

Abstract
Targeting cyclin-dependent kinases (CDKs) has recently emerged as a promising therapeutic approach against cancer. However, the anticancer mechanisms of different CDK inhibitors (CDKIs) are not well understood. Our recent study revealed that selective CDK4/6 inhibitors sensitize colorectal cancer (CRC) cells to therapy-induced apoptosis by inducing Death Receptor 5 (DR5) via the p53 family member p73. In this study, we investigated if this pathway is involved in anticancer effects of different CDKIs. We found that less-selective CDKIs, including flavopiridol, roscovitine, dinaciclib, and SNS-032, induced DR5 via p73-mediated transcriptional activation. The induction of DR5 by these CDKIs was mediated by dephosphorylation of p73 at Threonine 86 and p73 nuclear translocation. Knockdown of a common target of these CDKIs, including CDK1, 2, or 9, recapitulated p73-mediated DR5 induction. CDKIs strongly synergized with 5-fluorouracil (5-FU), the most commonly used CRC chemotherapy agent, in vitro and in vivo to promote growth suppression and apoptosis, which required DR5 and p73. Together, these findings indicate p73-mediated DR5 induction as a potential tumor suppressive mechanism and a critical target engaged by different CDKIs in potentiating therapy-induced apoptosis in CRC cells. These findings help better understand the anticancer mechanisms of CDKIs and may help facilitate their clinical development and applications in CRC.
Keywords: CDK inhibitor, DR5, p73, colorectal cancer, apoptosis
Introduction
Cyclin-dependent kinases (CDKs) are a group of serine/threonine kinases that play a key role in various physiological processes by phosphorylating their downstream substrates [1]. Upon binding to different cyclins, CDKs 1, 2, 4, and 6 directly promote cell cycle progression. CDKs 1 and 2 function as key regulators of mitosis and DNA replication in M and S phases, respectively, while CDKs 4 and 6 control G1/S transition. Other CDKs, in particular CDK9, are involved in transcriptional regulation [1]. As central regulators of cell division, CDKs’ functions are frequently altered in cancer, which drives hyperproliferation of cancer cells and contributes to other hallmarks of cancer [2, 3].
Targeting CDKs has recently emerged as a promising therapeutic approach against cancer. A number of small-molecule ATP-competitive CDK inhibitors (CDKIs) have been developed and some have shown promising anticancer activities. First-generation CDKIs, including flavopiridol (Alvocidib) and roscovitine, are pan-CDK inhibitors with relatively low specificity. Flavopiridol is a semi-synthetic flavonoid that inhibits CDKs 1, 2, 4, 5, and 9. It was tested in over 60 clinical trials and showed clinical efficacy in some patients with hematological malignancies such as acute myeloid leukemia (AML) [4]. Second-generation CDKIs such as dinaciclib and SNS-032 have increased selectivity for CDKs 1 and/or 2. More recently, selective CDK4/6 inhibitors (CDK4/6Is), including Palbociclib, Ribociclib, and Abemaciclib, have been approved by the United States Food and Drug Administration (FDA) for the treatment of hormone receptor positive and advanced breast cancer [5]. Anticancer effects of CDKIs include inhibition of the cell cycle, induction of cellular senescence, modulation of tumor cell metabolism and tumor microenvironment, and promotion of antitumor immunity [6]. However, the anticancer mechanisms of CDKIs, specifically first- and second-generation less-selective CDKIs, have remained poorly understood.
CDKIs can promote apoptosis especially when used in combination with other anticancer agents [7]. For example, flavopiridol was shown to potentiate apoptosis induced by radiation in colon cancer cells [8]. Nonetheless, it is unclear how CDK inhibition affects cell death signaling and if the apoptotic effect is essential for the anticancer activities of CDKIs. Apoptosis in mammalian cells is controlled by the extrinsic and/or intrinsic pathways. Engagement of the extrinsic pathway is mediated by activation of the tumor necrosis factor (TNF) family receptors, such as Death Receptor 5 (DR5; TRAILR2) and DR4, by their ligands such as TNF-related apoptosis-inducing ligand (TRAIL) [9], leading to activation of caspase 8 and effector caspases [10]. DR5 can also be activated via ligand-independent mechanisms, in particular transcriptional activation by p53, TAp73 (p73 thereafter), NF-κB, and C/EBP Homologous Protein (CHOP), in response to various stresses [11, 12]. The intrinsic pathway is governed by the Bcl-2 proteins via mitochondrial dysfunction [13]. The extrinsic and intrinsic pathways can crosstalk via the proapoptotic Bcl-2 family protein Bid.
CDKIs have shown promising activity against colorectal cancer (CRC) [14], the second leading cause of cancer-related deaths in the United States [15]. CRC patients are commonly treated with 5-fluorouracil (5-FU)-based chemotherapy, which is limited in efficacy and has substantial toxic side effects [16]. Our recent study revealed that CDK4/6Is sensitize CRC cells to chemotherapy-induced apoptosis by inducing DR5 via the p53 family member p73 [17]. In this study, we investigated other CDKIs and found that p73-dependent DR5 induction is a common effect of different CDKIs and plays a critical role in mediating the antitumor activity of CDK inhibition.
Results
CDKIs induce DR5 expression in CRC cells with different p53 status.
To investigate the effects of CDKIs in CRC cells, we treated p53-WT HCT116 CRC cells with first-generation CDKIs flavopiridol (FVP; CDK1/2/4/5/9 inhibitor) and roscovitine (CDK1/2/5/7/9 inhibitor), and second-generation CDKIs dinaciclib (CDK1/2/5/9 inhibitor) and SNS-032 (CDK1/2/4/7/9 inhibitor) [1]. We found that all agents at concentrations ≥ 0.1 μM induced DR5 expression (Fig. 1A). We focused on FVP in subsequent experiments due to its clinical applications and validated key findings using roscovitine and additional CDKIs. FVP and roscovitine treatments robustly upregulated DR5 protein and mRNA expression in a time-dependent manner (Fig. 1, B and C; Fig. S1, A and B). In contrast, other apoptosis regulators including the death receptor family and Bcl-2 family members were unchanged or modestly induced (Fig. 1, B and D; Fig. S1, A and C). Induction of DR5 by FVP and roscovitine was also observed in other CRC cell lines with WT or mutant p53 (Fig. 1, E and F; Fig. S1D). FVP treatment markedly increased the levels of cell-surface and nuclear DR5, but not cell-surface DR4, in HCT116 cells (Fig. 1G; Fig. S1E), suggesting DR5 activation by transcriptional upregulation. Along with our findings on CDK4/6Is [17], these results indicate that different CDKIs can induce DR5 expression in CRC cells independent of p53 status.
Figure 1. DR5 induction by CDKIs in CRC cells with different p53 status.
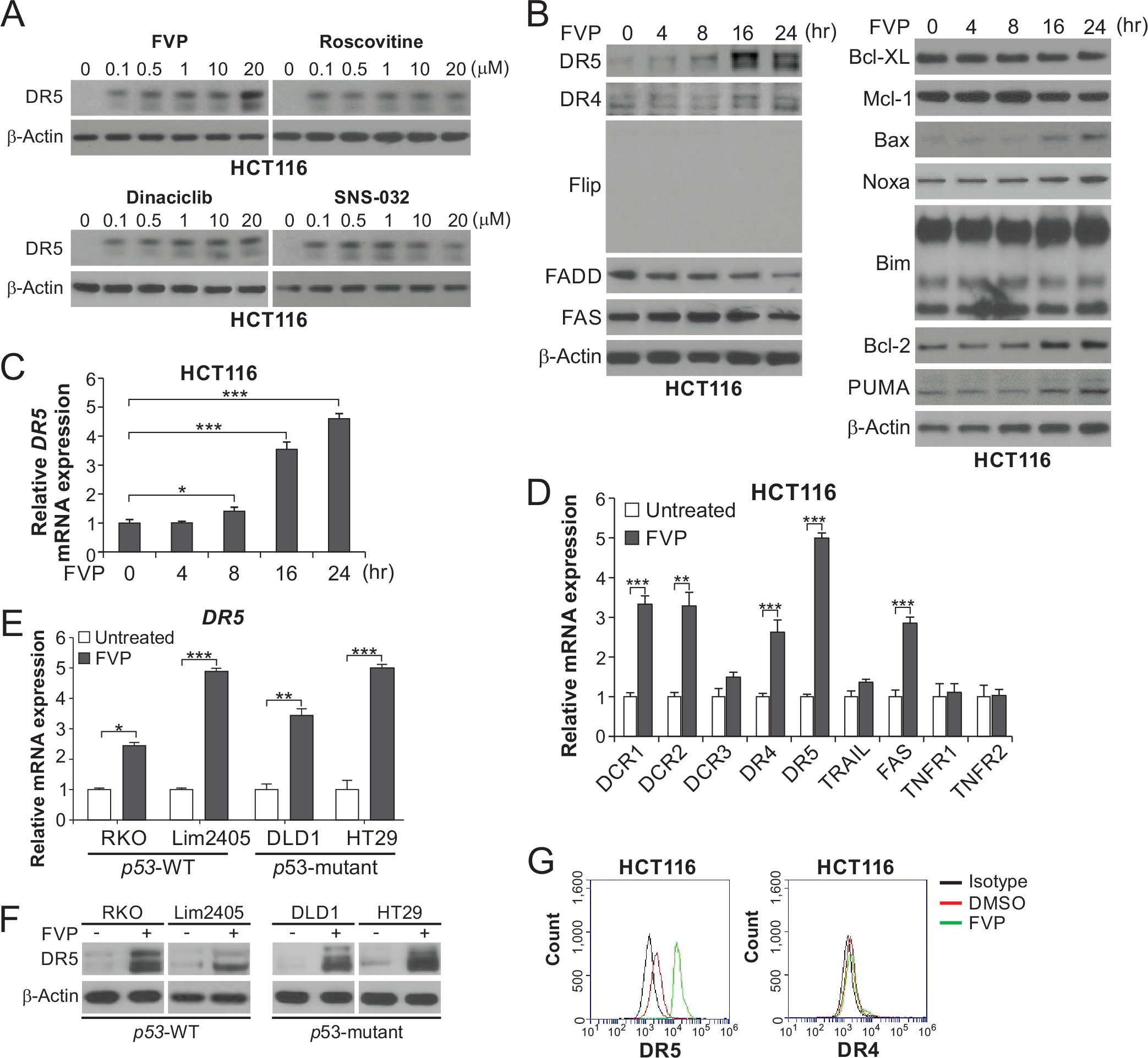
(A) Western blotting of DR5 in HCT116 cells treated with indicated CDKIs at indicated concentrations for 24 hours. (B) Western blotting of indicated proteins in HCT116 cells treated with 50 nM flavopiridol (FVP) at indicated time points. (C) Time course of DR5 mRNA induction in HCT116 cells treated with 50 nM FVP was analyzed by real-time reverse transcriptase (RT)-PCR. (D) HCT116 cells treated with 50 nM FVP for 24 hours were analyzed for mRNA expression of the indicated genes by real-time RT-PCR. (E), (F) DR5 expression in CRC cell lines with different p53 status and treated with 50 nM FVP for 24 hours was analyzed by (E) real-time RT-PCR and (F) Western blotting. (G) HCT116 cells treated with FVP as in (D) were analyzed for cell-surface DR5 and DR4 by flow cytometry. In (C)-(E), values were expressed as means ± SD of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
p73 transcriptionally activates DR5 upon CDKI-induced Thr86 dephosphorylation and nuclear translocation.
