

Abstract
Energy deprivation is a frequent adverse event in tumors caused by mutations, malperfusion, hypoxia, and nutrition deficit. The resulting bioenergetic stress leads to signaling and metabolic adaptation responses in tumor cells, secures survival, and adjusts migration activity. Recently, kinetic responses of cancer cells to energy deficit were identified, including a switch of invasive cancer cells to energy-conservative amoeboid migration and an enhanced capability for distant metastasis. We here review the energy programs employed by different cancer invasion modes, including collective, mesenchymal, and amoeboid migration, their interconversion in response to energy deprivation, and discuss the consequences for metastatic escape. Understanding the energetic requirements of amoeboid and other dissemination strategies offers rationales for improving therapeutic targeting of metastatic cancer progression.
Keywords: Cellular bioenergetics, migration plasticity, metabolic stress, amoeboid migration
Cancer invasion and nutrient deprivation
Cancer cell invasion and metastasis result from cytoskeletal activation in tumor cells to initiate migration and detach from the primary tumor followed by dissemination through interstitial tissue and across vessel walls [1,2]. As a consequence of migration, tumor cells can spread via blood and lymphatic vessels, to other organs and initiate metastatic regrowth. To migrate through tissues, tumor cells deploy migration strategies used by other cells during physiological processes, such as morphogenesis, wound healing, and inflammation. Three distinct but interconvertible migration programs have been identified, which differ in cell-cell and cell-matrix adhesion, cytoskeletal organization, and mechanochemical tissue interactions. Collective movement (see Glossary) depends on cell-cell adhesion of variable stability and represents an important migration mode in embryonic morphogenesis, vascular spouting, wound healing, and cancer cell metastasis [3–5]. Mesenchymal migration of individual cells involves prominent cell-matrix adhesions, actomyosin contractility, and proteolytic remodeling of the tissue, as detected in fibroblasts and tumor cells that have undergone the epithelial-to-mesenchymal transition [6]. Amoeboid migration is mediated by cortical actomyosin contractility, weak cell-matrix adhesion and pericellular proteolysis. This type of movement is mediated by changes in the cell shape, as detected in leukocytes, lymphoma cells and, rarely, solid tumors [7].
During cancer progression, cancer cells must adapt their energy production and energy consumption to local conditions of primary or metastatic tumor microenvironments. Rapidly growing tumors display high energy demands but simultaneously suffer from significant local perfusion deficits [8]. The resulting metabolic challenges include hypoxia and nutrient depletion, as well as an accumulation of cell-derived toxic metabolites [8,9]. Energy deprivation induces a cascade of adaptation responses in tumor cells, to reduce energy consumption and make use of alternative nutrient sources and metabolic pathways, to avoid cell death [10–12]. In addition, energy deprivation activates programs that induce migration and enable cell escape from perturbed tissue [13,14]. However, whether and by which mechanisms energy deprivation causes either arrest, activation, or switching of invasion programs has been unclear. Recent progress in applying live-cell microscopy in 3D tissue culture and tumor models in vivo has revealed the astounding adaptability of tumor cell migration programs in response to hypoxia and/or energy deprivation. As an outcome, an integrated program consisting of metabolic adaptation, growth control, and plasticity of tumor cell migration towards an energy-conserving amoeboid escape mode has been identified [15,16].
We here summarize the bioenergetic pathways engaged in cancer cell invasion, the commonalities, and differences of energy metabolism in collective, mesenchymal, and amoeboid modes, and their interconversions in response to energy deprivation. We discuss the adaptation of energy metabolism in response to oxygen nutrient deficiency and the resulting adaptation of migration strategies. Lastly, we highlight conversion to amoeboid dissemination as an integrated program securing both cancer cell dissemination through 3D tissue and survival. Understanding shared programs of energy metabolism and invasion mechanisms offers new perspectives for therapeutic intervention to combat metabolic resilience and metastatic escape.
Cell migration modes and metabolism
In moving cells, energy demands are tightly linked to cytoskeletal activity. Energy consumption results from the ATPase function of actin during actin filament formation, as well as cyclic protein phosphorylation and dephosphorylation of regulatory and adaptor proteins. These energy-consuming processes are required to build actin networks, regulate actin filament dynamics, and contract actin filaments by myosin motor activity [17]. Consequently, together with the enzymatic remodeling of extracellular matrix (ECM) structures, all actin-dependent steps of cell migration, including cell-cell and cell-matrix adhesion, change of cell shape, cell contraction, and force generation, consume significant amounts of the energy carriers adenosine triphosphate (ATP) and guanosine triphosphate (GTP) [17]. To effectively deliver energy at a subcellular scale to sites where the cytoskeleton is actively being rearranged, mitochondria and glycolytic enzymes interact with the actin cytoskeleton and ensure energy production near the site of consumption [18–22].
Actin filament formation and contraction.
Actin polymerization to filaments and turnover are critical to cell polarization and protrusion formation, including lamellipodia, filopodia, focal adhesions, and stress fibers [23]. A large amount of ATP is consumed by the ATPase activity of actin, to build actin filaments from monomers (Fig. 1A). Actin filament formation and actin network dynamics are regulated by actin-binding proteins (ABPs) [24]. Depending on upstream regulation, including Rho-family GTPases and cooperating kinases and phosphatases, ATP-consuming actin dynamics extend three protrusion types at the leading edge with different morphology, actin organization, and kinetics: lamellipodia, filopodia, blebs [24] (Fig. 1A). Actin stress fibers and actin filament networks become contracted by non-muscle myosin II [25], by ATP-dependent myosin motor activity, and are regulated by ATP-dependent kinases controlling Rho-regulatory light chain (RLC) activity (Fig. 1A).
Figure 1. Energy consuming processes during migration.
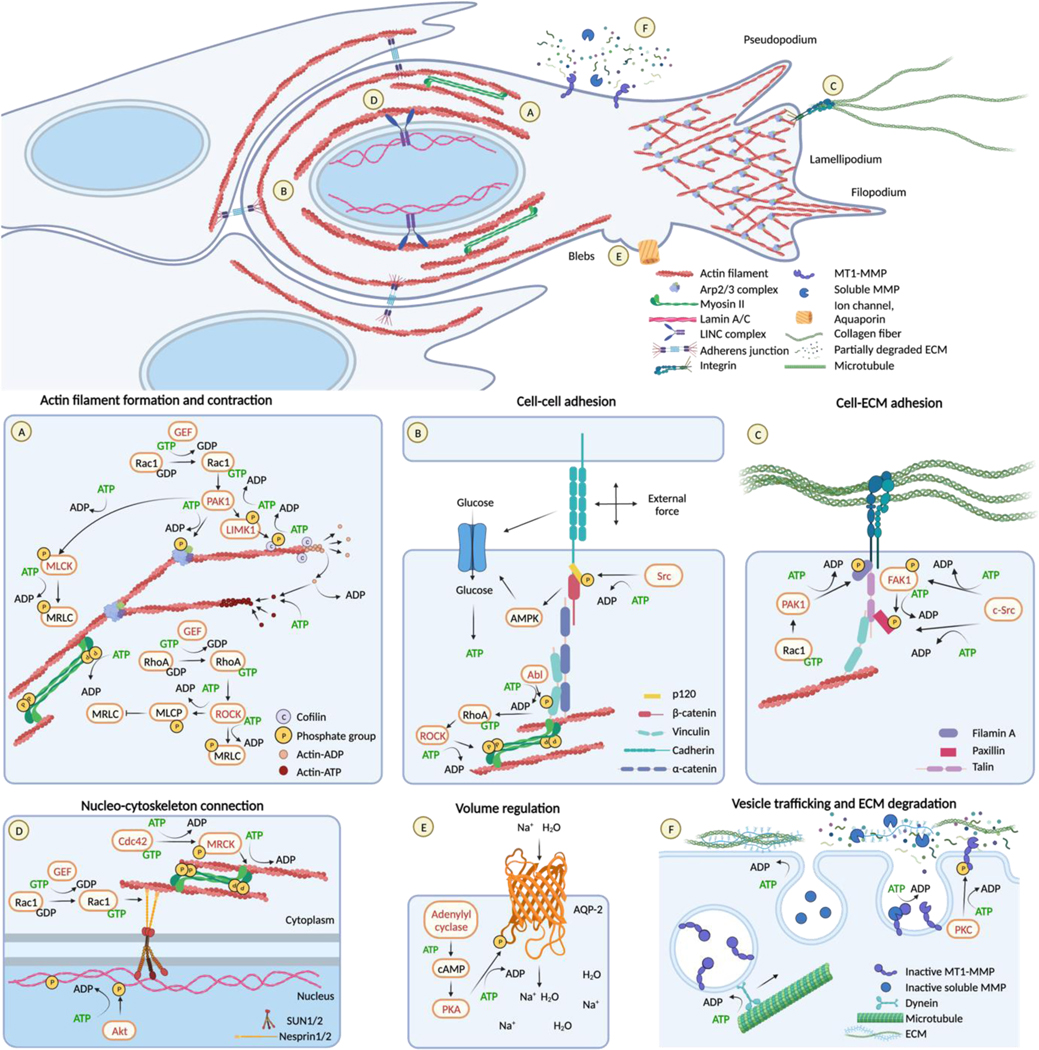
Overview (top panel) and individual ATP-consuming steps involved in cell movement (bottom panels, A-F).
