

Abstract
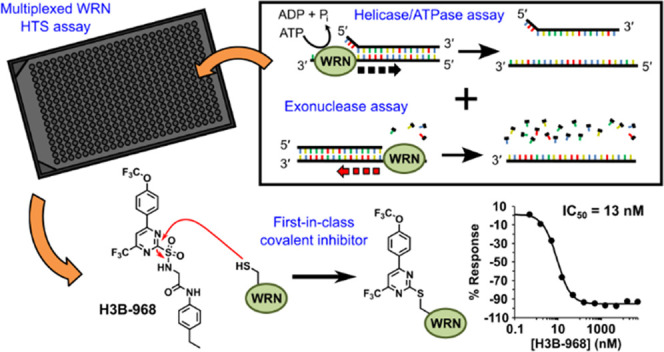
Werner syndrome protein (WRN) is a multifunctional enzyme with helicase, ATPase, and exonuclease activities that are necessary for numerous DNA-related transactions in the human cell. Recent studies identified WRN as a synthetic lethal target in cancers characterized by genomic microsatellite instability resulting from defects in DNA mismatch repair pathways. WRN’s helicase activity is essential for the viability of these high microsatellite instability (MSI-H) cancers and thus presents a therapeutic opportunity. To this end, we developed a multiplexed high-throughput screening assay that monitors exonuclease, ATPase, and helicase activities of full-length WRN. This screening campaign led to the discovery of 2-sulfonyl/sulfonamide pyrimidine derivatives as novel covalent inhibitors of WRN helicase activity. The compounds are specific for WRN versus other human RecQ family members and show competitive behavior with ATP. Examination of these novel chemical probes established the sulfonamide NH group as a key driver of compound potency. One of the leading compounds, H3B-960, showed consistent activities in a range of assays (IC50 = 22 nM, KD = 40 nM, KI = 32 nM), and the most potent compound identified, H3B-968, has inhibitory activity IC50 ∼ 10 nM. These kinetic properties trend toward other known covalent druglike molecules. Our work provides a new avenue for screening WRN for inhibitors that may be adaptable to different therapeutic modalities such as targeted protein degradation, as well as a proof of concept for the inhibition of WRN helicase activity by covalent molecules.
Introduction
The five human RecQ helicases play an important role in maintaining genomic integrity and stability, often being regarded as “guardians of the genome”.1 Indeed, three of these helicases have known inactivating mutations, any of which results in heritable disorders with characteristics that include genomic instability and a predisposition to developing cancer.2 WRN is one of these enzymes where inactivation results in the development of the progeroid disease Werner’s Syndrome. WRN and the other RecQ helicases translocate along single-stranded (ss) DNA and unwind a variety of double- or multistranded DNA structures with 3′ → 5′ directionality in an ATP-dependent manner.3 WRN also possesses an N-terminal domain with 3′ → 5′ exonuclease activity that releases deoxynucleoside monophosphates from 3′-recessed ends of double-stranded (ds) DNA.4,5 This activity is unique to WRN within the RecQ helicase family.
A sizable subset of endometrial, ovarian, colorectal, and gastric cancers is characterized by frequent insertion/deletion mutations in microsatellite regions of the genome that result from the inactivation of key genes in DNA mismatch repair pathways.6−9 While patients with MSI-H cancers generally respond well to immune checkpoint inhibitors (ICIs) with an overall response rate of ∼40%, there is still a significant population of nonresponders.10 Several groups identified WRN as a synthetic lethal vulnerability in MSI-H cell lines by mining the results of genome-wide RNAi or CRISPR gene inactivation studies in a large panel of cell lines.11−15 This vulnerability was linked to WRN’s helicase activity,12−14 which is required to unwind AT-rich DNA secondary structures that become more prevalent in MSI-H cells and are targets for MUS18-mediated fragmentation of the genome.16 Together with the observation that WRN knockdown is tolerated in microsatellite stable (MSS) cell lines,13 these results indicate that WRN helicase inhibitors may provide a new therapeutic option for MSI-H cancers.
These target validation data have placed WRN squarely in the crosshairs of drug discovery efforts in both academia and industry, and several chemical series have been reported that inhibit the helicase activity in the low micromolar range.17,18 Historically, helicases have been challenging targets to drug, with low hit rates from high-throughput screening campaigns and a bias toward finding undesired DNA intercalators.19 We therefore sought to screen full-length WRN using a multiplexed assay measuring its ATPase, helicase, and exonuclease activities simultaneously, which could lead to the identification of novel allosteric inhibitors that would otherwise be missed if screening the helicase domain alone. Furthermore, although WRN’s exonuclease activity is dispensable for MSI-H cancer viability, the domain is unique to WRN among the RecQ family of helicases and, therefore, potentially presents an opportunity to develop WRN-specific degraders from exonuclease inhibitors.
We report here the development and utilization of a catch-all, multiplexed activity assay to screen full-length WRN for inhibitors. The assay incorporates an optimized forked DNA substrate with a quenched fluorophore to measure DNA unwinding and ATPase activity in a single-readout, as well as a 3′-recessed RNA:DNA duplex that is degraded to ribonucleoside monophosphates for exonuclease activity detection using a commercially available fluorescence polarization-based assay. The properties of the fluorophores utilized for both readouts were chosen so that no overlap in signals would occur, allowing all WRN activities to be measured in a single-well reaction. Through our screening efforts, we identified 2-sulfonylpyrimidines as novel and selective covalent inhibitors of WRN helicase activity. Structure–activity relationship studies revealed that the 2-sulfonamide analogues of the primary hits were much more potent, and optimization of the substituents on the pyrimidine core and leaving group resulted in the tool compound H3B-968, which exhibited a potency of around 13 nM against WRN helicase activity. Kinetic studies indicate that the potency of 2-sulfonamide pyrimidines is largely driven by an increased binding affinity of WRN for the compounds rather than their chemical reactivity. Mechanistically, we show that these compounds appear to be competitive with ATP, but not with the DNA substrate. In summary, our work provides a proof of concept for the development of selective covalent WRN inhibitors.
Experimental Section
Protein Expression and Purification
Codon-optimized genes were synthesized and subcloned into the indicated vectors (Table S1). For insect cell expression of WRN and BLM, Sf21 cells (1.5–2.0 × 106 cells mL–1) were infected with baculovirus at an MOI of ∼2 and harvested after 72 h. Both full-length and helicase-domain WRN cultures were supplemented with sterile-filtered ZnSO4 to a final concentration of 50 μM prior to infection. WRN exonuclease domain and RecQL1 helicase domain were expressed in Escherichia coli BL21 (DE3) pLysE. Expression was induced at an OD of 0.6–0.9 with 0.2 mM IPTG overnight at 16 °C. For expression of RecQL1 helicase domain, cultures were also supplemented with ZnSO4 to 50 μM for 20 min at 37 °C prior to induction. In general, we find that supplementation of ZnSO4 during the expression of Zn-dependent proteins improves overall yields and specific activity, likely because the otherwise trace level of zinc in the media becomes a limiting reagent during expression. Full details of the purification can be found in the Supporting Information. The final purities for WRN and RecQL1 were ∼95% and for BLM ∼70% as judged by SDS-PAGE (Figure S1).
WRN Substrate Preparation
The oligonucleotides listed in Table S2 were purchased from IDT with standard desalting for unlabeled strands and HPLC purification for labeled strands. Forked helicase substrates and 3′-recessed exonuclease substrates were generated in 1× TE buffer (10 mM Tris pH 8.0, 1 mM EDTA) supplemented with 50 mM NaCl by mixing the coding and noncoding strands in a 1:1 (25 bp helicase and both exonuclease substrates) or 1.2:1 (10 bp substrate) molar ratio, heating to 95 °C for 5 min, and slowly cooling the oligo mixtures to room temperature overnight. Concentrations were estimated using a Nanodrop and the conversion factor of 1 OD260 equaling 33 ng/μL single-stranded DNA, 40 ng/μL single-stranded RNA, and 50 ng/μL for both double-stranded DNA and double-stranded RNA/DNA.
