

Abstract
Less than 5% of intravenously-injected nanoparticles (NPs) reach destined sites in the body due to opsonization and immune-based clearance in vascular circulation. By hitchhiking in situ onto specific blood components post-injection, NPs can selectively target tissue sites for unprecedentedly high drug delivery rates. Choline carboxylate ionic liquids (ILs) are biocompatible liquid salts <100X composed of bulky asymmetric cations and anions. This class of ILs has been previously shown to significantly extend circulation time and redirect biodistribution in BALB/c mice post-IV injection via hitchhiking on red blood cell (RBC) membranes. Herein, we synthesized & screened 60 choline carboxylic acid-based ILs to coat PLGA NPs and present the impact of structurally engineering the coordinated anion identity to selectively interface and hitchhike lymphocytes, monocytes, granulocytes, platelets, and RBCs in whole mouse blood for in situ targeted drug delivery. Furthermore, we find this nanoparticle platform to be biocompatible (non-cytotoxic), translate to human whole blood by resisting serum uptake and maintaining modest hitchhiking, and also significantly extend circulation retention over 24 hours in BALB/c healthy adult mice after IV injection. Because of their altered circulation profiles, we additionally observe dramatically different organ accumulation profiles compared to bare PLGA NPs. This study establishes an initial breakthrough platform for a modular and transformative targeting technology to hitchhike onto blood components with high efficacy and safety in the bloodstream post-IV administration.
INTRODUCTION
Cell-selective ionic liquid-coated polymeric nanoparticles may overcome intravenous drug delivery barriers in systemic circulation for more efficient organ targeting.
Despite their potential for targeted and non-toxic delivery of systemically-administered drugs, particularly chemotherapeutics1-5, very few intravenous (IV) nanoparticle (NP) delivery systems, and no polymer-based NPs, have successfully made it into the clinic6. A primary challenge remains the premature clearance of polymeric nanoparticles by the immune system soon after intravenous (IV) injection, initiated by mononuclear phagocytic system (MPS) components within the blood7,8. The mechanism of this clearance is driven by opsonization–the process of serum proteins rapidly adsorbing to NP surfaces and triggering the immune system via a series of monocyte and protein-signaling cascades9-12. In addition to phagocytosis, this system shuttles NPs to off-target accumulation sites, such as the kidneys, spleen, and liver, acting as filtration systems for rapid clearance13,14. This creates a significant challenge in designing bioresponsive and targeted IV NP systems, as, despite the surface functionalization strategy, fewer than 5% currently reach their destination after IV injection15-18. Hydrophilic PEGylation and lipid nanoparticles (LNPs) are two current strategies to extend circulation half-life time. However, most (~60%) patients are prone to develop anti-PEG antibodies in their bloodstream19-22, and PEGylation was previously shown to drive increased particle phagocytosis by neutrophils in human blood123. Additionally, depending on their formulation, LNPs are limited by range of targeting selectivity, sensitive storage conditions, instability in-vivo, and acute immune responses associated with toxicity caused by recent ionizable lipid technology23-25. Re-engineering of NP bio-interactions in the bloodstream via cellular hitchhiking is a new and recent approach that allows for biocompatible and facile re-directing of systemic NPs to desired tissues without significant interaction with the MPS 26-31. However, these bioinspired systems are also limited as they involve extraction of blood cells, combination, and re-injection back into the bloodstream, creating a challenge for streamlining accessible clinical translation 32.
Ionic liquids are liquid salts under 100 °C, composed of bulky and asymmetric cations and anions, which have novel potential to act as drug delivery biomaterials, seen in transdermal, antimicrobial, transbuccal, oral, and intravenous systems 33-43. With high structural tunability of either cationic or anionic partner, the resulting physicochemical and biophysical properties can be controlled both within the IL and with its interfacing environment 44-46. Furthermore, changing the length in alkyl chain structure of the cation of the IL has been shown to modulate interactions with albumin47, a primary serum protein in the bloodstream that participates in the protein adsorption and opsonization process48. A choline and trans-2-hexenoate IL has been used to electrostatically coat PLGA surfaces in order to repel serum proteins as well as direct affinity to RBC membranes in situ, thereby significantly extending circulation half-life and redirecting biodistribution to the first encountered capillary bed post-IV injection in both BALB/c mouse and Sprague-Dawley rat in vivo models49-50. We rationalize that by engineering the anion identity around that of trans-2-hexenoic acid in choline carboxylate ILs, we can identify new analog structures [ seen in Scheme 1] that drive selective affinity to white blood cell subpopulations, platelets, as well as RBCs, in whole blood. By engineering cellular hitchhiking affinity in whole blood, the targeting for diseased tissue sites can be significantly enhanced in an unprecedented way for virtually any disease manifesting at those locations, and in systemic circulation.
Ionic liquid engineering and nanoformulation of IL-PLGA NPs for redirected biodistribution via cellular hitchhiking in situ post-intravenous administration
Herein, we have synthesized and screened a library of biocompatible choline carboxylic acid-based ILs to coat PLGA nanoparticles, whereby we have structurally engineered the anion’s alkyl chain length, saturation degree, functionalization moieties (methyl and oxygen groups), and the cation: anion ionic ratio. We have physicochemically characterized the IL-coated particles (IL-NPs) by Dynamic Light Scattering (DLS), quantitative 1H NMR spectroscopy, and fluorescent encapsulation efficiency (%EE), and studied their biophysical interactions in whole mouse and human blood ex vivo, and their pharmacokinetic and biodistribution profile in vivo. We first synthesized carboxylic acid-terminated poly(lactic-co-glycolic) acid (PLGA [50:50])-based nanoparticles (NPs) via nanoprecipitation and solvent evaporation using an acetonitrile (ACN)-based organic phase with the far-red fluorescent dye 1,1'-dioctadecyl-3,3,3',3'- tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate (DiD, 2% wt/wt polymer) 51. Via DLS, bare DiD-loaded particles yielded a diameter of 57.83 5.61 nm, zeta potential of −22.94 3.85 mV, and PDI of 0.19 0.07 (n=4). We then synthesized ~60 different choline carboxylate ILs, which were coated on PLGA NPs and yielded a range of sizes below and above 200 nm, albeit with consistent anionic surface charge shifts and overall low PDI (< 0.3) (Table S1).
Bare & IL-coated NPs (with saline control) at 1 mg/mL were screened for hitchhiking potential by first being mixed with whole commercial BALB/c mouse blood (Bio-VT, NY, USA) at a 1:10 (v/v) ratio, approximately 100 NPs: RBC. After a brief mechanical inversion and 20 minutes of shaking incubation at 37 °C, the blood was centrifuged at 1000xg for 10 minutes, and each layer -- serum, white blood cells (WBCs), platelets, & red blood cells (RBCs) – was isolated carefully by micropipette using its cellular density gradient and placed into 1.5 mL microcentrifuge Eppendorf tubes, which were washed 3x with 1x PBS (pH 7.4) at 200xg for 5 minutes to remove unbound NPs. WBCs, platelets, and RBCs were then analyzed by Fluorescent Activated Cell Sorting (FACS) to identify the fraction of gated singlet cells colocalized with the NPs’ far-red dye.
After screening ~60 candidates by FACS, a structure-function relationship was found to drive affinity to certain cellular subpopulations within whole blood compared to others (Figure 1 and Fig.S1A&B). Particularly, anion chain length, saturation, and molar ionic ratio between cation: anion all play a role in redirecting cellular affinity in whole blood (full ranked heatmaps available in Fig.S1). For instance, with regard to RBCs (Fig.1C), saturated anions at a 1:1 ratio demonstrated a clear increasing approach towards a 7-carbon hotspot (~21.0); however, they showed no affinity from 8-10 carbon chain lengths. This may be due to stronger hydrophobic interactions possible with the longer saturated chains reducing the IL-NPs’ protein avoidance. Consistent with this theory, saturated anions at 1:2 maintained singular affinity at 5 carbons (15.6) with worse performance across the 6-10 carbon chains. Methylating the second carbon for saturated chains at 1:1 decreased their respective overall affinity but shifted the hotspot from 7 carbons to 6 carbons (7.8), favoring shorter chains. At a 1:2 cation: anion ratio, 2-methyl hexanoic acid maintained the best RBC performance (7.1).
Figure 1. IL-PLGA DiD NP top candidates demonstrate unique chemical compositions when coating PLGA NPs, varying but functional encapsulation efficiencies (EE, %), and magnitudes-greater hitchhiking affinities towards specific cell-types vs. PLGA NPs in whole blood.
