

Summary
Large-scale genomic studies have identified within-host adaptation as a hallmark of bacterial infections. However, the impact of physiological, metabolic, and immunological differences between distinct niches on the pathoadaptation of opportunistic pathogens remains elusive. Here, we profile the within-host adaptation and evolutionary trajectories of 976 isolates representing 119 lineages of uropathogenic Escherichia coli (UPEC) sampled longitudinally from both the gastrointestinal and urinary tracts of 123 patients with urinary tract infections. We show that lineages persisting in both niches within a patient exhibit increased allelic diversity. Habitat-specific selection results in niche-specific adaptive mutations and genes putatively mediating fitness in either environment. Within-lineage inter-habitat genomic plasticity mediated by mobile genetic elements (MGEs) provides the opportunistic pathogens with a mechanism to adapt to the physiological conditions of either habitat and lower MGE richness is associated with recurrence in gut-adapted UPEC lineages. Collectively, our results establish niche-specific adaptation as a driver of UPEC within-host evolution.
Keywords: Uropathogenic Escherichia coli, pathoadaptation, niche adaptation, evolution, genomic plasticity, mobile genetic elements
eTOC blurb
Thänert & Choi et al. show that lineages of uropathogenic E. coli (UPEC) persisting after resolution of symptomatic urinary tract infections adapt to the gastrointestinal and urinary environments. During this, mobile genetic elements facilitate the establishment of habitat-specific gene pools, providing UPEC with a mechanism to adapt to distinct physiological conditions.
Graphical Abstract
Introduction
During infection or colonization, bacterial pathogens adapt to their host by optimizing their ability to replicate, disseminate, and evade host immunity (Marvig et al., 2015; Sheppard et al., 2018). Under strong selection, mutations arise continuously within persisting strains but rarely sweep to fixation, resulting in lasting intraspecies allelic diversity that provides a record of the pressures encountered (Lieberman et al., 2014; Lourenço et al., 2016). Parallel signatures in unrelated hosts can identify pathoadaptive mutations in persisting pathogens, revealing common drivers of within-host adaptation (Lieberman et al., 2011). While a wealth of microbial whole genome sequencing (WGS) data has identified common patterns of pathogen adaptation (pathoadaptation) (Didelot et al., 2016; Gatt and Margalit, 2021; Rossi et al., 2020), studies of within-host evolution have, with few exceptions (Lees et al., 2017; Young et al., 2017), been limited to specific niches in the human body, potentially overlooking population dynamics of opportunistic pathogens occupying multiple body habitats. Accordingly, there is a limited understanding of how physiological barriers between habitats may impact pathoadaptation.
One in four women affected by a UTI will experience a recurrence (rUTI) within 6 months of initial infection (Foxman, 2014). Uropathogenic Escherichia coli (UPEC) are the most common cause of UTIs, accounting for approximately 75% of uncomplicated cases (Flores-Mireles et al., 2015). The recovery of UPEC from the gastrointestinal tract at asymptomatic time points before rUTI supports a model in which UPEC lineages can persist intestinally and re-seed the urinary tract (Chen et al., 2013; Nielsen et al., 2016; Thänert et al., 2019). Emergence of uro-adaptive mutations of the type 1 fimbrial adhesin FimH in urinary isolates that are rarely present in intestinal isolates suggests rapid adaptation to habitat-specific conditions (Chattopadhyay et al., 2007; Schwartz et al., 2013; Sokurenko, 2004; Weissman et al., 2007). In some patients, however, the absence of UPEC in the intestine and the recovery of UPEC from urine at asymptomatic timepoints (asymptomatic bacteriuria) highlight that patient-specific patterns of persistence may differentially shape UPEC pathoadaptation (Thänert et al., 2019). It is unclear how the distinct physiological, metabolic, immunologic, and microbial conditions of the gastrointestinal and urinary tract impact UPEC within-host adaptation. Evolutionary trade-offs between habitats pose the question as to which molecular mechanisms enable UPEC lineages to persist, adapt, and cause repeated episodes of UTI (Bricio-Moreno et al., 2018).
Here, we investigate the hypothesis that habitat-specific selection in the gastrointestinal and urinary tracts differentially shapes UPEC within-host evolution. To assess this hypothesis, we characterize colonization patterns of persisting UPEC lineages in a longitudinal, prospective cohort of UTI patients. We contrast the adaptation of lineages colonizing the gastrointestinal tract with those also recovered from the urinary tracts to identify habitat-specific adaptations of UPEC. By characterizing within-lineage mutational diversity, we identify distinct patterns of within-host adaptation between UPEC colonization types indicating that niche-adaptation shapes UPEC within-host adaptation. Finally, we identify mobile genetic elements (MGEs) as a major facilitator of within-lineage genomic plasticity associated with a pool of habitat-specific genes, putatively mediating UPEC fitness in either habitat and impacting recurrence in gut-adapted UPEC lineages.
Results
UPEC lineages persist in the gastrointestinal and urinary tracts
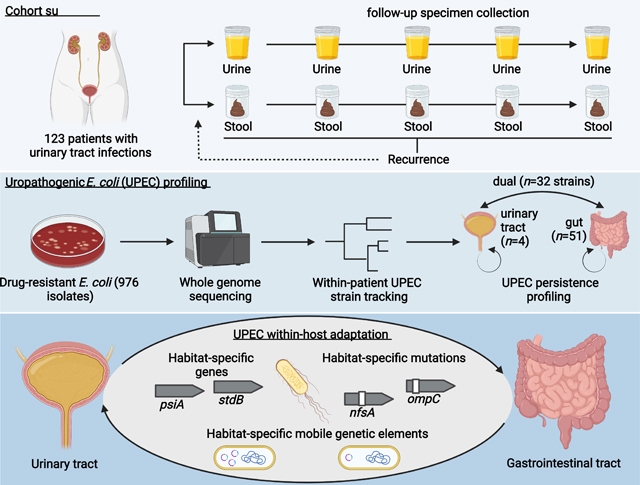
We collected 976 drug-resistant Escherichia coli isolates from a prospective, longitudinal cohort study of 123 patients presenting with symptomatic UTI caused by antibiotic resistant (AR) uropathogens. E. coli were cultured from 1,752 stool and urine specimens collected at study enrollment and subsequently at 10 asymptomatic time points over a 6-month follow-up period using a home shipment protocol (see Methods). Patients that experienced a rUTI within the follow-up period, were able to restart sample collection (42 patients, 34.15%).
To identify UPEC lineages persisting within patients, we characterized genomic relatedness of same-patient isolates using whole-genome sequencing (WGS) of all 976 E. coli isolates (average of 8.2 isolates/patient; Data S1). Following methodologies implemented in similar studies (Bronson et al., 2021; Coll et al., 2017), we profiled single nucleotide polymorphism (SNP) distances based on patient-specific core-genomes to differentiate isolates belonging to the same E. coli lineage as the causative agent of the index UTI from isolates representing distinct subspecies clusters. We observed that within-patient SNP distances followed a multimodal distribution (Fig S1A), with a notable paucity of within-patient pairwise isolate SNP distances between 500 and 10,000 SNPs. To assess plausibility of 500 SNPs as the upper limit of a UPEC lineage definition for this study, we estimated the average duration since last common ancestor (LCA) for each lineage. For each persistent lineage, we generated whole genome SNP trees based on lineage-specific reference assemblies and calculated the median branch length. We then divided this value by a previously reported estimated rate of E. coli base substitution (8.9 × 10−11 bp/generation) (Wielgoss et al., 2011). Importantly because our estimate is based on within-gut E. coli generation times, values for urinary persisters are likely less accurate. We estimated an average of ~0.33 (0–5.39, Fig S1B) years since the LCA, consistent with the reported history of recurrent UTIs in our patient cohort. Whole genome pairwise ANI values calculated between same-patient isolates further showed that isolates typed to the same lineage based on the 500 core genome SNPs cutoff exhibited high pairwise ANI values (99.991% (0.0127) - median (IQR)), while isolates from the same patient typed into distinct lineages and from distinct patients displayed lower, variable ANI values (97.288% (1.531), 97.268% (1.588), Fig S1C, D).
We applied the 500 core genome SNPs cutoff to all isolates cultured from the same patient and identified a total of 187 distinct subspecies clusters of E. coli (hereafter referred to as ‘lineages’ - Fig S1, Data S1). 702 isolates recovered at asymptomatic time points belonged to 119 lineages that were isolated as the causative agent of a UTI (diagnostic urinary isolate: DxU) and were defined as UPEC for the purpose of this study. The majority of these lineages belonged to the pandemic ExPEC sequence type complexes (STc) 131 (36.97%, Serotypes O25:H4 and O16:H5), predominately ST131-fimH30, and STc14 (21.85%, Serotype O75:H5, Data S1), predominately ST1193 (Table S1, Fig S2).
We characterized asymptomatic persistence of UPEC lineages based on longitudinal recovery of same-lineage E.coli from patient-matched urine and stool specimens, using standard-of-care clinical microbiology culturing methods (Fig 1A, Methods). We classified three distinct patterns of UPEC lineage persistence (see Methods): (1) gastrointestinal persistence (‘Gut colonizer’, 51 lineages, 46.4%), (2) persistence in both habitats (‘Dual colonizer’, 32 lineages, 29.1%), or (3) persistence in the urinary tract (‘Urinary colonizer’, 4 lineages, 3.6%, Fig 1A). Isolates belonging to these categories were used in downstream analysis to investigate UPEC within-host evolution. In 23 patients (20.9%) we did not find evidence for UPEC persistence in either the urinary or the gastrointestinal tract. While sequence type distribution did not differ between persistence types (Fig 1B), STs of non-persisting lineages differed significantly from that of persisters (Fig 1C, Fisher’s exact test P<0.001), with ST131 and ST1193 underrepresented among non-persisting lineages (Fisher’s exact test P<0.001). Interestingly, dual colonizers were associated with the majority of rUTI events attributable to a specific lineage during the 6-month follow-up period (57.9% (11/19 lineages), 36.8% (7/19) gut colonizer, 5.3% (1/19) urinary colonizer). Collectively, these observations suggest that colonization of the gut (Gut colonizer) or both environments (Dual colonizer) describe the majority of persistent UPEC.
Figure 1 |. Persistent UPEC lineages group into distinct colonization patterns.