Multiplexed WRN Activity Assay
WRN ATPase, helicase, and exonuclease activities were measured in single-well endpoint assays that combined a standard TAMRA-dequenching DNA unwinding readout17 with the Transcreener AMP2/GMP2 FP kit from Bellbrook Labs (Cat# 3015-100k) that detects ribonucleoside monophosphates (NMPs). FL-WRN (20 nM) was incubated alone or with varying amounts of compound in 3 μL of assay buffer (25 mM Tris pH 8.0, 5 mM NaCl, 2 mM MgCl2, 1 mM TCEP, 0.1 mg/mL BSA, 0.01% (v/v) Triton X-100) for 20 min in 384-well black nonbinding plates (Greiner Cat# 784900). Turnover was initiated by adding an equivalent volume of solution containing 120 μM ATP, 1 μM Hel-10bp substrate, 10 μM 10 bp trap, and 600 nM Exo-hybrid substrate in assay buffer, bringing the total reaction volume to 6 μL. After 30 min at room temperature, activity was quenched by the addition of 3 μL of a solution containing 60 mM EDTA, 2.25 μg/mL detection antibody, and 12 nM AMP/GMP Alexa Fluor 633 tracer in 12.5 mM Tris (pH 7.5), 0.1 mg/mL BSA, 0.01% (v/v) Triton X-100. TAMRA intensity was immediately measured using a Tecan Infinite M1000Pro plate reader with the following settings: λexcitation = 555 nm, λemission = 588 nm, bandwidths = 5 nm, gain = 150, 20 flashes, flash frequency = 400 Hz, integration time = 20 μs, lag time = 0 μs, settle time = 15 ms. Plates were subsequently incubated for 90 min at room temperature before reading the fluorescence polarization on the Tecan plate reader with the following settings: λexcitation = 635 nm, λemission = 675 nm, bandwidths = 5 nm, gain = 125, 10 flashes, settle time = 0 ms, G-factor = 0.9.
TAMRA-Dequenching DNA Unwinding Helicase Assay
The assay for measuring DNA unwinding activity alone was the same as described above for the multiplexed format except that the hybrid exonuclease substrate was omitted from the initiation solution. Endpoint reactions were quenched with 3 μL of 60 mM EDTA and immediately read on the Tecan plate reader. To examine the effect of glutathione (GSH) on compound potency, the order of reagent addition was altered from routine conditions. Varying amounts of compound were incubated in 3 μL of 2 mM GSH and 120 μM ATP in assay buffer for 20 min prior to adding 3 μL of 20 nM FL-WRN, 1 μM Hel-10bp substrate, and 10 μM Trap-10bp.
This assay was also utilized to measure DNA unwinding activity of WRN helicase domain in real time using the kinetic cycle module of the Tecan i-control software. Reactions were 6 μL in volume and consisted of 0.5 nM WRN helicase domain, 500 nM Hel-10bp substrate, 5 μM Trap-10bp, varying concentrations of ATP, and a half log dilution series of compound (top concentration = 50–75 μM) in assay buffer. Enzyme was added last to initiate turnover, and TAMRA intensity was measured every 10 s for 1 h using the same instrument settings as in the endpoint assays. For KI and kinact determination, the ATP concentration was held at 120 μM.
ADP-Glo ATPase assay
WRN ATPase activity was measured directly by ADP formation in an endpoint assay using the ADP-Glo kit from Promega (Cat# V9102). Under standard assay conditions, 20 nM FL-WRN or 1 nM WRN-hd500-946 was incubated alone or with varying amounts of compound in 3 μL of assay buffer for 20 min in 384-well black nonbinding plates. Reactions were initiated by adding 3 μL of a solution containing 120 μM ATP, 200 nM Hel-25bp substrate, and 200 nM 25bp trap in assay buffer (unless noted otherwise for individual experiments). After incubation for 30 min at room temperature, the reaction was terminated by adding 3 μL ADP-Glo reagent and incubating for 40 min at room temperature. Three μL of Kinase Detection reagent were then added, and the reactions were incubated for 30 min at room temperature prior to reading the luminescent signal with an Envision plate reader. In competition experiments utilizing a high final ATP concentration (500 μM to 1 mM), the ADP-Glo Max detection reagent from the ADP-Glo Max kit (Promega Cat# V7002) was utilized. Compound selectivity counter screens against other human RecQL helicases were completed with the ADP-Glo assay using final concentrations of 20 nM enzyme and 8 μM ATP for FL-BLM, and 25 nM enzyme and 500 μM ATP for RecQL1-hd.
Pyruvate Kinase/Lactate Dehydrogenase (PK/LDH) Coupled Assay
Full-length WRN ATPase activity was measured continuously using a PK/LDH coupled spectrophotometric assay. Assays were run in Corning 384-well clear-bottom plates (Cat# 3762) in a total volume of 15 μL containing 250 nM FL-WRN, 750 nM Hel_25bp substrate, 25 mM Tris (pH 8.0), 5 mM KCl, 15 mM MgCl2, 1 mM TCEP, 0.1 mg/mL BSA, 0.01% (v/v) Triton X-100, 1 mM NADH, 2.5 mM phosphoenolpyruvate, 2.6 U pyruvate kinase (174.2 U/mL), 4.1 U lactate dehydrogenase (275.8 U/mL), and ATP from 0 to 10 mM. WRN was added last to initiate turnover, and NADH consumption was followed spectrophotometrically at 340 nm for 20 min using a Tecan Infinite M1000Pro plate reader. Data were recorded every 20 s during the assay. The slopes of the linear portions of the progress curves were plotted against ATP concentration, and the data fit to the Michaelis–Menten equation to determine Km,ATP.
NMP Release Exonuclease Assay
The endpoint assay for measuring NMP release from the 3′-recessed RNA:DNA hybrid substrate was the same as described for the multiplexed format except that ATP, helicase substrate, and helicase trap were omitted from the initiation solution. FL-WRN and the exonuclease domain were assayed at final concentrations of 10 nM and 200 pM, respectively, and exonuclease domain assays were supplemented with MnCl2 to a final concentration of 500 μM.
2-Aminopurine Release Exonuclease Assay
WRN exonuclease activity on a 3′-recessed double-stranded DNA substrate was assessed in an endpoint assay monitoring 2-aminopurine-2′-deoxyribonucleoside monophosphate release. FL-WRN (5 nM) or exonuclease domain was incubated alone or with varying amounts of compound in 3 μL assay buffer for 20 min in 384-well black nonbinding plates. The reaction was initiated by the addition of 3 μL of a solution consisting of 400 nM Exo-2AP substrate and MnCl2 (FL-WRN = 500 μM, exonuclease domain = 2 mM) in assay buffer. After 30 min at room temperature, 3 μL of 60 mM EDTA was added to quench the reactions and the 2-aminopurine fluorescence intensity was measured on a Tecan plate reader with the following settings: λexcitation = 306 nm, λemission = 362 nm, bandwidths = 5 nm, gain = 150, 20 flashes, flash frequency = 400 Hz, integration time = 20 μs, lag time = 0 μs, settle time = 15 ms.
Intact Mass Spectrometry
Alkylated WRN helicase domain samples were prepared by incubating 10–60 μg protein with a 10-fold molar excess of compound overnight at 4 °C before storage at −20 °C until analysis. Samples were desalted using Zeba Spin desalting columns (7 kDa, 1000g for 2 min) prior to injecting 1–2 μg of protein for intact mass analysis on a Q Exactive mass spectrometer coupled to a Vanquish UPLC outfitted with an EASY-Spray Source (Thermo Fisher Scientific). Desalted proteins were separated on a 1 × 150 mm Waters BEH C4 (130 Å, 1.7 μm, 75 μm × 15 cm) and separated at a flow rate of 90 μL min–1. Solvent A was 0.1% formic acid in water and solvent B was 0.1% formic acid in acetonitrile. Column elution started at 10% B over 0–0.1 min and then increased linearly from 10 to 90% B over 0.1–8 min. The column was washed with 90% B for an additional 2 min before reequilibrating with 10% B for 7 min. Full scan data were acquired at a resolution of 35,000 (at 200 m/z) in the range of 600–2000 m/z with 10 microscans. The automatic gain control (AGC) target value was 3.00 × 106 and the maximum ion injection time was 100 ms. Mass spectra were deconvoluted using the Intact Protein Analysis module of BioPharma Finder 2.0. The ReSpect deconvolution algorithm was used with a mass range of 40–60 kDa, charge state range of 10–100, minimum adjacent charges between 6 and 10, and all other settings at their default values. The intact protein peak model was used with a 50,000 Da target mass. Source spectra were generated by averaging over manually selected retention times on the chromatogram and 95% confidence of noise rejection parameter was used. Charge state distributions were identified with a sequence matching mass tolerance of 20 ppm.
Isothermal Calorimetry (ITC)
ITC was performed with an ITC200 (Malvern Instruments) at 25 °C. WRN helicase domain was buffer-exchanged into assay buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 1% DMSO) prior to measurement. The cell was loaded with 300 μL of 15 μM protein, and the syringe was loaded with 40 μL of 150 μM H3B-960 formulated in identical assay buffer. Titration experiments consisted of sixteen 2.45 μL injections with 120 s of equilibration between injections. The final stoichiometry of compound and protein in the cell at the conclusion of the experiment was approximately 2:1. The enthalpy peaks were integrated and fit to a 1:1 binding model using Origin software.
