

Abstract
Dysregulation of the autotaxin (ATX, Enpp2)-lysophosphatidic acid (LPA) signaling in cancerous cells contributes to tumorigenesis and therapy resistance. We previously found that ATX activity was elevated in p53-KO mice compared to wild type (WT) mice. Here, we report that ATX expression was upregulated in mouse embryonic fibroblasts from p53-KO and p53R172H mutant mice. ATX promoter analysis combined with yeast one-hybrid testing revealed that WT p53 directly inhibits ATX expression via E2F7. Knockdown of E2F7 reduced ATX expression and chromosome immunoprecipitation showed that E2F7 promotes Enpp2 transcription through cooperative binding to two E2F7 sites (promoter region −1393 bp and second intron 996 bp). Using chromosome conformation capture, we found that chromosome looping brings together the two E2F7 binding sites. We discovered a p53 binding site in the first intron of murine Enpp2, but not in human ENPP2. Binding of p53 disrupted the E2F7-mediated chromosomal looping and repressed Enpp2 transcription in murine cells. In contrast, we found no disruption of E2F7-mediated ENPP2 transcription via direct p53 binding in human carcinoma cells. In summary, E2F7 is a common transcription factor that upregulates ATX in human and mouse cells but is subject to steric interference by direct intronic p53 binding only in mice.
Graphical Abstract
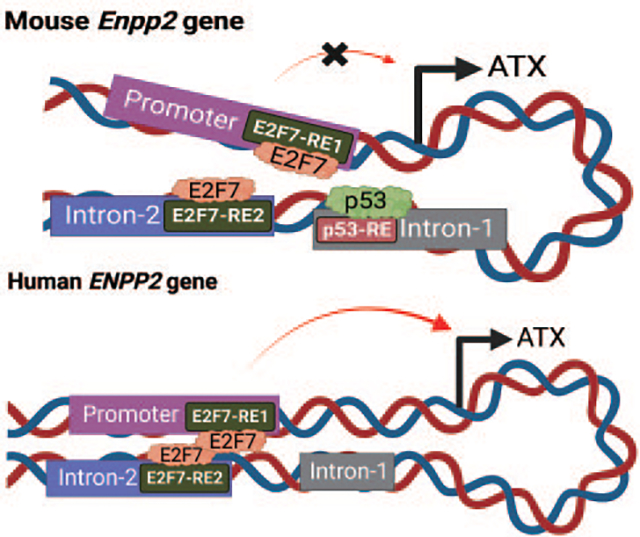
E2F7 drives Autotaxin/Enpp2 transcription via chromosome looping: Repression by p53 in murine but not human carciomas by K. Lin, S-C Lee, M. A. Dacheux, D. D. Norman, A. Balogh, H. Lee, G. Tigyi. The chromosomal loop in ENPP2 Caused by E2F7-dimerization is disrupted by direct binding of p53 in the murine Enpp2 gene. In human ENPP2 the lack of the intronic p53 site eliminates direct regulation by p53.
Introduction
The ectonucleotide pyrophosphatase/phosphodiesterase 2 (Enpp2) gene encodes the autotaxin (ATX) protein, a lysophospholipase D that converts lysophosphatidylcholine to lysophosphatidic acid (LPA). LPA functions through activation of multiple targets including LPA GPCR (LPAR) [1]. ATX is elevated in many types of carcinomas [2–5] and drives cancer progression by promoting invasion, angiogenesis, metastasis, therapy resistance, and inhibits antitumor immunity [6–9]. ATX/LPA is also secreted by cells of the tumor microenvironment (TME) [10–12] and regulates the communication between cancer cells and their TME [13]. ATX is the second most upregulated gene in therapy-resistant breast cancer stem-like cells, whereas the gene that encodes the lipid phosphate phosphatase 3 enzyme that metabolizes LPA is downregulated in these cells [14]. The ATX-LPAR axis also elicits chemo- and radiotherapy resistance of cells in vitro via LPAR2 [15, 16], and protects carcinomas from Taxol- and carboplatin-induced apoptosis [17, 18]. Because ATX plays a critical role in cancer progression and therapy resistance, understanding the molecular mechanisms responsible for its upregulation is of therapeutic significance.
Several transcription factors (TFs) have been reported to regulate Enpp2/ENPP2 expression. These include: AP-1, SP [19], c-Jun, Sp3 [19, 20], HOXA13 [21], β-catenin [22], FOX family TFs [23–25], SOX11, STAT3 [26], and NFAT1 [27, 28]. ENPP2 expression can also be regulated by histone deacetylases (HDAC3 and HDAC7) [29] and by methylation [30]. Moreover, post-translational modification by RNA-binding proteins HuR and AUF1 has been reported to promote ENPP2 mRNA stability [31]. Several microRNAs such as miR-29a/b/c and miR-101–3p have been identified as binding partners to ENPP2 mRNA in patients with chronic obstructive pulmonary disease [32, 33].
Previously, we demonstrated that the plasma ATX activity in p53-KO mice was significantly elevated relative to wild-type (WT) mice, and that myofibroblast derived from p53-KO mice showed ~16-fold higher ATX expression levels over WT. These data indicate that p53 negatively regulates murine ATX expression [34]. Enpp2 was reported to be one of the differentially expressed genes in p53-WT versus p53-KO mouse embryonic fibroblasts (MEF) [35]. Genome-wide ChIP-seq analysis showed that p53 occupies the regulatory domain of ENPP2 after p53 reactivation by Nutlin or induction of tumor cell apoptosis by chemotherapeutics in MCF-7 and HCT116 carcinomas [36]. In a genome-wide chromatin occupancy screen, 18,110 genes have been reported to contain p53-RE, of which 83 genes, including ENPP2, were validated experimentally [37]. These findings support the hypothesis that p53 interacts with ENPP2 and may transcriptionally augment or suppress its expression. However, the mechanism by which p53 represses Enpp2 has not been investigated previously. In this study, we aimed to dissect the transcriptional repression of Enpp2/ENPP2 by p53 in murine and human cells.
Material and Methods
Cell culture.
HEK293T, MDA-MB-231, SKOV3 cells, and B16-F10 cells were obtained from ATCC (Manassas, VA). WT and p53−/− HCT116 cells were from Dr. Sunny Wu (UTHSC, Memphis, TN). WT, p53-KO, and p53R172H MEF were from Dr. Gerard Zambetti (St. Jude Children’s Research Hospital, Memphis, TN). HEK293T, MDA-MB-231, and SKOV3 cells were cultured in DMEM (Corning; Corning, NY) supplemented with 10% FBS (R&D System; Minneapolis, MN) and 1% penicillin/streptomycin (Invitrogen; Waltham, MA). MEF and B16-F10 cells were cultured in DMEM (Lonza; Basel, Switzerland) supplemented with 10% FBS, 1% non-essential amino acids (Invitrogen), and 1mM L-glutamine (Invitrogen). Cells were starved in FBS-free DMEM for 2 h followed by treatment with either 20 μM of Nutlin-3A or (Sigma-Aldrich, St. Louis, MO) vehicle control 0.1% DMSO for 24 h.
Cloning of Enpp2 promoter and E2F7.
Mouse genomic DNA isolated from MEF were amplified with Q5 polymerase (New England Biolabs (NEB); Ipswich, MA) using primers in (Supplementary Table 1). The amplified Enpp2 promoter sequence was cloned into Dual-Luciferase Reporter Lentivectors (Applied Biological Materials; Richmond, British Columbia, Canada) using an In-Fusion cloning kit (Takara; San Jose, CA). The mouse E2F7 cDNA was cloned at the C’-terminus of yellow fluorescent protein (YFP) in the pEYFP-C1 plasmid.
Mutation of E2F7.
A site-directed mutagenesis kit (NEB) was used according to the manufacturer’s instructions to delete the E2F7-REs using primers in Supplementary Table 1 or to mutate the E2F7 DNA binding dimerization domains.
Yeast one-hybrid (Y1H) screening.
Y1H screen was performed as described previously [38, 39]. The coding region of suspected TFs (prey proteins) was cloned to pGLDT7-AD vector (Takara). Tandemly repeated ATX promoter regions (bait sequences) were amplified and cloned into the pABAi vector (Takara) with Aureobasidin A (AbA) resistance reporter gene (AbAr).
Cell transfection.
Cells were cultured in Opti-MEM (Invitrogen) and transfected using Lipofectamine 2000 or 3000 (Invitrogen). Mouse and human SMART Pool siRNAs were used to knock down (KD) NR1H3, E2F7, and p53 (sequences listed in Supplementary Table 2, Dharmacon, Cambridge, UK).
RT-qPCR.
500 ng of RNA was reverse transcribed to cDNA with the RevertAid Reverse Transcription kit (Invitrogen). qPCR was performed using the PowerUp-SYBR Green master mix in a QuantStudio-6 PCR machine (Applied Biosystems Inc (ABI), Waltham, MA). Primers are listed in Supplementary Table 3. The relative expression level of each gene was normalized to GAPDH using the ΔΔCt method.
Immunoblotting.
Equal amounts of protein were loaded to SDS-PAGE and transferred to nitrocellulose membranes as described in [40]. Primary antibodies used were: Anti-mE2F7 (PA5-68911, Thermo-Fisher; 1:2000), anti-hE2F7 (HPA064866, Millipore; Burlington, MA; 1:2000), monoclonal anti-ATX 4F1 (provided by Dr. Junken Aoki, Tokyo University, 1:1000), anti-mNR1H3 (ab176323, Abcam, Waltham, MA; 1:3000), and anti-Actin (MAB1501, Sigma-Aldrich; 1:600,000). Secondary antibodies applied were goat anti-mouse HRP (A4416, Sigma-Aldrich; 1:500), goat anti-rat HRP (A10549, ThermoFisher, 1:2000), or goat anti-rabbit HRP antibody (31460 ThermoFisher; 1:3000).
Luciferase assays.
Luciferase activity was measured using a Dual-luciferase assay kit (Promega; Madison, WI) according to the manufacturer’s instructions.
Chromatin immunoprecipitation (ChIP).
