

Abstract
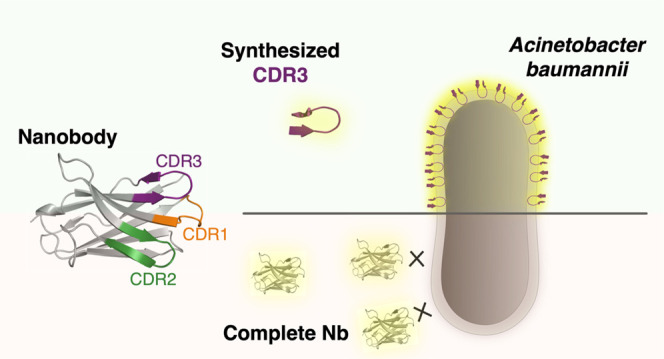
Membrane interaction constitutes to be an essential parameter in the mode of action of entities such as proteins, as well as cell-penetrating and antimicrobial peptides, resulting in noninvasive or lytic activities depending on the membrane compositions and interactions. Recently, a nanobody able to interact with the top priority, multidrug-resistant bacterial pathogen Acinetobacter baumannii was discovered, although binding took place with fixed cells only. To potentially overcome this limitation, linear peptides corresponding to the complementarity-determining regions (CDR) were synthesized and fluorescently labeled. Microscopy data indicated clear membrane interactions of the CDR3 sequence with living A. baumannii cells, indicating both the importance of the CDR3 as part of the parent nanobody paratope and the improved binding ability and thus avoiding the need for permeabilization of the cells. In addition, cyclization of the peptide with an additionally introduced rigidifying 1,2,3-triazole bridge retains its binding ability while proteolytically protecting the peptide. Overall, this study resulted in the discovery of novel peptides binding a multidrug-resistant pathogen.
Introduction
Biological membranes are indispensable cellular components functioning as physical barriers fundamental to the functioning and survival of cells. The composition and organization of these typical bilayer structures vary considerably between cell types and species,1 and determine the possible interaction with natural and synthetic entities. From a therapeutic perspective, membrane interaction is an important step in the cellular uptake of various bioactive substances in eukaryotic cells to allow access to intracellular targets.2 Although classical small-molecule drugs can be tweaked to allow cell internalization, membrane crossing of hydrophilic molecules like peptides, proteins, and nucleic acids is less straightforward. Significant research has been devoted to the development of cell-penetrating peptides (CPPs), such as TAT, penetratin, transportan, Pep-1, and oligoarginines, which allow the transport of cargoes inside a cell.3−5 The mode of action of CPPs has been vastly discussed, proposing the two main mechanisms for their membrane transfer to be direct transduction and energy-dependent endocytosis, which generally rely on the initial interaction of the peptides with the membrane.6 This membrane interaction is particularly dependent on the physicochemical properties of the CPPs, which frequently have cationic or amphipathic character due to the presence of multiple positively charged residues and/or hydrophobic (aromatic) residues.7,8
Curiously, some characteristics of CPPs match the ones of antimicrobial peptides (AMPs), and several CPPs show membrane-disrupting lytic activities in bacteria, sharply contrasting their rather noninvasive penetration of eukaryotic cells.9−12 These contrasting activities can be related to the highly differing membrane compositions: eukaryotic cells contain a single bilayer with a wide variety of mainly zwitterionic phospholipids combined with cholesterol. In contrast, bacterial cytoplasmic membranes rather contain the zwitterionic phosphatidylethanolamine and significantly higher amounts of anionic phospholipids, together with a peptidoglycan layer, supplemented in Gram-negative bacteria with a second outer asymmetric membrane containing lipopolysaccharides at the outside.13,14 The mode of action of positively charged AMPs mostly relies initially on electrostatic interactions with the negatively charged bacterial membranes, provoking their accumulation on the membrane surfaces, whereas hydrophobic residues assist in the membrane penetration and/or disruption, for which various models have been proposed.15,16
Peptides interacting with bacteria can be especially interesting when they target problematic bacteria, in particular antibiotic-resistant human pathogens. In the last few years, the increasing rise of antibiotic-resistant bacteria has rendered the need for alternative antibiotics urgent.17,18 The World Health Organization (WHO) officially published a “Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics”, designating carbapenem-resistant Acinetobacter baumannii as top priority of the critical bacterial pathogen group for which “research and development of new antibiotics is critically needed”.19,20 In addition, the Centers for Disease Control and Prevention (CDC) categorized A. baumannii as “an urgent threat to public health”.21 The bacterium also belongs to the ESKAPE group of most problematic nosocomial bacterial agents.22
A. baumannii is a Gram-negative, opportunistic human pathogen.23−25 It is a capsulated and nonflagellated bacterium that produces different types of lipooligosaccharides instead of the classical lipopolysaccharides. This bacterium most commonly causes ventilator-associated pneumonia, central-line-associated bloodstream, soft tissue, catheter-associated, and urinary tract infections.21,25 The factors favoring infections are often paired with disruption of the skin protective barriers such as open wounds and burns, colonization of mechanical devices such as catheters and ventilation equipment, prolonged hospital stays, and immunocompromised patients.17,24,26 A recent global study on multidrug-resistant (MDR) pathogens highlighted those infections by A. baumannii as the sixth highest mortality rate worldwide.18
The infamous pathogen gained its reputation due to an alarming combination of characteristics, environmental persistence, and antibiotic resistance, which makes it especially problematic in a clinical context. The environmental persistence of A. baumannii refers to its ability to survive prolonged periods of desiccation and resist disinfectants.27−29 The emergence of drug resistance in the bacterium is accelerated by its facile acquisition of exogenous DNA, and thus drug resistance genes, through horizontal gene transfer.22,30,31 The facilitated DNA uptake is also responsible for the rapidly evolving, dynamic genome of A. baumannii. Consequently, a high diversity is found in clinical isolates, with a strikingly small core genome of 16.5%.32,33 Moreover, 25% of the genome is unique in A. baumannii isolates.34 This represents a challenge in the identification of broadly active compounds recognizing the majority of A. baumannii isolates.