We then investigated the mechanism of DR5 induction by CDKIs. Treating HCT116 cells with FVP or roscovitine did not affect the expression of DR5-inducing transcription factors, including p53, p73, p65 subunit of NF-κB, E2F1, and CHOP (Fig. S2, A and B). Interestingly, DR5 induction was abrogated by p73 KD in HCT116 cells (Fig. 2, A–C), but not affected by p53 KO or CHOP KD (Fig. S2, C–E). HCT116 cells with knock-in (KI) of Flag-tagged p73 (Flag-KI) [18] were used to probe endogenous p73 without the interference of non-specific bands (Fig. 2, B and C). The requirement of p73 for DR5 induction by FVP was confirmed in other CRC cell lines with p73 KD by siRNA (Fig. 2D).
Figure 2. Transcriptional activation of DR5 by p73 upon CDKI-induced Thr86 dephosphorylation and nuclear translocation.
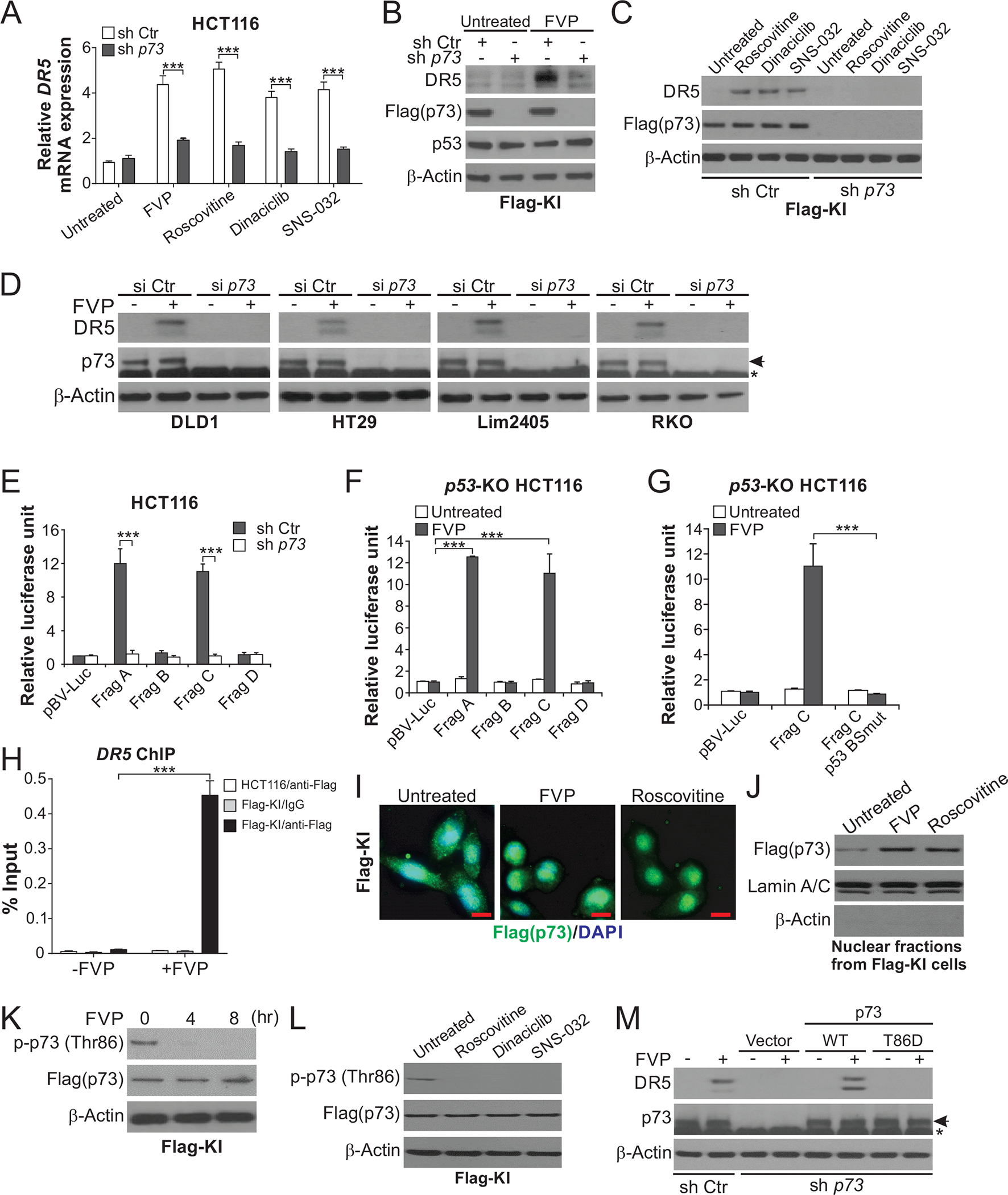
(A) HCT116 cells with stable p73 KD by shRNA (sh p73) or control scrambled shRNA (sh Ctr) were treated with 50 nM FVP, 1 μM roscovitine, 100 nM dinaciclib, or 100 nM SNS-032 for 24 hours. DR5 mRNA expression was analyzed by real-time RT-PCR. (B), (C) Western blotting of indicated proteins in sh p73 and sh Ctr Flag-KI cells treated with CDKIs as in (A). (D) Western blotting of DR5 in indicated CRC cell lines transfected with p73 siRNA (si p73) or control scrambled siRNA (si Ctr) and treated with 50 nM FVP for 24 hours. Arrows indicate p73 and stars indicate nonspecific bands. (E)-(G) Sh p73, sh Ctr, or p53-KO HCT116 cells were transfected with DR5 promoter luciferase reporters and then treated with 50 nM FVP for 24 hours. Reporter activities were measured and normalized to the untreated control samples. In (E) and (F), reporters contain DNA fragments (Frag A-D) covering different regions of the DR5 promoter, among which Frag A and Frag C contain the p53 binding site (Fig. S2F). In (G), reporters contain Frag C with WT or mutant p53 binding site (p53 BSmut) (Fig. S2F). (H) Parental and Flag-KI HCT116 cells with or without treatment with 50 nM FVP for 24 hours were analyzed for the binding of Flag-tagged p73 to the DR5 promoter by chromatin immunoprecipitation (ChIP) using anti-Flag-conjugated beads with IgG as negative control, followed by quantitative real-time PCR analysis of the DR5 promoter region covering the p53 binding site. (I), (J) Flag-KI HCT116 cells were treated with 50 nM FVP or 1 μM roscovitine. Flag-tagged p73 nuclear translocation was analyzed by (I) flag immunofluorescence (green) at 4 hours with DAPI (blue) for nuclear counterstaining (scale bars: 10 μm), and (J) Western blotting of nuclear fractions isolated from cells treated for 24 hours with Lamin A/C (nuclear) and β-actin (cytoplasmic) as loading and fractionation controls. (K), (L) Western blotting of indicated proteins in Flag-KI HCT116 cells treated with (K) 50 nM FVP at indicated time points, or (L) 1 μM roscovitine, 100 nM dinaciclib, or 100 nM SNS-032 for 8 hours. (M) Western blotting of indicated proteins in sh Ctr and sh p73 HCT116 cells transfected with indicated p73 expression constructs and treated with 50 nM FVP for 24 hours. In (A) and (E)-(H), values were expressed as means ± SD of three independent experiments. ***, P < 0.001.