The cells were suspended in cold PBS at 2×106 cells/mL, crosslinked with 1% formaldehyde (FA) for 10 min, quenched with 0.125M glycine for 5 min and twice with PBS. Next, 107 cells were suspended in ChIP buffer and lysed by sonication to obtain a fragment size distribution of 200 – 1000 bp. Lysate (20 μL) was mixed with TE buffer (80 μL) and digested with 4 μg RNase A at 65°C overnight. The eluted samples were incubated at 60°C for 30 min with 2 μg of Proteinase K (Sigma-Aldrich). Purified DNA (25 μg) was added to either 5 μg of anti-mE2F7 antibodies (A303-037A, Thermo-Fisher) or negative control rabbit IgG antibodies (ab172730, Abcam) to precipitate the protein of interest using the ChIP Kit-One step (Abcam). Isolated DNA segments were analyzed using qPCR with primers in Supplementary Table 3.
Chromatin conformation capture (3C)-qPCR assay.
The 3C-qPCR analysis was performed as previously described [41]. SDS-treated DNA was digested overnight with endonuclease DpnII or MaeI (NEB) and inactivated with 0.5% SDS at 65°C for 30 min. Digested chromatin (2 μg) was ligated with T4 DNA ligase (NEB) overnight at 16°C. The ligated DNA was reverse-crosslinked at 60°C overnight with 4 μg of RNase A (Sigma-Aldrich), and digested with 2 μg of Proteinase K for 30 min at 60°C. Seven DpnII or MaeI recognition sites flanking the target regions in the mouse or human genome, respectively, were analyzed with SYBR Green. Digestion efficiency was monitored by qPCR using primer pairs (Supplementary Table 4) that encompassed the DpnII or MaeI restriction sites.
Statistical analysis.
Data from three experiments are expressed as means ± SD. Statistical analysis was performed with GraphPad Prism 9.1.2 software (San Diego, CA). Significant differences were calculated using paired Student’s t-test (two groups) or one-way ANOVA with Tukey’s multiple comparison test.
Data Availability Statement.
The experimental data that support the findings of this study are openly available in the figshare website at the following link https://figshare.com/projects/E2F7_drives_ATX_transcription_via_chromosome_looping_Repression_by_p53_in_murine_but_not_human_carcinomas/160789 under reference number 160789.
Results
p53 suppresses ATX expression in MEF.
Previously, we demonstrated that ATX expression and ATX enzymatic activity in plasma are significantly increased in p53-KO mice [34]. Here, we obtained MEF isolated from WT, p53-KO, and p53R172H mice and analyzed the expression of p53 and Enpp2 by RT-qPCR (Figures 1A & 1C) and immunoblot (Figure 1D). We confirmed that p53-KO MEF do not express p53 mRNA or protein (Figures 1A & 1D). However, these MEF expressed higher levels of ATX mRNA and protein than WT MEF (Figures 1C & 1D). Furthermore, p53 MEF harboring the R172H mutation in its DNA binding domain also displayed elevated levels of ATX expression (Figures 1C & 1D). We also verified that the expression of the canonical p53 downstream target gene p21 was significantly reduced in the p53-KO and p53R172H MEF (Figures 1B & 1D). These results suggest that p53 represses ATX expression in MEF.
Figure 1. p53 negatively regulates ATX expression in MEF.
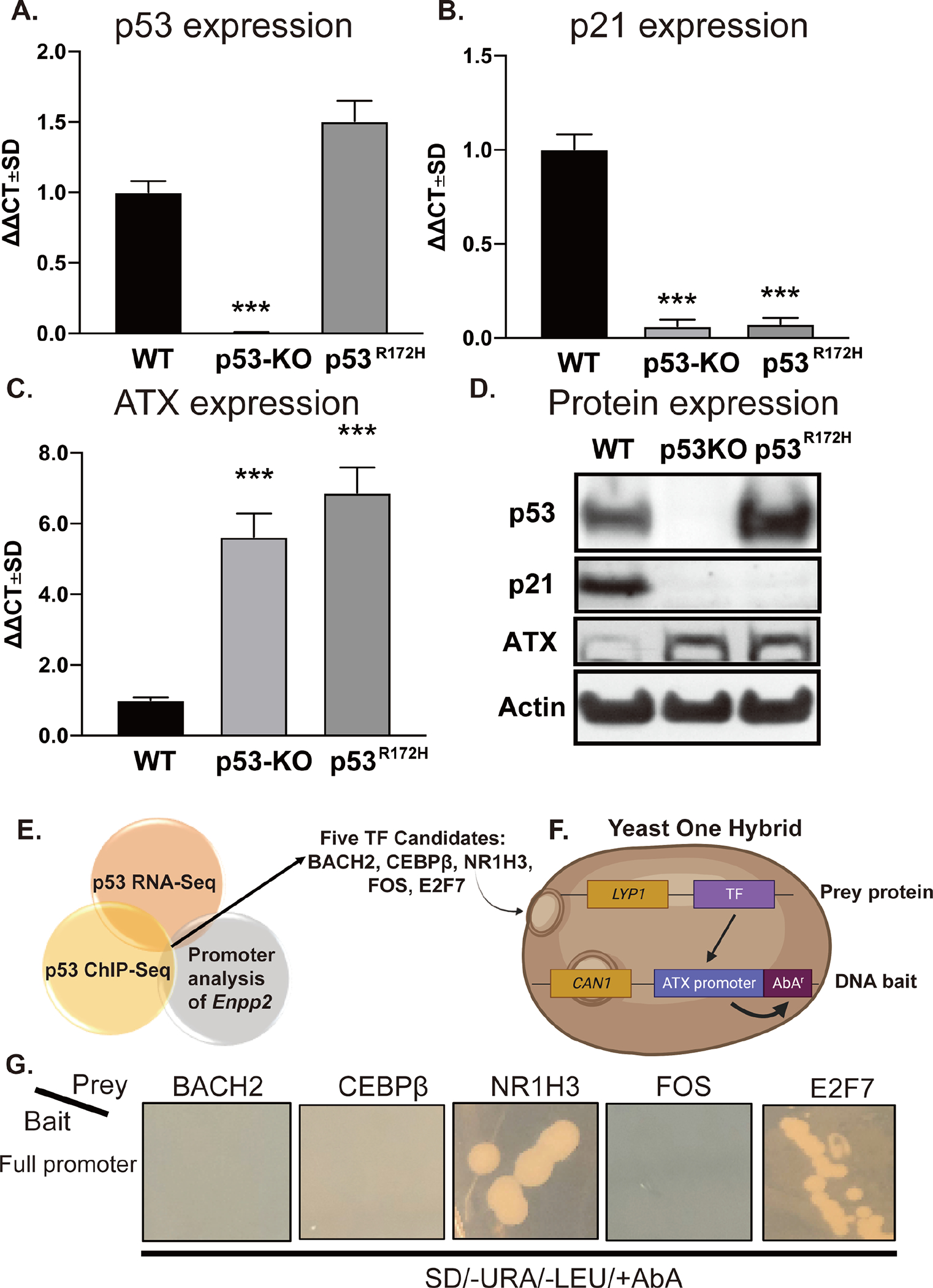
RNA was isolated from WT, p53-KO, and p53R172H MEF and p53 (A), p21 (B), and ATX (C) mRNA expression was evaluated by RT-qPCR. Fold changes in mRNA (ΔΔCt) were calculated, and data are represented as the mean ± SD of three independent experiments. *** indicates p < 0.001. (D) Expression of p53, p21, and ATX were determined by immunoblotting with antibodies in cell lysates prepared from WT, p53-KO, and p53R172H MEF. Note the robust upregulation of ATX protein in p53-KO and p53R172H MEF. (E) The results of the promoter analysis of Enpp2 were cross-referenced with published p53 ChIP-Seq and RNA-Seq databases. The dissimilarity threshold was set at 10% as the selection criterion. (F) Five TFs, BACH2, CEBPβ, NR1H3, FOS, and E2F7, were identified and cloned as “prey protein” into the pGLDT7-AD vector. The Enpp2 promoter sequence was cloned to “DNA bait” in pABAi vector, which contains an AbA resistance gene as described in the methods. (G) The pairs of prey and bait vectors were transformed to Y1HGold yeasts and grown on plates in the presence of SD-Leu-URA+AbA. The positive interaction between the prey protein and the DNA bait caused the expression of AbAr and growth in the plate. LYP1: lysine permease; CAN1: plasma membrane arginine permease; SD: synthetic defined medium.
The Enpp2 promoter contains binding sites for NR1H3 and E2F7.
The finding that ATX is increased in p53R172H mutant MEF suggests that the DNA binding domain of p53 is required for repressing ATX expression. Although p53 is known to activate gene expression directly, it has not been demonstrated to directly repress gene transcription [42]. We hypothesized that gene repression might occur with p53 acting as an indirect transcriptional repressor of (an) unknown TF(s) that become(s) disinhibited when p53 is knocked out or inactivated by mutation. To identify the putative TF(s), we used the TRANSFAC and ALGGEN-PROMO software to identify TFs with predicted binding sites in the Enpp2 gene. This result was cross-referenced with TFs that are known downstream targets of p53 that were selected from published p53 RNA-seq and p53 ChIP-seq databases [35, 36]. We identified five putative candidates: nuclear receptor subfamily 1 group H member 3 (NR1H3), BTB domain and CNC homolog 2 (BACH2), activator protein 1 (AP-1)/FOS proto-oncogene, CCAAT/enhancer binding protein beta (CEBPβ), and E2F transcription factor 7 (E2F7). These candidates, which met both criteria of being a p53 downstream target and occupying binding sites in the Enpp2 promoter (Figure 1E), were further evaluated using Y1H assay (Figure 1F). As shown in Figure 1G, we found that yeast cells transformed with plasmids containing tandem copies of the Enpp2 promoter with either an E2F7- or NR1H3-expression vector grew colonies in the presence of AbA, suggesting that both E2F7 and NR1H3 interact with the Enpp2 promoter.
Knockdown of E2F7 but not NR1H3 suppresses ATX expression.
To determine if E2F7 or NR1H3 regulates ATX expression, each TF knockdown (KD) with siRNA was generated in WT, p53-KO, or p53R172H MEF. The KD efficiency of NR1H3 siRNA was confirmed by mRNA expression of the Nr1h3 gene and its downstream target, the fructose transporter Glut5 (Supplementary Figure 1A and 1B). NR1H3 protein levels were also reduced by 40–60% at 48 h after transfection with NR1H3 siRNA in WT (Figure 2A), p53-KO (Figure 2B), and p53R172H (Figure 2C) MEF. We found that KD of NR1H3 did not alter ATX protein expression in all three cell lines, suggesting that it is unlikely to regulate ATX expression. On the contrary, siRNA-mediated KD of E2F7 decreased ATX protein expression by ~35–40% compared to MEF transfected with scrambled siRNA (Figures 2D–F), suggesting that E2F7, but not NR1H3, positively regulates Enpp2 transcription. The KD efficiency of E2F7 siRNA was confirmed by mRNA expression of the E2f7 gene, its downstream target cyclin B1 (Ccnb1; Supplementary Figure 1C and 1D), and E2F7 protein levels (Figures 2D–F).
