

SUMMARY
Despite the success of CAR-T cell cancer immunotherapy, challenges in efficacy and safety remain. Investigators have begun to enhance CAR-T cells with expression of accessory molecules to address these challenges. Current systems rely on constitutive transgene expression or multiple viral vectors, resulting in unregulated response and product heterogeneity. Here, we develop a genetic platform that combines autonomous antigen-induced production of an accessory molecule, with constitutive CAR expression in a single lentiviral vector called Uni-Vect. The broad therapeutic application of Uni-Vect is demonstrated in vivo by activation-dependent expression of (i) an immunostimulatory cytokine that improves efficacy, (ii) an antibody that ameliorates cytokine release syndrome, and (iii) transcription factors that modulate T cell biology. Uni-Vect is also implemented as a platform to characterize immune receptors. Overall, we demonstrate that Uni-Vect provides a foundation for a more clinically actionable next-generation cellular immunotherapy.
INTRODUCTION
Engineered T cell immunotherapies mediate unprecedented clinical responses in hematological malignancies1–6. However, their therapeutic efficacy is limited by immunosuppression or T cell dysfunction7. Furthermore, adoptive cellular immunotherapies can cause life-threatening complications such as cytokine release syndrome (CRS) and neurological toxicity8.
Attempts to enhance the efficacy of redirected T cells often involve armoring with soluble factors or engineered surface receptors9. In particular, IL-12 is being developed for delivery in a constitutive10,11 or inducible, nuclear factor of activated T cells (NFAT)-driven12–15 manner. Tumor infiltrating-lymphocytes (TILs) transduced with NFAT-driven IL-12 were previously tested in human clinical trials16. However, the trial was closed due to toxicities likely related to ectopic IL-12 expression by TILs, leading to the conclusion that further refinement would be necessary before this approach could be safely used in a clinical setting.
A key challenge is genetically integrating both an immune receptor and an accessory molecule in a clinically feasible and safe manner. Transient expression of the introduced effector molecule is preferred over constitutive expression to avoid systemic toxicities or malignant transformation of the cellular product. Furthermore, non-human derived genetic components should be avoided to reduce immunogenicity. The most common approach to achieve transient expression in CAR-T cells is a dual vector system in which separate viruses enable constitutive expression of a CAR and inducible expression of an accessory molecule12,13,16–18. These systems result in non-homogenous genetic modifications of the cellular product, which require costly cell sorting, and can limit translational potential.
Viral systems that combine NFAT-driven expression of one transgene with constitutive expression of a second transgene in a single vector are an improvement to two-vector systems14,15,19,20. As a route toward clinical translation, we developed and characterized unique genetically integrated systems that combine autonomous antigen-induced production of an accessory molecule, with constitutive immune receptor expression in a single vector, referred to as Uni-Vect. We first demonstrate the applicability of Uni-Vect as a research tool for developing immune receptors. Next, we focus on in vivo therapeutic applications and feature three approaches to upgrade CAR-T cell functions by (i) enhancing anti-tumor activity with human IL-12, (ii) ameliorating CRS with secretion of an IL-6 receptor alpha (IL-6Rα) blocking antibody, and (iii) programming T cell intrinsic features and expansion via transient transcription factor expression.
RESULTS
Design, Development and Validation of Uni-Vect in Primary Human T Cells
To develop Uni-Vect, a versatile platform for cellular immunotherapy, we combined a constitutive immune receptor (CAR or TCR) with inducible effector molecule expression in a single lentiviral vector system (Figure 1A). To achieve a transient response, we rewired endogenous T cell activation mediated NFAT signaling, triggered by an immune receptor’s recognition of its cognate target, to the expression of an accessory molecule. Using fluorescent reporter proteins, we tested several designs with varied orientation of the inducible and constitutive modules (forward or reverse), activation-inducible promoters (PCD69, PCD137, and PNFAT), and presence of SV40 polyadenylation signal (SV40 pA) (Figure S1A–E). Two configurations with NFAT driven inducible modules emerged as the most relevant candidates (Figure 1B).
Figure 1: Design and Validation of Uni-Vect in Primary Human T Cells.
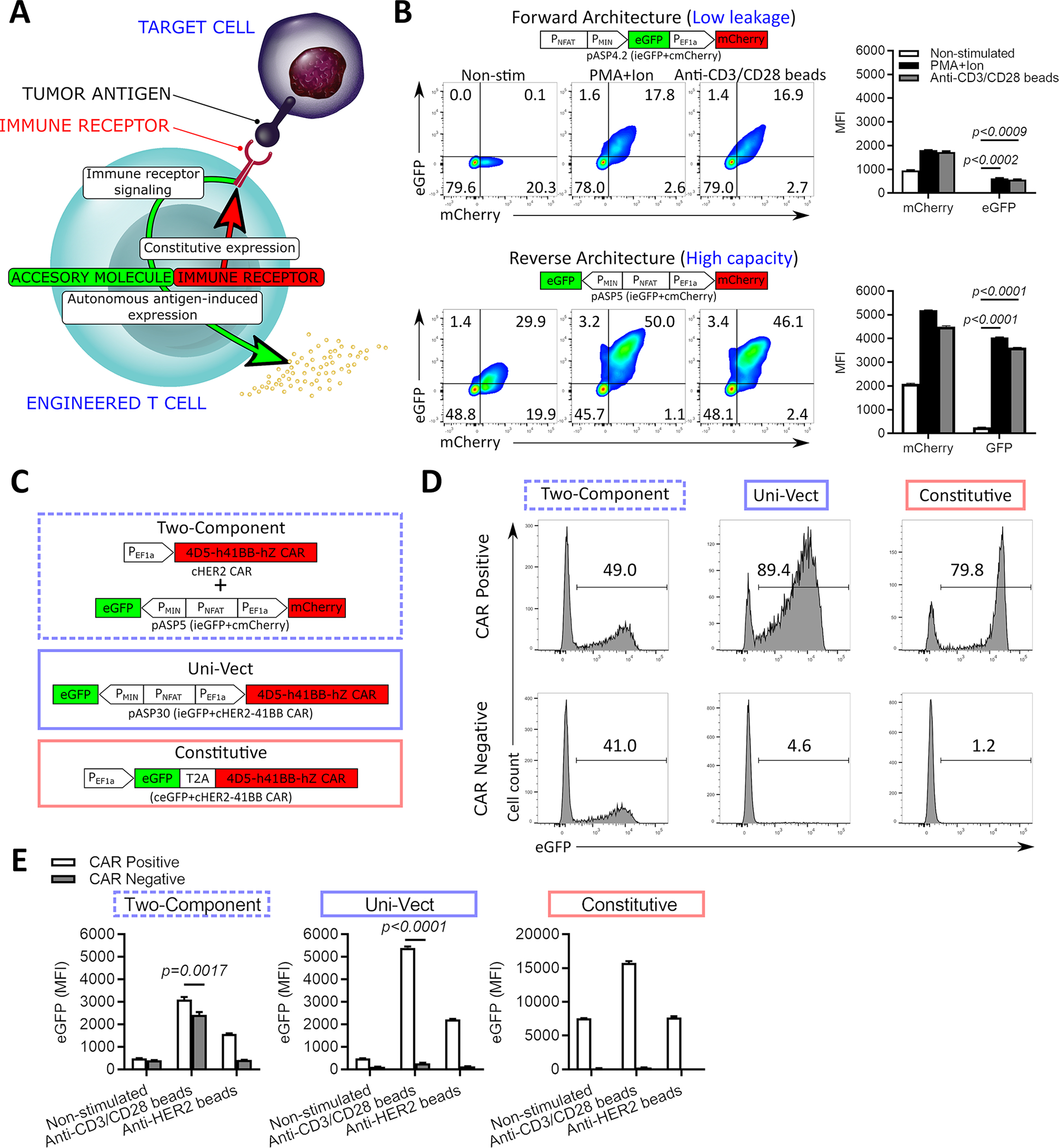
(A) Schematic representation of Uni-Vect recognizing a target antigen. (B) Primary human T cells were transduced with pASP4.2 or pASP5 at MOI5 and stimulated. Expression of reporter genes was monitored and quantified as MFI after 24 h. (n=3). (Welch ANOVA) (C) Schematic representation of compared constructs/experimental groups. (D-E) Primary human T cells were transduced with the vectors shown in C. Cells were sorted into CAR+ and CAR− fractions then stimulated. (D) eGFP expression of cells activated with PMA+ionomycin overnight. Histograms are shown for CAR+ and CAR− fractions. (E) Cells were left unstimulated or stimulated through TCRs or CARs for 24 h. MFI of eGFP is shown. (n=3). (Student’s T test). MFI; Mean fluorescence intensity. NS; not significant. All data with error bars are presented as mean ± SD. All data are representative of two or more experiments. See also Figures S1 and S2 and Table S1.
The first architecture, pASP4.2 (ieGFP+cmCherry, Table S1), places inducible eGFP upstream of a constitutive EF1α-driven mCherry module in a forward orientation. In both Jurkat (Figure S1C) and primary human T cells (Figure 1B) we observed coordinate expression of constitutive mCherry and inducible NFAT-driven expression of eGFP only upon activation, demonstrating that Uni-Vect functioned as designed. This architecture demonstrated tight regulation of the inducible module with no detectable expression of eGFP in an unstimulated state (Figure 1B), but resulted in relatively low virus titer (Figure S1C). To improve viral titers and output of the inducible module, we tested a second architecture, pASP5 (ieGFP+cmCherry, Table S1), where the modules as in the forward architecture were placed back-to-back in an anti-sense orientation relative to each other. This reverse architecture produced substantially higher virus titer (Figure S1E) and inducible expression of eGFP in the activated state, but also more basal eGFP expression (Figure 1B). We investigated if the upstream ATG start codon between the PMIN and the linker in the forward architecture influenced system performance and demonstrated no significant difference compared to a design where ATG was absent (Figure S2A). Priority was given to the version of Uni-Vect without upstream ATG. To further confirm our observations were due to differences in architecture, the amounts of lentiviral particles were adjusted to achieve comparable transduction efficiency. Under these conditions, the expression of inducible eGFP was higher in the reverse architecture in both non-stimulated (leakage) and stimulated states (Figure S2B) indicating that vector design influenced transgene expression. Furthermore, in comparison to PNFAT, the response of endogenous activation-inducible promoters PCD69 and PCD137 was inferior when integrated into Uni-Vect (Figure S1A–E) and similar results were observed in primary T cells (Figure S1F).
Response kinetics and the capability to respond to multiple rounds of activation are important aspects of inducible systems in T cells. Using live-cell imaging to monitor fluorescence in real-time, eGFP expression was observed 4 h post-stimulation in Jurkat cells engineered with each architecture, thus revealing the rapid kinetics of Uni-Vect (Figure S2C). Upon withdrawal of stimulation, eGFP decreased gradually over 4 days, due to its intrinsic half-life, then increased again after re-stimulation at day 5. As in previous assays, we observed differences in response amplitude between the two orientations.
Integration of a CAR in Uni-Vect and Comparison with a Two-Component Lentiviral System
Following optimization of our constructs, a second-generation HER2-specific CAR (clone 4D5;21) was exchanged into the constitutive module of the reverse architecture to generate pASP30 (ieGFP+cHER2−41BB CAR, Table S1). Primary human T cells engineered with pASP30 responded specifically to HER2+ SKOV3 cancer cells by upregulating the inducible module (Figure S2D) and lysing target cells (Figure S2E). Loss of surface CAR staining was observed in activated eGFP+ cells. We hypothesized that the loss was transient, due to either CAR internalization or masking after ligation with cognate antigens as is commonly observed in CAR-T cells activated through high density antigen stimulation22,23. To test this hypothesis, we sorted CAR+ cells before co-culture and demonstrated after 2 days that surface CAR expression was reduced after binding and lysing antigen positive target cells but was re-expressed by day 5 (Figure S2F). Because we observed basal expression of the inducible module in the reverse architecture, we tested whether tonic signaling was contributing factor. We found that dasatinib, an inhibitor of proximal CAR and TCR signaling24, did not affect basal expression of the inducible module, whereas it decreased expression in the activated state, suggesting that vector design, not tonic signaling, determines basal leakage (Figure S2G).
To compare the functionalities of Uni-Vect with currently utilized strategies for armoring CAR-T cells, we benchmarked pASP30 (ieGFP+cHER2−41BB CAR, Table S1) against co-transduction with two lentiviral vectors pASP5 (ieGFP+cmCherry, Table S1) and pP7 (constitutive HER2–41BB CAR, Table S1) and a constitutive single vector system (constitutive eGFP-2A-HER2-41BB CAR, Table S1) (Figure 1C). With Uni-Vect and the constitutive system, both the accessory and the CAR modules are expected to integrate into the same genomic locus at equimolar ratios, since they are delivered as a single DNA molecule. In stimulated human T cells, the CAR+ population homogenously expressed eGFP, while the CAR− population did not (Figure 1D). In contrast, the two-component system resulted in separate integration events of inducible and constitutive modules leading to a non-homogenous modification of the cellular product were both CAR+ and CAR− fractions partially expressed eGFP (Figure 1D). This heterogeneity also resulted in reduced output of the inducible module when CAR+ cells were activated through either CARs or TCRs (Figure 1E). Together, these results demonstrate how Uni-Vect delivers more accessory molecule expression in CAR+ cells and less dysregulated expression in CAR− cells than two-vector systems. Apart from a simplified modular assembly of lentiviral transfer plasmid and virus production where only one viral product is manufactured Uni-Vect also removes any need for sorting of the cellular product.
Like Uni-Vect, the constitutive system is expected to integrate accessory and CAR modules into the same genomic locus at equimolar ratios. While the constitutive system generated a homogenous product, its basal expression of eGFP in the non-stimulated state exceeded that of Uni-Vect T cells that were stimulated through the CAR via HER2 beads (Figure 1E). In contrast, the ability of Uni-Vect transiently achieve the levels of expression that are basal in constitutive systems demonstrates its potential to deliver potent immune modulators with an enhanced safety profile.
Endogenous TCR Knockout Renders CAR Signaling an Exclusive Activator of the System
CAR-T cells express endogenous TCRs that can recognize epitopes presented in pMHCs to trigger T cell activation, thus upregulating NFAT25. Because the inducible Uni-Vect module senses NFAT signaling, it may be activated by target antigen stimulation via the CAR, or through the T cell’s endogenous TCR upon recognition of specific pMHC (Figure 2A). Stimulation through the TCR causes undesired accessory molecule expression independent of the presence of a target tumor antigen.
Figure 2: T-Cell Receptor Knockout in Uni-Vect CAR-T cells.