(A) Schematic representation of UPEC colonization patterns (Left) as determined by recovery from stool (brown circles) and urine (yellow circles) from UTI patients with available DxU isolates. The definition for each colonization type is given below the schematic. UPEC lineages (n=119) are classified into four persistence types: gut colonizer, dual colonizer, urinary colonizer, and non colonizer. (Middle) UPEC lineage presence at follow-up sample collection points as determined by whole genome sequencing of isolates (Key: 1: enrollment; 2: 0–3 days post-antibiotic treatment (pAT); 3: 7–14 days pAT; 4: 30–60 days pAT; 5: 150–180 days pAT). Bars indicate the fraction of patient’s urine (yellow) and stool (brown) specimens positive for the disease causing UPEC lineage at each sampling point. Patients are grouped by UPEC lineage persistence type. Only data from the first episode caused by a UPEC lineage is shown. (Right) Number of UPEC lineages falling into each colonization category (gut colonizer=51, dual colonizer=32, urinary colonizer=4, and non colonizer=23). Boxes group together panels showing data of the same persistence type. (B) Sequence types (ST) are evenly distributed between UPEC persistence types. Prevalence of the two dominant STs, ST131 and ST1193, is color highlighted. (C) ST composition varies significantly between persisting and non-persisting lineages (n=110 lineages, Fisher’s exact test, P<0.001). ST131 (light purple) and ST1193 (dark purple) are significantly underrepresented in the set of non-persisting UPEC lineages (n=110 lineages, Fisher’s exact test, P<0.001). Prevalence of the two dominant STs, ST131 and ST1193, is color highlighted.
Urinary persistence is associated with increased allelic diversity of UPEC lineages
To assess the impact of environmental selection on UPEC within-host evolution, we profiled the within-host adaptation of UPEC lineages in their persistence habitats (i.e., gut colonizers in the gut, dual colonizers in gut and urinary tract, and urinary colonizers in the urinary tract). We identified all within-lineage SNPs by aligning sequenced reads against lineage-specific pseudo-assemblies, as previously described (Thänert et al., 2019; Zhao et al., 2019).
By inferring the ancestral sequence through maximum parsimony, we found that urinary persistence is associated with significantly increased distance to the most recent common ancestor (dMRCA) compared to gut colonizing lineages (Fig 2A, n=87 lineages, Kruskal-Wallis P=1.38e−05, Dunn post-hoc test gut vs dual colonizer P=2.39e−05, gut vs urinary colonizer P=3.32e−02). These observations are consistent with two potential explanations; First, urinary persistence may enable UPEC lineages to persist within a host for longer durations. Alternatively, considering that E. coli are native to the gut, disparate selective pressure in the urinary tract could result in habitat-specific fitness maxima distinct from those of the gastrointestinal tract and extend the spectrum of positively selected mutations, diversifying the allelic repertoire of persisting UPEC lineages.
Figure 2 |. Niche-specific adaptation shapes UPEC within-host adaptation.

(A) Boxplot of lineage dMRCA values (n=87 lineages, Kruskal-Wallis P=1.38e−05, Dunn post-hoc test gut vs dual colonizer P=2.39e−05, gut vs urinary colonizer P=3.32e−02). Outliers (outside 1.5x interquartile range) are depicted as points. Whiskers represent 1.5x interquartile range. Upper, middle, and lower box lines indicate 75th, 50th, and 25th percentiles, respectively. (B) Histogram of gene-wise dN/dS values with signatures of non-random mutation (Permutation test, P<0.05) mutated in parallel across more than two lineages (m≥2, top) or in one lineage (m=1, middle), and in genes non-significant in permutation test (bottom). Median and median absolute deviation (MAD) are given for both gene groups. Dashed vertical line indicates neutral selection at dN/dS=1. (C) Genes found to be mutated in parallel in ≥3 lineages, normalized by the total number of gene-carrying lineages. Hypothetical genes are not shown. Color of the bar corresponds to colonization type in which mutations were found (gut colonizer - blue, dual colonizer - maroon, urinary colonizer - light yellow). Color bar below the histogram provides GO category (as shown in Fig 2D) for all genes with GO terms annotation found to be significantly enriched in a colonization type. (D) Network visualization of GO terms significantly overrepresented in the pool of genes with non-random signature of selection within-lineages as defined by the permutation test. Bubble size represents number of mutations in genes categorized into each GO term. Color of bubbles corresponds to colonization type GO terms were enriched in (gut colonizer: blue; dual colonizer: maroon; urinary colonizer: light yellow; gut/dual colonizer: purple; gut/urinary colonizer: black). GO terms were clustered semantically into the 2D space using REVIGO. Circles group together semantically related GO terms.
UPEC niche-specific adaptation shapes within-host adaptation
To test the hypothesis that urinary persistence results in trajectories of within-host adaptation distinct from those observed in the gut, we annotated within-lineage allelic diversity (SNPs, insertions, deletions) at the gene level. We implemented permutation tests, randomly distributing the number of observed mutations over each lineage’s pseudo-assembly to generate a null distribution. We then compared observed against expected frequencies to identify genes with signatures of non-random evolution across lineages. Permutation tests were conducted independently for colonization types to characterize the effect of distinct persistence patterns.
Our analysis identified 253 genes with mutational signatures indicating non-random selection (n=87 lineages, Permutation test, confidence interval 95%). To validate that positive selection drives mutations in this gene set, we calculated per gene dN/dS ratios, a canonical metric for selection. We found a robust enrichment of elevated dN/dS values for both genes mutated in a single lineage (Fig 2B, m=1, median 11.57±11.41 median absolute deviation (MAD)) or in parallel across multiple lineages (m≥2, 11.52±10.78) compared to genes non-significant by permutation test (median 0.97±0.98). Consistent with this observation, the overall dN/dS value for all genes significant by permutation test and mutated in parallel across lineages, 1.34 (0.96–2.02, 95% confidence interval by binomial sampling), indicated that adaptation drives mutation in these genes. In contrast, genes carrying mutations but non-significant by permutation test were under purifying selection (dN/dS 0.32, 0.30–0.35), consistent with previous literature (Zhao et al., 2019).
Mutations of a single gene (wbbL) was observed in all colonization types, while 12 genes were shared between at least two groups (Data S2A, B, C). Virulence- and drug-associated genes were mutated in parallel frequently across colonization types (Fig 2C), including capsule-related genes neuC (dN/dS 7.3) and mprA (dN/dS 17.5), as well as wbbL (dN/dS 59.4), coding a rhamnosyl transferase critical for O-antigen synthesis. As both capsule and O-antigen directly affect UPEC fitness in vivo(Sarkar et al., 2014), these mutations may also affect UPEC persistence. Further, genes implicated in antibiotic resistance, including ompC (dN/dS 17.8), acrR (dN/dS 5.8), nfsA (dN/dS 17.8), and nfsB (dN/dS 10.9) (Choi and Lee, 2019; Osei Sekyere, 2018; Su et al., 2007), were found to be under positive selection across lineages. Interestingly, mutations of the biofilm suppressing antiterminator RfaH encoding gene (dN/dS 33.5) were exclusively found in lineages persisting within the urinary tract. Biofilms are critical UPEC colonization factors, enabling adhesion to abiotic (catheter) and biotic (urinary tract) surfaces (Beloin et al., 2006).
To assess functional adaptation of UPEC during persistence comprehensively, we performed Gene Ontology term overrepresentation analysis (GOOA) in the pool of all genes mutated within-lineages that exhibited a signature of non-random selection (Data S3A, B, C). Strikingly, functional categories under selection differed between colonization types, with only a small set of core-functions (sialic acid transport, membrane assembly, antibiotic resistance, negative regulation of transcription) found to be under selection in multiple colonization types (Fig 2D). Distinct transport capabilities, response to environmental stressors, metabolic processes, and regulatory functions were selected in gut-restricted and dual colonizers (Fig 2D), indicating that distinct persistence patterns differentially shape within-host adaptation of persisting UPEC lineages. Functions found to be under selection in dual colonizers, including iron ion transport, response to pH, response to nitric oxide, ornithine metabolism, or fumarate metabolism (Fig 2D), have been linked to urinary fitness of UPEC and likely direct adaptations towards the habitat-specific conditions of the urinary tract (Hibbing et al., 2020; Mann et al., 2017). Collectively, these results support the idea that niche-specific selection shapes the evolutionary trajectories of persisting UPEC, altering the landscape of positively selected functionalities for multi-habitat lineages.
Within-host adaptation of UPEC impacts resistance phenotypes
We observed that 79.4% of the within-lineage allelic diversity in genes mutated in parallel among dual colonizing lineages was structured by habitat, with mutations only occurring in a single habitat within a lineage (Fig 3A). Similarly, when including 71 additional urinary isolates from the 51 gut colonizing lineages and implementing our permutation test to identify genes under positive selection (Data S2D), we found that an even larger fraction of mutations in genes with parallel signature across lineages was only found in isolates cultured from one sample type (93.5%, Fisher’s exact test, P=0.001). As urinary colonizers had no representative gut isolates, they were not included in this analysis. We reasoned that this phenomenon could result from two potential processes: (1) a consequence of genetic bottlenecks upon habitat transition, or (2) habitat-specific selection resulting in divergent subpopulations within the same lineage in the gastrointestinal and urinary tract.
Figure 3 |. UPEC niche-specific adaptation impacts antibiotic resistance phenotypes.

(A) The majority of allelic diversity in genes found to be mutated in parallel within gut and dual colonizers is structured by habitat (Fisher’s exact test P=0.001). Color of the bar corresponds to either dual colonizer (maroon) or gut colonizers (blue). (B) (Top) Phylogeny of lineage WU-041_1 with annotated non-synonymous ompC mutation and corresponding phenotypic resistance to ampicillin/sulbactam. Black squares denote gene presence or antibiotic resistance. White squares indicate gene absence or drug susceptibility. Grey squares indicate intermediate drug susceptibility. Phylogeny is unrooted based on SNP distances. (Bottom) SNP locations on the ompC gene. The porin domain is annotated in grey. Circle size corresponds to number of isolates carrying that mutation. (C) Lineage WU-046_2 exhibited nonsynonymous barA and nfsA mutations in urinary isolates only, corresponding to phenotypic resistance to nitrofurantoin. Phylogeny is unrooted based on SNP distances. Labels as in (B).
