. Selective chemical inhibitors were used to target candidate components of the pathway. The constitutive activity was blocked by two independent inhibitors of IκBα phosphorylation, suggesting that aberrant activation occurs upstream of this point. NFκB inhibition resulted in a dose-dependent decrease in cell proliferation and viability and a dose-dependent increase in apoptosis. Inhibitors of earlier components of the pathway were without effect. Two independent inhibitors of inhibitor of kappaB kinase (IKK)β, a catalytic subunit of the NFκB activation complex, were unable to inhibit the constitutive activity, but surprisingly also ligand-induced activity. These findings suggest a central role for IKKβ; however, no mutations or altered expression of the IKKβ, IKKα, or IKKγ genes was observed in the cell lines or in a panel of human GCT samples. This study highlights unresolved issues in understanding the pathogenesis of GCT and in the use of the COV434 and KGN cells lines as model systems.
Electronic supplementary material
The online version of this article (doi:10.1007/s12672-013-0146-x) contains supplementary material, which is available to authorized users.
Keywords: Granulosa Cell, T47D Cell, Granulosa Cell Tumor, Normal Ovarian Tissue, Renilla Luciferase Construct
Introduction
Granulosa cell tumors of the ovary (GCT) are a specific subset of malignant ovarian cancers, believed to arise from the FSH-responsive, proliferating granulosa cells of the preovulatory follicle [1]. Some defining features of GCT include their hormonal activity, including inhibin and estrogen biosynthesis, and a high rate of recurrence in association with relatively long intervals to relapse [2]. Two subtypes of GCT have historically been classified based on clinical presentation and histological characteristics: the juvenile and the adult form, of which the latter accounts for 95 % of GCT [2]. More recently, the identification of a novel somatic missense mutation in the FOXL2 gene (C134W), which is present in ∼97 % of the adult GCT subtype and absent from the juvenile subtype, clearly defines these GCT subsets [3–6].
Two human GCT-derived cell lines, COV434, and KGN, have been utilized as in vitro model systems to investigate granulosa cell tumorigenesis [2]. The presence of the FOXL2 C134W mutation in the KGN cell line and the wild-type FOXL2 status of the COV434 cell line suggests they represent the adult and juvenile GCT subtypes, respectively [3]. Like human GCT specimens [7], the COV434 and KGN cell lines predominantly expressed both estrogen receptor (ER)β mRNA and protein, whilst no ERα protein expression is observed [8, 9]. Studies aimed at investigating the significance of abundant ERβ expression in the malignant phenotype of GCT have revealed that both COV434 and KGN cell lines display constitutive nuclear factor κB (NFκB) and activator protein 1 (AP-1) transcriptional activity [9]. Furthermore, whilst inhibition of AP-1 activity using mitogen-activated protein kinase (MAPK) inhibitors had no effect on ERβ transcriptional repression, inhibition of the NFκB pathway using the chemical compound BAY11-7082, which acts by inhibiting the phosphorylation of inhibitor of κBα (IκBα), restored ERβ-mediated transactivation [9]. More recently, Bilandzic et al. [10] have shown an impact of BAY11-7082 treatment on TGFβ signaling in KGN cells. These findings demonstrate that constitutive NFκB activity has a functional consequence; the transrepression of ERβ-mediated transcription in the COV434 and KGN cell lines [9]. It is suggested that the aberrant NFκB signaling may provide a pro-proliferative advantage in granulosa cells by (1) its direct anti-apoptotic effects and (2) the transrepression of inhibitory signaling pathways including ERβ [9].
Aberrant NFκB signaling has been reported in most major forms of human cancers, including those of the bone marrow, lymph nodes, breast, prostate, and pancreas [11]. NFκB is a family of five ubiquitously expressed transcription factors, RelA/p65, c-Rel, RelB, NFκB1 (p50), and NFκB2 (p52), which form various hetero- and homodimers upon activation to regulate the expression of more than 150 target genes [12]. The central regulator of NFκB activation is the inhibitor of kappaB kinase (IKK) complex which consists of two highly homologous catalytic kinase components, IKKα and IKKβ [13, 14], and an essential regulatory subunit, NFκB essential modulator (NEMO)/IKKγ [15, 16]. Phosphorylation of the IKK complex in turn phosphorylates the inhibitory molecule of NFκB, IκBα, targeting it for polyubiquitination and proteosomal degradation, thereby freeing up NFκB to enter the nucleus where it initiates transcription [17–19]. It is thought to contribute to malignant transformation by inappropriate activation of cellular functions that are commonly associated with tumor promotion, including stimulating cell growth and proliferation, inhibiting programmed cell death, and promoting angiogenesis, invasion, and metastasis [20, 21].
In this study, we sought to fully characterize the NFκB signaling pathway in two human GCT-derived cell lines using a targeted inhibitory approach and to determine the mechanism(s) and pathogenic significance of the constitutive NFκB signaling seen in these cell lines.
Materials and Methods
Human Tissue Specimens
GCT and normal ovarian tissue were obtained initially for a study of serum inhibin levels in ovarian tumors [22, 23] and subsequently for studies of the molecular pathogenesis of GCT [3, 7, 24–30]. Normal ovarian tissue was obtained from premenopausal women who had undergone elective hysterectomy with oophorectomy for a range of conditions not associated with ovarian malignancy (Table 1). RNA from the tissue and the two GCT-derived cell lines was extracted using the guanidine thiocyanate/caesium chloride method as described previously [25]. Tissue collection was approved by the Research and Ethics Committee of the Monash Medical Centre, Clayton, Australia, and all women gave written informed consent prior to tissue collection.
Table 1.
Clinical details and FOXL2 C134W mutation status for GCT, cell lines, and normal ovarian tissue samples
| Sample | Tissue type | FOXL2 C134W status | Age (years) | Menopausal status | Tumor stage |
|---|---|---|---|---|---|
| 1 | Adult GCT | Het | 31 | Pre | I |
| 2 | Adult GCT | Het | 43 | Pre | Ia |
| 3 | Adult GCT | Het | 53 | Pre | I |
| 4 | Adult GCT | Het | 54 | Pre | I |
| 5 | Adult GCT | Het | 50 | Post | Ic |
| 6 | Adult GCT | Het | N/A | N/A | N/A |
| 7 | Adult GCT | Het | 48 | Pre | Recurrent |
| 8 | Adult GCT | Het | 85 | Post | Recurrent |
| 9 | Adult GCT | Het | 66 | Post | Recurrent, metastatic |
| 10 | Adult GCT | Het | 58 | Post | Recurrent |
| 11 | Adult GCT | Hemi/Homozygous | 50 | Pre | Recurrent |
| 12 | Adult GCT | Het | 45 | Pre | Recurrent |
| 13 | Adult GCT | Het | 56 | Post | Recurrent |
| 14 | Juvenile GCT | WT | 4 | Pre | Juvenile |
| 15 | Juvenile GCT | WT | 17 | Pre | Juvenile, (metastatic?) II |
| 16 | Juvenile GCT | WT | ∼33 | Pre | Juvenile, recurrent |
| KGN | Cell line | Het | 73 | – | Recurrent |
| COV434 | Cell line | WT | 27 | – | Primary—metastatic |
| 17 | Normal ovary | WT | N/A | Pre | N/A |
| 18 | Normal ovary | WT | N/A | Pre | N/A |
| 19 | Normal ovary | WT | 41 | Pre | Endometriosis/adenomyosis |
| 20 | Normal ovary | WT | 43 | Pre | Cervical adenoma |
| 21 | Normal ovary | WT | 49 | Pre | Fibroid uterus |
| 22 | Normal ovary | WT | 33 | Pre | Intractable premenstrual tension |
| 23 | Normal ovary | WT | 30 | Pre | Endometrial carcinoma |
| 24 | Normal ovary | WT | 50 | Pre | Endometrial carcinoma |
| 25 | Normal ovary | WT | 48 | Pre | Endometriosis/adenomyosis |
| 26 | Normal ovary | WT | 48 | Pre | N/A |
Normal ovaries are from patients who have undergone total abdominal hysterectomy/bilateral salpingo-oophorectomy for the following conditions: endometriosis/adenomyosis (nos. 19 and 25), endometrial carcinoma (nos. 23 and 24), fibroid uterus (no. 21), cervical adenoma (no. 20), and intractable premenstrual tension (no. 22)
Cell Lines
Two human GCT-derived cell lines, COV434 [31] and KGN [32], and the human ductal breast epithelial tumor cell line, T47D, were used in this study. COV434 and T47D cells were maintained in DMEM (Invitrogen, Carlsbad, USA) and KGN cells in DMEM/F12 (Invitrogen), both supplemented with 10 % fetal bovine serum (Sigma-Aldrich, St. Louis, USA), 5 % penicillin/streptomycin (Invitrogen) and 1 % l-glutamine (Sigma-Aldrich), 37 °C in a 95 % air/5 % CO2 humidified incubator.