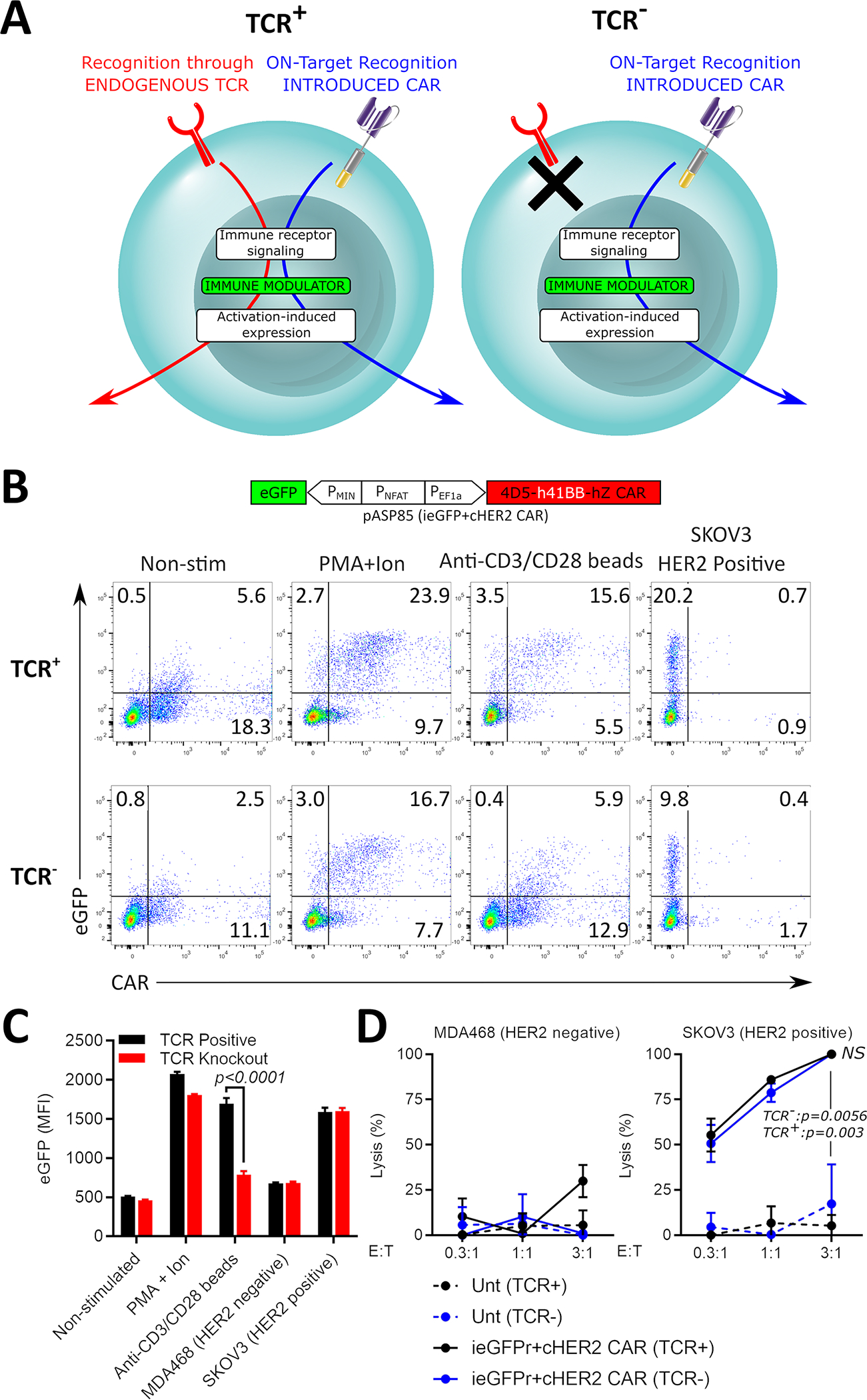
(A) Uni-Vect-CAR-T cells may be stimulated by either the introduced CAR or the endogenous TCR. Endogenous TCR knockout prevents expression of the accessory molecule induced by endogenous TCR signaling. (B) TCR knockout in pASP85-CAR-T cells. NFAT signaling induced by 24 h stimulation with PMA+ionomycin, anti-CD3/CD28 dynabeads, or target SKOV3 cells was monitored in TCR+ and TCR− CAR-T cells by flow cytometry via built-in NFAT-eGFP inducible module. (C) MFI quantification of NFAT-inducible eGFP in Q1 and Q2. (n=3) (Student’s T test) (D) Lysis of target and non-target cells by TCR+ and TCR− pASP85-CAR-T cells (n=3). (ANOVA multiple-comparison). NS; not significant. All data with error bars are presented as mean ± SD. All data are representative of three or more experiments. See also Figure S3 and Table S1.
To reduce potential toxicities related to unintentional triggering of NFAT-inducible accessory molecule expression, the endogenous TCR was knocked out using a CRISPR/Cas9 method compatible with lentiviral transduction of Uni-Vect (Figure S3A). Using sgRNAs targeting genes encoding TCRα and TCRβ,, the endogenous TCR was knocked out in primary human T cells with 80–90% efficiency (Figure S3B). These primary T cells were then transduced with pASP85 (ieGFP+cHER2−41BB CAR, Table S1) and the remaining TCR+ cells were removed by negative selection (Figure S3B). The expansion of edited cells was only partially impaired compared to mock, due to the negative selection step following TCR knockout during expansion (Figure S3C).
Following expansion, pASP85-transduced TCR+ and TCR− cells were stimulated through TCRs and analyzed by flow cytometry (Figure 2B). While unedited TCR+ cells produced a high eGFP mean florescent intensity (MFI), the response of TCR− cells remained comparable to that of non-stimulated controls, indicating functional knockout. Importantly, TCR− and TCR+ CAR-T cells responded similarly to a HER2+ cell line (Figure 2C). Cytotoxic T-cell function of the edited CAR-T cells was not affected (Figure 2D) indicating that TCR knockout Uni-Vect cells retain CAR-mediated effector functions. This data provides proof-of-concept for the knockout of an endogenous TCR when introducing Uni-Vect into primary human T cells for the creation of CAR-T cells with precise transgene expression. This may improve the safety of armored CAR-T therapies, especially when potent accessory molecules are utilized.
Uni-Vect Expression Cassettes for Characterization of T-cell Receptors
We predicted that a built-in NFAT-inducible reporter of T cell activation in the Uni-Vect system could provide a platform to functionally assess T-cell receptors. Therefore, we developed a Jurkat E6.1 cell line deficient in endogenous TCR expression and with introduced CD8αβ (to enable MHC class I recognition by the introduced TCR) and Uni-Vect system featuring NFAT-inducible eGFP in the forward (pASP89, Table S1) or reverse (pASP90, Table S1) architecture (Figure 3A). Cells were sorted for the TCR−/CD8αβ+/mCherry(Uni-Vect)+ population to generate JpASP90 or JpASP89cell lines. Melanoma neoantigen (PORCN H364Y)-specific TCRα and β chains26,27 were introduced into these cell lines and αβTCR expression was confirmed. Expression of the TCR conferred reactivity to a peptide (LLHGFSFYL)/HLA-A*02:01; p/HLA) multimer demonstrating assembly and antigen specificity (Figure 3B and Figure S4A). Next, we stimulated PORCN H364Y-specific JpASP90 or JpASP89 using titrated peptide concentrations in the presence of antigen presenting cells (monoallelic HLA-A*02:01-expressing K562). Increasing peptide concentrations led to increased eGFP expression demonstrating the Uni-Vect system’s utility as a platform to monitor TCR activation (Figure 3B, Figure S4A and S4B). Antigen stimulation of JpASP90 cells resulted in eGFP expression in up to 80% of cells accompanied with 5-fold increase in MFI. CD69 upregulation and production of GM-CSF, IL-2, TNFα and IL-8 correlated with eGFP expression as a readout of T cell activation (Figure S4C).
Figure 3: Uni-Vect Expression Cassettes for Characterization of T-cell Receptors.
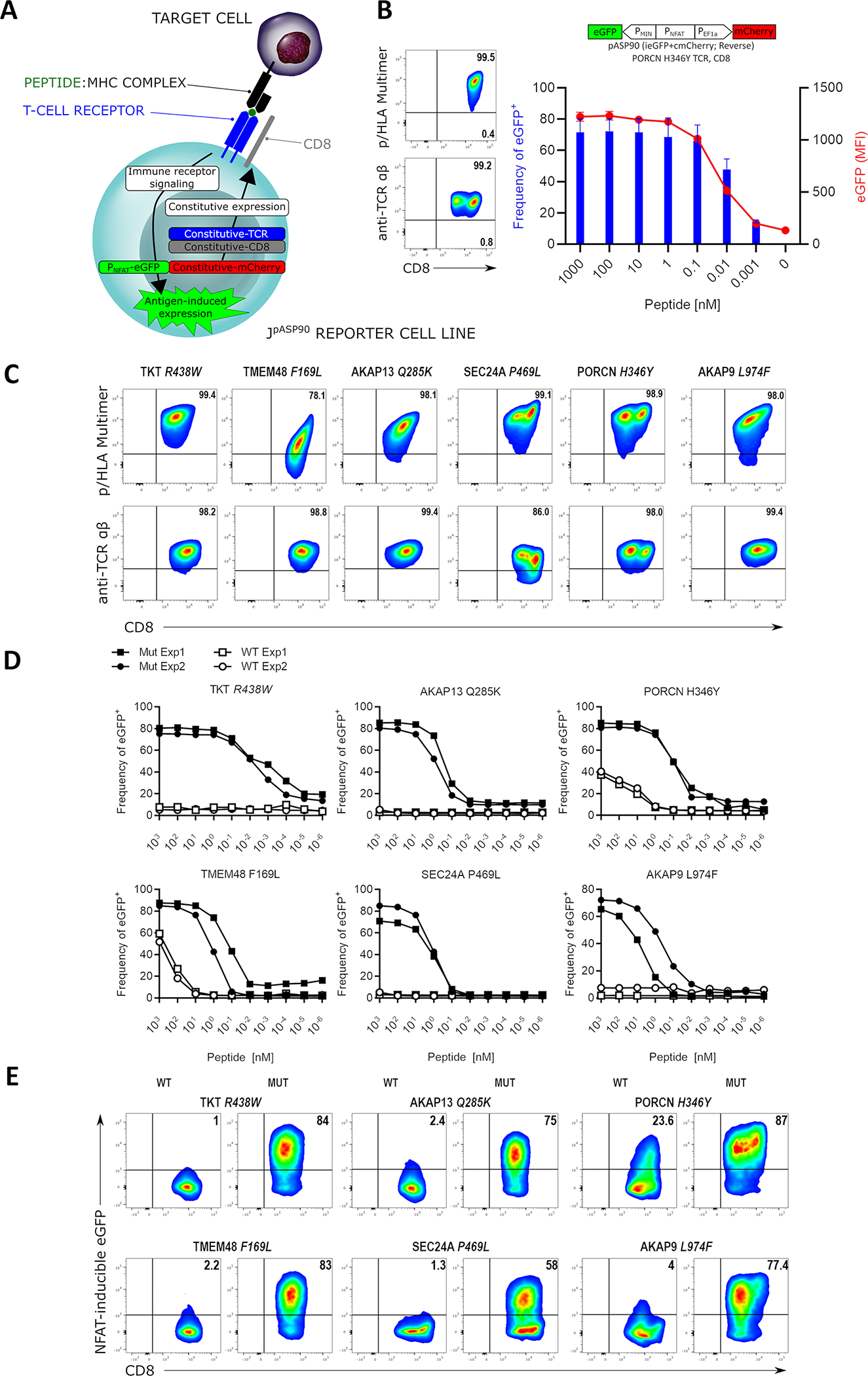
(A) Schematic representation of Uni-Vect-Jurkat reporter cell lines JpASP90 and JpASP89. (B) Transduction of TCR constructs into JpASP90 reporter cell lines. Titrated concentrations of peptide (LLHGFSFYL) antigen in the presence of antigen presenting cells led to upregulation of NFAT-inducible eGFP expression as a surrogate for activation via TCR signaling. Blue bars represent percentage of eGFP positive cells, red line represents MFI of eGFP at each concentration. (n=3). (C) Flow cytometry analysis of 6 neoantigen-specific/HLA-A*02:01 restricted TCRs engineered into the JpASP90 reporter cell line. (D) NFAT reporter EC50 curves for neoantigen-specific TCRs expressed in JpASP90 reporter cell line upon activation with mutated (blue) and wild-type (red) peptides. (E) JpASP90 reporter cell line expressing the various TCRs were cultured for 16 h with melanoma cell line DM6 expressing tandem mini-gene constructs encoding mutated or wild-type antigen and analyzed for inducible eGFP expression. All data are representative of two or more experiments. All data with error bars are presented as mean ± SD. See also Figure S4 and Tables S1 and S2.
To further evaluate our platform, 6 distinct TCRs directed at HLA-A*02:01-restricted neoantigen peptide complexes (Table S2) were introduced into JpASP90 cells. All, except SEC24A P469L-specific TCRs had similar expression (Figure 3C). However, a range of MFI intensities was observed when p/HLA multimers were used as staining reagents suggesting these TCRs may display different avidities for their corresponding p/HLA complexes. This was confirmed by titrations of neoantigen peptides that demonstrated a range of avidities among TCR-engineered JpASP90 cells, with those cells expressing SEC24A P469L TCR requiring the highest peptide concentration to achieved 50% cell activation (Figure 3D and Table S2). Additionally, two TCRs (PORCN H346Y and TMEM48 F169L) recognized wild type peptide at high peptide concentrations. This finding is consistent with our previous reports demonstrating cross-reactivity in primary PORCN H346Y- and TMEM48 F169L-specific CD8+ T cells with these receptors26,27.
Finally, for discovery of tumor reactive TCRs, the ability of this platform to be activated by endogenously processed and presented antigen is of critical importance. To this end, DM6 melanoma cells were engineered to express the tandem mini-gene constructs (TMC) encoding a mutated (MUT) or wild-type (WT) peptides inserted in a 27 amino acid framework allowing for minimal epitope processing and presentation. As shown in Figure 3E, JpASP90 cells expressing the various TCRs upregulate eGFP expression when co-cultured with DM6 cells expressing MUT but not WT TMC. Only cells expressing PORCN H346Y-specific TCR are activated by WT TMC, a finding consistent with prior results obtained when loaded peptides were used as antigens (Figure 3D). Overall, these results demonstrate that the Uni-Vect NFAT sensor represents a reliable platform to characterize expression, specificity, and functional avidity of TCRs.
IL-12 Enabled Uni-Vect Improves Antitumor Efficacy of CAR-T Cells In Vivo
IL-12 is a pleiotropic pro-inflammatory cytokine, but the severe side effects associated with systemic administration has limited its use in cancer patients12,28. However, CAR-T cells expressing constitutive IL-12 are currently under clinical investigation (NCT02498912) and is considered a current state of the art treatment. We hypothesized that localized CAR-inducible secretion of IL-12 would increase the efficacy of CAR-T cells without causing significant toxicity (Figure 4A). To test our hypothesis we first placed human IL-1229 into the low leakage forward architecture of Uni-Vect, to generate construct pASP18 (iIL-12+cmCherry, Table S1). To test for reversible expression of IL-12, pASP18 T cells were stimulated for 2 days, followed by 2 days of rest, and re-stimulated for 2 days, each time replacing media. Secreted hIL-12 was detected only when cells were stimulated, demonstrating a tightly regulated response that was also fully reversible and capable of responding to a second activation event (Figure 4B). Control CAR-T cells secreted no IL-12 after stimulation (Figure S5A).
Figure 4: iIL-12 Enabled Uni-Vect Improves Antitumor Efficacy of CAR-T Cells In Vivo.