Worldwide, multidrug- to extensively drug- and even pandrug-resistant A. baumannii strains have been reported.35 Treatments for A. baumannii infections are limited and few new therapeutics are in the pipeline. Currently, infections are treated with a combination of antibiotics, often with significant side effects.36 Several studies have proposed nonantibiotic, alternative treatments, such as phage therapy, antimicrobial peptides, and antibody-based therapeutics.36 Antibody-based therapeutics for bacterial infections mainly comprise monoclonal antibodies targeting surface-exposed entities on the bacterial cell or toxins, thereby eliciting opsonophagocytosis, complement-mediated killing, or, in the case of binding toxins, a neutralizing effect.37 Another type of antibody-based biomolecules are nanobodies. A few reports have been made on the use of nanobodies to target pathogenic bacteria, including Listeria monocytogenes, Ehrlichia chaffeensis, Legionella pneumophila, and Campylobacter jejuni.38−42
Nanobodies (Nb, VHH), the variable domains of camelid-derived heavy-chain only antibodies, have emerged in various fields of research, including, for example, anticancer treatments.43−47 Despite the 10-fold reduction in molecular size (ca. 15 vs 150 kDa), these recombinantly produced single domains can reach the favorable high affinities and specificities for antigens of classical heterotetrameric monoclonal antibodies. The size reduction of Nbs provides favorable properties over monoclonal antibodies, including lower production costs, enhanced thermal and chemical stabilities, improved tissue penetration, lowered immunogenic responses, and the ability to access cavities on the target surface.48,49 Although the antigen-binding paratope of a classical antibody is centered on six different complementarity-determining regions (CDRs), a Nb has only three CDRs available for interaction with the antigen. The lack of light-chain CDRs is frequently compensated by increased CDR lengths and higher amino acid variability, especially in CDR3, allowing the existence of CDR conformations deviating from the restricted canonical structures observed in conventional antibodies.50−52 In addition, the CDR3 of a Nb accounts, on average, for more than half of all binding interactions of the whole paratope, thereby playing a predominant role.53,54
The reduction of a paratope toward a synthetic peptide of limited molecular weight (1–2 kDa) might provide an alternative to nanobodies or antibodies. Numerous studies reported peptides based on the CDR sequences of monoclonal antibodies, although their successes were rather limited.55 Only in some rare cases where a dominant CDR loop was present, optimized CDR-based peptides could fully mimic the binding and cellular effects of an antibody.56 In the case of Nbs, peptide paratope mimicry has only scarcely been reported.55 Nonetheless, the high importance of the CDR3 in some Nbs is expected to provide an appropriate basis for such mimicry. In addition, CDR-derived peptides can provide insights into the significance of each individual CDR in the absence of structural data.
In the context of peptide paratope mimetics and membrane-interacting peptides, this study investigated the potential interaction of Nb CDR loops with A. baumannii strains. The CDR segments of an A. baumannii binding Nb were synthesized as linear peptides to evaluate their (potential) separate interaction with the bacterium. A particular importance of the CDR3 sequence was observed.
Results and Discussion
NbD4 Binds Fixed A. baumannii Cells
Prior to this study, the nanobody NbD4 (Reference number CA19404) was identified to bind the A. baumannii strain AB5075-VUB. This strain is capsulated, multidrug-resistant, and virulent.33 Therefore, the strain was also chosen in this study for the further characterization of the Nb-bacterium interaction by fluorescence microscopy. After incubating sulfoCy3-labeled NbD4 with the bacteria, the labeled nanobody allowed screening for its binding to bacterial cells. First, the interaction of NbD4 with fixed A. baumannii cells was tested. Under these conditions, distinct labeling of 100% of the cells was observed (Figure 1A). Moreover, the micrographs showed a labeling pattern indicating a typical membrane interaction of NbD4 with the bacterial cells (Figure S2). In a next step, NbD4 was also tested for interaction with living bacterial cells. Unfortunately, only 2.94 ± 0.65% of the bacteria were labeled by NbD4 (Figure 1A). Fixing bacteria with paraformaldehyde might alter the bacterial cell surface and cause permeabilization as has been described for Eukaryotic cells.57 Therefore, the permeabilization of bacteria might facilitate access of the nanobody to its target which potentially explains the observed binding behavior. The target itself, however, remains unknown as pull-down58 and mass spectrometry experiments were inconclusive.
Figure 1.

Nanobody-derived peptide 3 improves binding of living A. baumannii cells. The Nb and derived peptides are labeled with SulfoCy3 which was visualized by fluorescence microscopy. (A) Comparison of Nb binding on living and fixed (1% PFA) bacteria. The scale bar is 5 μm. (B) Comparison of binding of peptides 2 and 3. One cell was picked to illustrate the membrane labeling profile shown in (C) Graph generated by ImageJ and GraphPad Prism. All data were collected in biological triplicate.
NbD4 CDR Identification and Peptide Design
Following the identification of the ability of the Nb to interact with fixed A. baumannii bacteria, but only a minority of living A. baumannii cells, we explored whether reducing the size of the nanobody improves the accessibility and/or labeling of living bacteria. As the antigen-binding paratope of the nanobody is composed of 3 CDR sequences, and these CDR sequences contain the largest sequence variability, these CDR sequences were synthesized separately to test their own binding ability. Although structural data of the Nb is not available, the CDR sequences can be extracted by comparison to known sequences and structures, since excluding the CDR sequences, the nanobody sequences are rather structurally conserved framework regions.50 For this purpose, different numbering schemes and CDR definitions have been developed, such as the commonly used Kabat, Chothia, Martin, and IMGT schemes.59 However, these schemes do not consider the increased CDR lengths and sequence variability (at the level of both the CDR and framework regions) observed in Nbs, and some fail to correctly identify the full CDR sequences since they were developed based on monoclonal antibody structures. Based on the comparison with previously reported Nb structures, new extended CDR definitions have been applied.51,54,60 In this paper, we considered the definition used by Zavrtanik et al., which follows the IMGT CDR definitions extended with additional residues at the C-terminal end of CDR2 and CDR3, as these frequently interact with antigens.54 To increase the possibility of taking the full secondary loop structures (i.e., mostly β-hairpin structures for CDR2, and possibly CDR3), the Nb sequence was compared to known experimental Nb structures having partially overlapping CDR sequences (see Supporting Figure S1). Based on these comparisons, Thr55 and Trp118 were included in the CDR2 and CDR3 sequences.
The identified CDR sequences were synthesized by standard Fmoc/tBu-based solid-phase peptide synthesis (SPPS) protocols as C-terminal amides on a Rink amide AM or Sieber resin to obtain, respectively, side-chain-unprotected or -protected peptides. To allow visualization of the possible interaction of the CDR-representing peptides with the studied bacteria, they were extended with a spacing β-alanine at the N-terminus and labeled with a sulfonated cyanine 3 (SulfoCy3) fluorophore (Table 1). The dye was introduced by reaction of the free N-terminus of the peptides with an N-hydroxysuccinimide ester present on the labeling agent. This labeling was performed on purified unprotected peptides in the case of peptides 2 and 3. To allow orthogonality of the N-terminal amine with the ε-amine of a lysine present, peptide 1 was obtained by labeling a side-chain-protected precursor in solution, followed by standard cleavage with a TFA-based cleavage cocktail (see the Supporting Information for more details).