To test whether niche-specific adaptation may in fact play a role in shaping allelic breakdown along habitat lines in persisting UPEC lineages, we focused on a subset of mutations with a tractable phenotypic impact. We had previously observed strong selection for mutations in antibiotic-resistance associated genes during persistence (Fig 2D) and reasoned that niche-specific adaptation would result in niche-dependent resistance phenotypes. Therefore, we identified mutations in antibiotic resistance genes and profiled isolate resistance phenotypes for both dual and gut colonizing lineages. We found that the nonsynonymous ompC R191C mutation in dual colonizing lineage WU-041_1 was exclusively found in urinary isolates and coincided with the gain of ampicillin/sulbactam (Fig 3B). Importantly, we found that non-synonymous mutations of ompC, including another instance of R191C in lineage PN-004_1, were restricted to urinary isolates. Similarly, we found nfsA Q191* mutation in gut colonizing lineage WU-046_2 exclusively in isolates cultured from urine specimens during symptomatic disease and immediately preceding recurrence (Fig 3C), associated with the gain of phenotypic nitrofurantoin resistance. Moreover, identified resistance-conferring mutations of nfsA, including another premature stop codon in lineage PN-004_1 (nfsA W237*), were restricted to urinary isolates. Together, these findings indicate niche-dependent fitness benefits of mutations in these two genes and a role of niche-specific adaptation in shaping within-host adaptation of persisting UPEC lineages.
We further reasoned that if these observed mutations provide UPEC with direct fitness benefits, they may also be found in UPEC genomes sequenced in different studies. To test this, we downloaded a set of 703 UPEC genomes previously curated from multiple studies(Biggel et al., 2020) and profiled allelic identify of ompC and nfsA at all positions observed to be variable in this study. We found that for ompC and nfsA in 2/4 cases and 1/4 cases, respectively, the exact mutations identified in our study were observed in published UPEC genomes (Fig S3). This suggests that similar selective pressures to the ones characterized in this study are shaping adaptation of ompC and nfsA in the larger UPEC population.
Genomic plasticity facilitates UPEC niche adaptation
Differential abundance of genes within an otherwise clonal population, termed genomic plasticity, can facilitate rapid adaptation of bacterial pathogens to new environments (Darch et al., 2015; Gabrielaite et al., 2020; Hammond et al., 2020). The distinct physiological conditions of the gastrointestinal and urinary tracts are likely to require disparate metabolic and colonization factors. We therefore hypothesized that genomic plasticity may enable persisting UPEC lineages to maintain fitness in both the gastrointestinal and urinary environment.
Persisting gut populations of gut colonizers exhibited more homogenous gene profiles than dual colonizers (Fig 4A, n=87 lineages, Kruskal-Wallis test P=0.009, Dunn post-hoc test P=0.012), indicating that habitat diversification is associated with a larger pool of flexible genes. We hypothesized that this difference may be caused by greater inter-habitat heterogeneity in persisting dual colonizers not observed in lineages persisting in the gut. To test this hypothesis, we analyzed inter-habitat similarity of same-lineage isolate gene profiles, including all 71 urinary isolates from the 51 gut colonizing lineages. We found that isolates collected from the same sample type were significantly more likely to carry similar genes, while colonization types did not differ significantly (Fig 4B, n=87 lineages, Two-way ANOVA, habitat P=5.94e−4, colonization type P>0.05), suggesting that genomic plasticity contributes to niche adaptation of all persisting UPEC lineages.
Figure 4 |. Persisting UPEC lineages exhibit niche-specific genomic plasticity.

(A) Boxplot of average within-lineage Jaccard distances based on gene presence/absence data (n=87 lineages, Kruskal-Wallis test P=0.009, Dunn post-hoc test gut vs dual colonizer P=0.012). Outliers (outside 1.5x interquartile range) are depicted as points. Whiskers represent 1.5x interquartile range. Upper, middle, and lower box lines indicate 75th, 50th, and 25th percentiles, respectively. (B) Average between- and within-habitat lineage Jaccard distances based on gene presence/absence data of same-lineage isolates by colonization type (n=87 lineages, Two-way ANOVA, habitat P=5.94e−4, colonization type P>0.05). Outliers (outside 1.5x interquartile range) are depicted as points. Whiskers represent 1.5x interquartile range. Upper, middle, and lower box lines indicate 75th, 50th, and 25th percentiles, respectively. Colors correspond to within-lineage comparison (between habitats: grey; within gut: brown; within urinary tract: yellow). (C) (Top) Two-sided histogram of within-lineage habitat-specific genes of dual (maroon) and gut (blue) colonizers. Urinary-specific genes are shown towards the left. Gut-specific genes are shown towards the right. (Bottom) Genes most frequently found to be urine (left) or gut (right) specific across lineages, normalized by the total number of gene-carrying lineages. Bar color corresponds to the colonization type a gene was found in as habitat specific. Hypothetical genes are not shown. (D) Overrepresented GO terms associated with urine specific genes of dual (top - maroon) or gut colonizers (bottom - blue). Bubble size corresponds to the number of habitat-specific genes in each GO term. (E) Overrepresented GO terms associated with stool specific genes, using the same formatting as in (D).
1,553 genes were restricted to either urinary or stool isolates in the 83 UPEC gut and dual colonizing lineages and therefore may play a role in habitat adaptation (Fig 4C, Data S4). Interestingly, three plasmid-associated genes, psiA, yggR, and stbB, were found to be restricted to gut isolates in 5 independent lineages. To comprehensively profile functional selection on the variable genetic portion of each lineage in either habitat we performed GOOA on the pool of habitat-specific genes. We identified nitrogen compound and iron uptake mechanisms as key factors for urinary adaptation in both dual and gut colonizing lineages (Fig 4D, Fig S4A, Fisher’s exact test GO:0071705 P=0.018 - dual - and P=0.002 - gut, GO:0055072 P=1.81e−4 and P=2.51e−7, GO:0044718 P=0.024 and P=0.018, Data S3D, E). Specifically, systems facilitating the uptake of ferric-citrate complexes that are abundant in urine were found to be habitat-associated in gut as well as dual colonizers (Fig 4D) (Robinson et al., 2018).
Few functionalities were overrepresented in stool isolates of dual colonizing lineages (Fig 4E). Conversely, the gut-specific gene pool of gut colonizers exhibited enrichment of multiple functionalities implicated in E. coli gut colonization and virulence, including antibiotic resistance, fumarate transport, type IV secretion, and pilus assembly (Elhenawy et al., 2021; Jones et al., 2011; Spaulding et al., 2017). Notably, GO terms associated with plasmid maintenance genes were found to be enriched in intestinal isolates of gut colonizing lineages, commonly coinciding with presence/absence of virulence and resistance genes (Fig S4A, B, C, D, Fisher’s exact test GO:0030541 P=0.044, GO:0006276 P=1.77e−3, Data S3F, G). We therefore hypothesized that MGEs may facilitate niche adaptation in persisting UPEC lineages.
Heterogenous MGE carriage facilitates habitat-associated genomic plasticity
To evaluate the role of MGEs in the genomic plasticity of persisting UPEC lineages, we comprehensively identified regions of differential coverage in isolates of the same lineage as previously described(Zhao et al., 2019). These regions are candidate MGEs differentially abundant in isolates of the same lineage. We annotated the list of putative MGEs (Fig 5A, Data S5A), combining in silico detection of plasmidic contigs and database-driven annotation of de novo identified MGEs as previously described (see Methods, Fig S5)(Durrant et al., 2020; Thänert et al., 2019). 57.1% (887/1553 genes) of the habitat-specific gene pool mapped back to putative MGEs. As expected, we found antibiotic resistance genes (ARGs), proteolysis, and conjugation mechanisms associated with plasmidic MGEs (Fig 5B). Pathofunctions that were implicated as habitat-specific in our previous analysis, including iron import systems, type II and type IV secretion systems, and cell adhesion genes, were found to be enriched within MGE subcategories.
Figure 5 |. Mobile genetic elements drive niche-specific genomic plasticity of UPEC.

(A) Visualization of within-lineage MGEs. Element length (log-scale) is plotted against element count. IS, insertion sequence; CDS, coding sequence. (B) GO terms overrepresented in selected MGE subclasses. (C) Box plot of average within-lineage Jaccard distance based on MGE presence/absence data of same-lineage isolates between habitats (grey), within gut (brown), and within urine (yellow) grouped by colonization type. All comparisons are statistically significant (n=87 lineages, Two-way ANOVA P≤1.57e−05, Tukey post-hoc gut colonizer within-gut vs between habitats P<0.001, gut colonizer between habitat vs dual colonizer between habitat P=0.014). (D) MGE richness is larger in gut compared to urine isolates (n=87 lineages, Two-way ANOVA P=0.042). Outliers (outside 1.5x interquartile range) are depicted as points. Whiskers represent 1.5x interquartile range. Upper, middle, and lower box lines indicate 75th, 50th, and 25th percentiles, respectively. (E) Unrooted phylogeny of lineage PN-040_1 based on SNP distances annotated with selected habitat-specific genes. Relative short-read coverage over selected, habitat-specific MGEs harboring depicted genes is shown. (F) Unrooted phylogeny of lineage PN-004_1 based on SNP distances annotated with selected habitat-specific genes. Relative short-read coverage over selected, habitat-specific MGEs harboring depicted genes is shown.
To profile potential sharing of UPEC MGEs with other species we mapped all MGE contigs to the NCBI nucleotide database. We found that plasmidic MGEs had the broadest putative host range (Fig S6A). However, plasmidic MGEs exclusively identified in urinary isolates exhibited a trend towards a narrower host range compared to those found in the gut (Fig S6A, ANOVA P=0.053, Tukey post-hoc test vs gut-exclusive P=0.053, vs dual-habitat P=0.057). Moreover, these MGEs were significantly less likely to be mapped to common gut residents, including Salmonella enterica, Citrobacter freundii, or Enterobacter cloacae (Fig S6B, Fisher’s exact test, FDR corrected P<0.05), indicating that gut-associated plasmidic MGEs are more likely be shared with other gut residents.
Contrary to the high intra-habitat dissimilarity of lineage MGE profiles in urinary colonizers (Fig 5C), we observed homogenous within-habitat MGE carriage in dual and gut colonizing lineages. In gut colonizing lineages, heterogeneity of MGE carriage was significantly elevated across habitats compared to within-habitat, as well as significantly larger compared to dual colonizers (Fig 5C, n=87 lineages, Two-way ANOVA P≤1.57e−05, Tukey post-hoc P<0.001 and P=0.014, respectively, Data S5B). These results suggest that multi-habitat selection in dual colonizers may stabilize the MGE pool across habitat boundaries. Urinary isolates’ MGE pools were significantly smaller compared to intestinal isolates (Fig 5D, n=87 lineages, Two-way ANOVA P=0.042). Moreover, we found that habitat-specific genes from metabolic, antibiotic resistance, and virulence-associated functional categories were mapped to MGEs exclusively present in urinary or stool isolates (Fig 5E, F). These observations suggest that mobilization of key functions associated with adaptation to either habitat, such as iron acquisition or nitrogen compound uptake in the urinary tract (Fig 4D), may play a key role in UPEC niche adaptation.