General Synthetic Procedures
2-Amino-N-arylacetamides
The general synthetic route to afford 2-amino-N-arylacetamides is shown (Scheme S1). Step 1. Boc-Glycine (1.0 equiv), Ar2-NH2 (1.0 equiv), N,N-diisopropylethylamine (3.0 equiv), and N,N-dimethylformamide were added to a round-bottom flask. 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (1.1 equiv) was then added, and the mixture was allowed to stir at room temperature for 16 h. Upon completion of the reaction, water was added and the resulting white precipitate was collected by filtration. The solid was further washed with water and then dried under vacuum to form the desired product 1. Step 2. Dioxane in HCl (10.0 equiv) was added to a suspension of 1 (1.0 equiv) in dichloromethane (DCM). The reaction mixture was allowed to stir at room temperature for 16 h. Upon completion of the reaction, volatiles were removed under vacuum. The residue was redissolved in 5:1 DCM/methanol (MeOH) and washed twice by saturated sodium bicarbonate and once by brine. The organic layer was collected, dried over sodium sulfate, filtered, and condensed to afford the crude product. The crude product was purified by silica gel column with 0% to 20% MeOH in DCM. The desired fractions were combined and concentrated to afford the title compound 2.
2-Sulfonamide Pyrimidines
The general synthetic route followed to afford 2-sulfonamide pyrimidines is shown (Scheme S2). Step 1. 4-Chloro-2-(methylthio)-6-(trifluoromethyl)pyrimidine (1.0 equiv), Ar1 boronic acid (1.2 equiv), Pd(PPh3)2Cl2 (0.1 equiv), and K3PO4 (2.5 equiv) were added to a round-bottom flask. The flask was degassed before adding a 5:1 mixture of 1,4-dioxane:degassed water under nitrogen. The reaction mixture was allowed to stir at 100 °C for 16 h. Upon completion, the mixture was filtered through Celite and the filtrate was concentrated under vacuum to afford the crude product. The crude product was purified by a silica gel column with 0–30% ethyl acetate in hexanes. The desired fractions were combined and concentrated under reduced pressure to afford compound 3. Step 2. m-Chloroperoxybenzoic acid (3.0 equiv) was added to a stirred solution of 3 (1.0 equiv) in DCM. The reaction mixture was allowed to stir at room temperature for 16 h. Upon completion of the reaction, saturated sodium bicarbonate and saturated sodium thiosulfate were added to the product and the layers were allowed to separate. The organic layer was collected and washed once with saturated sodium thiosulfate and once with saturated sodium bicarbonate. The layer was then dried over sodium sulfate, filtered, and concentrated under vacuum to afford product 4. The crude product was carried on to the next step without further purification. Step 3. To a round-bottom flask with a stir bar were added 4 (1.0 equiv) in acetonitrile (MeCN), phenylmethanethiol (1.5 equiv), and triethylamine (3.0 equiv). The reaction mixture was allowed to stir at 80 °C for 16 h. Upon completion of the reaction, volatiles were removed under vacuum. The residue was purified by a silica gel column with 0% to 10% ethyl acetate in hexanes. The desired fractions were combined and concentrated to afford compound 5. Step 4. Sulfuryl chloride (2.0 equiv) was added to a stirred solution of 5 (1.0 equiv) in DCM/acetic acid/H2O (10:1:1). The mixture was allowed to stir at room temperature for 2 h. Upon completion of the reaction, volatiles were removed under vacuum to afford product 6. The crude residue was carried on to the next step without further purification. Step 5. 2-Amino-N-arylacetamide 2 (2.0 equiv) was added to a stirred solution of 6 (1.0 equiv) in ethyl acetate. The mixture was allowed to stir at room temperature for 16 h before removing the volatiles under vacuum. The residue was purified by silica gel column with 0% to 100% ethyl acetate in hexane or HPLC with 0–100% MeCN in water (modifier: 0.1% formic acid). The desired fractions were combined and concentrated to afford the title compound 7. Compound purity was assessed by LC-MS and NMR spectrometry and judged to be >90%.
Additional Experimental Material
Additional methodology for protein purification, DNA intercalator and protein aggregator counter screening, differential scanning fluorimetry, GSH stability, and peptide mapping LC-MS can be found in the Supporting Information.
Results
WRN Purification
Full-length WRN protein was produced from insect cells (Figure S1). Our initial pilot purifications had high absorbance at A260 relative to A280, indicative of contaminating DNA. We were able to disrupt the WRN-DNA interaction and normalize the A260/A280 ratio by employing on-column high-concentration salt washes prior to elution during the Ni-NTA chromatography step. We also observed severe protein loss when concentrating the protein using standard Amicon filtration units. Therefore, in our second Hi-Trap Q chromatography step, we endeavored to concentrate the protein by eluting in a step gradient of high salt. Using this method, we obtained 95% pure protein at ∼4 mg mL–1. Follow-up analytical size exclusion chromatography (SEC) showed the protein ran as a monomer, but due to on-column dilution, we did not pursue SEC at prep scale.
WRN Multiplexed Assay Optimization and Validation
To measure WRN helicase and ATPase activity, we utilized a DNA unwinding assay that monitors TAMRA fluorescence dequenching of a forked DNA substrate as a readout.17 In this format, helicase activity results in DNA strand separation and unmasking of a TAMRA fluorophore, resulting in a gain of fluorescent signal, the progress of which can be monitored over time. We initially started assay optimization with a 25 bp substrate and encountered two issues. First, we found the product ssDNAs generated from WRN helicase activity reannealed over time, either spontaneously or by WRN-mediated strand annealing activity,20 resulting in a gradual loss of TAMRA fluorescence intensity with incubation times longer than ∼5 min (Figure S2A). This requenching occurred regardless of the substrate length. The inclusion of an unlabeled ssDNA trap prevented the labeled product strands from reannealing and, thus, stabilized the TAMRA intensity for at least 30 min (Figure S2B). Titration of the trap revealed that five equivalents or more relative to the substrate concentration was sufficient for long-term signal stability; we chose to use 10 equiv in our multiplexed assay. The sequence of the trap needed to be complementary to only the double-stranded region of the forked substrates to prevent enzyme-independent strand swapping (Figure S2C). The second issue was determining the Km,ATP to inform appropriate screening conditions. We found that substrates with longer double-stranded regions required more ATP to power unwinding, leading to discrepancies in WRN’s apparent Km for ATP in different assay formats. We determined the Km,ATP for WRN to be 37 μM in a pyruvate kinase/lactate dehydrogenase coupled ATPase assay using the 25 bp substrate, which is consistent with prior literature (Figure S2D).21 In contrast, an apparent Km,ATP of ≥108 μM was measured using our DNA unwinding assay with the same substrate (Figure S2D). We were able to harmonize Km’s from the ATPase and unwinding assays by reducing the double-stranded region’s length to 10 bp. This shortening reduces the amount of ATP necessary to achieve unwinding and therefore lowers the apparent Km,ATP to 28 μM in the unwinding assay (Figure S2D). We therefore chose 60 μM ATP as our final concentration for screening. This optimization of substrate and ATP enabled us to monitor helicase and ATPase activity under single-readout near-balanced conditions, increasing the likelihood of identifying competitive, noncompetitive, or uncompetitive inhibitors during screening.
To incorporate the exonuclease readout, we adapted the high-throughput screen-ready Transcreener AMP2/GMP2 phosphodiesterase fluorescence polarization assay from Bellbrook Labs to exploit literature precedent for WRN-mediated degradation of RNA strands in RNA:DNA duplexes.22,23 WRN exonuclease activity on RNA releases AMP, GMP, and CMP, which displace an Alexa Fluor 633 tracer bound to an anti-NMP antibody, resulting in a loss of fluorescence polarization of the tracer. The fluorescent properties of Alexa Fluor 633 and TAMRA minimally overlap, thereby lowering the chances of crosstalk between the helicase and exonuclease readouts and enabling the assays to be multiplexed.
