

Abstract
Enhancer of zeste homolog 2 (EZH2) is an epigenetic regulator that controls the normal biology of germinal B cells. Overexpression or mutation of EZH2 is associated with malignant transformation in a number of B-cell malignancies; thus, EZH2 inhibitors are an attractive therapeutic option for these targets. Several EZH2 inhibitors have entered clinical trials, but there remains an important question as to how EZH2 inhibitor mechanism of action differs in patients with mutant and wild-type EZH2. This review discusses the EZH2-driven mechanisms that lead to the development of B-cell lymphomas and act as therapeutic targets. Another key area of investigation is whether EZH2 inhibitors will work synergistically with existing immunomodulatory drugs and chemotherapy regimens. In summary, EZH2 inhibitors show potential as treatment for a range of B-cell lymphomas, and numerous clinical evaluations are currently underway.
Keywords: B cell, epigenomics, EZH2 inhibitor, lymphoma, tazemetostat
1. Introduction
B-cell lymphoma is a common subtype of non-Hodgkin lymphoma (NHL), which is the seventh-most prevalent cancer in the United States and has the sixth-highest mortality rate [1]. The most frequently diagnosed B-cell lymphoma subtypes include diffuse large B-cell lymphoma (DLBCL; 31% of diagnosed lymphomas) and follicular lymphoma (FL; 22%) [1].
Although overall and progression-free survival among patients with FL have improved, with recent studies demonstrating that 48% and 35% of patients treated with immunochemotherapy remain progression-free for 8 and 10 years, respectively [4–7]., advanced FL is still commonly characterized by patterns of relapse of increasingly shortened duration. As patients progress and relapse through successive therapeutic regimens, survival outcomes decline with increasing lines of therapy [5,8].
Recent insights into the mutational landscape of FL indicate that it is a disease driven by epigenetic dysregulation, with mutations in genes (the most frequently mutated chromatin regulators include EZH2, CREBBP, and KMT2D) encoding for histone-modifying enzymes identified as early initiating drivers [11,12]. These findings raise the possibility that FL could be treated by altering the activity or restoring downstream effects of the affected enzymes. Among the available third-line treatments approved by the US Food and Drug Administration (FDA), enhancer of zeste homolog 2 (EZH2) inhibitors are a novel drug class used to treat relapsed or refractory (R/R) FL that target a component of these epigenetic pathways [13]. EZH2 is an epigenetic regulator that plays an essential role in the normal biology of germinal center (GC) B cells [14–17]. Gain-of-function EZH2 mutations can lead to aberrant EZH2 activity that drives malignant transformation linked to the formation of an abnormal immunologic niche [14,18]. EZH2 mutations are also commonly found in patients with DLBCL and are believed to drive oncogenesis [14,19]. By contrast, the more frequent KMT2D and CREBBP mutations are characterized by loss of function; therefore, they are less amenable to therapeutic targeting than EZH2 [12]. Since FL is derived from GC B cells, the malignant cells in FL may still be dependent on EZH2, even in the absence of gain-of-function EZH2 mutations [14,16].
Among aggressive B-cell lymphomas such as DLBCL and grade 3B FL, EZH2 is overexpressed in over 80% of patients studied, which suggests that EZH2-targeted therapies could also be used against aggressive B-cell lymphomas [20]. However, interplay between EZH2 overexpression and lymphomagenesis has not been fully elucidated.
With data demonstrating the efficacy of tazemetostat, in treating patients with R/R FL with or without EZH2 mutations and other EZH2 inhibitors being developed, it is important to illuminate how EZH2 inhibitors mechanistically provide therapeutic benefit to both of these populations and to those with other types of B-cell lymphoma [21]. As such, the purpose of this review article is to discuss the EZH2-driven mechanisms that lead to the development of B-cell lymphomas and act as therapeutic targets, to evaluate preclinical and clinical data for existing EZH2 inhibitors, and to provide new insights into this evolving drug class.
2. Germinal center reaction B-cell lymphoma pathogenesis
2.1. Germinal center reaction as a nidus for lymphomagenesis
Upon immunologic challenge, T cells induce antigen-dependent activation of mature B cells at the border of lymphoid follicles [22,23]. Some of these activated B cells then migrate to the interior of these follicles and undergo a proliferative burst during which they replicate at extremely rapid rates, thus forming transient GCs [24]. Proliferating GC B cells are called centroblasts, which simultaneously undergo somatic hypermutation of their immunoglobulin genes due to the actions of a GC-specific enzyme called activation-induced cytidine deaminase (AICDA) [25]. This process allows for stochastic genetic diversification of immunoglobulin genes within the GC reaction. After several rounds of replication, centroblasts presumably become exhausted and migrate from the dark zone of the GC to the light zone, which contains specialized T cells called T-follicular helper (TFH) cells, as well as specialized follicular dendritic cells (FDCs) [26]. These post-replicative GC B cells are called centrocytes, and have an absolute requirement for TFH help in order to survive [26,27]. Only those few centrocytes that express immunoglobulin with affinity for the relevant antigen are able to strongly interact with TFH cells [26–28]. T-cell help will then enable these centrocytes to return to the dark zone of the GC for further rounds of proliferative bursting [28,29]. After several rounds of these cycles, high-affinity GC B cells are formed that can exit the GC reaction as plasma cells and high- or intermediate-affinity memory B cells (Figure 1A) [30].
Figure 1.