Table 1. Sequences of Synthesized Nb CDR-Based Linear Peptides.
| code | corresponding Nb CDR | sequence |
|---|---|---|
| 1 | CDR1 | SulfoCy3-β-Ala-Gly-Ile-Ser-Lys-Ser-Ile-Thr-Ile-NH2 |
| 2 | CDR2 | SulfoCy3-β-Ala-Thr-Ile-Thr-Ser-Gly-Gly-Thr-Thr-Asn-NH2 |
| 3 | CDR3 | SulfoCy3-β-Ala-Asn-Ala-Arg-Arg-Leu-Arg-Glu-Tyr-Trp-NH2 |
Linear CDR3 Analogue Can Bind Living AB5075-VUB
To examine whether peptides 1–3 (corresponding to Nb CDR sequences 1–3, resp.) have improved binding properties on A. baumannii compared to the parent nanobody, fluorescence microscopy was used. After incubation of the peptides with living bacterial cells, the sulfoCy3-labeling of the peptides allowed screening for the presence of the peptides on A. baumannii bacteria. Due to the high hydrophobic nature of peptide 1, and its aggregation propensity in solution, no results could be obtained for this peptide. Additionally, no binding was observed on the bacterial cells for peptide 2, but gratifyingly, clear binding was observed on all bacterial cells by peptide 3 (Figure 2B). Moreover, the observed pattern on the micrographs indicated a membrane interaction of peptide 3, which was corroborated by the intensity profile of a cell intersection (Figure 1C).
Figure 2.

Nanobody-derived peptide 3 binds 6 A. baumannii clinical isolates and 2 frequently used lab strains. Phase contrast images show the different morphologies of the bacteria. The SulfoCy3-labeled peptide 3 is visualized by fluorescence microscopy and shows the membrane labeling of the bacteria. The scale bar is 5 μm. All data were collected in biological triplicate.
As the fluorescence assay was performed on living, nonfixed bacteria, this excludes unrelated binding events related to partial membrane permeabilization caused by the fixating agent, which has been observed before in investigations on cell-penetrating peptides in eukaryotic cells.61 Moreover, permeabilization seems to be required for binding of the parent nanobody as only a minority of living bacteria was bound by NbD4. Since there was no binding observed for peptide 2, and peptide 1 could not be tested, but clear membrane labeling was observed for peptide 3, the latter can be considered as the minimal required element for membrane binding. This result indicates that the CDR3 sequence comprises an important element in the membrane interaction of the parent nanobody, with the smaller size of the peptide allowing enhanced accessibility, and hence, the observed binding on living, capsulated A. baumannii bacteria.
Linear CDR3 Analogue Can Bind a Variety of A. baumannii Isolates
Due to the high diversity found in clinical isolates,33 it is not evident that a peptide interacting with the A. baumannii strain AB5075-VUB will also interact with different A. baumannii strains. Therefore, a selection of relevant clinical isolates was included to test for binding of peptide 3, namely, strains AB3-VUB, AB36-VUB, AB39-VUB, AB180-VUB, AB193-VUB, and AB213-VUB.33,62 These 6 clinical isolates have striking differences: they differ in capsule types and thicknesses, different antibiograms, and were isolated from Belgian hospitals in the last 8 years.33,34 In addition, 2 frequently used lab strains, ATCC19606 and ATCC17978, were included as well. By fluorescence microscopy, the binding ability of peptide 3 was investigated on these A. baumannii strains.
For all tested strains, membrane labeling is observed. The labeling is observed on 100% of the bacterial population of all strains, though with different intensities (Figure 2). Interestingly, these varying degrees of labeling are observed in between isolates, but also within the population of one strain. Due to the high diversity found among A. baumannii isolates, the varying degree of labeling in between isolates could be caused by a multitude of factors, ranging from a charge-based interaction with the surface of the bacterium to the availability of the target. The varying degree of labeling within one strain can potentially be explained by the growth phase of individual cells or the phenotypic heterogeneity found in A. baumannii strains.63,64
Proteolytically Protected Cyclic CDR3 Analogue Preserves Binding
The CDR sequences of a nanobody are positioned on a structurally stabilized β-sheet adopting a framework that directs and limits their conformational populations. Therefore, displaying CDR sequences solely in a linear peptide format may not provide sufficient rigidity to mimic the Nb paratope due to the flexible nature of linear peptides, as these can adopt a plethora of conformations. Various elements have been reported to allow stabilization of peptide conformations into, for example, secondary structures such as α-helices, turns, β-sheets, and other irregular conformations,65,66 wherein backbone macrocyclization constitutes a powerful strategy to reduce the flexibility of peptides and allow the preorganization of their backbone and side chains. Peptide cyclization was also shown to modulate the membrane interaction and activity of some antimicrobial and cell-penetrating peptides, next to improving their proteolytic stability.67−73
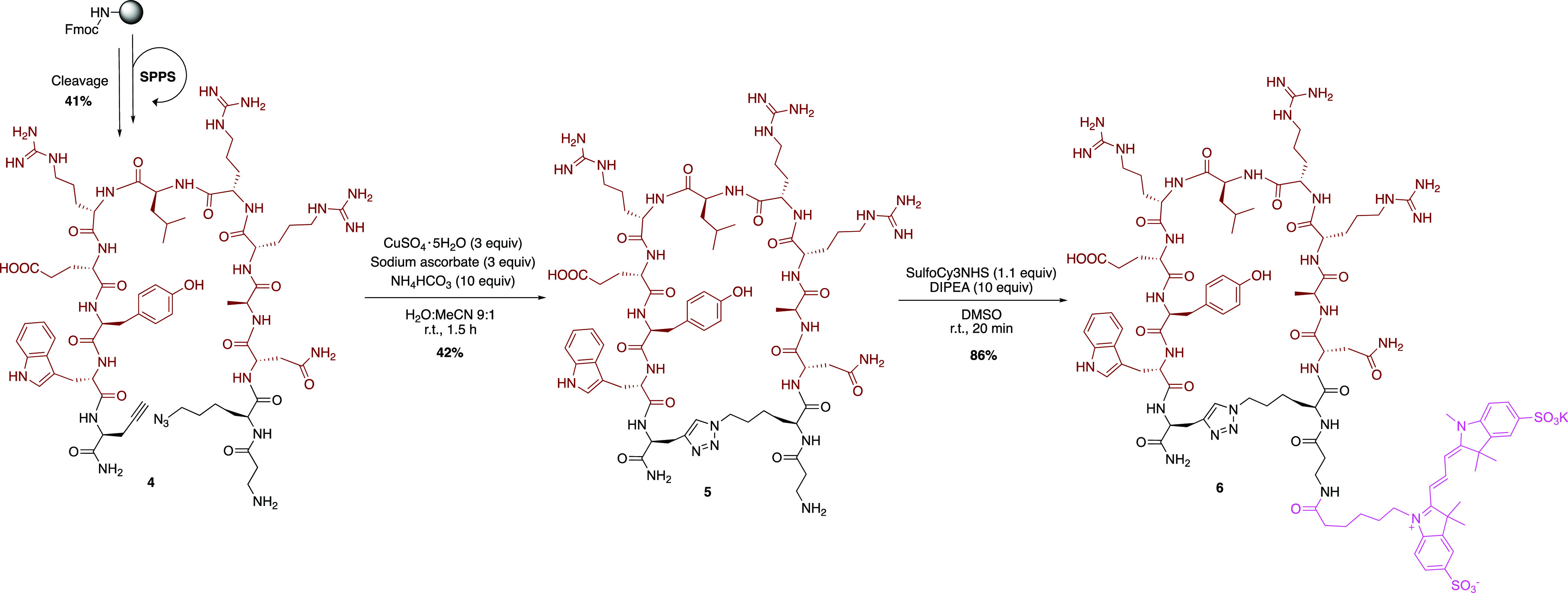
Therefore, we considered constraining the CDR3-presenting linear peptide 3 by the addition of a rigidifying 1,2,3-triazole bridge, which might assist in mimicking the conformational sampling of the parent Nb CDR3. For this purpose, linear precursor 4 containing additional propargylglycine and azidolysine residues at the terminal positions was synthesized by standard SPPS methods (Scheme 1). Subsequently, the peptide was cyclized in solution using a cycloaddition reaction between the alkyne and azide functions, in the presence of copper(I) to form a 1,4-disubstituted 1,2,3-triazole linkage. The obtained cyclic peptide 5 was labeled by the reaction of the free amine, present on the N-terminal β-alanine residue, with the activated N-hydroxysuccinimide ester of sulfonated cyanine 3 in basic conditions, resulting in fluorescent peptide 6 after preparative HPLC purification.