Successful multiplexing required careful optimization of the exonuclease and helicase assay conditions. One of the first considerations was the choice of divalent cofactors. Mn2+ and Mg2+ can both act as cofactors for WRN exonuclease activity.23,24 We chose to utilize Mg2+ at 2 mM in the multiplexed assay given that (1) Mg2+ is additionally required for WRN ATPase activity, (2) Mn2+ at concentrations above 500 μM attenuated WRN helicase activity (Figures S3A), and (3) Mn2+ stimulated low levels of helicase-independent TAMRA dequenching of the forked helicase substrate (Figure S3B). The latter activity was likely due to the reported ability of WRN exonuclease domain to cleave bases from the blunt end of a forked DNA substrate.25 As Mg2+-catalyzed exonuclease activity was slower than with Mn2+, we achieved a good signal window in the assay utilizing a minimal Transcreener antibody concentration of 3 μg mL–1. We additionally found that the use of ATP to power WRN helicase activity interfered with the exonuclease readout by artificially decreasing the fluorescence polarization of the tracer (Figure S3C). To circumvent this issue, we replaced ATP with dATP (Figure S3C), which is unable to displace the Alexa-633 probe but still able to power DNA translocation by WRN.21
After confirming that the substrates for the DNA unwinding and NMP release assays could be combined in a single-well reaction without any crosstalk, we proceeded to optimize reagent concentrations and scale down the reaction volume. Screening WRN near Km,dATP would increase the chances of identifying helicase/ATPase inhibitors with different mechanisms of action. An apparent Km of 24 μM for dATP was determined using the DNA unwinding assay with the 10 bp substrate (Figure S2D); we therefore fixed the dATP concentration in the multiplexed assay at 60 μM and adjusted enzyme and substrate concentrations as well as incubation time to find the optimal point with a good assay window and low level of substrate turnover. We achieved assay windows in both readouts of 3-fold or better incubating 10 nM FL-WRN, 500 nM 10 bp helicase substrate, 5 μM trap, and 300 nM hybrid exonuclease substrate for 30 min in a final volume of 6 μL. For the helicase readout, real-time progress curves were linear over the incubation period and reached a total substrate consumption of about 30–35% (Figure S4). For the exonuclease readout, the reagent concentrations and reaction time were sufficient to produce 70% of the maximal fluorescence polarization change, a condition recommended by the manufacturer for a robust assay. To quench the reactions, we spiked EDTA into the Bellbrook antibody/tracer solution after the reaction period expired. Since we used one plate reader to read both activities, we measured the TAMRA fluorescence intensity immediately after addition of the EDTA–Bellbrook reagent mixture and the Alexa Fluor 633 fluorescence polarization after a 90 min incubation time required for signal development. Validation studies using full 384-well plates yielded Z′-factors greater than 0.8 for both the helicase/ATPase and exonuclease activities, indicating that the assay was fully adapted for high-throughput screening.
High-Throughput Screening of Full-Length WRN
Primary screening was conducted at 25 μM compound concentration using a combination of commercially available and proprietary in-house libraries totaling just over 154,500 compounds. The respective mean signal-to-noise ratio and Z′-factor were 5.4 ± 1.2 and 0.77 ± 0.05 for the helicase/ATPase readout, and 4.4 ± 0.7 and 0.81 ± 0.02 for the exonuclease readout, respectively (Figure 1A). Screening hits were defined as compounds falling three standard deviations outside the mean, which corresponded to cutoffs of 34% for helicase inhibition and 13% for exonuclease inhibition (Figure S5). A total of 627 helicase/ATPase and 1027 exonuclease hits were identified with hit rates of 0.41 and 0.66%, respectively, and an overall hit rate of 1.07%. Of these compounds, 126 were considered hits in both readouts (Figure S6). Primary hits were evaluated in an 8-point dose response to confirm inhibitory activities and then subjected to counter screens to remove nonspecific aggregators and DNA intercalators (Figure 1B). We also screened the primary hits with orthogonal assays to eliminate false positives generated in the primary screen readouts: Promega’s ADP-Glo bioluminescence assay for helicase/ATPase activity and the fluorescence intensity increase resulting from the release of 2-aminopurine-2′-deoxyribonucleoside monophosphate from a 3′-recessed double-stranded DNA for exonuclease activity (Figure 1B).26 These assays enabled us to further triage our hits on the following criteria: (1) ≥ 50% response in both orthogonal assays with an IC50 < 30 μM, and (2) no response in the counter screens. This filter narrowed our chemical matter to 188 compounds, consisting of 81 helicase/ATPase, 86 exonuclease, and 21 pan-activity inhibitors. Assaying these compounds with recombinant helicase (residues 500–946)17 and exonuclease domain (residues 36–238)24 revealed none were selective for full-length WRN. For the pan-activity inhibitors, the results indicated that these compounds were behaving as PAINs molecules and, therefore, were excluded from further study.
Figure 1.

Summary of full-length WRN high-throughput screening campaign using a catch-all multiplexed assay to measure ATPase, helicase, and exonuclease activities from the same well. (A) Helicase (top) and exonuclease (bottom) readout Z′-factors across all plates from the primary screen. (B) Screening funnel utilized to filter primary hits and determine hit selectivity for FL-WRN versus the individual helicase and exonuclease domains.
Several exonuclease and helicase/ATPase inhibitor series with SAR, moderate potency (IC50 = ∼1 to 22 μM), and tractable structures emerged from triaging and cluster analysis of the screening hits. One cluster of helicase/ATPase hits contained compounds with a 2-sulfonylpyrimidine core; the representative member, H3B-859, exhibited IC50 values of 0.9 and 2.7 μM in the TAMRA-dequenching DNA unwinding and ADP-Glo assays, respectively (Figure 2A, Table 1) and no activity in the exonuclease assay. Initial biophysical characterization did not produce promising results. Titration of WRN helicase domain with H3B-859 produced a large negative Tm shift in differential scanning fluorimetry (DSF) experiments, but only at the top compound concentration of 100 μM (Figure S7). A related compound produced a positive Tm shift of less than 1 °C up to a concentration of about 32 μM (Figure S7). Additional 1H- and 19F-NMR ligand observed binding studies and isothermal titration calorimetry analyses were likewise inconclusive. Indeed, H3B-859 exhibited low solubility in aqueous buffer; a sample prepared with a nominal concentration of 150 μM had an actual concentration of approximately 17 μM. This solubility issue likely explains the challenges in obtaining biophysical data.
Figure 2.

Evidence for covalent modification of WRN by H3B-859. (A) Chemical structure for starting lead H3B-859. The pyrimidine ring is numbered according to convention. (B) Real-time DNA unwinding progress curves with 0.6 nM WRN helicase domain, 0.5 μM Hel-10bp, 5 μM Trap-10bp, 120 μM ATP, and the indicated concentrations of H3B-859. (C) Intact mass analysis of WRN helicase domain (expected MW = 51026.6 Da) with DMSO. (D) Intact mass analysis after overnight treatment with a 10-fold excess of H3B-859. Alkylation by H3B-859 is expected to shift the mass by 253 Da. Masses corresponding to unmodified, monoalkylated, and dialkylated proteins are indicated in the spectrum as 0×, 1×, and 2×, respectively. (E) Proposed mechanism of inhibition by 2-sulfonylpyrimidine. A cysteine from WRN undergoes nucleophilic aromatic substitution to form the indicated adduct with departure of the sulfinic acid leaving group.
Table 1. Activity of 2-Sulfonyl/Sulfonamide Pyrimidines as WRN Helicase Inhibitorsa.

Unless noted otherwise, the reported IC50 values are the average of at least two replicate measurements. ND = not determined.
2-Sulfonylpyrimidines Covalently Modify WRN
While these biophysical readouts were inconclusive, we were intrigued to observe curvature in the DNA unwinding progress curves with increasing compound concentration under steady-state conditions (Figure 2B). In this kinetic readout, the enzymatic reaction is initiated by the addition of WRN with no compound preincubation step. The observed time-dependent inhibition is a classic sign of a covalent mechanism of action, and a survey of the literature reveals precedent for cysteine alkylation by 2-sulfonylpyrimidines.27−29 Here, the cysteine attacks the 2 position of the pyrimidine ring in a nucleophilic aromatic substitution reaction (SNAr) resulting in alkylation and displacement of a sulfinic acid. To confirm covalent modification, we incubated the WRN helicase domain with DMSO or a 10-fold excess of H3B-859 overnight at 4 °C and then analyzed the intact protein mass by ESI-MS (Figure 2C,D). Comparison of the DMSO and compound-treated samples shows a mass shift of +252 Da, consistent with the addition of the expected 4-(4-methoxyphenyl)-6-(trifluoromethyl)pyrimidine adduct (Figure 2D,E). Relative quantification of the peaks indicated ∼86% of the protein was alkylated by H3B-859 on at least one cysteine residue. Two minor peaks (relative abundance < 10%) were also present corresponding to unmodified protein and two alkylation events. These data clearly show that WRN is covalently modified by 2-sulfonylpyrimidines and strongly suggest that this modification results in the inhibition of WRN helicase activity. Moreover, the comparatively minor secondary alkylation event suggests that H3B-859 exhibits a favorable selectivity profile.