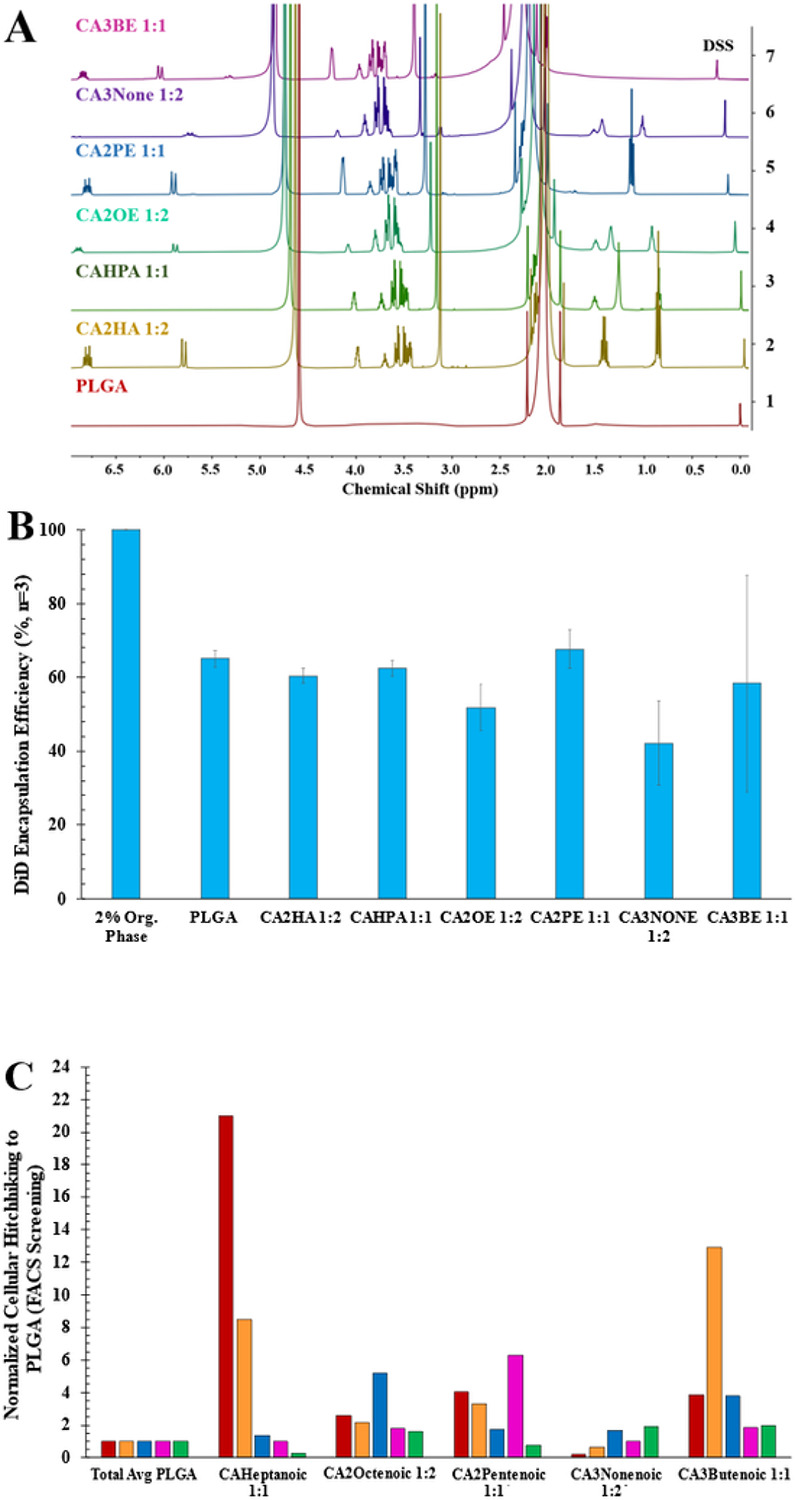
A) Quantitative 1H NMR spectroscopy (DSS, 0.00 ppm: 0.2 mg DSS/1 mg NPs in D2O) illustrates the impact of varying carboxylate anion structure (violet-gold) on the assembly of PLGA NP surfaces (red), quantified in Table S3. B) DiD Encapsulation Efficiency (EE%) by fluorescent plate reader shows the impact of each top candidate IL coating on the fluorescence of PLGA DiD NPs, ranging from 40-70%, when directly compared to the organic phase used for synthesis (Table S4). C) From a FACS screen of 60+ screened IL-PLGA DiD NPs in whole blood (normalized to bare PLGA DiD NPs), top IL candidates show unprecedented and selective cellular hitchhiking to RBCs (red), platelets (orange), lymphocytes (blue), monocytes (pink), and granulocytes (green).
While we previously demonstrated the RBC-hitchhiking capabilities of CA2HA 1:2, structural screening analysis conducted in this report concurred that adding a trans double bond to the anion generally increased its hitchhiking capability, but that placement drastically affected performance (notably, 8-carbon chains consistently performed the worst for RBCs in every structural category but directed the highest lymphocyte affinity when in the 2-unsaturated chains in Fig.S1E). For instance, at a 1:1 ratio, C2-unsaturation maximized performance at the shortest chain length (CA2BE 1:1, 17.6), drastically decreased at 5 carbons, and again increased from 6 carbons to 9 carbons (11.0). However, at a 1:2 ratio, 2-unsaturated anions all performed roughly the same (4-7 carbons, 9 carbons: ~6-11). In comparison, at a 1:1 ratio, 3-unsaturation showed a steep increase approaching the 6-carbon hot spot (15.1), with loss of affinity as the chain is increased to 9 carbons. However, at a 1:2 ratio, 3-unsaturation shifted the hotspot to 5 carbons (6.7), albeit reduced in comparative RBC affinity compared with the 1:1 ratio. When methylating the 2-unsaturated anions at the C2 position, RBC affinity was comparatively quenched relative to the unmethylated anion analogue, with redirection of CA2Me2PE affinity to lymphocytes. Interestingly, at a 1:2 ratio, methylating trans-2-pentenoic acid uniquely enhanced its RBC affinity (19.0). Hence, at a 1:1 ratio, the methylation position requires being staggered with the double bond with one degree of separation to enhance RBC affinity, while at 1:2, methylation enhances RBC affinity when on the same carbon position as the double bond.
In contrast, platelet hitchhiking affinity (Fig.S1D) followed almost the opposite relationship to RBCs. For instance, for 2-unsaturated carbon chains, a gradient relationship was observed between 4-8 carbons, but the hot spot was at 9 carbons for 1:1 (5.1) and at 7 (13.2) and 9 carbons (13.3) for 1:2, illustrating the longer carbon chains required for higher platelet affinity when the double bond is present at C2 (as opposed to the short-medium length end of the spectrum for RBCs). However, more rigid structures may perform best for platelet hitchhiking at shorter alkyl lengths: when the 2nd carbon is methylated on saturated anions, the 1:1 ratio shows a shift in hotspot to 6 carbons (CA2MeHexa 1:1), and to 5 carbons (CA2MePenta 1:2) at a 1:2 ratio, shorter than the 7 carbon (CAHPA 1:1) hotspot observed for RBCs. Additionally, while RBC affinity at 3-unsaturation is concentrated around medium-chain length anions, platelet hitchhiking shows the highest and most selective affinity at the most rigid and shortest length anion (CA3BE 1:1), drastically contrasted at its opposite relationship at 1:2 (highest affinity from 8-10 carbons).
Interestingly, screened candidates for granulocyte hitchhiking demonstrated either high but non-specific affinity or lower affinity and higher specificity. However, granulocytes showed the highest structural location-based contrast compared to monocyte hitchhiking (Fig.S1F and Fig.S1G). For example, shorter unsaturated structures CA3Me2HA 1:2 and CA2PE 1:1 (CA2PE 1:1 selected as the top candidate due to higher biocompatibility) performed the most selectively for monocytes, while longer unsaturated structure CA3None 1:2 comparatively had slightly less affinity than CA2None 1:2 or CA3DE 1:2 but was the most selective to granulocytes amongst the screened candidates when normalized to PLGA. Because of its selectivity, CA3None 1:2 was thus chosen as a top candidate to show proof-of-principle. However, if multiple targets were desired, other highly performing screened candidates could be chosen for study.
The DLS profile of the selected top in-situ hitchhiking candidates (Fig.1C) successfully remained under 200 nm within a size range of 130-170 nm, with surface charge ranges between −49 & −65 mV, and 0.06-0.20 PDI range (Table S2). We then studied the IL coating assembly using quantitative 1H NMR spectroscopy (Fig.1A). We were able to resolve anionic protons (5.5-7.0 ppm) reflecting the degree of alkyl chain unsaturation (violet, indigo, blue, green, and fully saturated gold), and we noted the formation of new multiplet patterns between choline peaks (3.5-4.0 ppm). This evidences a unique choline-PLGA surface “footprint” for each IL-PLGA coating assembly, while the negative surface charge indicates that the anion interfaces with the environment on the outermost layer of the coating (Table S1, Surface Charge). Additionally, we used a deuterated sodium trimethylsilylpropanesulfonate (DSS) standard to quantify the precise amount of total IL/mg PLGA and the molecular weights of the synthesized ILs, which allowed us to calculate a 24-hour IC50-biocompatible range 52-56 of approximate IL dosages (4.90 x 10−7 to 3.50 x 10−6 mol IL/100 μL dose for a ~25 mg mouse) for in-vivo administration (Table S3). Interestingly, anion hydrocarbon length played both a factor in the size of the coated nanoparticles (Table S2), as well as the amount of IL on the NP (Table S3), indicating that the bulkiness of the structure and likely sterics (from the location placement of double bond) played a role in the preferential assembly dynamics during nanoparticle coating. For example, the least number of molecules of the 9-carbon chain CA3None 1:2 (granulocyte candidate) was required (4.90 x 10−7 mol/mg PLGA) to created one of the largest coated NPs (164.1 37.2 nm), while the 5-carbon chain CA2PE 1:1 (monocyte candidate) required the highest number of molecules (3.50 x 10−6 mol/mg PLGA) at the lower range of IL-NP top candidate size (139.5 30.8 nm).
We next measured the percent encapsulation efficiency (EE) of DiD dye inside NPs coated with the top IL candidates (Fig.1B & Table S4) to be from 42.2 11.4 (CA3None 1:2) to 67.7 5.1% (CA2PE 1:1), consistent with the EE of our published CA2HA 1:2 IL-NP control at 60.43 2.03% 50. It is possible that the EE variation arises from the impact of a water solvation layer between PLGA and the coating, which is dependent on the anion structure and its degree of hydrophobicity, as it assembles with the cation, which creates the primary interface on the PLGA surface 57-62.
Structural composition directs engineered IL-PLGA NPs to drive different cellular membrane affinity in whole BALB/c mouse and human blood without inducing significant phagocytosis.
After FACS screening, top candidates were selected (Fig.1C) and the experiment was repeated using a fluorescent plate reader for quantitative evaluation in situ (n=4, SEM) as a percentage of cell-bound DiD-encapsulated NPs relative to amount mixed with whole BALB/c mouse blood (Fig.2A). It was revealed that, using a 1 x PBS-treated control baseline (Fig.S4), bare PLGA NPs were primarily found in the serum (30.4 ± 5.9%) with the next highest population likely phagocytosed by WBCs (11.9 ± 2.0%). In contrast, choline heptanoate 1:1 (CAHPA 1:1) bound nearly half of available RBCs (47.5 ± 7.0 %), choline 2-octenoate 1:2 (CA2OE 1:2) preferentially bound WBCs (lymphocytes, from the FACS screen) (26.3 ± 4.6 %), choline 2-pentenoate 1:1 (CA2PE 1:1) preferentially bound monocyte WBCs (33.1 ± 2.0 %), and choline 3-nonenoate 1:2 (CA3None 1:2) preferentially bound granulocyte WBCs (32.7 ± 2.6 %). Lastly, choline 3-butenoate 1:1 (CA3BE 1:1) favored platelets (20.8 ± 2.8 %) with some residual secondary affinity to RBCs (14.1 1.5%). Subsequently, all top candidates quantitatively evaluated in Fig.2A were immediately imaged by live-cell confocal microscopy to verify if the favored cellular populations bound were undergoing phagocytosis vs. hitchhiking (Fig.2B-M). Note that for each event of WBC cellular hitchhiking among top candidates (Fig.2.H-J), DiD colocalization (red) was clearly observed attaching/embedding into the membrane (green/yellow) as opposed to activation & direct cellular internalization observed by bare PLGA NPs (Fig.2.E-G). Additionally, the observed morphology of CA3BE 1:1-bound platelets suggested they were in a dormant state as opposed to activated, indicating stealth-like properties during hitchhiking.
Figure 2. IL-PLGA DiD NP hitchhiking candidates show prominent qualitative and quantitative cellular hitchhiking in whole mouse blood via live-cell confocal microscopy (37 °C; yellow: PE-CDllb+, green: FITC-Ly6G+, red: DiD NPs) and fluorescent plate reader, which translates modestly into human whole blood via generally reduced serum residence (gold) compared to PLGA, and significantly increased fluorescence in RBC (red), platelet (black), and WBC populations (blue).