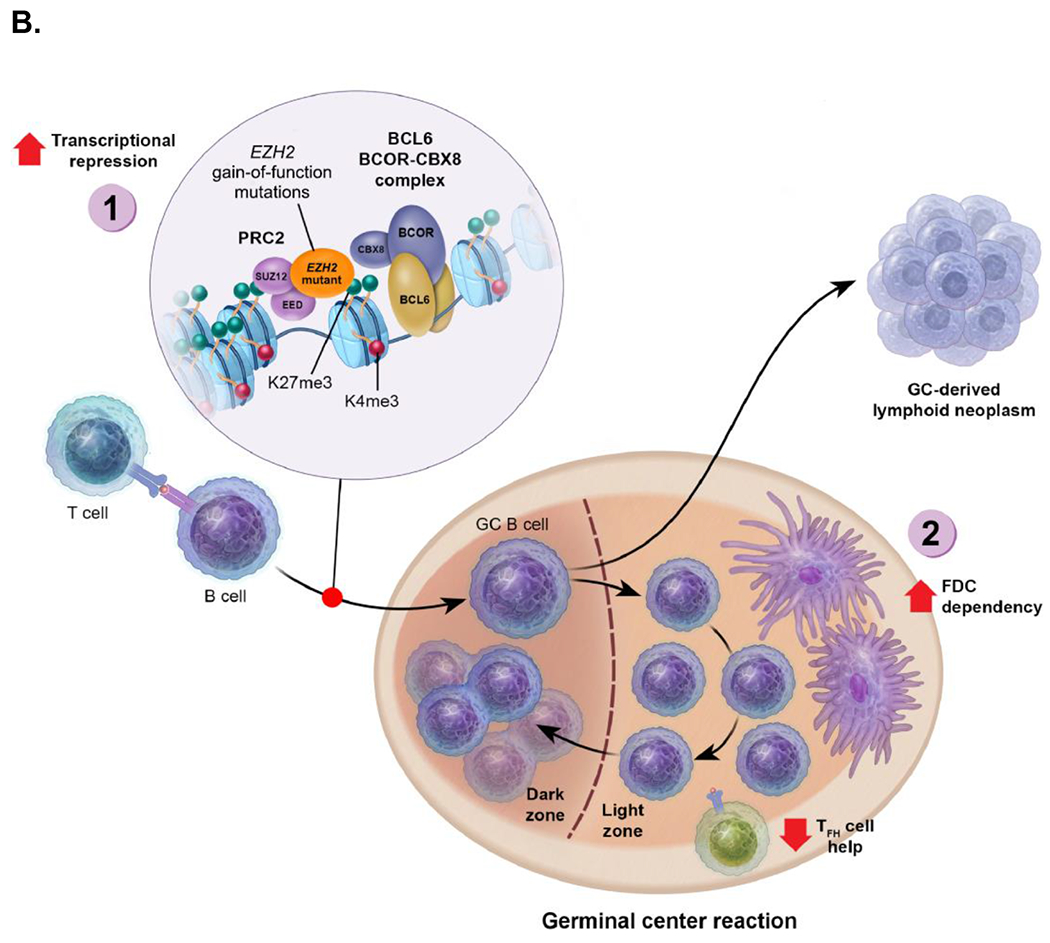
(A) Normal germinal center physiology. (1.) Upregulation of EZH2 leads to trimethylation of H3K27 and silencing of genes involved in cell cycle checkpoints, differentiation, and GC exit. (2.) Loss of BCL6 may lead to reversal of the germinal center-specific H3K27me3 program upon exit from the germinal center, which is accompanied by reduced EZH2 levels. (B) The implications of EZH2 gain-of-function mutations in the germinal center reaction and lymphomagenesis. (1.) Massive increases in H3K27me3 lead to aberrant repression of target genes involved in cell cycle checkpoints, differentiation, and germinal center exit. (2.) Reduced requirement for T-cell help and increased dependency on follicular dendritic cells, accompanied by expansion of the follicular dendritic cell networks.
BCOR-CBX8, BCL6 corepressor-chromobox homolog 8; EZH2, enhancer of zeste homolog 2; FDC, follicular dendritic cell; GC, germinal center; K4, lysine 4; K27, lysine 27; me3, trimethylated; PRC2, polycomb repressive complex 2; TFH, T-follicular helper.
[Figure panels should be in color online only]
During replication, centroblasts can experience extensive DNA damage due to off-target mutations induced by AICDA, as well as due to their rapid proliferation rates [31]. Moreover, DNA damage checkpoints are attenuated in centroblasts, allowing these cells to divide even while accumulating mutations [31]. This mutagenic GC process is the basis for the development of a majority of B-cell neoplasms, including FL [32].
2.2. EZH2, an essential driver of the GC reaction and rational therapeutic target in B-cell lymphomas
EZH2 is a histone methyltransferase that has the specific function of covalently adding methyl groups to lysine 27 of histone 3 (H3K27). Enzymatically, EZH2 is highly efficient at inducing H3K27 mono (H3K27me1) and dimethylation (H3K27me2), and less so at inducing H3K27 trimethylation (H3K27me3). However, H3K27me3 is the most critical effect, since this mark mediates the characteristic gene-silencing effect mediated by EZH2 [33]. Importantly, EZH2 is upregulated in B cells as they enter the GC reaction, and genetic ablation of EZH2 completely abrogates the ability of B cells to form GCs [14,15]. The role of EZH2 in GC B cells is to induce transient silencing of genes involved in cell cycle checkpoints, immune signaling pathways, and to restrain plasma cell differentiation through its H3K27me3 actions [14,16,17,34]. This involves cooperation with BCL6, a GC reaction master regulatory factor, to facilitate recruitment and combinatorial tethering of a BCL6 corepressor-chromobox homolog 8 (BCOR-CBX8) complex to gene promoters [16]. This dependency on cooperation with BCL6 (a DNA-sequence specific transcription factor) likely explains how it is that EZH2 is selectively active at genes relevant for the GC reaction in the context of GC B cells (Figure 1A). EZH2 is necessary for GC B cells to proliferate and undergo somatic hypermutation. EZH2 also plays a role in supporting terminal plasma cell differentiation at later time points, independent of GC B cells [14,16,17,34].
The essential role of EZH2-mediated trimethylation of cell cycle checkpoint genes was highlighted by the fact that deletion of key EZH2 target genes, such as CDKN1A and CDKN2A, restores GC formation in EZH2 knockout mice [17]. It is of interest that genetic ablation of AICDA also restores GC formation in EZH2 knockout mice, suggesting an as of yet unclear role for EZH2 in DNA damage response checkpoints [15]. These points are critical to appreciate when considering the role of EZH2 in GC-derived lymphomas, such as FL, since these tumor cells retain dependency on EZH2 to mediate these same crucial functions. Thus, EZH2 mutation is a well-defined, rational therapeutic target in FL and in GC DLBCL because it suppresses key cellular checkpoints and differentiation pathways to maintain oncogenic characteristics of GC B cells. There is still much to learn regarding mechanisms that cause wild-type lymphomas to retain dependency on EZH2.
2.3. Role of EZH2 somatic mutations in GC-derived lymphomas
Approximately 25% of patients with FL manifest somatic mutations of EZH2, the majority of which occur at tyrosine 641 (Y641), located within the catalytic SET domain that mediates H3K27me3 [35]. EZH2Y641 mutations are also present in approximately 22% of patients with GC DLCBL [36]. EZH2 mutations in lymphoma bear the signature of both AICDA–mediated cytosine deamination, as well as aging-associated DNA damage patterns [37]. The EZH2Y641 mutant proteins manifest an aberrant enzymatic profile consisting of severely impaired H3K27 monomethylation, but far more efficient rates of H3K27me3 [38]. Accordingly, EZH2Y641 mutant lymphoma cells feature increased abundance of H3K27me3 [14,38]. Mass spectrometry performed in primary GC B cells engineered for expression of EZH2Y641 mutant protein from the endogenous EZH2 locus revealed massive increases in H3k27me3, with corresponding reductions in H3K27me2 and, to a lesser extent, in H3K27me1, but no effect on methylation of other histone residues [18]. This increase in H3K27me3 manifests at the genomic level in both invasive spreading of this mark upstream and downstream of transcription start sites, and aberrant gain of H3K27me3 at gene promoters that do not normally contain this mark [18]. Gain of promoter H3K27me3 results in aberrant repression of the respective genes, whereas it is not clear if there is any transcriptional consequence to the spreading of H3K27me3 to other kinds of genomic elements such as gene enhancers (Figure 1B) [14,18]. However, it is worth noting that gene promoters that become aberrantly silenced by EZH2Y641 mutant proteins tend to be close to promoters typically marked by normal EZH2, and that H3K27me3 seems to spread from these normal EZH2 target promoters to the de novo mutant-specific promoters [18].