RT2 Profiler™ PCR Array
The Human NFκB Signaling Pathway RT2Profiler™ PCR Array (cat no. APHS-025C; SuperArray Bioscience, Frederick, USA) was used to profile the expression of a focused panel of genes related to NFκB-mediated signal transduction. Each well of the 96-well plate contained a gene-specific primer pair optimized to analyze the expression of 84 NFκB pathway-specific genes, plus five housekeeping genes and three RNA quality controls simultaneously using SYBR Green based real-time PCR detection. The controls were included to monitor genomic DNA contamination, RNA quality, and general PCR performance. One microgram pooled total RNA from three separate passages of each cell line, COV434 and KGN, was reverse-transcribed for 60 min at 50 °C in a total volume of 20 μl using SuperScript III reverse transcriptase (Invitrogen). First-strand synthesis was performed using 250 ng random hexamers. One hundred two microliters of diluted first-strand cDNA synthesis reaction was mixed with 1,275 μl of RT2 SYBR Green/ROX qPCR Master Mix (SuperArray Bioscience) and 1,173 μl of ddH2O. Twenty-five microliters of the master mix template cocktail was added to each well of the PCR array. Cycling conditions comprised of a 10-min initial denaturation step at 95 °C, then 40 cycles of denaturing (95 °C for 15 s) and annealing (60 °C for 1 min) using an Applied Biosystems 7900HT Fast Real-Time PCR System (Applied Biosystems, Carlsbad, USA). The threshold cycle (Ct) for each well was calculated using the instrument’s software and exported to the manufacturer’s RT2 Profiler PCR Array Data Analysis Template v2.0 for analysis. By considering the expression levels observed in the housekeeping genes in comparison to those observed in NFκB pathway-specific genes, an approximate indication of expression levels was derived.
Luciferase Reporter Assays
Initially COV434 and KGN cells were seeded in 12-well plates and co-transfected with 500 ng/well of either a NFκB-inducible reporter plasmid (pNFκB-Luc) containing four repeats of the κB cis-enhancer element (TGGGGACTTTCCGC) or the enhancerless reporter (pTAL-Luc) (Clontech, Mountain View, USA) along with 100 ng/well of a constitutively expressing Renilla luciferase construct (pRL-TK; Promega, Madison, USA) as a transfection efficiency control using TransIT®-LT1 Transfection Reagent (Mirus, Madison, USA) for COV434 cells and SuperFect Transfection Reagent (Qiagen, Hilden, Germany) for KGN cells, according to the manufacturers’ instructions. In fresh medium, cells were treated with either the appropriate vehicle control, tumor necrosis factor α (TNFα; Sigma; 10 ng/ml), interleukin-1α (IL-1α; Sigma; 10 ng/ml) and/or a selective chemical inhibitor at the appropriate concentration for 24 h. Cells were lysed using 1× Passive Lysis Buffer (Promega) and the activities of firefly (Photinus pyralis) and Renilla (Renilla reniformis) luciferases were measured using a Dual-Luciferase® Reporter Assay System (Promega) according to the manufacturer’s instructions on a PerkinElmer EnVision® 2103 Multilabel Reader controlled by Wallac EnVision® Manager software (version 1.12; PerkinElmer, Waltham, USA). Whilst the raw data counts for both firefly luciferase activity and Renilla luciferase activity were relatively consistent within each treatment group, when Renilla luciferase activity was compared between treatment groups, activity was highly variable. This argued against co-transfection with the Renilla luciferase construct as a transfection efficiency control in these cells so subsequent experiments were performed in the absence of the pRL-TK construct. Instead, replicates were performed in sextuplicate for each treatment group as a control for transfection variation. Firefly luciferase activity was measured using Luciferase Assay Reagent (Promega) according to the manufacturer’s instructions and activity was expressed as arbitrary units relative to pNFκB-Luc-transfected cells treated with vehicle. All experiments were carried out at least thrice with sextuplicate wells per point. All values represent the mean ± the standard error of the mean (SEM). Chemical inhibitors used were: BAY11-7082 (Calbiochem, La Jolla, USA), IMD-0354, interleukin-1 receptor-associated kinase (IRAK)-1/4 inhibitor, Necrostatin-1(Sigma-Aldrich), SC-514 (Calbiochem), and PS-1145 dihydrochloride (Sigma-Aldrich).
Cell Viability, Proliferation, and Apoptosis Assays
Cells were seeded in 96-well plates at ∼80 % confluency in the appropriate complete medium and incubated in fresh medium containing either the appropriate vehicle control or the specific chemical inhibitor for the specified length of time. Cell proliferation and cell viability was measured using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega) and CellTiter-Glo® Luminescent Cell Viability Assay (Promega), respectively, according to the manufacturer’s instructions. All experiments were carried out at least four times with triplicate wells per point. All values represent the mean ± SEM.
To measure apoptosis cells were incubated with 5 μl of 1:500 Hoechst 33342 (Invitrogen)/ 1:500 Yo-Pro®-1 iodide (Invitrogen) per well for 30 min. Media was removed and cells washed twice with 100 μl phosphate-buffered saline (PBS). In 100 μl PBS/well, plates were scanned on a Cellomics ArrayScan HCS Reader (Thermo Scientific, Waltham, USA). Cells were excited at 340 ± 50 nm (for Hoechst 33342) and 455 ± 40 nm (Yo-Pro®-1) and fluorescence emission measured at 515 ± 20 nm for both. The number of cells exhibiting Yo-Pro®-1fluorescence was counted and positive cells were expressed as the percentage of Yo-Pro®-1 to Hoechst 33342.
Direct Sequencing
One microgram of total RNA was reverse-transcribed for 60 min at 50 °C in a total volume of 20 μl using SuperScript III reverse transcriptase (Invitrogen). First-strand synthesis was performed using 250 ng random hexamers. The oligonucleotide primers and PCR conditions used to amplify the coding regions of IKKα, IKKβ, and NEMO are listed in Supplementary Table 1. VeritiTM 96-well Thermal Cycler (Applied Biosystems, Foster City, USA). In all cases, an amplicon of the predicted size when compared with the molecular weight standards was obtained. The PCR products were purified using a QIAquick Gel Extraction Kit (Qiagen). For each sample, forward and reverse sequences were determined using the primers described in Supplementary Table 1 as sequencing primers and a BigDye® Terminator Cycle Sequencing Ready Reaction Kit, v3.1 (Applied Biosystems), according to the manufacturer’s protocol. Analysis was carried out on an ABI 3130xl Genetic Analyzer (Applied Biosystems) and assembled using Sequencher 3.1.1 software.
Quantitative PCR Analysis
Quantitative PCR was performed with the following oligonucleotide primers: IKKα (IKBKA), sense 5′- AATGGATCACATTTTGAATTTGA-3′, antisense 5′- TAGCTTTCAGTTGTTGTGATGC-3′; IKKβ (IKBKB), Sense 5′- TGGAAGAGGTGGTGAGCTTAATG-3′, antisense 5′- CTAGCAGGGTGCAGAGGTTATGT-3′; NEMO (IKBKG), sense 5′- ACCAGCTCTTCCAAGAATACGAC-3′, antisense 5′- CGCTTCCTCATGTCCTCGAT-3′, using an Applied Biosystems 7900HT Fast Real-Time PCR System with all reactions performed in triplicate. Cycling conditions comprised of a 10-min initial denaturation step at 95 °C, then 40 cycles of denaturing (95 °C for 30 s), annealing (55 °C for 30 s) and extension (72 °C for 45 s). The β2-microglobulin (B2M) primers have been described previously [26]. Yields were converted to femtograms based on the standard curve for each PCR product, and the resultant mRNA levels were normalized to the β2M mRNA level per sample. The data were calculated from the results of three independent experiments.
Small Interfering RNA Transfection
Small interfering RNAs (siRNAs) were introduced into COV434 and KGN cells using Lipofectamine 2000 transfection reagent (Invitrogen), according to the manufacturer’s instructions. Briefly, cells were plated overnight in the appropriate growth medium without antibiotics. The medium was removed and cells were rinsed once with PBS. Cells were cultured in serum- and antibiotic-free medium containing lipofectamine (mock), lipofectamine plus non-targeting (control) siRNA, or lipofectamine plus siRNA for IKBKB (Thermo Scientific Dharmacon, Lafayette, USA) at a final concentration of 50 nM for 48 h. Gene knockdown efficiency was evaluated by Western blot analysis.
Western Blot Analysis
Whole cell extracts were prepared by lysing cells in NP-40 buffer (20 mM Tris pH 7.5, 100 mM NaCl, 0.5 % NP-40, 0.5 mM EDTA, 0.5 mM PMSF) with 0.5 % protease inhibitor cocktail (Sigma-Aldrich). Western blot analysis involved standard procedures with an AmershamTM ECL Plus Western Blot Detection System (GE Healthcare, Little Chalfont, UK). Equal amounts of cell lysates were subjected to SDS-PAGE and transferred to hydrophobic polyvinylidene difluoride (PVDF) membrane (GE Healthcare, Little Chalfont, UK). Proteins were probed with a primary anti-IKKβ rabbit monoclonal antibody (Sigma-Aldrich) and secondary polyclonal rabbit anti-goat HRP antibody (Dako, Glostrup, Denmark). Blots were stripped with 1X Re-Blot Plus Strong Antibody Stripping Solution (Millipore, Billerica, USA) and reprobed with an anti-β-Actin (13E5) HRP conjugate rabbit monoclonal antibody (Cell Signaling Technology, Danvers, USA) to ensure equal loading per well.