Scheme 1. Synthesis of the Cyclic Peptide 6.
To allow comparison with linear peptide 3, the membrane binding capacity of peptide 6 was also evaluated on living AB5075-VUB bacteria using fluorescence microscopy. For both peptides, clear membrane labeling is observed (Figure 3A). To determine whether peptide 6 can bind the bacterial cells as well as its linear version, the maximum observed fluorescence intensities of the bacterial cells after treatment with the labeled peptides were compared (Figure 3B). No statistically significant difference was found between the fluorescence intensities of the linear and cyclic peptides. However, the cyclic peptide 6 was expected to have a more stable nature as the cyclization proteolytically protects the peptide.73,74 A stability assay on selected peptides in human plasma validated that the half-life of the acetylated variant of peptide 6 is indeed doubled compared to the same variant of peptide 3 (55.74 ± 0.34 vs 36.25 ± 0.48 min, respectively; Figure S4). Therefore, cyclization of the linear CDR3 results in a preferred tool compound since it is proteolytically more stable, and can bind the bacterial cells equally well. Intriguingly, despite averages being nonsignificantly different, we observe a different distribution of the maximum intensity profile with the cyclic compared to the linear version (Figure 3B).
Figure 3.

Comparison of binding ability of linear peptide 3 and its cyclic variant peptide 6. (A) Living AB5075-VUB cells (seen in phase contrast images) bound by both peptides (labeled by sulfoCy3). (B) Comparison of maximum fluorescence intensity signal measured on AB5075-VUB cells for (n = 300/biological replicate) the cyclic and linear peptide. Scale bar 5 μm. All data were collected in biological triplicate.
Altogether, our fluorescence binding data indicate a clear membrane interaction of labeled linear and cyclic peptides displaying the parent Nb CDR3 sequence with the top priority pathogen A. baumannii. Although the elements involved in the binding interaction of the parent Nb with A. baumannii bacterial cells could not be clearly defined until now, the peptidomifmetic approach indicates that the CDR3 sequence is a minimal required element and important factor in the membrane interaction of the Nb paratope. The lowered, but herein beneficial, specificity of the CDR3-displaying peptides compared to the Nb might indicate a modulating contribution of CDR1 and CDR2 in the Nb paratope, although no direct interaction was observed in linear peptides displaying their corresponding sequence.
Conclusions
Starting from the discovery of a Nb showcasing binding of the multidrug-resistant pathogen A. baumannii, the significance of the individual CDR loops in this interaction was investigated by synthesis and evaluation of single linear peptides. Based on fluorescence binding experiments, clear membrane interactions of the Nb CDR3 with living, capsulated AB5075-VUB cells could be visualized, in contrast to the parent Nb which required cell fixation to obtain labeling of the whole bacterial population. Moreover, the Nb CDR3 can bind a variety of living, multidrug-resistant clinical A. baumannii isolates as well, despite their high genetic diversity. To increase the proteolytic stability of the linear CDR3-presenting peptide, it was constrained using a 1,2,3-triazole linking element between the side chains of terminally added unnatural amino acid residues, which reduces the conformational flexibility of the peptide. The introduced cyclization did not alter the binding ability but does offer a more stable version of the peptide. Altogether, the followed peptide mimicry approach allowed us to identify the CDR3 as a minimal required element in the membrane interaction of the parent Nb with the investigated A. baumannii strains. Moreover, the CDR3-derived peptide avoids the need for permeabilization to be able to bind the bacterial cells. Due to their specificity, the peptides can potentially serve as holds targeting tags in, for example, peptide–drug conjugates since it was demonstrated that they can function as a nontoxic, specific label for A. baumannii. The smaller size of the peptide (ca. 1–2 kDa) compared to the Nb (ca. 14 kDa) might allow easier membrane permeabilization and visualization of membrane binding on living capsulated bacteria. In addition, the straightforward chemical accessibility of peptides provides a clear advantage over the recombinantly produced nanobodies.
Materials and Methods
Bacterial Strains, Growth Conditions, and Fixation
A collection of 3 classically used (AB5075-VUB - CP070362, Tn7::Pst-sfGFP; ATCC19606; ATCC17978) and 6 clinically isolated A. baumannii strains (AB3-VUB; AB36-VUB; AB39-VUB; AB180-VUB; AB193-VUB; AB213-VUB) were used in this study. The bacterial cultures were started from a single clone and were grown for 17 h at 37 °C under agitation (165 rpm) in low-salt broth (Luria-Bertani formulation, Duchefa Biochemie). Cells were collected by centrifugation (8000g) and normalized to OD600 = 3 (109 CFU/ml) in phosphate-buffered saline (PBS) for further steps. Fixation of the cells was by 1 h incubation of the cells in PBS, supplemented with 1% paraformaldehyde in PBS (Thermo Scientific Chemicals).