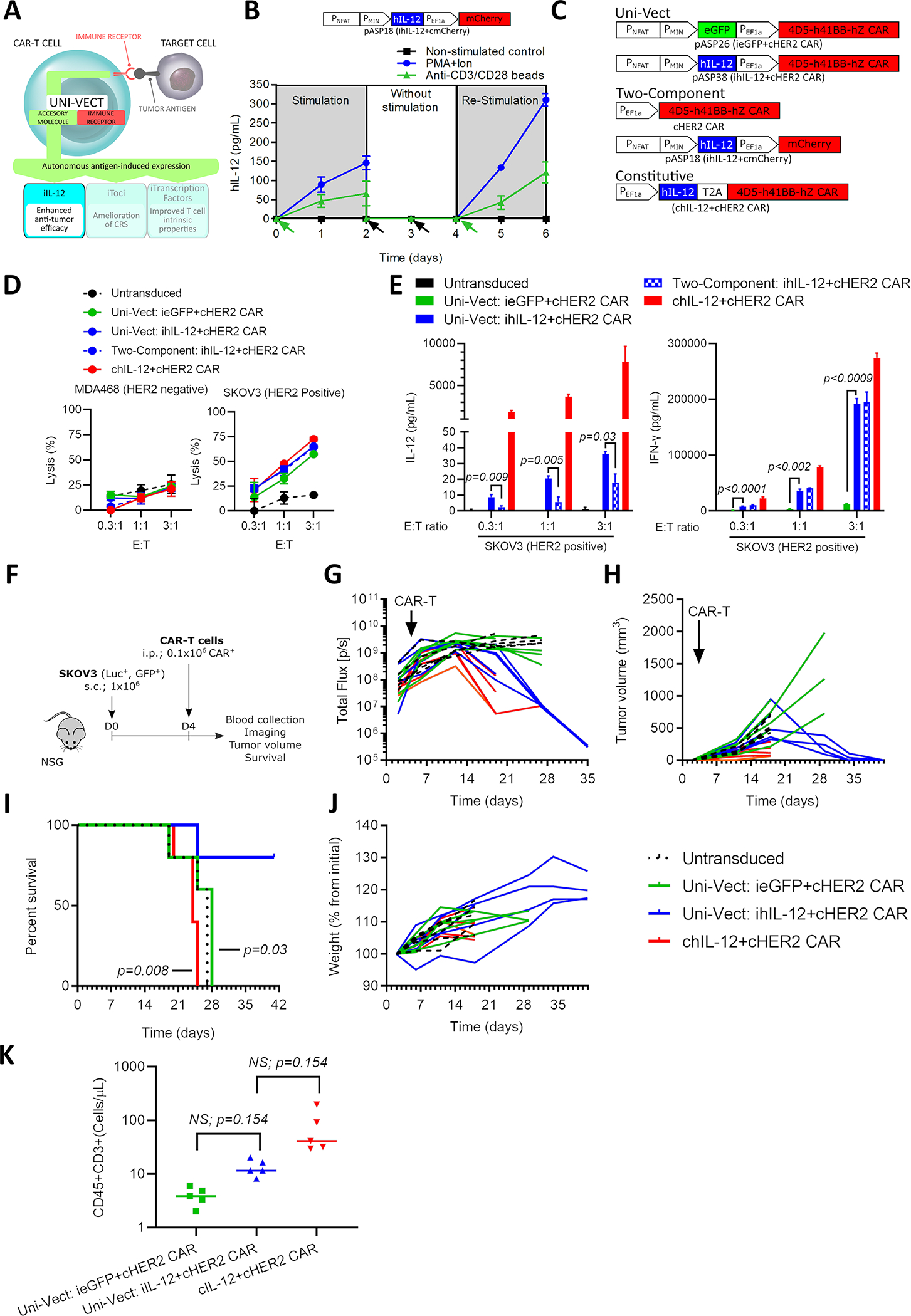
(A) Schematics of iIL-12 integrated into Uni-Vect. (B) Primary T cells transduced with pASP18 were stimulated for 2 days, followed by 2 days of rest, and then re-stimulated for another 2 days. IL-12 secretion was measured at 24 h increments. (n=3). Green arrow represents stimulation and black arrow represents media exchange. (C) Schematic representation of compared constructs/experimental groups. (D) CAR-T cells were co-cultured with HER2+ and HER2− target cell lines. Lysis was measured by luciferase assay. (n=3) (E) IL-12 and IFN-γ secretion from co-cultures in (D). (Welch ANOVA) (F) In vivo study design. CAR-T cells were injected i.p. at 0.1 × 106 CAR-T per mouse in NSG mice with established HER2+ SKOV3 ovarian cancer tumors. (n = 5 mice per group) (G) Quantified tumor luciferase activity. (H) Tumor volume as measured by caliper. (I) Percent survival in each group (Log-rank Mantel-Cox test). (J) Plot of mouse weight versus time by group (n=5). (K) CD3+ T cell counts in peripheral blood at day 17. (Kruskal-Wallis test). i.p; Intraperitoneal, s.c.; Subcutaneous. Each line in G, H, and J and each dot in K represents individual mice. NS; not significant. All data are representative of two or more experiments. All data with error bars are presented as mean ± SD. See also Figure S5 and Table S1.
We generated iIL-12-CAR-T cells by combining inducible hIL-12 with a constitutive HER2 targeting CAR in the construct pASP38 (iIL-12+cHER2-41BB CAR, Table S1). As controls, we generated HER2-targeting CAR-T cells with ieGFP (pASP26, ieGFP+cHER2−41BB CAR, Table S1), constitutive hIL-12 expression (chIL-12, chIL-12+cHER2-41BB CAR, Table S1) and a two-vector system (Figure 4C). Following in vitro expansion, we found that the phenotypic effector memory compartment was consistently enriched in cellular products that comprised engineered IL-12 either in inducible or constitutive mode of expression (Figure S5B), presumably due to previously described mechanisms30. Similar lysis was observed between all cellular products in co-culture with a target cell line (Figure 4D). Inducible expression of hIL-12 in the presence of HER2+ SKOV3 cells was higher in Uni-Vect cells compared to the two-component system, demonstrating greater on-target delivery. When left unstimulated or in the presence of an antigen negative cell line inducible hIL-12 secretion was at or below our detection limit (Figure S5C). Marked increase in IFN-γ was observed in ihIL-12 compared to control ieGFP CAR-T cells (Figure 4E), demonstrating the function of inducible hIL-12 as consistent with its endogenous role in inducing Th1 responses and promoting IFN-γ secretion.
To test iIL-12-CAR-T cells in vivo, we first determined the efficacy of iIL-12-CAR-T cells at a dose of 0.5 million cells per mouse (Figure S5D). iIL-12-CAR-T cells were able to eliminate tumors, unlike control HER2 CAR-T cells lacking iIL-12, as demonstrated by imaging (Figure S5E), tumor volume (Figure S5F), and survival (Figure S5G). CD8 and CD4 T cells in peripheral blood persisted only in mice receiving iIL-12-CAR-T cells (Figure S5H). Circulating IFN-γ was detected in the serum only in this group, while IL-12 was detectable at low levels in 50% of mice (Figure S5I). There was no signs of toxicity or detectable differences in weight (Figure S5J).
To demonstrate improved safety with respect to currently available constitutive IL-12 alternatives we performed similar experiment as above (Figure 4F) which recapitulated improved efficacy data at a dose as low as 0.1 million cells per mouse (Figure 4G–4I). We found that 80% of mice receiving Uni-Vect ihIL-12+cHER2 CAR-T cells cleared tumors (Figure 4G and 4H) and survived (Figure 4I) while all mice with chIL-12 expression died due to toxicities related to unregulated release of IL-12. In contrast, ihIL-12 did not cause observable toxicities or weight loss in surviving mice (Figure 4J). Although not significant, T cells in peripheral blood were higher in ihIL-12 compared to control eGFP CAR-T cells, and T cell counts were elevated in chIL-12 compared to inducible setting, which may have contributed to toxicity (Figure 4K).
Importantly, control iIL-12-CAR-T cells targeting an irrelevant antigen, CD20 (pASP36; iIL-12+cCD20−41BB CAR, Table S1), were unable to control HER2+ tumor cell growth, indicating that xenoreactivity is not responsible for tumor clearance in our model (Figure S5K). To further address this possibility we tested whether TCR knockout iIL-12-CAR-T cells were also able to control tumor growth. In the same model, edited TCR knockout iIL-12-CAR-T cells were effective at a dose of 0.15 million CAR+ per mouse (Figure S5L). Together, these results demonstrate how potent immune modulators delivered with Uni-Vect fit within a tight therapeutic window, which is essential for their use in armored CAR-T cell therapies.
Antigen-inducible Secretion of Tocilizumab-based scFv-Fc from CAR-T Cells to Ameliorate CRS In Vivo
Systemic administration of the IL-6Rα blocking antibody tocilizumab is the predominant clinical strategy to control CRS associated with CAR-T cell therapy31. However, controlling neurological toxicities is difficult because the blood-brain barrier restricts entry of tocilizumab into the central nervous system, in some cases necessitating invasive intra-thecal administration8,32. Therefore, we utilized Uni-Vect to develop CAR-T cells that autonomously deliver a tocilizumab-based antibody (Figure 5A).
Figure 5: Antigen-inducible Secretion of Biologically Active Tocilizumab-based scFv-Fc from CAR-T cells.
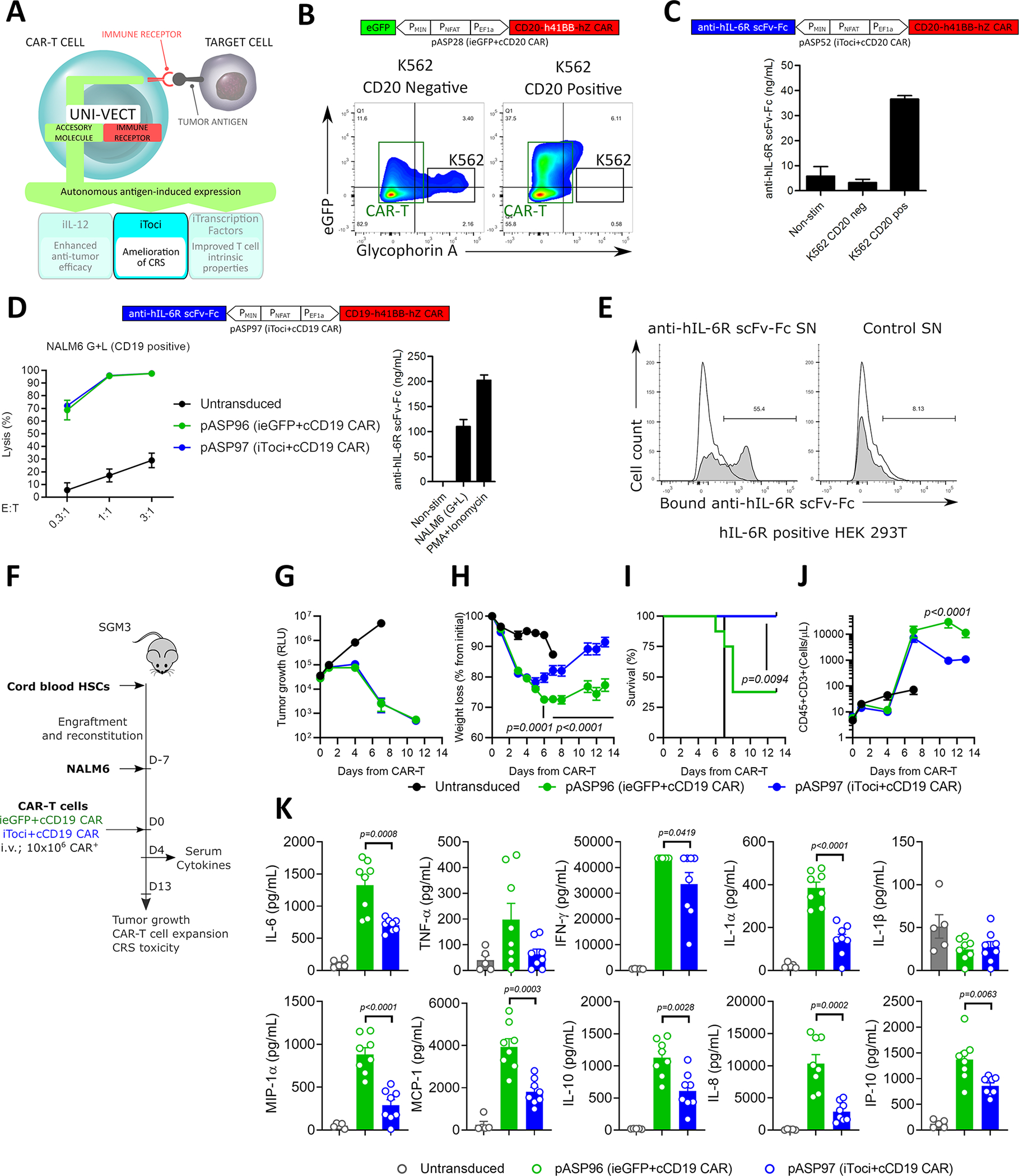
(A) Schematics of iToci integrated into Uni-Vect. (B) pASP28-CAR-T cells were co-cultured with CD20+ or CD20− K562 cells at 5:1 effector to target ratio for 24 h. Flow graph represents inducible eGFP, concomitant with target cell lysis. K562 cell lines (black rectangle) were stained for Glycophorin A expression and appear as Glycophorin+eGFP− population. CAR-T cells appear in Glycophorin− population (green rectangle). (C) pASP52-CAR-T cells were co-cultured with CD20+ or CD20− K562 cells at 3:1 effector to target ratio for 72 h. Antigen-dependent secretion of iToci was measured. (n=3) (D) pASP97-CAR-T cells or control pASP96-CAR-T cells were co-cultured with CD19+ NALM6 cell line for 48 h. Lysis was measured by luciferase assay (Left) and concomitant secretion of iToci was measured by ELISA (Right). (n=3) (E) pASP52-CAR-T cells or control pASP28-CAR-T cells were co-cultured with CD20+ cell line. Supernatants from these co-cultures were added to the HEK 293T cells engineered to express hIL-6Rα and binding of iToci was examined. (F) In vivo study design. pASP97-CAR-T cells or control pASP96-CAR-T cells were injected in humanized SGM3 mice engrafted with CD19+ NALM6 cell line. (n = 8 mice per group for iToci+cCD19 CAR and ieGFP+cCD19 CAR; n = 5 for untransduced group) Experiment was performed once. We monitored (G) tumor growth control, (H) weight loss (Two-Way ANOVA), (I) survival of mice (Log-rank Mantel-Cox test) and (J) hCD45+CD3+ T cell numbers in peripheral blood (Two-Way ANOVA). (K) Inflammatory cytokines were measured in serum 4 days after CAR-T cells injection (Two-Way ANOVA). SN, supernatant. NS; not significant. Error bars are presented as mean ± SD in C and D and as mean ± SEM in G, H, J and K. See also Figure S6 and Table S1.
First, we evaluated the functionality of Uni-Vect in the context of hematological malignancies, where treatment with CAR-T cells is associated with CRS. A CD20 CAR was integrated in the reverse architecture plasmid to achieve high levels of inducible expression, generating pASP28 (ieGFP+cCD20−41BB CAR, Table S1). Robust eGFP expression was concurrent with target cell lysis when pASP28-CAR-T cells were co-cultured with CD20-transduced K562, but not with the CD20− parental line (Figure 5B and Figure S6A).