Statistical Analysis
Statistical analysis for each data set was performed using tools within the GraphPad Prism software package (version 5.03; GraphPad Software Inc., San Diego, USA). Means were compared using a one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for multiple comparisons. A p value of less than 0.05 was considered statistically significant. The data is presented as means ± standard error of the mean (SEM). p values are annotated as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***) throughout.
Results
COV434 Cells Are Resistant to IL-1-Induced NFκB Activity
The overall aim of this study was to characterize the NFκB signaling pathway in the COV434 and KGN cell lines using a targeted inhibitory approach. The proinflammatory cytokines tumor necrosis factor α (TNFα) and interleukin-1 (IL-1) are well known stimulants of the classical NFκB pathway and were used as positive controls. COV434 and KGN cells were transfected with the NFκB reporter construct and treated with either vehicle, TNFα or IL-1. As previously [9], at baseline luciferase activity with the NFκB reporter was approximately fivefold that of the enhancerless reporter, reflecting the constitutive activity (Fig. 1). Treatment of both COV434 and KGN cells with TNFα resulted in a further induction of luciferase activity above that seen for vehicle-treated cells (Fig. 1), as also previously reported [9]. KGN cells treated with IL-1 exhibited a further induction of luciferase activity above that seen for both vehicle and TNFα treated cells, whilst COV434 cells showed no further increase in luciferase activity above constitutive output, indicating COV434 cells are resistant to IL-1 stimulation of NFκB activation (Fig. 1). As a result, in subsequent experiments TNFα was used as a positive control for both COV434 and KGN cells whilst IL-1 was used as a positive control for KGN cells only.
Fig. 1.

TNFα and IL-1 as positive controls for NFκB pathway activation. a KGN and b COV434 cells were transiently transfected with 0.5 μg of either pTAL-Luc (gray bar) or pNFκB-Luc (black bar). Twenty-four hours post transfection cells were treated with either vehicle, TNFα (10 ng/ml) or IL-1α (10 ng/ml) for 24 h. Firefly luciferase activity was measured and expressed as arbitrary units relative to vehicle-treated cells. The results from four separate experiments, each of which was performed in sextuplicate, are shown as mean ± SEM. ***p < 0.001; ns, not significant; EV, empty vector
Blocking IκBα Phosphorylation Inhibits Constitutive NFκB Activity
Before proceeding with targeted NFκB pathway inhibition, we confirmed the finding that constitutive NFκB signaling could be inhibited in COV434 and KGN cells upon treatment with the chemical compound BAY11-7082 [9], which acts by blocking phosphorylation of the NFκB inhibitory molecule, IκBα. BAY11-7082 inhibited both constitutive and TNFα-induced NFκB activity in both cell lines as well as IL-1-induced activity in KGN cells (Fig. 2a, b). An independent inhibitor of IκBα phosphorylation, IMD-0354, also inhibited both constitutive and ligand-induced NFκB activity in a dose-dependent manner (Fig. 2c, d), confirming the finding that the point of aberrant activation must be occurring upstream of IκBα phosphorylation in the NFκB signaling pathway. The functional consequence of NFκB inhibition has not previously been reported. In the presence of BAY-117082 (Fig. 3a, b) and IMD-0354 (Fig. 3c, d), COV434 and KGN cells exhibited a dose-dependent decrease in cell viability and cell proliferation and a dose-dependent increase in apoptosis.
Fig. 2.
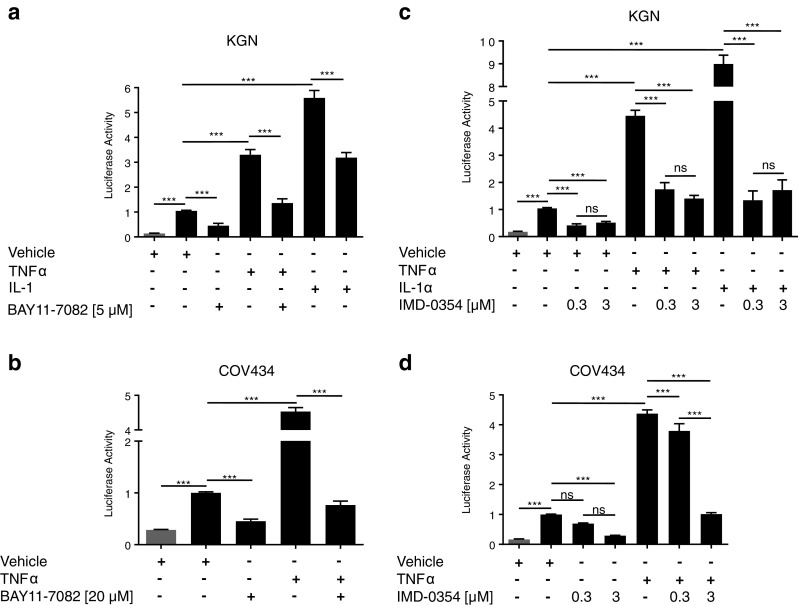
IκBα-specific inhibitors block NFκB activity in KGN and COV434 cells. KGN and COV434 cells were transiently transfected with 0.5 μg of either pTAL-Luc (gray bar) or pNFκB-Luc (black bar) for 24 h. Transfected cells were treated with either vehicle, TNFα (10 ng/ml), IL-1α (10 ng/ml), and/or a, b BAY11-7082 or c, d IMD-0357 at the indicated concentrations for 24 h. Firefly luciferase activity was measured and expressed as arbitrary units relative to vehicle-treated cells. The results from four separate experiments, each of which was performed in sextuplicate, are shown as mean ± SEM. ***p < 0.001; ns, not significant
Fig. 3.
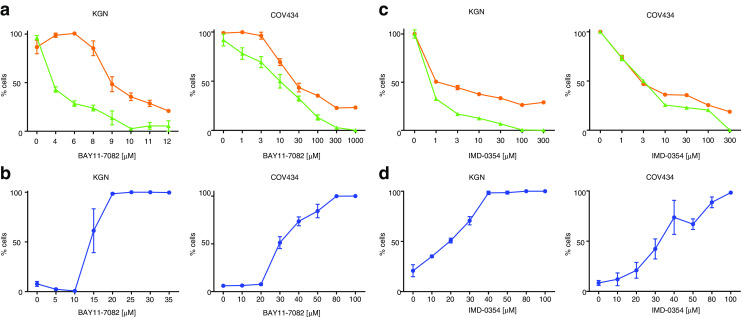
Effects of IκBα-specific inhibitors on KGN and COV434 cell viability, cell proliferation, and apoptosis. Subconfluent KGN and COV434 cells were treated with incremental concentrations of BAY11-7082 (a, b) or IMD-0354 (c, d) for 24 h. Cell proliferation was examined by quantifying the formazan product of MTS reduction after 24 h (orange circle). Cell viability was examined by measuring ATP present after 24 h (green triangle). Each point represents the mean ± SEM of four separate experiments performed in triplicate, relative to the baseline activity detected in those cells treated with vehicle alone. To assess apoptosis, 30 min prior to the end of treatment, cells were stained with the nuclear dyes, Hoechst 33342 and Yo-Pro-1. Live cell fluorescent microscopy imaging was performed, exciting cells at 340 ± 50 nm (for Hoechst 33342) and 455 ± 40 nm (Yo-Pro®-1) and measuring fluorescence emission at 515 ± 20 nm (for both). Cell mortality was quantified by expressing the number of Yo-Pro®-1-positive cells as a percentage of the number of Hoechst 33342-positive cells (blue circle). Each point represents the mean ± SEM of triplicate measurements within one representative assay performed in triplicate
Effect of Interleukin-1 Receptor-Associated Kinase 1 (IRAK-1) Targeted Inhibition
That the NFκB constitutive activity observed in COV434 and KGN cell lines can be inhibited by blocking IκBα phosphorylation implies that aberrant activation must be occurring at a point upstream of IκB in the pathway. In order to rationally identify candidates for selective targeted inhibition, we examined mRNA expression levels of a focused panel of genes related to NFκB-mediated signal transduction (Supplementary Table 2). Given that NFκB pathway signaling is constitutively activated in both cell lines, we chose to focus on genes that were highly expressed in both lines, as it is those which are likely to be the cause of the aberrant activity. Expression levels of the NFκB pathway-specific genes were determined for the two lines using the housekeeping genes to facilitate the comparison. Expression levels were largely consistent across the two cell lines (Supplementary Table 2). Genes of interest, that is, genes abundantly expressed in both cell lines, included, IRAK-1 (interleukin-1 receptor-associated kinase 1), RAF1, IKKβ (IκB kinase β), and CFLAR (CASP8 and FADD-like apoptosis regulator). IRAK-1 was the most abundantly expressed gene in both cell lines. IRAK-1 encodes one of two serine/threonine kinases, the other being IRAK-4, that become associated with the activated IL-1 receptor to initiate a downstream signaling cascade involving the IKK complex, ultimately resulting in NFκB transcriptional activation [33, 34].