(A) Actin-ATP monomers polymerize to filaments. Filament dynamics are controlled by actin-binding proteins, including cofilin, under the control of LIMK and phosphatases, under the control by Rac1. Rac 1 further controls PAK1- Arp2/3 engagement for filament branching. NM II mediates actin filaments contraction, under the control of MRLC phosphorylation regulated by MLCK and MLCP controlled by Rac1 and RhoA, respectively.
(B) ATP consumption is involved in cadherin adaptor molecules regulation via phosphorylation as p120 is under Src kinases activity. In response to external forces, E-cadherin stimulates AMPK signaling. AMPK stimulates increased glucose uptake and its conversion into ATP. AMPK further acts on kinases (Abl) to phosphorylate vinculin and the RhoA–ROCK-myosin II axis.
(C) Integrins activity, clustering and turn-over require energy as they are regulated by cycles of phosphorylation and dephosphorylation of their cytoplasmatic tail and adaptor proteins. The examples show paxillin phosphorylation by FAK and Src kinases and filamin A controlled by Rac1-PAK1 axis.
(D) ATP is engaged in MRCK activity and regulation by the Rho GTPase Cdc42, which leads to myosin contractility around the nucleus. ATP is further required for LINC complex activity and actin cable dynamics. For example, Rac1 interacts with Nesprin-2 to connect the LINC complex to actin. Src-mediated phosphorylation of Lamin A causes lamin A disassembly from the inner nuclear lamina.
(E) ATP-dependent phosphorylation and regulation of ion channel pumping into the cytoplasm and aquaporin activity. For example, AQP-2 is phosphorylated by PKA, and activated by cAMP.
(F) ATP-consuming steps during ECM degradation, including kinesin and dynein-mediated vesicle transport of proteases, endo/exocytic protease transport, autocatalytic activation of the zymogen; zymogen cleavage by activating protease. MT1-MMP activity can further be regulated through LIMK-mediated phosphorylation of the cytoplasmic tail. Created with BioRender.com
Lamellipodia and filopodia are actin polymerization-driven extensions of the plasma membrane that require respective branching and bundling of actin filaments and myosin contractility to engage with the extracellular substrate [24]. Alternatively, blebs form when the intracellular hydrostatic pressure increases, and the plasma membrane locally detaches from the actin cortex [26]. Lamellipodia and filopodia depend upon actin turnover, whereas blebs form without actin polymerization [26]. Consequently, energy demands incurred by each protrusion type may differ (BOX 1) [24].
BOX 1 | Analysis of energy metabolism with single-cell and subcellular resolution.
A range of classical methods allows analysis of energy production and consumption in cell populations in bulk culture, including detection of oxygen consumption, production of lactate, ATP concentration (luciferase assay), and extracellular acidification rate [143]. However, linking mechanisms of cell migration to cell metabolism is technically challenging, as it depends on co-registering cell migration and energy fluxes in live-cell culture over time image analysis of individual cells in cell populations to account for inter-individual heterogeneity during mixed responses.
At the single-cell level, fluorescent reporters allow to detect the oxidative state of mitochondria (e.g., JC-1 [144]), the oscillation of glycolysis [145], the ATP:ADP ratio (PercevalHR), the glucose uptake (2-NBDG probe), NADH:NAD+ redox state (Peredox probe), and H2O2 gradients (e.g., HyPer 7) [146]. To link the metabolic state to cytoskeletal action, energy flux reporting needs to be linked to molecular intervention of defined pathways, and effects on metabolism caused by the intervention recorded. For example, inhibition of RhoA reduces the oscillating activity of glycolysis in adherent endothelial cells, indicating that glycolysis fuels RhoA-mediated actomyosin contractility [145]. However, mapping the role of metabolic pathways for the generation of individual actin-based structures, such as protrusion types or cortical versus perinuclear actin filaments, and the respective energy needs of these structures, will require spatially defined live-cell measurements with a subcellular resolution to report locally produced and consumed ATP equivalents or local enzyme activity in metabolic pathways. In addition, the co-registration of several metabolic pathways in the same cell is currently limited due to the spectral overlap of available fluorescent reporters. Thus, in-depth information linking the relative weight and cooperation of energy pathways to migration modes and individual actin-based structures will require the development of functionally and spectrally complementary single-cell reporter systems. In addition, combining spectrally unmixed multi-channel recordings of energy states with molecular-based in silico modeling on ATP consumption will enable predictions on the energy needs of individual cellular substructures and the changes associated with plasticity responses [133].
Cell-cell adhesion.
Adherens junctions support cell-cell adhesion between collectively migrating tumor cells and mediate tumor cell interaction with stromal fibroblasts and/or macrophages during invasion [27,28]. Cadherins transmit force to the actin and microtubule cytoskeleton through adaptor molecules and ABPs, with ATP consumed by upstream regulators Abelson (Abl) tyrosine kinase, Src, RhoA, and Rac1 [29]. For example, Abl phosphorylates vinculin which then binds actin, activates RhoA, and increases actomyosin contractility at cell-cell interactions [30]. Cadherin mechano-coupling activates metabolic signaling and energy production, including increased glucose uptake and ATP production [31].
Cell-ECM adhesion.
Integrin activation, clustering, and mechano-coupling are mediated by adaptor proteins, including talin, vinculin, paxillin, filamin A and α-actinin [32], under the control of ATP-consuming upstream kinases and phosphatases [33]. In concert, focal adhesion kinase (FAK), Src-family kinases, integrin-linked kinase (ILK), PAK Ser/Thr kinases, and tyrosine phosphatases SHP2 [34] and PTP-PEST [35] control integrin engagement, adhesion turnover and migration [36]. Integrin interactions with actin filaments and mechano-coupling further depend on the localized activation of Rac and RhoA and engagement of myosin-II, in an ATP-dependent manner [33].
Nucleo-cytoskeleton connection.
The nucleus is the largest and stiffest organelle, which becomes deformed, moved, and mechanically protected in moving cells by peri-nuclear actomyosin networks, in an ATP-dependent manner [37]. Rho GTPases Cdc42 and RhoA control the nucleo-actin connection by myosin-II-mediated crosslinking and contraction (Fig. 1D) [38–40]. Perinuclear actin couples to the nuclear envelope via the adaptors nesprin and SUN proteins under the control of Rac1 [41]. ATP is further required to assemble, deform, and disassemble the nuclear lamina consisting of a filamentous network of A/C- and B-type lamins, under the control of kinases including protein kinase B (PKB/Akt) [42,43] and Src kinase [44]. Src further regulates nuclear stiffness via phosphorylation of inner nuclear membrane proteins (e.g., emerin) [45].
Volume regulation.
Intracellular water content and cell volume during cell migration are regulated by ion transporters (e.g., Na+-H+ exchanger, Na+-K+-2Cl−cotransporters) and aquaporins (AQPs), which jointly control the intracellular water content [46,47]. ATP is consumed by the ion channel pump activity and by phosphorylation to regulate aquaporin activity regulation through phosphorylation (e.g., the cAMP-PKA axis) (Fig. 1E) [46,48,49]. Aquaporins cooperate with cytoskeletal proteins and support protrusion formation at the leading edge (e.g., AQP-1, −4, and −5) or local shrinkage and detachment at the cell rear [50].
Proteolytic tissue remodeling.