Interestingly, the association of MGEs with ARGs resulted in a pool of ‘hidden’ ARGs not observed in the DxU isolate but present in other isolates of the same lineage (Fig S7). Isolates harboring ‘hidden’ ARGs frequently showed concordant variation in their replicon profile compared to the DxU isolate (66/78 cases, 84.6%), corroborating differential resistance plasmid carriage as a potential driver of within-lineage plasticity of ARGs.
Decreased MGE richness is associated with rUTI in gut-colonizing UPEC lineages
Based on our observation of decreased urinary richness of MGEs, we hypothesized that MGE richness may hamper urinary fitness of gut-adapted lineages of UPEC resulting in an inverse relationship between MGE richness and the likelihood of a lineage causing a rUTI during our follow-up period. In fact, we found that gut colonizer lineages causing rUTI exhibited significantly lower average MGE richness per isolate compared to their non-rUTI counterparts (Fig 6A, n=43 lineages, Welch’s t-test, FDR corrected P=0.001). Notably, no such relationship was observed for dual colonizers (n=26 lineages, Welch’s t-test, FDR corrected P=0.884).
Figure 6 |. Gut colonizing UPEC lineages causing rUTI exhibit decreased MGE richness.

(A) MGE richness of lineages causing rUTI during the follow-up period and non-rUTI lineages parsed by colonization type (n=73 lineages, Welch’s t-test, FDR corrected gut colonizer P=0.001, dual and urinary colonizer FDR corrected P>0.05). Outliers (outside 1.5x interquartile range) are depicted as points. Whiskers represent 1.5x interquartile range. Upper, middle, and lower box lines indicate 75th, 50th, and 25th percentiles, respectively. (B) (Left) Pseudo enrichment score of GO terms in the pool of MGEs absent or stable in urinary isolates of gut colonizing UPEC lineages. Top 19 GO categories by P-value are visualized. Pink bars indicate gene associated GO terms overrepresented in the urine instable MGE pool, black bars indicate GO terms enriched in the pool of MGEs stable in urinary isolates. Pseudo enrichment score was calculated by adding one count to all GO categories. (Middle) P-values for each GO category determined from overrepresentation analysis using hypergeometric distribution. (Right) Proportion of each visualized GO term in the MGE associated gene pool of rUTI and non-rUTI causing lineages of gut colonizing UPEC. Grey tiles indicate absence of a GO term in the MGE gene pool.
Despite considerable variability in the functional composition of their mobilized gene pool, no functional category was significantly enriched after correcting for multiple hypothesis testing in either rUTI or non-rUTI lineages (Fig S8A, n=69 lineages, Fisher’s exact test, all FDR corrected P>0.05). However, we observed a trend towards lower mobilized ARG richness in rUTI lineages compared to non-rUTI lineages (Fig S8B, C, n=69 lineages, Wilcoxon rank-sum test P=0.055). We found no difference between the mobilized ARG richness of UPEC persistence types (Fig S8D, E, n=87 lineages, Kruskal-Wallis P=0.231).
To identify mobilized functions negatively impacting urinary fitness of gut-adapted UPEC lineages, we characterized the habitat association of each putative MGE for all gut colonizer lineages. We identified a large gut-specific MGE pool (238/457, 52.08%) absent from any urinary isolate. GOOA of genes present on these gut-specific MGEs identified 9 out of 94 GO categories significantly depleted in urinary isolates (Fig 6B, Fisher’s exact test, FDR-corrected P-value<0.05, Data S3H), including DNA-related, lipid biosynthetic, and type-IV secretion system processes. Interestingly, while some gut-specific GO categories were absent from the MGE pool of rUTI-causing gut colonizers (e.g., antibiotic biosynthesis, tryptophan biosynthesis), these GO terms were in general not underrepresented in their MGE pool (Fig 6B).
Discussion
Invasion and colonization of the urinary from the gastrointestinal tract is the first step in the infectious cascade of the majority of UTIs caused by UPEC (Kaper et al., 2004). While the affordable implementation of WGS in longitudinal cohort studies has uncovered adaptive patterns of various species to specific host environments(Didelot et al., 2016; Gatt and Margalit, 2021), the within-host pathoadaptation of multi-habitat pathogens remains understudied. Here, we characterize the pathoadaptation of UPEC, one of the most common bacterial pathogens recovered from multiple body sites. Viewing UPEC within-host evolution in the context of their respective niche is key to understanding the origins of urovirulence in inherently intestinal E. coli, particularly in light of the lack of a defining genomic signature of UPEC (Schreiber et al., 2017).
Our results support three distinct models of UPEC persistence: exclusive persistence in the gastrointestinal tract (gut colonizer), persistence in both the gastrointestinal and urinary tracts (dual colonizer), and exclusive persistence in the urinary tract (urine colonizer). We find that these distinct patterns of persistence differentially shape UPEC within-host pathoadapation. While development of antibiotic resistance is strongly selected for in all persisting UPEC lineages, as previously reported for other pathogens (Fajardo-Lubián et al., 2019; Khademi et al., 2019; Rossi et al., 2020), we find that distinct functions are under selection in gut and dual colonizers. Specifically, signatures of positive selection in distinct transport functions indicate that niche specific adaptation directly impacts evolutionary trajectories of pathoadaptive traits(Tang and Saier, 2014). Further adaptation to multiple habitats diversifies allelic profiles of persisting UPEC lineages. Intriguingly, potential inter-habitat transfer resulting in the influx of uroadaptive mutations back into gut populations may consequentially lower the fitness boundaries for urinary re-colonization by intrinsically gut-adapted E. coli. Experimental evidence has shown that virulence factors critical for uro-colonization are similarly beneficial in the intestinal reservoir (Chen et al., 2013; Russell et al., 2018; Spaulding et al., 2017), mitigating theoretical evolutionary trade-offs. These observations suggest that urovirulence may be a direct consequence of the generalist properties of the E. coli virulence repertoire (Brown et al., 2012), which is, as we show, fine-tuned by habitat-specific adaptations in the urinary tract.
Our observations support the hypothesis that persistent pathogen colonization requires within-lineage genotypic heterogeneity originating from both in situ adaptation as well as genomic plasticity (Hammond et al., 2020). The prevalence of habitat-restricted mutations and genomic plasticity between urine and stool isolates provides strong evidence that niche-specific adaptation dictates within-host evolution during UPEC persistence. We find that habitat-specific genes are associated with functions that increase E. coli fitness in the intestinal or urinary habitat, such as piliation, iron acquisition, nitrogen import, or anaerobic respiration (Elhenawy et al., 2021; Jones et al., 2011; Robinson et al., 2018; Spaulding et al., 2017). Persisting pathogen lineages require mechanisms that facilitate rapid rearrangements of large genomic regions to adapt to the distinct selective regimes of each habitat. Requirements for rapid genomic plasticity have been described for other pathogens, specifically during early stages of habitat colonization (Gabrielaite et al., 2020; Rau et al., 2012). Our results support the hypothesis that those genomic rearrangements are in part facilitated by MGEs (Sokurenko et al., 2006). Intriguingly, we observed that functions related to DNA repair were depleted in the MGE gene pool of urinary isolates from gut-adapted UPEC. This observation is consistent with the concept that stress-induced mutagenesis enables maladapted bacteria to evolve rapidly to their environment and may therefore be beneficial following urinary inoculation with gut-adapted lineage of UPEC (Shee et al., 2011). Heterogenous MGE carriage provides opportunistic pathogens with a unique mechanism to maintain fitness in multiple habitats. In vitro experiments have shown that complex environments result in discontinuous plasmid distribution in clonal populations, potentially resulting in fitness benefits in changing environments (Rodríguez-Beltrán et al., 2021; Slater et al., 2008, 2010). Our results support the hypothesis that MGE-mediated plasticity in bacterial populations is a key mechanism for habitat adaptation and may directly impact bacterial fitness upon habitat transition. Our data further suggest that a pool of gut-specific MGEs shared with other gut resident species may be lost in the urinary environment. Moreover, we find that gut colonizing lineages causing rUTI during our follow-up period have significantly lower MGE richness compared to their non-rUTI counterparts, suggesting an inverse relationship between MGE richness and likelihood of rUTI in gut-adapted lineages of UPEC. Consistent with predictions from in vitro work (Harrison et al., 2018), the absence of a similar trend in dual colonizers suggests that multi-habitat colonization stabilizes plasmid carriage under spatially heterogenous selection, potentially via mechanisms like compensatory mutations (Hall et al., 2021; Harrison et al., 2015).
However, important questions remain to be investigated. This study could not address the topic of directionality and inter-habitat transfer, the frequency of which may impact adaptative trajectories of persisting UPEC lineages. Moreover, given the apparent importance of genomic plasticity for UPEC fitness, localization of functions on either the chromosome or MGEs may determine the uropathogenic potential of intestinal E. coli lineages. The mosaic structure of plasmids poses the question which functions determine plasmid spread, evolution and persistence in UPEC lineages. While our study represents one of the largest genomic databases of UPEC to date, a number of patients were lost due to drop-out limiting the number of available isolates from follow-up episodes, specifically diagnostic isolates from outpatient settings. Similarly, our study lacked a representative number of lineages persisting exclusively in the urinary tract, that are potentially uniquely adapted to the urinary environment. Large multi-episode sampling efforts from patients at risk for rUTI are required to support rarity of this persistence type and the novel genomic predictions of our study.
This study, harnessing an expansive, longitudinal patient cohort sampled at multiple habitats, provides a framework for future investigations, studying the role of both in vivo mutations and genomic plasticity in the within-host adaptation of bacterial pathogens across niches. Similar investigations in other species may reveal further mechanisms of colonization and aid targeted decolonization of persisting human pathogens.
STAR Methods
RESOURCE AVAILABLITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Gautam Dantas (dantas@wustl.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Raw sequencing data has been deposited at the NCBI SRA database and are publicly available as of the date of publication. Accession numbers are listed in the Key Resource table. Relevant raw data and metadata can be found as supplementary data spreadsheets.