Although the use of kinase-inactive constructs as a means of targeted inhibition had initially been proposed, the “squelching” effect observed upon co-transfection of the Renilla luciferase construct (pRL-TK) and the NFκB reporter construct in the GCT-derived cell lines meant an alternative inhibitory approach was necessary, and as a result, chemical inhibitors were utilized. An IRAK1/4 selective inhibitor with a published IC50 of 300 and 200 nM for IRAK-1 and IRAK-4, respectively (Merck Inhibitor Source Book, second edition), was used to block IRAK-1 activation. The inhibitor had little to no effect on cell proliferation or cell viably up to a concentration of ∼3 μM (Fig. 4). In the range of 3–10 μM, there was a rapid decline in both cell viability and proliferation, however, given the known IC50, it is likely to be the result of an “off-target” or toxic effect of the compound at high concentrations (Fig. 4). The IRAK-1/4 inhibitor had no effect on constitutive NFκB activity in both COV434 and KGN cells (Fig. 5). However, it did inhibit IL-1-induced activity in KGN cells, demonstrating that the compound was appropriately active and functional (Fig. 5a).
Fig. 4.

Effects of IRAK-1/4-specific inhibitor on KGN and COV434 cell viability and cell proliferation. To examine the effect of the IRAK-1/4-specific inhibitor on cell viability and proliferation subconfluent KGN and COV434 cells were treated with incremental concentrations of the IRAK-1/4 inhibitor for 24 h. Cell proliferation was examined by quantifying the formazan product of MTS reduction after 24 h (orange circle). Cell viability was examined by measuring ATP present after 24 h (green triangle). Each point represents the mean ± SEM of four separate experiments performed in triplicate, relative to the baseline activity detected in those cells treated with vehicle alone
Fig. 5.

IRAK-1/4 inhibition does not block NFκB constitutive activity in KGN and COV434 cells. to assess IRAK-1/4 blockade on NFκB activity a KGN and b COV434 cells were transiently transfected with 0.5 μg of either pTAL-Luc (gray bar) or pNFκB-Luc (black bar) for 24 h. Transfected cells were treated with either vehicle, TNFα (10 ng/ml), IL-1α (10 ng/ml), and/or IRAK-1/4 inhibitor (5 μM) for 24 h. Firefly luciferase activity was measured and expressed as arbitrary units relative to vehicle-treated cells. The results from four separate experiments, each of which was performed in sextuplicate, are shown as mean ± SEM. ***p < 0.001; ns, not significant
Effect of Receptor-Interacting Protein 1 (RIP1) Targeted Inhibition
As no effect was observed following IRAK-1/4 inhibition, we chose to examine the effect of blocking TNFα-mediated activation by targeting a component of the pathway specific to TNFα-induced activity. When bound by its ligand, the TNF receptor (TNFR1) activates several receptor-associated proteins which in turn recruit receptor-interacting protein 1 (RIP1). RIP1 binds to NEMO, the regulatory component of the IKK complex, and has been shown to play an essential role in NFκB activation in response to TNFα, but not IL-1 [35–37]. Necrostatin-1, a selective allosteric inhibitor of RIP1 [38], had little to no effect on cell proliferation or cell viably, nor did it inhibit NFκB constitutive or ligand-induced activity (data not shown).
Effect of IKKβ Targeted Inhibition
Given the results seen for pathway inhibition using the IRAK-1/4 and RIP1 inhibitors, and considering that both IL-1 and TNFα-mediated pathways of NFκB activation converge at the IKK complex, a chemical inhibitor was selected to specifically target IKKβ, a catalytic component of the IKK complex that has been shown to be essential for activation of NFκB in response to proinflammatory stimuli [39–41].
The compound SC-514 has been shown to act as a highly selective inhibitor of IKKβ and to have no inhibitory effect on other IKK isoforms or other serine–threonine and tyrosine kinases [42]. SC-514 had no effect on cell proliferation or cell viability in COV434 and KGN cells (Fig. 6a), nor did it have an effect on the constitutive activity in either cell line (Fig. 7a, b). Surprisingly, however, it also had no effect on TNFα and IL-1-induced activity in both cell lines (Fig. 7a, b).
Fig. 6.

Effects IKK complex inhibition on KGN and COV434 cell viability and cell proliferation. To examine the effect of IKK complex inhibition on cell viability and proliferation subconfluent KGN and COV434 cells were treated with incremental concentrations of either a SC-514 or b PS-1145 for 24 h. Cell proliferation was examined by quantifying the formazan product of MTS reduction after 24 h (orange circle). Cell viability was examined by measuring ATP present after 24 h (green triangle). Each point represents the mean ± SEM of four separate experiments performed in triplicate, relative to the baseline activity detected in those cells treated with vehicle alone
Fig. 7.
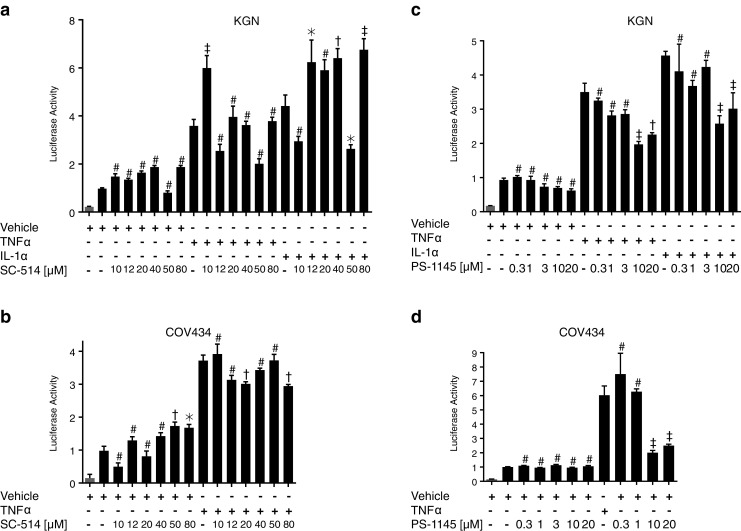
Inhibition of the IKK complex does not block NFκB constitutive activity in KGN and COV434 cells. To assess IKK complex blockade on NFκB activity, KGN and COV434 cells were transiently transfected with 0.5 μg of either pTAL-Luc (gray bar) or pNFκB-Luc (black bar) for 24 h. Transfected cells were treated with either vehicle, TNFα (10 ng/ml), IL-1α (10 ng/ml), and/or SC-514 (a, b) and/or PS-1145 (c, d) at increasing concentrations as indicated for 24 h. Firefly luciferase activity was measured and expressed as arbitrary units relative to vehicle-treated cells. The results from two to four separate experiments, each of which was performed in sextuplicate, are shown as mean ± SEM. #, not significant; *p < 0.05; †p < 0.01; ‡p < 0.001 when the inhibitor in the presence of vehicle, TNFα or IL-1α was compared to vehicle, TNFα or IL-1α alone, respectively
Given this unexpected result, the effect of an alternative IKKβ inhibitor was also examined. PS-1145 is reported to be a potent inhibitor of IKKβ [43]; however, its exact specificity is not well-defined and it possibly targets both catalytic components of the IKK complex [44]. PS-1145 had little or no effect on cell proliferation and cell viably in COV434 cells; however, it did inhibit cell proliferation and cell viably in a largely parallel, dose-dependent manner in KGN cells (Fig. 6b). In addition, PS-1145 had no effect on constitutive activity in COV434 cells; however, it did inhibit TNFα-induced activity at concentrations of 10 and 20 μM in the COV434 cells (Fig. 7d). In accordance with the effect seen on cell proliferation and cell viability, PS-1145 inhibited constitutive activity at concentrations ≥3 μM in KGN cells, as well as TNFα and IL-1-induced activity, at concentrations ≥10 μM (Fig. 7c). Surprisingly however, the effect observed on ligand-induced activity was not as marked as that seen in COV434 cells.
Given the unexpected lack of response for SC-514 and the attenuated response to PS-1145 on ligand-induced NFκB, the efficacy of the two compounds was tested by examining their ability to inhibit TNFα-induced NFκB activation in the T47D breast cancer cell line (Fig. 8). T47D cells are estrogen receptor positive and do not display constitutive NFκB activity [45]. Both SC-514 and PS-1145, as well as BAY11-4708, inhibited TNFα-induced activity in a dose-dependent manner indicating the IKK-specific inhibitors were capable of exerting their predicted biological effect.
Fig. 8.