EZH2Y641 mutations are always heterozygous, which is intuitively logical because the wild-type enzyme is still needed to mediate the first step in H3K27 methylation (H3K27me1) as a substrate for the mutant protein. This principle was confirmed in experiments showing that mice engineered for homozygous expression of EZH2Y641 mutants in GC B cells actually phenocopy homozygous EZH2 knockout [16]. In contrast, heterozygous expression of EZH2Y641 mutants caused massive expansion of GCs, a classical manifestation of oncogenes that drive lymphomagenesis [14,16,18]. A small fraction of patients with FL also feature somatic mutations at a minor hotspot at residue A677 adjacent to the SET domain [39]. The EZH2A677 mutant protein also drives massive gains in H3K27me3 activity, but in marked contrast to EZH2Y641 mutants, is still able to drive H3K27me1 [39]. Sporadic non-synonymous mutations in other residues are reported as well, but are of unknown significance [35].
Of note, EZH2 is only active when interacting with other core subunits of the polycomb repressive complex 2 (PRC2), such as EED and SUZ12 [40]. Since these proteins are expressed at stoichiometric levels, excess expression of EZH2 would theoretically be inactive in the absence of gain in expression of the other PRC2 subunits. Indeed, experimental data show that overexpression of wild-type EZH2 does not accelerate lymphomagenesis in mice bearing a BCL2 transgene [14]. In contrast, both ectopic or endogenous expression of EZH2Y641 yields more rapid development of lymphomas in mice in cooperation with either BCL2 or BCL6 (Figure 1B) [14,16,41].
The role of mutant EZH2 in B-cell lymphomagenesis is complex and incompletely understood. Effects of EZH2 mutation at early stages of transformation and FL may be somewhat different than its role in more advanced lymphomas and DLBCL. It is also noteworthy that, although EZH2 gain-of-function mutations are common in lymphomas, myeloid malignancies are often characterized by loss-of-function mutations. However, the reasons as to why EZH2 has such different biological effects in myeloid versus GC B cells remain unclear [42]. Detailed analysis of EZH2Y641 effects in B cells showed that the observed GC hyperplasia can be specifically attributed to expansion of centrocytes [18]. The reason for this was traced to the aberrant de novo H3K27me3 of promoters for genes linked to interaction with TFH cells (eg, ICAM1), which results in their aberrant repression [18]. This was concordant with the upregulation of genes (perhaps through an indirect mechanism) that favor interaction and response to FDCs, such as Ltb and Tnfrsf13c (B-cell activating factor [BAFF] receptor) [18]. As a consequence, EZH2Y641 GC B cells were no longer strictly dependent on T-cell help to maintain their survival. Instead, GCs containing EZH2Y641 GC B cells featured massive expansion of FDC networks, which were essential to maintain survival and proliferation of EZH2Y641 GC B cells (Figure 1B) [18]. A primary effect of EZH2Y641 mutation is thus to reprogram the immune niche to allow persistent growth of centrocytes through preferential interaction with dendritic cells. Strikingly, mutant EZH2 FL in humans is significantly associated with extensive FDC networks interdigitated throughout the malignant lymphoid follicles [18]. Hence the role of EZH2 mutations at least in early-stage FL may be linked specifically to its role in establishing a favorable tumor microenvironment. Although mutant EZH2 GC B cells featured FDC network expansion, the FDC networks were reduced or lost in mutant EZH2 DLBCL mouse models [43].
It was shown recently that both human and murine EZH2Y641 mutant GC B-cell DLBCLs feature strong downregulation of major histocompatibility complex (MHC) class I and MHC class II expression due to aberrant H3K27me3, and that this was reversible with EZH2 inhibitors [44]. However, this effect is not seen in EZH2Y641 GC B cells or human FLs [18]. Thus, it is possible that the aberrant H3K27 methyltransferase function of mutant EZH2 results in ongoing stochastic silencing of genes that, over time, results in selection for a maximally immune silent state. In this way, malignant B cells become progressively more autonomous from interactions and interdependency from other components of the immune microenvironment (Figure 1B). A majority of preclinical studies of EZH2 inhibitors have focused on cell autonomous effects on DLBCL cells in vitro or in immune-compromised mice. These responses generally consist of proliferation arrest and partial plasma cell differentiation [14,45]. Importantly, such results are seen both in wild-type and mutant EZH2 lymphoma cells and are linked to derepression of cell cycle checkpoint and differentiation genes that are repressed by EZH2, regardless of its mutation status [14]. However, the important immune effect of mutant EZH2 suggests that EZH2 inhibitors also may induce prominent anti-lymphoma immune effects in these patients, which would have been missed in preclinical studies published to date. Moreover, wild-type EZH2 plays a role in limiting differentiation and plasticity in T cells and may favor evasion from immune surveillance because of T-cell autonomous effects on T-regulatory cells (reviewed elsewhere [46,47]). These are important points to consider when interpreting the outcomes of EZH2 inhibitor clinical trials, and when designing correlative science protocols.
3. EZH2 inhibitors
3.1. EZH2 inhibitor monotherapy, preclinical data
EZH2 inhibitors CPI-0209 [48], PF-06821497 [49], valemetostat [50], and tazemetostat [51,52] have demonstrated preclinical efficacy in cancer cell lines, xenograft tumor models, or both, as found in the published literature (Table 1). All of these agents have decreased H3K27me3 levels in cancer cell lines, with some inhibiting cell proliferation as well (Table 1). Of note, tazemetostat and valemetostat have published preclinical activity in wild-type EZH2 DLBCL cell lines, and tazemetostat has demonstrated preclinical activity in wild-type EZH2 mantle cell lymphoma (MCL) cell lines resistant to Bruton tyrosine kinase inhibitors [51,53,54]. In mutant EZH2Y641N KARPAS-422 xenograft DLBCL tumor models, all agents exerted antitumor effects (Table 1). Tazemetostat also demonstrated efficacy in mutant EZH2Y641F and mutant EZH2A682G lymphoma tumors [51]. These promising preclinical results are starting to translate into encouraging antitumor benefits in the clinic.
Table 1.