Nanobody Discovery, Purification, and Labeling
A llama (lama glama) was immunized weekly with a mix of fixed A. baumannii cells (approximately 1.6 108 CFU). After the final injection, total RNA was extracted from peripheral blood mononuclear cells from which cDNA was synthesized, as described earlier.75,76 The cDNA was cloned into the phagemid vector pMESy4, enabling expression of Nbs with an N-terminal periplasmic leader sequence and a C-terminal 6-His-EPEA tag. The resulting phage library, consisting of approximately 4.108 clones, was enriched by two phage display selection rounds using fixed A. baumannii cells (approximately 2.56 109 CFU). Out of the enriched library, clones were transformed to E. coli WK6. Cells were grown in Terrific Broth (Duchefa Biochemie) and then induced by IPTG for periplasmic expression and purified as described elsewhere.77 Labeling of the nanobody was done by incubation with molar 3-fold of sulfo-Cyanine3 NHS ester (ex:548/em:563, Lumiprobe) for 1 h, quenching of the reaction with 1 mM Tris buffer, followed by overnight dialysis in PBS.
Peptide Synthesis
Peptides were synthesized by Fmoc/tBu-based solid-phase peptide synthesis using Rink Amide AM or Sieber amide resins (0.10 mmol scales) on a CEM Liberty Lite Automated Microwave Peptide Synthesizer or through manual synthesis. On the “Liberty Lite peptide synthesizer, couplings were performed using 4 equiv of amino acids (0.5 M solution in DMF), N,N′-diisopropylcarbodiimide (DIC, 0.5 M solution in DMF), and Oxyma Pure (1.0 M solution in DMF). Although standard couplings were performed for 2.1 min at 90 °C, Fmoc-Arg(Pbf)-OH was coupled twice at 75 °C. Fmoc deprotections were carried out for 1.05 min at 90 °C using 20% (v/v) 4-methylpiperidine in DMF. Between every step, the resin was washed with DMF (3 times). Manual Fmoc deprotections were performed using 20% (v/v) 4-methylpiperidine in DMF for 5 and 15 min. Amino acid residues were manually coupled using 3 equiv of protected amino acid, 3 equiv of HBTU and 6 equiv. of N,N-diisopropylethylamine (DIPEA) in DMF for 1 h at room temperature. Between every step, the resin was washed thoroughly with DMF (3 times) and DCM (3 times). Peptide cleavages from a Rink Amide AM resin were performed with a freshly prepared cocktail of TFA/TIS/H2O 90/5/5 (v/v/v) for 3 h at room temperature. After precipitation with cold diethyl ether and lyophilization, the crude peptides were purified by preparative RP-HPLC yielding the pure peptides under their TFA salt form as white powders.
Fluorophore linkage on unprotected pure peptides (0.004–0.01 mmol scale) was performed in a glass vial sealed form light using sulfonated cyanine 3 NHS (1.1 equiv) and DIPEA (10 equiv) in DMSO (2 mL). After full conversion observed by analytical RP-HPLC (typically less than 20 min), the pink solution was directly purified by preparative RP-HPLC, yielding the pure labeled peptides as pink powders after lyophilization.
Peptide 1 was synthesized under its unlabeled fully protected form on a Sieber amide resin, followed by cleavage using 1% TFA in DCM (5 times 1 min). The combined filtrates were pooled and directly evaporated under reduced pressure, followed by direct labeling on a small fraction (5.70 mg, 0.0078 mmol) using sulfonated cyanine 3 NHS (1.0 equiv) and DIPEA (10 equiv) in DMSO:DCM 1:3 (0.8 mL). After overnight reaction, the side-chain-protecting groups were removed by stirring the peptide in a solution of TFA/TIS/H2O 90/5/5 (v/v/v) for 2 h at room temperature. After concentration in vacuo, the peptide was directly purified by preparative RP-HPLC.
Cyclic peptide 6 was synthesized as an unlabeled linear peptide as outlined above with manual coupling of Fmoc-propargylglycine and Fmoc-azidolysine. After cleavage and purification, the linear peptide (0.03 mmol, 0.75 mM) was cyclized in solution using copper(II) sulfate pentahydrate (3.2 equiv), sodium ascorbate (3.4 equiv), and ammonium hydrogen carbonate (10.0 equiv) in H2O:MeCN 9:1 for 2 h at room temperature. After lyophilization and purification, the cyclic peptide was labeled and purified as outlined above.
Light and Fluorescence Microscopy
To allow binding, 100 μL of the peptide or the labeled NbD4 (100 μM) was incubated with 100 μL of fixed or living bacterial cells (approximately 105 CFU) for 30 min at 37 °C, under agitation (165 rpm). To remove unbound Nb or peptide, the cells were centrifugated (8000g). Finally, the bacteria were spotted on a 1.5% agarose pad (Thermo Scientific Gene Frame). Microscopy images were acquired using a Leica DMi8 fluorescence microscope with a DFC7000 GT camera (Leica Microsystems CMS GmbH). The GFP expressed in the cytoplasm of the AB5075-VUB strain was used to determine the right focal plane, after which phase contrast and fluorescent images were acquired. The fluorescent images were acquired with a Leica RHOD filter set (ex:546/em:585), using the following parameters: exposure = 512 ms; gain = 1. The raw data was processed by using ImageJ software where brightness was adjusted equally for all fluorescence micrographs.
The labeling degree of the cells was determined by the ratio of the labeled cells and all of the cells in the phase contrast micrograph. Intensity profiles of the cells (n = 300/biological replicate) were obtained using ImageJ software, and the maximum gray value of each intensity profile was plotted using GraphPad Prism. The statistical significance of peptides 3 and 6 was tested using a nonparametric Mann–Whitney U-test.
Acknowledgments
K.V.h. and S.B. thank the Research Foundation–Flanders (FWO Vlaanderen) for providing a PhD fellowship to K.V.h (FWOTM931). K.V.h., M.M., and S.B. thank the Research Council of the Vrije Universiteit Brussel for financial support through the spearhead (SRP50) program. A.B., C.V.d.H., and H.R. thank the Research Foundation–Flanders (FWO Vlaanderen) for providing a PhD SB fellowship to A.B. (File number 77258). C.V.d.H. acknowledges the Vlaams Instituut voor Biotechnologie (VIB) for their support. E.P. and J.S. acknowledge the support and the use of resources of Instruct-ERIC, part of the European Strategy Forum on Research Infrastructures (ESFRI), and the Research Foundation–Flanders (FWO) for their support to the Nanobody discovery and Nele Buys for the technical assistance during Nanobody discovery.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.3c00116.