Key resources table
This paper does not report original code. We use well-established computational and statistical analysis software and packages. These are fully referenced in the Method section and Key Resource table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient cohort
Subjects for this prospective, multi-center cohort study were recruited from patients with positive clinically indicated urine cultures at Barnes-Jewish Hospital/Washington University in St. Louis (WU), St. Louis, Missouri, Duke University Hospital (DK), Durham, North Carolina, the Hospital of the University of Pennsylvania (PN), Philadelphia, Pennsylvania and Rush University Medical Center (RH), Chicago, Illinois. This study was approved by the Washington University Human Research Protection Office as the single IRB; local IRB approval was obtained as necessary. Patients with a symptomatic UTI diagnosed and treated by a physician and a urine culture that yielded E. coli with one of the following resistances were included in the current analysis: (1) resistance to ciprofloxacin or levofloxacin, (2) resistance to any third generation cephalosporin, (3) resistance to ertapenem and susceptible to meropenem, imipenem, and/or doripenem, (4) resistance to >2 of the following antimicrobial classes: carbapenems, aminoglycosides, fluoroquinolones, fourth generation cephalosporins, piperacillin/tazobactam, or (5) identification of any of the following resistance mechanisms: ESBL, CRE, KPC, NDM-1, OXA-48, IMP, IMP-1, or VIM.
Patients were excluded if they were younger than 18 years, if more than one organism was detected by the clinical laboratory at or above the clinical significance threshold, had any chronic indwelling urinary device, or any medical or surgical condition leading to intestinal or urinary system disease or anatomic alteration. Written, informed consent was obtained from all patients. Patients age averaged 56.26 years (range: 18–94, median: 59). 93.5% of patients were female, and 6.50% of patients male. 58.54% of patients self-reported their race as White, and 37.40% as Black. 4.07% of patients reported their ethnicity as Hispanic. Pearson’s chi-square tests indicated no significant association of age, gender, or race with UTI recurrence or UPEC colonization.
123 of 127 enrolled patients had at least one biological specimen yielding E. coli and were included in the current study. This total includes data from 12 patients enrolled at WU reported in a pilot study(Thänert et al., 2019). In total, 41 patients were enrolled at WU, 22 at DK, 12 at RH and 48 at PN.
METHOD DETAILS
Sample collection and processing
Enrolled subjects submitted stool and urine specimens to the study team at eleven sampling points over a 6-month follow-up period; enrollment (sampling point 01); the end of UTI antimicrobial treatment (02); days 3 (03), 7 (04), 14 (05), 30 (06), 60 (07), 90 (08), 120 (09), 150 (10), and 180 (11) post-treatment. If patients experienced rUTI during the 6-month follow-up period, they were invited to continue to participate with a new follow-up period. Visual schematic of the study design was created with BioRender.com. Samples were kept on ice immediately after production and during transport by courier. Upon arrival to the lab, samples were immediately cultured or prepared for long-term storage and frozen at −80 °C.
Stool and urine samples collected at sampling points 01, 02, 04, 06, and 11 were selectively cultured to assess asymptomatic uropathogen persistence. For stool culturing, ~1 g of stool sample was supplemented with an equal amount of PBS (w/v) and vortexed to homogenize the samples. Ten, 10-fold serial dilutions of the homogenate were prepared in PBS and 10μl of the first 10 dilutions were streaked on selective agar using a 10 μL calibrated loop. For urine culture, urines were directly plated onto selective agar using a 10 μL calibrated loop using a cross-streak pattern. After 20–30 hours of incubation, agar plates were examined for growth of the putative pathogen.
Selective agars were selected to be specific to each patient’s identified UPEC. MacConkey agar (MAC) supplemented with ciprofloxacin was used for ciprofloxacin-resistant E. coli, while ESBL E. coli was cultured on Hardy Diagnostic’s ESBL agar and MAC agar supplemented with cefotaxime. A single, representative colony of each distinct colony morphology present on a given culture plate was selected for further processing and sequenced-based analysis. The identity of the cultured pathogens was confirmed using MALDI-TOF MS (VITEK MS, bioMérieux, Durham, NC, USA). Single colonies were diluted in TSB/glycerol and stored at −80°C for later sequencing-based and phenotypic analysis. If patients were unable to submit a specimen at a predetermined sampling point samples collected at the next closest available time point were selected for analysis. Additionally, pre-recurrence specimens of rUTI patients and time-matched samples from non-rUTI were further processed. Non-rUTI patients were matched to rUTI patients based on (1) colonization status (defined below) and (2) treatment antibiotic during the first episode.
Antimicrobial susceptibility testing
Antimicrobial susceptibility testing of pathogens was performed on Mueller Hinton agar (Hardy Diagnostics, Santa Maria, CA, USA) using Kirby Bauer disk diffusion with antibiotic disks purchased from Hardy Diagnostics (Santa Maria, CA, USA) and Becton Dickinson (Franklin Lakes, NJ, USA). Results were interpreted according to consensus-based medical laboratory standards as provided in the Clinical and Laboratory Standards Institute (CLSI) guidelines for antimicrobial susceptibility testing (Melvin P. Weinstein, 2018), which provide species-specific breakpoint definitions for determining susceptibility or resistance.
DNA extraction, short-read sequencing, and quality filtering
Isolates were streaked onto blood agar (Hardy Diagnostics, Santa Maria, CA, USA) and incubated at 35°C overnight. Genomic DNA was extracted using the QIAamp Bacteremia DNA kit (Qiagen, Germantown, MD, USA). Sequencing libraries from both isolate gDNA and fecal metagenomic DNA were prepared using the Nextera kit (Illumina, San Diego, CA, USA) (Baym et al., 2015). Libraries were pooled and sequenced (2 ×150 bp) to a depth of ~2.5 million reads on the NextSeq 500 HighOutput platform (Illumina, San Diego, CA, USA). The resulting reads were trimmed of adapters using Trimmomatic v.36 (parameters: LEADING:10 TRAILING:10 SLIDINGWINDOW:4:15 MINLEN:60)(Bolger et al., 2014).
QUANTIFICATION AND STATISTICAL ANALYSIS
Isolate genome assembly and annotation
Draft genomes were assembled using SPAdes v.3.11.0 (parameters: -k 21,33,55,77 - careful)(Bankevich et al., 2012). The resulting scaffolds.fasta files were used for analysis. The quality of draft genomes was assessed by calculating assembly statistics using QUAST v5.0.2 and checkM v.1.0.13 (Gurevich et al., 2013; Parks et al., 2015). High-quality assemblies (<300 contigs, >90% of genome in contigs >1000bp, completeness >90%, contamination <5%) were annotated for open reading frames with Prokka v.1.12 (default parameters, contigs > 500 bp) (Seemann, 2014). Twenty-four publicly available E. coli genomes of known phylogroup were downloaded from NCBI to use as reference and annotated as described above (Data S6A). These genomes were used to assign phylogroups to the isolates sequenced in this study based on core-genome relatedness to the set of references. ARGs were annotated in silico using RGI-CARD v.5.1.0 (95% identity, 100% coverage) and Resfinder v.4.0 (95% identity, 100% coverage) (Jia et al., 2017; Zankari et al., 2012).
Phylogenetic analysis and lineage definition
MLST were annotated in silico using mlst v2.11 (default parameter) and serotypes were assigned using serotypefinder v2.0.1 (parameters: -mp blast −l 0.8 -t 0.90) (Joensen et al., 2015; Larsen et al., 2012). Core-genome alignments were generated using Roary v3.8.0 (default parameters, -cd 100)(Page et al., 2015). For sequence type-specific phylogenetic analysis core-genomes were constructed using all isolates typed to ST 131 or 1193, respectively (Figure S2). To define lineages, all E. coli isolates from the same patient were used for core-genome construction. Patient-specific core-genome sizes are provided in Data S7A. Newick trees of the core genome phylogenies were generated using FastTree v.2.1.10 (parameters: -gtr -nt) and visualized using iTOL v.4 (Letunic and Bork, 2019; Price et al., 2009).
To define E. coli lineages, patient-specific pairwise core-genome SNP distances were determined from the patient-specific Roary core-genome alignments via snp-sites v.2.4.0 (default parameter) (Page et al., 2016). Output files were converted into SNP distance matrices using custom R and python scripts. Based on the distribution of pairwise SNP distances (Fig S1, Data S7B), E. coli lineages were herein defined to have <500 SNPs. Lineages were defined to be UPEC for the purpose of this study if they were isolated as the causative agent (DxU isolate) of a UTI. Pairwise ANI values between same-patient isolates were calculated using fastANI v1.3 (parameters: --fragLen 3,000, --minFraction 0.5) (Jain et al., 2018).
Determination of colonization patterns, lineage persistence, and rUTI causing UPEC
To understand colonization dynamics of UPEC and assess the impact of inter-habitat transfer on UPEC within-host adaptation, each UPEC lineage was categorized into one of four distinct persistence patterns: urinary tract colonization, intestinal colonization, dual, and uncolonized. Lineages were characterized as colonizing a given habitat (1) if the UPEC lineage was recovered from a habitat- specific specimen (stool/urine) at >1 collection point, or (2) if all habitat-specific specimens (stool/urine) from a UTI episode were positive for the UPEC lineage. DxU urine specimens were not considered for classification purposes. Lineages for which either type of specimen from their corresponding patient was unavailable were left unclassified. Lineages were further classified as rUTI if (1) the patient of isolation experienced a recurrence during the follow-up period and either (2) the same lineage was isolated as the DxU isolate of a rUTI or (3) no other lineage of E. coli was isolated at any point during follow-up. Lineages without follow-up DxU isolates or when multiple lineages of E. coli were isolated from a rUTI patient were left unclassified. Lineages from non-rUTI patients were classified as non-rUTI.