To determine if p73 directly activates DR5 transcription, we utilized a series of DR5 promoter luciferase reporters (Fig. S2F). FVP treatment strongly activated reporters containing the p53 binding site (Frag A and C; Fig. S2F), which was abolished by p73 KD but not p53 KO (Fig. 2, E and F). Mutations in 2 nucleotides in the p53 binding site (Fig. S2F) inactivated the Frag C reporter (Fig. 2G). We then used Flag-KI HCT116 cells to analyze the binding of p73 to the DR5 promoter by chromatin immunoprecipitation (ChIP). FVP treatment markedly enhanced the binding of Flag-tagged p73 to the DR5 promoter (Fig. 2H). p73 is regulated by nuclear import to transcriptionally activate p53 targets [19, 20]. Treating Flag-KI cells with FVP or roscovitine induced p73 nuclear translocation as shown by immunostaining (Fig. 2I) and enriched p73 in nuclear fractions (Fig. 2J).
p73 subcellular localization and activity are regulated by post-translational modifications such as threonine (Thr) phosphorylation [21]. Thr86 phosphorylation (p-Thr86) of p73 is suppressed by CDK4/6Is, which promotes p73 nuclear translocation as shown by us recently [17]. We found that treating Flag-KI cells with FVP or other CDKIs also inhibited p73 p-Thr86 (Fig. 2, K and L). Expression of WT, but not phospho-mimic T86D mutant, restored DR5 induction by FVP in p73-KD cells (Fig. 2M). Together, these results demonstrate a critical role of p73 and the p53 binding site for DR5 induction by different CDKIs, which is mediated through p73 nuclear translocation upon loss of p-Thr86.
CDK1, 2 or 9 depletion recapitulates p73-mediated DR5 induction.
We previously showed that CDK4 or 6 KD induced DR5 via p73 [17]. To determine a potential role of other CDKs in suppressing the p73-DR5 axis, we used siRNA to knock down CDK1, 2, and 9, which are commonly inhibited by CDKIs. CDK1, 2, or 9 KD markedly induced DR5 protein and mRNA expression in p53-WT HCT116 and p53-mutant DLD1 cells (Fig. 3, A and B; Fig. S3, A and B). In contrast, other death receptor family members and DR5-inducing transcription factors were unaffected or slightly induced (Fig. 3, A and B). p73 KD, but not p65 or CHOP KD, abrogated DR5 induction by CDK1, 2, or 9 KD in HCT116 cells (Fig. 3C; Fig. S3, C and D). Similar to CDK4 KD, CDK1, 2, or 9 KD inhibited p73 p-Thr86 (Fig. 3A), and strongly activated DR5 protomer reporters in a p73-dependent but p53-independent manner (Fig. 3, D and E), which also required WT p53 binding site (Fig. 3F). Furthermore, CDK1, 2, 4, or 9 KD markedly enhanced the binding of p73 to the DR5 promoter (Fig. 3G). These results indicate that on-target CDK inhibition by CDKIs promotes p73-mediated DR5 induction.
Figure 3. Knockdown of CDK1, 2, or 9 recapitulates p73-mediated DR5 induction.
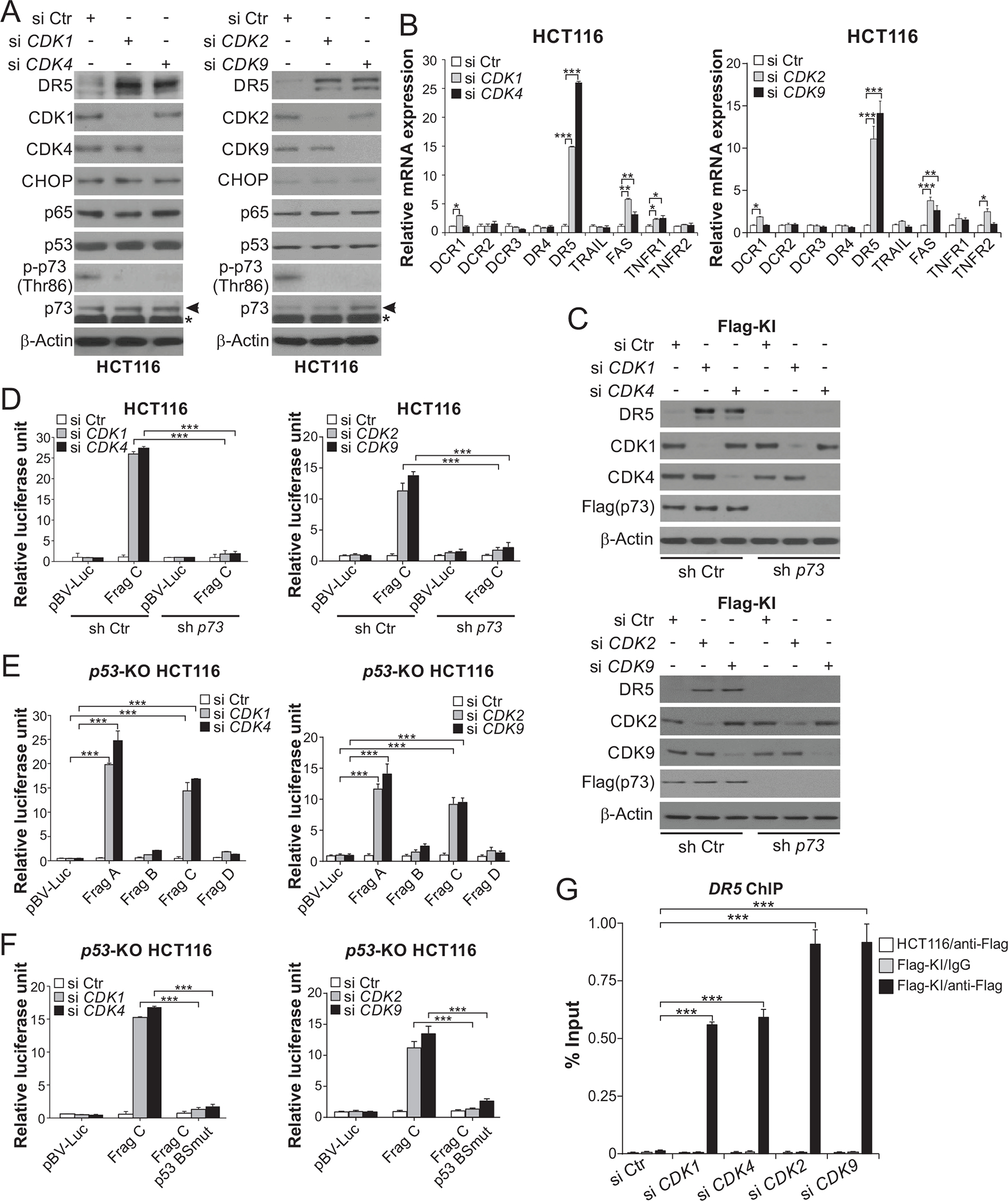
(A), (B) HCT116 cells transfected with control scrambled (Ctr), CDK1, 2, 4, or 9 siRNA for 24 hours. (A) Western blotting of indicated proteins. Arrows indicate p73 and stars indicate nonspecific bands. (B) Real-time RT-PCR analysis of mRNA expression of indicated apoptosis regulators. The results were normalized to cells transfected with si Ctr. (C) Western blotting of indicated proteins in stable p73 KD (sh p73) and control (sh Ctr) Flag-KI HCT116 cells transfected with indicated siRNA for 24 hours. (D)-(F) Sh p73, sh Ctr, or p53-KO HCT116 cells were transfected with the indicated DR5 promoter reporters along with indicated siRNA for 24 hours. Reporter activities were measured and normalized to the untreated control samples. In (D) and (E), reporters contain different regions of the DR5 promoter (Frag A-D). The p53 binding site is present in Frag A and Frag C (Fig. S2F). In (F), reporters contain Frag C with WT or mutant p53 binding site (p53 BSmut) (Fig. S2F). (G) Parental and Flag-KI HCT116 cells were transfected with indicated siRNA for 24 hours. The binding of Flag-tagged p73 to the DR5 promoter was analyzed by ChIP as in Fig. 2H. In (B) and (D)-(G), values were expressed as means ± SD of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DR5 mediates the apoptotic and chemosensitization effects of CDKIs via crosstalk of the intrinsic and extrinsic pathways.
CDKIs are often used as chemosensitizers in combination with cytotoxic chemotherapy. FVP combined with 5-FU across different concentrations were found to be highly synergistic in HCT116 and DLD1 cells (Fig. 4, A–C; Fig. S4, A–C). CDKIs combined with 5-FU markedly enhanced DR5 induction and apoptosis in different CRC cell lines, compared to either agent alone (Fig. 4, D–G; Fig. S5, A and B). The enhanced DR5 induction can be explained by p53-dependent and -independent mechanisms. In DR5-KO HCT116 cells previously established [22], the synergy of the CDKI/5-FU combinations in apoptosis induction, caspase activation, and inhibition of colony formation was blocked (Fig. 4, D–I; Fig. S5, C and D). Apoptosis was restored by DR5 expression in DR5-KO cells (Fig. S5E). p73 KD, but not TRAIL KD, also suppressed apoptosis and caspase activation induced by the FVP/5-FU combination (Fig. 4, J and K; Fig. S6A). Apoptosis was enhanced by TRAIL, but not affected by DR5 blockade with antibody (Fig. S6B). Consistent with DR5-mediated death-inducing signaling complex (DISC) formation/activation [10], transfection with c-FLIPs, which is expressed in CRC cells [23], partially inhibited apoptosis and caspase activation induced by the FVP/5-FU combination (Fig. S6, C and D). Furthermore, perturbing DR5 downstream apoptosis regulators, including FADD, caspase 8, and Bid [10], abrogated apoptosis and caspase activation induced by the FVP/5-FU combination (Fig. 5, A–C; Fig. S6E), supporting crosstalk of the intrinsic and extrinsic apoptotic pathways. Indeed, apoptosis and caspase activation induced by the combination required the intrinsic pathway component Bax (Fig. 5D) and involved DR5-dependent Bax conformational change and cytochrome c release (Fig. 5, E and F). Cell death induced by the FVP/5-FU combination was not affected by the necroptosis inhibitor necrostatin-1 or necrosulfonamide (Fig. S6, F and G), suggesting lack of DR5-mediated necroptosis [24].