Compound Optimization
We next focused on improving the biochemical potency of H3B-859 (Table 1). We initially designed H3B-219 by changing the sulfone to the corresponding sulfonamide. We reasoned after nucleophilic attack, the resulting sulfuramidous acid would likely be both electronically delocalized and unstable, making it a better leaving group than the corresponding sulfinic acid from H3B-859. In fact, we were gratified to observe that H3B-219 had an 8-fold improvement in activity in the DNA unwinding assay (Table 1). Methylation of the sulfonamide (8) showed similar activity to H3B-859 (Table 1). These activity data would be consistent with our leaving group hypothesis or could also suggest the sulfonamide forms either an H-bond interaction with the protein or an intramolecular H-bond to one of the pyrimidine nitrogens, which may help preconfigure or rigidify the compound for more optimal binding or adduct formation. Interestingly, while H3B-219 demonstrated marked improvement in the DNA unwinding assay, there was modest improvement in the ATPase assay (Table 1), only reaching a percent response of about 55–60%. We confirmed that the apparent incomplete inhibition did not stem from the interference of the ADP-Glo assay, as identical results were obtained utilizing the Transcreener ADP2 fluorescence polarization kinase assay. The lack of full inhibition by H3B-219 implies that under the assay conditions, some ATPase activity remains.
With this more potent compound H3B-219, we next sought to make the electrophile less reactive. Pyridine cores are reported to be tunable electrophiles for cysteine alkylation;30 therefore, we explored pyrimidine-to-pyridine core replacement. Unfortunately, compounds 9 and 10 were inactive in both assays, and attempts to tune the reactivity of the pyrimidine by changing the 6-trifluoromethyl to the methyl group (11) also proved detrimental to potency. We next sought to further characterize the structural constraints of the binding pocket through modifications at the 4- and 2-positions of the pyrimidine ring. Substituting the methoxyphenyl group in H3B-219 with a trifluoromethoxy phenyl group (H3B-960) was well tolerated, improving the potency ∼15-fold and ∼6-fold in the ATPase and DNA unwinding assays, respectively (Table 1). Moreover, H3B-960 shows a full response (100% inhibition) in ADP-Glo assay. Larger substitutions on the 4-position, such as a naphthalene group (12), showed similar activity to H3B-219 (Table 1). We also found that interactions off the pyrimidine 2-position (the leaving group) were important for activity. Indeed, elimination of the methyl aniline group (13) and linker (14) resulted in complete or significant loss in activity, respectively. Masking of the amide nitrogen atom with a methyl group (15) resulted in diminished potency as well. We substituted the glycine linker of H3B-219 with a variety of l- and d-amino acids to rigidify the structure and introduce substituents to gain additional interactions with the protein. Of these, only small side chains in the l-configuration were tolerated but still exhibited a loss in potency (Table S3). Interestingly, replacement of the glycine linker with (S)-3-amino-2-pyrrolidinone (16) resulted in similar potency to H3B-219 in both assay formats, whereas the (R)-enantiomer (17) exhibited a much larger discrepancy in potency between the two assays. Relative to H3B-219, replacing the methyl aniline with ethyl aniline (18) results in a 3-fold increase in potency in DNA unwinding assay while showing similar potency in the ADP-Glo assay. More polar fused ring systems like indazole (19) were not tolerated. Combining the optimal groups from the 4- and 2- positions resulted in H3B-968, bearing the trifluoromethoxy phenyl group at the 4-position of the pyrimidine ring and the ethyl aniline on the leaving group 2-position. This compound displays the greatest biochemical potency with an IC50 of 41 nM in the ADP-Glo assay and 13 nM in the DNA unwinding assay (Table 1).
Encouraged by these biochemical potency data, we next characterized their reactivity using a glutathione (GSH) stability assay. The initial HTS hit H3B-859 and active compounds H3B-219 and H3B-960 were found to be unstable in the presence of physiological concentrations (5 mM) of GSH, with decomposition half-lives of less than 10 min. Furthermore, preincubating the compounds with 2 mM GSH resulted in a complete loss of potency in both the DNA unwinding and ADP-Glo assays. In contrast, the inactive compounds containing either pyridine cores (9 and 10) or a methyl substituent at the 6-position of the pyrimidine core (11) were stable in the presence of GSH (t1/2 > 360 min).
Characterization of WRN Inhibition by 2-Sulfonamide Pyrimidines
With these potent chemical probes in hand, we set about to characterize their MOA in our biochemical assays. Like H3B-859, both H3B-219 and H3B-960 imparted significant deviations from linearity in the kinetic progress curves for DNA unwinding under steady-state conditions, which is consistent with a covalent mechanism of action (Figure 3A,B). Mass shifts corresponding to the expected adducts (253 Da for H3B-219 and 307 Da for H3B-960) were observed confirming alkylation of WRN (Figure 3C,D). For H3B-960, a second peak was observed (relative abundance ∼25%) consistent with dialkylation. With the more potent compound H3B-960, we were also able to obtain biophysical evidence for binding by ITC (Figure 4A). The individual titration peaks were relatively broad, consistent with a relatively slow binding, but showed very strong enthalpy (∼11 kcal/mol) as expected for covalent bond formation. Importantly, the data are consistent with ∼1:1 stoichiometry and a KD of 40 ± 15 nM, in excellent agreement with the biochemical data (Table 1). Of note, the time scale for each ITC injection is 30 s with 2 min equilibration periods between injections, while the mass spec timescales were overnight incubations in 10-fold molar excess, which likely explains the 1:1 stoichiometry by ITC but a secondary alkylation event by mass spectrometry.
Figure 3.

Evidence for covalent modification of WRN by 2-sulfonamide pyrimidines. The top panels show real-time DNA unwinding progress curves with varying concentrations of H3B-219 (A) and H3B-960 (B). Assays contained 0.6 nM WRN helicase domain, 0.5 μM Hel-10bp, 5 μM Trap-10bp, 120 μM ATP, and the indicated compound concentrations. The bottom panels show the intact mass spectra of WRN helicase domain (expected MW = 50939.5 Da) treated overnight with 10-fold excess of H3B-219 (C) or H3B-960 (D). Alkylation by H3B-219 and H3B-960 is expected to shift the mass by 253 or 307 Da adduct, respectively. For H3B-960, the indicated masses at 1× and 2× correspond to the mono- and dialkylated protein.
Figure 4.

Binding and selectivity data for 2-sulfonyl/sulfonamide pyrimidines. (A) Evidence for binding of H3B-960 to WRN helicase domain by isothermal titration calorimetry. Selectivity of H3B-859 (B) and H3B-219 (C) for WRN (black) versus BLM (red) and RecQL1 helicase domain (blue). Eleven-point dose responses were evaluated with the ADP-Glo assay as described in the Experimental Section. WRN IC50 values were 865 and 171 nM for H3B-859 and H3B-219, respectively, whereas IC50 values for BLM and RecQL1 helicase domain were greater than 50 μM.
Selectivity toward WRN
We next sought to understand the selectivity of this chemical series to WRN compared to other RecQL helicases. This family of helicases has an overall low sequence conservation (∼30% identity). We expressed and purified full-length Bloom Syndrome helicase (BLM) and RecQL1 helicase domain and assayed them in 11-point dose responses with H3B-859 and H3B-219 using the ADP-Glo assay. RecQL1 was unresponsive to inhibition by either compound, whereas BLM exhibited weak inhibition by both at concentrations of around 4 μM or higher (Figure 4B,C). These concentrations were at least 5-fold higher than the IC50 values observed for WRN (Table 1). The results indicate that 2-sulfonyl/sulfonamide pyrimidines have a promising selectivity profile for WRN.
Mode of Inhibition
To gain insight into whether the compounds were behaving competitively with respect to ATP, we utilized the ADP-Glo assay to determine how their potency and response vary at ATP concentrations approximately 20-fold higher than our measured KM,ATP. Running ADP-Glo assays with H3B-859, H3B-219, and H3B-960 in the presence of 1 mM ATP resulted in a notable weakening in their potency (Table 2) and response (Figure S8) compared to assays run near Km,ATP. Likewise, we utilized the DNA unwinding assay to perform a similar competition experiment with varying concentrations of DNA substrate; the results clearly showed that the potency (Table 3) and response (Figure S9) of FL-WRN to the H3B-859, H3B-219, and H3B-960 were unaffected by these changes. The results of these studies strongly suggest that alkylation of WRN by 2-sulfonyl/sulfonamide pyrimidines is competitive with respect to ATP but noncompetitive with respect to DNA.