Characterization of within-lineage allelic diversity
To determine the allelic diversity between isolates from the same lineage, “pseudo-assemblies” were constructed for each UPEC lineage, as previously described (Thänert et al., 2019; Zhao et al., 2019). Equal proportions of reads from each isolate of a given lineage were pooled, assembled into a draft genome using SPAdes v.3.11.0 (parameters: -k 21,33,55,77 -careful), and annotated using Prokka v.1.12 (default parameters, contigs > 500 bp) (Bankevich et al., 2012; Seemann, 2014). These pseudo-assemblies were used as high-resolution reference genomes to characterize within-lineage allelic variation. Isolate reads were mapped to their respective pseudo-assemblies using Bowtie2 v.2.3.4 (parameters: -X 2000 --no-mixed --very-sensitive --n-ceil 0,0.01) (Langmead et al., 2019). SNPs and insertions/deletions were annotated using SAMtools v.1.9 and BCFtools v.1.9 (parameters: bcftools call -c -I ‘DP>10 & QS>0.95’, bcftools view -i ‘FQ<−85’) (Danecek and McCarthy, 2017; Li et al., 2009). SNPs were further filtered for major allele frequency >90% and gene presence in >60% of isolates from a given lineage, to exclude SNPs in potential MGEs. Mutated loci were mapped back to the reference GFF file (from Prokka) to identify corresponding coding sequences. Pairwise SNP distance matrices were used to construct unrooted lineage-specific phylogenetic trees, using the ape package in R v.3.6.3 (Paradis and Schliep, 2019). Time to last common ancestor (LCA) was estimated using median branch lengths of the resulting tree (determined via ape function ‘edge.length’) and dividing it by the estimated rate of E. coli evolution of 8.9 × 10−11 per base-pair per generation (Wielgoss et al., 2011), given an intestinal generation time of 80 minutes (Poulsen et al., 1995; Rang et al., 1999).
dMRCA estimation
To estimate dMRCA for each lineage, we generated parsimonious SNP trees using PHYLIP v3.697 (Felsenstein, 1989) to infer the ancestral sequence. VCF files resulting from within-lineage SNP characterization above were merged (bcftools merge --merge snps) including an isolate from the closest-related lineage according to ANI as an outgroup. The resulting VCF files were converted to ‘.phy’ format using the s_vcf2phylip.py script published by Ortiz et al on Github (https://github.com/edgardomortiz/vcf2phylip/blob/master/vcf2phylip.py). Files were used as input in the PHYLIP dnapars program (default parameters). Isolate dMRCA values were determined based on variable positions to the ancestral allele and used to calculate lineage averages. Lineage dMRCA values were compared between colonization types using Kruskal-Wallis with Dunn post-hoc test. P-values were adjusted for multiple comparisons using the Benjamini-Hochberg method (FDR).
Permutation test for non-random distribution of mutations
To identify non-random parallel evolution in UPEC lineages separate permutation tests were implemented for the two main colonization types; gut colonizers (gut isolates only) and dual colonizers. Mutations were randomly distributed across the lineage-specific pseudo-reference assemblies (i.e., if a lineage exhibited 10 SNPs total, 10 random SNPs were assigned in the genome). This process was repeated 1000 times for all lineages. The overall simulated distribution was used as the expected (neutral) distribution to test significance. The P-value was calculated as the top percentile of the neutral distribution at which the observed lineage count was present. To profile UPEC within-host adaptation, gut colonizers’ pseudo-reference assemblies were generated using only gut isolate reads. To profile inter-habitat, within-lineage mutations, 71 urinary isolates from the 51 gut colonizing lineages were added and permutations were re-run.
Estimation of dN/dS
To determine signatures of positive selection at specific genes, isolate gene sequences were aligned using Snippy v4.3.8, using as a reference the corresponding pseudo-assembly .ffn file as annotated by Prokka v3.8.0. STOP codons were masked from the Snippy snps.consensus.fa output files using a custom script. dN/dS values for each gene’s lineage-specific alignment were determined in Genomegamap v1.0.1 using the Maximum Likelihood estimation (Wilson, 2021). Overall dN/dS values for gene groups were estimated by generating a codon-based library of all possible mutations and calculating expected N/S ratios for each gene in the gene group. Overall dN/dS values were then calculated by summarizing the observed non-synonymous and synonymous mutations over all genes within the gene group. 95% confidence intervals were calculated by sampling from a binomial distribution as done previously (Zhao et al., 2019). Insertions/deletions as well as genes of plasmidic origin, due to their increased genetic variability(Rodríguez-Beltrán et al., 2021), were masked for group-wise dN/dS calculations.
Identification of within-lineage genomic plasticity
The accessory gene content of each UPEC lineage was identified based on a collapsed set of non-redundant genes. Therefore, clusters homologous genes were identified using CD-HIT(Fu et al., 2012), clustering translated gene sequences clustering at >90% amino acid identity. Within-lineage Jaccard dissimilarities (distances) of accessory gene content were calculated using the VEGAN package in R v.3.6.3 (Dixon, 2003). Average values for each lineage were used in comparisons. Dissimilarities of gene content were compared between colonization types, between and within habitat using ANOVA and Kruskal-Wallis with Dunn post-hoc. P-values were adjusted for multiple comparisons using the Benjamini-Hochberg method (FDR).
GO overrepresentation analysis (GOOA)
To gain insights into the functions under selection during UPEC persistence, we annotated GO terms of genes with non-random mutational signatures (as per the permutation test above) or habitat-specific within-lineage abundance patterns using blast2go (Götz et al., 2008). We compared gene-set associated GO terms frequencies to their expected value as determined using a fully GO-annotated colonization-type specific background (i.e., pangenome of each colonization type). To reduce redundancy in the GO term list associated with habitat-specific genes, we clustered overlapping GO terms using REVIGO prior to analysis allowing small similarity (<0.5) (Supek et al., 2011). Functional categories under selection during UPEC within-host persistence were identified using one-sided Fisher’s exact test (hypergeometric distribution) in R v.3.6.3. P-values were adjusted for multiple comparisons using the Benjamini-Hochberg method (FDR). Fold-changes (enrichment scores) were calculated comparing observed vs expected values. For GO network analysis significant GOOA results were clustered semantically using REVIGO and visualized using Cytoscape (Shannon et al., 2003; Supek et al., 2011).
Comparison with published UPEC genomes
We downloaded raw reads for 703 UPEC genomes previously curated from multiple studies (Data S7B) from NCBI(Biggel et al., 2020). We assembled genomes using SPAdes v.3.11.0 and assemblies using Prokka v.1.12 (default parameters). We extracted the amino acid sequences of OmpC and NsfA, found to be under positive selection and associated with the gain of phenotypic antibiotic resistance in this study, from all assemblies containing these genes. We queried the mutations (SNPs and INDELs) identified in this study against the set of reference sequences and extracted sequences from UPEC genomes containing the same mutations. We performed multiple sequence alignment between variable regions from our study and UPEC genomes using Clustal Omega and visualized alignments using MView (Madeira et al., 2019). OmpC and NfsA sequences from UTI89 were used as a reference.
MGE identification, annotation and characterization
We identified putative MGEs differentially abundant in isolates of the same lineage by aligning short reads to the pseudo-reference assembly. Candidate regions of at least 500bp length and <0.2X relative coverage in at least one isolate were considered for further analysis. Candidate MGEs in closed genomic proximity (<1 read pair - 300bp apart) were clustered to account for sporadic read mapping into conserved genomic regions interrupting continuous MGE identification. If candidate MGEs covered >90% of a contig in the pseudo-assembly, the whole contig was defined as a candidate MGE. Coverage for all putative within-lineage MGEs was determined for all isolates and a MGE presence/absence matrix was generated based on the average relative coverage for putative MGEs in each isolate’s short read alignment. <0.2X relative coverage over the complete length of the MGE equaled absence and >0.8X relative coverage equaled presence in a given isolate. Intermediate values were defined to be unclear evidence of MGE presence/absence. Within-lineage similarity of isolate MGE profiles was assessed using Jaccard dissimilarities (distances) calculated using the VEGAN package in R v.3.6.3 (Dixon, 2003). Comparison of MGE profiles was performed using ANOVA with Tukey post-hoc test and Welch’s t-test. P-values were adjusted for multiple comparisons using the Benjamini-Hochberg method (FDR).
MGEs were annotated similarly to a previously published protocol for de novo MGE identification (Durrant et al., 2020). The pool of within-lineage MGEs was queried for prophages using PHASTER (Arndt et al., 2016). MGE contigs of plasmidic origin were identified combining replicon typing using ‘Plasmid MLST’ with mapping within-lineage MGE contigs to the complete pool of plasmidic contigs identified de novo in the draft assemblies of all isolate as previously described (Jolley et al., 2018; Thänert et al., 2019). This “lineage-plasmidome” was identified using plasmidSPAdes v.3.11.0 (parameters: --plasmid -k 21,33,55,77 –careful), Recycler v.0.6.2 (parameters: -k 77 -i True), and PlasmidFinder v.4.0 (parameters: -p enterobacteriaceae -k 95.00) (Antipov et al., 2016; Carattoli et al., 2014; Rozov et al., 2017). A non-redundant list of putative plasmidic contigs was validated against the NCBI plasmid database using ncbi-blast v.2.6.0+ (McGinnis and Madden, 2004). Contigs with >90% identity and >90% coverage of plasmid in the database were retained. This total “lineage-plasmidome” was annotated using Prokka v.1.12 (default parameters), the eggnog-mapper v.6.8 (parameters: -m diamond --query-cover 0.9), RGI-CARD v.5.1.0 (95% identity, 100% coverage), and Resfinder v.4.0 (95% identity, 100% coverage) (Huerta-Cepas et al., 2019; Jia et al., 2017; Seemann, 2014; Zankari et al., 2012). MGEs were determined to be plasmidic if they (1) had an exact replicon match in the Plasmid MLST database or (2) if they aligned to a contig of de novo identified plasmidic origin at >80% coverage and 99% identity using ncbi-blast v.2.6.0+ (McGinnis and Madden, 2004). Insertion sequences (IS) and transposases were identified in MGEs by blasting against the ISfinder database(Siguier et al., 2006). As the repetitive nature of IS frequently causes short-read assemblies to break, incomplete IS are often found at the edge of contigs. To account for this, IS were determined to be present if either (1) a partial IS match was identified at the edge of contig with >95% identity or (2) an IS was identified at >90% identity and >80% coverage. IS elements were defined as elements that only contained an IS/Transposase and no other genes. Lastly, recombinases were identified in the Prokka annotations of the MGE pool.
Consistent with previous methods (Durrant et al., 2020), the final annotation for each MGE was assigned hierarchically from specific to general as follows; (1) Intact phages, (2) Plasmid, (3) IS element, (4) CDS+Transposase, (5) Recombinase, (6) Questionable/Incomplete phage, (7) Contains CDS, and (8) No CDS. Habitat-specific genes were identified in the MGE pool using ncbi-blast v.2.6.0+ and determined to be present if (1) coverage >90% at 99% identity or (2) coverage >10% at 100% identify and the gene was determined to be located at the edge of a contig (McGinnis and Madden, 2004).
To reduce the likelihood of false positives, GOOA of mobilized functions between rUTI and non-rUTI lineages (Fig 6B) was performed after filtering out GO-terms present in less than 5% of all analyzed lineages. GO term overrepresentation in the mobilized gene pool of either rUTI or non-rUTI lineages was assessed using Fisher’s exact test. P-values were adjusted for multiple comparisons using the Benjamini-Hochberg method (FDR). Pseudo enrichment scores were calculated comparing observed GO term abundances between compared groups adding the minimal value in the array as a pseudocount.