Nb CDR3 determination, membrane interaction image analysis, and interaction of peptide 3 with E. coli; peptide synthesis and characterization: general methods, general SPSS protocols, synthesis & characterization of linear peptides 1–3, synthesis of cyclic peptide 6, synthesis of acetylated analogues, copies of HPLC spectra, proteolytic stability, and cytotoxicity on living A. baumannii cells; and quantification of labeled cells (PDF)
Author Contributions
# A.B., K.V.h., S.B. and C.V.d.H contributed equally to this work. A.B., K.V.h., C.V.d.H., and S.B. designed the experiments and wrote the manuscript. C.V.d.H., E.P., J.S., N.B., and H.R. generated the nanobody library. A.B. and C.V.d.H identified the binding nanobody and performed microscopy experiments. K.V.h., M.M., and S.B. designed and synthesized the peptides. C.M. performed the peptide stability assay.
The authors declare no competing financial interest.
Supplementary Material
References
- Harayama T.; Riezman H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. 10.1038/nrm.2017.138. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Qin X.; Kong F.; Chen P.; Pan G. Improving cellular uptake of therapeutic entities through interaction with components of cell membrane. Drug Delivery 2019, 26, 328–342. 10.1080/10717544.2019.1582730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti G.; Brambilla L.; Rossi D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. 10.1016/j.tips.2017.01.003. [DOI] [PubMed] [Google Scholar]
- Kristensen M.; Birch D.; Morck Nielsen H. Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. Int. J. Mol. Sci. 2016, 17, 185 10.3390/ijms17020185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J.; Bi Y.; Zhang H.; Dong S.; Teng L.; Lee R. J.; Yang Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. 10.3389/fphar.2020.00697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruseska I.; Zimmer A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. 10.3762/bjnano.11.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujals S.; Fernandez-Carneado J.; Lopez-Iglesias C.; Kogan M. J.; Giralt E. Mechanistic aspects of CPP-mediated intracellular drug delivery: relevance of CPP self-assembly. Biochim. Biophys. Acta, Biomembr. 2006, 1758, 264–279. 10.1016/j.bbamem.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Kalafatovic D.; Giralt E. Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules 2017, 22, 1929 10.3390/molecules22111929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahnsen J. S.; Franzyk H.; Sandberg-Schaal A.; Nielsen H. M. Antimicrobial and cell-penetrating properties of penetratin analogs: effect of sequence and secondary structure. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 223–232. 10.1016/j.bbamem.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Rodriguez Plaza J. G.; Morales-Nava R.; Diener C.; Schreiber G.; Gonzalez Z. D.; Lara Ortiz M. T.; Ortega Blake I.; Pantoja O.; Volkmer R.; Klipp E.; et al. Cell penetrating peptides and cationic antibacterial peptides: two sides of the same coin. J. Biol. Chem. 2014, 289, 14448–14457. 10.1074/jbc.M113.515023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobone S.; Piazzon A.; Orioni B.; Pedersen J. Z.; Nan Y. H.; Hahm K. S.; Shin S. Y.; Stella L. The thin line between cell-penetrating and antimicrobial peptides: the case of Pep-1 and Pep-1-K. J. Pept. Sci. 2011, 17, 335–341. 10.1002/psc.1340. [DOI] [PubMed] [Google Scholar]
- Splith K.; Neundorf I. Antimicrobial peptides with cell-penetrating peptide properties and vice versa. Eur. Biophys. J. 2011, 40, 387–397. 10.1007/s00249-011-0682-7. [DOI] [PubMed] [Google Scholar]
- Sani M. A.; Separovic F. How Membrane-Active Peptides Get into Lipid Membranes. Acc. Chem. Res. 2016, 49, 1130–1138. 10.1021/acs.accounts.6b00074. [DOI] [PubMed] [Google Scholar]
- Epand R. M.; Epand R. F. Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 289–294. 10.1016/j.bbamem.2008.08.023. [DOI] [PubMed] [Google Scholar]
- Teixeira V.; Feio M. J.; Bastos M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. 10.1016/j.plipres.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Brogden K. A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria?. Nat. Rev. Microbiol. 2005, 3, 238–250. 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- Weiner L. M.; Webb A. K.; Limbago B.; Dudeck M. A.; Patel J.; Kallen A. J.; Edwards J. R.; Sievert D. M. Antimicrobial-Resistant Pathogens Associated With Healthcare-Associated Infections: Summary of Data Reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2011-2014. Infect Control Hosp Epidemiol 2016, 37, 1288–1301. 10.1017/ice.2016.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborators G. B. D. A. R. Global mortality associated with 33 bacterial pathogens in 2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 2023, 400, 2221–2248. 10.1016/S0140-6736(22)02185-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organization W. H.Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. Organization, W. H., Ed.; Geneva, Switzerland, 2017.
- Tacconelli E.; Carrara E.; Savoldi A.; Harbarth S.; Mendelson M.; Monnet D. L.; Pulcini C.; Kahlmeter G.; Kluytmans J.; Carmeli Y.; et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. 10.1016/S1473-3099(17)30753-3. [DOI] [PubMed] [Google Scholar]
- Department of Health and Human Services, CDC. Antibiotic Resistance Threats in the United States. Atlanta, GA: U.S., 2019, 10.15620/cdc:82532. [DOI]
- Kyriakidis I.; Vasileiou E.; Pana Z. D.; Tragiannidis A. Acinetobacter baumannii Antibiotic Resistance Mechanisms. Pathogens 2021, 10, 373 10.3390/pathogens10030373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard A.; O’Donoghue M.; Feeney A.; Sleator R. D. Acinetobacter baumannii: an emerging opportunistic pathogen. Virulence 2012, 3, 243–250. 10.4161/viru.19700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteway C.; Breine A.; Philippe C.; Van der Henst C. Acinetobacter baumannii. Trends Microbiol. 2022, 30, 199–200. 10.1016/j.tim.2021.11.008. [DOI] [PubMed] [Google Scholar]
- Ayoub Moubareck C.; Hammoudi Halat D. Insights into Acinetobacter baumannii: A Review of Microbiological, Virulence, and Resistance Traits in a Threatening Nosocomial Pathogen. Antibiotics 2020, 9, 119 10.3390/antibiotics9030119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D.; Nielsen T. B.; Bonomo R. A.; Pantapalangkoor P.; Luna B.; Spellberg B. Clinical and Pathophysiological Overview of Acinetobacter Infections: a Century of Challenges. Clin Microbiol Rev 2017, 30, 409–447. 10.1128/CMR.00058-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding C. M.; Hennon S. W.; Feldman M. F. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat. Rev. Microbiol. 2018, 16, 91–102. 10.1038/nrmicro.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler S.; Muller V. Coping with low water activities and osmotic stress in Acinetobacter baumannii: significance, current status and perspectives. Environ. Microbiol. 2019, 21, 2212–2230. 10.1111/1462-2920.14565. [DOI] [PubMed] [Google Scholar]