EZH2 Inhibitor Preclinical Efficacy
| EZH2 Inhibitor | Effects on H3K27me3 Levels | Effects on Cell Line Proliferation | Effects on Tumor Growth |
|---|---|---|---|
| CPI-0209 | Reduced H3K27me3 levels in HeLa cells [48] | None documented | Demonstrated antitumor effects against KARPAS-422a tumor xenografts [48] |
| PF-06821497 | Reduced H3K27me3 levels with an IC50 of 4 nmol/L in KARPAS-422a cell lines [49] | Inhibited cell proliferation with an IC50 of 6 nmol/L in KARPAS-422a cell lines [49] | Demonstrated tumor growth inhibition and tumor regression in KARPAS-422a tumor xenografts [49] |
| Valemetostat | Reduced H3K27me3 levels in KARPAS-422a cell lines [50] | Inhibited in vitro growth of KARPAS-422a cell lines with an IC50 of 4.8 nmol/L [50] | Demonstrated antitumor effects against KARPAS-422a tumor xenografts |
| Tazemetostat | Reduced H3K27me3 levels in both wild-type and mutant EZH2 DLBCL cell lines after 4 days with an IC50 value ranging from 2-90 nmol/L [51] | Inhibited cell proliferation of both wild-type and mutant EZH2 DLBCL cell lines after 11 days, with an IC50 ranging from 0.00049–7.6 μmol/L [51] Exerted a cytotoxic response and induced cell apoptosis in KARPAS-422a cell lines and reduced cell proliferation among wild-type EZH2 SU-DHL-5, Farage, and TMD8 B-cell lymphoma cell lines [52] |
Demonstrated tumor growth inhibition of mutant EZH2Y641F WSU-DLCL2 xenograft tumors and tumor eradication of KARPAS-422a and mutant EZH2A682G Pfeiffer xenograft tumors in vivo [51] |
Mutant EZH2Y641N DLBCL cells.
DLBCL, diffuse large B-cell lymphoma; IC50, half-maximal inhibitory concentration.
3.2. EZH2 inhibitor monotherapy, clinical data
The demonstrated link between EZH2 activity and lymphomagenesis has prompted interest in investigating the clinical benefit of EZH2 inhibitors for the treatment of lymphomas. With the clinical failure of GSK2816126 [55], 5 investigational (CPI-0209, HH2853, PF-06821497, SHR2554, and valemetostat [a dual EZH1/2 inhibitor]) and 1 FDA–approved (tazemetostat) EZH2 inhibitors remain actively studied in patients with lymphoma as of April 2021. Because of their promising preclinical activity (see Section 3.3), these agents have progressed to clinical trials. However, each EZH2 inhibitor has a unique clinical development program (Table 2), and only valemetostat and tazemetostat have published clinical data for patients with lymphoma.
Table 2.
Ongoing EZH2 Inhibitor Clinical Studies in Lymphomas
| EZH2 Inhibitor | NCT Number | Phase | Therapeutic Area | Interventional Arms |
|---|---|---|---|---|
| CPI-0209 | NCT04104776 | Phase 1/2 | Advanced solid tumors and DLBCL | Two arms: • CPI-0209 alone (RP2D) • CPI-0209 plus irinotecan (RP2D) |
| HH2853 | NCT04390737 | Phase 1 | R/R NHL and advanced solid tumors | One arm: HH2853 alone (50–1000 mg BID) |
| PF-06821497 | NCT03460977 | Phase 1 | SCLC, FL, CRPC, and DLBCL | One arm: PF-06821497 alone (75–625 mg BID) |
| SHR2554 | NCT03603951 | Phase 1 | R/R mature lymphoid neoplasms | One arm: SHR2554 alone (RP2D) |
| Valemetostat | NCT04102150 | Phase 2 | R/R ATL | One arm: valemetostat alone (200 mg QD) |
| NCT02732275 | Phase 1 | R/R PTCL and R/R ATL | One arm: valemetostat alone (150–300 mg QD) | |
| NCT04703192 | Phase 2 | R/R PTCL and R/R ATL | One arm: valemetostat alone (200 mg QD) | |
| NCT04842877 | Phase 2 | R/R B-cell lymphoma (5 subtypes) | One arm: valemetostat alone (200 mg QD) | |
| Tazemetostat | NCT04224493 | Phase 1b/3 | R/R FL | Two arms: • Tazemetostat (RP3D) plus R2 • Placebo plus R2 |
| NCT02889523 | Phase 1b/2 | DLBCL and FL | One arm: tazemetostat (800 mg BID) plus R-CHOP |
ATL, adult T-cell leukemia/lymphoma; BID, twice daily; CRPC, castration-resistant prostate cancer; DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; NHL, non-Hodgkin lymphoma; QD, once daily; R2, lenalidomide plus rituximab; R-CHOP, cyclophosphamide, doxorubicin, vincristine, prednisone, plus rituximab; RP2D, recommended phase 2 dose; RP3D, recommended phase 3 dose; R/R, relapsed/refractory; SCLC, small cell lung cancer.
A phase 1 study of valemetostat in patients with R/R B-cell and T-cell lymphomas reported a preliminary objective response rate (ORR) of 47.4% (1 complete response [CR] and 8 partial responses [PRs]) in 19 patients with B-cell lymphomas [56]. Among the patients with B-cell lymphomas, 47.4% had a best percentage reduction in tumor size of more than 50% [56]. Safety data were reported in an earlier data cut (n=15), and common treatment-related adverse events included decreased platelet count (73%), anemia (47%), decreased lymphocyte count (40%), and decreased neutrophil count (40%) [57]. A larger phase 2 study is examining valemetostat in up to 141 patients diagnosed with aggressive B-cell lymphomas (n=40), FL (n=40), MCL (n=20), indolent lymphomas (n=20), and Hodgkin lymphoma (n=20) (NCT04842877). Given the demonstrated activity of valemetostat, its efficacy is being further evaluated in various subtypes of R/R T-cell lymphoma and in R/R B-cell lymphomas in ongoing and proposed clinical trials (Table 2).