We further assessed MGE host ranges by aligning putative MGE contigs against the NCBI nucleotide database using ncbi-blast v.2.6.0+ (McGinnis and Madden, 2004), filtering for hits with >95% identity and 95% query coverage. Uncultured bacteria, eukaryotes, synthetic constructs/vectors, and mixed communities were filtered from the resulting hits. Taxa IDs were converted to species-level annotations and the number of species-level blast hits was summarized per MGE category. Statistical comparisons were performed using ANOVA and species underre-presented in the urinary MGE pool were determined using one-sided Fisher’s exact test. The 25 species most abundant in the blast hitlist were considered for statistical analysis. P-values were corrected for multiple-hypothesis testing using the Benjamini-Hochberg method (FDR).
General statistical approaches
Statistical comparisons were performed using ANOVA with Tukey post-hoc, Kruskal-Wallis with Dunn post-hoc, Welch’s t-test, and Fisher’s exact test as outlined above. Parametric or nonparametric tests were selected for a given comparison based on whether the underlying data approximated a normal distribution (Shapiro-Wilk test). When multiple-hypothesis were investigated, P-values were corrected for multiple-hypothesis testing using the Benjamini-Hochberg method (FDR). P-values <0.05 were considered ‘significant’. Statistical details, including the statistical test used for each comparison, the number of observations (n), definition of center, dispersion and precision can be found in the Results section, the figure legends, and figures.
Supplementary Material
Data S4 | Gene presence/absence matrix, Related to Figure 4
Data S6 | Reference genomes used in this study, Related to STAR Methods and Figure 1
Data S7 | Patient-specific core-genome assemblies and SNP distances, Related to STAR Methods
Highlights.
UPEC lineages persist within the gastrointestinal and urinary tracts of UTI patients
Habitat-specific selection impacts UPEC within-host adaptation
Genomic plasticity facilitates UPEC niche adaptation
Within-lineage genomic plasticity is facilitated by mobile genetic elements
Acknowledgements
The authors thank Eric Keen, Drew J. Schwartz, Bejan Mahmud, Alaric D’Souza, Kimberley Sukhum, Manish Boolchandani, and Mary K. Hayden for providing scientific discussions and support. We thank the staff at the Edison Family Center for Genome Sciences and Systems Biology at Washington University School of Medicine: Bonnie Dee, Kathleen Matheny, and Keith Page for administrative support, Jessica Hoisington-Lopez and MariaLynn Crosby for managing the high-throughput sequencing core, and Eric Martin and Brian Koebbe for computational support. We thank Carrie Crook, Tony James and Emily Reesey for providing study coordination support. This work was supported in part by awards to the authors from the U.S. Centers for Disease Control and Prevention Epicenter Prevention Program (grant U54CK000482; principal investigator, Victoria Fraser); to J.H.K. from the Longer Life Foundation (an RGA/Washington University partnership), the National Center for Advancing Translational Sciences (grants KL2TR002346 and UL1TR002345), and the National Institute of Allergy and Infectious Diseases (NIAID) (grant K23A1137321) of the National Institutes of Health (NIH); and to G.D. from NIAID (grant R01AI123394) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (grant R01HD092414) of NIH. R.T.’s research was funded by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation; grant 402733540). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antipov D, Hartwick N, Shen M, Raiko M, Lapidus A, and Pevzner PA. (2016). plasmidSPAdes: assembling plasmids from whole genome sequencing data. Bioinformatics 32, btw493. [DOI] [PubMed] [Google Scholar]
- Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, and Wishart DS. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. (2012). SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, and Kishony R. (2015). Inexpensive Multiplexed Library Preparation for Megabase-Sized Genomes. PLoS One 10, e0128036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloin C, Michaelis K, Lindner K, Landini P, Hacker J, Ghigo JM, and Dobrindt U. (2006). The Transcriptional Antiterminator RfaH Represses Biofilm Formation in Escherichia coli. J. Bacteriol. 188, 1316–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggel M, Xavier BB, Johnson JR, Nielsen KL, Frimodt-Møller N, Matheeussen V, Goossens H, Moons P, and Van Puyvelde S. (2020). Horizontally acquired papGII-containing pathogenicity islands underlie the emergence of invasive uropathogenic Escherichia coli lineages. Nat. Commun. 2020 111 11, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricio-Moreno L, Sheridan VH, Goodhead I, Armstrong S, Wong JKL, Waters EM, Sarsby J, Panagiotou S, Dunn J, Chakraborty A, et al. (2018). Evolutionary trade-offs associated with loss of PmrB function in host-adapted Pseudomonas aeruginosa. Nat. Commun. 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronson RA, Gupta C, Manson AL, Nguyen JA, Bahadirli-Talbott A, Parrish NM, Earl AM, and Cohen KA. (2021). Global phylogenomic analyses of Mycobacterium abscessus provide context for non cystic fibrosis infections and the evolution of antibiotic resistance. Nat. Commun. 2021 121 12, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SP, Cornforth DM, and Mideo N. (2012). Evolution of virulence in opportunistic pathogens: Generalism, plasticity, and control. Trends Microbiol. 20, 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carattoli A, Zankari E, García-Fernández A, Voldby Larsen M, Lund O, Villa L, Møller Aarestrup F, and Hasman H. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Feldgarden M, Weissman SJ, Dykhuizen DE, Van Belle G, and Sokurenko EV. (2007). Haplotype diversity in “source-sink” dynamics of Escherichia coli urovirulence. J. Mol. Evol. 64, 204–214. [DOI] [PubMed] [Google Scholar]
- Chen SL, Wu M, Henderson JP, Hooton TM, Hibbing ME, Hultgren SJ, and Gordon JI. (2013). Genomic diversity and fitness of E. coli strains recovered from the intestinal and urinary tracts of women with recurrent urinary tract infection. Sci. Transl. Med. 5, 184ra60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi U, and Lee CR. (2019). Distinct Roles of Outer Membrane Porins in Antibiotic Resistance and Membrane Integrity in Escherichia coli. Front. Microbiol. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll F, Harrison EM, Toleman MS, Reuter S, Raven KE, Blane B, Palmer B, Kappeler ARM, Brown NM, Török ME, et al. (2017). Longitudinal genomic surveillance of MRSA in the UK reveals transmission patterns in hospitals and the community. Sci. Transl. Med. 9, 953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, and McCarthy SA. (2017). BCFtools/csq: Haplotype-aware variant consequences. Bioinformatics 33, 2037–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darch SE, McNally A, Harrison F, Corander J, Barr HL, Paszkiewicz K, Holden S, Fogarty A, Crusz SA, and Diggle SP. (2015). Recombination is a key driver of genomic and phenotypic diversity in a Pseudomonas aeruginosa population during cystic fibrosis infection. Sci. Rep. 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Walker AS, Peto TE, Crook DW, and Wilson DJ. (2016). Within-host evolution of bacterial pathogens. Nat. Rev. Microbiol. 14, 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon P. (2003). VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930. [Google Scholar]
- Durrant MG, Li MM, Siranosian BA, Montgomery SB, and Bhatt AS. (2020). A Bioinformatic Analysis of Integrative Mobile Genetic Elements Highlights Their Role in Bacterial Adaptation. Cell Host Microbe 27, 140–153.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhenawy W, Hordienko S, Gould S, Oberc AM, Tsai CN, Hubbard TP, Waldor MK, and Coombes BK. (2021). High-throughput fitness screening and transcriptomics identify a role for a type IV secretion system in the pathogenesis of Crohn’s disease-associated Escherichia coli. Nat. Commun. 12, 2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo-Lubián A, Ben Zakour NL, Agyekum A, Qi Q, and Iredell JR. (2019). Host adaptation and convergent evolution increases antibiotic resistance without loss of virulence in a major human pathogen. PLoS Pathog. 15, e1007218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. (1989). PHYLIP - Phylogeny Inference Package (Version 3.2). Cladistics 5, 164–166. [Google Scholar]
- Flores-Mireles AL, Walker JN, Caparon M, and Hultgren SJ. (2015). Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 13, 269–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foxman B. (2014). Urinary tract infection syndromes. Occurrence, recurrence, bacteriology, risk factors, and disease burden. Infect. Dis. Clin. North Am. 28, 1–13. [DOI] [PubMed] [Google Scholar]
- Fu L, Niu B, Zhu Z, Wu S, and Li W. (2012). CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielaite M, Johansen HK, Molin S, Nielsen FC, and Marvig RL. (2020). Gene loss and acquisition in lineages of pseudomonas aeruginosa evolving in cystic fibrosis patient airways. MBio 11, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatt YE, and Margalit H. (2021). Common Adaptive Strategies Underlie Within-Host Evolution of Bacterial Pathogens. Mol. Biol. Evol. 38, 1101–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talón M, Dopazo J, and Conesa A. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, and Tesler G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JPJ, Wright RCT, Harrison E, Muddiman KJ, Wood AJ, Paterson S, and Brockhurst MA. (2021). Plasmid fitness costs are caused by specific genetic conflicts enabling resolution by compensatory mutation. PLOS Biol. 19, e3001225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond JA, Gordon EA, Socarras KM, Mell JC, and Ehrlich GD. (2020). Beyond the pan-genome: Current perspectives on the functional and practical outcomes of the distributed genome hypothesis. Biochem. Soc. Trans. 48, 2437–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison E, Guymer D, Spiers AJ, Paterson S, and Brockhurst MA. (2015). Parallel Compensatory Evolution Stabilizes Plasmids across the Parasitism-Mutualism Continuum. Curr. Biol. 25, 2034–2039. [DOI] [PubMed] [Google Scholar]
- Harrison E, Hall JPJ, and Brockhurst MA. (2018). Migration promotes plasmid stability under spatially heterogeneous positive selection. Proc. R. Soc. B Biol. Sci. 285, 20180324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbing ME, Dodson KW, Kalas V, Chen SL, and Hultgren SJ. (2020). Adaptation of arginine synthesis among uropathogenic branches of the escherichia coli phylogeny reveals adjustment to the urinary tract habitat. MBio 11, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen LJ, et al. (2019). EggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, and Aluru S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, Lago BA, Dave BM, Pereira S, Sharma AN, et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joensen KG, Tetzschner AMM, Iguchi A, Aarestrup FM, and Scheutz F. (2015). Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J. Clin. Microbiol. 53, 2410–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley KA, Bray JE, and Maiden MCJ. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications [version 1; referees: 2 approved]. Wellcome Open Res. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SA, Gibson T, Maltby RC, Chowdhury FZ, Stewart V, Cohen PS, and Conway T. (2011). Anaerobic Respiration of Escherichia coli in the Mouse Intestine. Infect. Immun. 79, 4218–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaper JB, Nataro JP, and Mobley HLT. (2004). Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004 22 2, 123–140. [DOI] [PubMed] [Google Scholar]