Figure 2. Knockdown of E2F7 decreases ATX expression.
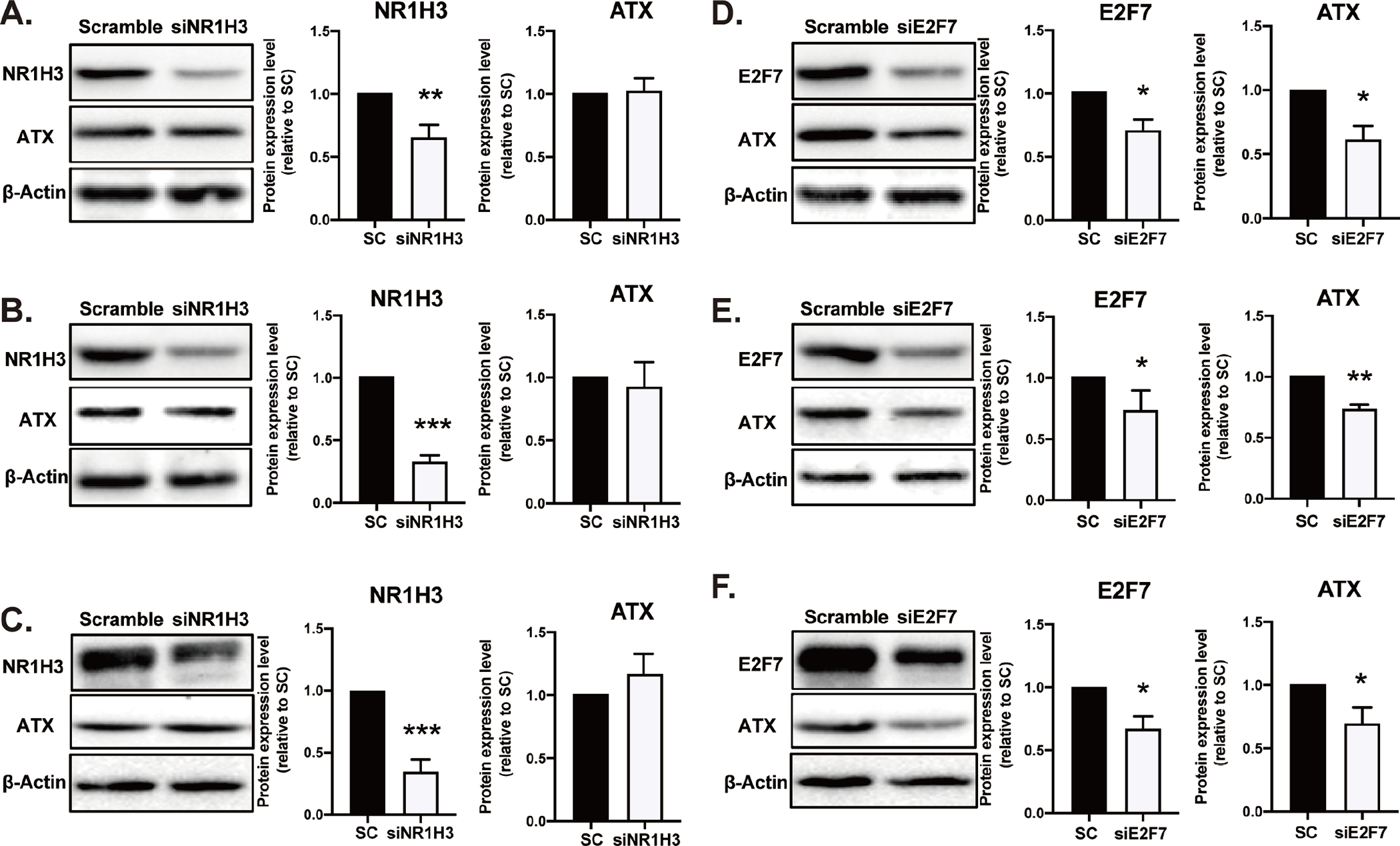
100 nM of NR1H3 siRNA was transfected in (A) WT, (B) p53-KO, and (C) p53R172H MEF for 48 h. On the other hand, 150 nM siRNA targeting E2F7 was applied to (D) WT, (E) p53-KO, and (F) p53R172H MEF for 24 h. Protein was extracted for immunoblotting with antibodies against NR1H3, E2F7, ATX, and β-actin. Densitometry analysis represents the percent of NR1H3, E2F7, and ATX band intensity normalized to β-actin. The data represents the mean ± SD of three independent experiments. *p < 0.05 and **p<0.01 indicate significant differences. SC: scramble.
The transcriptional activity of Enpp2 promoter is upregulated by E2F7.
Because KD of E2F7 downregulated ATX expression in MEF, we hypothesized that ATX is a transcriptional target of E2F7. Using the TRANSFAC and ALLGEN-PROMO softwares, we identified two E2F7-response elements (E2F7-RE) in the promoter (E2F7-RE1 at −1393 – −1385) and in the second intron (E2F7-RE2, at 996 – 1009) of murine Enpp2. We also identified a p53-RE in the first intron (p53-RE at 162 – 171, Figure 3A). To evaluate the role of these E2F7-RE in Enpp2 transcription, we cloned the Enpp2 promoter into a dual-luciferase reporter plasmid system. This Enpp2 dual-luciferase reporter plasmid was co-transfected with the YFP-E2F7 construct in HEK293T cells. We showed that Enpp2 transcription was increased by two-fold when 100–200 ng of E2F7-expression plasmid was co-transfected with the reporter plasmid (Figure 3B, black bars).
Figure 3. E2F7 enhances the transcriptional activity of the ATX promoter.
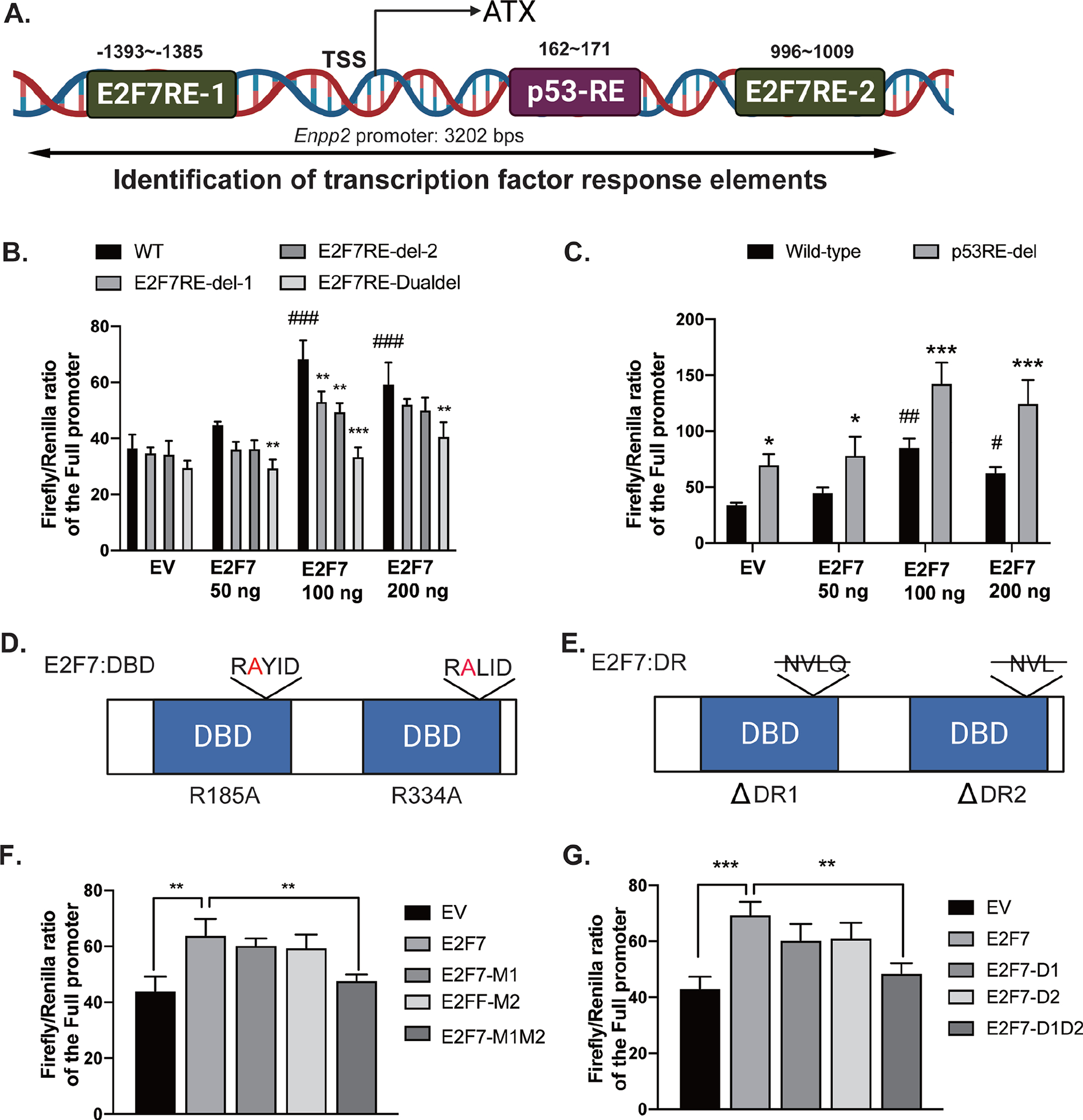
(A) A 3202 bp Enpp2 promoter was retrieved from Ensembl database and analyzed by TRANSFAC and ALGGEN-PROMO online software. Two potential E2F7-RE (−1393~−1385, CCCGCTCTG, E2F7-RE1; 996~1009, TGTCTCCCCGGGAA, E2F7-RE2) and one p53-RE (162~171, TTGGCAGGAT) were identified. (B) Single deletion (E2F7RE-del-1 or E2F7RE-del-2) or double deletion (E2F7RE-Dualdel) of E2F7-RE and (C) deletion of p53-RE were introduced into the reporter plasmids. Co-transfection of E2F7-expression vector and luciferase reporter was carried out in HEK293T cells as described in methods. (D-E) The diagrams show mutations in the DNA binding domain (DBD) and dimerization residues (DR) of E2F7, respectively. Single arginine-to-alanine mutation (M1 or M2) or double mutations (M1M2) of E2F7 were generated in the E2F7-expression vector. Likewise, single deletion of DR (D1 or D2) and double deletions (D1D2) were generated in the E2F7-expression vector. (F) The E2F7-DBD mutation and (G) the E2F7-DR mutation vectors were co-transfected with Enpp2 promoter reporter plasmid into HEK cells. Luciferase activity was measured after transient co-transfection of these plasmids. Luciferase activity was normalized to Renilla luciferase activity. Data are represented as the mean ± SD of three independent experiments. *, #p < 0.05, **, ##p<0.01, and ***, ###p < 0.001 indicate significant differences. * indicates the comparison between the values from wild-type promoter and E2F7-RE deletion promoter plasmids. # indicates the comparison between EV and E2F7-expression vectors. EV: empty vector; DBD: DNA binding domain; DR: dimerization residue.