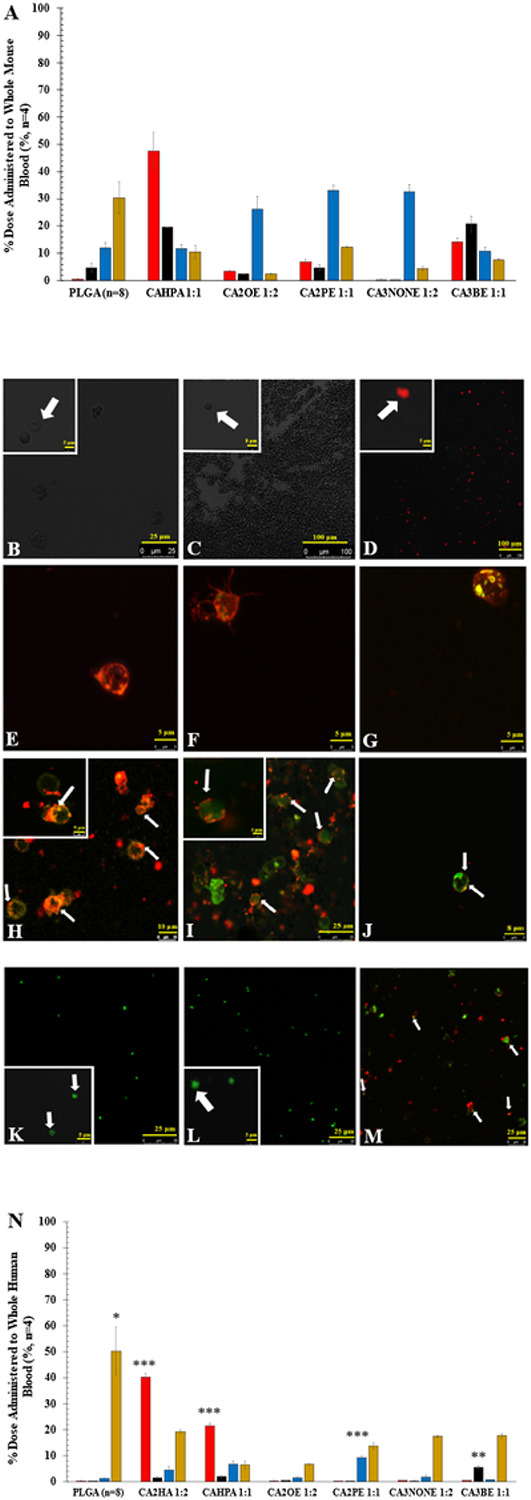
A) Demonstrates cellular affinity of each top IL-PLGA DiD NP candidate in whole BALB/c mouse blood by fluorescent plate reader. Shown: quantified average % NP distribution per cell type for each top candidate in whole mouse BALB/c blood (n=4, SEM). Color key: Gold (Serum) , Blue (WBC), Black (Platelet), Red (RBC). Pictured by live cell confocal microscopy are the following isolated live cell populations (zoomed-in insets in white boxes) from whole blood treated with: B) 1x PBS-treated RBCs, C) PLGA-treated RBCs, D) CAHPA 1:1-treated RBCs; E) PLGA-treated Lymphocyte WBCs, F) PLGA-treated Dendritic Monocyte WBCs, G) PLGA-treated Granulocyte WBCs; H) CA2OE 1:2-treated Lymphocyte WBCs, I) CA2PE 1:1-treated Monocyte WBCs; J)CA3None 1:2-treated Granulocyte WBCs; K) 1x PBS-treated platelets, L)PLGA-treated platelets, M) CA3BE 1:1-treated platelets. N)Demonstrates translational cellular affinity of each top IL-PLGA DiD NP candidate (n=4) vs. bare PLGA (n=8) in human whole blood. Significance against bare PLGA was performed as a two-tailed t-test of unequal variance, with serum uptake screened across all candidates and bare PLGA via ANOVA (*** = p<0.001, ** = p<0.01, & * = p<0.05).
Translational affinity of cellular hitchhiking was then evaluated by re-measuring quantitative distribution of the NP dose to whole blood in adult human female whole blood (n=4, SEM). As human whole blood demonstrates higher immunogenicity than mouse whole blood, bare PLGA NPs showed negligible affinity/uptake to other cell types and instead significantly enhanced residence in the serum (50.30 9.3%, ANOVA, p = 0.000128), likely from serum protein adsorption. Interestingly, positive control CA2HA 1:2 only slightly decreased in RBC hitchhiking capabilities in human whole blood (40.2 1.5 %), although with increased serum uptake (19.1 0.8%), indicating some weakened resistance in human whole blood. In contrast, top RBC candidate CAHPA 1:1 demonstrated half its RBC hitchhiking potential (21.4 1.04%) with drastically reduced serum uptake (6.41.5%) in human blood. Compared to platelet candidate CA3BE 1:1 (p = 0.0025, 5.3 0.5%) & monocyte candidate CA2PE 1:1 (p = 2.3 x , 9.3 0.6 %), a non-significant increase was seen in binding for lymphocyte candidate CA2OE 1:2 (1.4 0.2%) and granulocyte candidate CA3None 1:2 (1.6 0.8%). However, the lowered rates of serum uptake indicates that in human blood these NP candidates are more likely to free-circulate, instead of becoming directly opsonized. Importantly, to illustrate both translational affinity in human, and also to show that choice of anticoagulation (k2EDTA) was not artificially hindering phagocytosis, experimentation was replicated with heparin anticoagulant, and both BALB/c and human WBCs were then imaged by live-cell confocal microscopy, successfully indicating varying degrees of membrane penetration as opposed to phagocytic internalization (Fig. S2 & S3).
Hitchhiking by engineered IL-PLGA NPs operates with stealth and biocompatibility in both whole BALB/c mouse and human blood.
To establish that cellular affinity was not inducing cytotoxicity in both BALB/c and human whole blood during the hitchhiking process, cellular biocompatibility was measured for RBCs (hemolysis), WBCS (PI/Annexin V), RBCs, and platelets (%CD62P+) by either plate reader or flow cytometry (FACS). Including CA2HA 1:2-coated reference NPs, all hitchhiking candidates remained under 10% hemolysis, exhibiting general RBC biocompatibility across human and mouse for IV injection. However, each IL-coated candidate interestingly exhibits different hemolytic properties that may be aligned to cell-type and cell-affinity-related variance in hitchhiking properties across platforms. For example, while CAHPA 1:1 (p = 0.88) shows negligible differences in hemolytic properties across mice and human RBCs, CA2PE 1:1 (p = 0.0014) and CA3None 1:2 (p = 0.00087) candidates showed significantly less hemolysis in human vs. mouse, as opposed to bare PLGA (p = 0.034), CA3BE 1:1(p = 0.02), and CA2OE 1:2-coated NPs (p = 3.8 x 10−6), which show significantly more hemolysis in human vs. mouse blood.
While all BALB/c and human WBCs show a total apoptosis in the range of under 10%, indicating high biocompatibility, the pattern of apoptosis differs between mouse and human lymphocytes, monocytes, and granulocytes. BALB/c lymphocytes show more late apoptosis than early, albeit no difference across treatments in both early (p = 0.054, ANOVA) or late apoptosis (p = 0.89, ANOVA). BALB/c monocytes instead show the opposite trend, additionally with a significant difference in only early apoptosis (p = 0.0066, ANOVA), occurring specifically between PLGA & CA3None 1:2 (p = 0.0077, two-tail t-test), but not across all treatments during late apoptosis (p = 0.17, ANOVA). Interestingly, bare PLGA NPs demonstrated significantly higher granulocyte early apoptosis than all other treatments (p = 8.4E-05, ANOVA), likely from both short-range electrostatic adsorption63 as well as phagocytic activity64 in response to the PLGA. During late granulocyte apoptosis, IL-coated NPs demonstrated generally lower levels compared to controls (p = 9.9 x 10−6, ANOVA) with granulocyte candidate CA3None 1:2-coated NPs notably demonstrating significant protective effects compared to bare PLGA (p = 0.0062, two-tail t-test). In contrast, human WBCs show negligible levels of late apoptosis across all treatments for lymphocytes (early: CA2PE 1:1 with the least relative to PLGA, p = 0.012; late:CA2PE 1:1 with the most relative to PLGA, p = 0.016) , monocytes (ANOVA early: p = 0.06; ANOVA late: p = 0.14) and granulocytes (CA2PE 1:1 with the least relative to PLGA: early (p = 0.027) & late (p = 0.03)), with a common trend of apoptosis occurring during the early phase.
Lastly, platelet activation was measured on the percentage of both BALB/c pooled gender and human adult female CD41+ platelets that co-stained for CD62P+ by FACS. Compared to BALB/c platelets, human platelets were found to naturally tend towards aggregation and activation when treated with the Adenosine Diphosphate (ADP) platelet activation positive control alone (two-tail t-test of means, p = 0.00026). Given this, the significance of platelet activation between mouse and human after bare PLGA NP treatment (p = 0.004), is surprising when directly contrasted with the lack of significant difference from CA3BE 1:1-coated PLGA NP treatment (p = 0.17). This finding not only confirms the presence of the IL coating on the PLGA NPs, but also indicates that membrane attachment facilitated by CA3BE 1:1 is performed without G-protein-coupled receptor surface activation65-67. Instead, the 3-Butenoic acid anion on the outermost layer of the PLGA coating is likely instead either noncovalently tethering/hydrogen-bonding to platelet membrane glycoproteins, to or within amino/choline phospholipid rafts, or engaging with flippase-mediated pump transport into the membrane 68-71. Additionally, because phospholipid-transport pathways are also present in RBCs, the mechanism of CA3BE 1:1 transport into the membrane may also regulate its secondary affinity to RBCs 72,73.
Hitchhiking of RBCs, WBCs, and platelets by engineered IL-PLGA NPs in BALB/c mice significantly extends circulation half-life in vivo and drives dramatically altered biodistribution 24 hours post tail-vein injection.