We then designed an IL-6Rα blocking antibody by linking the tocilizumab-derived heavy and light chains33 in a scFv format fused with human IgG1 Fc to obtain anti-hIL6Rα scFv-Fc referred to as iToci (Table S1). This molecule was cloned into the inducible module of pASP28 to generate pASP52 (iToci+cCD20−41BB CAR, Table S1). CAR-T cells expressed iTociin the presence of CD20 antigen (Figure 5C) and had similar phenotype compared to control ieGFP CAR-T cells following ex vivo expansion (Figure S6B). Next we validated iToci-CAR-T cells in the context of a second hematological antigen system using a clinically applicable CD19 CAR1,5, using the newly generated pASP97 (iToci+cCD19−41BB CAR, Table S1) and control pASP96 (ieGFP+cCD19−41BB CAR, Table S1). In primary T cells, both constructs lysed a CD19+ cell line at similar efficacy, concomitant with inducible antibody secretion (Figure 5D). Retention of CAR-mediated lysis demonstrated that secretion of iToci did not impair the cytotoxic activity of engineered cells in vitro. To demonstrate functionality of the autonomously secreted IL-6Rα-blocking antibody from primary T cells, we investigated its binding capacity to human IL-6Rα expressed on the cell surface. Supernatant from iToci producing, but not control CAR-T cells, stained IL-6Rα expressing cells demonstrating specific binding activity (Figure 5E).
Uni-Vect is modularly designed, which makes it amenable for customized design regarding selection of the receptors and effectors. Additionally, by exchanging the DNA response element, it can be engineered to sense customized inputs. Here, Uni-Vect was reconfigured (Figure S6C,D) to develop a cell-based assay to interrogate IL-6 signaling and determine whether anti-hIL6Rα scFv-Fc is biologically active as an IL-6 blocking agent (see STAR Methods). Specific and dose dependent blocking of IL-6 signaling was observed, albeit at a level less efficient than recombinant tocilizumab (Figure S6E). These results demonstrate that the engineered Fc-based IL-6Rα blocking antibody is biologically active.
Finally, we tested if iToci-CAR-T cells can ameliorate CRS in vivo in a humanized mouse model34,35. Manufactured iToci (pASP97; iToci+cCD19−41BB CAR, Table S1) and control ieGFP (ieGFP+cCD19−41BB CAR, Table S1) CAR-T cells demonstrated similar in vitro expansion (Figure S6F). After verifying engraftment of human hematopoietic stem cells in SGM3 mice, they were challenged with NALM6 cells (Figure 5F). 7 days later, iToci or control ieGFP CAR-T cells were injected and mice were monitored for CRS manifestation over the next 13 days. We found that both, iToci and control iGFP CAR-T cells fully controlled tumor growth (Figure 5G). In contrast to control CAR-T cells, CRS was ameliorated in iToci group as indicated by only transient weight loss (Figure 5H) and survival of 100% compared to 37% in control mice (Figure 5I). Of note, although similar at day 4, T cell expansion was lower in iToci compared to control group as CRS progressed at later time points (Figure 5J). Key inflammatory cytokines associated with CRS severity were significantly decreased in the iToci group on day 4 after infusion (Figure 5K). This includes IL-6, which counters clinical observations following tocilizumab administration36. Notably, 2/8 instances of hemorrhages were observed with control CAR-T while none were observed in the iToci group (Figure S6G). In one mouse with multifocal micro-hemorrhages, a parenchymal hemorrhage of the cerebellum (Figure S6G, left), and a parenchymal hemorrhage of the cortex (Figure S6G, right) were observed.
In a repeated experiment the amelioration of CRS was tested in a more stringent model with rapid weight loss and higher levels of secreted cytokines. The explanation for such challenging conditions is not clear and may have to do with donor-intrinsic factors of the cord-blood HSCs or CAR-T cells themselves. Even under these challenging conditions, iToci was still effective at providing a significant advantage in terms of survival (Figure S6H) and decreased release of cytokines (Figure S6I). Overall, these data demonstrate that Uni-Vect can be implemented for autonomous antigen-triggered secretion of biologically active IL-6Rα blocking antibody by CAR-T cells in situ to ameliorate CRS.
Inducible Transcription Factors Program Therapeutically Relevant CAR-T Cell States and Improve Expansion In Vivo
Transcription factors are master regulators of T cell differentiation, expansion, fitness, and anti-tumor responses37–39. These features, which we refer to as therapeutically relevant T cell states (TRTS), are highly present in successful CAR-T therapies. Here, we explored the implementation of Uni-Vect to install the transcription factors FOXO1 and TCF7, which are associated with early differentiation states, T cell memory, and a functional phenotype, to modulate TRTS. Using Uni-Vect we combined NFAT-inducible TCF7 in the construct pASP72 (iTCF7+cHER2−41BB CAR, Table S1) or FOXO1 in construct pASP73 (iFOXO1−3A+cHER2−41BB CAR, Table S1) with a constitutive HER2-targeting CAR (Figure 6A). For the latter, we created a human FOXO1 version called FOXO1–3A that is resistant to AKT-mediated phosphorylation and hence insensitive to nuclear export40,41.
Figure 6: Inducible Transcription Factors Program Therapeutically Relevant CAR-T Cell States and Improve Expansion In Vivo.
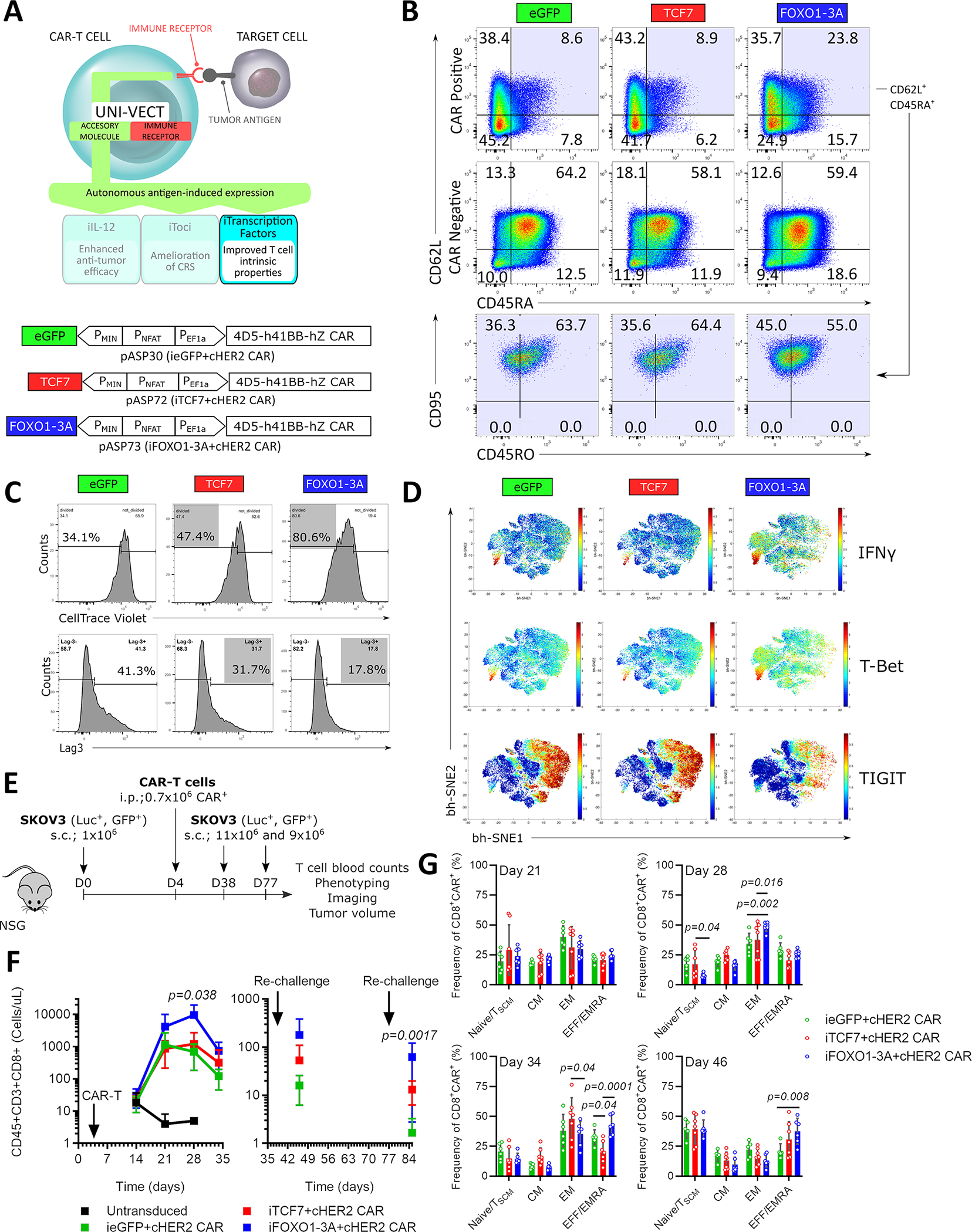
(A) Schematics of iTFs integrated into Uni-Vect; pASP30, pASP72, and pASP73 Uni-Vect constructs. (B) iTF-CAR-T cells were exposed to SKOV3 cells twice (Detailed in Figure S7A). Phenotype was analyzed 3 days after second stimulation. Data is representative of 2 experiments performed. (C) iTF-CAR-T cells were sorted for CAR+ population and exposed to repeated stimulations with SKOV3 cells (Detailed in Figure S7A). Upper: After 2nd stimulation, iTF-CAR-T cells were labeled with CellTrace Violet and re-exposed to SKOV3 cells for 5 days. Proliferation was measured. Lower: Lag3 expression after 3rd stimulation. (D) 3 days after 4th round of stimulation described in (C), T cell phenotype markers were analyzed by CyTOF. (E) In vivo study design. n = 7 mice per experimental group. After initial tumor clearance, mice were re-challenged twice with SKOV3. (F) CD8+ T cell counts in peripheral blood. Left: initial response and Right: after re-challenges. (Kruskal-Wallis test) (G) Memory phenotype of CAR+ T cells from peripheral blood. For F and G, all data with error bars are presented as mean ± SD. (Two-way ANOVA). In vitro data are representative of three experiments. Data from in vivo study are representative of two experiments performed with two independent iTF-CAR-T cell products. See also Figure S7 and Table S1.
In CAR-T cells exposed to repeated stimulations with HER2+ SKOV3 target cancer cells in vitro (Figure S7A upper panel), we found that antigen-dependent FOXO1–3A expression preserved a less differentiated phenotype, as determined by the increased frequency of CD62L+, CD45RA+ CAR-T cells compared to control cells. This population was CD95+, indicating a T memory stem cell phenotype. These effects were observed in CAR+ but not in CAR− populations (Figure 6B).
In experiments where sorted CAR+ iTF-CAR-T cells were re-stimulated multiple times with SKOV3 targets in vitro (Figure S7A lower panel), inducible expression of both TCF7 and FOXO1–3A enhanced antigen-dependent proliferation of iTF-CAR-T cells and decreased expression of the inhibitory marker Lag3 (Figure 6C). CyTOF analysis of iTF-CAR-T cells revealed that FOXO1–3A favorably affected T cell markers associated with effector function, including increased expression of IFN-γ and T-Bet, and a reduction in the inhibitory marker TIGIT (Figure 6D). Incorporation of the iTFs did not impair the cytolytic capacity of HER2-targeting CAR-T cells at the time of manufacturing or after the generated infusion product was subjected to several rounds of antigen exposure (Figure S7B). To reduce possibility that the transduction efficiency and/or levels of CAR expression would affect the results, prior to functional assays we normalized the frequency of CAR+ cells based on the initial transduction and monitored MFI as a measure of CAR expression. Although the transduction efficiency varied depending on the genetic construct, the MFI of CAR+ cells was similar among the groups for all in vitro and in vivo experiments. (Figure S7C).
We then tested the in vivo activity of iTCF7 and iFOXO1–3A compared to control ieGFP-CAR-T cells. Mice harboring an established subcutaneous HER2+ SKOV3 ovarian tumor recieved 0.7 million CAR+ iTF-CAR-T cells (Figure 6E). At the dose administered, all three cellular products with exception of two mice in ieGFP and one in iFOXO1–3A groups cleared tumors and survived after re-challenge with SKOV3 (Figure S7D and S7E). We noticed minor differences in kinetics where mice in iFOXO1–3A group seemed to have the fastest response to re-challenges (Figure S7D). We simultaneously investigated how inducible TCF7 and FOXO1–3A affected CAR-T cell expansion and persistence in vivo. Expansion of T cells in peripheral blood on day 28 was significantly increased in the iFOXO1−3A-CAR-T cell group (14-fold) when compared to control CAR-T cells, while the effect of activation induced TCF7 expression was less prominent (Figure 6F and Figure S7F for individual mice). Importantly, iFOXO1−3A-CAR-T cell expansion was transient, and ultimately contracted after the tumor was cleared. Upon a second re-challenge on day 77, iFOXO1−3A CAR-T cells again expanded significantly more than control cells. We did not observe toxicities or weight loss in any responding mice even at the point of peak expansion in the iFOXO1–3A CAR-T cell group (Figure S7G).
The improved in vitro expansion and accompanying differences in the phenotype of the manufactured iFOXO1–3A cellular product led us to investigate what differentiation states and phenotypes are associated with increased in vivo expansion. We analyzed the phenotype of CAR+ T cells from mice in different treatment cohorts (Figure S7H for gating strategy). A consistent increase in the effector/EMRA T cell compartment (CD62L−, CD45RA+) was observed in the iFOXO1−3A-CAR-T cell cohort compared to control CAR-T cells at the later stages of experiment (Figure 6G). At the time of peak expansion on day 28 that was not the case, but we did observe significant increase in EM compartment (CD62L−, CD45RA−). Together these results demonstrate that inducible FOXO1–3A expression in CAR-T cells improves TRTS, which translated into improved antigen-dependent and transient expansion in vivo, while the favorable phenotypic profile may support improved fitness.
DISCUSSION
T cell-intrinsic dysfunctions and the immunosuppressive tumor microenvironment are key factors limiting the clinical efficacy of CAR-T immunotherapies. Even when efficacious, significant CAR-T cell toxicities can limit their therapeutic potential. These shortcomings emphasize a dire need for strategies to safely outfit T cells with critical attributes required for effective therapy. We addressed this challenge through development of the Uni-Vect platform: a single lentiviral vector that combines activation-induced production of an accessory molecule and constitutive expression of an immune receptor.
When developing the system, two distinct configurations emerged. A forward architecture demonstrated low leakage for tight control of potent immune modulators, and a reverse architecture achieved high levels of inducible expression. The observed increase in transcriptional activity of a non-activated inducible module in the reverse orientation (Figure S2B) is unlikely due to tonic signaling (Figure S2G) and may be explained by bidirectional activity of the EF1α promoter in a combination with PMIN. Indeed, bi-directional promoters have been previously developed and characterized in lentiviral vectors42. The proximity of the EF1α promoter with the NFAT response element, might also increase accessibility of transcriptional machinery to enhance inducible output in the reverse architecture.
The limitations in clinical activity and safety of CAR-T cells highlights the need to accessorize, or “armor,” CAR-T cells. However, current strategies to engineer T cells with inducible factors using activation-triggered promoters are at risk of dysregulated endogenous TCR driven expression of the immune modulator. By knocking out the endogenous TCR, we rendered CAR signaling an exclusive activator of the inducible system (Figure 2); a refinement that may reduce adverse events mediated by dysregulated accessory molecule expression. Our approach integrates endogenous TCR knockout in a CAR-T cell manufacturing procedure similar to one used in clinical trials43. Although TCR knock-out results in a modest reduction in transduction efficiency, small numbers of TCR knock-out, IL-12 secreting CAR-T cells can safely mediate cancer regression (Figure S5L). Moreover, clinical trial results demonstrate that cancer remission can occur with transduction efficiency as low as 5%, and alternative protocols for TCR knockout may improve transduction efficiency5.