To determine whether one or both E2F7-RE was required for Enpp2 transcription, we deleted the two binding sites either individually (E2F7RE-del-1, E2F7RE-del-2) or in combination (E2F7RE-Dualdel). When single deletion mutations were co-transfected with the E2F7-expression plasmid, the transcriptional activity of Enpp2 appears to be partially reduced, although statistical significance was observed only when 100ng of E2F7 plasmid was co-transfected (Figure 3B, dark gray bars). In contrast, double deletion of E2F7-RE significantly reduced luciferase activity by 60% (Figure 3B, light gray bars), suggesting that both E2F7-RE are required for Enpp2 transcription at the same time. Furthermore, we deleted p53-RE in the first intron and found that it significantly increased luciferase activity by two-fold (Figure 3C, gray bars), suggesting that binding of p53 in the first intron inhibits Enpp2 transcription.
The E2F7 protein has two DNA-binding domains (DBD) and two dimerization residues (DR), all of which are required for its transcriptional activity [43]. We generated the series of DBD and DR mutants shown in Table 1 and evaluated their relative transcriptional activity of Enpp2 (Figures 3D & 3E). Whereas single mutations in either the DBD or DR did not significantly affect its transcriptional activity, overexpression of the E2F7-DBD double mutation reduced Enpp2 transcriptional activity to the basal reporter activity detected in empty vector-transfected cells (Figure 3F, M1M2). Likewise, Enpp2 promoter activity was reduced to basal level in cells transfected with the double mutant of E2F7-DR, relative to E2F7-WT (Figure 3G, D1D2). These data suggest that impairing either the DBD or DR residues of E2F7 abolishes the transcriptional upregulation of Enpp2.
Table 1.
E2F7 DBD and DR mutations and plasmids designations
| Plasmid name | Abbreviation |
|---|---|
| YFP-E2F7-R185A | M1 |
| YFP-E2F7-R334A | M2 |
| YFP-E2F7- R185A/R334A | M1M2 |
| YFP-E2F7- ΔDR1 | D1 |
| YFP-E2F7- ΔDR2 | D2 |
| YFP-E2F7- ΔDR1/ΔDR2 | D1D2 |
E2F7 physically binds to the Enpp2 promoter.
We used ChIP assays to determine whether E2F7 binds directly to the Enpp2 regulatory region. Chromatin fragments prepared from WT, p53-KO, and p53R172H MEF were immunoprecipitated with either anti-E2F7 or control mouse IgG antibodies. The isolated DNA was amplified by qPCR to quantify the enrichment of DNA that encompasses the predicted E2F7-RE in the Enpp2 regulatory region. Multiple primers were designed to amplify seven DNA sequences (ChIP 1–7) that encompass E2F7-RE1 and E2F7-RE2 (Figure 4A). In WT-MEF, the regions of the Enpp2 regulatory domain 1600~1000 bps upstream of the transcription start site (TSS), which encompasses E2F7-RE1 (ChIP-2) and two nearby regions (ChIP-1 and ChIP-3) were significantly enriched after immunoprecipitation with anti-E2F7 antibodies (Figure 4B). Likewise, E2F7 robustly occupied the same region upstream of the TSS in p53-KO (Figure 4C) and p53R172H MEF (Figure 4D), to the same extent detected in WT MEF. Interestingly, the region that encompasses E2F7-RE2 at +1000 bp (ChIP-7) was also amplified after enrichment with E2F7 antibodies in WT, p53-KO, and p53R172H MEF (Figures 4B–4D), confirming that E2F7 upregulated Enpp2 transcription by directly binding to both E2F7-RE in the promoter and second intron. We also note that p53R172H MEF displayed a different ChIP distribution from that of WT or p53-KO MEF, where a significant interaction of E2F7 with ChIP-5 was observed only in p53R172H MEF.
Figure 4. E2F7 binds to the promoter and second intron of Enpp2.
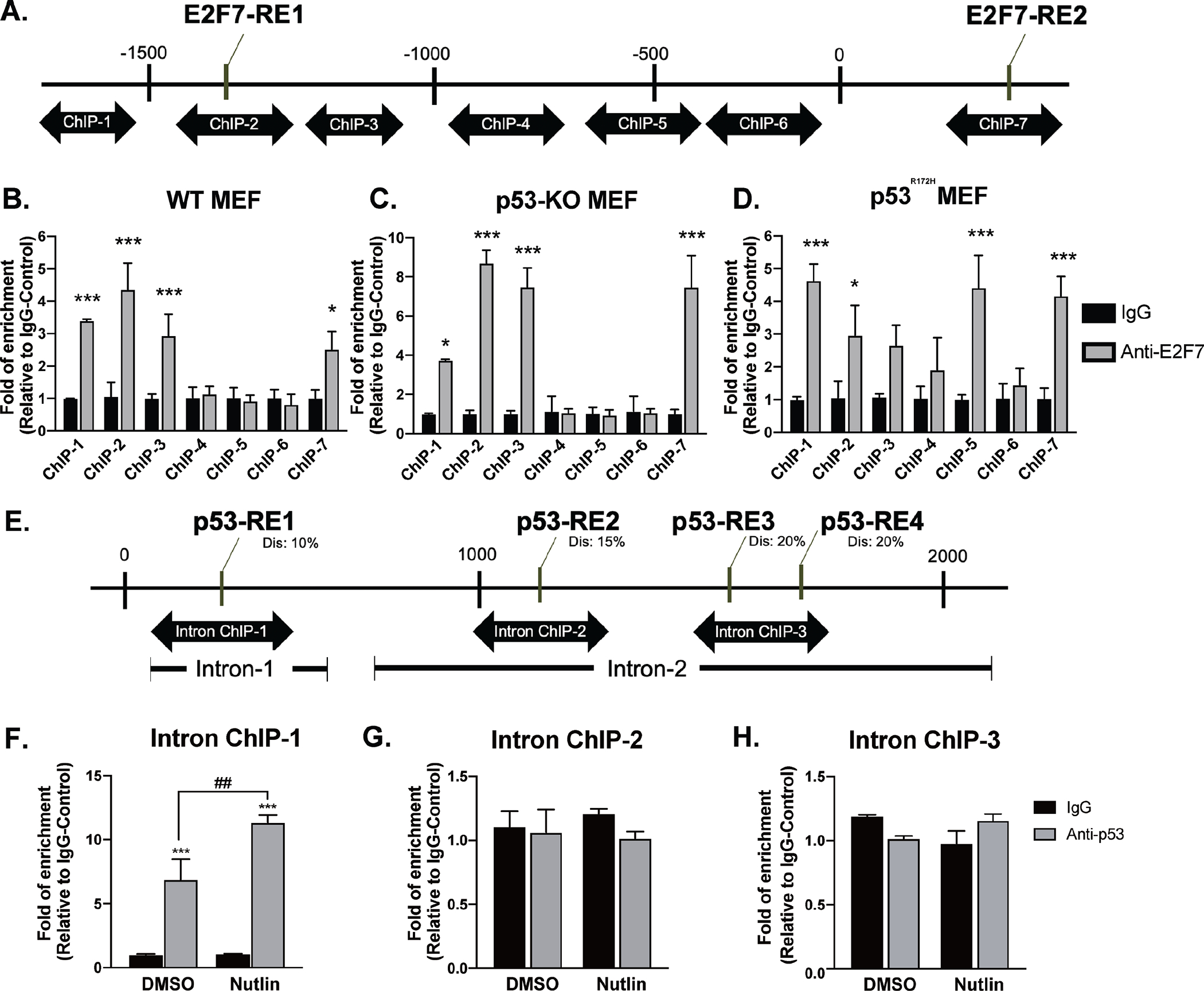
(A) Diagram of the ChIP-qPCR primers targeting the E2F7-RE. (B)WT, (C) p53-KO, and (D) p53R172H cells were subjected to ChIP-qPCR analysis with anti-E2F7 and nonspecific control IgG antibodies. The E2F7-bound DNA was quantified by qPCR using specific primers (ChIP-1~ChIP-7) targeting the promoter and intron sequences of Enpp2. (E) Diagram of the ChIP-qPCR primers targeting potential p53-RE. WT cells were starved for 1h followed by incubating with 20 μM Nutlin 3A for 24 h. The cells were further subjected to ChIP-qPCR analysis with anti-p53 or nonspecific control IgG antibodies. The primers targeting (F) p53-RE1 in the first intron, (G) p53-RE2, (H) p53-RE3 and p53-RE4 in the second intron were used to quantify the p53-bound DNA. Enrichment of E2F7- and p53-binding was normalized to non-specific control IgG. Analyses represent the mean of three independent experiments. One-way ANOVA was performed to compare the data from multiple experiments to determine the significance of E2F7 binding at a given site. *p < 0.05 and ***p < 0.001 indicate significant differences. Dis: dissimilarity.
Next, we applied anti-p53 antibody in the ChIP assay combined with the treatment of Nutlin-3A to monitor the occupancy of p53 in the intron of mouse Enpp2. Primer pairs (Supplementary table 3) were designed to target the potential p53-RE in the first and second intron of mouse Enpp2 (Figure 4E). The ChIP results indicated that the DNA region encompassing p53-RE in the first intron was significantly enriched after IP with anti-p53 antibody (Figure 4F). Furthermore, two additional p53-RE in the second intron were identified when the stringency of dissimilarity was reduced from 10% to 20% (Figure 4E). However, ChIP results showed that p53 does not bind to these two sites (Figure 4G & H). These results confirm that p53 represses Enpp2 transcription by binding to the p53-RE in the first intron rather than the second intron of Enpp2.