Figure 4. DR5 is required for apoptosis induced by CDKI/5-FU combinations in CRC cells.
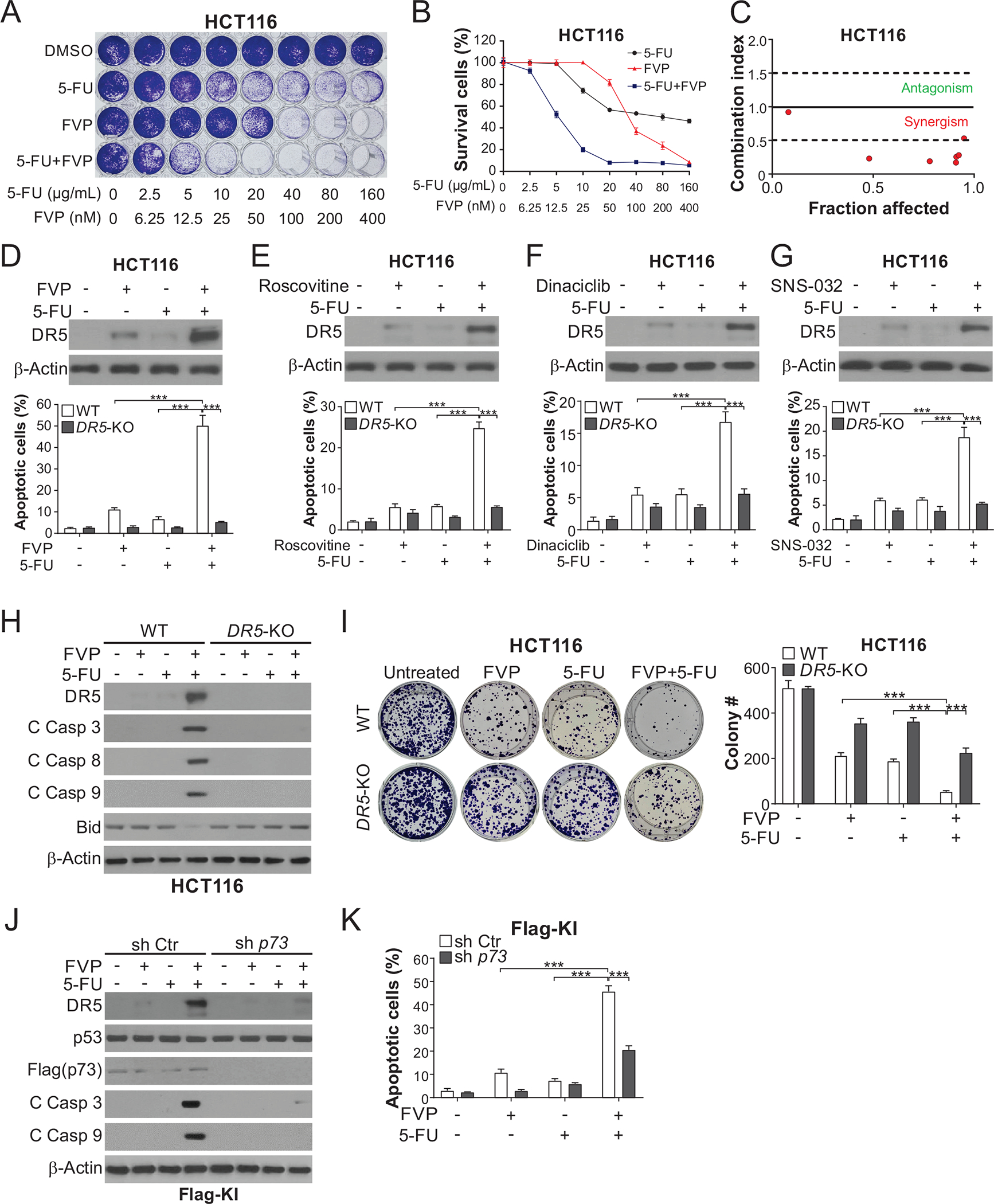
(A)-(C) HCT116 cells treated with FVP alone or in combination with 5-FU at indicated concentrations for 48 hours were analyzed by (A) crystal violet staining of attached cells, (B) MTS analysis of viability, and (C) combination indexes calculated by CompuSyn. (D)-(G) HCT116 cells were treated with 15 μg/mL 5-FU +/− (D) 20 nM FVP, (E) 1 μM roscovitine, (F) 100 nM dinaciclib, or (G) 100 nM SNS-032 for 24 hours. Upper panels: Western blotting of indicated proteins; lower panels: analysis of apoptosis by counting cells with condensed and fragmented nuclei after nuclear staining with Hoechst 33258. (H), (I) WT and DR5-KO HCT116 cells were treated with 20 nM FVP, 15 μg/mL 5-FU, or their combination for 24 hours. (H) Western blotting of indicated proteins. (I) Analysis of long-term cell viability by colony formation assay, which was done by seeding an equal number of treated cells in 6-well plates and staining attached cells with crystal violet on day 14. Left, representative pictures of colonies; right, quantification of colony numbers. (J), (K) Flag-KI HCT116 cells with sh p73 or sh Ctr were treated with 20 nM FVP, 15 μg/mL 5-FU, or their combination for 24 hours. (J) Western blotting of indicated proteins. (K) Analysis of apoptosis by nuclear fragmentation as in (D)-(G). In (B), (D)-(G), (I), and (K), values were expressed as means ± SD of three independent experiments. ***, P < 0.001.
Figure 5. DR5 mediates apoptosis induced by CDKIs combined with 5-FU via crosstalk of the intrinsic and extrinsic pathways.
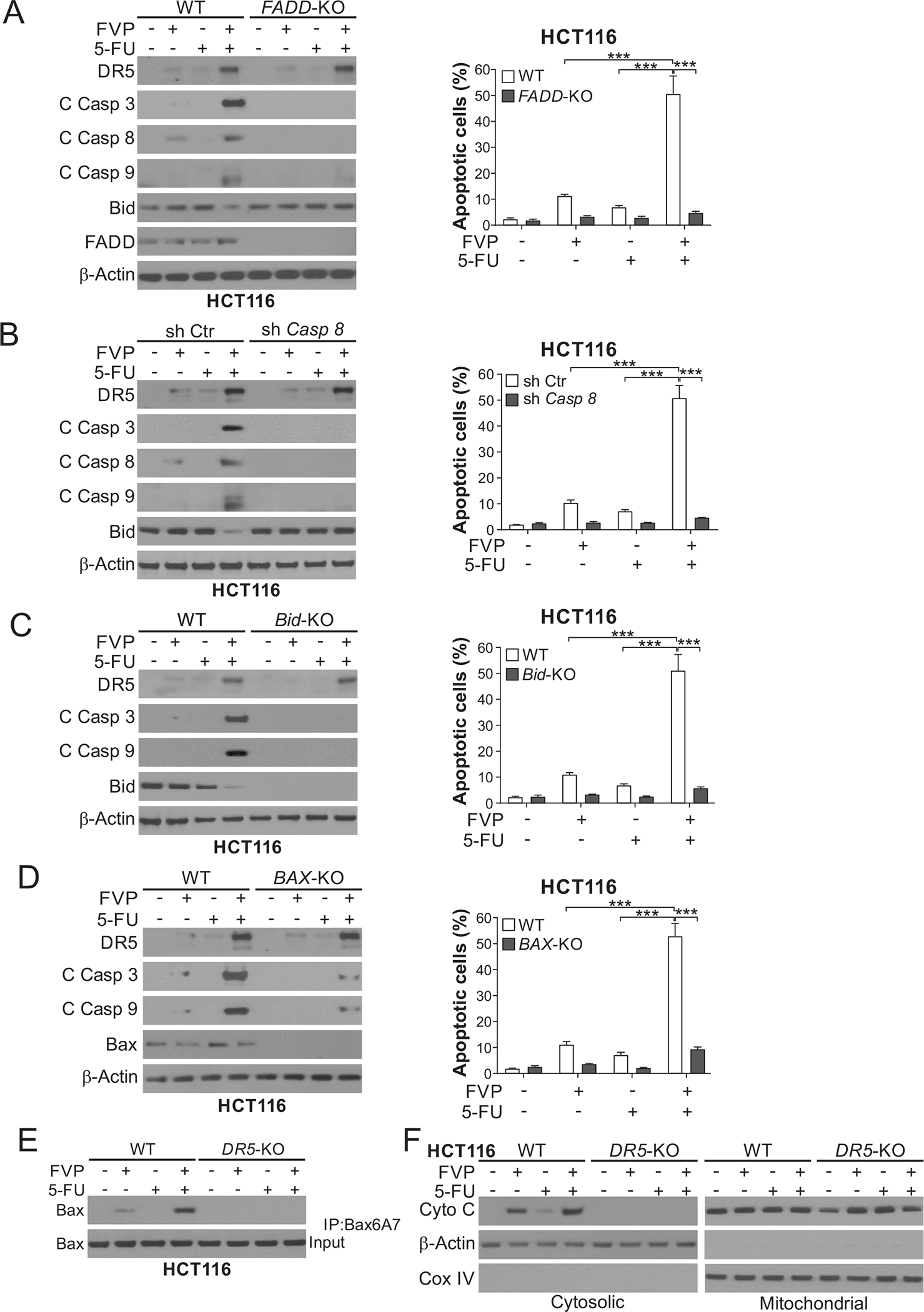
(A)-(D) WT HCT116 cells along with isogenic (A) FADD-KO, (B) stable caspase 8 shRNA knockdown (Casp 8-KD) and shRNA control (sh Ctr), (C) Bid-KO, and (D) BAX-KO HCT116 cells were treated with 20 nM FVP, 15 μg/mL 5-FU, or their combination for 24 hours. Left panels: Western blotting of indicated proteins; right panels: analysis of apoptosis by counting cells with condensed and fragmented nuclei after nuclear staining with Hoechst 33258. (E), (F) WT and DR5-KO HCT116 cells were treated as in (A)-(D). (E) Bax conformational change was analyzed by immunoprecipitation (IP) with anti-Bax 6A7 (activated) antibody followed by Western blotting. (F) Cytochrome c release was analyzed by Western blotting of mitochondrial and cytosolic fractions isolated from treated cells. β-Actin and cytochrome oxidase subunit IV (COX IV) were used as controls for loading and fractionation. In (A)-(D), values were expressed as means ± SD of three independent experiments. ***, P < 0.001.