Alternatively, endogenous TCRs can be edited by integration of a gene encoding an immune receptor into the TRAC locus utilizing CRISPR-Cas9 and either viral44 or non-viral45,46 donor template delivery. In contrast to Uni-Vect, these approaches tested only constitutive immune receptor expression. Another strategy employed TALEN technology with virally delivered donor template to place a CAR into the TRAC locus and IL-12p70 into either IL2Rα or PDCD1 genes. This approach enabled simultaneous TCR knockout and activation-inducible expression of an effector molecule47 but, in contrast to Uni-Vect, led to non-homogenous modification of the cellular product. Integration of Uni-Vect in these genome targeting platforms could thus enable inducible effector molecule and constitutive immune receptor expression in one step for a homogenous T cell product while knocking out the endogenous TCR or other molecules.
For potential therapeutic implementations of Uni-Vect, we featured three approaches to upgrade CAR-T cell functions. We developed iIL-12-CAR-T cells and demonstrated improved efficacy and safety compared to current alternatives. iIL-12, but not control ieGFP, CAR-T cells were capable of eradicating solid tumors in vivo at low doses. Importantly, iIL-12 did not cause lethal toxicity as was the case when IL-12 was expressed constitutively (Figure 4). In addition to the transient nature of IL-12 secretion by iIL-12-CAR-T cells, a lower overall level of IL-12 may have contributed to the reduced toxicity in this preclinical model, compared with constitutive IL-12. Liu et al. recently developed a single vector system where inducible and constitutive modules were placed into face-to-face orientation and demonstrated that inducible IL-12 improved the anti-tumor activity of CAR-T cells14. We did not include this architecture when optimizing Uni-Vect. We hypothesized that such a configuration would lead to interference during transcription. However, their study demonstrated that this configuration is also feasible, even with SV40 pA being placed in the reverse orientation, which in our system, compromised viral titer. Comparison of both systems may reveal new principles in engineering complex vector systems.
In our second therapeutic implementation, we addressed CAR-T cell toxicities through autonomous secretion of tocilizumab-based antibody to suppress CRS. To validate the biological activity of iToci, we adapted Uni-Vect to detect IL-6 signaling which demonstrated modularity that enabled detection of customized signals (Figure S6C). We anticipate that this STAT3-responsive system can also be therapeutically applied in iToci-CAR-T cells to quantitatively respond to IL-6 levels characteristic of CRS. We demonstrated that iToci-CAR-T cells enabled autonomous in situ expression of iToci that ameliorated CRS-related toxicity in a humanized mouse model. A recent study demonstrated that constitutive expression of an anti-inflammatory IL-1 receptor antagonist in CAR-T cells protected mice from severe CRS48. In contrast, iToci-CAR-T cells are capable of autoregulation to self-sufficiently alleviate CRS manifestations with tocilizumab-based antibody secretion. Interestingly, we observed lower in vivo expansion of iToci-CAR-T cells compared to control CAR-T cells (Figure 5J), implying that multiple factors may have contributed to CRS amelioration. IL-6 signaling blockade may have limited expansion at later time points by reducing the overall inflammatory milieu. This hypothesis is supported by the decreased levels of inflammatory cytokines on day 4 when T-cell numbers in the blood were equivalent. Conversely, we cannot exclude that iToci-CAR-T cells can be qualitatively different compared to control CAR-T cells in function in addition to secreting iToci, although in vitro expansion, phenotype and in vitro and in vivo cytotoxic activity was similar (Figure 5 and Figure S6). Even though further studies are necessary to better understand the underlying mechanisms, we conclude that iToci-CAR-T cells were able to reach the right balance of in vivo expansion and cytokine secretion to be effective in tumor clearance without lethal toxicity (Figure 5G–K).
Our third therapeutic implementation programs T cell differentiation states, functionalities, and expansion via inducible TF expression. Analysis of iTF-CAR-T cell function and phenotype revealed that antigen-inducible expression of a single TF can favorably affect TRTS (Figure 6B–D). iFOXO1−3A-CAR-T cells demonstrated remarkably increased peak expansion in peripheral blood when compared to control CAR-T cells (Figure 6F). iTF-CAR-T cell expansion was transient due to Uni-Vect, and T cells ultimately contracted after tumors were cleared. This finding underscores the importance of inducible TF expression to reduce the risk of malignant transformation or autoimmunity. No antigen-independent proliferation or sustained growth was observed in vitro or in vivo. FOXO1–3A was recently tested in the context of cellular immunotherapies where nanocarriers were used to deliver FOXO1–3A mRNA in vitro for transient expression during the manufacturing process to program CAR-T cells in a “hit-and-run” manner41. Uni-Vect enables permanent genetic modification of the cellular product while the expression of FOXO1–3A remains transient. This expands the activity of FOXO1–3A beyond T cell manufacturing to autonomously function after infusion. This was demonstrated through an in vitro re-challenge model, where iFOXO1−3A-CAR-T cells developed less differentiated T cell states, increased TSCM pools (Figure 6B) and also increased expression of molecules that indicate efficacy (Figure 6D). Induced differences in T cell states were aligned with improved in vitro and in vivo expansion. Unexpectedly, there was a slight shift to a phenotype associated with effector functions of iFOXO1−3A-CAR-T cells in vivo (Figure 6G). Further in-depth studies are needed to investigate the underlying mechanisms. In our models, inducible TCF7 resulted in modest phenotypic change and increased expansion. In a recent study46, TCF7 was identified as one of the lead candidates that increased T cell proliferation but not tumor clearance by TCR engineered T cells. A comparison between inducible and constitutive expression of FOXO1–3A and TCF7 in CAR-T is an important next step. However, the potential for uncontrolled proliferation and malignant transformation due to constitutive TF expression may be an unnecessary risk given the improvements already evident when using Uni-Vect.
Engineered T cells expressing transgenic receptors have immense potential as therapeutic agents, but extensive characterization of potential immune receptors is necessary to increase therapeutic efficacy and reduce the risk of adverse events32. Although testing receptors in primary T cells is indispensable, a Uni-Vect cell-based system that can assess NFAT activation as a readout of TCR signaling provides a rapid screening assay to qualify receptors for further studies. We utilized Uni-Vect to develop JASP90 or JASP89, and demonstrated how TCRs engineered into these reporter cell lines can be characterized for antigen specificity and avidity (Figure 3) with a pattern of recognition identical to T cells from which the receptors were isolated26,27. Compared to other antigen-responsive reporter cell lines that use two-component systems49–51, Uni-Vect enables homogenous modification and straight-forward selection of cells that eliminates the need for clonal selection and activation to reveal the signal. Furthermore, Uni-Vect allows for rapid query of cell line panels or tumors for processing and presentation of specific tumor antigens (Figure 3) and, for screening of antigen expression on non-targeted tissues to avoid toxicities. This system may be complementary to other recently developed platforms52,53. Uni-Vect lays the foundation for a single vector system with fixed stoichiometry for researchers who need to characterize the activity of candidate immune receptors in either cell lines or primary cells.
Our findings demonstrate that Uni-Vect is a broadly applicable platform for upgrading T cells in a user-friendly plug-and-play manner (Figure 7) and enables the manufacturing of well-defined cell products with homogenous transgene distribution. We demonstrate three therapeutic implementations of Uni-Vect in developing CAR-T cells with upgraded functionalities. Additionally, Uni-Vect can be utilized to screen and characterize antigen-specific immune receptors. While these implementations are significant and have therapeutic potential, the downstream applications of Uni-Vect extend well beyond what could be covered in this paper. As the field of immunotherapy progresses, we predict that the need for a system that efficiently couples inducible and constitutive gene expression will expand. With these contributions, we have established a foundation for more effective and clinically actionable next-generation cellular immunotherapies.
Figure 7: Schematic Representation of the Uni-Vect Platform and Implementations.
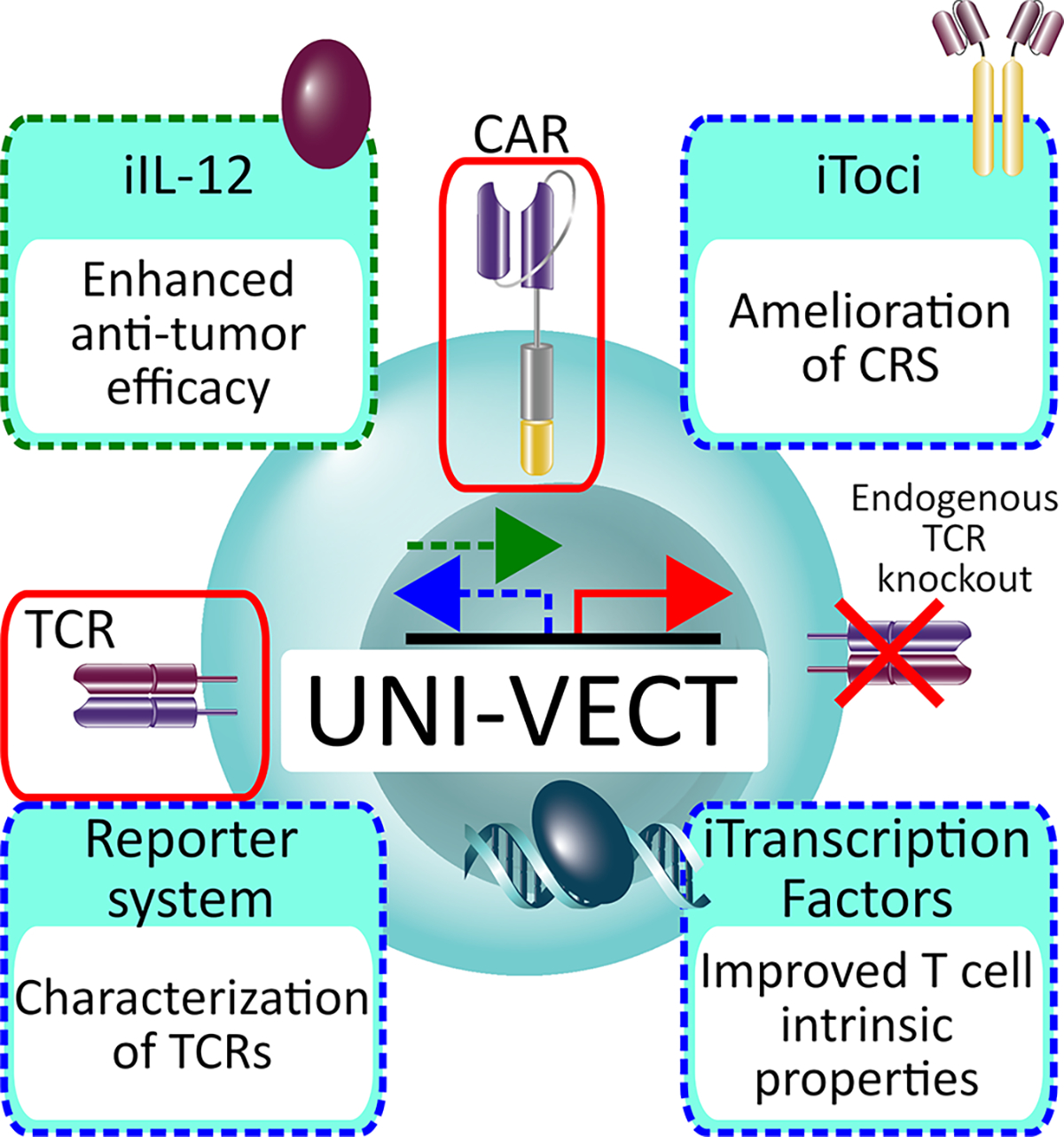
Uni-Vect is a modular platform to create upgraded T cells in a user-friendly plug-and-play manner. Two architectures of inducible module are represented by green (forward) and blue (reverse) dashed arrows. The constitutive module is represented by red arrow. Three therapeutic and one research tool implementations are shown and are color-coded according to the architecture used.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel J. Powell Jr. (poda@pennmedicine.upenn.edu).
Materials Availability
All genetic constructs and sequences are listed in the Table S1. Plasmids generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and Code Availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell Lines and Primary Human T Cells
Human CAR-T cells were generated from normal donor T cells provided by the University of Pennsylvania Human Immunology Core. All primary T cell studies were covered through approval by the University of Pennsylvania Institutional Review Board (IRB). All de-identified donors signed approved consent forms. Cells were cultured in complete medium (CM) comprising of RPMI 1640 (GIBCO) supplemented with 10% FBS (VWR), 100 U/mL of penicillin, and 100 μg/mL of streptomycin at 37 °C and 5% CO2. All cell lines were routinely tested for mycoplasma. Details are listed in Methods Details (Cell lines section). For the in vivo iToci study, the human primary samples provided are human cord blood CD34+ cells. Cells were purchased from a commercially available vendor (Lonza; product number “Dn39444” for the experiment in Figure 5F–K, “CB21/0017/35” for the experiment in Figure S6H,I). Normal donor T cells: T cells were isolated from healthy volunteers who previously signed an informed consent, which authorized use for general research purposes. Samples were centrally collected and sent to the laboratory for processing after complete anonymization. The leukemia cell line NALM-6 was purchased from the American Type Culture Collection (ATCC) and cultured in RPMI 1640 (BioWhittaker) supplemented with 10% FBS (Lonza), 100 IU/ml penicillin/streptomycin and glutamine. The following cell lines were gift from the Gill lab at the University of Pennsylvania: Parental and engineered CBG+GFP+ human HER2+ ovarian cancer cell line SKOV3, parental and engineered CBG+GFP+ human HER2− breast cancer cell line MDA-MB-468, CD20+ and engineered CBG+GFP+ Raji Burkitt’s lymphoma and CD19+ and engineered CBG+GFP+ NALM6 human acute lymphoblastic leukemia. These cells were further sorted for uniform protein expression using fluorescence-activated cell sorting (FACS). Parental and engineered CD20+ K562 erythro-megakaryoblastic leukemia cell lines were a gift from the Posey lab at the University of Pennsylvania54. HEK 293T and Jurkat cell lines were purchased from the American Type Culture Collection (ATCC). Cells were cultured in complete medium (CM) comprising of RPMI 1640 (GIBCO) supplemented with 10% FBS (VWR), 100 U/mL of penicillin, and 100 μg/mL of streptomycin at 37 °C and 5% CO2. All cell lines were routinely tested for mycoplasma.
Animal Model Details
NOD/SCID/IL2rγnull (NSG) mice were purchased from the Stem Cell and Xenograft Core (University of Pennsylvania). Female mice (6–12 weeks old) were kept in a pathogen-free environment within individually ventilated cages. Animal studies were performed in full accordance with the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC protocol n. 805773). The specific designs of in vivo studies are indicated at each experiment in the manuscript.
The iToci study: All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of IRCSS San Raffaele Scientific Institute and by the Italian Governmental Health Institute (Rome, Italy). Mice were kept in a specific-pathogen-free (SPF) facility within individually ventilated cages and were given irradiated food and water ad libitum. All experiments were approved by the Institutional Animal Care and Use Committee (IACUC 646) of IRCSS San Raffaele Scientific Institute and by the Italian Governmental Health Institute (Rome, Italy). 6 to 8-week-old SGM3 (NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ; The Jackson Laboratory) were used for experiments. Both male and female animals were employed in all experiments.