Invasive cells can facilitate their movement through the ECM by its proteolytic degradation through matrix metalloproteinases (MMPs) and other proteases, by an energy-dependent multi-step process (Fig. 1F) [51]. ATP and GTP are consumed for MMP expression, protein folding, transport of MMP-containing vesicles via microtubules and motor proteins [52], MMP activation [53], and exocytosis [54]. Cell-surface-localized proteolysis depends on the delivery and recycling of transmembrane proteases (e.g., membrane-type I matrix metalloproteinase, MT1-MMP) as well as phosphorylation of the cytoplasmic tail (Fig. 1F) [55].
Metabolic pathways fueling cell migration
The metabolic pathways delivering energy for basic cell functions, including cytoskeletal dynamics and cell movement, depends on intracellular ATP, nicotinamide adenine dinucleotide phosphate (NAD(P)H), and flavin adenine dinucleotide (FADH). In response to energy deprivation, cells upscale nutrient uptake and adapt the metabolic pathways (reviewed in [56]). We here briefly summarize key metabolic pathways and their interdependence with cell migration.
Energy production under normal conditions.
When oxygen and glucose supply is unperturbed, invading cancer cells balance their energy homeostasis mostly between oxidative phosphorylation and glycolysis, to maintain migration activity in response to mechanical and chemical cues in the microenvironment [57]. Oxidative phosphorylation (OxPhos) provides localized energy production to the most energy-demanding regions of the cell. Mitochondrial trafficking to the leading edge of the cell support cytoskeletal dynamics, membrane protrusion, and focal adhesion assembly [19–22]. Localized glycolysis occurs near sites of cytoskeletal activity and supports migration dynamics by ATP production [58,59]. For example, phosphofructokinase-1 (PFK-1), the rate-limiting enzyme of glycolysis, binds actin in active form, thus controlling glycolysis near cytoskeletal activity [17]. Other glycolytic enzymes, including aldolase and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), bind actin filaments as inactive enzymes, which, after release, undergo activation near sites of cytoskeletal dynamics [17]. Under both normal conditions and metabolic deprivation, the bioenergetic functions of OxPhos and glycolysis are complementary, cooperative, and respond to nutrient availability [60]. Glycolysis can occur without oxygen involvement but is rapidly adaptive in delivering ATP, whereas OxPhos depends upon oxygen availability and constitutively produces high amounts of ATP [60].
Energy deprivation.
Under conditions of metabolic stress, including acidosis, hypoxia, and nutrient deprivation, additional mechanisms for ATP production become activated, including autophagy, amino acid and creatine metabolism, and lipid oxidation [61,62]. Within minutes after energy deprivation, cancer cells can adapt ATP production by activating pathways regulating cell metabolism, including AMP-activated protein kinase (AMPK), hypoxia-inducible factor, and calpain, which enable an acute bioenergetic response [63,64]. AMPK stimulates glucose uptake (through the glucose transporters Glut1 and Glut4) and ATP production through glycolysis [64]. AMPK further promotes the use of alternative energy sources, including lipid import into mitochondria for ß-oxidation and autophagy [64]. In addition, AMPK reduces energy expenditure by inhibiting the mTOR pathway, which delays RNA translation and cell cycle progression [65]. Hypoxia-inducible factor 1 (HIF-1), mediates the transition from oxidative to glycolytic metabolism [66] as well as autophagy [114], to maintain ATP levels. In response to hypoxia, HIF-signaling upregulates the expression of glycolytic proteins (e.g., GLUT1, PFKFB, lactate dehydrogenase A) [66,69] and inactivates the TCA cycle [66]. Besides adapting their energy metabolism, tumor cells further broaden the spectrum of sources of rate-limiting metabolites and molecules able to fuel the TCA cycle, by degradation of products that are typically present in the metabolically perturbed microenvironment (BOX 2). This includes proline from degraded ECM sources [70], amino acids (e.g., glutamine, glutamate) by intracellular biosynthesis or from the extracellular space [71], extracellular creatine [72], and lactate which is produced by glycolysis in metabolically perturbed tumors [73]. Extracellular nutrients become internalized via molecular transporters (GLUT1, MCT1, LAT1), whereas multi-molecular aggregated proteins and lipids as well as cell fragments become internalized via macropinocytosis [62,74].
BOX 2 | Intersection of energy deprivation, metabolic stress, and toxic waste.
In the metabolically perturbed tumor microenvironment, adaptations of energy production often coincide with metabolic stress responses induced by non-toxic and toxic extracellular metabolites and inflammation, but also stress responses to therapy. Oxygen deprivation increases the intracellular production of reactive oxygen species (ROS) by mitochondrial complexes I and III. In addition, extracellular ROS is produced by activated neutrophils and macrophages in the tumor microenvironment [147]. ROS can oxidize protein thiols, lipids, and DNA and directly perturb cell integrity [148] and activate pathways of cell adhesion and migration, by cysteine oxidation of signaling proteins (e.g., mitogen-activated protein kinase (MAPK) and NF-kB), ABPs, actin [149] or upstream receptors (e.g., EGFR) [148]. Ultimately, excessive ROS production can impair cancer cell migration and survival and limit metastatic spread [150]. Non-toxic metabolites including purine nucleotides (e.g., adenosine) can activate G protein-coupled adenosine receptors (ADORAs), which promote cytoskeletal activation and invasion, as well as proliferation and angiogenesis [4]. Intracellular and extracellular products which accumulate in metabolically stressed tissue, including H+, lactate, and ammonia, can perturb the metabolism, viability, and migration of cells. For example, extracellular acidosis leads to the activation of RhoA, downregulates cell-cell adhesions, and upregulates MMP expression, ultimately favoring EMT and invasive properties [114]. Lactate, besides lowering the extracellular pH, can also act as a signaling metabolite increasing the HIF-1α-dependent hypoxia response and leading to proliferation, dissemination, and escape from the immune system mediated by the lactate-activated G protein-coupled receptor GPR81 [73]. In concert, pathways stress pathways and bioenergetic adaptation mediate integrated metabolic stress responses toward tumor cell invasion and metastatic escape.
The type and extent of adaptation of the energy metabolism depend on the severity of oxygen and energy deprivation and occurs in a cell- and tumor type-dependent manner, in order to secure intracellular glucose, ATP, and NADPH production for cell survival and migration [56].
Interdependence of energy consumption and migration strategy
Cells can migrate individually, without cell-cell adhesion, or collectively when cell-cell adhesions are retained [75,76]. The ATP consumption involved in individual or collective migration depends on the engagement of the adhesive, cytoskeletal, and proteolytic activities, resulting in differing energy demands.
Collective migration.
Collective movement depends on actin dynamics in coordination with cadherin-based cell-cell adhesion and gap-junctional intercellular communication, in concert with integrin-mediated mechano-coupling to ECM and proteolytic ECM remodeling [75,77]. Due to its mechanochemical complexity, the energy demands of collective migration in cancer cells are high (Fig. 2) [16,78].
Fig. 2. Interdependence of energy consumption and migration strategy.

Collective migration depends on strong cell-cell adhesion, Rac1-mediated actin dynamics, Rho-A mediated contractility, integrin-mediated ECM adhesion and deformation together with pericellular proteolysis. Because of its molecular and mechanical complexity, collective migration is energetically costly, particularly for the leader cells that must overcome substrate resistance. Collective-to-mesenchymal (CMT) single-cell transition is mediated by the downregulation of intercellular adhesions. Losing cell-cell junctions allows mesenchymal single cells to save some energy, even though their elongated morphology still requires actin activity at the leading edge, cytoskeletal contractility, ECM-adhesion, and proteolysis. Mesenchymal-to-amoeboid transition (MAT) results from lowering adhesion to the substrate and pericellular proteolysis. The pseudopodal amoeboid mode retains actin-rich protrusions while the blebbing mode completely relies on Rho-mediated actomyosin contractility. By lowering most of the ATP-consuming steps of motility, the amoeboid mode seems to minimize the energy demands of migration. The lower panel shows the hypothetical coupling of migration modes and metabolic reprogramming. Created with BioRender.com
Leader cells directing and paving the way for collective invasion require a higher level of intracellular ATP/ADP compared to follower cells [78]. High ATP demands may result from Rac1-mediated protrusion formation, high activity of integrins, adhesion-regulating kinases, RhoA-mediated actomyosin contractility, and pericellular proteolysis (e.g., MMP-14, cathepsin B) [79]. Leader cells further depend upon connexin-43-dependent extracellular release of purine derivatives, including ATP, ADP, and adenosine, which activate the adenosine receptor 1 (ADORA1), Akt, and leader cell function in an autocrine manner [4]. To remove tissue barriers, leader cells perform MMP-mediated proteolysis and realign ECM structures creating trails of least resistance [80]. In concert, these mechanical and molecular activities of leader cells result in high energy demands.