The 24-hour pharmacokinetic and biodistribution profiles of the top IL-PLGA DiD NP candidates were evaluated in adult, healthy female BALB/c mice after intravenous tail-vein injection (approximate dosage in Table S3). No adverse physiological symptoms were observed for RBC, lymphocyte, granulocyte, or platelet candidates. However, monocyte candidate CA2PE 1:1 demonstrated some bowel disruption, likely from micro/lymph vessel NP shear during the homeostatic influx of cell-hitchhiked nanoparticles into lymph nodes located in the GI tract (such as intestine and jejunum)74,75, as well as the possible added effect of enterohepatic recycling76, aggregating the majority of CA2PE 1:1 PLGA DiD NPs in the intestine and pancreas with minimal clearance (Fig.S5). However, as shown in Figure S5, circulating monocytes that are bound by CA2PE 1:1 first traffic to the intestines from the tail vein at 2 hours (2 hours: intestine: 35.1%, pancreas: 1.5%) before migrating to the pancreas, with additional brain accumulation at 24 hours (24 hours: intestine: 14.9%, pancreas: 34.0%, brain: 6.3%). However, minimal accumulation in the remaining blood circulating organs, including the liver and spleen, indicates that the monocytes are not being triggered for phagocytic clearance or replenishing of inflammatory hepatic monocytes77,78. It is possible that initial monocyte trafficking to the intestines represents homeostatic entry of the lymphatic vasculature to join resident macrophages79, with either eventual differentiation into resident macrophages in the pancreas, complemented by the induction of inflammatory reactions such as inflammatory bowel syndrome (IBS) or mesenteric lymphadenitis80,81, or even possibly acute pancreatitis82,83. As such, these possible phenomena is likely responsible for the death of one mouse at the 2-hour timepoint, and another at the 6-hour timepoint. Issues such as this can likely be resolved via a further dosing study, and this concern may not pose such an issue within a diseased mouse model, where the monocytes are being actively transported to disease sites.
Encouragingly, the remaining white blood cell (WBC) hitchhiking candidates demonstrate notable biocompatibility over the 24-hour circulation period (n=3, average of injected dose (%) ± standard error of the mean (SEM)). In contrast with the negligible pharmacokinetic (PK) behavior of bare PLGA NPs alone at 1 hour (0.5 ± 0.07%) and 24 hours (0.13 ± 0.02%), lymphocyte candidate CA2OE 1:2 demonstrated an initial drop in circulating NPs to 42.2 ± 1.2% (p = 0.0007) at 1 hour with 10.8 ± 2.1% (p = 0.037) retained in the bloodstream at 24 hours. Granulocyte candidate CA3None 1:2 fared similar to CA2OE 1:2 at both 1 hour (46.4 ± 4.1%, p = 0.39) and 6 hours (29.0 ± 5.7%, p = 0.67) but had a non-significant higher retention in the bloodstream at 24 hours than CA2OE 1:2 (18.6 ± 1.0%, p = 0.13). Complementing their similar circulation PK profile, the intestine is the primary biodistribution site for both CA2OE 1:2 (39.8 ± 4.3%) and CA3None 1:2 (66.5 ± 9.5%) at 24 hours –however, granulocyte candidate CA3None 1:2 is much more highly selective to the intestines (secondary accumulation in liver: 10.6 ± 1.4%) than CA2OE 1:2, which has a secondary accumulation in the spleen (12.0 ± 4.5 %), tertiary accumulation in the heart (6.8 ± 0.6%), and lastly the liver (6.6 ± 1.5%).
Post tail-vein injection, both NP-hitchhiked lymphocyte and granulocyte populations would likely initially traffic from circulation to the first available lymph nodes in the intestine (for example: via T-cell recruitment and B-cell homing vs. chemokine-driven stimulated CCR7-expressing membrane-hitchhiked neutrophils84,85. Interestingly, the route of lymphatic entry to the intestine for lymphocytes (high endothelial venules to Peyer’s patches) differs from that of mesenteric entry of monocytes (afferent lymphatic vessels), and GI-tract entry of granulocytes (L-selectin-mediated entry to lymph interstitium): this may be a significant determinant in the perceived similarity in PK, but difference in BD and physiological response 86-91.
Additionally, after encountering the first point of shear (intestinal/gut lymph nodes), CA2OE 1:2-hitchhiked lymphocytes can re-enter the bloodstream circulation via exit through the efferent lymph and shearing through endothelial-modulated postcapillary venules, at which point lymphocytes can migrate downstream and access the cardiovascular and circulatory system through the thoracic duct, as well as may more easily activate, and enter the spleen, both of which were observed notable accumulation sites92-94. However, the vast majority of CA3None 1:2-coated NPs were observed to primarily remain in the intestines at 24 hours. The noticeable lack of travel to any other tissue may be due to weaker forces of membrane adhesion, potentially from the bulkiness of the anionic structure, allowing the IL-PLGA NPs to immediately shear onto endothelial cell linings at the first point of gut lymph entry from the tail-vein (with likely the remaining ~11% to be cleared from the liver) 27. However, the placement of the double bond at the anion’s tertiary carbon offers structural rigidity, which may offer protein-phobic protection (by sterics and reduced hydrogen-bonding) of sheared CA3None 1:2-PLGA NPs from further tissue clearance 95.
In comparison, red blood cell candidate CAHPA 1:1 and platelet candidate CA3BE 1:1 both demonstrated similarly robust circulation profiles. CAHPA 1:1 was observed to reduce only about 50 % at 1 hour (53.9 ± 12.2%) and then taper to a stabilized retention of 20.2 ± 5.8% at 24 hours. Its initial drop and plateau between 6-24 hrs notably differs from the established PK profile of CA2HA 1:2, which had higher retention at every timepoint but a steeper decreasing curve. The structural difference between trans-2-hexenoic acid (2HA) and heptanoic acid (HPA) anions may play several roles in their IL-coating PK variation, as well as their observed BD (Fig.4D). Firstly, the alkyl chain of HPA is longer than 2HA by one carbon and is not restricted (6 free carbon centers) at the center of the chain by a trans double bond (3 free carbon centers). Because of this, HPA has more flexibility in the length of its free interacting alkyl chain tail, which potentially contributes different strengths in initial docking interactions with choline-rich phospholipids and the bilayer within the RBC membrane than 2HA, increasing the number of NPs adsorbed per RBC96,97,31. Additionally, because of the anionic structural difference, the hitchhiking pathway post-docking may also be different; for example, 2HA may interact with the short/medium chain monocarboxylate transport pathway98-100, while HPA may interact with the Band-3/Anion Exchanger (AE1) pathway 101-104.
Figure 4. IL-PLGA NP candidates with selective cellular affinity in whole blood significantly extend BALB/c mouse pharmacokinetics (PK) and redirect organ biodistribution (BD) 24 hours after IV-tail-vein injection.

A-C) PK curves & D) BD represented as average (n=3) of injected dose ± SEM. Bare PLGA NPs (black) functionalized at the surface with only carboxylic acid (black) are almost completely cleared from circulation 1 hour after injection and mainly accumulate in the liver & spleen. In contrast, all candidates have significantly higher retention at 24 hours, with WBC candidates bearing similar PK profiles to a PEGylated control49. Two-samples evaluated at a time via paired two-tail t-test):*=p<0.05, **=p<0.01. BD: Lymphocyte candidate CA2OE 1:2 (purple) and granulocyte candidate CA3None 1:2 (green) primarily accumulate in the intestine lymphatic vasculature, while platelet candidate CA3BE 1:1 (blue) redirects BD to the pancreas and spleen. RBC candidate CAHPA 1:1 (red) shears selectively in the liver, but with negligible accumulation in the spleen compared to bare PLGA. For BD, * indicates significantly less or greater organ accumulation at p<0.05 relative to bare PLGA NPs (black) per organ type.
When examining the biodistribution of CAHPA 1:1, 47.7 ± 6.4% accumulated in the liver, but only 4.2 ± 3.3% was directed to the spleen, far less than bare PLGA NPs (26.8 ± 0.65%). This suggests not direct protein-avoidance but stronger RBC membrane hitchhiking to avoid opsonization. This is because the first site of accumulation is not a gradual distribution of shear within chambers of the first pulmonary capillary bed (CA2HA 1:2), but likely a sudden NP “drop-off” of oxygenated RBCs from the lung circulation into the hepatic artery at 1 hour after IL-NP:RBC weakening from enduring multiple levels of matrix stiffness, shear stress, and arterial pressure105-109. Additionally, the location of liver accumulation is likely in the parenchyma and portal triad ducts, as some enterohepatic recycling from the liver to the bloodstream and intestines was observed (2.4 ± 1.1%), without further CAHPA 1:1 PLGA NP loss from circulation 6-24 hours13.
Despite a similar circulation PK profile to CAHPA 1:1 at 1 hour (p = 0.67), 6 hours (p = 0.26), and 24 hours (p = 0.11), CA3BE 1:1 is better retained in circulation at all timepoints over 24 hours in line with platelet circulation (24 hrs: 27.8 ± 3.0%)110. Respectively, the primary tissue accumulation sites of the platelet vs. RBC candidate was found to drastically differ. CA3BE 1:1-coated PLGA NPs were the only treatment that accumulated in the pancreas at 24 hours (31.4 ± 2.0%). Equivalent accumulation was also found in the spleen (27.5 ± 3.3%), with tertiary accumulation in the kidneys (8.5 ± 2.0%). Interestingly, however, and opposite to CAHPA 1:1, very little accumulated in the liver (3.2 ± 0.5%). With regards to the second site of accumulation, the spleen is a central organ responsible for platelet storage and driving interactions such as dynamic exchange with circulating platelets in the bloodstream111-114. This accumulation profile may also be partially contributed to by the secondary affinity to RBCs where the NP-surface binding density is much lower/weaker than CAHPA 1:1115,116. Rather than direction to clearance, pancreas accumulation may then reasonably follow from the splenic artery which shuttles blood to vascularize the pancreas from at least 4 entry sites117.
While the precise mechanism of biodistribution (ie. exact NP accumulation sites within tissue-specific cells) and anion-mediated cellular binding on a molecular level are still the subject of active investigation, this technology can be directly harnessed to use WBCs, RBCs, and platelets and access organs with high bioavailability and bioactivity for applications such as intestinal diseases (IBS, polyps, Crohn’s), pancreatic cancer, diabetes, sickle cell disease, renal disease, and liver diseases.
Conclusions
Herein we describe, for the first time, biological ionic liquids that can be used as biocompatible polymeric nanoparticle coatings to interface with and selectively hitchhike onto different blood components in situ. While hitchhiking-mediated PK and BD are subject to change under disease conditions, which are not yet known118, this mode of cellular hitchhiking demonstrates the principle that the tissue locations of nanoparticle accumulation can be drastically redirected by structurally engineering the ionic liquid composition comprising the PLGA NP surface coating.While fundamental understanding of ionic liquid-membrane interactions is yet to be elucidated, the ability to target specific subpopulations of the blood across both mouse and human platforms allows for its use in a broad range of technologies, including immunotherapies and gene therapies. As such, this work opens the field towards further chemical, biological, and physical exploration of in situ cellular hitchhiking.
Figure 3. IL-PLGA NP candidates show high levels of biocompatibility with their respective cellular-hitchhiking affinity-types via RBC hemolysis, WBC apoptosis, and platelet activation assays.