METHOD DETAILS
Design and molecular cloning of lentiviral constructs
The lentiviral constructs designed and used in this study are listed and explained in detail in Table S1 including their architecture and annotated sequences of relevant elements. The basic Uni-Vect genetic cassettes comprise NFAT-inducible eGFP and constitutive mCherry modules in the single lentiviral transfer plasmid either in forward (pASP4.2) or reverse (pASP5) orientation (Table S1). The Uni-Vect cassette was synthesized (GeneArt) and cloned into 3rd generation self-inactivating (SIN) lentiviral transfer vector pELNS55 or pTRPE56 that The University of Pennsylvania uses for clinical CAR-T cell manufacturing. In the forward architecture design, antigen-inducible eGFP and downstream constitutive EF1α driven mCherry modules are placed in the forward 5’-3’ orientation relative to the 5’LTR. The inducible module is composed of a synthetic NFAT response element that comprises 6 repetitions of the consensus NFAT binding site derived from the pGL4.30 (Promega) (PNFAT; Table S1) placed directly upstream of the previous validated57 minimal promoter PMIN, which is derived from the pGL4.23 (Promega). A linker region placed downstream of the PMIN creates the optimized distance between transcription and translation start sites, followed by the Kozak sequence and a start codon for eGFP. To further characterize the system, we investigated if an upstream ATG start codon between the PMIN and the linker in pASP4.2 influenced system performance and demonstrated no significant difference compared to pASP4.2.1 where ATG was absent (Figure S2A). Priority was given to the version of Uni-Vect without upstream ATG. The reverse architecture is composed of the same modules as in the forward architecture but placed in the back-to-back orientation. The inducible module was therefore placed in a reverse 3’−5’ orientation relative to the 5’LTR. hIL-12 fusion protein, iToci (anti-hIL6Rα scFv-Fc-Myc tag), hIL-6Rα, TCF7 and FOXO1–3A were synthesized (GeneArt) and cloned into the Uni-Vect as described in Table S1. All genetic constructs were prepared using standard digest and ligate restriction enzymes-based molecular cloning techniques and were sequence verified. All cloning and plasmid propagation steps were performed in XL1-Blue Supercompetent Cells (Agilent Technologies, Cat# 200236) to maintain integrity of lentiviral transfer plasmids.
Lentivirus production
The human embryonic kidney (HEK) 293 T cell line (American Type Culture Collection, Manassas, VA, USA) were grown to 70% confluency in CM at 37 °C and 5% CO2. Uni-Vect transfer plasmid and lentiviral packaging plasmids pRSV.REV (Rev expression vector), pMDLg/p.RRE (Gag/Pol expression plasmid) and pVSV-G (VSV glycoprotein expression vector) were added to Opti-MEM (Thermo Cat. 31985088) at a 15:18:18:7 mass-unit ratio. This mixture was added to a 10:1 Opti-MEM and Lipofectamine 2000 (Thermo Cat# 11668019) mixture at 1:1 volume ratio and let incubate at RT for 10 min before addition to HEK 293T cells in fresh culturing media. Supernatants containing the lentivirus were collected at 24 h and 48 h and passed through 0.45 μm filters. Filtered lentivirus product was concentrated by ultracentrifugation at 25 000 rpm for 2.5 h and stored at −80 °C. The viral titer was determined by transducing HEK 293T cells and expressed as Infection Units per mL (IU/mL).
Generation of CAR-T cells: T-Cell activation, transduction and expansion
Healthy donor primary human T cells were purchased from the Human Immunology Core (University of Pennsylvania). CD4+ and CD8+ T cells were combined at 1:1 ratio and stimulated with anti-CD3/CD28 dynabeads (Invitrogen) in a 3:1 ratio on Day 0. After 24 h (Day 1), lentivirus was added at a multiplicity of infection (MOI) of 0.3–10 (depending on the experiment). CM volume was doubled daily until Day 6 when Dynabeads were removed by magnetic separation. Cells were then counted daily by Coulter Counter (Beckman Coulter) and maintained at 0.75 × 106 cells/mL by addition of fresh IL-2 (100 IU/mL, Prometheus Therapeutics and Diagnostics) supplemented CM until day 10 when media was switched to non-supplemented CM. Cells were either frozen or used experimentally once the cell volume reached 280–330 fL at around Day 14. In some experiments as stated in the manuscript, CAR-T cells were sorted for CAR+ using FACS. During manufacturing, T cells were maintained at 37 °C and 5% CO2. The transduction efficiency was determined by flow cytometry for the expression of relevant gene (mCherry, eGFP or immune receptor). For experiments with Jurkat cell line, cells were transduced with Uni-Vect lentiviruses at various MOIs as stated in the manuscript and used in experiments 2–5 days later. For evaluation of the Uni-Vect in primary human T cells they were transduced with constructs at MOIs indicated in the figures and expanded according to the standard protocol described above. Rested cells were stimulated with PMA+ionomycin (Cell Stimulation Cocktail, eBioscience™ Cat# 00-4970-93), antiCD3/CD28 dynabeads, HER2 beads (Acro Biosystems Cat# K006) or with HER2+ SKOV3 ovarian cancer and HER2− MDA468 cell lines at effector to target (E:T) ratios indicated in the figures with or without dasatinib (Sigma-Aldrich). Inducible eGFP was monitored using flow cytometry. Expression of reporter genes was monitored by flow cytometry and quantified as MFI after 24 h and by fluorescence microscopy. In reversibility experiments, transduced Jurkat cells were FACS sorted for mCherry+.
To compare the functionalities of Uni-Vect with a two-component lentiviral system, we benchmarked pASP30 (ieGFP+cHER2−41BB CAR, Table S1) against a system with modules split into two lentiviral vectors pASP5 (ieGFP+cmCherry, Table S1) and pP7 (constitutive HER2–41BB CAR, Table S1). We transduced primary T cells with either pASP30 or a combination of pASP5 and pP7 viruses, each at MOI3. In principle, the number of integrated inducible and constitutive modules should be similar between Uni-Vect and the two-component system but with the total amount of virus added being two times higher in the latter.
TCR Knockout in CAR-T cells using CRISPR/Cas9 RNPs
Healthy donor primary human T cells were purchased from the Human Immunology Core (University of Pennsylvania). CD4+ and CD8+ T cells were combined at 1:1 number ratio. On Day 0 T-cells were centrifuged and resuspended in fresh CM supplemented with IL-7 and IL-15 (25 ng/mL each, R&D Systems). On Day 1 T-cells were washed 1x with PBS. 5μg TRAC (5’-TGTGCTAGACATGAGGTCTA-3’) and 5μg TRBC (5’-GGAGAATGACGAGTGGACCC-3’) sgRNA per sample were incubated with 10μg TrueCut Cas9 (Thermo Cat# A36498) for 10 min. PBS was aspirated from washed T-cells and the 50e6/ml (5e6 in 100uL) cells were electroporated using the Amaxa P3 Primary Cell kit and protocol (Lonza Cat# V4XP-3012) with a Lonza 4-D Nucleofector electroporation device (Cat# AAF-1002B model). After electroporation, cells were resuspended in IL-7/IL-15 (25 ng/mL each) supplemented CM for 48 h before use in the CAR-T cell production protocol as described in “Generation of CAR-T cells: T-Cell activation, transduction and expansion”. Residual TCR expression following knockout was sufficient for activation by Dynabeads in the CAR-T cell production protocol. Following expansion, edited TCR− CAR-T cell products were purified by negative selection with anti-biotin microbeads (Miltenyi Cat# 130−105-637) and biotin conjugated anti-CD3 (Biolegend, Cat# 317320) on a MACS separation column (Miltenyi, Cat# 130−042-201) where the remaining CD3 positive, and therefore TCR+, cells were removed. Flow through containing TCR− cells was collected and cells were placed in fresh CM. This pipeline is schematically represented in Figure S3A.
Characterization of T-cell Receptors
A Jurkat TCRα/β negative cell line was generated from the Jurkat E6.1 cell line using Cas9 protein and TRAC and TRBC1/TRBC2 sgRNA as described above. This TCR α/β negative Jurkat cell line was transduced with human CD8α/β heterodimer and pASP89 or pASP90 Uni-Vect/mCherry constructs to generate JASP89 or JASP90 reporter lines. JASP89 or JASP90 reporter cells were flow cytometry sorted to 99% purity based on TCR negative (anti-human TCRα/β antibody clone IP26, Biolegend, San Diego, CA), CD8 positive (anti-CD8 antibody, ThermoFisher Scientific, Waltham, MA), mCherry positive expression and low basal NFAT (eGFP+) signal. Melanoma neoantigen-specific TCRs (Table S2) were cloned into pTRPE, and produced lentivirus was used for transduction of JASP89 or JASP90 reporter cells. Neoantigen-specific TCR-expressing cells were sorted using custom p/HLA multimers (Proimmune, Sarasota, FL) and expanded for use in functional assays. For antigen recognition assays, titrated concentrations of mutated or wild-type peptide (10 μM – 1 pM) were used to pulse monoallelic HLA-A*02:01-expressing K562 cells for 1 h at 37 °C. TCR signaling via NFAT activation was monitored by eGFP expression. CD69 (anti-CD69 antibody, Biolegend, San Diego, CA) expression was monitored after stimulation by cell surface staining. Culture supernatants were collected after activation and tested in multi-plex assay for cytokine production.
Peptide titration data was fitted to a dose response curve by linear non-regression analysis using GraphPad Prism version 7.0. TCR-expressing JASP89 or JASP90 cells activated with PMA (50 ng/mL) and Ionomycin (750 ng/mL) were included as positive controls. For recognition of processed and presented antigen, TCR-expressing JASP89 or JASP90 cells were mixed at a 1:1 ratio with TMC-expressing DM6, incubated for 16–24 h. eGFP expression was monitored by flow cytometry. TMC consisting of 9 minigenes (Table S2) were cloned into pTRPE, expressed as lentivirus and used to transduce DM6 cells. TMC expressing cells were selected by sorting for RFP+ cells expressing cell surface HLA-A*02:01/SVG9 peptide complexes as detected by a T-cell receptor mimic (TCRm) monoclonal antibody58. MUT- and WT-TMC reactivity with the HLA-A*02:01/SVG9 peptide complex specific TCRm monoclonal antibody validated expression of the mini-gene constructs.
In vitro analysis of CAR-T cell function
Cytotoxicity assays. Lytic function of CAR-T cells was measured using the LucScreen Extended-Glow Luciferase Reporter Gene Assay System (Applied Biosystems). Depending on the experiment, Uni-Vect-engineered CAR-T cells and tumor cells expressing click beetle green luciferase (CBG) were combined at 3:1, 1:1, 0.5:1 and 0.3:1 Effector to Target (E:T) ratios in a total of 200 μL CM in an white opaque walled flat bottom plate and co-cultured overnight at 37 °C and 5% CO2. Plates were then centrifuged at 1200 rpm for 5 min, 100 μL of medium was removed from each well for further analysis, and luciferase buffer was added directly into the plate according to the manufacturer’s protocol. Luciferase readings were obtained using a microplate reader (BioTek Synergy H4). Cytotoxicity was calculated using the following equation where: (1 − luminescence(CAR-T cells + target cells)/luminescence(Target cells)) × 100. Cytokine release. CAR-T cells were stimulated according to specific experiment either with PMA+ionomycin or through co-culture with target cells. Cytokine levels were measured by a sandwich ELISA according to the manufacturer’s instructions (BioLegend) as follows: human hIFN-γ was detected by hIFN-γ (BioLegend, Cat# 430104), human hIL-2 was detected by hIL-2 (BioLegend, Cat# 431804) and hIL-12 was hIL-12 (BioLegend, Cat# 431704). Live-cell imaging. Jurkat cells were transduced with pASP4.2 or pASP5 and sorted 5 days later for mCherry+. Next day the cells were plated in 96-well plates and stimulated with PMA+ionomycin for 24 h and then washed, left unstimulated for 120 h, and then re-stimulated. eGFP was monitored in a real-time manner using IncuCyte live-cell imaging (Incucyte® Systems for Live-Cell Imaging and Analysis).
Flow cytometry
Samples were spun down at 1300 RPM, then washed once with staining buffer. Samples collected from mice had 2 mL of Ack Lysis buffer added for 5 min to lyse red blood cells then washed once with PBS. Samples were stained for CAR surface expression for 30 minutes at room temperature with 100 μL solution containing the goat polyclonal anti-human IgG (Sigma-Aldrich, Cat# I1886–2ML) conjugated with Lightning-Link APC (Expedeon). After CAR staining, samples were washed three times with staining buffer and then incubated at 4°C for 30 minutes in 50 μL of an antibody cocktail to label human surface markers. The following anti-human antibodies were used: CD4 Pacific Blue (BioLegend Cat# 300521), CD45RA APCCy7 (BioLegend Cat# 304128), CD45RO BV570 (BioLegend Cat# 304226), CD62L BV605 (BD Biosciences Cat# 562719), CD95 PE (BioLegend Cat# 305608), CD8 BV786 (BD Biosciences Cat# 563823). Samples were washed three times and incubated with Live/Dead Aqua (Thermo-Fischer Scientific Cat# L-34966) for 10 minutes to discriminate live and dead cells. Samples were washed twice to remove Live/Dead Aqua before being run on a Fortessa.
Fluorescent-Activated Cell Sorting (FACS)
Samples were spun down at 1300 RPM, then washed once with staining buffer (phosphate-buffered saline, 2% fetal bovine serum). Samples were stained for CAR surface expression for 30 minutes at room temperature with 100 μL solution containing the goat polyclonal anti-human IgG (Sigma-Aldrich, Cat# I1886–2ML) conjugated with Lightning-Link APC (Expedeon). After CAR staining, samples were washed three times with staining buffer then stained with Live/Dead Aqua (Thermo-Fischer Scientific Cat# L-34966) for 10 minutes to discriminate live and dead cells. Samples were washed twice to remove Live/Dead Aqua. Samples were sent to the The Penn Cytomics and Cell Sorting Resource Laboratory at UPenn for fluorescent-activated cell sorting (FACS) on an Aria to sort for CAR positive cells. For sorting parameters, singlets were detected using FSC-H versus FSC-A followed by SSC-H versus SSC-A. Cells negative for Live/Dead Aqua, were identified as live cells.
Fluorescence microscopy
Uni-Vect-engineered CAR-T cells or Jurkat cells were stimulated according to specific experiment either with PMA+ionomycin or target cells. To directly observe Uni-Vect performance, eGFP and mCherry were visualized using EVOS Cell Imaging System (ThermoFisher Scientific).
Biological activity of iToci
To analyze biological activity of iToci (anti-hIL6Rα scFv-Fc-Myc tag), we first developed a cell-based reporter assay to interrogate IL-6 signaling. We adapted the reverse architecture of Uni-Vect by exchanging NFAT with the STAT3 response element and integrating IL-6Rα as a constitutive module to generate pASP59 (Table S1). To validate the reporter system, HEK 293T cells were transduced with pASP59 and stimulated with increasing amounts of human IL-6 and eGFP expression was monitored via flow cytometry.