Follower cells maintain actin-based connections with the leader and the neighboring cells through cadherins [81] and simultaneously generate force transmission via integrins to the ECM substrate by lateral lamellipodia [3]. They further reinforce cadherin-mediated junctions in response to pulling forces [82] and contribute to proteolytic ECM degradation [80,83]. Follower cells maintain moderately reduced ATP levels, possibly due to reduced mechanical work required to move along a path initially build by leader cells [78].
Leader and follower cells are interconvertible. As leader cells invade, their energy gradually depletes, leading to leader-follower cell transition allowing a follower cell to become a new leader cell [79]. Metabolic shifts, detectable as increased mitochondrial respiration, or upregulation of the glucose transporter 1 (GLUT1) support the energy required during collective migration in both leader and follower cells [78,84]. Thus, cell positioning and function in moving cell groups are reflected by differing energy consumption, yet the mechanochemical activities and subcellular structures underlying the correlation between metabolic programs and cell positioning during collective migration remain to be identified (BOX 1).
Individually migrating cells.
Depending on the adhesive strength of cell-matrix interaction and the extent of proteolytic remodeling of the ECM, individually moving cells deploy mesenchymal, or amoeboid migration strategies with differing energy consumption. When compared to collective-migrating cells, single cells lack cell-cell adhesion, cadherin-mediated response to forces and signaling, and accordingly move with reduced energy demands.
Mesenchymal single cells resemble leader cells during collective migration [6,85], although their energy demands are lower due to the lack of cell-cell junctions. ATP consumption secures protrusive actin polymerization at the leading edge, strong adhesive interaction, spindle-shaped cell extension, and deformation of ECM by substantial actomyosin contraction (Fig. 2) [16,86]. During migration, mesenchymal cells further remodel the ECM by proteolytic degradation and deposition of ECM molecules [6]. The energy demands reflect the amount of actin-mediated cell protrusion and mechanical work executed by the cell. For example, lamellipodia and filopodia in moving cancer cells are disabled after the inhibition of oxidative phosphorylation or glucose metabolism [16]. Furthermore, glucose uptake and ATP:ADP ratio, an indicator of energy production, are increased when cells exert force on the matrix and/or interact with denser matrices [86,87]. Likewise, when confronted with ECM substrates of high stiffness, moving cells upscale integrin engagement, F-actin bundling, and stress fibers formation and, concomitantly, maintain high levels of glycolysis, through tripartite motif-containing protein 21 (TRIM21)-mediated upregulation of PFK-1 [59]. Thus, in mesenchymal cells, adhesion and contractility are coregulated with energy metabolism (Fig. 2).
Amoeboid-moving cells develop weak adhesion to ECM substrate and move via small pseudopodia or lamellipodia formed by protrusive actin polymerization or blebs-induced hydrostatic pressure towards the front [88,89]. The pseudopodal amoeboid type of migration occurs in cells generating actin-rich protrusions at the leading edge which generate weak adhesions towards ECM [90–92]. Amoeboid-moving cancer cells are sustained by low levels of mitochondrial activity and, hence, are considered energetically efficient [16,93]. The amoeboid movement relies on rear-polarized myosin II activity controlled by the Rho-ROCK pathway [94], which drives the retrograde flow of the actin cytoskeletal cortex and generates frictional forces as well as non-adhesive mechanical intercalation with the substrate [95,96]. Depending on the cell type and environmental condition, amoeboid migration may or may not cause proteolytic modification of the ECM [97–100]. Although proteolytic activity costs energy, it creates a path of least resistance that may save actin efforts for cell deformation thus, arguably, resulting in reduced net energy demands. Thus, actin flow and actomyosin contractility are retained in amoeboid-moving cells, but energy demands resulting from strong adhesions and force transmission, stress fibers, cell-cell interactions, and proteolytic ECM remodeling are reduced.
Reprogramming of cancer cell invasion by bioenergetic stress
Invading tumor cells, when confronted with metabolic challenges, can undergo a bioenergetic adaptation response which secures cell survival and persisting migration. Plasticity of invasion programs can be induced by hypoxia and nutrient deprivation, and result in epithelial-to-mesenchymal transition (EMT), mesenchymal-to-amoeboid transition, and collective-to-amoeboid transition [16,101].
Intersection of energy metabolism and migration programs.
Bioenergetic programs and mechanisms of invasion are interconnected. In parallel, energy metabolism programs cooperate with cellular responses to metabolic stress evoked by toxic metabolic products to secure survival and migration (BOX 2). Deprivation of oxygen and/or nutrients can directly impact the efficiency and/or mode of cell migration, and the ability of cancer cells to rewire their metabolism and exploit different energy sources is critical to sustaining migration. Pharmacological interference with either OxPhos or glycolysis results in the conversion of collective to single-cell migration (discussed below) [16]. Hypoxia and HIFs signaling support Rho GTPase-mediated actomyosin contractility and cell migration through activation of glycolysis [102]. Restriction of glutamate availability inhibits pseudopod formation and migration of tumor cells [103]. Likewise, the inhibition of glutaminase, which catalyzes the hydrolysis of glutamine to glutamate, has been shown to block the oncogenic transformation induced by at least three different Rho GTPases (Cdc42, Rac1, RhoC) in fibroblasts [104], invasion cancer and lymphoma cells [104], and the expression and activity of metalloproteinases (e.g., MMP2 and MMP9) [105].
Whereas adaptive nutrient uptake secures energy fueling for migration activity, autophagy additionally impacts the migration machinery directly by the degradation of proteins involved in cell adhesion and cytoskeletal dynamics. In moving fibroblasts, autophagosomes become polarized toward the cell front [106], where they degrade Rho guanine nucleotide exchange factors (e.g., guanine nucleotide exchange factor H1, GEF-H1) [128]. This, in turn, reduces RhoA activity and favors mesenchymal migration [128]. Autophagy further degrades cytoskeletal adapter proteins, including paxillin and talin, which disassembles focal adhesions [107,108] as well as adherens junction proteins [109], which weakens cell-cell cohesion [110]. In cancer cells, autophagy either inhibits RhoA and migration in 2D culture [111] or activates RhoA and enhances migration through transwell filters [112]; thus, the contribution of autophagy to cancer invasion may depend on the cell type and migration model. The impact of other energy sources, e.g., creatine, and macropinocytosis [62,74], remains to be established.
EMT is induced by microenvironmental cytokine and growth factor signaling, as well as by hypoxia, acidosis, and nutrient deprivation [113–116]. By transcriptional control, EMT downregulates cell-cell adhesions and includes cell elongation by cytoskeletal reorganization, so cells can detach from the epithelium and move individually [117]. This transition enhances invasion alongside changes in energy consumption and production, including a switch from OxPhos to glycolytic energy metabolism [116,118]. The degree of EMT and type of metabolic reprogramming are connected. In vitro and in vivo evidence [119,120], further confirmed by in silico modeling [121], show that tumor cells undergoing partial EMT increase glycolysis levels and reach a hybrid state with both epithelial and mesenchymal traits as well as high activity of both glycolysis and OxPhos. This hybrid state may give rise to a fully mesenchymal phenotype, with decreased glycolytic levels (Fig. 2, asterisk) or, when glycolysis and OxPhos are both decreased, transition into a quiescent mesenchymal-like state [116]. Additional bioenergetic programs implicated in EMT induction or maintenance of EMT include the pentose phosphate pathway to support gluconeogenesis [122], and proline and glutamine metabolism [123].
Amoeboid plasticity.