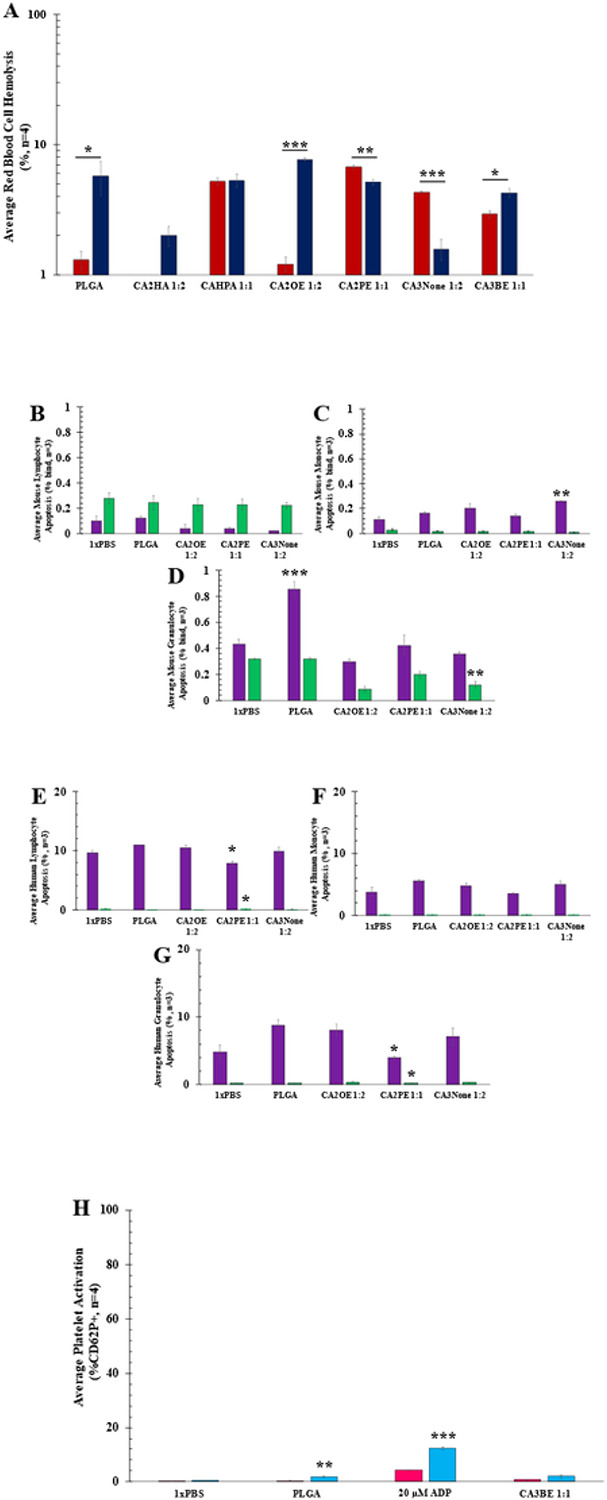
Data represented as average with standard error of mean (SEM). Significance determined by paired two-tailed t-test for two samples at a time between mouse and human samples: *=p<0.05, **=p<0.01, ***=p<0.001. A) Red Blood Cell Hemolysis Assay by fluorescent plate reader of all top candidates and bare PLGA NPs in BALB/c mouse RBCs (red) and Human RBCs (navy) (n=4, SEM). B-G) PI/Annexin V WBC early (purple) vs. late (green) apoptosis assayed by FACS of top lymphocyte (CA2OE 1:2), monocytes (CA2PE 1:1), and granulocyte (CA3None 1:2) hitchhiking candidates from: B-D)whole BALB/c mouse blood or E-G) whole human blood treatment (n=3, ± SEM). H) Platelet activation response by FACS (% activation, CD62P+ from CD41-labeled platelets) to platelet candidate CA3BE 1:1 in whole BALB/c mouse (pink) or human (blue) whole blood (n=4, SEM).
Acknowledgements
EELT acknowledges the PhRMA Foundation, the National Science Foundation (#2236629, #1757220 and #2204193), the National Institutes of Health (1R01EB034086), and the College of Liberal Arts at the University of Mississippi for funding. CMH acknowledges Sigma Xi student Grant-In-Aid-Of-Research (GIAR) #G0315202198510166. OE-A acknowledges NIH (R01 HL145709) for funding. TAW and MH acknowledge support from the NIH through grant #P20 GM130460-01A17937. Images reported in this publication were supported by the GlyCORE Imaging Core and an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under award P20GM103460. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number P30GM122733. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
All animal studies were conduction under the supervision and approval of the University of Mississippi IACUC under protocol 21-004.
COI Statement: EELT and CMH are named as inventors on intellectual property disclosures describing this work
Contributor Information
Christine M. Hamadani, University of Mississippi
Gaya S. Dasanayake, University of Mississippi
Claylee M. Chism, University of Mississippi
Meghan E. Gorniak, University of Mississippi
Wake G. Monroe, University of Mississippi
Anya Merrell, University of Mississippi.
Mercedes C. Pride, University of Mississippi
Rebekah Heintz, University of Mississippi.
Karen Wong, University of Mississippi.
Mehjabeen Hossain, University of Mississippi.
George Taylor, University of Mississippi.
Sara X. Edgecomb, University of Mississippi
Deauntaye Jones, University of Mississippi.
Joy Dhar, University of Mississippi.
Alison Banka, University of Michigan.
Gagandeep Singh, University of Mississippi.
Priyavrat Vashisth, University of Mississippi.
Joh'nis Randall, University of Mississippi.
Donovan S. Darlington, University of Mississippi
Jaylon Everett, University of Mississippi.
Ethan Jarrett, University of Mississippi.
Thomas A. Werfel, University of Mississippi
Omolola Eniola-Adefeso, University of Michigan.
Eden E. L. Tanner, University of Mississippi
References
- 1.Palanikumar L., Al-Hosani S., Kalmouni M. et al. pH-responsive high stability polymeric nanoparticles for targeted delivery of anticancer therapeutics. Commun Biol 3, 95 (2020). 10.1038/s42003-020-0817-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Senapati S., Mahanta A.K., Kumar S. et al. Controlled drug delivery vehicles for cancer treatment and their performance. Sig Transduct Target Ther 3, 7 (2018). 10.1038/s41392-017-0004-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Enlow EM, Luft JC, Napier ME, DeSimone JM. Potent engineered PLGA nanoparticles by virtue of exceptionally high chemotherapeutic loadings. Nano Lett. 2011. Feb 9;11(2):808–13. doi: 10.1021/nl104117p. Epub 2011 Jan 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitchell M.J., Billingsley M.M., Haley R.M. et al. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov 20, 101–124 (2021). 10.1038/s41573-020-0090-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calzoni E, Cesaretti A, Polchi A, Di Michele A, Tancini B, Emiliani C. Biocompatible Polymer Nanoparticles for Drug Delivery Applications in Cancer and Neurodegenerative Disorder Therapies. J Funct Biomater. 2019. Jan 8;10(1):4. doi: 10.3390/jfb10010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anselmo AC, Mitragotri S. Nanoparticles in the clinic: An update. Bioeng Transl Med. 2019. Sep 5;4(3):e10143. doi: 10.1002/btm2.10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheffey VV, Siew EB, Tanner EEL, Eniola-Adefeso O. PLGA's Plight and the Role of Stealth Surface Modification Strategies in Its Use for Intravenous Particulate Drug Delivery. Adv Healthc Mater. 2022. Apr;11(8):e2101536. doi: 10.1002/adhm.202101536. Epub 2022 Jan 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008. Jul-Aug;5(4):505–15. doi: 10.1021/mp800051m.Epub 2008 Aug 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Visalakshan RM, MacGregor MN, Sasidharan S, Ghazaryan A, Mierczynska-Vasilev AM, Morsbach S, Mailänder V, Landfester K, Hayball JD, Vasilev K. Biomaterial Surface Hydrophobicity-Mediated Serum Protein Adsorption and Immune Responses. ACS Appl Mater Interfaces. 2019. Aug 7;11(31):27615–27623. doi: 10.1021/acsami.9b09900. [DOI] [PubMed] [Google Scholar]
- 10.Zanganeh S, Spitler R, Erfanzadeh M, Alkilany AM, Mahmoudi M. Protein corona: Opportunities and challenges. Int J Biochem Cell Biol. 2016. Jun;75:143–7. doi: 10.1016/j.biocel.2016.01.005. Epub 2016 Jan 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 2012. Feb 1;134(4):2139–47. doi: 10.1021/ja2084338. [DOI] [PubMed] [Google Scholar]
- 12.Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H. Nanoparticle Uptake: The Phagocyte Problem. Nano Today. 2015. Aug;10(4):487–510. doi: 10.1016/j.nantod.2015.06.006. Epub 2015 Sep 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-liver interactions: Cellular uptake and hepatobiliary elimination. J Control Release. 2016. Oct 28;240:332–348. doi: 10.1016/j.jconrel.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 14.Milligan JJ, Saha S. A Nanoparticle's Journey to the Tumor: Strategies to Overcome First-Pass Metabolism and Their Limitations. Cancers (Basel). 2022. Mar 29;14(7):1741. doi: 10.3390/cancers14071741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilhelm S., Tavares A., Dai Q. et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater 1, 16014 (2016). 10.1038/natrevmats.2016.14 [DOI] [Google Scholar]
- 16.Zein R, Sharrouf W, Selting K. Physical Properties of Nanoparticles That Result in Improved Cancer Targeting. J Oncol. 2020. Jul 13;2020:5194780. doi: 10.1155/2020/5194780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sykes EA, Dai Q, Sarsons CD, Chen J, Rocheleau JV, Hwang DM, Zheng G, Cramb DT, Rinker KD, Chan WC. Tailoring nanoparticle designs to target cancer based on tumor pathophysiology. Proc Natl Acad Sci U S A. 2016. Mar 1;113(9):E1142–51. doi: 10.1073/pnas.1521265113. Epub 2016 Feb 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang L, Ye Y, Cai L. Above the dose threshold: a simple way to improve the delivery efficiency of nanomedicine. Signal Transduct Target Ther. 2020. Nov 20;5(1):272. doi: 10.1038/s41392-020-00400-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ju Y., Carreño J.M., Simon V. et al. Impact of anti-PEG antibodies induced by SARS-CoV-2 mRNA vaccines. Nat Rev Immunol 23, 135–136 (2023). 10.1038/s41577-022-00825-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.d'Avanzo N., Celia C., Barone A., Carafa M., Di Marzio L., Santos H.A. and Fresta M. (2020), Immunogenicity of Polyethylene Glycol Based Nanomedicines: Mechanisms, Clinical Implications and Systematic Approach. Adv. Therap., 3: 1900170. 10.1002/adtp.201900170 [DOI] [Google Scholar]
- 21.Chen BM, Cheng TL, Roffler SR. Polyethylene Glycol Immunogenicity: Theoretical, Clinical, and Practical Aspects of Anti-Polyethylene Glycol Antibodies. ACS Nano. 2021. Sep 28;15(9):14022–14048. doi: 10.1021/acsnano.1c05922. Epub 2021 Sep 1. [DOI] [PubMed] [Google Scholar]
- 22.Zhang P, Sun F, Liu S, Jiang S. Anti-PEG antibodies in the clinic: Current issues and beyond PEGylation. J Control Release. 2016. Dec 28;244(Pt B):184–193. doi: 10.1016/j.jconrel.2016.06.040. Epub 2016 Jun 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tenchov R, Bird R, Curtze AE, Zhou Q. Lipid Nanoparticles–From Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano. 2021. Nov 23;15(11):16982–17015. doi: 10.1021/acsnano.1c04996. Epub 2021 Jun 28. [DOI] [PubMed] [Google Scholar]