SC-514 and PS-1145 inhibit TNFα-induced NFκB activity in T47D cells. T47D cells were transiently transfected with 0.5 μg pNFkB-Luc for 24 h. Transfected cells were treated with TNFα (10 ng/ml) ± BAY11-7082 (5 and 20 μM), SC-514 (10 and 50 μM), and PS-1145 (3 and 10 μM) for 24 h. Firefly luciferase activity was measured and expressed as arbitrary units relative to vehicle-treated cells. The results from two separate experiments, each of which was performed in sextuplicate are shown as mean ± SEM. ***, p < 0.001; ns, not significant
Gene Sequencing and Mutation Analysis of Components of the IKK Complex
It is the catalytic components of the IKK complex which phosphorylate IκBα, thus freeing NFκB to enter the nucleus where it initiates transcription of its target genes. The lack of response seen to SC-514 and PS-1145 in COV434 and KGN cells, despite the proven efficacy of these compounds in T47D cells, led us to speculate that aberrant activation of IKKβ, or other components of the IKK complex, may play a role in the constitutive activation of the NFκB pathway. RT-PCR of total RNA was performed to amplify overlapping amplicons encompassing the entire coding region of each of the IKKβ (IKBKB), IKKα (IKBKA), and NEMO/IKKγ (IKBKG) genes in COV434, KGN, and T47D cells (Supplementary Table 1). All amplicons were of the expected size when visualized on an ethidium bromide stained gel (data not shown). For each amplicon, forward and reverse sequences were determined using the PCR primers as sequencing primers. The sequence generated from T47D cells was used as a reference control. A nonconservative, single-nucleotide change was identified in IKBKB in the KGN cells, in which a G was substituted for an A at nucleotide position 1,577 (c.G1577A; p.R526Q). Although this heterozygous change lies in a highly conserved region of the IKKβ protein (Supplementary Fig. 1), it was subsequently identified in a single-nucleotide polymorphism (SNP) database. Interestingly, this SNP has been reported to occur at a low frequency in two Asian populations (Han Chinese, 0.178 and Japanese, 0.114) and to be absent from Sub-Saharan African and Western and Northern European populations (NCBI Reference SNP Cluster Report: rs2272736; www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?rs=2272736). The panel of human GCT samples was also examined and the R526Q change was not found to be present in any of the tumors (n = 22; data not shown).
IKKα and IKKβ share 50 % overall sequence identity and 70 % protein similarity and both associate with NEMO/IKKγ to form the IKK complex [46]. Although NEMO has no catalytic function and it is commonly referred to as the “scaffold” protein of the IKK complex, it has been shown to play an essential role in the canonical NFκB pathway [16, 47]. Given the lack of evidence of mutations in IKBKB in the GCT-derived cell lines and tumor panel, the IKBKA and IKBKG genes were also sequenced. A nonconservative, single-nucleotide change was identified in IKBKA in the COV434 and KGN cells, in which a G was substituted for an A at nucleotide position 802, corresponding to a valine to isoleucine substitution at amino acid 267 in the protein product (c.G802A; p.V267I) (data not shown). The panel of human GCT samples was also examined for the V267I change, however in all cases they were homozygous for a valine at this position (n = 22; data not shown).
Gene Expression Analysis of IKK Complex Subunits in Cell Lines and Ovarian Tissue Samples
Quantitative real-time RT-PCR analysis was employed to examine the expression profile of the IKBKB, IKBKA, and IKBKG genes in the KGN and COV434 cell lines as well as a panel of human GCT samples (n = 16) and whole normal ovaries (n = 10) (Fig. 9). The clinical details of the GCT and normal ovarian samples examined are listed in Table 1. Only histopathologically classified adult and juvenile tumor specimens that were FOXL2 mutation positive and negative, respectively, were included in the analysis. IKBKB expression levels were remarkably constant across the panel of adult GCT (samples 1–15). Of the three histopathologically classified juvenile GCT samples, two exhibited lower levels of IKKβ expression compared to adult GCT and normal ovarian samples. The two cell lines showed a slight increase in expression when compared to levels across the adult GCT panel of samples however, in comparison to the relatively heterogeneous levels seen in the panel of normal ovaries, they were not remarkable. IKBKA was apparently expressed at much lower levels compared to IKBKB and was somewhat variable across both the GCT and normal ovarian tissue panels. IKBKG gene expression levels were in a similar range to that of IKBKB and again, expression levels were consistent across both the GCT and normal ovarian tissue samples. None of the three genes examined showed an increased level of expression in tumors when compared to that of the normal ovaries. The apparent dramatically higher expression of these genes in the COV434 cell line needs to be interpreted with some caution as, in contrast to the tumor panel and the KGN cells, they have rather low levels of the β2-microglobulin gene, in retrospect, it is probably not the optimal “housekeeping gene” in these cells.
Fig. 9.

Gene expression analysis of the IKK complex subunits in ovarian tissue samples. Reverse-transcribed cDNA was amplified using primers specific to each of the human IKBKB (a), IKBKA (b), and IKBKG (c) genes. Real-time PCR analysis was performed to determine mRNA expression levels in adult (n = 15) and juvenile (n = 3) GCT, two human GCT-derived cell lines (KGN and COV434) and normal ovarian tissue (n = 10). Each bar represents the mean ± SEM of three independent experiments performed in triplicate
siRNA Silencing of IKBKB Expression in COV434 and KGN Cells
To further elucidate the role, if any, of IKKβ in the aberrant activation of the NFκB pathway, small interfering RNA (siRNA) was used to examine the effect of abolishing IKKβ protein expression on NFκB activity in the COV434 and KGN cell lines. Silencing of IKKβ expression had no effect on constitutive activity in either COV434 or KGN cells (Fig. 10).
Fig. 10.

siRNA silencing of IKKβ gene expression does not inhibit NFκB constitutive activity in COV434 and KGN cells. a KGN and COV434 cells were transfected with 50 nM control-siRNA or IKKβ-siRNA for 48 h. Knockdown was confirmed by Western blot analysis. b Following 48 h of siRNA transfection, cells were transfected with 0.5 μg of pNFκB-Luc for a further 24 h. Firefly luciferase activity was measured and expressed as arbitrary units relative to Mock-siRNA-transfected cells. The results from three separate experiments, each of which was performed in sextuplicate, are shown as mean ± SEM. EV, empty vector
Discussion
Granulosa cell tumors of the ovary (GCT) represent a specific subset of malignant ovarian tumors and exhibit a molecular phenotype and gene expression profile that is consistent with proliferating granulosa cells of the late preovulatory follicle [1, 24]. Consistent with normal FSH-responsive granulosa cells, GCT exhibit expression of the FSH receptor gene [30], FSH binding [48, 49], estrogen synthesis [50], ERβ expression [8], and expression of the inhibin subunit genes with synthesis of biologically active inhibin [23, 25, 51]. Given their molecular phenotype, it is our hypothesis that GCT arise from granulosa cells through a limited set of molecular events which result in the aberrant activation of specific signaling pathways known to be important in granulosa cell proliferation.
The KGN cell line, derived from a recurrent metastatic GCT, expresses aromatase activity that can be further stimulated by FSH [32]. Similarly, the COV434 cell line is also FSH responsive, with increased estradiol secretion observed upon treatment with FSH [52, 53]. Using the GCT-derived cell lines as experimental models for the analysis of GCT, we sought to gain insight into ERβ function in granulosa cells and its contribution to the pathogenesis of GCT [9]. The results revealed that both the NFκB and AP-1 transcription factors are constitutively activated in COV434 and KGN cell lines and that the functional consequence of NFκB but not AP-1 activation is the transrepression of ERβ-mediated activation in these cells [9]. Furthermore, inhibition of NFκB signaling downregulated the constitutive activity [9]. To date, little is known about the function of NFκB in normal granulosa cells. Wang et al. [54] reported that the NFκB pathway mediates the FSH-induced up-regulation of X-linked inhibitor of apoptosis protein (XIAP) expression in rodent granulosa cells, contributing to follicular growth. Recently, Woods et al. [55] have shown that the Toll-like receptor, TLR4, is located on the surface of both COV434 and KGN cells; when TLR4 is stimulated by lipopolysaccharide there is an increase in NFκB activity.
Several subsequent studies contribute to our understanding of the possible mechanism of constitutive NFκB pathway activation. ERK1 and ERK2 were shown to be constitutively active in KGN cells with siRNA silencing of ERK1/2 protein expression resulting in the suppression of cell proliferation [56]. This suggests that the constitutive AP-1 activation observed by Chu et al. [9] is being driven via constitutive activation of an upstream component of the classical MAPK/ERK pathway. Indeed, Chu et al. [9] also showed that targeting ERK with a specific chemical inhibitor (PD98059) fully abrogated the constitutive activity of AP-1 in both COV434 and KGN cell lines. There is cross-talk between the MAPK/ERK pathway and the NFκB signaling pathway; thus, for instance, in melanoma cells the activating mutation of BRAF (V600E) results in activation of NFκB signaling with a consequent impact on tumorigenesis [57]. When taken together, these various studies and the known biology argue for a role for NFκB constitutive activity contributing to the unopposed proliferation of granulosa cells and the pathogenesis of granulosa cell tumors. In light of these findings, we sought to determine the mechanism(s) of constitutive NFκB pathway activation in the COV434 and KGN cell lines and to examine the functional consequence of targeted pathway inhibition.
In establishing positive controls in these cell systems, it was observed that whilst IL-1 was able to activate the NFκB signaling pathway in the KGN cell line, COV434 cells were IL-1 resistant. This differential response to IL-1 was observed despite the two cell lines exhibiting equivalent expression of the IL-1 receptor gene (Supplementary Table 2), suggesting that the COV434 cells’ resistance originates from a mechanism downstream of the receptor. It is possible that the loss of the IL-1 response within the COV434 cells is due to the phenomenon of genetic drift, whereby frequent rounds of passaging can lead to the loss and/or gain of anomalous cellular properties. Alternatively however, in light of the differential FOXL2 mutation status of the KGN cells compared to COV434 [3], it may be that the loss of the IL-1 response is part of the pathogenesis of juvenile GCT which may warrant full characterization. Thus, the expression of the IL-1 receptor at a protein level needs to be determined as well as exploring its cellular localization and the phosphorylation status of its downstream targets.