Chromosomal looping of E2F7 drives Enpp2 transcription.
Our ChIP and luciferase reporter analyses suggest that E2F7 binds to E2F7-RE1 and E2F7-RE2, both of which are required for E2F7-dependent Enpp2 transcription. We hypothesized that binding of two E2F7 to both sites causes chromosomal looping mediated by E2F7 dimerization. Therefore, we applied the 3C-qPCR technique to determine whether the two E2F7-RE interact with one another.
We selected Dpn II as a restriction enzyme that cuts near the predicted TF binding sites and designed a series of primers that would allow us to quantify the ligation frequency between the anchor fragment (anchor 1) located at E2F7-RE2 in the second intron and the Dpn II restriction fragments that flank the Enpp2 promoter upstream of the TSS (Figure 5A). The assay was performed in WT, p53-KO, and p53R172H MEF. We found that the interaction frequencies between the two E2F7-RE (Primer-3 and Anchor-1) are significantly higher in p53-KO and p53R172H MEF relative to WT MEF (Figure 5A). The interaction frequency gradually decreased when using Dpn II fragments further away from E2F7-RE1 because the likelihood of a specific interaction decreases with distance. In contrast, Anchor-2 (corresponding to p53-RE in the first intron, Figure 5B) displayed a significantly higher interaction frequency with Primer 3 only in WT MEF compared to p53-KO and p53R172H MEF (Figure 5B), suggesting that the binding of WT p53 to p53-RE in the first intron of Enpp2 can abolish the interaction between the two E2F7-RE. Taken together, these data suggest a mechanism whereby three-dimensional chromosomal looping is established between the two E2F7-RE to regulate Enpp2 transcription. To confirm these results, we repeated the 3C-qPCR analysis after first performing siRNA-mediated KD of E2F7 in MEF. E2F7-KD significantly decreased the interaction frequency between the two E2F7-RE sites (Primer#3 and Anchor-2) in p53-KO and p53R172H MEF, relative to the frequency observed with scrambled siRNAs (Figures 5D & 5E). However, KD of E2F7 did not appear to affect the DNA interaction frequencies in WT MEF (Figure 5C).
Figure 5. Chromatin conformation capture (3C) reveals chromosomal looping of the two E2F7 binding sites in MEF.
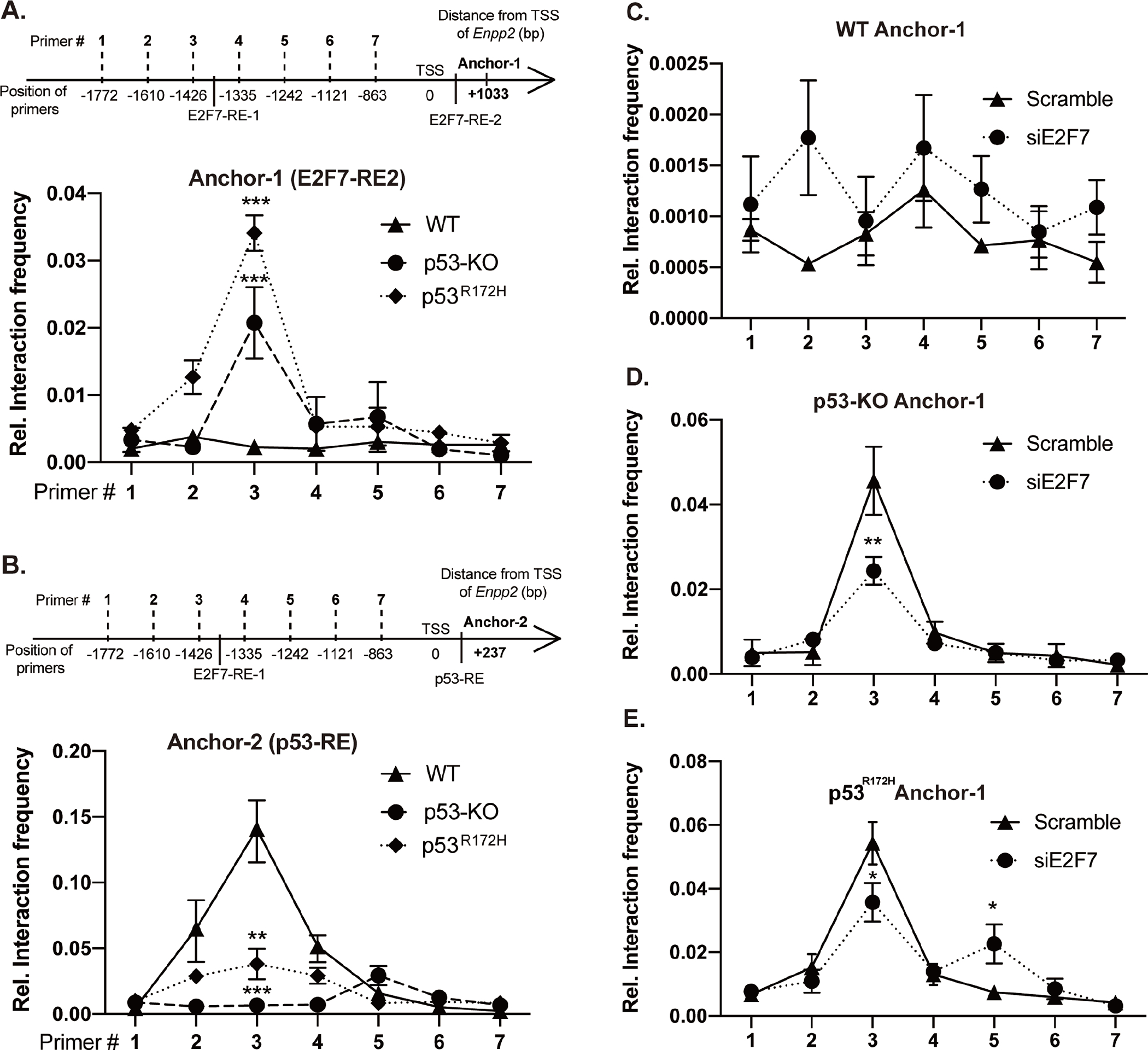
(A) Top: Schematic of 3C-qPCR primers targeting the DpnII restriction sites and E2F7 binding sites in the mouse Enpp2. Bottom: 3C-qPCR was performed in WT, p53-KO, and p53R172H cells using Anchor-1 (associated with the E2F7-RE2) and forward primers targeting E2F7-RE1 and promoter fragments. (B) Top: The schematic shows the primers targeting p53-RE and E2F7 binding sites in the mouse Enpp2 gene. Bottom: 3C-qPCR was applied to measure the interaction frequency between Anchor-2 (associated with the intronic p53-RE) and forward primers targeting E2F7-RE1 and promoter fragments. 3C assay measuring the crosslinking frequency in (C) WT, (D) p53-KO, and (E) p53R172H MEF transfected with E2F7 siRNA. Relative interaction frequency was normalized to GAPDH and plotted as the mean ± SD (how many independent experiments?). *p < 0.05, **p<0.01, and ***p < 0.001 indicate significant differences using ANOVA (n=3).
E2F7 is a novel transcriptional regulator of Enpp2 in non-transformed and malignant murine cells.
Our ChIP and 3C-qPCR results showed that E2F7 binds to the promoter and intronic region of Enpp2, resulting in chromosomal looping that is required for Enpp2 transcription in MEF. Previous studies suggest that intronic enhancers interact with their target promoters to form a chromosomal loop, which promotes transcriptional activation of various human and mouse genes [44, 45]. We examined whether this steric mechanism prevails in the regulation of Enpp2 in mouse B16-F10 melanoma cells that secretes high amounts of ATX. SiRNA-mediated KD of E2F7 significantly decreased ATX protein expression in B16-F10 (Figure 6A). Furthermore, ChIP assay confirmed that E2F7 physically binds to E2F7-RE1 (ChIP-3) and E2F7-RE2 (Figure 6B, ChIP-7). In addition, the interaction frequency between both E2F7-RE was also increased significantly in B16-F10 as determined by 3C-qPCR (Figure 6C). These results show that E2F7 promotes Enpp2 transcription in both non-transformed and malignant murine cells via a common mechanism.
Figure 6. E2F7 promotes the transcription of Enpp2 in mouse cancer cells.