- 24.Schoenmaker L, Witzigmann D, Kulkarni JA, Verbeke R, Kersten G, Jiskoot W, Crommelin DJA. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int J Pharm. 2021. May 15;601:120586. doi: 10.1016/j.ijpharm.2021.120586. Epub 2021 Apr 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han X., Zhang H., Butowska K. et al. An ionizable lipid toolbox for RNA delivery. Nat Commun 12, 7233 (2021). 10.1038/s41467-021-27493-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu J., Gao X., Wang S., He Y., Ma X., Zhang T., Liu X., Exploration 2023, 3, 20220045. 10.1002/EXP.20220045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brenner J.S., Pan D.C., Myerson J.W. et al. Red blood cell-hitchhiking boosts delivery of nanocarriers to chosen organs by orders of magnitude. Nat Commun 9, 2684 (2018). 10.1038/s41467-018-05079-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Z, Ukidve A, Gao Y, Kim J, Mitragotri S. Erythrocyte leveraged chemotherapy (ELeCt): Nanoparticle assembly on erythrocyte surface to combat lung metastasis. Sci Adv. 2019. Nov 13;5(11):eaax9250. doi: 10.1126/sciadv.aax9250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anselmo AC, Gupta V, Zern BJ, Pan D, Zakrewsky M, Muzykantov V, Mitragotri S. Delivering nanoparticles to lungs while avoiding liver and spleen through adsorption on red blood cells. ACS Nano. 2013. Dec 23;7(12):11129–37. doi: 10.1021/nn404853z. Epub 2013 Nov 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang E, Phan P, Algarni HA, Zhao Z. Red Blood Cell Inspired Strategies for Drug Delivery: Emerging Concepts and New Advances. Pharm Res. 2022. Nov;39(11):2673–2698. doi: 10.1007/s11095-022-03328-5. Epub 2022 Jul 7. [DOI] [PubMed] [Google Scholar]
- 31.Ukidve A, Zhao Z, Fehnel A, Krishnan V, Pan DC, Gao Y, Mandal A, Muzykantov V, Mitragotri S. Erythrocyte-driven immunization via biomimicry of their natural antigen-presenting function. Proc Natl Acad Sci U S A. 2020. Jul 28;117(30):17727–17736. doi: 10.1073/pnas.2002880117. Epub 2020 Jul 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lenders V., Escudero R., Koutsoumpou X. et al. Modularity of RBC hitchhiking with polymeric nanoparticles: testing the limits of non-covalent adsorption. J Nanobiotechnol 20, 333 (2022). 10.1186/s12951-022-01544-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu C, Chen B, Shi W, Huang W, Qian H. Ionic Liquids for Enhanced Drug Delivery: Recent Progress and Prevailing Challenges. Mol Pharm. 2022. Apr 4;19(4):1033–1046. doi: 10.1021/acs.molpharmaceut.1c00960. Epub 2022 Mar 11. [DOI] [PubMed] [Google Scholar]
- 34.Agatemor C, Ibsen KN, Tanner EEL, Mitragotri S. Ionic liquids for addressing unmet needs in healthcare. Bioeng Transl Med. 2018. Jan 19;3(1):7–25. doi: 10.1002/btm2.10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanner EEL, Curreri AM, Balkaran JPR, Selig-Wober NC, Yang AB, Kendig C, Fluhr MP, Kim N, Mitragotri S. Design Principles of Ionic Liquids for Transdermal Drug Delivery. Adv Mater. 2019. Jul;31(27):e1901103. doi: 10.1002/adma.201901103. Epub 2019 May 21. [DOI] [PubMed] [Google Scholar]
- 36.Zakrewsky M, Lovejoy KS, Kern TL, Miller TE, Le V, Nagy A, Goumas AM, Iyer RS, Del Sesto RE, Koppisch AT, Fox DT, Mitragotri S. Ionic liquids as a class of materials for transdermal delivery and pathogen neutralization. Proc Natl Acad Sci U S A. 2014. Sep 16;111(37):13313–8. doi: 10.1073/pnas.1403995111. Epub 2014 Aug 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vaidya A, Mitragotri S. Ionic liquid-mediated delivery of insulin to buccal mucosa. J Control Release. 2020. Nov 10;327:26–34. doi: 10.1016/j.jconrel.2020.07.037. Epub 2020 Jul 29. [DOI] [PubMed] [Google Scholar]
- 38.Nurunnabi M, Ibsen KN, Tanner EEL, Mitragotri S. Oral ionic liquid for the treatment of diet-induced obesity. Proc Natl Acad Sci U S A. 2019. Dec 10;116(50):25042–25047. doi: 10.1073/pnas.1914426116. Epub 2019 Nov 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Curreri AM, Kim J, Dunne M, Angsantikul P, Goetz M, Gao Y, Mitragotri S. Deep Eutectic Solvents for Subcutaneous Delivery of Protein Therapeutics. Adv Sci (Weinh). 2023. Mar;10(7):e2205389. doi: 10.1002/advs.202205389. Epub 2023 Jan 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Curreri AM, Mitragotri S, Tanner EEL. Recent Advances in Ionic Liquids in Biomedicine. Adv Sci (Weinh). 2021. Sep;8(17):e2004819. doi: 10.1002/advs.202004819. Epub 2021 Jul 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banerjee A, Ibsen K, Brown T, Chen R, Agatemor C, Mitragotri S. Ionic liquids for oral insulin delivery. Proc Natl Acad Sci U S A. 2018. Jul 10;115(28):7296–7301. doi: 10.1073/pnas.1722338115. Epub 2018 Jun 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Albadawi H, Zhang Z, Altun I, Hu J, Jamal L, Ibsen KN, Tanner EEL, Mitragotri S, Oklu R. Percutaneous liquid ablation agent for tumor treatment and drug delivery. Sci Transl Med. 2021. Feb 10;13(580):eabe3889. doi: 10.1126/scitranslmed.abe3889. [DOI] [PubMed] [Google Scholar]
- 43.Fernandes MM, Carvalho EO, Correia DM, Esperança JMSS, Padrão J, Ivanova K, Hoyo J, Tzanov T, Lanceros-Mendez S. Ionic Liquids as Biocompatible Antibacterial Agents: A Case Study on Structure-Related Bioactivity on Escherichia coli. ACS Appl Bio Mater. 2022. Nov 21;5(11):5181–5189. doi: 10.1021/acsabm.2c00615. Epub 2022 Oct 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pedro SN R Freire CS, Silvestre AJD, Freire MG. The Role of Ionic Liquids in the Pharmaceutical Field: An Overview of Relevant Applications. Int J Mol Sci. 2020. Nov 5;21(21):8298. doi: 10.3390/ijms21218298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reddy RR, Shanmugam G, Madhan B, Phani Kumar BVN. Selective binding and dynamics of imidazole alkyl sulfate ionic liquids with human serum albumin and collagen - a detailed NMR investigation. Phys Chem Chem Phys. 2018. Apr 4;20(14):9256–9268. doi: 10.1039/C7CP08298C. [DOI] [PubMed] [Google Scholar]
- 46.Chism Claylee M., Plash Samuel, Zuckerman Daniel, Dasanayake Gaya S., Bennett Maria, Tripathi Siddharth K., Pedigo Susan D., and Tanner Eden E. L.. Antimicrobial Effects of Anion Manipulation with Biocompatible Choline Carboxylic Acid-Based Ionic Liquids. ACS Applied Engineering Materials 2023. 1(1), 23–31. DOI: 10.1021/acsaenm.2c00004 [DOI] [Google Scholar]
- 47.Zhang M, Wang Y, Zhang H, Cao J, Fei Z, Wang Y. Impact of the alkyl chain length on binding of imidazolium-based ionic liquids to bovine serum albumin. Spectrochim Acta A Mol Biomol Spectrosc. 2018. May 5;196:323–333. doi: 10.1016/j.saa.2018.02.040. Epub 2018 Feb 14. [DOI] [PubMed] [Google Scholar]
- 48.Vincent M.P., Bobbala S., Karabin N.B. et al. Surface chemistry-mediated modulation of adsorbed albumin folding state specifies nanocarrier clearance by distinct macrophage subsets. Nat Commun 12, 648 (2021). 10.1038/s41467-020-20886-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamadani CM, Goetz MJ, Mitragotri S, Tanner EEL. Protein-avoidant ionic liquid (PAIL)-coated nanoparticles to increase bloodstream circulation and drive biodistribution. Sci Adv. 2020. Nov 25;6(48):eabd7563. doi: 10.1126/sciadv.abd7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hamadani CM, Mahdi F, Merrell A, Flanders J, Cao R, Vashisth P, Pride MC, Hunter AN, Singh G, Roman G, Paris JJ, Tanner EEL. Ionic Liquid Coating-Driven Nanoparticle Delivery to the Brain: Applications for NeuroHIV. Res Sq [Preprint]. 2023. Feb 14:rs.3.rs–2574352. doi: 10.21203/rs.3.rs-2574352/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu CM, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci U S A. 2011. Jul 5;108(27):10980–5. doi: 10.1073/pnas.1106634108. Epub 2011 Jun 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dung NT, Giang LNT, Thu PH, et al. Synthesis and Cytotoxic Evaluation of Carboxylic Acid-Functionalized Indenoisoquinolines. Natural Product Communications. 2019;14(5). doi: 10.1177/1934578X19849787 [DOI] [Google Scholar]
- 53.Nawshad Muhammad M. Ismail Hossain, Zakaria Man, Mohanad El-Harbawi, Bustam M. Azmi, Yousr Abdulhadi Noaman, Noorjahan Banu Mohamed Alitheen, Mei Kee Ng, Glenn Hefter, and Chun-Yang Yin. Synthesis and Physical Properties of Choline Carboxylate Ionic Liquids. Journal of Chemical & Engineering Data 2012. 57(8), 2191–2196. DOI: 10.1021/je300086w [DOI] [Google Scholar]
- 54.Klebeko J, Ossowicz-Rupniewska P, Świątek E, Szachnowska J, Janus E, Taneva SG, Krachmarova E, Guncheva M. Salicylic Acid as Ionic Liquid Formulation May Have Enhanced Potency to Treat Some Chronic Skin Diseases. Molecules. 2021. Dec 30;27(1):216. doi: 10.3390/molecules27010216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grassiri B, Mezzetta A, Maisetta G, Migone C, Fabiano A, Esin S, Guazzelli L, Zambito Y, Batoni G, Piras AM. Betaine- and L-Carnitine-Based Ionic Liquids as Solubilising and Stabilising Agents for the Formulation of Antimicrobial Eye Drops Containing Diacerein. Int J Mol Sci. 2023. Feb 1;24(3):2714. doi: 10.3390/ijms24032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakkarach A., Foo H.L., Song A.AL. et al. Anti-cancer and anti-inflammatory effects elicited by short chain fatty acids produced by Escherichia coli isolated from healthy human gut microbiota. Microb Cell Fact 20, 36 (2021). 10.1186/s12934-020-01477-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ichikawa T, Yoshio M, Hamasaki A, Mukai T, Ohno H, Kato T. Self-organization of room-temperature ionic liquids exhibiting liquid-crystalline bicontinuous cubic phases: formation of nano-ion channel networks. J Am Chem Soc. 2007. Sep 5;129(35):10662–3. doi: 10.1021/ja0740418. Epub 2007 Aug 15. [DOI] [PubMed] [Google Scholar]