Given that Chu et al. [9] observed inhibition of NFκB activity upon treatment of cells with the IκBα-specific chemical inhibitor, BAY11-7082, the functional consequence of this inhibition, was examined and found to be a dose-dependent decrease in cell proliferation and cell viability and a dose-dependent increase in apoptosis. These data were also confirmed with an independent inhibitor of IκBα phosphorylation, IMD-0354. These results not only confirmed that the mechanism of constitutive pathway activation is likely to originate upstream of IκBα phosphorylation in the GCT-derived cell lines but also demonstrate that unopposed NFκB signaling mediates the properties of oncogenic transformation in granulosa cells, that is, enhanced growth activity and protection from apoptotic cell death, and that inhibition of this pathway attenuates these cellular functions.
In order to further characterize the NFκB pathway, and given the high IRAK-1 mRNA expression levels observed in both cell lines, an IRAK-1/4-specific chemical inhibitor was employed. Although the IRAK-1/4 inhibitor had no effect on constitutive activity in either cell line, it did inhibit IL-1-induced activity in the KGN cells. Similarly, in order to specifically target the TNFα-mediated pathway of NFκB activation, a RIP1 kinase-specific chemical inhibitor, Necrostatin-1, was employed but shown to have no effect.
As both the TNFα and IL-1 pathways of NFκB activation converge at the IKK complex, and the complex itself is considered to be the core component of the NFκB cascade, IKKβ, the catalytic subunit most commonly involved in the activation of the canonical pathway was targeted [58]. Surprisingly however, whilst two independent inhibitors of IKKβ, SC-514, and PS-1145, had no effect on constitutive NFκB activity in both cell lines, they also had no effect on TNFα-induced activity in both the COV434 and KGN cell lines and IL-1-induced activity in the KGN cells. Both compounds were able to inhibit TNFα-induced NFκB activity in a dose-dependent manner in the T47D cell line, in which NFκB activity is known to be intact [45]. Together these data suggest that the lack of response observed in the GCT-derived cell lines was specific to these cells and points to a difference in the IKK complex that might be relevant to the constitutive activation of the NFκB pathway. Although no mutations were identified by direct sequencing of the coding regions of the IKKβ, IKKα, and NEMO genes, a single-nucleotide polymorphism was identified in the IKKβ gene in the KGN cell line (c.G1577A; p.R526G). This polymorphism occurs with uncommon frequency in certain Asian populations, and was present in the KGN cell line which was generated from a tumor removed from a Japanese woman, yet absent in the COV434 cells which were generated in the Netherlands. Whilst unlikely to be the cause of the constitutive activity, further investigation into whether the polymorphism may have contributed to the KGN cells’ lack of response to the two IKKβ-specific inhibitors was warranted (Supplementary Figs. 2 and 3). Moreover, the presence of the FOXL2 C134W mutation in the KGN cells [59, 60] and absence from the COV434 cells [3] strongly suggests that these cell lines are derived from adult and juvenile GCT, respectively. Given that they represent different GCT subtypes [2, 3], it is possible that the mechanism of NFκB constitutive activation and/or resistance to the IKKβ inhibitors may differ between the two cell lines. Preliminary experiments utilizing an IKKβ null mouse embryonic fibroblast cell line suggest that the polymorphism does not contribute to constitutive NFκB activity or resistance to IKKβ-specific inhibitors (Supplementary Figs. 2 and 3).
Although a nonconservative single-nucleotide polymorphism was also identified in IKKα in the COV434 and KGN cells, it is unlikely that it is contributing to the pathway’s constitutive activation, but remains to be formally excluded. Moreover, quantification of the mRNA levels of the three components of the IKK complex does not suggest that overexpression of these genes are a contributing factor in aberrant pathway activation.
Given that no evidence of mutation and/or overexpression of the components of the IKK complex was observed in either cell line, and that the IKKβ R526Q SNP identified in the KGN cell line did not appear to contribute to either constitutive activity or resistance to IKKβ-specific chemical inhibitors, it may be that signaling is occurring independently of IKKβ or the IKK complex. Although we are unable to distinguish between the two mechanisms, the results of the IKKβ-siRNA knockdown experiments suggest that constitutive NFκB signaling may be occurring independently of IKKβ.
Whether constitutively activated NFκB signaling also occurs in vivo in GCT remains to be determined. Whilst pathway activation would ideally be evaluated by examining protein–DNA binding and/or phosphorylation status of the NFκB subunits in tumor extracts, the relative infrequency with which GCT occur in the population and the low accessible numbers of tumor tissue renders these approaches unfeasible. As an alternative, a gene expression signature induced by NFκB activation in the COV434 and KGN cells might be identified to enable comparison with the gene expression profiles from a panel of human GCT, as has been reported for serous ovarian carcinoma [61]. In a small cohort of GCT examined, we have found RelA/p65 immunostaining to be nuclear, a surrogate marker of activation (A. Drummond, unpublished observation). In addition, expression of the NFκB regulated gene, XIAP, is upregulated in GCT (S. Chu, unpublished observation). Both of these findings are consistent with, but do not prove, NFκB activation in vivo.
It should be noted that the KGN and COV434 cell lines are commonly used as models of normal human granulosa cells and do not factor into analysis the constitutive NFκB activity. It seems a timely reminder that these cell lines are derived from granulosa cell tumors and clearly exhibit malignant properties.
The identification of the FOXL2 C134W somatic mutation has, in some respects, “reset” the field of GCT research. The universal presence of the mutation in adult GCT also argues for a role in tumor initiation rather than disease progression. Moreover, the absence of the mutation in juvenile GCT confirms that this subtype is a distinct disease and that its molecular etiology is different to that of adult GCT. Therefore, we hypothesize that if the FOXL2 mutation is the oncogenic trigger, constitutive activation of the NFκB pathway promotes tumor progression and recurrence/metastasis. This study has furthered our understanding of the role constitutively activated NFκB plays in the pathogenesis of GCT and has suggested a possible role for IKKβ, another component of the IKK complex, or kinase(s) directly involved in the phosphorylation of the IKK complex subunits. Activation via a non-classical pathway, however, remains a possibility. Characterization of the NFκB signaling pathway in the context of two GCT-derived cell lines provides insight into the mechanisms of aberrant pathway activation, and also prompts further investigation into its role in GCT pathogenesis.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(DOC 52 kb)
(DOC 90 kb)
Identification of a heterozygous single-nucleotide change c.G1577A; p.R526Q in the IKBKB gene in the KGN cell line (PPTX 55.8 kb)
Effect of IKKβ R526Q SNP on NFκB signaling. IKKβ−/− mouse embryonic fibroblasts were transiently transfected with 0.5 μg of the NFκB reporter gene plasmid alone (pNFκB-Luc black bar) and then with the addition of 50 ng of either vector control (pRK5 green bar), wild-type IKKβ (pRK5-C-Flag-IKKβ blue bar) or IKKβ containing the R526Q SNP (pRK5-C-Flag-IKKβR526Q orange bar) ± IL-1α for 24 h. Firefly luciferase activity was measured and expressed as arbitrary units relative to cells transfected with pNFκB-Luc alone and treated with vehicle. An equivalent increase in luciferase activity in cells transfected with wild-type IKKβ and cells transfected with IKKβ containing the R526Q SNP following treatment with both vehicle or IL-1α demonstrated that the R526Q SNP was not constitutively active, nor altered the response under these conditions, from that observed for the wild-type sequence. The results from three separate experiments, each of which was performed in sextuplicate, are shown as mean ± SEM. ***p < 0.001; ns, not significant (PPTX 72 kb)