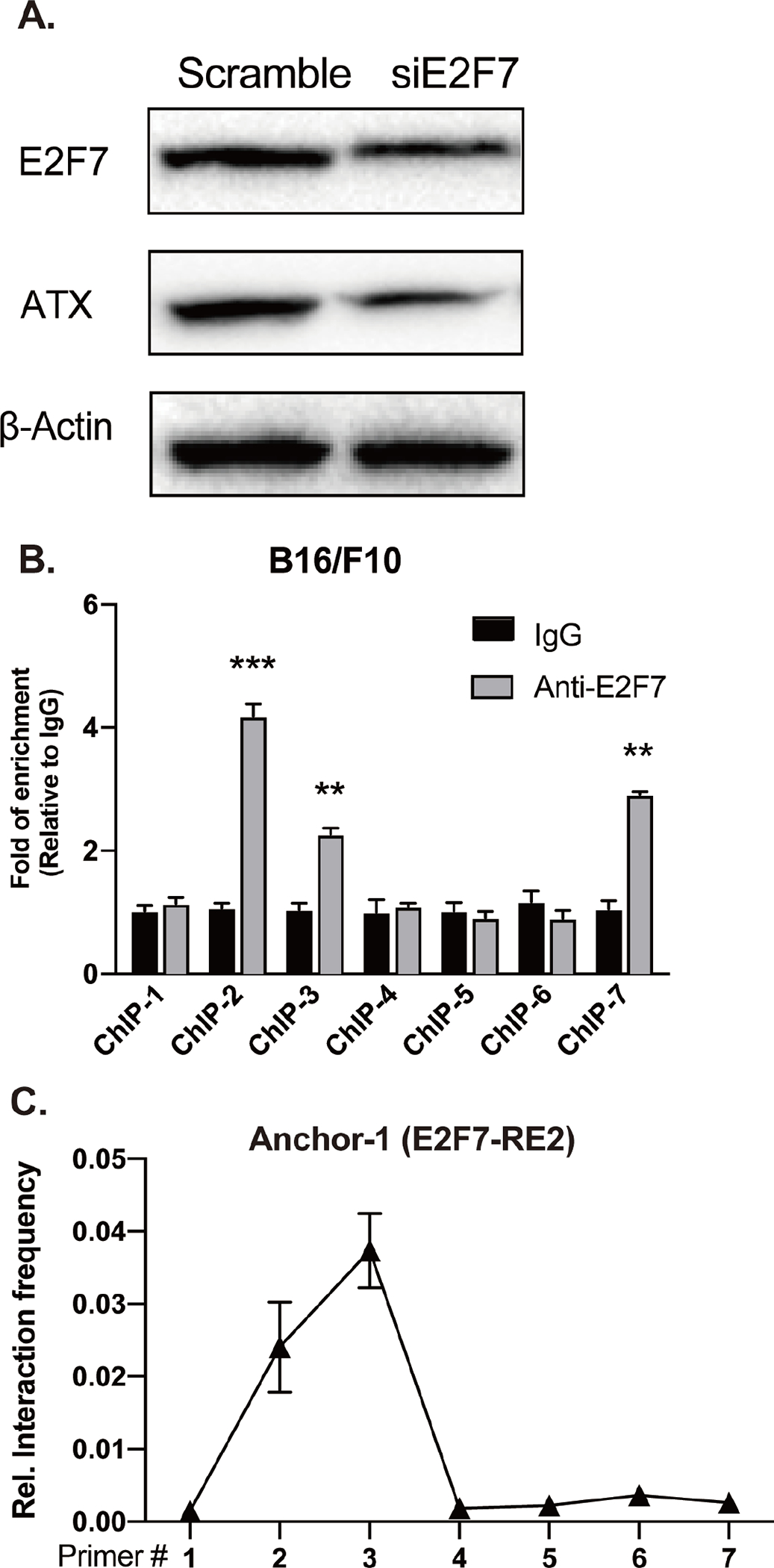
(A) B16-F10 melanoma cells were transfected with E2F7 siRNA and immunoblotting was applied to measure the E2F7, ATX, and β-actin protein expression. (B) B16-F10 cells were crosslinked with 4% (V/V) formaldehyde and total protein was extracted for ChIP-qPCR analysis. ChIP primers were used to target the E2F7-RE in the mouse Enpp2 gene. (C) 3C-qPCR was applied to measure the interaction frequency between Anchor-1 (associated with the intronic E2F7-RE2) and forward primers targeting E2F7-RE1 and promoter fragments in B16-F10 cells. The quantitative data are represented as the mean ± SD based on at least three independent experiments. *p < 0.05, **p<0.01, and ***p < 0.001 indicate significant differences.
Dissimilarity in the modulation of E2F7 activation by p53 in human versus murine carcinomas.
We probed whether this mechanism occurs in human carcinomas. Primer pairs targeting the recognition sites for MaeI rather than DpnII were used because the DpnII sites in the human genome are different from that in mouse (Figure 7A). WT or p53-KO human colon HCT116 carcinomas were tested in parallel to validate the effect of p53 on the E2F7 interaction with the human ENPP2 gene. 3C-qPCR analysis revealed an increased interaction frequency between E2F7-RE1 (Primer#2) and E2F7-RE2 of the human ENPP2 (Anchor-H), consistent with the results observed in mouse cells.
Figure 7. E2F7 regulates the expression of ATX in human cancer cell lines.
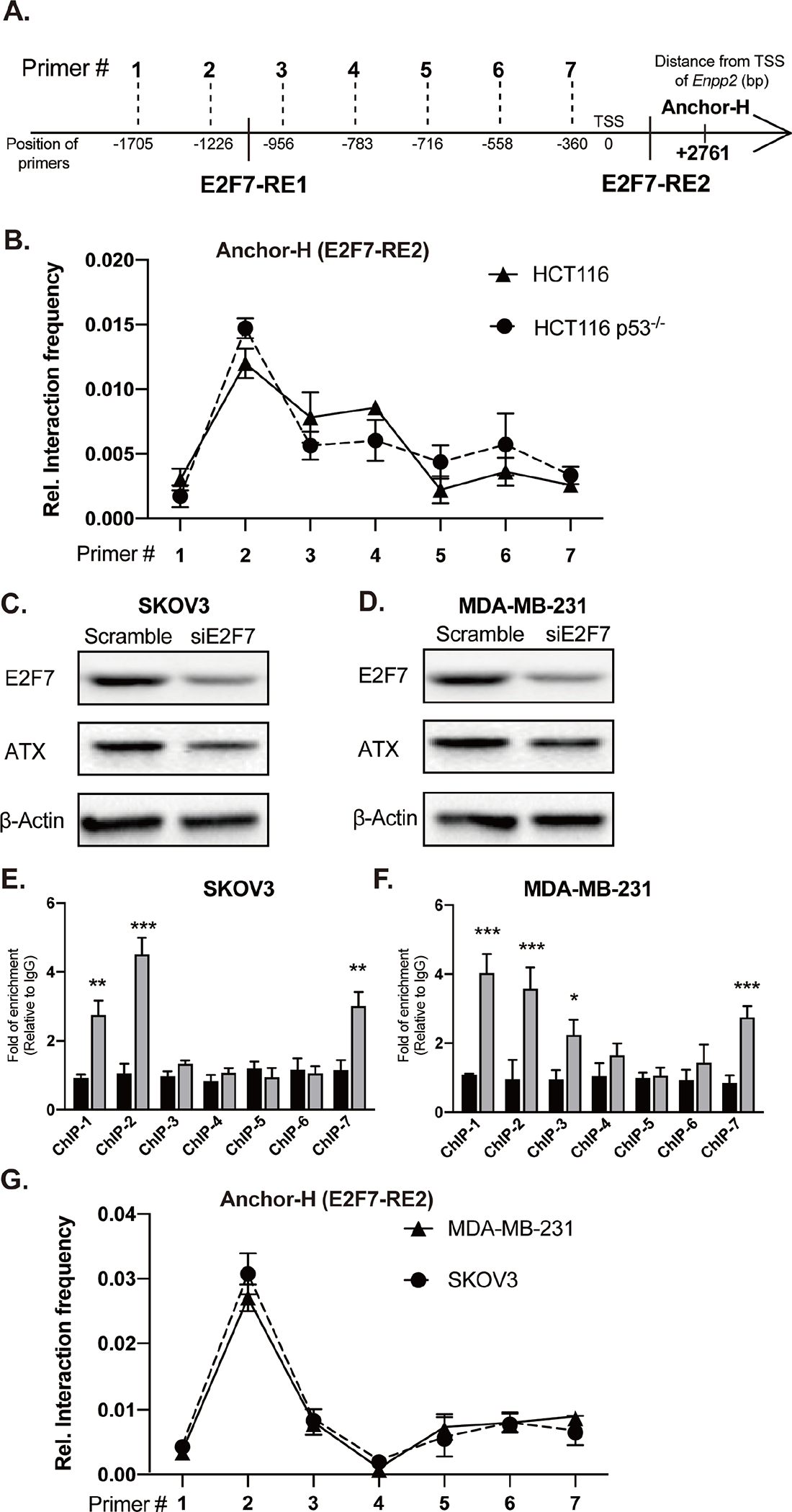
(A) Schematic of 3C-qPCR primers targeting the MaeI restriction sites and TF binding sites in the human ENPP2 gene. (B) 3C-qPCR was performed in WT and p53-KO HCT116 cells using Anchor H (associated with the E2F7-RE2) and seven forward primers targeting E2F7-RE1 and promoter fragments. Relative interaction frequency was normalized to GAPDH and plotted as the mean ± SD. E2F7 siRNA was applied to (C) SKOV3 and (D) MDA-MB-231 cells. Cells were transfected with 150 nM of scramble or E2F7 siRNA for 24 h. Total protein was used for immunoblotting with antibodies against E2F7, ATX, and β-actin. Total cell lysates were fixed and subjected to ChIP-qPCR analysis with anti-E2F7 and nonspecific control IgG antibodies. Enrichment of E2F7-binding was normalized to the nonspecific control IgG. ChIP-qPCR was used to verify the interaction between E2F7 and ENPP2 promoter in the (E) SKOV3 and (F) MDA-MB-231 cells. (G) 3C-qPCR was applied to measure the interaction frequency between Anchor-H (associated with the intronic E2F7-RE2) and forward primers targeting E2F7-RE1 and promoter fragments in human SKOV3 and MDA-MB-231. The quantitative data are represented as the mean ± SD of three independent experiments. *p < 0.05, **p<0.01, and ***p < 0.001 indicate significant differences.
In contrast, no significant differences in the interaction frequencies between WT and p53-KO HCT116 cells were detected (Figure 7B), suggesting that p53 does not disrupt the interaction between the two E2F7-RE leading to the repression of ENPP2 transcription. We analyzed the human ENPP2 sequence using the ALLGEN software. Surprisingly, no p53-RE were found in the first intron of ENPP2 using a 10% dissimilarity threshold, underlining differences in the regulatory regions between mouse and human Enpp2/ENPP2 [46]. However, reducing the stringency of dissimilarity from 10% to 20% identified six additional putative p53-RE in the first and second intron of human ENPP2. Using ChIP assays, we confirmed that p53 does not bind to any of the six p53-RE (Supplementary Figure 2). These results could explain why WT p53 failed to disrupt E2F7 chromosomal looping in WT HCT116 cells and did not result in ATX repression in human cells.
We expanded this observation to include human ovarian SKOV3 and breast MDA-MB-231 carcinomas, which differ in their p53 status (SKOV3: p53-KO; MDA-MB-231: p53R280K). SiRNA-mediated KD of E2F7 significantly decreased ATX protein levels in both cell lines (Figures 7C & 7D). Using ChIP, we detected significantly higher binding of E2F7 to the two E2F7-RE (ChIP-2 and ChIP-7) in SKOV3 (Figure 7E) and MDA-MB-231 cells (Figure 7F) relative to nonspecific control IgG. In addition, 3C-qPCR assay revealed a robust interaction between Primer#2 and Anchor-H in SKOV3 and MDA-MB-231 cells (Figure 7G), suggesting that a similar chromosomal loop develops between the two E2F7-RE. Taken together, our results indicate that E2F7 regulates the transcription of ENPP2 in human carcinomas, regardless of their p53 status.