When challenged by metabolic stress, including severe hypoxia or experimental induction of HIF signaling, collectively invading cancer cells abandon cell-cell interactions and transit to amoeboid movement, termed collective-to-amoeboid transition [15,124]. This plasticity response differs from EMT, as cells deactivate integrin-mediated cell-matrix adhesion and develop low-adhesive bleb-mediated movement [16]. This adaptation of migration mode depends on activation of the cysteine protease calpain-2, which cleaves talin and thereby weakens adhesion to ECM [125] (Fig. 3). The amoeboid transition concurs with repression of oxidative respiration and glycolysis to very low levels (Fig. 2), indicating that amoeboid dissemination of cancer cells movement can occur with very low energy consumption [126]. Amoeboid plasticity can further be induced when autophagy is inhibited, which leads to RhoA activation, actomyosin contractility, and rounding of otherwise mesenchymal fibroblastic cells [127]. Likewise, pharmacologic inhibition of oxidative phosphorylation or glycolysis causes both collective-to-amoeboid and mesenchymal-toamoeboid transition in cancer cells [128,129]. After culture in hypoxia, amoeboid-migrating cytotoxic T cells retain the full capability to accumulate in tumors as well as their anti-tumor effector function [130], indicating remarkable metabolic tolerance of amoeboid movement in leukocytes. Preconditioning of tumor cells by hypoxia is sufficient to strongly enhance experimental lung metastasis [16,132]. This may indicate that the metabolic programs are sufficiently sustained during the phase of circulation and impact early organ colonization. However, it remains to be established how long bioenergetic reprogramming remains active at the metastatic site.
Fig. 3. Amoeboid cancer cell migration – an “eco-mode”.

Hypoxic stress triggers the collective-to-amoeboid transition. This switch in migration mode relies on HIF-1a-mediated activation of calpain-2, a protease that cleaves Talin-1 and therefore decreases b1-integrin activity. This weakening of interactions with the ECM causes cell rounding and the formation of polarized membrane blebs. This transition to an amoeboid and more cost-effective type of migration might secure cell evasion from challenging microenvironments. Created with BioRender.com
Arguably, resulting from the constitutive lack of adherens junctions and low cell-matrix adhesion, amoeboid movement may represent an energetically low-demanding “eco-mode” of cell migration which is maintained by actin flow and hydrostatic regulation but lacks energy-consuming cell-cell interactions and occurs with minimal ECM deformation and remodeling [16,131]. Because of its low mechanical and bioenergetic complexity, amoeboid movement may be particularly suited to securing evasion from perturbed tissue sites with limited nutrient requirements, and this may increase the cell fitness towards enhanced metastasis [7,16,101]. The bioenergetic pathways, which support either integrin-mediated adhesion and actin-based treadmilling or poorly adhesive, ion- and water-channel-dependent migration modes in 3D environments remain to be clarified [16,133].
Concluding Remarks
Understanding the intersection of cancer energy metabolism and adaptive cancer invasion programs is necessary for categorizing types, plasticity, and vulnerability of cancer metastasis. Metabolic stress-induced EMT and amoeboid programs may occur independently or as overlapping programs in favor of local dissemination, intra- and extravasation, and organ colonization [7]. Consequently, discriminating cell-intrinsic and microenvironmental mechanisms of amoeboid cancer cell dissemination and metastasis may be important to tailor suitable interference strategies. Targeting options may include upstream regulators controlling migration mode switching, including mechanical stress and cytokine networks [134,135], as well as metastasis-enhancing pathways engaged by energy deprivation.
Limiting the transition to migration modes with lowered energy consumption may (i) reduce the migration speed and cell dissemination through the tissue and/or (ii) increase the energy deficit and, hence, cell survival. Calpain may emerge as a master regulator of cell migration plasticity in different contexts. Pharmacological inhibition of calpain, which releases cancer cells from adhesive interactions with ECM, abrogates the metastatic ability of cancer cells in response to hypoxia in experimental metastasis [16]. In addition, interfering with energy uptake and broadening of energy sources may delay invasion [56]. Pharmacological interference with AMPK, which secures energy production by glycolysis and other programs during periods of metabolic stress, may reduce the ability of tumor cells to adapt their metabolism at any step of the stressful metastatic cascade [64,136]. This is in line with recent in silico simulations which predict the therapeutic efficacy of AMPK inhibitors only when tumor cells maintain metabolic stress signaling [136]. Preventing the export of lactate derived from glycolysis, by inhibiting monocarboxylate transporters (MCTs), limits extracellular lactate as an alternative source of energy, prevents EMT development, and reduces the efficacy of invasion [73].
Interference with other overlapping pathways supporting tumor cells in both survival and migration, including the heat-shock response, EMT pathways (e.g., transforming growth factor ß, TGFß/tumor necrosis factor α, TNFα/interleukin 6, IL-6), interference with Rho/ROCK pathways activated in amoeboid movement [137], and targeted reversion of autophagy induction [138] may allow to further sensitize tumor cells to metabolic stress and decrease metastatic escape.
To identify patient subsets and define personalized targets, biomarkers that indicate an upregulation of metabolic stress signaling in combination with a high load of circulating tumor cells may be detected in liquid biopsies and/or circulating tumor cells to identify engaged metabolic stress programs in a tumor-type and -stage-dependent manner. Biomarkers indicating metabolic stress may be based on transcriptomic analysis in circulating tumor cells indicating a metabolically silent state (e.g., AXL, GLUT1), autophagy (e.g., repression of miR-205), and/or hypoxia response (e.g., HIF-1) [139–141] or metabolomic analysis reflecting the balance of oxidative and glycolytic programs [141].
Future avenues may include the identification of minimal metabolic deprivation stresses which can elicit reprogramming of metastasis. Likewise, the duration and mechanisms of persistence of metabolic stress signaling after evasion from the perturbed microenvironment remain to be identified, including epigenetic reprogramming involved in EMT and amoeboid programs. Both, elongated and amoeboid-rounded migratory modes can contribute to the EMT spectrum [7,142], consistent with potentially broad adaptability of both metabolic pathways and migration strategies in response to nutrient deprivation and stress by toxic metabolites. Thus, the molecular intervention of migration programs alone may not suffice to combat metastasis. Instead, the combined intervention of pathways supporting migration, cancer cell survival and, as discussed here, the response to energy deprivation may require targeting by combined approaches [123].
Highlights.
Mechanochemical strategies deployed by cancer cells to invade tissues depend upon different amounts of energy production and consumption.
Whereas collective and mesenchymal migration are bioenergetically demanding, amoeboid migration is energetically favorable.
Under conditions of energy deprivation, invading cancer cells adapt both cytoskeletal activity and metabolism in order to save energy and secure migration.
Recently identified transitions in response to oxygen and energy deprivation include the collective-to-amoeboid and mesenchymal-to-amoeboid transition
Understanding the crosstalk of bioenergetic and cell migration pathways will aid the identification of intervention points to interfere with tumor cell dissemination and metastasis.
Outstanding questions.
How are programs of energy production and migration modes jointly regulated, to optimize both energy demands and cell movement?
Which energy-conserving mechanisms are relevant for metastatic dissemination and can be detected in circulating tumor cells in cancer patients?
Which metabolic vulnerabilities are particularly suited for molecular intervention and in which phase during the metastatic cascade?
Which biomarkers and samples are best suited to identify and monitor patient subsets with adaptive metabolic stress response?
Which epigenetic alterations result from short- and/or long-lived metabolic stress and how do these alterations affect metastatic programs, including amoeboid behaviors?
How do metabolic stress programs cooperate with other programs of cancer progression, including EMT and stemness?
Acknowledgements
We thank Konstantinos Konstantopoulos for expert discussion and Mirjam Zegers for critical reading and helpful comments on the manuscript. The work of the laboratory was supported by grants from the European Research Council (ERC-AdG-2021-101054921), NIH (U54 CA210184-01; U54 CA261694-01) and the European Union (ITN-Inflanet EU-H2020-ITN 955576).