Contribution of IKKβ R526Q SNP to GCT-derived cell lines’ resistance to IKKβ specific inhibitors. IKKβ−/− MEFs were transiently co-transfected with 0.5 μg of pNFκB-Luc (<) and 50 ng of either vector control (pRK5<), wild-type IKKβ (pRK5-C-Flag-IKKβ <) or IKKβ containing the R526Q SNP (pRK5-C-Flag-IKKβR526Q<). Twenty-four hours post transfection, cells were treated with either vehicle or IL-1α (10 ng/ml) ± SC-514 (50 µM) and incubated at 37 °C for 24h. Firefly luciferase activity was measured and expressed as arbitrary units relative to cells transfected with pNFκB-Luc alone and treated with vehicle. No difference in luciferase activity was observed between cells transfected with wild-type IKKβ and IKKβ containing the R526Q SNP when treated with SC-514, either in the presence or absence of IL-1α, suggesting the R526Q SNP is not responsible for the resistance of the KGN cells to the IKKβ specific chemical inhibitors. The results from three separate experiments, each of which was performed in sextuplicate, are shown as mean ± SEM. #, not significant; *p<0.05; when wild-type IKKβ was compared to empty vector or IKKβ containing the R526Q SNP was compared to wild-type IKKβ in the presence of vehicle, IL-1α and/or SC-514 (PPTX 65.4 kb)
Acknowledgments
We thank Dr. Simon Chu for helpful discussions and Maria Alexiadis for outstanding technical assistance. Direct sequencing was performed by the Gandel Charitable Trust Sequencing Centre at the Monash Health Translation Precinct, Clayton 3168, Australia. The IKKβ-/- MEF cells were kindly provided by Associate Professor Paul Ekert, Walter and Eliza Hall Institute, Parkville 3052, Australia, and the pRK5-C-Flag-IKKβ construct by Dr. Ashley Mansell, Monash Institute of Medical Research, Clayton 3168, Australia. This work was supported by grants-in-aid from Cancer Council Victoria, the National Australia Bank Ovarian Cancer Research Foundation, the Granulosa Cell Tumor of the Ovary Foundation, and the National Health and Medical Research Council of Australia through a Senior Principal Research Fellowship to PJF (no. 1002559) and a Dora Lush Biomedical Postgraduate Research Scholarship to SJ (no. 441132). SJ was also in receipt of a Faculty of Medicine Postgraduate Excellence Award from Monash University. Prince Henry’s Institute is supported by the Victorian Government's Operational Infrastructure Support program. PHI Internal Data Audit no. 13-04.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Fuller PJ, Chu S, Fikret S, Burger HG. Molecular pathogenesis of granulosa cell tumours. Mol Cell Endocrinol. 2002;191(1):89–96. doi: 10.1016/S0303-7207(02)00059-X. [DOI] [PubMed] [Google Scholar]
- 2.Jamieson S, Fuller PJ. Molecular pathogenesis of granulosa cell tumors of the ovary. Endocr Rev. 2012;33(1):109–144. doi: 10.1210/er.2011-0014. [DOI] [PubMed] [Google Scholar]
- 3.Jamieson S, Butzow R, Andersson N, Alexiadis M, Unkila-Kallio L, Heikinheimo M, Fuller PJ, Anttonen M. The FOXL2 C134W mutation is characteristic of adult granulosa cell tumors of the ovary. Mod Pathol. 2010;23(11):1477–1485. doi: 10.1038/modpathol.2010.145. [DOI] [PubMed] [Google Scholar]
- 4.Kim MS, Hur SY, Yoo NJ, Lee SH. Mutational analysis of FOXL2 codon 134 in granulosa cell tumour of ovary and other human cancers. J Pathol. 2010;221(2):147–152. doi: 10.1002/path.2688. [DOI] [PubMed] [Google Scholar]
- 5.Kim T, Sung CO, Song SY, Bae DS, Choi YL. FOXL2 mutation in granulosa-cell tumours of the ovary. Histopathology. 2010;56(3):408–410. doi: 10.1111/j.1365-2559.2010.03487.x. [DOI] [PubMed] [Google Scholar]
- 6.Shah SP, Kobel M, Senz J, Morin RD, Clarke BA, Wiegand KC, Leung G, et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N Engl J Med. 2009;360(26):2719–2729. doi: 10.1056/NEJMoa0902542. [DOI] [PubMed] [Google Scholar]
- 7.Alexiadis M, Eriksson N, Jamieson S, Davis M, Drummond AE, Chu S, Clyne CD, Muscat GE, Fuller PJ. Nuclear receptor profiling of ovarian granulosa cell tumors. Horm Cancer. 2011;2(3):157–169. doi: 10.1007/s12672-011-0069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu S, Mamers P, Burger HG, Fuller PJ. Estrogen receptor isoform gene expression in ovarian stromal and epithelial tumors. J Clin Endocrinol Metab. 2000;85(3):1200–1205. doi: 10.1210/jc.85.3.1200. [DOI] [PubMed] [Google Scholar]
- 9.Chu S, Nishi Y, Yanase T, Nawata H, Fuller PJ. Transrepression of estrogen receptor beta signaling by nuclear factor-κB in ovarian granulosa cells. Mol Endocrinol. 2004;18(8):1919–1928. doi: 10.1210/me.2004-0021. [DOI] [PubMed] [Google Scholar]
- 10.Bilandzic M, Chu S, Wang Y, Tan HL, Fuller PJ, Findlay JK, Stenvers KL. Betaglycan alters NFkappaB-TGFbeta2 cross talk to reduce survival of human granulosa tumor cells. Mol Endocrinol. 2013;27(3):466–479. doi: 10.1210/me.2012-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. NF-kappaB addiction and its role in cancer: ‘one size does not fit all’. Oncogene. 2011;30(14):1615–1630. doi: 10.1038/onc.2010.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18(49):6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 13.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278(5339):860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 14.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388(6642):548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 15.Rothwarf DM, Zandi E, Natoli G, Karin M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395(6699):297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 16.Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, Kirk HE, Kay RJ, Israel A. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell. 1998;93(7):1231–1240. doi: 10.1016/S0092-8674(00)81466-X. [DOI] [PubMed] [Google Scholar]
- 17.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267(5203):1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 18.Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 1995;14(12):2876–2883. doi: 10.1002/j.1460-2075.1995.tb07287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scherer DC, Brockman JA, Chen Z, Maniatis T, Ballard DW. Signal-induced degradation of I kappa B alpha requires site-specific ubiquitination. Proc Natl Acad Sci U S A. 1995;92(24):11259–11263. doi: 10.1073/pnas.92.24.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 21.Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25(51):6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 22.Jobling T, Mamers P, Healy DL, MacLachlan V, Burger HG, Quinn M, Rome R, Day AJ. A prospective study of inhibin in granulosa cell tumors of the ovary. Gynecol Oncol. 1994;55(2):285–289. doi: 10.1006/gyno.1994.1291. [DOI] [PubMed] [Google Scholar]
- 23.Healy DL, Burger HG, Mamers P, Jobling T, Bangah M, Quinn M, Grant P, Day AJ, Rome R, Campbell JJ. Elevated serum inhibin concentrations in postmenopausal women with ovarian tumors. N Engl J Med. 1993;329(21):1539–1542. doi: 10.1056/NEJM199311183292104. [DOI] [PubMed] [Google Scholar]
- 24.Chu S, Rushdi S, Zumpe ET, Mamers P, Healy DL, Jobling T, Burger HG, Fuller PJ. FSH-regulated gene expression profiles in ovarian tumours and normal ovaries. Mol Hum Reprod. 2002;8(5):426–433. doi: 10.1093/molehr/8.5.426. [DOI] [PubMed] [Google Scholar]
- 25.Fuller PJ, Chu S, Jobling T, Mamers P, Healy DL, Burger HG. Inhibin subunit gene expression in ovarian cancer. Gynecol Oncol. 1999;73(2):273–279. doi: 10.1006/gyno.1999.5356. [DOI] [PubMed] [Google Scholar]
- 26.Jamieson S, Alexiadis M, Fuller PJ. Expression status and mutational analysis of the ras and B-raf genes in ovarian granulosa cell and epithelial tumors. Gynecol Oncol. 2004;95(3):603–609. doi: 10.1016/j.ygyno.2004.07.062. [DOI] [PubMed] [Google Scholar]
- 27.Alexiadis M, Mamers P, Chu S, Fuller PJ. Insulin-like growth factor, insulin-like growth factor-binding protein-4, and pregnancy-associated plasma protein-A gene expression in human granulosa cell tumors. Int. J. Gynecol. Cancer. 2006;16(6):1973–1979. doi: 10.1111/j.1525-1438.2006.00724.x. [DOI] [PubMed] [Google Scholar]
- 28.Bittinger S, Alexiadis M, Fuller PJ. Expression status and mutational analysis of the PTEN and P13K subunit genes in ovarian granulosa cell tumors. Int. J. Gynecol. Cancer. 2009;19(3):339–342. doi: 10.1111/IGC.0b013e3181a1cdfd. [DOI] [PubMed] [Google Scholar]
- 29.Chu S, Alexiadis M, Fuller PJ. Expression, mutational analysis and in vitro response of imatinib mesylate and nilotinib target genes in ovarian granulosa cell tumors. Gynecol Oncol. 2008;108(1):182–190. doi: 10.1016/j.ygyno.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 30.Fuller PJ, Verity K, Shen Y, Mamers P, Jobling T, Burger HG. No evidence of a role for mutations or polymorphisms of the follicle-stimulating hormone receptor in ovarian granulosa cell tumors. J Clin Endocrinol Metab. 1998;83(1):274–279. doi: 10.1210/jc.83.1.274. [DOI] [PubMed] [Google Scholar]
- 31.van den Berg-Bakker CA, Hagemeijer A, Franken-Postma EM, Smit VT, Kuppen PJ, van Ravenswaay Claasen HH, Cornelisse CJ, Schrier PI. Establishment and characterization of 7 ovarian carcinoma cell lines and one granulosa tumor cell line: growth features and cytogenetics. Int J Cancer. 1993;53(4):613–620. doi: 10.1002/ijc.2910530415. [DOI] [PubMed] [Google Scholar]
- 32.Nishi Y, Yanase T, Mu Y, Oba K, Ichino I, Saito M, Nomura M, et al. Establishment and characterization of a steroidogenic human granulosa-like tumor cell line, KGN, that expresses functional follicle-stimulating hormone receptor. Endocrinology. 2001;142(1):437–445. doi: 10.1210/en.142.1.437. [DOI] [PubMed] [Google Scholar]