- Zeidler S.; Muller V. The role of compatible solutes in desiccation resistance of Acinetobacter baumannii. MicrobiologyOpen 2019, 8, e00740 10.1002/mbo3.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva G. J.; Domingues S. Insights on the Horizontal Gene Transfer of Carbapenemase Determinants in the Opportunistic Pathogen Acinetobacter baumannii. Microorganisms 2016, 4, 29 10.3390/microorganisms4030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; He L.; Tao X.; Meng F.; Zhang J. High DNA Uptake Capacity of International Clone II Acinetobacter baumannii Detected by a Novel Planktonic Natural Transformation Assay. Front. Microbiol. 2019, 10, 2165. 10.3389/fmicb.2019.02165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperi F.; Antunes L. C.; Blom J.; Villa L.; Iacono M.; Visca P.; Carattoli A. The genomics of Acinetobacter baumannii: insights into genome plasticity, antimicrobial resistance and pathogenicity. IUBMB Life 2011, 63, 1068–1074. 10.1002/iub.531. [DOI] [PubMed] [Google Scholar]
- Valcek A.; Philippe C.; Whiteway C.; Robino E.; Nesporova K.; Bové M.; Coenye T.; De Pooter T.; De Coster W.; Strazisar M.; et al. Phenotypic Characterization and Heterogeneity among Modern Clinical Isolates of Acinetobacter baumannii. Microbiol. Spectrum 2023, 11, e03061-22 10.1128/spectrum.03061-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcek A.; Nesporova K.; Whiteway C.; De Pooter T.; De Coster W.; Strazisar M.; Van der Henst C. Genomic Analysis of a Strain Collection Containing Multidrug-, Extensively Drug-, Pandrug-, and Carbapenem-Resistant Modern Clinical Isolates of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2022, 66, 9. 10.1128/aac.00892-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiorakos A. P.; Srinivasan A.; Carey R. B.; Carmeli Y.; Falagas M. E.; Giske C. G.; Harbarth S.; Hindler J. F.; Kahlmeter G.; Olsson-Liljequist B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]
- Bartal C.; Rolston K. V. I.; Nesher L. Carbapenem-resistant Acinetobacter baumannii: Colonization, Infection and Current Treatment Options. Infect. Dis. Ther. 2022, 11, 683–694. 10.1007/s40121-022-00597-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler S.; Hubloher J.; Konig P.; Ngu N. D.; Scholz A.; Averhoff B.; Muller V. Salt induction and activation of MtlD, the key enzyme in the synthesis of the compatible solute mannitol in Acinetobacter baumannii. MicrobiologyOpen 2018, 7, e00614 10.1002/mbo3.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Y.; Chen Y.; Sivaccumar J. P.; An Z.; Xia N.; Luo W. Research progress and applications of nanobody in human infectious diseases. Front. Pharmacol. 2022, 13, 963978 10.3389/fphar.2022.963978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanmarsenille C.; Díaz del Olmo I.; Diaz Del Olmo I.; Elseviers J.; Hassanzadeh Ghassabeh G.; Moonens K.; Vertommen D.; Martel A.; Haesebrouck F.; Pasmans F.; Hernalsteens J. P. Nanobodies targeting conserved epitopes on the major outer membrane protein of Campylobacter as potential tools for control of Campylobacter colonization. Vet. Res. 2017, 48, 86 10.1186/s13567-017-0491-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M. T.; Huh I.; Shenai A.; Brooks T. M.; Brooks C. L. Structural basis of V(H)H-mediated neutralization of the food-borne pathogen Listeria monocytogenes. J. Biol. Chem. 2018, 293, 13626–13635. 10.1074/jbc.RA118.003888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Lin M.; Yan Q.; Budachetri K.; Hou L.; Sahni A.; Liu H.; Han N. C.; Lakritz J.; Pei D.; Rikihisa Y. An intracellular nanobody targeting T4SS effector inhibits Ehrlichia infection. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2024102118 10.1073/pnas.2024102118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff P.; Zimmermann I.; Arnold F. M.; Hutter C. A. J.; Morger D.; Opitz L.; Poveda L.; Keserue H. A.; Panse C.; Roschitzki B.; Seeger M. A. Engineered peptide barcodes for in-depth analyses of binding protein libraries. Nat. Methods 2019, 16, 421–428. 10.1038/s41592-019-0389-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyldermans S. Applications of Nanobodies. Annu. Rev. Anim. Biosci. 2021, 9, 401–421. 10.1146/annurev-animal-021419-083831. [DOI] [PubMed] [Google Scholar]
- Steeland S.; Vandenbroucke R. E.; Libert C. Nanobodies as therapeutics: big opportunities for small antibodies. Drug Discovery Today 2016, 21, 1076–1113. 10.1016/j.drudis.2016.04.003. [DOI] [PubMed] [Google Scholar]
- De Meyer T.; Muyldermans S.; Depicker A. Nanobody-based products as research and diagnostic tools. Trends Biotechnol. 2014, 32, 263–270. 10.1016/j.tib-tech.2014.03.001. [DOI] [PubMed] [Google Scholar]
- Beghein E.; Gettemans J. Nanobody Technology: A Versatile Toolkit for Microscopic Imaging, Protein-Protein Interaction Analysis, and Protein Function Exploration. Front. Immunol. 2017, 8, 771. 10.3389/fimmu.2017.00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram J. R.; Schmidt F. I.; Ploegh H. L. Exploiting nanobodies’ singular traits. Annu. Rev. Immunol. 2018, 36, 695–715. 10.1146/annurev-immunol-042617-053327. [DOI] [PubMed] [Google Scholar]
- Ji F.; Ren J.; Vincke C.; Jia L.; Muyldermans S.. Nanobodies: From Serendipitous Discovery of Heavy Chain-Only Antibodies in Camelids to a Wide Range of Useful Applications. In Single-Domain Antibodies, Methods and Protocols; Hussack G.; Henry K. A., Eds.; Methods in Molecular Biology; Humana Press, 2022; pp 3–18 10.1007/978-1-0716-2075-5. [DOI] [PubMed] [Google Scholar]
- De Genst E.; Silence K.; Decanniere K.; Conrath K.; Loris R.; Kinne J.; Muyldermans S.; Wyns L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 4586–4591. 10.1073/pnas.0505379103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyldermans S.; Cambillau C.; Wyns L. Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends Biomed. Sci. 2001, 26, 230–235. 10.1016/S0968-0004(01)01790-X. [DOI] [PubMed] [Google Scholar]