Initial phase 1 efficacy results with tazemetostat in 21 patients with R/R B-cell NHL (ORR 38% [3 CRs and 5 PRs]; 95% CI: 18.1–61.6) from a phase 1/2 study provided sufficient evidence to continue further phase 2 clinical investigations [58]. Recently, phase 2 results of the wild-type (n=54) and mutant (n=45) EZH2 FL cohorts were published [21]. Patients in the wild-type and mutant EZH2 FL cohorts achieved an ORR of 35% (2 CRs and 17 PRs; 95% CI: 23–49) and 69% (6 CRs and 25 PRs; 95% CI: 53–82), respectively [21]. The responses in patients with wild-type EZH2 FL suggest that wild-type EZH2 contributes to lymphomagenesis, but the disparity in ORR between the cohorts suggests that mutant EZH2 may be a main oncogenic driver in those patients with EZH2 mutations. Thus, mutant EZH2 lymphomas may have a dependency on aberrant EZH2 activity. These findings seem to mirror those observed in the preclinical setting, although interpretation of such results is limited given the paucity of preclinical studies in primary lymphomas (Table 1). Interestingly, median duration of response (wild-type EZH2 [n=19]: 13.0 months [95% CI: 5.6–not evaluable (NE)]; mutant EZH2 [n=31]: 10.9 months [95% CI: 7.2–NE]) and median progression-free survival (wild-type EZH2: 11.1 months [95% CI: 3.7–14.6]; mutant EZH2: 13.8 months [95% CI: 10.7–22.0]) were similar for the wild-type and mutant EZH2 FL cohorts [21], supporting the use of tazemetostat independent of EZH2 mutation status. Furthermore, responses appear to be durable in patients, regardless of their EZH2 mutational status. Additionally, patients with R/R FL who received tazemetostat had low rates of discontinuation (8%), dose reduction (9%), and dose interruption (27%) for toxicities, and experienced a low incidence of grade 3/4 adverse events (the most common being grade 3/4 neutropenia [3%], thrombocytopenia [3%], and anemia [2%]) [21]. Common treatment-related adverse events included nausea (19%), alopecia (14%), asthenia (14%), and diarrhea (12%) [21].
Based on the results of this phase 1/2 trial, tazemetostat received accelerated FDA approval for the treatment of adults with mutant EZH2 R/R FL who have received 2 prior therapies, and for the treatment of adults with R/R FL who have no satisfactory alternative treatment options [13]. Despite demonstrating preclinical effectiveness against human DLBCL cells in mice, tazemetostat is less effective in patients with DLBCL than in those with FL.
3.3. EZH2 inhibitor in combination therapies, preclinical data
Several preclinical studies show that dual EZH1/EZH2 inhibitors possess synergistic activity against acute myeloid leukemia, DLBCL, and multiple myeloma (MM) cell lines versus selective EZH2 inhibitors [50,59].
When EZH2 mutations occur in the context of B cells that already harbor BCL2 translocations [37,60], the combined effect of aberrant survival and immune reprogramming that would occur in GC B cells due to BCL2 and EZH2 mutations yields a potent oncogenic effect. Indeed, preclinical studies administering EZH2 and BCL2 inhibitors show dramatic synergy against EZH2 mutant lymphomas in vitro and in vivo [14,61]. This result may be partly due to EZH2 inhibitors lowering apoptotic thresholds in lymphoma cells, pointing to a rationale for combining such inhibitors in clinical trials [61]. In addition, the fact that deregulated BCL6 expression cooperates with mutant EZH2 and requires wild-type EZH2 for its oncogenic effects explains the synergistic activity of combination therapy with BCL6 and EZH2 inhibitors and justifies exploration of the clinical utility of such combinations in the future [16]. Other preclinical data demonstrate that HDAC3 inhibition reduces the growth of xenograft DLBCL tumor models and leads to the induction of genes silenced by BCL6, suggesting that treatment with HDAC3 and EZH2 inhibitors could be a promising regimen [62]. These preclinical data are also consistent with study results suggesting activity of EZH2 inhibitors with HDAC inhibitors [63]. Another preclinical study found that treatment of mutant EZH2Y641N with the single agents prednisone and doxorubicin and the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) restored MHC-I/II expression, whereas bendamustine treatment had no effect on MHC expression [64]. The latter finding is hypothesis generating, knowing that a retrospective analysis of gene mutations and clinical risk factors in patients with FL found that those with EZH2 mutations in the high-risk Follicular Lymphoma International Prognostic Index subset experienced longer progression-free survival following treatment with R-CHOP or with rituximab plus cyclophosphamide, vincristine, and prednisone (R-CVP) [65], but not following treatment with bendamustine [66]. Given the tumor-modifying and microenvironment-modifying effects of EZH2 inhibition, it is possible that combination with appropriate immunotherapies may be beneficial [46], and several rituximab-based combination therapy regimens are being explored (see Section 3.4).
Several preclinical studies in MM cell lines have shown that synergy exists between EZH2 inhibitors and drugs inducing cereblon-mediated degradation of proteins, such as Ikaros family zinc finger 1 (IKZF1) (eg, lenalidomide, pomalidomide). Simultaneous inhibition of wild-type EZH2 and DNA methyltransferase resensitized lenalidomide-resistant MM cells to lenalidomide and pomalidomide [67]. Another study found that tazemetostat pretreatment resensitized drug-resistant MM cells to lenalidomide and that IKZF1, IRF4, and MYC protein levels in those cells were lowered more by the combination treatment than by lenalidomide monotherapy [68]. In particular, EZH2 and transcription regulators IKZF1 and IKZF3 share a set of target genes that includes IRF4, providing further evidence that these overlapping pathways may mediate the synergy between EZH2 inhibitors and these cereblon-targeting agents [69]. A recent study found that IKZF1-knockout DLBCL cells were sensitized to the EZH2 inhibitor tazemetostat and that the combination of an EZH2 inhibitor and lenalidomide triggered apoptosis in DLBCL cells independent of EZH2 mutational status. Based on these findings, the authors hypothesized that EZH2 inhibition in wild-type EZH2 cells may act by controlling increases in H3K27 acetylation, and the effects of EZH2 inhibition with lenalidomide seem synergistic [70]. In addition to cellular studies, a xenograft study found that MM tumor growth was suppressed better with an EZH2 inhibitor and pomalidomide combination therapy than with either agent as monotherapy [69]. These studies with wild-type EZH2 MM cells emphasize the importance of the wild-type EZH2 gene in several key signaling pathways that can be exploited by combination therapies. It is worth investigating whether similar synergistic effects between EZH2 inhibitors and cereblon-targeting degraders are observable in patients with FL.
3.4. EZH2 inhibitor combination therapy, clinical data
To verify the clinical efficacy of tazemetostat and its synergy in combination with immunomodulatory imide drugs, a confirmatory phase 1b/3 trial of tazemetostat (dosed twice daily [BID] for 12 months, then given as a maintenance dose for 2 years) in combination with R2 for R/R FL was initiated (Table 2). Preliminary efficacy data are encouraging, with 13 complete responses and 19 partial responses in the evaluable population of 35 patients. The safety run-in analysis shows that this combination is safe and tolerable, and that patients are able to maintain a high dose intensity. There was no clear dose response for treatment-emergent adverse events or dose modifications. Recruitment for the phase 3 portion of the trial is ongoing [71].
Another ongoing phase 1b/2 trial is examining the efficacy of tazemetostat in combination with rituximab plus CHOP (R-CHOP) in patients with newly diagnosed high-risk DLBCL (aged 60–80 years, age-adjusted International Prognostic Index 2–3) or transformed FL (NCT02889523) [72]. At the time of interim analyses (median follow-up, 20.6, 14.5, and 7.5 months at tazemetostat dose levels of 400, 600, and 800 mg BID respectively), the metabolic CR rate was 76.5% (n=13/17) [72]. The phase 2 portion of the trial will enroll up to 184 patients with DLBCL or FL at the recommended phase 2 dose of tazemetostat; patients with DLBCL or FL will receive tazemetostat 800 mg BID plus R-CHOP for 8 × 21-day treatment cycles, then patients with FL will be given tazemetostat 800 mg BID for 6 months and rituximab every 2 months for 2 years, as maintenance therapy.