Apoptosis can be induced by multiple CDKIs alone at relatively high concentrations, which was also dependent on DR5, p73, caspase 8, and Bid (Fig. S7, A–D). DR5 KO or p73 KD, but not CHOP KD or TRAIL KD, suppressed roscovitine-induced apoptosis, caspase activation, and inhibition of colony formation in HCT116 and DLD1 cells (Fig. S7, E–M). These results demonstrate a critical role of p73-mediated DR5 induction in the killing and chemosensitizing effects of CDKIs in CRC cells.
DR5 and p73 mediate the in vivo antitumor effects of FVP combined with 5-FU.
To determine the role of DR5 in mediating therapeutic effects in vivo, we established WT and DR5-KO HCT116 xenografts in athymic nude mice. Tumor bearing mice were treated with FVP alone or in combination with 5-FU. At relatively low doses, FVP or 5-FU alone had little effect on WT HCT116 xenografts (Fig. 6, A and B), and only induced DR5 slightly (Fig. 6C). The combination of FVP and 5-FU strongly inhibited tumor growth (Fig. 6, A and B), and markedly induced DR5 expression and apoptosis as detected by TUNEL and active caspase 3 immunostaining in the tumors (Fig. 6, C–E). The therapeutic efficacy and apoptosis were abolished in DR5-KO tumors (Fig. 6, A, B, D, and E).
Figure 6. DR5 is required for the in vivo antitumor effects of FVP combined with 5-FU.
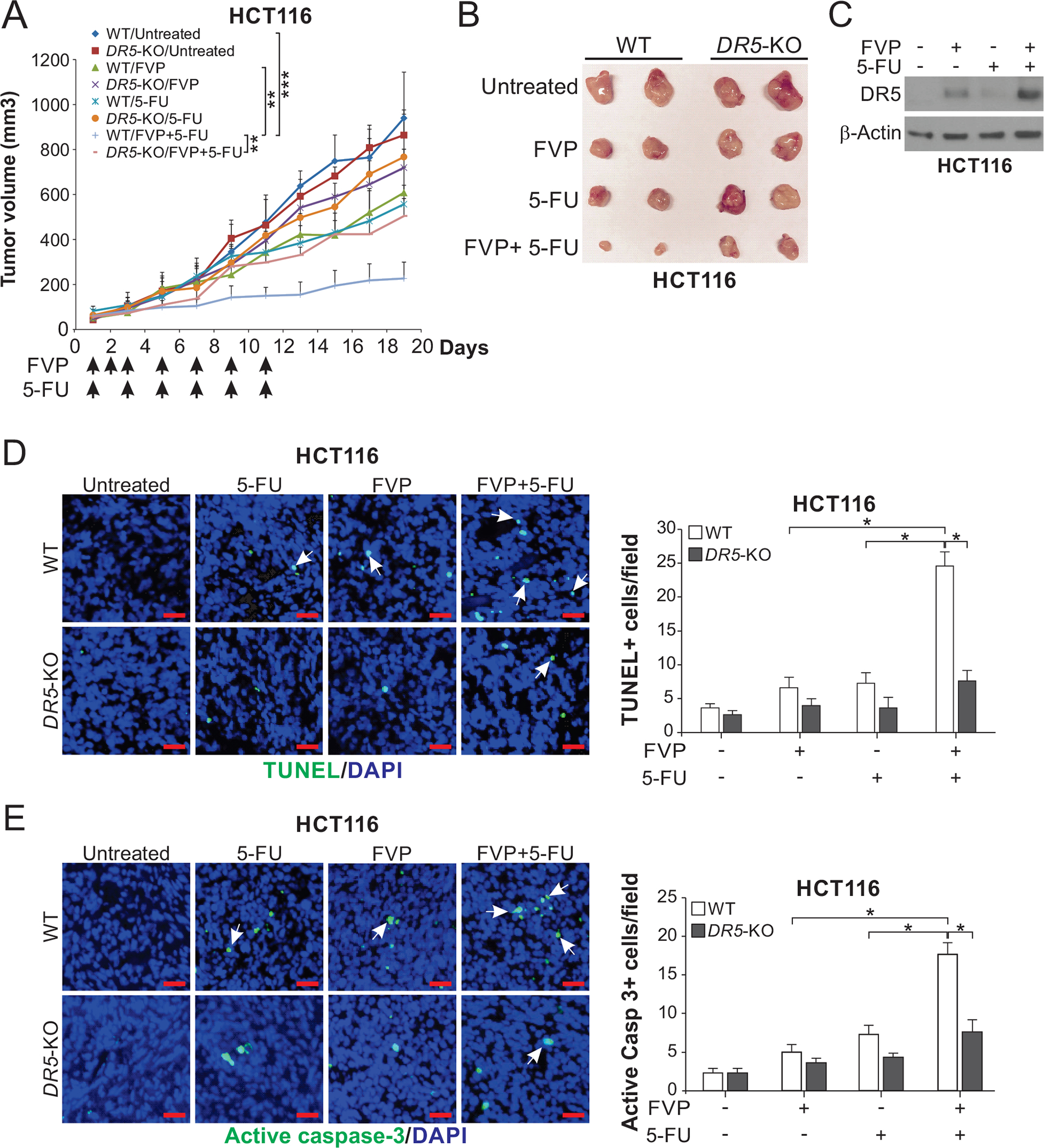
(A) Nude mice were injected s.c. with 4×106 WT or DR5-KO HCT116 cells. After tumor growth for 7 days, mice were treated with FVP (i.p.; 5 mg/kg every day for 3 days and then every other day), 5-FU (i.p.; 25 mg/kg every other day), or their combination as indicated for 11 days. Tumor volumes were calculated and plotted with P values for indicated comparisons (n=6 in each group; **, P < 0.01; ***, P < 0.001). (B) Representative tumors at the end of the experiment in (A). (C)-(E) Mice with WT and DR5-KO HCT116 xenograft tumors were treated as in (A) for 4 consecutive days. (C) Western blotting of DR5 in randomly selected WT tumors. Apoptosis was analyzed by (D) TUNEL and (E) active caspase 3 staining of tumor sections. Left, representative staining pictures with arrows indicating example positive signals (scale bar: 25 μm); right, quantification of positive signals. In (D) and (E), values were expressed as means ± SD of 3 independent experiments. *, P < 0.05.
We also determined the role of p73 similarly using p73-KD Flag-KI cells. Similar to DR5 KO, p73 KD suppressed the antitumor activity of the FVP/5-FU combination (Fig. 7, A and B), and abolished the induction of DR5, apoptosis, and caspase 3 activation (Fig. 7, C–E). These results indicate a critical role of p73-mediated DR5 induction and apoptosis in chemosensitization by FVP in vivo.
Figure 7. p73 is required for DR5 induction and the in vivo antitumor effects of the FVP/5-FU combination.