- 58.Atkin Rob and Warr Gregory G.. Structure in Confined Room-Temperature Ionic Liquids. The Journal of Physical Chemistry C 2007. 111 (13), 5162–5168. DOI: 10.1021/jp067420g. [DOI] [Google Scholar]
- 59.Atkin R, Craig VS, Wanless EJ, Biggs S. Mechanism of cationic surfactant adsorption at the solid-aqueous interface. Adv Colloid Interface Sci. 2003. May 30;103(3):219–304. doi: 10.1016/S0001-8686(03)00002-2. [DOI] [PubMed] [Google Scholar]
- 60.van der Vegt NFA, Nayar D. The Hydrophobic Effect and the Role of Cosolvents. J Phys Chem B. 2017. Nov 2;121(43):9986–9998. doi: 10.1021/acs.jpcb.7b06453. Epub 2017 Oct 7. [DOI] [PubMed] [Google Scholar]
- 61.Woodhead JL, Hall CK. Encapsulation Efficiency and Micellar Structure of Solute-Carrying Block Copolymer Nanoparticles. Macromolecules. 2011. Jun 14;44(13):5443–5451. doi: 10.1021/ma102938g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Otte A, Sharifi F, Park K. Interfacial tension effects on the properties of PLGA microparticles. Colloids Surf B Biointerfaces. 2020. Dec;196:111300. doi: 10.1016/j.colsurfb.2020.111300. Epub 2020 Aug 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pakulska MM, Elliott Donaghue I, Obermeyer JM, Tuladhar A, McLaughlin CK, Shendruk TN, Shoichet MS. Encapsulation-free controlled release: Electrostatic adsorption eliminates the need for protein encapsulation in PLGA nanoparticles. Sci Adv. 2016. May 27;2(5):e1600519. doi: 10.1126/sciadv.1600519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yin T., Li Y., Ren Y. et al. Phagocytosis of polymeric nanoparticles aided activation of macrophages to increase atherosclerotic plaques in ApoE−/− mice. J Nanobiotechnol 19, 121 (2021). 10.1186/s12951-021-00863-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Offermanns S. Activation of platelet function through G protein-coupled receptors. Circ Res. 2006. Dec 8;99(12):1293–304. doi: 10.1161/01.RES.0000251742.71301.16. [DOI] [PubMed] [Google Scholar]
- 66.Gurbel PA, Kuliopulos A, Tantry US. G-protein-coupled receptors signaling pathways in new antiplatelet drug development. Arterioscler Thromb Vasc Biol. 2015. Mar;35(3):500–12. doi: 10.1161/ATVBAHA.114.303412. Epub 2015 Jan 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stalker TJ, Newman DK, Ma P, Wannemacher KM, Brass LF. Platelet signaling. Handb Exp Pharmacol. 2012;(210):59–85. doi: 10.1007/978-3-642-29423-5_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li CQ, Vindigni A, Sadler JE, Wardell MR. Platelet glycoprotein Ib alpha binds to thrombin anion-binding exosite II inducing allosteric changes in the activity of thrombin. J Biol Chem. 2001. Mar 2;276(9):6161–8. doi: 10.1074/jbc.M004164200. Epub 2000 Oct 9. [DOI] [PubMed] [Google Scholar]
- 69.Seaman GV. Electrochemical features of platelet interactions. Thromb Res. 1976. May;8(2 suppl):235–46. doi: 10.1016/0049-3848(76)90066-9. [DOI] [PubMed] [Google Scholar]
- 70.Wang J., Yu C., Zhuang J. et al. The role of phosphatidylserine on the membrane in immunity and blood coagulation. Biomark Res 10, 4 (2022). 10.1186/s40364-021-00346-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lhermusier T, Chap H, Payrastre B. Platelet membrane phospholipid asymmetry: from the characterization of a scramblase activity to the identification of an essential protein mutated in Scott syndrome. J Thromb Haemost. 2011. Oct;9(10):1883–91. doi: 10.1111/j.1538-7836.2011.04478.x. [DOI] [PubMed] [Google Scholar]
- 72.Schroit AJ, Zwaal RF. Transbilayer movement of phospholipids in red cell and platelet membranes. Biochim Biophys Acta. 1991. Nov 13;1071(3):313–29. doi: 10.1016/0304-4157(91)90019-s. [DOI] [PubMed] [Google Scholar]
- 73.Geldwerth D, Kuypers FA, Bütikofer P, Allary M, Lubin BH, Devaux PF. Transbilayer mobility and distribution of red cell phospholipids during storage. J Clin Invest. 1993. Jul;92(1):308–14. doi: 10.1172/JCI116568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van den Broeck W, Derore A, Simoens P. Anatomy and nomenclature of murine lymph nodes: Descriptive study and nomenclatory standardization in BALB/cAnNCrl mice. J Immunol Methods. 2006. May 30;312(1-2):12–9. doi: 10.1016/j.jim.2006.01.022. Epub 2006 Mar 6. [DOI] [PubMed] [Google Scholar]
- 75.Miura S, Hokari R, Tsuzuki Y. Mucosal immunity in gut and lymphoid cell trafficking. Ann Vasc Dis. 2012;5(3):275–81. doi: 10.3400/avd.ra.12.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ibarra M., Trocóniz I.F. & Fagiolino P. Enteric reabsorption processes and their impact on drug pharmacokinetics. Sci Rep 11, 5794 (2021). 10.1038/s41598-021-85174-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Srinivas M, Sharma P, Jhunjhunwala S. Phagocytic Uptake of Polymeric Particles by Immune Cells under Flow Conditions. Mol Pharm. 2021. Dec 6;18(12):4501–4510. doi: 10.1021/acs.molpharmaceut.1c00698. Epub 2021 Nov 8. [DOI] [PubMed] [Google Scholar]
- 78.Lai SM, Sheng J, Gupta P, Renia L, Duan K, Zolezzi F, Karjalainen K, Newell EW, Ruedl C. Organ-Specific Fate, Recruitment, and Refilling Dynamics of Tissue-Resident Macrophages during Blood-Stage Malaria. Cell Rep. 2018. Dec 11;25(11):3099–3109.e3. doi: 10.1016/j.celrep.2018.11.059. [DOI] [PubMed] [Google Scholar]
- 79.Robinson A, Han CZ, Glass CK, Pollard JW. Monocyte Regulation in Homeostasis and Malignancy. Trends Immunol. 2021. Feb;42(2):104–119. doi: 10.1016/j.it.2020.12.001. Epub 2021 Jan 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guan X, Morris ME. Pharmacokinetics of the Monocarboxylate Transporter 1 Inhibitor AZD3965 in Mice: Potential Enterohepatic Circulation and Target-Mediated Disposition. Pharm Res. 2019. Dec 10;37(1):5. doi: 10.1007/s11095-019-2735-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Qi S., Wang X., Chang K. et al. The bright future of nanotechnology in lymphatic system imaging and imaging-guided surgery. J Nanobiotechnol 20, 24 (2022). 10.1186/s12951-021-01232-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Manohar M, Jones EK, Rubin SJS, Subrahmanyam PB, Swaminathan G, Mikhail D, Bai L, Singh G, Wei Y, Sharma V, Siebert JC, Maecker HT, Husain SZ, Park WG, Pandol SJ, Habtezion A. Novel Circulating and Tissue Monocytes as Well as Macrophages in Pancreatitis and Recovery. Gastroenterology. 2021. Dec;161(6):2014–2029.e14. doi: 10.1053/j.gastro.2021.08.033. Epub 2021 Aug 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hu F, Lou N, Jiao J, Guo F, Xiang H, Shang D. Macrophages in pancreatitis: Mechanisms and therapeutic potential. Biomed Pharmacother. 2020. Nov;131:110693. doi: 10.1016/j.biopha.2020.110693. Epub 2020 Sep 1. [DOI] [PubMed] [Google Scholar]
- 84.Habtezion A, Nguyen LP, Hadeiba H, Butcher EC. Leukocyte Trafficking to the Small Intestine and Colon. Gastroenterology. 2016. Feb;150(2):340–54. doi: 10.1053/j.gastro.2015.10.046. Epub 2015 Nov 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Beauvillain C, Cunin P, Doni A, Scotet M, Jaillon S, Loiry ML, Magistrelli G, Masternak K, Chevailler A, Delneste Y, Jeannin P. CCR7 is involved in the migration of neutrophils to lymph nodes. Blood. 2011. Jan 27;117(4):1196–204. doi: 10.1182/blood-2009-11-254490. Epub 2010 Nov 4. [DOI] [PubMed] [Google Scholar]
- 86.Jakovija A, Chtanova T. Neutrophil Interactions with the Lymphatic System. Cells. 2021. Aug 17;10(8):2106. doi: 10.3390/cells10082106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bogoslowski A, Wijeyesinghe S, Lee WY, Chen CS, Alanani S, Jenne C, Steeber DA, Scheiermann C, Butcher EC, Masopust D, Kubes P. Neutrophils Recirculate through Lymph Nodes to Survey Tissues for Pathogens. J Immunol. 2020. May 1;204(9):2552–2561. doi: 10.4049/jimmunol.2000022. Epub 2020 Mar 23. [DOI] [PubMed] [Google Scholar]