To produce iToci, pASP52-CAR-T cells (iToci+cCD20−41BB CAR) were co-cultured with CD20+ cell line for 72 h to induce antigen-dependent expression and secretion. iToci was also expressed in a constitutive manner by transducing HEK 293T cells with the construct pASP75 (EF1α-iToci). Supernatants from cell cultures were collected and iToci levels were measured using human IgG1 Platinum ELISA (eBioscience, Cat# BMS2092TWO). First, supernatants from activated iToci-CAR-T cells were added to the HEK 293T cells engineered to express hIL-6Rα and incubated for 1 h. Binding iToci was detected by secondary stain against Myc tag (Cell Signaling Technology, Cat#2233S). Second, the biological activity of the anti-hIL6Rα scFv-Fc (iToci) was further analyzed by competitive inhibition of IL-6 signaling. To that end, Uni-Vect was reconfigured to develop a cell-based assay system to interrogate IL-6 signaling. We adapted the reverse architecture of Uni-Vect by exchanging NFAT with the STAT3 response element and integrating IL-6Rα into a constitutive module to generate pASP59 (STAT3-eGFP+chIL-6Rα, Figure S6C left and Table S1). To demonstrate functionality of this reporter system, HEK 293T cells were transduced with pASP59 and exposed to increasing amounts of human IL-6 which triggered a dose dependent response observed by eGFP MFI (Figure S6C right). To avoid a need of stimulating iToci-CAR-T cells to produce iToci, which would interfere with the developed cell-based assay because of factors that activate the STAT3-responsive promoter, we constitutively expressed iToci in HEK 293T cells and verified its molecular mass (Figure S6D). Supernatant containing constitutively expressed iToci or control antibody from HEK 293T cells was added to the generated IL-6-responsive reporter cell line 15 min before stimulation with hIL-6 (7.5 ng/mL, R&D Systems Cat# 206-IL-050/CF). After 24 h, the molar ratio-dependent decrease in eGFP expression was monitored via flow cytometry. Recombinant tocilizumab (Actemra) was used as a control.
Western blot
Supernatant (30 μL) from constitutively expressed iToci (anti-hIL6Rα scFv-Fc-Myc tag) was mixed with loading buffer (Lammeli buffer; BioRad) with or without 5% β-mercaptoethanol (BioRad), boiled for 5 min at 95 °C and centrifuged at 14000g for 3 min. Samples were resolved on 4−15% Minigels protean TGX (BioRad) and run at 150 V for 1 h. A protein ladder (BioRad) was run along with the samples. Protein samples were transferred to a PVDF membrane (Millipore) at 100 V for 1 h. The membranes were washed with TBST (1% Tween; BioRad) and incubated overnight at 4 °C with Myc-Tag (9B11) Mouse mAb HRP Conjugate (1:1000) (Cell Signaling Technology, Cat# 2040S). Membranes were washed four times in between the primary and secondary antibody incubation steps. Membranes were developed using the ECL prime Western blotting detection reagent (GE Healthcare, RPN2236) and imaged (GE ImageQuant LAS 4000 series imaging system). Irrelevant constitutively expressed protein was used as a control.
Mass cytometry staining and analysis
All mass cytometry reagents were either bought from Fluidigm or conjugated in house using MAXPAR kits (Fluidigm). Antibody information is in Key resource table. Cell identifier stain Iridium191/193, identifier 127Idu, and Cisplatin 195 were purchased from Fluidigm. All antibodies were titrated to determine optimal concentrations for staining samples. For downstream intracellular effector molecule detection, samples were treated with GolgiStop (BD Biosciences) 4–5 hours before staining. For staining, single-cell suspensions of ieGFP (pASP30), iTCF7 (pASP72), and iFOXO1–3A (pASP73) CAR-T cells were spun down, and incubated with 127IdU (5-Iodo-2’ -deoxyuridine) and cisplatin 195 in PBS for 10 minutes at room temperature to identify live/dead cells. Next, cells were washed in the staining buffer provided by Fluidigm and incubated for 30 minutes at room temperature with an antibody cocktail containing all surface antibodies. Following surface marker incubation, cells were washed three times in staining buffer. To detect intracellular markers, cells were fixed using 30 minutes with nuclear antigen staining buffer (Fluidigm), washed twice with Maxpar Nuclear Antigen Staining Perm (Fluidigm), and then incubated with the intracellular staining cocktail for 2hrs at room temperature. Cells were washed three times before being fixed in 1.6% PFA with 125nM Iridium 191/193 overnight at 4C. The next day, cells were washed 2x in PBS, then 1x in dH2O. Data acquisition was performed on a CyTOF Helios (Fluidigm) by the CyTOF Mass Cytometer Core at the University of Pennsylvania. The CyTOF core performed bead-based normalization for all samples. To perform traditional biaxial analysis, Flowjo version 10 software was used. Intact single cells were identified using Iridium 191/193 and event-length. Live cells were identified using 127IdU and cisplatin, where dead cells are positive for cisplatin. CD3 and CD45 positivity identified T-cells. High-dimensional analysis was performed using viSNE from cyt a tool written in Matlab (R2016b). Single, live, CD3+CD45+ exported FCS data from ieGFP (pASP30), iTCF7 (pASP72), and iFOXO1–3A (pASP73) Uni-Vect samples were imported into cyt, arcsinh5-transformed, randomly subsampled by cyt for 25,000 cells per sample, then bh-SNE mapped as described59. Bh-SNE mapping used the following parameters: CD95, CTLA-4, CD62L, PD-1, CD45RO, CD244, CD4, CD8, EOMES, Lag-3, TIGIT, CCR7, Granzyme B, CD57. All viSNE plots were created using cyt.
KEY RESOURCES TABLE.
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Anti-Myc tag (Alexa Fluor® 647 Conjugate) | Cell Signaling Technology | Cat# 2233S |
| Goat polyclonal anti-human IgG | Sigma-Aldrich | Cat# I1886-2ML |
| CD4 Pacific Blue | BioLegend | Cat# 300521 |
| CD45RA APCCy7 | BioLegend | Cat# 304128 |
| CD45RO BV570 | BioLegend | Cat# 304226 |
| CD62L BV605 | BD Biosciences | Cat# 562719 |
| CD95 PE | BioLegend | Cat# 305608 |
| CD8 BV786 | BD Biosciences | Cat# 563823 |
| anti-Myc-Tag (9B11) Mouse mAb HRP Conjugate | Cell Signaling Technology | Cat# 2040S |
| anti-CD95 Clone DX2 164Dy | Fluidigm | Cat# 3164008B |
| anti-CTLA-4 Clone 14D3 170Er | Fluidigm | Cat# 3170005B |
| anti-CD62L Clone DREG56 153Eu | Fluidigm | Cat# 3153004B |
| anti-PD-1 Clone EH12.2H7 155Gd | Fluidigm | Cat# 3155009B |
| anti-CD45RO Clone UCHL1 165Ho | Fluidigm | Cat# 3165011B |
| anti-CD244 polyclonal 142Nd | Novus Biologicals | Cat# AF1039 |
| anti-CD4 Clone RPAT4 145Nd | Fluidigm | Cat# 3145001B |
| anti-CD8 Clone RPAT8 146Nd | Fluidigm | Cat# 3146001B |
| anti-EOMES clone 644730 148Nd | Novus Biologicals | Cat# MAB6166 |
| anti-Lag-3 Clone 11C3C65 150Nd | Fluidigm | Cat# 3150030B |
| anti-CD3 Clone UCHT1 170Er | Fluidigm | Cat# 3170001B |
| anti-TIGIT Clone MBSA43 154Sm | Fluidigm | Cat# 3154016B |
| anti-CCR7 Clone G043H7 159Tb | Fluidigm | Cat# 3159003A |
| anti-CD45 Clone HI30 89Y | Fluidigm | Cat# 3089003B |
| anti-Granzyme B Clone GB11 171Yb | Fluidigm | Cat# 3171002B |
| anti-CD57 Clone HCD57 172Yb | Fluidigm | Cat# 3172009B |
| T-cell receptor mimic (TCRm) monoclonal antibody | Kim, S. et al.58 | NA |
| Biological Samples | ||
| Primary T cells | University of Pennsylvania Human Immunology Core. | NA |
| CD34+ Cells | Lonza | Product number “Dn39444” for the experiment in Figure 5F–K, “CB21/0017/35” for the experiment in Figure S6H,I |
| Chemicals, peptides, and recombinant proteins | ||
| anti-CD3/CD28 dynabeads | Invitrogen | Cat# 402.03D |
| anti-biotin microbeads | Miltenyi | Cat# 130-105-637 |
| HER2 beads | Acro Biosystems | Cat# K006 |
| PMA+ionomycin | eBioscience | Cat# 00-4970-93 |
| Live/Dead Aqua | Thermo-Fischer Scientific | Cat# L-34966 |
| RPMI 1640 | GIBCO | Cat# 72400-047 |
| Foetal bovine serum (FBS) | VWR | Cat# S170G |
| Peptides | This manuscript | Please see Table S2 |
| Custom p/HLA multimers | Proimmune, Sarasota, FL | NA |
| Critical commercial assays | ||
| Amaxa P3 Primary Cell kit and protocol | Lonza | Cat# V4XP-3012 |
| Experimental Models: Cell Lines | ||
| HEK 293T | ATCC | CRL-3216 |
| Jurkat E6.1 | ATCC | TIB-152 |
| CD19+ NALM6 human acute lymphoblastic leukemia | ATCC | CRL-3273 |
| Engineered CBG+GFP+ NALM6 human acute lymphoblastic leukemia | University of Pennsylvania, Dr. Saar Gill | NA |
| Parental human HER2+ ovarian cancer cell line SKOV3 | ATCC | HBT-77 |
| Engineered CBG+GFP+ human HER2+ ovarian cancer cell line SKOV3 | University of Pennsylvania, | NA |
| Dr. Saar Gill | ||
| Parental HER2− breast cancer cell line MDA-MB-468 | ATCC | HBT-132 |
| Engineered CBG+GFP+ HER2− breast cancer cell line MDA-MB-468 | University of Pennsylvania, Dr. Saar Gill | NA |
| CD20+ Raji Burkitťs lymphoma | ATCC | CCL-86 |
| Engineered CBG+GFP+ Raji Burkitťs lymphoma | University of Pennsylvania, Dr. Saar Gill | NA |
| K562 erythro-megakaryoblastic leukemia cell line | ATCC | CRL-3343 |
| Engineered CD20+ K562 erythro-megakaryoblastic leukemia cell line | University of Pennsylvania, Dr. Avery D. Posey, Jr.; Panjwani, M.K. et al.54 | NA |
| XL1-Blue Supercompetent Cells | Agilent Technologies | Cat# 200236 |
| Experimental Models: Organisms/Strains | ||
| NOD/scid/IL2rg | University of Pennsylvania Stem Cell and Xenograft Core | RRID:IMSR_JAX:005557 |
| SGM3 (NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ | The Jackson Laboratory | RRID:IMSR_JAX:013062 |
| Recombinant DNA | ||
| Lentiviral construct pASP4.2 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP4.2.1 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP5 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP7 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP8 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP9 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP18 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP26 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP28 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP30 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP36 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP38 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP52 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP59 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP72 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP73 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP75 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP85 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP89 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP90 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP96 | This manuscript | Please see Table S1 |
| Lentiviral construct pASP97 | This manuscript | Please see Table S1 |
| Lentiviral construct ceGFP | This manuscript | Please see Table S1 |
| Lentiviral construct chIL-12 | This manuscript | Please see Table S1 |
| Software and algorithms | ||
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/ |
| FlowJo | BD | https://www.flowjo.com/ |
| Cyt tool (R2016b) | Columbia University | https://www.c2b2.columbia.edu/danapeerlab/html/cyt-download.html |
Animal studies
NOD/SCID/IL2rγnull (NSG) mice were purchased from the Stem Cell and Xenograft Core (University of Pennsylvania). Female mice (6−12 weeks old) were kept in a pathogen-free environment within individually ventilated cages following protocols approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC protocol n. 805773). The specific designs of in vivo studies are indicated at each experiment in the manuscript. For solid human HER2+ ovarian cancer tumor models, mice were subcutaneously (s.c.) injected in the flank with engineered CBG+GFP+ human HER2+ ovarian cancer cell line SKOV3 (numbers used in specific experiments are indicated in the figure). After 4 days when tumors were established and confirmed by imaging, CAR+ T cells targeting HER2 at doses indicated in the specific experiments in the figure, were injected intraperitoneally (i.p.). For the iTF-CAR-T cell study, mice were re-challenged with the same cell line at doses and times indicated in the figure. Tumor progression/clearance was measured via (i) bioluminescence imaging and quantified as the total flux per second and (ii) tumor volume as determined by caliper measurement using the formula volume = 3.14/6 × (length × width2), with length being the longest diameter and width being the shortest diameter. Mice were sacrificed upon losing 20% body weight or reaching more than 20 mm in diameter. Blood samples were collected via retro-orbital bleeds. Number of peripheral blood T cells was quantified using Trucount Tubes following the provided protocol (BD Biosciences). The staining panel consisted of anti-human CD3-brilliant violet 605 (BioLegend), anti-human CD45-PE (eBiosciences), and anti-human CD8− APC-H7 (BD Biosciences).
In vivo iToci-CAR-T cell study in humanized mice
All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of IRCSS San Raffaele Scientific Institute and by the Italian Governmental Health Institute (Rome, Italy). Mice were kept in a specific-pathogen-free (SPF) facility within individually ventilated cages. 6 to 8-week-old female and male SGM3 (NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ; The Jackson Laboratory) mice were sub-lethally irradiated (200 cGy) and infused with 1×105 human cord blood CD34+ cells (Lonza). Upon hematopoietic reconstitution, mice were injected intravenously (i.v.) with 0.5×106 NALM6 acute lymphoblastic leukemia cells expressing a secreted luciferase and the LNGFR marker gene. Seven days later, mice received 1×107 ieGFP+cCD19 CAR-T cells, iToci+cCD19 CAR-T cells or untransduced T cells by i.v. infusion. Tumor growth was monitored by bioluminescence assay using the QUANTI-Luc detection reagent (InvivoGen, rep-qlc1) and expressed as relative light units (RLUs). Circulating human T cells were analyzed by FACS and counted using Flow-Count Fluorospheres (BeckmanCoulter). For evaluating CRS toxicity, mice were daily monitored for weight loss and analyzed for human cytokine concentrations at day 4 by LegendPLEX (Biolegend). Mice were euthanized when RLU was ≥ 1.6×106 or when signs of inhumane suffering manifested. For evaluating signs of neurotoxicity, brains from the experimental mice were collected at necropsy, fixed in 4% buffered formalin, embedded in paraffin, cut and stained. Haematoxylin and eosin stained 3-μm paraffin sections were examined for histopathological analysis, blindly and independently by two pathologists. Sections were acquired by a digital scanner Aperio Scanscope and photomicrographs were taken using Aperio Image Scope v10.2.1.2315.