4. Conclusions and future considerations
EZH2 plays a pivotal role in the normal biology of GC B cells, particularly as a driver for the GC reaction via the silencing of key genes that regulate cell proliferation, immune signaling, and differentiation. Although only approximately 25% of FL tumors are characterized by gain-of-function EZH2 mutations, most FL tumors are likely dependent on EZH2 for their growth and survival, regardless of their EZH2 mutational status. Even with recent studies further elucidating the role of EZH2 in lymphomagenesis and the tumor microenvironment, the pleiotropic mechanisms by which EZH2 mutations can drive the pathogenesis of FL and other B-cell lymphomas still are not fully understood.
As seen with the clinical data of valemetostat and tazemetostat, EZH2 inhibition has demonstrated efficacy in treating patients with FL, and the favorable safety profiles of EZH2 inhibitors suggest that they are promising candidates for combination treatment regimens. These data, along with preclinical studies showing the potential efficacy of EZH2 inhibitors in MCL and in combination with BCL6 and BCL2 inhibitors in FL. The study combining tazemetostat with the immunotherapy R2 (NCT04224493) is the furthest along in terms of clinical development, with high response rates and no new safety signals. Clinical trials offer the possibility of exploring novel multimodal approaches to treating a variety of GC-derived B-cell malignancies [14,16,46,61]. As our understanding of the role of EZH2 in GC-derived lymphomas evolves, the applicability of EZH2-directed therapies across different malignancies will continue to expand.
Practice points
Overexpression or mutation of EZH2 in patients with B-cell lymphoma is associated with growth of malignancies.
Therapies targeting EZH2 may offer a novel way of overcoming chemoresistance mechanisms in patients with relapsed or refractory lymphoma
The current indication for tazemetostat allows the agent to be prescribed in the absence of satisfactory alternative treatment options, regardless of EZH2 mutation status or line of therapy. Clinical trials are ongoing in both wild-type and mutant EZH2 tumors to assess the efficacy of EZH2 inhibitors, alone and in combination, in earlier lines of treatment.
EZH2 inhibitors work through different mechanisms of action in patients with mutant versus wild-type EZH2.
Research agenda
Phase 1b/2 or 1b/3 studies in FL and DLBCL examining safety and efficacy are currently underway for several EZH2 inhibitors.
Better understanding of biomarkers of response in the wild-type EZH2 FL population.
Defining the dose, tolerability, and optimal treatment duration of tazemetostat in combination with immunomodulatory agents and chemotherapy regimens is a high priority, particularly with rituximab-based regimens.
A major clinical objective will be determining the efficacy of EZH2 inhibitors in B-cell lymphomas and hematological malignancies other than R/R FL, such as DLBCL.
HIGHLIGHTS.
Both wild-type and mutant EZH2 contribute to lymphomagenesis
EZH2 inhibitors are promising agents for the treatment of B-cell lymphomas
Preclinical and clinical data show favorable tolerability of EZH2 inhibitors
EZH2 inhibitors are effective in combination therapy regimens
Acknowledgments
Medical writing and editorial assistance was provided by Claire L. Jarvis, PhD, and Andrew Scott, PharmD, of Peloton Advantage, an OPEN Health company, and funded by Epizyme, Inc.
Role of the funding source
This research was funded by Epizyme, Inc. Medical writing and editorial support were provided by Andrew Scott, PharmD, Claire Jarvis, PhD, and Duprane Young, PhD, of Peloton Advantage, LLC, an OPEN Health company, and funded by Epizyme, Inc.
Declaration of competing interest
FM: Honoraria from Janssen. Consultant or advisory role with AbbVie, Bristol Myers Squibb, Celgene, Epizyme, Genmab, Gilead Sciences, Kymera, and Roche. Expert testimony for Roche.
GS: Honoraria from AbbVie, Celgene, Gilead Sciences, Janssen, MorphoSys, Novartis, and Roche/Genentech. Consulting or advisory role with Alimera Sciences, BeiGene, Bristol Myers Squibb, Celgene, Debiopharm Group, Epizyme, Genmab, Genmab, Gilead Sciences, Incyte, Janssen, Miltenyi Biotec, MorphoSys, Novartis, Roche/Genentech, and VelosBio.
CLB: Research funding from Autolus, Bayer, Epizyme, Inc., Janssen, Novartis, Roche, and Xynomic. Consulting or advisory role with ADC Therapeutics, GLG, Juno/Celgene, Karyopharm, Kite, Life Sci, Seattle Genetics, and TG Therapeutics. Honoraria from DAVA Oncology, Medscape, and touchIME. Stock or ownership in Bristol Myers Squibb, Moderna, Novavax, Pfizer, Regeneron, and Viatris.
HT: Honoraria from Bristol-Myers Squibb and Servier. Consulting or advisory role with Janssen, Karyopharm Therapeutics, and Roche. Travel, accommodations, and expenses from Roche.
AC: Honoraria from Celgene and Takeda. Consulting or advisory role with Roche. Travel, accommodations, and expenses from Takeda, Janssen, and Gilead.
TP: Honoraria from Bayer, Genentech, Gilead Sciences, Incyte, Pharmacyclics, and Seattle Genetics. Consulting or advisory role with Bayer, Celgene, Curis, Genentech, Gilead Sciences, Gilead Sciences, Incyte, Kite/Gilead, Pharmacyclics, and Seattle Genetics. Speakers’ bureau for Genmab. Research funding from AbbVie, Bayer, and Pharmacyclics/Janssen.
JB: Consulting or advisory role with AbbVie, Adaptive Biotechnologies, AstraZeneca, Bayer, BeiGene, Epizyme, Genentech/Roche, Kura Oncology, Kymera, MorphoSys, Novartis, Seattle Genetics, and Verastem. Speakers’ bureau for BeiGene and Seattle Genetics.
AM: Consulting or advisory role: AstraZeneca, Bristol Myers-Squibb, Constellation, Daiichi Sankyo, Epizyme, Exo Therapeutics, Janssen, and KDAC. Research funding from AstraZeneca, Daiichi Sankyo, Epizyme, Janssen, and Sanofi.
References
- 1.Thandra KC, Barsouk A, Saginala K, Padala SA, Barsouk A, Rawla P. Epidemiology of non-Hodgkin’s lymphoma. Med Sci (Basel) 2021; 9. 10.3390/medsci9010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bakhshi TJ, Georgel PT. Genetic and epigenetic determinants of diffuse large B-cell lymphoma. Blood Cancer J 2020; 10: 123. 10.1038/s41408-020-00389-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer stat facts: NHL — diffuse large b-cell lymphoma (DLBCL). National Cancer Institute, 2022. Available at: https://seer.cancer.gov/statfacts/html/dlbcl.html. Accessed: 12 May 2022.