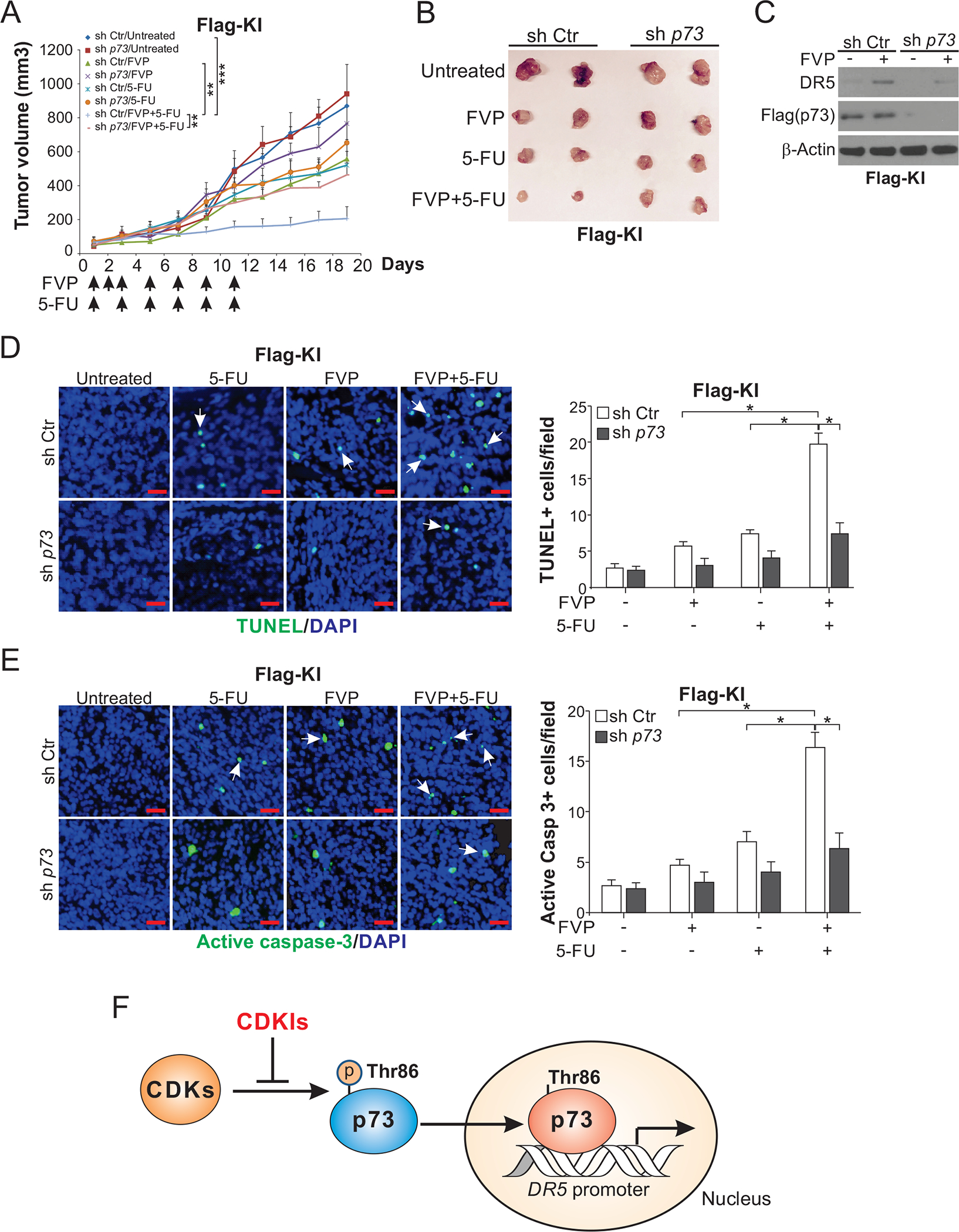
(A) Nude mice were injected s.c. with 4×106 sh p73 or sh Ctr Flag-KI HCT116 cells. After tumor growth for 7 days, mice were treated with FVP (i.p.; 5 mg/kg every day for 3 days and then every other day), 5-FU (i.p.; 25 mg/kg every other day), or their combination as indicated for 11 days. Tumor volume at indicated time points after treatment was calculated and plotted with p values for indicated comparisons (n=6 in each group; **, P < 0.01; ***, P < 0.001). (B) Representative tumors at the end of the experiment in (A). (C)-(E) Mice with sh p73 or sh Ctr Flag-KI HCT116 xenograft tumors were treated as in (A) for 4 consecutive days. (C) Western blotting of DR5 in randomly selected WT tumors. Apoptosis was analyzed by (D) TUNEL and (E) active caspase 3 staining of paraffin-embedded sections of sh Ctr and sh p73 tumor tissues. Left, representative staining pictures with arrows indicating example positive signals (scale bar: 25 μm); right, quantification of positive signals. (F) Schematic diagram of a model depicting p73-mediated DR5 induction by CDKIs. In (D) and (E), values were expressed as means ± SD of 3 independent experiments. *, P < 0.05.
Discussion
Our results from the current and previous studies demonstrate that both selective CDK4/6Is and less-selective CDKIs induce p73-dependent transcriptional activation of DR5. Similar to CDK4/6Is [17], less-selective CDKIs also reduce p-Thr86 of p73, and increase p73 nuclear translocation, binding to the DR5 promoter, and DR5 transcription in CRC cells (Fig. 7F). CDK 1, 2, 4, or 9 KD recapitulates suppression of p73 p-Thr86 and p73-mediated DR5 induction. DR5 is activated by CDKIs in a ligand-independent fashion through transcriptional upregulation. Furthermore, both DR5 and p73 are indispensable for the anticancer activity of less-selective CDKIs. Together, these results demonstrate that p73-mediated DR5 induction is a common and on-target effect of different CDKIs in potentiating therapy-induced apoptosis in CRC cells.
Unlike p53-dependent DR5 induction by DNA damage [11], DR5 induction by CDKIs is p53-independent while requiring the same p53 binding site in the promoter, consistent with the distinct functions of p73 and p53 in regulating common target genes but in response to different stress signals. p73 is rarely mutated in cancer, while its activity and expression are subjected to regulation by post-translational modifications. p73 subcellular localization and activity are regulated via p-Thr86 by different CDKs, including CDKs 1, 2, 4, 6, and 9, suggesting a key role of p73 in keeping the activities of different CDKs in check. In addition to DR5 induction, p73 p-Thr86 has a pro-proliferation function in the suppression of p21 induction by CDK1/2-cyclin A/B kinases [25]. p73 can also be phosphorylated at other sites such as Tyr99 and Ser289 by c-Abl, PKCδ, and other kinases, which regulate p73 stability and pro-apoptotic activity [21]. Deregulated cell cycle and CDK activity are a hallmark caused by p53 deficiency as well as other oncogenic events [3]. p73-dependent DR5 induction may therefore represent a tumor suppressive mechanism. The p53 family members and their roles in coordinating the response to CDK inhibition in different cancer types remain to be further elucidated.
Our data showed that CDKIs at a concentration as low as 0.1 μM were sufficient to robustly induce DR5 expression (Fig. 1A). This finding is consistent with previously described nanomolar inhibitory concentrations 50 (IC50s) of FVP, dinaciclib, and SNS-032 in CDK inhibition [1]. However, roscovitine was shown to have an IC50 of 0.2–0.8 μM on different CDKs [1], which are higher than the lowest concentration (0.1 μM) we used. This discrepancy could be explained by different assays used for analyzing CDK inhibition by roscovitine. Previous studies utilized in vitro, cell-free kinase assays to measure CDK inhibition by roscovitine [26, 27]. These biochemical assays analyzed CDK/cyclin complexes purified from normal cells of model systems and canonical CDK substrates such as Histone H1 [26, 27]. In contrast, our study analyzed the effects of roscovitine in cultured human cancer cells on CDKs that phosphorylate p73, a non-canonical CDK substrate. Kinase inhibitors often have different IC50s in cell-free biochemical assays and cell-based assays. Interestingly, knockdown of just one CDK was sufficient to cause p73 dephosphorylation despite the presence of other CDKs that can phosphorylates p73 (Fig. 3A). It is possible that multiple CDKs work in concert in one complex to promote p73 p-Thr86. Therefore, it may require less CDKI such as roscovitine to suppress p73 p-Thr86 due to its inhibitory effects on multiple CDKs, which may accumulatively promote p73 dephosphorylation and DR5 induction. It is also possible that overall CDK activity in cancer cells is tightly regulated due to oncogenic addiction. Depletion of one CDK may affect other CDKs by perturbing the balance between CDKs and CDK regulators such as cyclins and endogenous CDK inhibitors. For example, CDK1, 2, or 9 KD may affect the activities of CDK4 and CDK6, which can directly bind to and phosphorylate p73 [17].
Our results are consistent with the critical and versatile functions of CDKs in cancer. CDKs not only control cell cycle progression, but also regulate gene transcription, translation, DNA, RNA and energy metabolism, intracellular transport, protein degradation, signal transduction, and other cellular processes [28]. Accumulating evidence supports that cancer cells become addicted to non-canonical functions and targets of CDKs during tumor development [29, 30]. For example, both CDK1 and CDK9 phosphorylate RNA polymerase II, which promotes transcription elongation [28]. CDK9 has been explored as a therapeutic target to inhibit dysregulated transcription in cancer. CDK9 inhibition suppresses the expression of the antiapoptotic protein Mcl-1 to induce apoptosis in some cancer cells [31]. The role of non-cell cycle targets of CDKs and their functions in cancer cells are not well understood and remain to be further explored for developing CDK-targeted therapies.
At present, there are at least 40 CDKIs that are in various stages of research and development. Compared with FDA-approved selective CDK4/6Is, less-selective CDKIs have largely been unsuccessful in clinical studies. FVP was extensively tested in many clinical trials and only showed limited efficacy on some hematological malignancies, but not solid tumors. The general failure of less-selective CDKIs in clinical studies can be explained by several reasons, including insufficient understanding of the mechanism of action, lack of appropriate patient selection, toxicity, and poor therapeutic window [1]. Different combination strategies have been explored to improve the clinical applications of CDKIs, including less-selective CDKIs. Our in vitro and in vivo data suggest that p73-mediated DR5 induction may be a common mechanism and a useful biomarker for assessing potential therapeutic efficacy of CDKI combinations.
5-FU-based chemotherapy regimens, such as FOLFOX and FORFIRI, often have limited efficacy on CRC patients with advanced diseases [32, 33]. The activation of p73-DR5 axis might be particularly relevant in CRC. The enhanced DR5 induction in CRC cells by the combination of 5-FU and FVP or other CDKIs may be explained by simultaneous activation of DR5 transcription by p73, p53, and other transcription factors. Enhanced DR5 induction by 5-FU and CDKI combinations can lower the minimum level of apoptotic signal required for triggering cell death, underlying their synergism in CRC cells. In addition to 5-FU, CDKIs have been combined with other antitumor agents to improve therapeutic efficacy in other tumor types. For example, FVP combined with the proteasome inhibitor drug carfilzomib elicited a robust therapeutic response in adrenocortical carcinoma [34]. FVP synergized with the Bcl-2 inhibitor venetoclax to promote AML cell death [35]. It would be interesting to test if p73-mediated DR5 induction plays a role in the therapeutic efficacy of these agents by engaging both the extrinsic and intrinsic apoptotic pathways.