Glossary
- Actin-binding proteins
mediators of actin filament organization and dynamics, including elongation (by formins), branching to actin networks (by Actin Related Protein 2/3 complex, Arp2/3), severing (cofilin) and disassembly to monomers (cofilin)
- Actin filament
flexible and thin microfilaments formed by polymerization and depolymerization of actin monomers, which determine cell adhesion, shape, stability, and movement
- Adherens junction
initiator and stabilizer of cell-cell adhesion, composed of cadherin adhesion receptors, –intracellular adaptors (e.g., β-catenin, α-catenin, p120-catenin) and actin filaments
- Ameboid migration
migration mode driven by rounded cell shape, blebbing or pseudopodal protrusions and weak or absent cell-matrix adhesions
- AMP-activated protein kinase (AMPK)
central sensor of low intracellular ATP or high ADP and AMP levels which responds to energy deficiency by inhibiting ATP consumption and inducing ATP production by favoring glycose uptake
- Autophagy
catabolic process by which cellular components are engulfed in autophagosomes, degraded to sugars, fatty acids, and amino acids, and recycled to pyruvate and glucose to secure glycolysis and oxidative phosphorylation under conditions of energy deprivation
- Blebs
poorly adhesive roundish membrane protrusions that form due to hydrostatic pressure, contain cortical actin, and frequently support amoeboid movement
- Collective movement
motility mode of groups, sheets or strands of cells that preserve cell–cell junctions and synchronize their intracellular signaling and actin dynamics
- Epithelial-mesenchymal transition
multi-step activation and differentiation process by which epithelial cells achieve mesenchymal phenotypes, activation of migration and a delay of cell cycle progression
- Fatty acid oxidation
primary mitochondrial aerobic pathway of fatty acid catabolism to acetyl-CoA to produce proteins, carbohydrates, and lipid
- Filopodia
thin, spindle-shaped, and dynamic actin-rich protrusions at the leading edge that adhere to and probe the environment during cell migration
- Glycolysis
rapidly adaptive metabolic pathway (100-fold faster than OxPhos), which yields 2 moles of ATP per mole of glucose and can occur under oxygen-dependent or -independent, aerobic, or non-aerobic conditions
- Hypoxia-inducible factor 1 (HIF-1)
central regulator of cell response to hypoxia
- Integrin
adhesion receptors that connect the actin cytoskeleton with extracellular ligands through affinity regulation and clustering, and thereby form transient ECM interactions and generate traction forces required for migration
- Lamellipodia
flat membrane protrusion at the front of moving cells which extends by actin network dynamics, adheres to substrate by integrins, and pulls the cell forward by myosin II motors
- Macropinocytosis
internalization of extracellular proteins and necrotic cell debris (necrocytosis), often followed by degradation in phagolysosomes
- Mesenchymal migration
migration mode characterized by fibroblast-like morphology, focalized interactions to ECM, and protease-dependent ECM degradation
- Non-muscle myosin II
actin-binding protein involved in actin cross-linking and contraction of actin filaments
- Oxidative phosphorylation
energy-producing pathway in mitochondria that depends on oxygen and converts glucose via the tricarboxylic acid cycle into ATP (36 ATP per glucose molecule)
- Pentose phosphate pathway
alternative pathway of glucose metabolism that provides metabolites for nucleotide synthesis, cell survival and growth, including nicotinamide adenine dinucleotide phosphate and ribose 5-phosphate
- Pseudopodia
actin-rich short-lived membrane protrusions involved in cell migration and chemotaxis
- Rho-family GTPases
family of signaling G proteins that function as regulators of cytoskeletal dynamics, cell polarity, adhesion, and migration
- Stress fibers
filamentous actin bundled in parallel and contracted by myosin II motors in cooperation with actin-binding proteins (e.g., α-actinin)
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fares J. et al. (2020) Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct Target Ther 5, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Margarido AS et al. (2020) Cellular Plasticity during Metastasis: New Insights Provided by Intravital Microscopy. Cold Spring Harb Perspect Med 10, a037267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayor R. and Etienne-Manneville S. (2016) The front and rear of collective cell migration. Nat Rev Mol Cell Biol 17, 97–109 [DOI] [PubMed] [Google Scholar]
- 4.Khalil AA et al. (2020) Collective invasion induced by an autocrine purinergic loop through connexin-43 hemichannels. J Cell Biol 219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung KJ and Ewald AJ (2016) A collective route to metastasis: Seeding by tumor cell clusters. Science (1979) 352, 167–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedl P. and Wolf K. (2009) Proteolytic interstitial cell migration: a five-step process. Cancer and Metastasis Reviews 28, 129–135 [DOI] [PubMed] [Google Scholar]
- 7.Graziani V. et al. (2022) The amoeboid state as part of the epithelial-tomesenchymal transition programme. Trends Cell Biol 32, 228–242 [DOI] [PubMed] [Google Scholar]
- 8.Jing X. et al. (2019) Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer 18, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertout JA et al. (2008) The impact of O2 availability on human cancer. Nat Rev Cancer 8, 967–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKeown SR (2014) Defining normoxia, physoxia and hypoxia in tumours—implications for treatment response. Br J Radiol 87, 20130676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pavlova NN and Thompson CB (2016) The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.vander Heiden MG and DeBerardinis RJ (2017) Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caino MC et al. (2013) Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. Journal of Clinical Investigation 123, 2907–2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nobre AR et al. (2018) The Different Routes to Metastasis via HypoxiaRegulated Programs. Trends Cell Biol 28, 941–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lehmann S. et al. (2017) Hypoxia Induces a HIF-1-Dependent Transition from Collective-to-Amoeboid Dissemination in Epithelial Cancer Cells. Curr Biol 27, 392–400 [DOI] [PubMed] [Google Scholar]
- 16.te Boekhorst V. et al. (2022) Calpain-2 regulates hypoxia/HIF-induced plasticity toward amoeboid cancer cell migration and metastasis. Current Biology 32, 412–427.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeWane G. et al. (2021) Fueling the cytoskeleton – links between cell metabolism and actin remodeling. J Cell Sci 134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kishore M. et al. (2018) Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity 48, 831–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cunniff B. et al. (2016) AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol Biol Cell 27, 2662–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuler M-H et al. (2017) Miro1-mediated mitochondrial positioning shapes intracellular energy gradients required for cell migration. Mol Biol Cell 28, 2159–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Horssen R. et al. (2009) Modulation of cell motility by spatial repositioning of enzymatic ATP/ADP exchange capacity. J Biol Chem 284, 1620–7 [DOI] [PubMed] [Google Scholar]
- 22.Daniel Redaet et al. (2019) Mitochondria tether to Focal Adhesions during cell migration and regulate their size. bioRxiv [Google Scholar]
- 23.Caswell PT and Zech T. (2018) Actin-Based Cell Protrusion in a 3D Matrix. Trends Cell Biol 28, 823–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ridley AJ (2011) Life at the Leading Edge. Cell 145, 1012–1022 [DOI] [PubMed] [Google Scholar]
- 25.Vicente-Manzanares M. et al. (2009) Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 10, 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charras G. and Paluch E. (2008) Blebs lead the way: how to migrate without lamellipodia. Nat Rev Mol Cell Biol 9, 730–736 [DOI] [PubMed] [Google Scholar]
- 27.Ortiz-Otero N. et al. (2020) Chemotherapy-induced release of circulating-tumor cells into the bloodstream in collective migration units with cancer-associated fibroblasts in metastatic cancer patients. BMC Cancer 20, 873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Labernadie A. et al. (2017) A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol 19, 224–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mège RM and Ishiyama N. (2017) Integration of Cadherin Adhesion and Cytoskeleton at Adherens Junctions. Cold Spring Harb Perspect Biol 9, a028738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bays JL et al. (2014) Vinculin phosphorylation differentially regulates mechanotransduction at cell-cell and cell-matrix adhesions. J Cell Biol 205, 251–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bays JL et al. (2017) Linking E-cadherin mechanotransduction to cell metabolism through force-mediated activation of AMPK. Nat Cell Biol 19, 724–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Z. et al. (2016) Integrin-mediated mechanotransduction. Journal of Cell Biology 215, 445–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parsons JT et al. (2010) Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 11, 633–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartman ZR et al. (2013) The tyrosine phosphatase SHP2 regulates focal adhesion kinase to promote EGF-induced lamellipodia persistence and cell migration. Mol Cancer Res 11, 651–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Z. et al. (2018) PTPN12/PTP-PEST Regulates Phosphorylation-Dependent Ubiquitination and Stability of Focal Adhesion Substrates in Invasive Glioblastoma Cells. Cancer Res 78, 3809–3822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huttenlocher A. and Horwitz AR (2011) Integrins in cell migration. Cold Spring Harb Perspect Biol 3, a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graham DM et al. (2018) Enucleated cells reveal differential roles of the nucleus in cell migration, polarity, and mechanotransduction. J Cell Biol 217, 895–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Versaevel M. et al. (2012) Spatial coordination between cell and nuclear shape within micropatterned endothelial cells. Nat Commun 3, 671. [DOI] [PubMed] [Google Scholar]