- 33.Cao Z, Henzel WJ, Gao X. IRAK: a kinase associated with the interleukin-1 receptor. Science. 1996;271(5252):1128–1131. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- 34.Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science. 1997;278(5343):1612–1615. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- 35.Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4(4):387–396. doi: 10.1016/S1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 36.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8(3):297–303. doi: 10.1016/S1074-7613(00)80535-X. [DOI] [PubMed] [Google Scholar]
- 37.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell. 2006;22(2):245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 38.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999;284(5412):321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 40.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J. Exp. Med. 1999;189(11):1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chu WM, Ostertag D, Li ZW, Chang L, Chen Y, Hu Y, Williams B, Perrault J, Karin M. JNK2 and IKKbeta are required for activating the innate response to viral infection. Immunity. 1999;11(6):721–731. doi: 10.1016/S1074-7613(00)80146-6. [DOI] [PubMed] [Google Scholar]
- 42.Kishore N, Sommers C, Mathialagan S, Guzova J, Yao M, Hauser S, Huynh K, et al. A selective IKK-2 inhibitor blocks NF-kappa B-dependent gene expression in interleukin-1 beta-stimulated synovial fibroblasts. J Biol Chem. 2003;278(35):32861–32871. doi: 10.1074/jbc.M211439200. [DOI] [PubMed] [Google Scholar]
- 43.Castro AC, Dang LC, Soucy F, Grenier L, Mazdiyasni H, Hottelet M, Parent L, Pien C, Palombella V, Adams J. Novel IKK inhibitors: beta-carbolines. Bioorg. Med. Chem. Lett. 2003;13(14):2419–2422. doi: 10.1016/S0960-894X(03)00408-6. [DOI] [PubMed] [Google Scholar]
- 44.Yemelyanov A, Gasparian A, Lindholm P, Dang L, Pierce JW, Kisseljov F, Karseladze A, Budunova I. Effects of IKK inhibitor PS1145 on NF-kappaB function, proliferation, apoptosis and invasion activity in prostate carcinoma cells. Oncogene. 2006;25(3):387–398. doi: 10.1038/sj.onc.1209066. [DOI] [PubMed] [Google Scholar]
- 45.Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ, Jr, Sledge GW., Jr Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol Cell Biol. 1997;17(7):3629–3639. doi: 10.1128/mcb.17.7.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006(357):re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 47.Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, Elia AJ, Mak TW. Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev. 2000;14(7):854–862. [PMC free article] [PubMed] [Google Scholar]
- 48.Stouffer RL, Grodin MS, Davis JR, Surwit EA. Investigation of binding sites for follicle-stimulating hormone and chorionic gonadotropin in human ovarian cancers. J Clin Endocrinol Metab. 1984;59(3):441–446. doi: 10.1210/jcem-59-3-441. [DOI] [PubMed] [Google Scholar]
- 49.Graves PE, Surwit EA, Davis JR, Stouffer RL. Adenylate cyclase in human ovarian cancers: sensitivity to gonadotropins and nonhormonal activators. Am J Obstet Gynecol. 1985;153(8):877–882. doi: 10.1016/0002-9378(85)90693-3. [DOI] [PubMed] [Google Scholar]
- 50.Amsterdam A, Selvaraj N. Control of differentiation, transformation, and apoptosis in granulosa cells by oncogenes, oncoviruses, and tumor suppressor genes. Endocr Rev. 1997;18(4):435–461. doi: 10.1210/er.18.4.435. [DOI] [PubMed] [Google Scholar]
- 51.Lappöhn RE, Burger HG, Bouma J, Bangah M, Krans M, de Bruijn HW. Inhibin as a marker for granulosa-cell tumors. N Engl J Med. 1989;321(12):790–793. doi: 10.1056/NEJM198909213211204. [DOI] [PubMed] [Google Scholar]
- 52.Zhang H, Vollmer M, De Geyter M, Litzistorf Y, Ladewig A, Durrenberger M, Guggenheim R, Miny P, Holzgreve W, De Geyter C. Characterization of an immortalized human granulosa cell line (COV434) Mol Hum Reprod. 2000;6(2):146–153. doi: 10.1093/molehr/6.2.146. [DOI] [PubMed] [Google Scholar]
- 53.Havelock JC, Rainey WE, Carr BR. Ovarian granulosa cell lines. Mol Cell Endocrinol. 2004;228(1–2):67–78. doi: 10.1016/j.mce.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 54.Wang Y, Chan S, Tsang BK. Involvement of inhibitory nuclear factor-kappaB (NFkappaB)-independent NFkappaB activation in the gonadotropic regulation of X-linked inhibitor of apoptosis expression during ovarian follicular development in vitro. Endocrinology. 2002;143(7):2732–2740. doi: 10.1210/en.143.7.2732. [DOI] [PubMed] [Google Scholar]
- 55.Woods DC, White YA, Dau C, Johnson AL. TLR4 activates NF-kappaB in human ovarian granulosa tumor cells. Biochem Biophys Res Commun. 2011;409(4):675–680. doi: 10.1016/j.bbrc.2011.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Steinmetz R, Wagoner HA, Zeng P, Hammond JR, Hannon TS, Meyers JL, Pescovitz OH. Mechanisms regulating the constitutive activation of the extracellular signal-regulated kinase (ERK) signaling pathway in ovarian cancer and the effect of ribonucleic acid interference for ERK1/2 on cancer cell proliferation. Mol Endocrinol. 2004;18(10):2570–2582. doi: 10.1210/me.2004-0082. [DOI] [PubMed] [Google Scholar]
- 57.Liu J, Suresh Kumar KG, Yu D, Molton SA, McMahon M, Herlyn M, Thomas-Tikhonenko A, Fuchs SY. Oncogenic BRAF regulates beta-Trcp expression and NF-kappaB activity in human melanoma cells. Oncogene. 2007;26(13):1954–1958. doi: 10.1038/sj.onc.1209994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26(3):203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benayoun BA, Caburet S, Dipietromaria A, Georges A, D'Haene B, Pandaranayaka PJ, L'Hote D, et al. Functional exploration of the adult ovarian granulosa cell tumor-associated somatic FOXL2 mutation p.Cys134Trp (c.402C>G) PLoS One. 2010;5(1):e8789. doi: 10.1371/journal.pone.0008789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schrader KA, Gorbatcheva B, Senz J, Heravi-Moussavi A, Melnyk N, Salamanca C, Maines-Bandiera S, et al. The specificity of the FOXL2 c.402C>G Somatic mutation: a survey of solid tumors. PLoS One. 2009;4(11):e7988. doi: 10.1371/journal.pone.0007988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hernandez L, Hsu SC, Davidson B, Birrer MJ, Kohn EC, Annunziata CM. Activation of NF-kappaB signaling by inhibitor of NF-kappaB kinase beta increases aggressiveness of ovarian cancer. Cancer Res. 2010;70(10):4005–4014. doi: 10.1158/0008-5472.CAN-09-3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 52 kb)
(DOC 90 kb)
Identification of a heterozygous single-nucleotide change c.G1577A; p.R526Q in the IKBKB gene in the KGN cell line (PPTX 55.8 kb)
Effect of IKKβ R526Q SNP on NFκB signaling. IKKβ−/− mouse embryonic fibroblasts were transiently transfected with 0.5 μg of the NFκB reporter gene plasmid alone (pNFκB-Luc black bar) and then with the addition of 50 ng of either vector control (pRK5 green bar), wild-type IKKβ (pRK5-C-Flag-IKKβ blue bar) or IKKβ containing the R526Q SNP (pRK5-C-Flag-IKKβR526Q orange bar) ± IL-1α for 24 h. Firefly luciferase activity was measured and expressed as arbitrary units relative to cells transfected with pNFκB-Luc alone and treated with vehicle. An equivalent increase in luciferase activity in cells transfected with wild-type IKKβ and cells transfected with IKKβ containing the R526Q SNP following treatment with both vehicle or IL-1α demonstrated that the R526Q SNP was not constitutively active, nor altered the response under these conditions, from that observed for the wild-type sequence. The results from three separate experiments, each of which was performed in sextuplicate, are shown as mean ± SEM. ***p < 0.001; ns, not significant (PPTX 72 kb)
Contribution of IKKβ R526Q SNP to GCT-derived cell lines’ resistance to IKKβ specific inhibitors. IKKβ−/− MEFs were transiently co-transfected with 0.5 μg of pNFκB-Luc (<) and 50 ng of either vector control (pRK5<), wild-type IKKβ (pRK5-C-Flag-IKKβ <) or IKKβ containing the R526Q SNP (pRK5-C-Flag-IKKβR526Q<). Twenty-four hours post transfection, cells were treated with either vehicle or IL-1α (10 ng/ml) ± SC-514 (50 µM) and incubated at 37 °C for 24h. Firefly luciferase activity was measured and expressed as arbitrary units relative to cells transfected with pNFκB-Luc alone and treated with vehicle. No difference in luciferase activity was observed between cells transfected with wild-type IKKβ and IKKβ containing the R526Q SNP when treated with SC-514, either in the presence or absence of IL-1α, suggesting the R526Q SNP is not responsible for the resistance of the KGN cells to the IKKβ specific chemical inhibitors. The results from three separate experiments, each of which was performed in sextuplicate, are shown as mean ± SEM. #, not significant; *p<0.05; when wild-type IKKβ was compared to empty vector or IKKβ containing the R526Q SNP was compared to wild-type IKKβ in the presence of vehicle, IL-1α and/or SC-514 (PPTX 65.4 kb)