In conclusion, we demonstrate p73-mediated DR5 induction as an on-target and common mechanism of different CDKIs to potentiate apoptosis and the antitumor effects of chemotherapy in CRC cells in vitro and in vivo. These findings might help better understand the anticancer mechanisms of CDKIs and facilitate their further clinical development and applications.
Materials and methods
Cell culture and drug treatment
The human colorectal cancer cell lines, including HCT116, RKO, DLD1, and HT29, were purchased from the American Type Culture Collection. Lim2405 was obtained from Dr. Alberto Bardelli (University of Torino, Italy). Authentication of parental cell lines was performed by analyzing DNA mismatch repair status and p53, KRAS, BRAF, PIK3CA, FBW7, and other mutations as described [36]. Previously described isogenic HCT116 derivative cell lines included p53-knockout (KO), BAX-KO [37], FADD-KO, Bid-KO, DR5-KO, p73-KO, Caspase 8 knockdown (KD) by shRNA (sh Casp 8) [17], p73 KD by shRNA (sh p73), and Flag-p73 knock-in (Flag-KI) cells [18]. Authentication of isogenic derivative cell lines was done by sequencing targeted genomic regions and by analyzing protein expression. All cell lines were frequently examined for mycoplasma contamination by PCR. Cell lines within 15 passages from the original stocks were used.
Reagents used for cell culture, including chemicals, cell culture media, and supplements, are described in Table S1. All cell lines were cultured in McCoy’s 5A modified media (Invitrogen) containing 10% defined FBS (HyClone), 100 units/mL penicillin, and 100 μg/mL streptomycin (Invitrogen), and maintained at 37°C in 5% CO2. Cells plated in 12-well plates at 20–30% density were used for drug treatment. DMSO stocks of FVP, roscovitine, dinaciclib, SNS-032, 5-FU, and inhibitors of cell death pathways were prepared and further diluted to appropriate concentrations with cell culture media.
Western blotting
Western blotting was done using antibodies listed in Table S1 as described [38].
Real-time reverse transcriptase (RT) PCR
Isolation of total RNA from cells was performed using the Mini RNA Isolation II kit (Zymo Research) according to the manufacturer’s protocol. cDNA was generated from total RNA (1 μg) by using SuperScript III reverse transcriptase (Invitrogen). PCR was carried out using previously described conditions [39] and primers listed in Table S2.
Flow cytometry analysis of cell-surface DR5 and DR4
HCT116 cells were seeded at a density of 5×105 cells/well in a 12-well plate. After FVP treatment for 24 hours, cells were stained with anti-DR5 or anti-DR4 antibody for 2 hours, followed by detection with FITC-conjugated goat anti-rabbit IgG (1:1000) for 2 hours. Isotype-matched IgG antibodies were used as a control. Cell-surface DR5 and DR4 were measured by flow cytometry with CytoFlex (BD Biosciences).
Transfection and siRNA knockdown
Plasmid constructs and siRNAs are listed in Table S1. Lipofectamine 2000 (Invitrogen) was used to transfect constructs and siRNA according to the manufacturer’s instructions. Transfection of siRNA was done at 24 hours before drug treatment using 200 pmol of siRNA.
Analysis of luciferase reporter activity
DR5 luciferase reporter constructs were previously described [17]. To measure reporter activities, cells were transfected with WT or mutant DR5 reporters together with the transfection control β-galactosidase reporter pCMVβ (Promega). Collection of cell lysates, measurement of the luciferase activities, and normalization to pCMVβ were carried out as previously described [40]. Reporter experiments were performed in triplicates and repeated three times.
Chromatin immunoprecipitation (ChIP)
The binding of p73 to DR5 promoter was analyzed by ChIP using Flag-KI HCT116 cells with parental HCT116 cells as control. Chromatin Immunoprecipitation Assay Kit (EMD Millipore) was used for ChIP according to manufacturer’s instructions. Immunoprecipitation (IP) was done by using EZview Red Anti-Flag M2 Affinity Gel (Sigma), followed by PCR analysis of the DR5 promoter as previously described [40].
Analysis of p73 and DR5 nuclear translocation
Flag-KI HCT116 cells were treated with FVP or roscovitine and analyzed for p73 nuclear translocation by immunofluorescence and nuclear fractionation. For immunofluorescence, cells plated and treated in chamber slides were subject to primary staining with anti-Flag (Cell Signaling) at 4°C overnight, and then secondary staining with the anti-rabbit AlexaFluor 488-conjugated secondary antibody (Invitrogen) for 1 hour, as previously described [41]. Olympus IX71 microscope was used for image acquisition. For nuclear fractionation, nuclear extracts were isolated from cells plated and treated in 75-cm2 flasks using NE-PER Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher) according to the manufacturer’s instructions, followed by p73 Western blotting. Nuclear extracts isolated from FVP-treated HCT116 cells were analyzed for DR5 by Western blotting.
Combination treatment
Cells were seeded in 96-well plates at a concentration of 3.0×104 cells/mL in 100 μL of medium per well, and then treated with FVP alone or in combination with 5-FU for 48 hours. Cell viability was analyzed by crystal violet staining and 3-(4,5-dimethylthiazol-2-yl)-5-(3- carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (Promega) as described [22]. Chemiluminescence was measured on a Wallac Victor 1420 Multilabel Counter (Perkin Elmer, Waltham, MA). Calculation of combination index (CI) was done by using the CompuSyn program (ComboSyn Inc). Each measurement was conducted in triplicates and repeated three times.
Analysis of apoptosis, cytochrome c release, and Bax conformational change
Apoptosis was assayed by staining adherent and floating cells with Hoechst 33258 (Invitrogen) and enumerating cells containing condensed and fragmented nuclei [39]. A minimum of 300 cells were counted for each sample. Apoptosis was also analyzed by flow cytometry of cells stained with Annexin V/Propidium Iodide (PI) (Invitrogen) as described [40]. Colony formation assays were used to determine long-term cell survival as described [42]. After drug treatment, equal numbers of cells were plated into 6-well plates in drug-free medium at appropriate dilutions. After 14 days, colonies were detected by crystal violet staining. Each assay was done in triplicates and repeated three times.
For analysis of the cytosolic release of mitochondrial cytochrome c, cytoplasmic and mitochondrial fractions were prepared by using Mitochondrial Fractionation Kit (Active Motif) according to the manufacturer’s instructions. Western blotting was used to detect cytochrome c in both cytoplasmic and mitochondrial fractions.
To detect Bax conformational change, cells were collected and lysed in CHAPS buffer (150 mM NaCl, 10 mM Hepes, pH 7.4, and 1% CHAPS) containing a protease inhibitor cocktail, incubated on ice for 30 minutes, and then centrifuged at 15,000×g for 15 minutes to precipitate nuclei and cell debris. Cell lysates were incubated with 2 μg of anti-Bax 6A7 monoclonal antibody (Sigma) at 4°C for 1 hour, and then with 30 μL of EZview Red protein G affinity gel (Sigma) at 4°C overnight. Immunoprecipitants were collected by a brief spin, washed three times with 1 mL CHAPS buffer, solubilized with 1×Laemmli buffer, and subjected to Western blotting.
Xenograft tumor experiments
The University of Pittsburgh Institutional Animal Care and Use Committee approved all animal experiments. Sample size was estimated based on previous experience [38, 40]. All treated animals were analyzed and there was no randomization and blinding to the group allocation. Female 5–6-week-old Nu/Nu mice (Charles River) were housed in micro isolator cages and maintained in a sterile environment. Mice were allowed access to water and chow ad libitum. Establishment of xenograft tumors was done by subcutaneously injecting 4×106 WT, DR5-KO, sh control, or sh p73 HCT116 cells in both flanks. After tumor growth for 7 days, mice were treated with FVP (i.p.; 5 mg/kg every day for 3 days and then every other day), 5-FU (i.p.; 25 mg/kg every other day), or their combination for 11 days. Six mice were analyzed in each group. Tumor volumes were measured by calipers and calculated using the formula ½ × length × width2. Mice were sacrificed after tumors reached ~1.0 cm3 in size. Tumors were dissected and fixed in 10% formalin and embedded in paraffin. Five-μm paraffin-embedded tumor sections were used for immunostaining for terminal deoxynucleotidyl transferase-mediated dUTP Nick End Labeling (TUNEL; EMD Millipore) and active caspase 3, with signal detection by an AlexaFluor 488-conjugated secondary antibody (Invitrogen) and nuclear counterstaining by 4’ 6-Diamidino-2-phenylindole (DAPI).
Statistical Analysis
Prism 9 software (GraphPad) was used for statistical analysis. For the results of cell culture and immunostaining experiments, Student’s t-test was used to calculate P values. Means ± standard deviation (SD) are shown in the figures. For the results of animal treatment experiments, ANOVA with Fisher’s LSD post-hoc test was used to calculate P values for tumor volume analysis. Means ± standard error of the means (SEMs) are shown in the figures. Differences were considered to be significant if P < 0.05.
Supplementary Material
Acknowledgments
The authors thank our lab members for their discussion and critical reading. This work was supported by the U.S. National Institutes of Health grants (R01CA203028, R01CA217141, R01CA236271, R01CA247231, and R01CA248112 to L. Zhang; R01CA215481 and R01CA260900 to J. Yu; T32GM133332 to K. Ermine). This project used the UPMC Hillman Cancer Center Animal Facility, Cytometry Facility, and Tissue and Research Pathology Services, which are supported in part by P30CA047904.