- Khademi SMH, Sazinas P, and Jelsbak L. (2019). Within-Host Adaptation Mediated by Intergenic Evolution in Pseudomonas aeruginosa. Genome Biol. Evol. 11, 1385–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Wilks C, Antonescu V, and Charles R. (2019). Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 35, 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, Jelsbak L, Sicheritz-Ponten T, Ussery DW, Aarestrup FM, et al. (2012). Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 50, 1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees JA, Kremer PHC, Manso AS, Croucher NJ, Ferwerda B, Serón MV, Oggioni MR, Parkhill J, Brouwer MC, van der Ende A, et al. (2017). Large scale genomic analysis shows no evidence for pathogen adaptation between the blood and cerebrospinal fluid niches during bacterial meningitis. Microb. Genomics 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, and Bork P. (2019). Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman TD, Michel JB, Aingaran M, Potter-Bynoe G, Roux D, Davis MR, Skurnik D, Leiby N, Lipuma JJ, Goldberg JB, et al. (2011). Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat. Genet. 43, 1275–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman TD, Flett KB, Yelin I, Martin TR, McAdam AJ, Priebe GP, and Kishony R. (2014). Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nat. Genet. 46, 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenço M, Ramiro RS, Güleresi D, Barroso-Batista J, Xavier KB, Gordo I, and Sousa A. (2016). A Mutational Hotspot and Strong Selection Contribute to the Order of Mutations Selected for during Escherichia coli Adaptation to the Gut. PLOS Genet. 12, e1006420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann R, Mediati DG, Duggin IG, Harry EJ, and Bottomley AL. (2017). Metabolic adaptations of Uropathogenic E. coli in the urinary tract. Front. Cell. Infect. Microbiol. 7, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvig RL, Sommer LM, Molin S, and Johansen HK. (2015). Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 47, 57–64. [DOI] [PubMed] [Google Scholar]
- McGinnis S, and Madden TL. (2004). BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melvin P, Weinstein M. (2018). M100Ed29 | Performance Standards for Antimicrobial Susceptibility Testing, 29th Edition. [Google Scholar]
- Nielsen KL, Stegger M, Godfrey PA, Feldgarden M, Andersen PS, and Frimodt-Møller N. (2016). Adaptation of Escherichia coli traversing from the faecal environment to the urinary tract. Int. J. Med. Microbiol. 306, 595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osei Sekyere J. (2018). Genomic insights into nitrofurantoin resistance mechanisms and epidemiology in clinical Enterobacteriaceae. Futur. Sci. OA 4, FSO293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, Fookes M, Falush D, Keane JA, and Parkhill J. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page AJ, Taylor B, Delaney AJ, Soares J, Seemann T, Keane JA, and Harris SR. (2016). SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genomics 2, e000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, and Schliep K. (2019). Ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. [DOI] [PubMed] [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, and Tyson GW. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen LK, Licht TR, Rang C, Krogfelt KA, and Molin S. (1995). Physiological state of Escherichia coli BJ4 growing in the large intestines of streptomycin-treated mice. J. Bacteriol. 177, 5840–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP. (2009). Fasttree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rang CU, Licht TR, Midtvedt T, Conway PL, Chao L, Krogfelt KA, Cohen PS, and Molin S. (1999). Estimation of growth rates of Escherichia coli BJ4 in streptomycin- treated and previously germfree mice by in situ rRNA hybridization. Clin. Diagn. Lab. Immunol. 6, 434–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau MH, Marvig RL, Ehrlich GD, Molin S, and Jelsbak L. (2012). Deletion and acquisition of genomic content during early stage adaptation of Pseudomonas aeruginosa to a human host environment. Environ. Microbiol. 14, 2200–2211. [DOI] [PubMed] [Google Scholar]
- Robinson AE, Heffernan JR, and Henderson JP. (2018). The iron hand of uropathogenic Escherichia coli: The role of transition metal control in virulence. Future Microbiol. 13, 813–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Beltrán J, DelaFuente J, León-Sampedro R, MacLean RC, and San Millán Á. (2021). Beyond horizontal gene transfer: the role of plasmids in bacterial evolution. Nat. Rev. Microbiol. 1–13. [DOI] [PubMed] [Google Scholar]
- Rossi E, La Rosa R, Bartell JA, Marvig RL, Haagensen JAJ, Sommer LM, Molin S, and Johansen HK. (2020). Pseudomonas aeruginosa adaptation and evolution in patients with cystic fibrosis. Nat. Rev. Microbiol. 19, 331–342. [DOI] [PubMed] [Google Scholar]
- Rozov R, Brown Kav A, Bogumil D, Shterzer N, Halperin E, Mizrahi I, and Shamir R. (2017). Recycler: an algorithm for detecting plasmids from de novo assembly graphs. Bioinformatics 33, 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell CW, Fleming BA, Jost CA, Tran A, Stenquist AT, Wambaugh MA, Bronner MP, and Mulvey MA. (2018). Context-dependent requirements for FimH and other canonical virulence factors in gut colonization by extraintestinal pathogenic Escherichia coli. Infect. Immun. 86, e00746–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Ulett GC, Totsika M, Phan MD, and Schembri MA. (2014). Role of capsule and O antigen in the virulence of uropathogenic Escherichia coli. PLoS One 9, e94786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber HL, Spaulding CN, Dodson KW, Livny J, and Hultgren SJ. (2017). One size doesn’t fit all: Unraveling the diversity of factors and interactions that drive E. coli urovirulence. Ann. Transl. Med. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DJ, Kalas V, Pinkner JS, Chen SL, Spaulding CN, Dodson KW, and Hultgren SJ. (2013). Positively selected FimH residues enhance virulence during urinary tract infection by altering FimH conformation. Proc. Natl. Acad. Sci. 110, 15530–15537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T. (2003). Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shee C, Gibson JL, Darrow MC, Gonzalez C, and Rosenberg SM. (2011). Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proc. Natl. Acad. Sci. 108, 13659–13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard SK, Guttman DS, and Fitzgerald JR. (2018). Population genomics of bacterial host adaptation. Nat. Rev. Genet. 19, 549–565. [DOI] [PubMed] [Google Scholar]
- Siguier P, Perochon J, Lestrade L, Mahillon J, and Chandler M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater FR, Bruce KD, Ellis RJ, Lilley AK, and Turner SL. (2008). Heterogeneous selection in a spatially structured environment affects fitness tradeoffs of plasmid carriage in pseudomonads. Appl. Environ. Microbiol. 74, 3189–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater FR, Bruce KD, Ellis RJ, Lilley AK, Turner SL, Slater FR, Lilley AK, Turner SL, Bruce KD, Ellis RJ, et al. (2010). Determining the Effects of a Spatially Heterogeneous Selection Pressure on Bacterial Population Structure at the Sub-millimetre Scale. Microb. Ecol. 60, 873–884. [DOI] [PubMed] [Google Scholar]
- Sokurenko EV. (2004). Selection Footprint in the FimH Adhesin Shows Pathoadaptive Niche Differentiation in Escherichia coli. Mol. Biol. Evol. 21, 1373–1383. [DOI] [PubMed] [Google Scholar]
- Sokurenko EV, Gomulkiewicz R, and Dykhuizen DE. (2006). Source-sink dynamics of virulence evolution. Nat. Rev. Microbiol. 4, 548–555. [DOI] [PubMed] [Google Scholar]
- Spaulding CN, Klein RD, Ruer S, Kau AL, Schreiber HL, Cusumano ZT, Dodson KW, Pinkner JS, Fremont DH, Janetka JW, et al. (2017). Selective depletion of uropathogenic E. coli from the gut by a FimH antagonist. Nature 546, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Rutherford DJ, and Yu EW. (2007). Characterization of the multidrug efflux regulator AcrR from Escherichia coli. Biochem. Biophys. Res. Commun. 361, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, and Šmuc T. (2011). REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS One 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, and Saier MH. (2014). Transport proteins promoting Escherichia coli pathogenesis. Microb. Pathog. 71–72, 41–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thänert R, Reske KA, Hink T, Wallace MA, Wang B, Schwartz DJ, Seiler S, Cass C, Burnham C-AD, Dubberke ER, et al. (2019). Comparative Genomics of Antibiotic-Resistant Uropathogens Implicates Three Routes for Recurrence of Urinary Tract Infections. MBio 10, e01977–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman SJ, Beskhlebnaya V, Chesnokova V, Chattopadhyay S, Stamm WE, Hooton TM, and Sokurenko EV. (2007). Differential stability and trade-off effects of pathoadaptive mutations in the Escherichia coli FimH adhesin. Infect. Immun. 75, 3548–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielgoss S, Schneider D, Barrick JE, Tenaillon O, Cruveiller S, Chane-Woon-Ming B, Médigue C, and Lenski RE. (2011). Mutation rate inferred from synonymous substitutions in a long-term evolution experiment with escherichia coli. G3 Genes, Genomes, Genet. 1, 183–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DJ. (2021). GenomegaMap: Within-Species Genome-Wide dN=dS Estimation from over 10,000 Genomes. Mol. Biol. Evol. 37, 2450–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young BC, Wu CH, Gordon NC, Cole K, Price JR, Liu E, Sheppard AE, Perera S, Charlesworth J, Golubchik T, et al. (2017). Severe infections emerge from commensal bacteria by adaptive evolution. Elife 6, e30637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, and Larsen MV. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha S, Lieberman TD, Poyet M, Groussin M, Xavier RJ, Alm EJ, Kauffman KM, and Gibbons SM. (2019). Adaptive Evolution within Gut Microbiomes of Healthy People Article Adaptive Evolution within Gut Microbiomes of Healthy People. Cell Host Microbe 25, 656–667.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S4 | Gene presence/absence matrix, Related to Figure 4
Data S6 | Reference genomes used in this study, Related to STAR Methods and Figure 1
Data S7 | Patient-specific core-genome assemblies and SNP distances, Related to STAR Methods
Data Availability Statement
Raw sequencing data has been deposited at the NCBI SRA database and are publicly available as of the date of publication. Accession numbers are listed in the Key Resource table. Relevant raw data and metadata can be found as supplementary data spreadsheets.
Key resources table
This paper does not report original code. We use well-established computational and statistical analysis software and packages. These are fully referenced in the Method section and Key Resource table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.