- 39.Khatau SB et al. (2009) A perinuclear actin cap regulates nuclear shape. Proc Natl Acad Sci U S A 106, 19017–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomes ER et al. (2005) Nuclear Movement Regulated by Cdc42, MRCK, Myosin, and Actin Flow Establishes MTOC Polarization in Migrating Cells. Cell 121, 451–463 [DOI] [PubMed] [Google Scholar]
- 41.Schwartz C. et al. (2017) Lamins and nesprin-1 mediate inside-out mechanical coupling in muscle cell precursors through FHOD1. Sci Rep 7, 1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bertacchini J. et al. (2013) The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression. FASEB J 27, 2145–55 [DOI] [PubMed] [Google Scholar]
- 43.Bell Emily S. et al. (2021) Low lamn A levels enhance confined cell migration and metastatic capacity in breast cancer. biRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chu C-T et al. (2021) Tyrosine phosphorylation of lamin A by Src promotes disassembly of nuclear lamina in interphase. Life Sci Alliance 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guilluy C. et al. (2014) Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat Cell Biol 16, 376–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stroka KM et al. (2014) Water permeation drives tumor cell migration in confined microenvironments. Cell 157, 611–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morishita K. et al. (2019) Cell volume regulation in cancer cell migration driven by osmotic water flow. Cancer Sci 110, 2337–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eto K. et al. (2010) Phosphorylation of aquaporin-2 regulates its water permeability. J Biol Chem 285, 40777–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y. et al. (2019) On the energy efficiency of cell migration in diverse physical environments. Proc Natl Acad Sci U S A 116, 23894–23900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Ieso ML and Yool AJ (2018) Mechanisms of Aquaporin-Facilitated Cancer Invasion and Metastasis. Front Chem 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Castro-Castro A. et al. (2016) Cellular and Molecular Mechanisms of MT1-MMP-Dependent Cancer Cell Invasion. Annu Rev Cell Dev Biol 32, 555–576 [DOI] [PubMed] [Google Scholar]
- 52.Bravo-Cordero JJ et al. (2007) MT1-MMP proinvasive activity is regulated by a novel Rab8-dependent exocytic pathway. EMBO J 26, 1499–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Itoh Y. (2001) Homophilic complex formation of MT1-MMP facilitates proMMP-2 activation on the cell surface and promotes tumor cell invasion. EMBO J 20, 4782–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Niland S. et al. (2021) Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int J Mol Sci 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lagoutte E. et al. (2016) LIMK Regulates Tumor-Cell Invasion and Matrix Degradation Through Tyrosine Phosphorylation of MT1-MMP. Sci Rep 6, 24925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martínez-Reyes I. and Chandel NS (2021) Cancer metabolism: looking forward. Nat Rev Cancer 21, 669–680 [DOI] [PubMed] [Google Scholar]
- 57.Jia D. et al. (2019) Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proceedings of the National Academy of Sciences 116, 3909–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shiraishi T. et al. (2015) Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 6, 130–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park JS et al. (2020) Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 578, 621–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.ZHENG J. (2012) Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol Lett 4, 1151–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Poillet-Perez L. and White E. (2019) Role of tumor and host autophagy in cancer metabolism. Genes Dev 33, 610–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nikolaou S. and Machesky LM (2020) The stressful tumour environment drives plasticity of cell migration programmes, contributing to metastasis. J Pathol 250, 612–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Louie MC et al. (2020) Total Cellular ATP Production Changes With Primary Substrate in MCF7 Breast Cancer Cells. Front Oncol 10, 1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Herzig S. and Shaw RJ (2018) AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19, 121–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saxton RA and Sabatini DM (2017) mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nagao A. et al. (2019) HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int J Mol Sci 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu H. et al. (2014) Upregulation of autophagy by hypoxia-inducible factor-1α promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol Rep 32, 935–42 [DOI] [PubMed] [Google Scholar]
- 68.Peng Y-F et al. (2013) Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 9, 2056–68 [DOI] [PubMed] [Google Scholar]
- 69.Minchenko O. et al. (2004) Hypoxia induces transcription of 6-phosphofructo2-kinase/fructose-2,6-biphosphatase-4 gene via hypoxia-inducible factor-1alpha activation. FEBS Lett 576, 14–20 [DOI] [PubMed] [Google Scholar]
- 70.Liu Y. et al. (2020) Cancer progression is mediated by proline catabolism in non-small cell lung cancer. Oncogene 39, 2358–2376 [DOI] [PubMed] [Google Scholar]
- 71.Cluntun AA et al. (2017) Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 3, 169–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Loo JM et al. (2015) Extracellular Metabolic Energetics Can Promote Cancer Progression. Cell 160, 393–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baltazar F. et al. (2020) Lactate Beyond a Waste Metabolite: Metabolic Affairs and Signaling in Malignancy. Front Oncol 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jayashankar V. and Edinger AL (2020) Macropinocytosis confers resistance to therapies targeting cancer anabolism. Nat Commun 11, 1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.te Boekhorst V. et al. (2016) Plasticity of Cell Migration In Vivo and In Silico. Annu Rev Cell Dev Biol 32, 491–526 [DOI] [PubMed] [Google Scholar]
- 76.Yamada KM and Sixt M. (2019) Mechanisms of 3D cell migration. Nat Rev Mol Cell Biol 20, 738–752 [DOI] [PubMed] [Google Scholar]
- 77.Haeger A. et al. (2015) Collective cell migration: guidance principles and hierarchies. Trends Cell Biol 25, 556–66 [DOI] [PubMed] [Google Scholar]
- 78.Zhang J. et al. (2019) Energetic regulation of coordinated leader-follower dynamics during collective invasion of breast cancer cells. Proc Natl Acad Sci U S A 116, 7867–7872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vilchez Mercedes SA et al. (2021) Decoding leader cells in collective cancer invasion. Nat Rev Cancer 21, 592–604 [DOI] [PubMed] [Google Scholar]
- 80.Wolf K. et al. (2007) Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol 9, 893–904 [DOI] [PubMed] [Google Scholar]
- 81.Friedl P. and Mayor R. (2017) Tuning Collective Cell Migration by Cell-Cell Junction Regulation. Cold Spring Harb Perspect Biol 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bazellières E. et al. (2015) Control of cell-cell forces and collective cell dynamics by the intercellular adhesome. Nat Cell Biol 17, 409–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Friedl P. and Wolf K. (2008) Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer Res 68, 7247–9 [DOI] [PubMed] [Google Scholar]
- 84.Commander R. et al. (2020) Subpopulation targeting of pyruvate dehydrogenase and GLUT1 decouples metabolic heterogeneity during collective cancer cell invasion. Nat Commun 11, 1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang J. et al. (2019) Energetic regulation of coordinated leader–follower dynamics during collective invasion of breast cancer cells. Proceedings of the National Academy of Sciences 116, 7867–7872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zanotelli MR et al. (2018) Regulation of ATP utilization during metastatic cell migration by collagen architecture. Mol Biol Cell 29, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zanotelli MR et al. (2019) Energetic costs regulated by cell mechanics and confinement are predictive of migration path during decision-making. Nat Commun 10, 4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bergert M. et al. (2012) Cell mechanics control rapid transitions between blebs and lamellipodia during migration. Proc Natl Acad Sci U S A 109, 14434–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Paluch E. et al. (2005) Cortical Actomyosin Breakage Triggers Shape Oscillations in Cells and Cell Fragments. Biophys J 89, 724–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Barros-Becker F. et al. (2017) Live imaging reveals distinct modes of neutrophil and macrophage migration within interstitial tissues. J Cell Sci 130, 3801–3808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lorentzen A. et al. (2011) An ezrin-rich, rigid uropod-like structure directs movement of amoeboid blebbing cells. J Cell Sci 124, 1256–1267 [DOI] [PubMed] [Google Scholar]
- 92.Tolde O. et al. (2018) Quantitative phase imaging unravels new insight into dynamics of mesenchymal and amoeboid cancer cell invasion. Sci Rep 8, 12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Eva Crosas-Molist et al. (2021) AMPK is a mechano-metabolic sensor linking cell adhesion and mitochondrial dynamics to Myosin II dependent cell migration. Dev Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ruprecht V. et al. (2015) Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell 160, 673–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reversat A. et al. (2020) Cellular locomotion using environmental topography. Nature 582, 582–585 [DOI] [PubMed] [Google Scholar]