- 4.Tan D, Horning SJ, Hoppe RT, Levy R, Rosenberg SA, Sigal BM, et al. Improvements in observed and relative survival in follicular grade 1-2 lymphoma during 4 decades: the Stanford University experience. Blood 2013; 122: 981–7. 10.1182/blood-2013-03-491514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Batlevi CL, Sha F, Alperovich A, Ni A, Smith K, Ying Z, et al. Follicular lymphoma in the modern era: survival, treatment outcomes, and identification of high-risk subgroups. Blood Cancer J 2020; 10: 74. 10.1038/s41408-020-00340-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bachy E, Seymour JF, Feugier P, Offner F, López-Guillermo A, Belada D, et al. Sustained progression-free survival benefit of rituximab maintenance in patients with follicular lymphoma: long-term results of the PRIMA Study. J Clin Oncol 2019; 37: 2815–24. 10.1200/jco.19.01073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luminari S, Ferrari A, Manni M, Dondi A, Chiarenza A, Merli F, et al. Long-term results of the FOLL05 trial comparing R-CVP versus R-CHOP versus R-FM for the initial treatment of patients with advanced-stage symptomatic follicular lymphoma. J Clin Oncol 2018; 36: 689–96. 10.1200/jco.2017.74.1652. [DOI] [PubMed] [Google Scholar]
- 8.Link BK, Day BM, Zhou X, Zelenetz AD, Dawson KL, Cerhan JR, et al. Second-line and subsequent therapy and outcomes for follicular lymphoma in the United States: data from the observational National LymphoCare Study. Br J Haematol 2019; 184: 660–3. 10.1111/bjh.15149. [DOI] [PubMed] [Google Scholar]
- 9.NCCN Clinical Practice Guidelines in Oncology. B-Cell Lymphomas v5.2021. NCCN, 2021. Available at: https://www.nccn.org/professionals/physician_gls/default_nojava.aspx. Accessed: July 12, 2021.
- 10.Salles G How do I sequence therapy for follicular lymphoma? Hematology 2020; 2020: 287–94. 10.1182/hematology.2020000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okosun J, Bödör C, Wang J, Araf S, Yang CY, Pan C, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet 2014; 46: 176–81. 10.1038/ng.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lunning MA, Green MR. Mutation of chromatin modifiers; an emerging hallmark of germinal center B-cell lymphomas. Blood Cancer J 2015; 5: e361. 10.1038/bcj.2015.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tazverik [package insert]. Cambridge, MA: Epizyme, 2020. [Google Scholar]
- 14.Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013; 23: 677–92. 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest 2013; 123: 5009–22. 10.1172/jci70626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Béguelin W, Teater M, Gearhart MD, Calvo Fernández MT, Goldstein RL, Cárdenas MG, et al. EZH2 and BCL6 cooperate to assemble CBX8-BCOR complex to repress bivalent promoters, mediate germinal center formation and lymphomagenesis. Cancer Cell 2016; 30: 197–213. 10.1016/j.ccell.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Béguelin W, Rivas MA, Calvo Fernández MT, Teater M, Purwada A, Redmond D, et al. EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nat Commun 2017; 8: 877. 10.1038/s41467-017-01029-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Béguelin W, Teater M, Meydan C, Hoehn KB, Phillip JM, Soshnev AA, et al. Mutant EZH2 induces a pre-malignant lymphoma niche by reprogramming the immune response. Cancer Cell 2020; 37: 655–73.e11. 10.1016/j.ccell.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011; 476: 298–303. 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian X, Pelton A, Shahsafaei A, Dorfman DM. Differential expression of enhancer of zeste homolog 2 (EZH2) protein in small cell and aggressive B-cell non-Hodgkin lymphomas and differential regulation of EZH2 expression by p-ERK1/2 and MYC in aggressive B-cell lymphomas. Mod Pathol 2016; 29: 1050–7. 10.1038/modpathol.2016.114. [DOI] [PubMed] [Google Scholar]
- 21.Morschhauser F, Tilly H, Chaidos A, McKay P, Phillips T, Assouline S, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol 2020; 21: 1433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, Jenkins MK. Visualization of specific B and T lymphocyte interactions in the lymph node. Science 1998; 281: 96–9. 10.1126/science.281.5373.96. [DOI] [PubMed] [Google Scholar]
- 23.Okada T, Miller MJ, Parker I, Krummel MF, Neighbors M, Hartley SB, et al. Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PLoS Biol 2005; 3: e150. 10.1371/journal.pbio.0030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coffey F, Alabyev B, Manser T. Initial clonal expansion of germinal center B cells takes place at the perimeter of follicles. Immunity 2009; 30: 599–609. 10.1016/j.immuni.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000; 102: 553–63. 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 26.Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 2010; 143: 592–605. 10.1016/j.cell.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwickert TA, Victora GD, Fooksman DR, Kamphorst AO, Mugnier MR, Gitlin AD, et al. A dynamic T cell-limited checkpoint regulates affinity-dependent B cell entry into the germinal center. J Exp Med 2011; 208: 1243–52. 10.1084/jem.20102477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature 2014; 509: 637–40. 10.1038/nature13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gitlin AD, Mayer CT, Oliveira TY, Shulman Z, Jones MJ, Koren A, et al. HUMORAL IMMUNITY. T cell help controls the speed of the cell cycle in germinal center B cells. Science 2015; 349: 643–6. 10.1126/science.aac4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weisel FJ, Zuccarino-Catania GV, Chikina M, Shlomchik MJ. A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity 2016; 44: 116–30. 10.1016/j.immuni.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ranuncolo SM, Polo JM, Melnick A. BCL6 represses CHEK1 and suppresses DNA damage pathways in normal and malignant B-cells. Blood Cells Mol Dis 2008; 41: 95–9. 10.1016/j.bcmd.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mlynarczyk C, Fontán L, Melnick A. Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol Rev 2019; 288: 214–39. 10.1111/imr.12755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee CH, Holder M, Grau D, Saldaña-Meyer R, Yu JR, Ganai RA, et al. Distinct stimulatory mechanisms regulate the catalytic activity of polycomb repressive complex 2. Mol Cell 2018; 70: 435–48.e5. 10.1016/j.molcel.2018.