- 88.Lok LSC, Dennison TW, Mahbubani KM, Saeb-Parsy K, Chilvers ER, Clatworthy MR. Phenotypically distinct neutrophils patrol uninfected human and mouse lymph nodes. Proc Natl Acad Sci U S A. 2019. Sep 17;116(38):19083–19089. doi: 10.1073/pnas.1905054116. Epub 2019 Sep 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hampton HR, Chtanova T. Lymphatic Migration of Immune Cells. Front Immunol. 2019. May 28;10:1168. doi: 10.3389/fimmu.2019.01168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ager A, May MJ. Understanding high endothelial venules: Lessons for cancer immunology. Oncoimmunology. 2015. May 7;4(6):e1008791. doi: 10.1080/2162402X.2015.1008791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kobayashi N, Takahashi D, Takano S, Kimura S, Hase K. The Roles of Peyer's Patches and Microfold Cells in the Gut Immune System: Relevance to Autoimmune Diseases. Front Immunol. 2019. Oct 9;10:2345. doi: 10.3389/fimmu.2019.02345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Haig DM, Hopkins J, Miller HR. Local immune responses in afferent and efferent lymph. Immunology. 1999. Feb;96(2):155–63. doi: 10.1046/j.1365-2567.1999.00681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ratnayake CBB, Escott ABJ, Phillips ARJ, Windsor JA. The anatomy and physiology of the terminal thoracic duct and ostial valve in health and disease: potential implications for intervention. J Anat. 2018. Jul;233(1):1–14. doi: 10.1111/joa.12811. Epub 2018 Apr 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell. 4th edition. New York: Garland Science; 2002. Lymphocytes and the Cellular Basis of Adaptive Immunity. Available from: https://www.ncbi.nlm.nih.gov/books/NBK26921/. [Google Scholar]
- 95.Eger EI 2nd, Laster MJ. The effect of rigidity, shape, unsaturation, and length on the anesthetic potency of hydrocarbons. Anesth Analg. 2001. Jun;92(6):1477–82. doi: 10.1097/00000539-200106000-00025. [DOI] [PubMed] [Google Scholar]
- 96.Chen CH, Tian CA, Chiu CC. The Effects of Alkyl Chain Combinations on the Structural and Mechanical Properties of Biomimetic Ion Pair Amphiphile Bilayers. Bioengineering (Basel). 2017. Oct 11;4(4):84. doi: 10.3390/bioengineering4040084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vanni S, Riccardi L, Palermo G, De Vivo M. Structure and Dynamics of the Acyl Chains in the Membrane Trafficking and Enzymatic Processing of Lipids. Acc Chem Res. 2019. Nov 19;52(11):3087–3096. doi: 10.1021/acs.accounts.9b00134. Epub 2019 Jul 31. [DOI] [PubMed] [Google Scholar]
- 98.Vijay N, Morris ME. Role of monocarboxylate transporters in drug delivery to the brain. Curr Pharm Des. 2014;20(10):1487–98. doi: 10.2174/13816128113199990462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Koho NM, Väihkönen LK, Pösö AR. Lactate transport in red blood cells by monocarboxylate transporters. Equine Vet J Suppl. 2002. Sep;(34):555–9. doi: 10.1111/j.2042-3306.2002.tb05482.x. [DOI] [PubMed] [Google Scholar]
- 100.Szabó E., Kulin A., Korányi L. et al. Alterations in erythrocyte membrane transporter expression levels in type 2 diabetic patients. Sci Rep 11, 2765 (2021). 10.1038/s41598-021-82417-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kalli AC, Reithmeier RAF. Interaction of the human erythrocyte Band 3 anion exchanger 1 (AE1, SLC4A1) with lipids and glycophorin A: Molecular organization of the Wright (Wr) blood group antigen. PLoS Comput Biol. 2018. Jul 16;14(7):e1006284. doi: 10.1371/journal.pcbi.1006284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jennings ML. Cell physiology and molecular mechanism of anion transport by erythrocyte band 3/AE1. Am J Physiol Cell Physiol. 2021. Dec 1;321(6):C1028–C1059. doi: 10.1152/ajpcell.00275.2021. Epub 2021 Oct 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stubbs CD, Smith AD. The modification of mammalian membrane polyunsaturated fatty acid composition in relation to membrane fluidity and function. Biochim Biophys Acta. 1984. Jan 27;779(1):89–137. doi: 10.1016/0304-4157(84)90005-4. [DOI] [PubMed] [Google Scholar]
- 104.Revskij D, Haubold S, Viergutz T, Kröger-Koch C, Tuchscherer A, Kienberger H, Rychlik M, Tröscher A, Hammon HM, Schuberth HJ, Mielenz M. Dietary Fatty Acids Affect Red Blood Cell Membrane Composition and Red Blood Cell ATP Release in Dairy Cows. Int J Mol Sci. 2019. Jun 5;20(11):2769. doi: 10.3390/ijms20112769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Paudel SS, deWeever A, Sayner S, Stevens T, Tambe DT. Substrate stiffness modulates migration and local intercellular membrane motion in pulmonary endothelial cell monolayers. Am J Physiol Cell Physiol. 2022. Sep 1;323(3):C936–C949. doi: 10.1152/ajpcell.00339.2021. Epub 2022 Aug 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xia T, Zhao R, Feng F, Yang L. The Effect of Matrix Stiffness on Human Hepatocyte Migration and Function-An In Vitro Research. Polymers (Basel). 2020. Aug 24;12(9):1903. doi: 10.3390/polym12091903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zheng J, Lu C, Ding Y, Zhang J, Tan F, Liu J, Yang G, Wang Y, Li Z, Yang M, Yang Y, Gong W, Gao C. Red blood cell-hitchhiking mediated pulmonary delivery of ivermectin: Effects of nanoparticle properties. Int J Pharm. 2022. May 10;619:121719. doi: 10.1016/j.ijpharm.2022.121719. Epub 2022 Apr 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang Y, Pisapati AV, Zhang XF, Cheng X. Recent Developments in Nanomaterial-Based Shear-Sensitive Drug Delivery Systems. Adv Healthc Mater. 2021. Jul;10(13):e2002196. doi: 10.1002/adhm.202002196. Epub 2021 Jun 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sinha A, Shaporev A, Nosoudi N, Lei Y, Vertegel A, Lessner S, Vyavahare N. Nanoparticle targeting to diseased vasculature for imaging and therapy. Nanomedicine. 2014. Jul;10(5):1003–12. doi: 10.1016/j.nano.2014.02.002. Epub 2014 Feb 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Watson S.P., Morgan N.V. and Harrison P. (2015). The Vascular Function of Platelets. In Postgraduate Haematology (eds Hoffbrand A.V., Higgs D.R., Keeling D.M. and Mehta A.B.). 10.1002/9781118853771.ch37 [DOI] [Google Scholar]
- 111.Layendecker SJ, McDonald TP. The relative roles of the spleen and bone marrow in platelet production in mice. Exp Hematol. 1982. Apr;10(4):332–42. [PubMed] [Google Scholar]
- 112.Nicolai L, Leunig A, Pekayvaz K, Esefeld M, Anjum A, Rath J, Riedlinger E, Ehreiser V, Mader M, Eivers L, Hoffknecht ML, Zhang Z, Kugelmann D, Rossaro D, Escaig R, Kaiser R, Polewka V, Titova A, Petzold T, Spiekermann K, Iannacone M, Thiele T, Greinacher A, Stark K, Massberg S. Thrombocytopenia and splenic platelet-directed immune responses after IV ChAdOx1 nCov-19 administration. Blood. 2022. Aug 4;140(5):478–490. doi: 10.1182/blood.2021014712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Holinstat M. Normal platelet function. Cancer Metastasis Rev. 2017. Jun;36(2):195–198. doi: 10.1007/s10555-017-9677-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Penny R, Rozenberg MC, Firkin BG. The splenic platelet pool. Blood. 1966. Jan;27(1):1–16. [PubMed] [Google Scholar]
- 115.Pivkin IV, Peng Z, Karniadakis GE, Buffet PA, Dao M, Suresh S. Biomechanics of red blood cells in human spleen and consequences for physiology and disease. Proc Natl Acad Sci U S A. 2016. Jul 12;113(28):7804–9. doi: 10.1073/pnas.1606751113. Epub 2016 Jun 27. Erratum in: Proc Natl Acad Sci U S A. 2017 May 30;114(22):E4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.CROSBY WH. Normal functions of the spleen relative to red blood cells: a review. Blood. 1959. Apr;14(4):399–408. [PubMed] [Google Scholar]
- 117.Kulenović A, Sarac-Hadzihalilović A. Blood vessels distribution in body and tail of pancreas- a comparative study of age related variation. Bosn J Basic Med Sci. 2010. May;10(2):89–93. doi: 10.17305/bjbms.2010.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Silva Lima B, Videira MA. Toxicology and Biodistribution: The Clinical Value of Animal Biodistribution Studies. Mol Ther Methods Clin Dev. 2018. Jan 31;8:183–197. doi: 10.1016/j.omtm.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nichols I. R., Hamadani C. M., Chism C. M., Hunter A. N., Ahmad H., Wontor K., Williams A. E., Vashisth P, Hammer N. I., Kundu S., Tanner E. E. L., The Impact of Water on Choline-2-Octenoic Ionic Liquid-Facilitated Transdermal Transport. Adv. Therap. 2022, 6, 2200096. 10.1002/adtp.202200096. [DOI] [Google Scholar]
- 120.Hamadani C, et al. Development of Ionic Liquid-Coated PLGA Nanoparticles for Applications in Intravenous Drug Delivery. Nature Protocols, 2022. (Accepted; in proof). [DOI] [PubMed] [Google Scholar]
- 121.Hamadani C.M., Chandrasiri I., Yaddehige M.L., Dasanayake G.S., Owolabi I., Flynt A., Hossain M., Liberman L., Lodge T.P., Werfel T.A., Watkins D.L., and Tanner E.E.L., 2022. Improved nanoformulation and bio-functionalization of linear-dendritic block copolymers with biocompatible ionic liquids. Nanoscale, 14(16), pp.6021–6036. [DOI] [PubMed] [Google Scholar]
- 122.Evans BC, Nelson CE, Yu SS, Beavers KR, Kim AJ, Li H, Nelson HM, Giorgio TD, Duvall CL. Ex vivo red blood cell hemolysis assay for the evaluation of pH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs. J Vis Exp. 2013. Mar 9;(73):e50166. doi: 10.3791/50166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kelley WJ, Fromen CA, Lopez-Cazares G, Eniola-Adefeso O. PEGylation of model drug carriers enhances phagocytosis by primary human neutrophils. Acta Biomater. 2018. Oct 1;79:283–293. doi: 10.1016/j.actbio.2018.09.001. Epub 2018 Sep 6. [DOI] [PMC free article] [PubMed] [Google Scholar]