Abbreviations:
- AML
acute myeloid leukemia
- CDK
cyclin-dependent kinase
- CDKI
CDK inhibitor
- CDK4/6I
CDK4/6 inhibitor
- CHOP
C/EBP homologous protein
- ChIP
chromatin immunoprecipitation
- CI
combination index
- COX IV
cytochrome oxidase subunit IV
- CRC
colorectal cancer
- DAPI
4’ 6-Diamidino-2-phenylindole
- DISC
death-inducing signaling complex
- DR5
death receptor 5
- FDA
Food and Drug Administration
- 5-FU
5-fluorouracil
- IC50
inhibitory concentration 50
- IP
immunoprecipitation
- KD
knockdown
- KO
knockout
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- Nec-1
necrostatin-1
- NSA
necrosulfonamide
- PI
Propidium Iodide
- p-Thr86
Thr86 phosphorylation
- RT
reverse transcriptase
- SD
standard deviation
- SEM
standard error of the mean
- shRNA
small hairpin RNA
- siRNA
small interfering RNA
- Thr
threonine
- TNF
tumor necrosis factor
- TRAIL
TNF-related apoptosis-inducing ligand
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP Nick End Labeling
- WT
wild-type
- z-VAD
z-VAD-fmk
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References:
- 1.Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 2015; 14: 130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009; 9: 153–166. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 4.Zeidner JF, Foster MC, Blackford AL, Litzow MR, Morris LE, Strickland SA et al. Final results of a randomized multicenter phase II study of alvocidib, cytarabine, and mitoxantrone versus cytarabine and daunorubicin (7 + 3) in newly diagnosed high-risk acute myeloid leukemia (AML). Leuk Res 2018; 72: 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du Q, Guo X, Wang M, Li Y, Sun X, Li Q. The application and prospect of CDK4/6 inhibitors in malignant solid tumors. J Hematol Oncol 2020; 13: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein ME, Kovatcheva M, Davis LE, Tap WD, Koff A. CDK4/6 Inhibitors: The Mechanism of Action May Not Be as Simple as Once Thought. Cancer Cell 2018; 34: 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J, Zhou L, Zhao S, Dicker DT, El-Deiry WS. The CDK4/6 inhibitor palbociclib synergizes with irinotecan to promote colorectal cancer cell death under hypoxia. Cell Cycle 2017; 16: 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jung C, Motwani M, Kortmansky J, Sirotnak FM, She Y, Gonen M et al. The cyclin-dependent kinase inhibitor flavopiridol potentiates gamma-irradiation-induced apoptosis in colon and gastric cancer cells. Clin Cancer Res 2003; 9: 6052–6061. [PubMed] [Google Scholar]
- 9.Yuan X, Gajan A, Chu Q, Xiong H, Wu K, Wu GS. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Rev 2018; 37: 733–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashkenazi A Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat Rev Drug Discov 2008; 7: 1001–1012. [DOI] [PubMed] [Google Scholar]
- 11.Wu GS, Burns TF, McDonald ER 3rd, Jiang W, Meng R, Krantz ID et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet 1997; 17: 141–143. [DOI] [PubMed] [Google Scholar]
- 12.Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem 2004; 279: 45495–45502. [DOI] [PubMed] [Google Scholar]
- 13.Bhola PD, Letai A. Mitochondria-Judges and Executioners of Cell Death Sentences. Mol Cell 2016; 61: 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov 2016; 6: 740–753. [DOI] [PubMed] [Google Scholar]
- 15.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin 2021; 71: 7–33. [DOI] [PubMed] [Google Scholar]
- 16.Chu E An update on the current and emerging targeted agents in metastatic colorectal cancer. Clin Colorectal Cancer 2012; 11: 1–13. [DOI] [PubMed] [Google Scholar]
- 17.Tong J, Tan X, Song X, Gao M, Risnik D, Hao S et al. CDK4/6 Inhibition Suppresses p73 Phosphorylation and Activates DR5 to Potentiate Chemotherapy and Immune Checkpoint Blockade. Cancer Res 2022; 82: 1340–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen D, Ming L, Zou F, Peng Y, Van Houten B, Yu J et al. TAp73 promotes cell survival upon genotoxic stress by inhibiting p53 activity. Oncotarget 2014; 5: 8107–8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoue T, Stuart J, Leno R, Maki CG. Nuclear import and export signals in control of the p53-related protein p73. J Biol Chem 2002; 277: 15053–15060. [DOI] [PubMed] [Google Scholar]
- 20.Fontemaggi G, Kela I, Amariglio N, Rechavi G, Krishnamurthy J, Strano S et al. Identification of direct p73 target genes combining DNA microarray and chromatin immunoprecipitation analyses. J Biol Chem 2002; 277: 43359–43368. [DOI] [PubMed] [Google Scholar]
- 21.Rosenbluth JM, Pietenpol JA. The jury is in: p73 is a tumor suppressor after all. Genes Dev 2008; 22: 2591–2595. [DOI] [PubMed] [Google Scholar]
- 22.Tong J, Tan X, Risnik D, Gao M, Song X, Ermine K et al. BET protein degradation triggers DR5-mediated immunogenic cell death to suppress colorectal cancer and potentiate immune checkpoint blockade. Oncogene 2021; 40: 6566–6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leibowitz B, Qiu W, Buchanan ME, Zou F, Vernon P, Moyer MP et al. BID mediates selective killing of APC-deficient cells in intestinal tumor suppression by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A 2014; 111: 16520–16525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen D, Yu J, Zhang L. Necroptosis: an alternative cell death program defending against cancer. Biochim Biophys Acta 2016; 1865: 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaiddon C, Lokshin M, Gross I, Levasseur D, Taya Y, Loeffler JP et al. Cyclin-dependent kinases phosphorylate p73 at threonine 86 in a cell cycle-dependent manner and negatively regulate p73. J Biol Chem 2003; 278: 27421–27431. [DOI] [PubMed] [Google Scholar]
- 26.De Azevedo WF, Leclerc S, Meijer L, Havlicek L, Strnad M, Kim SH. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur J Biochem 1997; 243: 518–526. [DOI] [PubMed] [Google Scholar]
- 27.Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N et al. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem 1997; 243: 527–536. [DOI] [PubMed] [Google Scholar]
- 28.Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer 2017; 17: 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hydbring P, Malumbres M, Sicinski P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat Rev Mol Cell Biol 2016; 17: 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frank DA. Cyclin-Dependent Kinase 4/6 Inhibitors: Is a Noncanonical Substrate the Key Target? Cancer Res 2022; 82: 1170–1171. [DOI] [PubMed] [Google Scholar]
- 31.Cidado J, Boiko S, Proia T, Ferguson D, Criscione SW, San Martin M et al. AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin Cancer Res 2020; 26: 922–934. [DOI] [PubMed] [Google Scholar]
- 32.Meyerhardt JA, Mayer RJ. Systemic therapy for colorectal cancer. N Engl J Med 2005; 352: 476–487. [DOI] [PubMed] [Google Scholar]
- 33.Chen ML, Fang CH, Liang LS, Dai LH, Wang XK. A meta-analysis of chemotherapy regimen fluorouracil/leucovorin/oxaliplatin compared with fluorouracil/leucovorin in treating advanced colorectal cancer. Surg Oncol 2010; 19: 38–45. [DOI] [PubMed] [Google Scholar]
- 34.Nilubol N, Boufraqech M, Zhang L, Gaskins K, Shen M, Zhang YQ et al. Synergistic combination of flavopiridol and carfilzomib targets commonly dysregulated pathways in adrenocortical carcinoma and has biomarkers of response. Oncotarget 2018; 9: 33030–33042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bogenberger J, Whatcott C, Hansen N, Delman D, Shi CX, Kim W et al. Combined venetoclax and alvocidib in acute myeloid leukemia. Oncotarget 2017; 8: 107206–107222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tong J, Tan S, Zou F, Yu J, Zhang L. FBW7 mutations mediate resistance of colorectal cancer to targeted therapies by blocking Mcl-1 degradation. Oncogene 2017; 36: 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science 2000; 290: 989–992. [DOI] [PubMed] [Google Scholar]
- 38.Tong J, Zheng X, Tan X, Fletcher R, Nikolovska-Coleska Z, Yu J et al. Mcl-1 Phosphorylation without Degradation Mediates Sensitivity to HDAC Inhibitors by Liberating BH3-Only Proteins. Cancer Res 2018; 78: 4704–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tong J, Wang P, Tan S, Chen D, Nikolovska-Coleska Z, Zou F et al. Mcl-1 Degradation Is Required for Targeted Therapeutics to Eradicate Colon Cancer Cells. Cancer Res 2017; 77: 2512–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan X, Tong J, Wang YJ, Fletcher R, Schoen RE, Yu J et al. BET inhibitors potentiate chemotherapy and killing of SPOP-mutant colon cancer cells via induction of DR5. Cancer Res 2019; 79: 1191–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yue W, Sun Q, Dacic S, Landreneau RJ, Siegfried JM, Yu J et al. Downregulation of Dkk3 activates beta-catenin/TCF-4 signaling in lung cancer. Carcinogenesis 2008; 29: 84–92. [DOI] [PubMed] [Google Scholar]
- 42.Dudgeon C, Wang P, Sun X, Peng R, Sun Q, Yu J et al. PUMA induction by FoxO3a mediates the anticancer activities of the broad-range kinase inhibitor UCN-01. Mol Cancer Ther 2010; 9: 2893–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.