- Sircar A.; Sanni K. A.; Shi J.; Gray J. J. Analysis and modeling of the variable region of camelid single-domain antibodies. J. Immunol. 2011, 186, 6357–6367. 10.4049/jimmunol.1100116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell L. S.; Colwell L. J. Comparative analysis of nanobody sequence and structure data. Proteins 2018, 86, 697–706. 10.1002/prot.25497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell L. S.; Colwell L. J. Analysis of nanobody paratopes reveals greater diversity than classical antibodies. Protein Eng., Des. Sel. 2018, 31, 267–275. 10.1093/protein/gzy017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavrtanik U.; Lukan J.; Loris R.; Lah J.; Hadzi S. Structural basis of epitope recognition by heavy-chain camelid antibodies. J. Mol. Biol. 2018, 430, 4369–4386. 10.1016/j.jmb.2018.09.002. [DOI] [PubMed] [Google Scholar]
- Van holsbeeck K.; Martins J. C.; Ballet S. Downsizing antibodies: Towards complementarity-determining region (CDR)-based peptide mimetics. Bioorg. Chem. 2022, 119, 105563 10.1016/j.bioorg.2021.105563. [DOI] [PubMed] [Google Scholar]
- Kadam R. U.; Juraszek J.; Brandenburg B.; Buyck C.; Schepens W. B. G.; Kesteleyn B.; Stoops B.; Vreeken R. J.; Vermond J.; Goutier W.; et al. Potent peptidic fusion inhibitors of influenza virus. Science 2017, 358, 496–502. 10.1126/science.aan0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng R.; Zhang F.; Li M.; Wo X.; Su Y. W.; Wang W. Influence of Fixation and Permeabilization on the Mass Density of Single Cells: A Surface Plasmon Resonance Imaging Study. Front Chem. 2019, 7, 588. 10.3389/fchem.2019.00588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louche A.; Salcedo S. P.; Bigot S. P. Protein-Protein Interactions: Pull-Down Assays. Methods Mol. Biol. 2017, 1615, 247–255. [DOI] [PubMed] [Google Scholar]
- Dondelinger M.; Filee P.; Sauvage E.; Quinting B.; Muyldermans S.; Galleni M.; Vandevenne M. S. Understanding the Significance and Implications of Antibody Numbering and Antigen-Binding Surface/Residue Definition. Front. Immunol. 2018, 9, 2278. 10.3389/fimmu.2018.02278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilton E. E.; Opyr M. P.; Kailasam S.; Kothe R. F.; Wieden H. J. sdAb-DB: The Single Domain Antibody Database. ACS Synth. Biol. 2018, 7, 2480–2484. 10.1021/acssynbio.8b00407. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Melikov K.; Vives E.; Ramos C.; Verbeure B.; Gait M. J.; Chernomordik L. V.; Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- Valcek A.; Collier J.; Botzki A.; Van der Henst C. Acinetobase: the comprehensive database and repository of Acinetobacter strains. Database 2022, baac099. 10.1093/database/baac099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes Ruiz L. M.; Williams C. L.; Tamayo R. Enhancing bacterial survival through phenotypic heterogeneity. PLoS Pathog. 2020, 16, e1008439 10.1371/journal.ppat.1008439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteway C.; Valcek A.; Philippe C.; Strazisar M.; De Pooter T.; Mateus I.; Breine A.; Van der Henst C. Scarless excision of an insertion sequence restores capsule production and virulence in Acinetobacter baumannii. ISME J 2022, 16, 1473–1477. 10.1038/s41396-021-01179-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelay-Gimeno M.; Glas A.; Koch O.; Grossmann T. N. Structure-based design of inhibitors of protein-protein interactions: mimicking peptide binding epitopes. Angew. Chem., Int. Ed. 2015, 54, 8896–8927. 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu Q. N.; Young R.; Sudhakar H. K.; Gao T.; Huang T.; Tan Y. S.; Lau Y. H. Cyclisation strategies for stabilising peptides with irregular conformations. RSC Med. Chem. 2021, 12, 887–901. 10.1039/d1md00098e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty P. G.; Sahni A.; Pei D. Understanding Cell Penetration of Cyclic Peptides. Chem. Rev. 2019, 119, 10241–10287. 10.1021/acs.chemrev.9b00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lättig-Tünnemann G.; Prinz M.; Hoffmann D.; Behlke J.; Palm-Apergi C.; Morano I.; Herce H. D.; Cardoso M. C. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat. Commun. 2011, 2, 453 10.1038/ncomms1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z.; Martyna A.; Hard R. L.; Wang J.; Appiah-Kubi G.; Coss C.; Phelps M. A.; Rossman J. S.; Pei D. Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. 10.1021/acs.biochem.6b00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mika J. T.; Moiset G.; Cirac A. D.; Feliu L.; Bardaji E.; Planas M.; Sengupta D.; Marrink S. J.; Poolman B. Structural basis for the enhanced activity of cyclic antimicrobial peptides: the case of BPC194. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 2197–2205. 10.1016/j.bbamem.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Oddo A.; Thomsen T. T.; Britt H. M.; Lobner-Olesen A.; Thulstrup P. W.; Sanderson J. M.; Hansen P. R. Modulation of Backbone Flexibility for Effective Dissociation of Antibacterial and Hemolytic Activity in Cyclic Peptides. ACS Med. Chem. Lett. 2016, 7, 741–745. 10.1021/acsmedchemlett.5b00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards I. A.; Elliott A. G.; Kavanagh A. M.; Zuegg J.; Blaskovich M. A.; Cooper M. A. Contribution of Amphipathicity and Hydrophobicity to the Antimicrobial Activity and Cytotoxicity of beta-Hairpin Peptides. ACS Infect. Dis 2016, 2, 442–450. 10.1021/acsinfecdis.6b00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Z.; Yuan X.; Chen H.; Zhu Y.; Dong N.; Shan A. Strategies employed in the design of antimicrobial peptides with enhanced proteolytic stability. Biotechnol. Adv. 2022, 59, 107962 10.1016/j.biotechadv.2022.107962. [DOI] [PubMed] [Google Scholar]
- Park J. H.; Waters M. L. Positional effects of click cyclization on beta-hairpin structure, stability, and function. Org. Biomol. Chem. 2013, 11, 69–77. 10.1039/c2ob26445e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domanska K.; Vanderhaegen S.; Srinivasan V.; Pardon E.; Dupeux F.; Marquez J. A.; Giorgetti S.; Stoppini M.; Wyns L.; Bellotti V.; et al. Atomic structure of a nanobody-trapped domain-swapped dimer of an amyloidogenic beta2-microglobulin variant. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 1314–1319. 10.1073/pnas.1008560108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrath K. E.; Lauwereys M.; Galleni M.; Matagne A.; Frere J. M.; Kinne J.; Wyns L.; Muyldermans S. Beta-lactamase inhibitors derived from single-domain antibody fragments elicited in the camelidae. Antimicrob. Agents Chemother. 2001, 45, 2807–2812. 10.1128/AAC.45.10.2807-2812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardon E.; Laeremans T.; Triest S.; Rasmussen S. G.; Wohlkonig A.; Ruf A.; Muyldermans S.; Hol W. G.; Kobilka B. K.; Steyaert J. A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 2014, 9, 674–693. 10.1038/nprot.2014.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.