Table 2. 2-Sulfonyl/Sulfonamide Pyrimidine Potency with Low and High ATP Concentrations.
| compound | 60 μM ATP; IC50 (nM) | 1 mM ATP; IC50 (nM) |
|---|---|---|
| H3B-859 | 775 | >50,000 |
| H3B-219 | 881 | >50,000 |
| H3B-960 | 225 | 894 |
Table 3. 2-Sulfonyl/Sulfonamide Pyrimidine Potency with Low and High DNA Concentrations.
| compound | 50 nM DNA; IC50 (nM) | 500 nM DNA; IC50 (nM) |
|---|---|---|
| H3B-859 | 534 | 235 |
| H3B-219 | 313 | 385 |
| H3B-960 | 57 | 36 |
Compound Binding and Reactivity Contribute to Potency
We sought to understand more about the kinetic mechanism of WRN covalent inactivation by H3B-859, −219, and −960 using the real-time DNA unwinding assay with the WRN helicase domain. Progress curves (Figures 2 and 3) were fit to eq 1, where νi is the initial linear rate and t is time, to determine observed rate constants kobs at a given concentration of inhibitor. Plotting kobs versus inhibitor concentration revealed readily apparent saturation behavior for all three compounds (Figure 5), consistent with a simple two-step mechanism involving an initial reversible binding event prior to adduct formation. The data were fit by nonlinear regression to eq 2 to determine KI and kinact for each compound (Table 4). All compounds showed a similar rate of inactivation of WRN; however, KI decreased, suggesting increased binding affinity as the chemical series progressed. The efficiency of inactivation (kinact/KI) by H3B-960 approached values similar to those observed for FDA-approved or druglike inhibitors of EGFR,31 KRAS,32 and BTK.33 Importantly, these data supporting a two-step mechanism show that the chemical series engages a binding pocket on WRN and is not behaving simply as an indiscriminate Cys-labeling reagent, such as iodoacetamide.
| 1 |
| 2 |
Figure 5.

Covalent inactivation of WRN helicase domain. (A) Proposed two-step mechanism where the initial E–I* complex is a reversible binding event (KI) prior to the inactivation step that forms the covalent E–I complex (kinact). The compounds H3B-859 (B), H3B-219 (C), and H3B-960 (D) follow this two-step mechanism. Real-time DNA unwinding assays were run with 0.5 nM WRN helicase domain, 120 μM ATP, 500 nM Hel-10bp, and 5 μM Trap-10bp.
Table 4. Kinetic Parameters of 2-Sulfonyl/Sulfonamide Pyrimidines.
| compound | kinact (s–1) | KI (nM) | kinactKI–1 (M–1 s–1) |
|---|---|---|---|
| H3B-859 | 5.6 × 10–4 | 3090 | 180 |
| H3B-219 | 3.5 × 10–4 | 630 | 562 |
| H3B-960 | 4.5 × 10–4 | 32 | 14,000 |
Peptide Mapping
To determine the site of modification on WRN, we attempted peptide mapping LC-MS utilizing a battery of different proteases. Full-length WRN has 36 cysteine residues and the helicase domain has 19. Unfortunately, none of the observed peptides exhibited the expected mass shifts that would allow us to definitively map the binding site. We suspected that our inability to see the modified peptide directly was an artifact of detection, possibly because the modified peptide was retained on the column under all gradient conditions tested or the peptide-adduct fragmented under the ionization conditions. Therefore, we sought to quantify depleted peptides from the treated vs untreated samples (Figure 6). Interestingly, we observed clear and consistent depletion of the peptides containing C727 under all proteolytic conditions with all compounds tested (Figure 6). This difference is not due to protein concentration differences during injection because the total protein quantities were similar between the treated and untreated samples (Figure 6). C727 is located opposite the ATP binding pocket, and its alkylation could therefore impact ATP binding, which would be consistent with the observed ATP competitive behavior (Figure 7).
Figure 6.

Quantification of the abundance of WRN peptides containing C727 in DMSO controls versus samples treated with a 10-fold excess of H3B-219 (left) or H3B-960 (right) reveals significant depletion of the peptides treated with compound under multiple proteolytic conditions. Total protein quantification (representative example for samples treated with chymotrypsin shown) indicated that the depletion was not due to differences in sample concentration, suggesting that peptides containing C727 become alkylated upon compound treatment.
Figure 7.
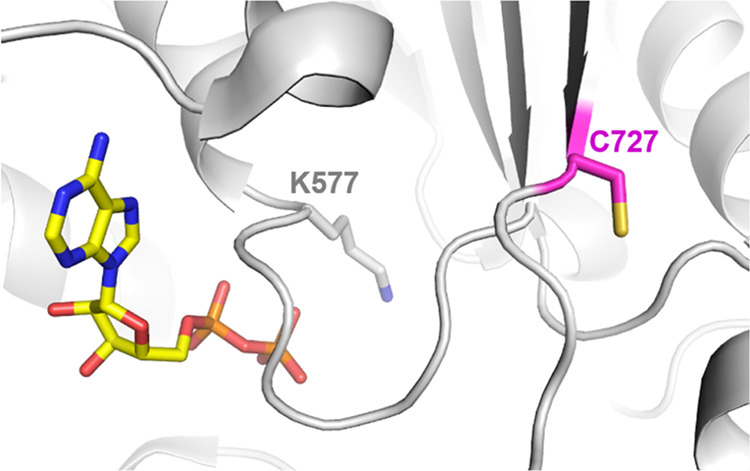
Location of proposed site of modification, C727, relative to ADP and the Walker motif K577 in the WRN crystal structure (PDB 6YHR). Covalent modification of the C727 allosteric site could impact the conformation of key residues involved in ATP binding and hydrolysis.
Discussion
Inhibitors of WRN helicase activity present a new therapeutic opportunity for patients with MSI-H cancers. WRN is a large multidomain protein with additional activities and, therefore, could be the target of allosteric inhibitors or heterobifunctional degraders that bind outside of the helicase domain. The catch-all, multiplexed assay that we present here has been carefully optimized to measure WRN’s ATPase, helicase, and exonuclease activities simultaneously under near-balanced conditions. We believe such a screening strategy is an efficient way to interrogate all of the various conformational states along the reaction coordinate of such complicated systems in an unbiased data-driven way. Indeed, the screening assay allows for multiple inhibitory modes of action to be explored in a single-well reaction. Our initial high-throughput screen highlighted 2-sulfonyl/sulfonamide pyrimidines as a novel series of covalent WRN ATPase/helicase inhibitors. Expansion of the SAR culminated in H3B-968 with ∼10 nM activity in our biochemical assays, demonstrating the successful application of our approach. Therefore, it’s possible that screening larger compound libraries could yield additional WRN inhibitors targeting the more physiologically relevant full-length form of the protein.
Our studies of the inhibition of WRN by 2-sulfonyl/sulfonamide pyrimidines revealed a discrepancy in the results of our DNA unwinding assay versus the orthogonal ADP-Glo assay for some of compounds generated early on in our SAR exploration (e.g., H3B-219). In general, the potency and maximum percent response were stronger in the DNA unwinding assay (Table 1), although the differences largely disappeared as we generated the more potent chemical probe compounds H3B-960 and H3B-968. The ability to reproduce the weaker potencies and responses by measuring ADP concentrations in orthogonal assay formats clearly indicated that the difference between ATP consumption and DNA unwinding was real and not an artifact of the assays. The source of this discrepancy is unclear given that a shorter forked helicase substrate was utilized to better match the amounts of unwinding products and ADP. One explanation for the discrepancy may stem from the lower levels of total alkylation observed with H3B-859 and H3B-219. Both compounds alkylated ∼85% of WRN helicase domain in our intact mass experiments. Given that WRN ATPase activity is significantly stimulated by DNA34 and that the kinact values for H3B-859 and H3B-219 are slow, it is likely that WRN undergoes several rounds of ATP hydrolysis before being inactivated by alkylation, resulting in a greater amount of ADP produced versus unwound forked substrate. A population of nonalkylated WRN molecules will further contribute to the product imbalance. In contrast, we were unable to detect any nonalkylated protein in our intact mass samples prepared with H3B-960 and H3B-968, and both exhibit faster kinact values. Therefore, it may be possible that inactivation of WRN in the presence of these compounds occurs more readily and, consequently, the product distribution of ADP and unwound DNA is more even. Further studies are required to understand the discrepancy between the two assay formats.