Discussion
ATX expression is amplified in various human carcinomas as well as in different cell types of the TME [2–5]. Our initial observation that p53-KO mice have elevated Enpp2 expression prompted us to investigate if p53 can regulate the transcription of Enpp2 expression. [40]. In the present study, we showed that MEF derived from p53-KO or p53R172H mice indeed express more ATX than MEF derived from WT mice, suggesting that WT p53 represses Enpp2 transcription. Because of the general consensus that p53 does not directly repress gene transcription [47], we tested the hypothesis that the repression of Enpp2 transcription by p53 is mediated indirectly via the regulation of (an) unknown TF(s) that is either regulated directly or indirectly by p53. We identified five TF candidates that could repress Enpp2 transcription. Among these candidates, only E2F7 interacted with the Enpp2 promoter (Figures 1 & 2) by binding to two E2F7-RE in the promoter region upstream of the TSS and in the second intron (Figure 3A). We found that direct binding to both E2F7-RE was required to drive Enpp2 transcription (Figure 3B & 4B). Furthermore, both DNA binding and dimerization abilities of E2F7 were also required for the upregulation of Enpp2 transcription (Figures 3F & 3G). Chromosomal looping has been shown to upregulate the transcription of several human and mouse genes [44, 45]. We examined whether E2F7-mediated transcription of Enpp2 might occur via a similar mechanism. Indeed, we found that E2F7 dimerization facilitates chromosomal looping between the two E2F7-RE sites (Figure 5A).
The presence of a unique p53 binding site in first intron of murine Enpp2 (Figure 3A) prompted us to examine if the mechanism by which p53 represses Enpp2 transcription occurs by binding to this site. Our data indicated that binding of p53 to the intronic p53-RE in the Enpp2 gene interferes with E2F7-mediated chromosomal looping in WT MEF but not p53-KO or p53R172H MEF (Figure 4F & 5B). Interestingly, the human ENPP2 gene lacks a p53-RE in its first intron (Supplementary Figure 2). Therefore, we examined the role of p53 in human carcinoma cells. We found that p53 had no effect on E2F7-dependent ENPP2 expression and ATX production in all of the human carcinoma cell lines we examined, regardless of their p53 status – WT, KO or p53R280K (Figure 7). Therefore, the difference between murine and human Enpp2/ENPP2 sequences may provide an explanation for the lack of p53-dependent direct repression of ENPP2 transcription via E2F7 in human carcinomas.
E2F7 is an atypical member of the E2F TF family and is well recognized for its tumor-suppressive roles via transcriptional repression of genes involved in S-phase entry [48]. E2F7 has been reported to exert pro-tumorigenic effects via direct binding to the promoter of EZH2 in glioblastoma [49]. Furthermore, E2F7 has been shown to mediate p53-dependent gene repression. Specifically, DNA damage caused activation of p53 was accompanied by increased expression of E2F7, which inhibited cellular proliferation by directly binding to the promoters of G1/S target genes and repressing their expression [47, 50]. Multiple studies have also reported the pro-tumorigenic activity of E2F7 [49, 51, 52], indicating the complex role of E2F7 in cancer that is now broadened by its role in the regulation of ATX transcription, which through increased LPA production can mediate or at least drive cancer progression. A unique characteristic of E2F7 is that it possesses two DBD and two DR important for both DNA-binding and dimerization (Figure 3D & E). The binding of E2F7 to its DNA target is completely blocked by mutation of either of the two DBD [53], and E2F7 homodimerization completely depends on the integrity of the two DR [54]. Our studies support the role of E2F7 homodimerization in its transcriptional activity, at least in the case of Enpp2 (Figure 3F & G). Additional studies focused on the mutations affecting E2F7 dimerization might unveil pro-tumorigenic and/or anti-tumor-promoting effects.
In our ChIP experiments, we found that ChIP-1 and ChIP-3 contain binding sites for NFκB that are present in both murine and human Enpp2/ENPP2. [40]. Previous study showed that irradiation of tumor-bearing mice significantly increased TNF-α levels [55], which has been shown to upregulate ATX expression via NFκB [56]. We showed that irradiation of HCT116 cells increased the mRNA expression of both E2F7 and ENPP2. Moreover, siRNA-mediated KD of E2F7 abrogates radiation-induced ENPP2 expression (Supplementary Figure 4), suggesting that E2F7 may play a role in regulating ENPP2 expression under genotoxic stress. Our findings raise the question of whether a potential interaction between E2F7 with NFκB could coregulate ENPP2 transcription under genotoxic stress. In addition, we found that E2F7 interacted with ChIP-5 only in p53R172H MEF, but not in WT or p53-KO MEF. Mutation of the R172H residue in p53 alters the tertiary structure of p53 and is regarded as a conformational mutant. Although p53R172H mutant together with other known p53 mutants (e.g. R273H, G245S, R248P) are not able to bind to consensus p53 binding sequences, they have been reported to bind to non-canonical DNA structures and exert gain-of-function properties [57, 58]. Another alternative mechanism for p53R172H gain-of-function is via interaction with other transcription factors such as p63, p73, and NF-Y, which results in altered gene transcription [59, 60]. More recently, studies have shown that p53R172H mutant is able to transcriptionally induce NFκB2 gene expression and to prolong NFκB response to TNF-α in human lung cancer cells [61, 62]. In this regard, we note that ChIP-5 also contains binding sites for NFκB. Thus, whether the interaction of E2F7 with ChIP-5 is mediated by the binding of p53R172H mutant to this DNA region or via its interaction with other transcription factors such as NFkB remains unknown. This hypothesis will have to be examined in future experiments.
In conclusion, we showed for the first time that E2F7 is a novel TF regulating Enpp2/ENPP2 transcription by directly binding to two E2F7-RE sites located in the promoter and second intron. Dimerization of DNA-bound E2F7 molecules results in chromosomal looping that brings the intronic E2F7-RE2 in proximity with the E2F7-RE1 in the Enpp2/ENPP2 promoter. Furthermore, our results identified mouse-human differences in the regulation of Enpp2/ENPP2 expression by p53 that are likely to complicate the interpretation of the role of p53-dependent ATX regulation in murine versus human carcinomas and of human explant models in SCID/NSG mice. It is conceivable that p53-reprograming-mediated non-cell-delimited production of ATX from stromal cells in the TME in murine carcinoma models may also prove to be profoundly different from that in human patients. Although we initially set out to investigate the role of p53 in regulating E2F7-mediated Enpp2 transcription, it is possible that E2F7 may also recruit other TFs such as NFκB to the transcriptional complex to promote the expression of ATX in the cells.
Supplementary Material
Acknowledgements
The authors thank Dr. Ying-Chung Lin from NTU for advice on the Y1H experiment, and Kyle Johnson-Moore from the UTHSC Office of Scientific Writing.
This research was funded by grants from the NCI (CA092160, G.J.T.), the Van Vleet Oncologic Research Fund (G.J.T.), the UTHSC Office of Research (G.J.T.), the William and Ella Owens Foundation (S.C.L.), and the Ministry of Science and Technology of Taiwan (MOST-109-2917I-564-029, K.H.L) and the Hungarian National Research, Development, and Innovation Office (OTKA K-125174, K-139230 and PD-132851, A.B.).
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Kihara Y, Maceyka M, Spiegel S, Chun J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br J Pharmacol 2014; 171: 3575–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdul Rahman M, Tan ML, Johnson SP, Hollows RJ, Chai WL, Mansell JP et al. Deregulation of lysophosphatidic acid metabolism in oral cancer promotes cell migration via the up-regulation of COX-2. PeerJ 2020; 8: e10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014; 511: 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dimova I, Raitcheva S, Dimitrov R, Doganov N, Toncheva D. Correlations between c-myc gene copy-number and clinicopathological parameters of ovarian tumours. Eur J Cancer 2006; 42: 674–679. [DOI] [PubMed] [Google Scholar]
- 5.Tigyi GJ, Yue J, Norman DD, Szabo E, Balogh A, Balazs L et al. Regulation of tumor cell – Microenvironment interaction by the autotaxin-lysophosphatidic acid receptor axis. Advances in Biological Regulation 2019; 71: 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu PY, Lin YC, Huang YL, Chen WM, Chen CC, Lee H. Mechanisms of Lysophosphatidic Acid-Mediated Lymphangiogenesis in Prostate Cancer. Cancers (Basel) 2018; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer 2003; 3: 582–591. [DOI] [PubMed] [Google Scholar]
- 8.Seo EJ, Kwon YW, Jang IH, Kim DK, Lee SI, Choi EJ et al. Autotaxin Regulates Maintenance of Ovarian Cancer Stem Cells through Lysophosphatidic Acid-Mediated Autocrine Mechanism. Stem Cells 2016; 34: 551–564. [DOI] [PubMed] [Google Scholar]
- 9.Lee D, Suh DS, Lee SC, Tigyi GJ, Kim JH. Role of autotaxin in cancer stem cells. Cancer Metastasis Rev 2018; 37: 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee SC, Dacheux MA, Norman DD, Balazs L, Torres RM, Augelli-Szafran CE et al. Regulation of Tumor Immunity by Lysophosphatidic Acid. Cancers (Basel) 2020; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dacheux MA, Lee SC, Shin Y, Norman DD, Lin KH, E S et al. Prometastatic Effect of ATX Derived from Alveolar Type II Pneumocytes and B16-F10 Melanoma Cells. Cancers (Basel) 2022; 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng W, Morokoff A. Lysophospholipid Signalling and the Tumour Microenvironment. Adv Exp Med Biol 2021; 1270: 123–144. [DOI] [PubMed] [Google Scholar]
- 13.Aiello S, Casiraghi F. Lysophosphatidic Acid: Promoter of Cancer Progression and of Tumor Microenvironment Development. A Promising Target for Anticancer Therapies? Cells 2021; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009; 138: 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng W, Shuyu E, Tsukahara R, Valentine WJ, Durgam G, Gududuru V et al. The lysophosphatidic acid type 2 receptor is required for protection against radiation-induced intestinal injury. Gastroenterology 2007; 132: 1834–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng W, Wang DA, Gosmanova E, Johnson LR, Tigyi G. LPA protects intestinal epithelial cells from apoptosis by inhibiting the mitochondrial pathway. Am J Physiol Gastrointest Liver Physiol 2003; 284: G821–829. [DOI] [PubMed] [Google Scholar]