- 96.O’Neill PR et al. (2018) Membrane Flow Drives an Adhesion-Independent Amoeboid Cell Migration Mode. Dev Cell 46, 9–22.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wolf K. et al. (2003) Amoeboid shape change and contact guidance: Tlymphocyte crawling through fibrillar collagen is independent of matrix remodeling by MMPs and other proteases. Blood 102, 3262–3269 [DOI] [PubMed] [Google Scholar]
- 98.Yan S. et al. (2016) MMP inhibitor Ilomastat induced amoeboid-like motility via activation of the Rho signaling pathway in glioblastoma cells. Tumor Biology 37, 16177–16186 [DOI] [PubMed] [Google Scholar]
- 99.Orgaz JL et al. (2014) Diverse matrix metalloproteinase functions regulate cancer amoeboid migration. Nat Commun 5, 4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Welf Erik S. et al. (2021) Mechanical worrying drives cell migration in crowded environments. bioRxiv [Google Scholar]
- 101.te Boekhorst V. and Friedl P. (2016) Plasticity of Cancer Cell Invasion—Mechanisms and Implications for Therapypp. 209–264 [DOI] [PubMed] [Google Scholar]
- 102.Zanotelli MR et al. (2021) Mechanoresponsive metabolism in cancer cell migration and metastasis. Cell Metab 33, 1307–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rzeski W. et al. (2001) Glutamate antagonists limit tumor growth. Proc Natl Acad Sci U S A 98, 6372–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang J-B et al. (2010) Targeting Mitochondrial Glutaminase Activity Inhibits Oncogenic Transformation. Cancer Cell 18, 207–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Prasad P. and Roy SS (2021) Glutamine regulates ovarian cancer cell migration and invasion through ETS1. Heliyon 7, e07064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Singh MK et al. (2021) Autophagy Is Polarized toward Cell Front during Migration and Spatially Perturbed by Oncogenic Ras. Cells 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sharifi MN et al. (2016) Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Rep 15, 1660–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kenific CM et al. (2016) NBR1 enables autophagy-dependent focal adhesion turnover. J Cell Biol 212, 577–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Santarosa M. and Maestro R. (2021) The Autophagic Route of E-Cadherin and Cell Adhesion Molecules in Cancer Progression. Cancers (Basel) 13, 6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Damiano V. et al. (2020) The Autophagy Machinery Contributes to E-cadherin Turnover in Breast Cancer. Front Cell Dev Biol 8, 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.El-Mais N. et al. (2021) Human recombinant arginase I [HuArgI (Co)-PEG5000]-induced arginine depletion inhibits ovarian cancer cell adhesion and migration through autophagy-mediated inhibition of RhoA. J Ovarian Res 14, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ren T. et al. (2019) Osteosarcoma cell intrinsic PD-L2 signals promote invasion and metastasis via the RhoA-ROCK-LIMK2 and autophagy pathways. Cell Death Dis 10, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tam SY et al. (2020) Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and Beyond. Front Oncol 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Boedtkjer E. and Pedersen SF (2020) The Acidic Tumor Microenvironment as a Driver of Cancer. Annu Rev Physiol 82, 103–126 [DOI] [PubMed] [Google Scholar]
- 115.García-Jiménez C. and Goding CR (2019) Starvation and Pseudo-Starvation as Drivers of Cancer Metastasis through Translation Reprogramming. Cell Metab 29, 254–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jia D. et al. (2021) Towards decoding the coupled decision-making of metabolism and epithelial-to-mesenchymal transition in cancer. Br J Cancer 124, 1902–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nieto MA (2011) The Ins and Outs of the Epithelial to Mesenchymal Transition in Health and Disease. Annu Rev Cell Dev Biol 27, 347–376 [DOI] [PubMed] [Google Scholar]
- 118.Lai X. et al. (2020) Epithelial-Mesenchymal Transition and Metabolic Switching in Cancer: Lessons From Somatic Cell Reprogramming. Front Cell Dev Biol 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dupuy F. et al. (2015) PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab 22, 577–589 [DOI] [PubMed] [Google Scholar]
- 120.Luo M. et al. (2018) Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab 28, 69–86.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jia D. et al. (2017) Distinguishing mechanisms underlying EMT tristability. Cancer Converg 1, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kim NH et al. (2017) Snail reprograms glucose metabolism by repressing phosphofructokinase PFKP allowing cancer cell survival under metabolic stress. Nat Commun 8, 14374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Weiss F. et al. (2022) Towards targeting of shared mechanisms of cancer metastasis and therapy resistance. Nat Rev Cancer 22, 157–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yang W. et al. (2015) A novel microfluidic platform for studying mammalian cell chemotaxis in different oxygen environments under zero-flow conditions. Biomicrofluidics 9, 044121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Strömblad S. (2022) Cancer biology: Hypoxia-induced talin tail-docking sparks cancer metastasis. Current Biology 32, R79–R81 [DOI] [PubMed] [Google Scholar]
- 126.Hecht I. et al. (2015) Tumor Invasion Optimization by Mesenchymal-Amoeboid Heterogeneity. Sci Rep 5, 10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yoshida T. et al. (2016) Autophagy suppresses cell migration by degrading GEF-H1, a RhoA GEF. Oncotarget 7, 34420–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jungbluth A. et al. (1994) Strong increase in the tyrosine phosphorylation of actin upon inhibition of oxidative phosphorylation: correlation with reversible rearrangements in the actin skeleton of Dictyostelium cells. J Cell Sci 107 ( Pt 1), 117–25 [DOI] [PubMed] [Google Scholar]
- 129.Bacci M. et al. (2016) miR-155 Drives Metabolic Reprogramming of ER + Breast Cancer Cells Following Long-Term Estrogen Deprivation and Predicts Clinical Response to Aromatase Inhibitors. Cancer Res 76, 1615–1626 [DOI] [PubMed] [Google Scholar]
- 130.Gropper Y. et al. (2017) Culturing CTLs under Hypoxic Conditions Enhances Their Cytolysis and Improves Their Anti-tumor Function. Cell Rep 20, 2547–2555 [DOI] [PubMed] [Google Scholar]
- 131.Paluch EK and Raz E. (2013) The role and regulation of blebs in cell migration. Curr Opin Cell Biol 25, 582–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Reiterer M. et al. (2019) Acute and chronic hypoxia differentially predispose lungs for metastases. Sci Rep 9, 10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li Y. et al. (2019) On the energy efficiency of cell migration in diverse physical environments. Proc Natl Acad Sci U S A 116, 23894–23900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.van Helvert S. et al. (2018) Mechanoreciprocity in cell migration. Nat Cell Biol 20, 8–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Odenthal J. et al. (2016) Plasticity of tumor cell invasion: governance by growth factors and cytokines. Carcinogenesis DOI: 10.1093/carcin/bgw098 [DOI] [PubMed] [Google Scholar]
- 136.Sadria M. et al. (2022) The mixed blessing of AMPK signaling in Cancer treatments. BMC Cancer 22, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tognoli ML et al. (2021) RASSF1C oncogene elicits amoeboid invasion, cancer stemness, and extracellular vesicle release via a SRC/Rho axis. EMBO J 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Amaravadi RK et al. (2019) Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov 9, 1167–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wu L. et al. (2021) Identification of a novel six autophagy-related genes signature for the prognostic and a miRNA-related autophagy predictor for antiPD-1 therapy responses in prostate cancer. BMC Cancer 21, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Peiró CHF et al. (2021) Diagnostic potential of hypoxia-induced genes in liquid biopsies of breast cancer patients. Sci Rep 11, 8724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Li Z. et al. (2019) Liquid biopsy-based single-cell metabolic phenotyping of lung cancer patients for informative diagnostics. Nat Commun 10, 3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Laird DW and Lampe PD (2022) Cellular mechanisms of connexin-based inherited diseases. Trends Cell Biol 32, 58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Garde A. and Sherwood DR (2021) Fueling Cell Invasion through Extracellular Matrix. Trends Cell Biol 31, 445–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sivandzade F. et al. (2019) Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio Protoc 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wu D. et al. (2021) Single-cell metabolic imaging reveals a SLC2A3-dependent glycolytic burst in motile endothelial cells. Nat Metab 3, 714–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mosier JA et al. (2021) Recent advances in understanding the role of metabolic heterogeneities in cell migration. Fac Rev 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Weinberg F. et al. (2019) Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers (Basel) 11, 1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lushchak VI and Storey KB (2021) Oxidative stress concept updated: Definitions, classifications, and regulatory pathways implicated. EXCLI J 20, 956–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Balta E. et al. (2021) Redox Regulation of the Actin Cytoskeleton in Cell Migration and Adhesion: On the Way to a Spatiotemporal View. Front Cell Dev Biol 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cheung EC et al. (2020) Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 37, 168–182.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]