QUANTIFICATION AND STATISTICAL ANALYSIS
Information on specific statistical tests used is provided in the figure legends and/or Method Details. Statistical analysis was performed in GraphPad Prism 8.0 (GraphPad). Each figure legend denotes the statistical test used. All central tendencies indicate the mean, and all error bars indicate standard deviation unless otherwise indicated. Statistical analysis and tests performed included 2-tailed Student t test, Two-way ANOVA or ANOVA multiple-comparison with P values generated using Tukey’s or Welch multiple-comparisons test. For non-parametric comparisons, we used Kruskal-Wallis tests. All t-tests were two-sided. In figures, p values are shown. Log-Rank Mantel-Cox test was used for survival analysis. Technical replicates were assumed to be normally distributed, otherwise no assumptions of normality were made. Statistical details of all experiments can be found in figure legends.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the University of Pennsylvania (UPenn) Stem Cell and Xenograft Core, Small Animal Imaging Facility, Human Immunology Core, Flow Cytometry Core, and Wistar Institute Flow Cytometry for their technical support. The authors thank the UPenn CyTOF Core, specifically Takuya Ohtani for running mass cytometry samples. The authors thank Saar Gill (UPenn) for the provision of tumor cell lines, and CD19 and HER2 targeting CARs. We acknowledge John E. Wherry (UPenn), J. Joseph Melenhorst (UPenn), Robert Vonderheide (UPenn), Nicola Mason (UPenn), Saar Gill (UPenn), Miroslaw Kozlowski (UPenn), Marco Ruella (UPenn), Olga Schestova (UPenn), Mauro Castellarin (UPenn), and Jie Wang (Duke University) for insights and feedback. We thank Dalia Omran (UPenn) for coordinating distribution of samples. We thank Alfredo Tovar (NIBR) for help with molecular cloning of pASP4.2.1. We thank Incucyte® Systems for Live-Cell Imaging and Analysis for providing test equipment. Funding was provided by Parker Institute for Cancer Immunotherapy (PICI) (A.S., D.J.P., B.M.C, G.P.L., C.H.J.), R01EB026892 (D.J.P.) R21CA205794 (G.P.L.), R01CA204261 (B.M.C., G.P.L.). A.B. received funding from a pharmacology T32 training grant (T32GM008076). M.C. received funding from EU H2020 (project CARAMBA). C.B. received funding from AIRC (IG 18458 and IG 2020 ID 24965) and Italian Ministry of Research and University (PRIN 2017WC8499). C.B. and M.C. received funding from the Italian Ministry of Health (Ricerca Corrente CAR-T project: RCR-2019-23669115), EU IMI (project T2Evolve) and AIRC (AIRC 5 per Mille Rif. 22737). A.S. received funding from Slovenian Research Agency (ARRS) for Project J3-3084 and Program P1-0245 and from Research fund of the National Institute of Biology for Project 10ICIGEN (ICI). Graphics were created with Inkscape and GIMP.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
A.S., A.D.P., C.H.J. and D.J.P. are co-inventors on PCT International Patent Applications by The Trustees of the University of Pennsylvania, which incorporate discoveries and inventions described here. Tmunity Therapeutics has licensed non-exclusive rights to commercialize licensed cytokine secreting products, such as CAR or TCR-modified T-cells engineered to express a transgene-encoded cytokine. C.H.J is a co-scientific founder of Tmunity Therapeutics and Capstan Therapeutics and is a member of the scientific advisory boards of Alaunos, BluesphereBio, Cabaletta, Carisma, Cellares, Poseida, Verismo, and viTToria bio. C.H.J. is an inventor on patents and/or patent applications licensed to Novartis Institutes of Biomedical Research and Tmunity Therapeutics and receives license revenue from such licenses. D.J.P. is a member of the scientific advisory boards for Bellicium Pharmaceuticals and InsTIL Bio. D.J.P. is inventor on patents and/or patent applications licensed to Prescient Therapeutics, Tmunity Therapeutics, and Miltenyi Biotec and receives license revenue from those licenses.
REFERENCES
- 1.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al. (2013). Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 368, 1509–1518. 10.1056/nejmoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al. (2015). T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 385, 517–528. 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DAN, Lanier BJ, et al. (2010). Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 116, 4099–4102. 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, et al. (2011). Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 118, 4817–4828. 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porter DL, Levine BL, Kalos M, Bagg A, and June CH (2011). Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 365, 725–733. 10.1056/nejmoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, Brogdon JL, Pruteanu-Malinici I, Bhoj V, Landsburg D, et al. (2017). Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 28, 2545–2554. 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, Boesteanu AC, Wang Y, O’connor RS, Hwang WT, et al. (2018). Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 24, 563–571. 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, Komanduri KV, Lin Y, Jain N, Daver N, et al. (2017). Chimeric antigen receptor T-cell therapy — assessment and management of toxicities. Nat. Rev. Clin. Oncol. 15, 47–62. 10.1038/nrclinonc.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milone MC, Xu J, Chen SJ, Collins MKA, Zhou J, Powell DJ, and Melenhorst JJ (2021). Engineering-enhanced CAR T cells for improved cancer therapy. Nat. Cancer 2, 780–793. 10.1038/s43018-021-00241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z, Palmer DC, Reger RN, Borman ZA, Zhang L, et al. (2010). Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 70, 6725–6734. 10.1158/0008-5472.CAN-10-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeku OO, Purdon TJ, Koneru M, Spriggs D, and Brentjens RJ (2017). Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 7, 10541. 10.1038/s41598-017-10940-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, Rosenberg SA, and Morgan RA (2011). Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol. Ther. 19, 751–759. 10.1038/mt.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chmielewski M, Kopecky C, Hombach AA, and Abken H (2011). IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 71, 5697–5706. 10.1158/0008-5472.CAN-11-0103. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Di S, Shi B, Zhang H, Wang Y, Wu X, Luo H, Wang H, Li Z, and Jiang H (2019). Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3–Targeted Chimeric Antigen Receptor–Engineered T Cells in Hepatocellular Carcinoma. J. Immunol. 203, 198–207. 10.4049/jimmunol.1800033. [DOI] [PubMed] [Google Scholar]
- 15.Zimmermann K, Kuehle J, Dragon AC, Galla M, Kloth C, Rudek LS, Sandalcioglu IE, Neyazi B, Moritz T, Meyer J, et al. (2020). Design and characterization of an “all-in-one” lentiviral vector system combining constitutive anti-gd2 car expression and inducible cytokines. Cancers (Basel). 12, 1–22. 10.3390/cancers12020375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, Nahvi AV, Ngo LT, Sherry RM, Phan GQ, et al. (2015). Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res. 21, 2278–2288. 10.1158/1078-0432.CCR-14-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chmielewski M, and Abken H (2017). CAR T Cells Releasing IL-18 Convert to T-Bet high FoxO1 low Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 21, 3205–3219. 10.1016/j.celrep.2017.11.063. [DOI] [PubMed] [Google Scholar]
- 18.Roybal KT, Williams JZ, Morsut L, Walker WJ, Mcnally KA, and Lim WA (2016). Engineering T Cells with Customized Therapeutic Article Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 167, 419–432. 10.1016/j.cell.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kulemzin SV, Matvienko DA, Sabirov AH, Sokratyan AM, Chernikova DS, Belovezhets TN, Chikaev AN, Taranin AV, and Gorchakov AA (2019). Design and analysis of stably integrated reporters for inducible transgene expression in human T cells and CAR NK-cell lines. BMC Med. Genomics 12, 87–95. 10.1186/s12920-019-0489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel P, Bhatnagar P, Beviglia L, Repellin CE, Javitz H, and Sambucetti L (2018). Modular Antigen-Specific T-cell Biofactories for Calibrated In Vivo Synthesis of Engineered Proteins. Adv. Biosyst. 2, 1800210. 10.1002/adbi.201800210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, Sadelain M, Eshhar Z, Rosenberg SA, and Morgan RA (2009). A Herceptin-Based Chimeric Antigen Receptor with Modified Signaling Domains Leads to Enhanced Survival of Transduced T Lymphocytes and Antitumor Activity. J. Immunol. 183, 5563–5574. 10.4049/jimmunol.0900447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song D, Ye Q, Poussin M, Liu L, Figini M, and Jr DJP (2015). A fully human chimeric antigen receptor with potent activity against cancer cells but reduced risk for off-tumor toxicity. Oncotarget 6, 21533–21546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, Mi T, Switzer K, Singh H, Huls H, et al. (2015). Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 75, 3505–3518. 10.1158/0008-5472.CAN-15-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, Good Z, Belk JA, Daniel B, Klysz D, et al. (2021). Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 372. 10.1126/science.aba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feldman SA, Assadipour Y, Kriley I, Goff SL, and Rosenberg SA (2015). Adoptive Cell Therapy - Tumor-Infiltrating Lymphocytes, T-Cell Receptors, and Chimeric Antigen Receptors. Semin. Oncol. 42, 626–639. 10.1053/j.seminoncol.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, et al. (2015). A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science (80-. ). 348, 803–808. 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linette GP, Becker-Hapak M, Skidmore ZL, Baroja ML, Xu C, Hundal J, Spencer DH, Fu W, Cummins C, Robnett M, et al. (2019). Immunological ignorance is an enabling feature of the oligo-clonal T cell response to melanoma neoantigens. Proc. Natl. Acad. Sci. U. S. A. 116, 23662–23670. 10.1073/pnas.1906026116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, and Ryan JL (1997). Effects of Single-Dose Interleukin-12 Exposure on Interleukin-12 – Associated Toxicity and Interferon-g Production. 90, 2541–2548. [PubMed] [Google Scholar]
- 29.Lieschke GJ, Rao PK, Gately MK, and Mulligan RC (1996). Bioactive murine and human interleukin-12 fusion proteins which retain antitumor activity in vivo. Nat. Biotechnol. 15, 35–40. [DOI] [PubMed] [Google Scholar]
- 30.Chowdhury FZ, Ramos HJ, Davis LS, Forman J, and Farrar JD (2011). IL-12 selectively programs effector pathways that are stably expressed in human CD8+ effector memory T cells in vivo. Blood 118, 3890–3900. 10.1182/blood-2011-05-357111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh N, Hofmann TJ, Gershenson Z, Levine BL, Grupp SA, Teachey DT, and Barrett DM (2017). Monocyte lineage–derived IL-6 does not affect chimeric antigen receptor T-cell function. Cytotherapy 19, 867–880. 10.1016/j.jcyt.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson LA, and June CH (2016). Driving gene-engineered T cell immunotherapy of cancer. Cell Res. 27, 38–58. 10.1038/cr.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsuchiya M, Koh S, Bendig MM, Jones ST, and Saldanha JW (1998). Reshaped human antibody to human interleukin-6 receptor. [Google Scholar]
- 34.Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, Sanvito F, Ponzoni M, Doglioni C, Cristofori P, et al. (2018). Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 24, 739–748. 10.1038/s41591-018-0036-4. [DOI] [PubMed] [Google Scholar]
- 35.Arcangeli S, Bove C, Mezzanotte C, Camisa B, Falcone L, Manfredi F, Bezzecchi E, Khoury R El, Norata, R., Sanvito, F., et al. (2022). CAR T cell manufacturing from naive/stem memory T lymphocytes enhances antitumor responses while curtailing cytokine release syndrome. J. Clin. Invest. 132. 10.1172/JCI150807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, and Kakehi T (2008). Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood 112, 3959–3964. 10.1182/blood-2008-05-155846. [DOI] [PubMed] [Google Scholar]
- 37.Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, Johnson J, Staupe RP, Bengsch B, Xu C, et al. (2019). TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity 51, 840–855.e5. 10.1016/j.immuni.2019.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, Li PJ, Diolaiti ME, Ashworth A, Marson A, et al. (2018). Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 175, 1958–1971. 10.1016/j.cell.2018.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, et al. (2019). TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218. 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kodama S, Koike C, Negishi M, and Yamamoto Y (2004). Nuclear Receptors CAR and PXR Cross Talk with FOXO1 To Regulate Genes That Encode Drug-Metabolizing and Gluconeogenic Enzymes. Mol. Cell. Biol. 24, 7931–7940. 10.1128/MCB.24.18.7931-7940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moffett HF, Coon ME, Radtke S, Stephan SB, McKnight L, Lambert A, Stoddard BL, Kiem HP, and Stephan MT (2017). Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat. Commun. 8, 389. 10.1038/s41467-017-00505-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amendola M, Venneri MA, Biffi A, Vigna E, and Naldini L (2005). Coordinate dual-gene transgenesis by lentiviral vectors carrying synthetic bidirectional promoters. Nat. Biotechnol. 23, 108–116. 10.1038/nbt1049. [DOI] [PubMed] [Google Scholar]
- 43.Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F, et al. (2020). CRISPR-engineered T cells in patients with refractory cancer. Science (80-. ). 367, 1–12. 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eyquem J, Mansilla-Soto J, Giavridis T, Van Der Stegen SJC, Hamieh M, Cunanan KM, Odak A, Gönen M, and Sadelain M (2017). Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117. 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, Hiatt J, Saco J, Krystofinski P, Li H, et al. (2018). Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405–409. 10.1038/s41586-018-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roth TL, Li PJ, Blaeschke F, Nies JF, Apathy R, Mowery C, Yu R, Nguyen MLT, Lee Y, Truong A, et al. (2020). Pooled Knockin Targeting for Genome Engineering of Cellular Immunotherapies. Cell 181, 728–744. 10.1016/j.cell.2020.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sachdeva M, Busser BW, Temburni S, Jahangiri B, Gautron AS, Maréchal A, Juillerat A, Williams A, Depil S, Duchateau P, et al. (2019). Repurposing endogenous immune pathways to tailor and control chimeric antigen receptor T cell functionality. Nat. Commun. 10, 5100. 10.1038/s41467-019-13088-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, and Sadelain M (2018). CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 24, 731–738. 10.1038/s41591-018-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rydzek J, Nerreter T, Peng H, Jutz S, Leitner J, Steinberger P, Einsele H, Rader C, and Hudecek M (2019). Chimeric Antigen Receptor Library Screening Using a Novel NF- k B / NFAT Reporter Cell Platform. Mol. Ther. 27, 4–6. 10.1016/j.ymthe.2018.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hooijberg E, Bakker AQ, Ruizendaal JJ, and Spits H (2000). NFAT-controlled expression of GFP permits visualization and isolation of antigen-stimulated primary human T cells. Blood 96, 459–466. 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 51.Müller TR, Schuler C, Hammel M, Köhler A, Jutz S, Leitner J, Schober K, Busch DH, and Steinberger P (2020). A T-cell reporter platform for high-throughput and reliable investigation of TCR function and biology. Clin. Transl. Immunol. 9, 1–16. 10.1002/cti2.1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gejman RS, Jones HF, Klatt MG, Chang AY, Oh CY, Chandran SS, Korontsvit T, Zakahleva V, Dao T, Klebanoff CA, et al. (2020). Identification of the Targets of T-cell Receptor Therapeutic Agents and Cells by Use of a High-Throughput Genetic Platform. Cancer Immunol. Res. 8, 672–684. 10.1158/2326-6066.cir-19-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li G, Bethune MT, Wong S, Joglekar AV, Leonard MT, Wang JK, Kim JT, Cheng D, Peng S, Zaretsky JM, et al. (2019). T cell antigen discovery via trogocytosis. Nat. Methods 16, 183–190. 10.1038/s41592-018-0305-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Panjwani MK, Smith JB, Schutsky K, Gnanandarajah J, O’Connor CM, Powell DJ, and Mason NJ (2016). Feasibility and safety of RNA-transfected CD20-specific chimeric antigen receptor T cells in dogs with spontaneous B cell lymphoma. Mol. Ther. 24, 1602–1614. 10.1038/mt.2016.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, et al. (2009). Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 17, 1453–1464. 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJD, Scholler J, Song D, Porter DL, Carroll M, et al. (2015). CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 29, 1637–1647. 10.1038/leu.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smole A, Lainšček D, Bezeljak U, Horvat S, Jerala R, Article O, Smole A, Lainšček D, Bezeljak U, Horvat S, et al. (2017). A Synthetic Mammalian Therapeutic Gene Circuit for Sensing and Suppressing Inflammation. Mol. Ther. 25, 102–119. 10.1016/j.ymthe.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim S, Li L, McMurtrey CP, Hildebrand WH, Weidanz JA, Gillanders WE, Diamond MS, and Hansen TH (2010). Single-Chain HLA-A2 MHC Trimers That Incorporate an Immundominant Peptide Elicit Protective T Cell Immunity against Lethal West Nile Virus Infection. J. Immunol. 184, 4423–4430. 10.4049/jimmunol.0903955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amir EAD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, and Pe’Er D (2013). ViSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 31, 545–552. 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.