Analyzing the depleted peptides in our peptide mapping experiments highlighted the near-complete disappearance of C727-containing peptides relative to the other peptides. This indirectly implicates C727 as the site of covalent modification. Interestingly, C727 had also been observed to be modified by 4-hydroxy-2-nonenal (HNE), an unsaturated aldehyde produced during oxidative stress in the cell. This modification was shown to inhibit WRN and is thought to contribute to cellular senescence.35 Structurally, C727 is located at an allosteric site within 15 Å of the ATP binding site (Figure 7). The location of this residue is intriguing given that our preliminary mechanism of action studies suggest that alkylation by our compounds behaves competitively toward ATP binding (Table 2). The side chain of C727 is oriented away from the ATP site, suggesting that the alkyl group will not directly compete with ATP binding (Figure 7). Interestingly, hydrogen deuterium exchange experiments clearly show a moderate decrease in proton exchange in β-strand 6 upon the binding of the nonhydrolyzable ATP analog AMP-PNP,36 suggesting that changes in the strand’s conformation will propagate into the ATP binding site. Alkylation of C727, therefore, may alter the local structural conformation to block ATP binding or reposition essential residues for ATP hydrolysis into a catalytically incompetent state. The latter mechanism may be more relevant given that WRN’s Walker A motif, containing the catalytically essential lysine 577, is spatially juxtaposed to C727 (Figure 7). The lack of conservation of C727 in other human RecQL helicases also provides a good explanation for the selectivity of our compounds for WRN. Further studies are warranted to confirm C727 in WRN as the target of alkylation by 2-sulfonyl/sulfonamide pyrimidines.
Our work provides a proof of concept that targeting WRN helicase activity with covalent inhibitors is possible and may be a viable therapeutic approach for MSI-H cancers. To the best of our knowledge, 2-sulfonyl/sulfonamide pyrimidines are the first reported class of covalent inhibitors for WRN. Based on the SAR study and GSH stability assay results, the CF3 group at the 6-position of the pyrimidine ring is responsible for both the reactivity of the 2-sulfonyl/sulfonamide pyrimidines toward WRN and their instability toward low-molecular-weight thiols. Our initial attempts to tune the reactivity with different regioisomers of pyridine (9 and 10, Table 1) as replacements of the pyrimidine core resulted in a loss of activity. We envision that synthetic efforts to modify the core with various heterocycles and electron-withdrawing groups may lead to compounds with reasonable stability and biochemical and cellular activity. Such modifications may provide a promising future for these first-in-class covalent inhibitors of WRN.
Acknowledgments
The authors thank all employees of H3 Biomedicine for their helpful discussions related to this project.
Glossary
Abbreviations
- BLM
Bloom syndrome helicase
- DSF
differential scanning fluorimetry
- ESI-MS
electrospray ionization–mass spectrometry
- GSH
reduced glutathione
- HTS
high-throughput screen
- ICI
immune checkpoint inhibitor
- IMAC
immobilized metal affinity chromatography
- MSI-H
high microsatellite instability
- MSS
microsatellite stable
- PAINs
pan-assay inhibitory compounds
- PMSF
phenylmethylsulfonyl fluoride
- SAR
structure–activity relationship
- TAMRA
5-carboxytetramethylrhodamine
- TCEP
tris(2-carboxyethyl)phosphine
- WRN
Werner syndrome protein
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.2c00599.
Detailed experimental procedures, additional tables and figures, and schemes of synthetic routes (PDF)
Author Contributions
M.J.P.; H.L.; P.H.; H.Z.; D.M.B.; P.Z.; P.D.; M.K.; and N.A.L. designed research. M.J.P.; H.L.; S.Y.; P.H.; H.Z.; K.B.; S.H.; S.I.; X.P.; R.S.; L.Y.; P.G.; S.M.; and S.W.M. performed research. All authors contributed to the data analysis and writing and have given approval to the final version of the manuscript.
This work was funded by H3 Biomedicine, Inc.
The authors declare the following competing financial interest(s): The authors are former employees of H3 Biomedicine.
Supplementary Material
References
- Larsen N. B.; Hickson I. D.. RecQ Helicases: Conserved Guardians of Genomic Integrity. In Advances in Experimental Medicine and Biology, 2013; Vol. 767, pp 161–184. [DOI] [PubMed] [Google Scholar]
- Mohaghegh P.; Hickson I. D. DNA helicase deficiencies associated with cancer predisposition and premature ageing disorders. Hum. Mol. Genet. 2001, 10, 741–746. 10.1093/hmg/10.7.741. [DOI] [PubMed] [Google Scholar]
- Croteau D. L.; Popuri V.; Opresko P. L.; Bohr V. A. Human RecQ helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 2014, 83, 519–552. 10.1146/annurev-biochem-060713-035428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.; Li B.; Gray M. D.; Oshima J.; Mian I. S.; Campisi J. The premature ageing syndrome protein, WRN, is a 3′-->5′ exonuclease. Nat. Genet. 1998, 20, 114–116. 10.1038/2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath-Loeb A. S.; Shen J. C.; Loeb L. A.; Fry M. Werner syndrome protein. II. Characterization of the integral 3′ --> 5′ DNA exonuclease. J. Biol. Chem. 1998, 273, 34145–34150. 10.1074/jbc.273.51.34145. [DOI] [PubMed] [Google Scholar]
- Kim T. M.; Laird P. W.; Park P. J. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell 2013, 155, 858–868. 10.1016/j.cell.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunitomi H.; Banno K.; Yanokura M.; Takeda T.; Iijima M.; Nakamura K.; Iida M.; Adachi M.; Watanabe K.; Matoba Y.; Kobayashi Y.; Tominaga E.; Aoki D. New use of microsatellite instability analysis in endometrial cancer. Oncol. Lett. 2017, 14, 3297–3301. 10.3892/ol.2017.6640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal T.; Permuth-Wey J.; Kumar A.; Sellers T. A. Systematic review and meta-analysis of ovarian cancers: estimation of microsatellite-high frequency and characterization of mismatch repair deficient tumor histology. Clin. Cancer Res. 2008, 14, 6847–6854. 10.1158/1078-0432.CCR-08-1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P.; Li L.; Jiang X.; Li Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. 10.1186/s13045-019-0738-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemery S.; Keegan P.; Pazdur R. First FDA Approval Agnostic of Cancer Site - When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. 10.1056/NEJMp1709968. [DOI] [PubMed] [Google Scholar]
- Behan F. M.; Iorio F.; Picco G.; Gonçalves E.; Beaver C. M.; Migliardi G.; Santos R.; Rao Y.; Sassi F.; Pinnelli M.; Ansari R.; Harper S.; Jackson D. A.; McRae R.; Pooley R.; Wilkinson P.; van der Meer D.; Dow D.; Buser-Doepner C.; Bertotti A.; Trusolino L.; Stronach E. A.; Saez-Rodriguez J.; Yusa K.; Garnett M. J. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. 10.1038/s41586-019-1103-9. [DOI] [PubMed] [Google Scholar]