- 17.Samadi N, Gaetano C, Goping IS, Brindley DN. Autotaxin protects MCF-7 breast cancer and MDA-MB-435 melanoma cells against Taxol-induced apoptosis. Oncogene 2009; 28: 1028–1039. [DOI] [PubMed] [Google Scholar]
- 18.Vidot S, Witham J, Agarwal R, Greenhough S, Bamrah HS, Tigyi GJ et al. Autotaxin delays apoptosis induced by carboplatin in ovarian cancer cells. Cell Signal 2010; 22: 926–935. [DOI] [PubMed] [Google Scholar]
- 19.Farina AR, Cappabianca L, Ruggeri P, Di Ianni N, Ragone M, Merolle S et al. Constitutive autotaxin transcription by Nmyc-amplified and non-amplified neuroblastoma cells is regulated by a novel AP-1 and SP-mediated mechanism and abrogated by curcumin. FEBS Lett 2012; 586: 3681–3691. [DOI] [PubMed] [Google Scholar]
- 20.Sioletic S, Czaplinski J, Hu L, Fletcher JA, Fletcher CD, Wagner AJ et al. c-Jun promotes cell migration and drives expression of the motility factor ENPP2 in soft tissue sarcomas. J Pathol 2014; 234: 190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCabe CD, Innis JW. A genomic approach to the identification and characterization of HOXA13 functional binding elements. Nucleic Acids Res 2005; 33: 6782–6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sah JP, Hao NTT, Han X, Tran TTT, McCarthy S, Oh Y et al. Ectonucleotide pyrophosphatase 2 (ENPP2) plays a crucial role in myogenic differentiation through the regulation by WNT/β-Catenin signaling. Int J Biochem Cell Biol 2020; 118: 105661. [DOI] [PubMed] [Google Scholar]
- 23.Cao P, Walker NM, Braeuer RR, Mazzoni-Putman S, Aoki Y, Misumi K et al. Loss of FOXF1 expression promotes human lung-resident mesenchymal stromal cell migration via ATX/LPA/LPA1 signaling axis. Sci Rep 2020; 10: 21231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He L, Yang Y, Chen J, Zou P, Li J. Transcriptional activation of ENPP2 by FoxO4 protects cardiomyocytes from doxorubicin-induced toxicity. Mol Med Rep 2021; 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie D, Yu S, Li L, Quan M, Gao Y. The FOXM1/ATX signaling contributes to pancreatic cancer development. Am J Transl Res 2020; 12: 4478–4487. [PMC free article] [PubMed] [Google Scholar]
- 26.Azare J, Doane A, Leslie K, Chang Q, Berishaj M, Nnoli J et al. Stat3 mediates expression of autotaxin in breast cancer. PLoS One 2011; 6: e27851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braeuer RR, Zigler M, Kamiya T, Dobroff AS, Huang L, Choi W et al. Galectin-3 Contributes to Melanoma Growth and Metastasis via Regulation of NFAT1 and Autotaxin. Cancer Research 2012; 72: 5757–5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen M, O’Connor KL. Integrin α6β4 promotes expression of autotaxin/ENPP2 autocrine motility factor in breast carcinoma cells. Oncogene 2005; 24: 5125–5130. [DOI] [PubMed] [Google Scholar]
- 29.Li S, Wang B, Xu Y, Zhang J. Autotaxin is induced by TSA through HDAC3 and HDAC7 inhibition and antagonizes the TSA-induced cell apoptosis. Mol Cancer 2011; 10: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Panagopoulou M, Drosouni A, Fanidis D, Karaglani M, Balgkouranidou I, Xenidis N et al. ENPP2 Promoter Methylation Correlates with Decreased Gene Expression in Breast Cancer: Implementation as a Liquid Biopsy Biomarker. Int J Mol Sci 2022; 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun S, Zhang X, Lyu L, Li X, Yao S, Zhang J. Autotaxin Expression Is Regulated at the Post-transcriptional Level by the RNA-binding Proteins HuR and AUF1. J Biol Chem 2016; 291: 25823–25836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naz S, Kolmert J, Yang M, Reinke SN, Kamleh MA, Snowden S et al. Metabolomics analysis identifies sex-associated metabotypes of oxidative stress and the autotaxin-lysoPA axis in COPD. Eur Respir J 2017; 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murph MM. MicroRNA Regulation of the Autotaxin-Lysophosphatidic Acid Signaling Axis. Cancers (Basel) 2019; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee SC, Lin KH, Balogh A, Norman DD, Bavaria M, Kuo B et al. Dysregulation of lysophospholipid signaling by p53 in malignant cells and the tumor microenvironment. Cell Signal 2021; 78: 109850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev 2013; 27: 1016–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikulenkov F, Spinnler C, Li H, Tonelli C, Shi Y, Turunen M et al. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ 2012; 19: 1992–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sbisa E, Catalano D, Grillo G, Licciulli F, Turi A, Liuni S et al. p53FamTaG: a database resource of human p53, p63 and p73 direct target genes combining in silico prediction and microarray data. BMC Bioinformatics 2007; 8 Suppl 1: S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeh CS, Wang Z, Miao F, Ma H, Kao CT, Hsu TS et al. A novel synthetic-genetic-array-based yeast one-hybrid system for high discovery rate and short processing time. Genome Res 2019; 29: 1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ke YD, Huang YW, Viswanath KK, Hu CC, Yeh CM, Mitsuda N et al. NbNAC42 and NbZFP3 Transcription Factors Regulate the Virus Inducible NbAGO5 Promoter in Nicotiana benthamiana. Front Plant Sci 2022; 13: 924482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balogh A, Shimizu Y, Lee SC, Norman DD, Gangwar R, Bavaria M et al. The autotaxin-LPA2 GPCR axis is modulated by gamma-irradiation and facilitates DNA damage repair. Cell Signal 2015; 27: 1751–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.López-Fuentes E, Hernández-Hernández G, De Las Peñas A, Castaño I. Subtelomeric Chromatin Structure by Chromosome Conformation Capture (3C)-qPCR Methodology in Candida glabrata. Methods Mol Biol 2022; 2542: 71–89. [DOI] [PubMed] [Google Scholar]
- 42.Sullivan KD, Galbraith MD, Andrysik Z, Espinosa JM. Mechanisms of transcriptional regulation by p53. Cell Death & Differentiation 2018; 25: 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lammens T, Li J, Leone G, De Veylder L. Atypical E2Fs: new players in the E2F transcription factor family. Trends in Cell Biology 2009; 19: 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deshane J, Kim J, Bolisetty S, Hock TD, Hill-Kapturczak N, Agarwal A. Sp1 Regulates Chromatin Looping between an Intronic Enhancer and Distal Promoter of the Human Heme Oxygenase-1 Gene in Renal Cells Journal of Biological Chemistry 2010; 285: 16476–16486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ott CJ, Suszko M, Blackledge NP, Wright JE, Crawford GE, Harris A. A complex intronic enhancer regulates expression of the CFTR gene by direct interaction with the promoter. Journal of Cellular and Molecular Medicine 2009; 13: 680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Conservation Fischer M. and divergence of the p53 gene regulatory network between mice and humans. Oncogene 2019; 38: 4095–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carvajal LA, Hamard PJ, Tonnessen C, Manfredi JJ. E2F7, a novel target, is up-regulated by p53 and mediates DNA damage-dependent transcriptional repression. Genes Dev 2012; 26: 1533–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan R, Liu Q, Segeren HA, Yuniati L, Guardavaccaro D, Lebbink RJ et al. Cyclin F-dependent degradation of E2F7 is critical for DNA repair and G2-phase progression. Embo j 2019; 38: e101430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang R, Wang M, Zhang G, Bao Y, Wu Y, Li X et al. E2F7-EZH2 axis regulates PTEN/AKT/mTOR signalling and glioblastoma progression. Br J Cancer 2020; 123: 1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitxelena J, Apraiz A, Vallejo-Rodríguez J, García-Santisteban I, Fullaondo A, Alvarez-Fernández M et al. An E2F7-dependent transcriptional program modulates DNA damage repair and genomic stability. Nucleic Acids Research 2018; 46: 4546–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang F, Guo C, Cao X, Yan Y, Zhang J, Lv S. Gastric cancer cell-derived extracellular vesicles elevate E2F7 expression and activate the MAPK/ERK signaling to promote peritoneal metastasis through the delivery of SNHG12. Cell Death Discov 2022; 8: 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreno E, Pandit SK, Toussaint MJM, Bongiovanni L, Harkema L, van Essen SC et al. Atypical E2Fs either Counteract or Cooperate with RB during Tumorigenesis Depending on Tissue Context. Cancers (Basel) 2021; 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Stefano L, Jensen MR, Helin K. E2F7, a novel E2F featuring DP-independent repression of a subset of E2F-regulated genes. Embo j 2003; 22: 6289–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zalmas LP, Zhao X, Graham AL, Fisher R, Reilly C, Coutts AS et al. DNA-damage response control of E2F7 and E2F8. EMBO Rep 2008; 9: 252–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang N, Zhou Y, Zhu M, Zhang J, Cao M, Lei H et al. Optimization and evaluation of novel tetrahydropyrido[4,3-d]pyrimidine derivatives as ATX inhibitors for cardiac and hepatic fibrosis. Eur J Med Chem 2020; 187: 111904. [DOI] [PubMed] [Google Scholar]
- 56.Wu JM, Xu Y, Skill NJ, Sheng H, Zhao Z, Yu M et al. Autotaxin expression and its connection with the TNF-alpha-NF-kappaB axis in human hepatocellular carcinoma. Mol Cancer 2010; 9: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gohler T, Jager S, Warnecke G, Yasuda H, Kim E, Deppert W. Mutant p53 proteins bind DNA in a DNA structure-selective mode. Nucleic Acids Res 2005; 33: 1087–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koga H, Deppert W. Identification of genomic DNA sequences bound by mutant p53 protein (Gly245-->Ser) in vivo. Oncogene 2000; 19: 4178–4183. [DOI] [PubMed] [Google Scholar]
- 59.Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004; 119: 861–872. [DOI] [PubMed] [Google Scholar]
- 60.Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006; 10: 191–202. [DOI] [PubMed] [Google Scholar]
- 61.Scian MJ, Stagliano KE, Anderson MA, Hassan S, Bowman M, Miles MF et al. Tumor-derived p53 mutants induce NF-kappaB2 gene expression. Mol Cell Biol 2005; 25: 10097–10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weisz L, Damalas A, Liontos M, Karakaidos P, Fontemaggi G, Maor-Aloni R et al. Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res 2007; 67: 2396–2401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The experimental data that support the findings of this study are openly available in the figshare website at the following link https://figshare.com/projects/E2F7_drives_ATX_transcription_via_chromosome_looping_Repression_by_p53_in_murine_but_not_human_carcinomas/160789 under reference number 160789.