- Chan E. M.; Shibue T.; McFarland J. M.; Gaeta B.; Ghandi M.; Dumont N.; Gonzalez A.; McPartlan J. S.; Li T.; Zhang Y.; Bin Liu J.; Lazaro J. B.; Gu P.; Piett C. G.; Apffel A.; Ali S. O.; Deasy R.; Keskula P.; Ng R. W. S.; Roberts E. A.; Reznichenko E.; Leung L.; Alimova M.; Schenone M.; Islam M.; Maruvka Y. E.; Liu Y.; Roper J.; Raghavan S.; Giannakis M.; Tseng Y. Y.; Nagel Z. D.; D’Andrea A.; Root D. E.; Boehm J. S.; Getz G.; Chang S.; Golub T. R.; Tsherniak A.; Vazquez F.; Bass A. J. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 2019, 568, 551–556. 10.1038/s41586-019-1102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kategaya L.; Perumal S. K.; Hager J. H.; Belmont L. D. Werner Syndrome Helicase Is Required for the Survival of Cancer Cells with Microsatellite Instability. iScience 2019, 13, 488–497. 10.1016/j.isci.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb S.; Blaha-Ostermann S.; Kamper E.; Rippka J.; Schwarz C.; Ehrenhöfer-Wölfer K.; Schlattl A.; Wernitznig A.; Lipp J. J.; Nagasaka K.; van der Lelij P.; Bader G.; Koi M.; Goel A.; Neumüller R. A.; Peters J. M.; Kraut N.; Pearson M. A.; Petronczki M.; Wöhrle S. Werner syndrome helicase is a selective vulnerability of microsatellite instability-high tumor cells. eLife 2019, 8, e43333. 10.7554/eLife.43333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald E. R. 3rd; de Weck A.; Schlabach M. R.; Billy E.; Mavrakis K. J.; Hoffman G. R.; Belur D.; Castelletti D.; Frias E.; Gampa K.; Golji J.; Kao I.; Li L.; Megel P.; Perkins T. A.; Ramadan N.; Ruddy D. A.; Silver S. J.; Sovath S.; Stump M.; Weber O.; Widmer R.; Yu J.; Yu K.; Yue Y.; Abramowski D.; Ackley E.; Barrett R.; Berger J.; Bernard J. L.; Billig R.; Brachmann S. M.; Buxton F.; Caothien R.; Caushi J. X.; Chung F. S.; Cortés-Cros M.; deBeaumont R. S.; Delaunay C.; Desplat A.; Duong W.; Dwoske D. A.; Eldridge R. S.; Farsidjani A.; Feng F.; Feng J.; Flemming D.; Forrester W.; Galli G. G.; Gao Z.; Gauter F.; Gibaja V.; Haas K.; Hattenberger M.; Hood T.; Hurov K. E.; Jagani Z.; Jenal M.; Johnson J. A.; Jones M. D.; Kapoor A.; Korn J.; Liu J.; Liu Q.; Liu S.; Liu Y.; Loo A. T.; Macchi K. J.; Martin T.; McAllister G.; Meyer A.; Mollé S.; Pagliarini R. A.; Phadke T.; Repko B.; Schouwey T.; Shanahan F.; Shen Q.; Stamm C.; Stephan C.; Stucke V. M.; Tiedt R.; Varadarajan M.; Venkatesan K.; Vitari A. C.; Wallroth M.; Weiler J.; Zhang J.; Mickanin C.; Myer V. E.; Porter J. A.; Lai A.; Bitter H.; Lees E.; Keen N.; Kauffmann A.; Stegmeier F.; Hofmann F.; Schmelzle T.; Sellers W. R. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, 577–592.e10. 10.1016/j.cell.2017.07.005. [DOI] [PubMed] [Google Scholar]
- van Wietmarschen N.; Sridharan S.; Nathan W. J.; Tubbs A.; Chan E. M.; Callen E.; Wu W.; Belinky F.; Tripathi V.; Wong N.; Foster K.; Noorbakhsh J.; Garimella K.; Cruz-Migoni A.; Sommers J. A.; Huang Y.; Borah A. A.; Smith J. T.; Kalfon J.; Kesten N.; Fugger K.; Walker R. L.; Dolzhenko E.; Eberle M. A.; Hayward B. E.; Usdin K.; Freudenreich C. H.; Brosh R. M. Jr.; West S. C.; McHugh P. J.; Meltzer P. S.; Bass A. J.; Nussenzweig A. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 2020, 586, 292–298. 10.1038/s41586-020-2769-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommers J. A.; Kulikowicz T.; Croteau D. L.; Dexheimer T.; Dorjsuren D.; Jadhav A.; Maloney D. J.; Simeonov A.; Bohr V. A.; Brosh R. M. Jr. A high-throughput screen to identify novel small molecule inhibitors of the Werner Syndrome Helicase-Nuclease (WRN). PLoS One 2019, 14, e0210525 10.1371/journal.pone.0210525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal M.; Sommers J. A.; Shoemaker R. H.; Brosh R. M. Jr. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 1525–1530. 10.1073/pnas.1006423108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadrick W. R.; Ndjomou J.; Kolli R.; Mukherjee S.; Hanson A. M.; Frick D. N. Discovering new medicines targeting helicases: challenges and recent progress. SLAS Discovery 2013, 18, 761–781. 10.1177/1087057113482586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muftuoglu M.; Kulikowicz T.; Beck G.; Lee J. W.; Piotrowski J.; Bohr V. A. Intrinsic ssDNA annealing activity in the C-terminal region of WRN. Biochemistry 2008, 47, 10247–10254. 10.1021/bi800807n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J. C.; Gray M. D.; Oshima J.; Loeb L. A. Characterization of Werner syndrome protein DNA helicase activity: directionality, substrate dependence and stimulation by replication protein A. Nucleic Acids Res. 1998, 26, 2879–2885. 10.1093/nar/26.12.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.; Beresten S.; Li B.; Oshima J.; Ellis N. A.; Campisi J. Characterization of the human and mouse WRN 3′-->5′ exonuclease. Nucleic Acids Res. 2000, 28, 2396–2405. 10.1093/nar/28.12.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N.; Shiratori M.; Goto M.; Furuichi Y. Werner syndrome helicase contains a 5′-->3′ exonuclease activity that digests DNA and RNA strands in DNA/DNA and RNA/DNA duplexes dependent on unwinding. Nucleic Acids Res. 1999, 27, 2361–2368. 10.1093/nar/27.11.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry J. J. P.; Yannone S. M.; Holden L. G.; Hitomi C.; Asaithamby A.; Han S.; Cooper P. K.; Chen D. J.; Tainer J. A. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat. Struct. Mol. Biol. 2006, 13, 414–422. 10.1038/nsmb1088. [DOI] [PubMed] [Google Scholar]
- Opresko P. L.; Laine J. P.; Brosh R. M. Jr.; Seidman M. M.; Bohr V. A. Coordinate action of the helicase and 3′ to 5′ exonuclease of Werner syndrome protein. J. Biol. Chem. 2001, 276, 44677–44687. 10.1074/jbc.M107548200. [DOI] [PubMed] [Google Scholar]
- Teklemariam T. A.; Rivera O. D.; Nelson S. W.. Kinetic Analysis of the Exonuclease Activity of the Bacteriophage T4 Mre11-Rad50 Complex. In Methods in Enzymology, 2018; Vol. 600, pp 135–156. [DOI] [PubMed] [Google Scholar]
- Barthels F.; Meyr J.; Hammerschmidt S. J.; Marciniak T.; Rader H. J.; Ziebuhr W.; Engels B.; Schirmeister T. 2-Sulfonylpyrimidines as Privileged Warheads for the Development of S. aureus Sortase A Inhibitors. Front. Mol. Biosci. 2021, 8, 804970 10.3389/fmolb.2021.804970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M. R.; Joerger A. C.; Fersht A. R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E5271–E5280. 10.1073/pnas.1610421113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengler D. G.; Harikumar K. G.; Pollari S.; Sun Q.; Brown B. T.; Shinoki-Iwaya A.; Ardecky R.; Miller L. J.; Sergienko E. A. Discovery of small molecule positive allosteric modulators of the secretin receptor. Biochem. Pharmacol. 2021, 185, 114451 10.1016/j.bcp.2021.114451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambaldo C.; Vinogradova E. V.; Qi X.; Iaconelli J.; Suciu R. M.; Koh M.; Senkane K.; Chadwick S. R.; Sanchez B. B.; Chen J. S.; Chatterjee A. K.; Liu P.; Schultz P. G.; Cravatt B. F.; Bollong M. J. 2-Sulfonylpyridines as Tunable, Cysteine-Reactive Electrophiles. J. Am. Chem. Soc. 2020, 142, 8972–8979. 10.1021/jacs.0c02721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz P. A.; Kuzmic P.; Solowiej J.; Bergqvist S.; Bolanos B.; Almaden C.; Nagata A.; Ryan K.; Feng J.; Dalvie D.; Kath J. C.; Xu M.; Wani R.; Murray B. W. Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 173–178. 10.1073/pnas.1313733111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes M. R.; Zhang J.; Li L. S.; Hansen R.; Peters U.; Guo X.; Chen Y.; Babbar A.; Firdaus S. J.; Darjania L.; Feng J.; Chen J. H.; Li S.; Long Y. O.; Thach C.; Liu Y.; Zarieh A.; Ely T.; Kucharski J. M.; Kessler L. V.; Wu T.; Yu K.; Wang Y.; Yao Y.; Deng X.; Zarrinkar P. P.; Brehmer D.; Dhanak D.; Lorenzi M. V.; Hu-Lowe D.; Patricelli M. P.; Ren P.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589. 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Hopper M.; Gururaja T.; Kinoshita T.; Dean J. P.; Hill R. J.; Mongan A. Relative Selectivity of Covalent Inhibitors Requires Assessment of Inactivation Kinetics and Cellular Occupancy: A Case Study of Ibrutinib and Acalabrutinib. J. Pharmacol. Exp. Ther. 2020, 372, 331–338. 10.1124/jpet.119.262063. [DOI] [PubMed] [Google Scholar]
- Orren D. K.; Brosh R. M. Jr.; Nehlin J. O.; Machwe A.; Gray M. D.; Bohr V. A. Enzymatic and DNA binding properties of purified WRN protein: high affinity binding to single-stranded DNA but not to DNA damage induced by 4NQO. Nucleic Acids Res. 1999, 27, 3557–3566. 10.1093/nar/27.17.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerwińska J.; Poznanski J.; Debski J.; Bukowy Z.; Bohr V. A.; Tudek B.; Speina E. Catalytic activities of Werner protein are affected by adduction with 4-hydroxy-2-nonenal. Nucleic Acids Res. 2014, 42, 11119–11135. 10.1093/nar/gku783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman J. A.; Gavard A. E.; Lieb S.; Ravichandran M. C.; Hauer K.; Werni P.; Geist L.; Böttcher J.; Engen J. R.; Rumpel K.; Samwer M.; Petronczki M.; Gileadi O. Structure of the helicase core of Werner helicase, a key target in microsatellite instability cancers. Life Sci. Alliance 2021, 4, e202000795. 10.26508/lsa.202000795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.