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo M, Price MJ, Patterson DG, Barwick BG, Haines RR, Kania AK, et al. EZH2 represses the B cell transcriptional program and regulates antibody-secreting cell metabolism and antibody production. J Immunol 2018; 200: 1039–52. 10.4049/jimmunol.1701470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bödör C, Grossmann V, Popov N, Okosun J, O’Riain C, Tan K, et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 2013; 122: 3165–8. 10.1182/blood-2013-04-496893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 2010; 42: 181–5. 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 2018; 24: 679–90. 10.1038/s41591-018-0016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A 2010; 107: 20980–5. 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A 2012; 109: 2989–94. 10.1073/pnas.1116418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiao L, Liu X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 2015; 350: aac4383. 10.1126/science.aac4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Souroullas GP, Jeck WR, Parker JS, Simon JM, Liu JY, Paulk J, et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat Med 2016; 22: 632–40. 10.1038/nm.4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rinke J, Chase A, Cross NCP, Hochhaus A, Ernst T. EZH2 in Myeloid Malignancies. Cells 2020; 9. 10.3390/cells9071639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Isshiki Y, McNally D, Melnick AM, Béguelin W. Evolution of the tumor microenvironment throughout progression and transformation of EZH2 mutant follicular lymphoma [abstract 622]. Blood 2021; 138: 446-. 10.1182/blood-2021-148996. [DOI] [Google Scholar]
- 44.Ennishi D, Takata K, Béguelin W, Duns G, Mottok A, Farinha P, et al. Molecular and genetic characterization of MHC deficiency identifies EZH2 as therapeutic target for enhancing immune recognition. Cancer Discov 2019; 9: 546–63. 10.1158/2159-8290.cd-18-1090. [DOI] [PubMed] [Google Scholar]
- 45.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 2012; 8: 890–6. 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 46.Kim HJ, Cantor H, Cosmopoulos K. Overcoming immune checkpoint blockade resistance via EZH2 inhibition. Trends Immunol 2020; 41: 948–63. 10.1016/j.it.2020.08.010. [DOI] [PubMed] [Google Scholar]
- 47.Otsuka Y, Nishikori M, Arima H, Izumi K, Kitawaki T, Hishizawa M, et al. EZH2 inhibitors restore epigenetically silenced CD58 expression in B-cell lymphomas. Mol Immunol 2020; 119: 35–45. 10.1016/j.molimm.2020.01.006. [DOI] [PubMed] [Google Scholar]
- 48.Keller PJ, Meyer R, Zhao F, Mia L, Greenwald E, Han X, et al. Targeting epigenetic dysregulation in bladder cancer through inhibition of EZH2 [poster]. Presented at: Annual Meeting of the American Association for Cancer Research, Atlanta, GA, March 29-April 3, 2019. [Google Scholar]
- 49.Kung PP, Bingham P, Brooun A, Collins M, Deng YL, Dinh D, et al. Optimization of orally bioavailable enhancer of zeste homolog 2 (EZH2) inhibitors using ligand and property-based design strategies: identification of development candidate (R)-5,8-Dichloro-7-(methoxy(oxetan-3-yl)methyl)-2-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (PF-06821497). J Med Chem 2018; 61: 650–65. 10.1021/acs.jmedchem.7b01375. [DOI] [PubMed] [Google Scholar]
- 50.Honma D, Kanno O, Watanabe J, Kinoshita J, Hirasawa M, Nosaka E, et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci 2017; 108: 2069–78. 10.1111/cas.13326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther 2014; 13: 842–54. 10.1158/1535-7163.mct-13-0773. [DOI] [PubMed] [Google Scholar]
- 52.Brach D, Johnston-Blackwell D, Drew A, Lingaraj T, Motwani V, Warholic NM, et al. EZH2 Inhibition by tazemetostat results in altered dependency on B-cell activation signaling in DLBCL. Mol Cancer Ther 2017; 16: 2586–97. 10.1158/1535-7163.mct-16-0840. [DOI] [PubMed] [Google Scholar]
- 53.Honma D, Nosaka E, Shiroishi M, Takata Y, Hama Y, Yamamoto Y, et al. CHOPDS-3201, a Potent EZH1/2 Dual Inhibitor, Demonstrates Antitumor Activity in Non-Hodgkin Lymphoma (NHL) Regardless of EZH2 Mutation [poster]. Presented at: Annual Meeting of the American Society of Hematology, San Diego, CA, December 1-4, 2018. [Google Scholar]
- 54.Keats J, Lee A, Estanck V, Cunniff J, Chen W, Mehovic R, et al. EZH2 inhibitor tazemetostat demonstrates activity in preclinical models of Bruton’s tyrosine kinase inhibitor–resistant relapsed/refractory mantle cell lymphoma [poster EP859]. Presented at: Annual Congress of the European Hematology Association, June 9-17, 2021. [Google Scholar]
- 55.Yap TA, Winter JN, Giulino-Roth L, Longley J, Lopez J, Michot JM, et al. Phase I study of the novel enhancer of zeste homolog 2 (EZH2) inhibitor GSK2816126 in patients with advanced hematologic and solid tumors. Clin Cancer Res 2019; 25: 7331–9. 10.1158/1078-0432.ccr-18-4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morishima S, Ishitsuka K, Izutsu K, Kusumoto S, Makiyama J, Utsunomiya A, et al. First-in-human study of the EZH1/2 dual inhibitor valemetostat in relapsed or refractory non-hodgkin lymphoma (NHL) – updated results focusing on adult T-cell leukemia-lymphoma (ATL) [poster]. Presented at: Annual Meeting and Exposition of the American Society of Hematology, Orlando, FL, December 7-10, 2019. [Google Scholar]
- 57.Maruyama D, Tobinai K, Makita S, Ishida T, Kusumoto S, Ishitsuka K, et al. First-in-human study of the EZH1/2 dual inhibitor DS-3201b in patients with relapsed or refractory non-Hodgkin lymphomas — Preliminary Results [abstract]. Blood 2017; 130: 4070. 10.1182/blood.V130.Suppl_1.4070.4070. [DOI] [Google Scholar]
- 58.Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 2018; 19: 649–59. 10.1016/s1470-2045(18)30145-1. [DOI] [PubMed] [Google Scholar]
- 59.Fujita S, Honma D, Adachi N, Araki K, Takamatsu E, Aoyama K, et al. Novel leukemia stem cell-targeted therapy for acute myeloid leukemia based on dual inhibition of Ezh1/Ezh2 [abstract]. Blood 2015; 126: 457-. 10.1182/blood.V126.23.457.457. [DOI] [Google Scholar]
- 60.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med 2018; 378: 1396–407. 10.1056/NEJMoa1801445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scholze H, Stephenson RE, Reynolds R, Shah S, Puri R, Butler SD, et al. Combined EZH2 and Bcl-2 inhibitors as precision therapy for genetically defined DLBCL subtypes. Blood Adv 2020; 4: 5226–31. 10.1182/bloodadvances.2020002580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mondello P, Tadros S, Teater M, Fontan L, Chang AY, Jain N, et al. Selective inhibition of HDAC3 targets synthetic vulnerabilities and activates immune surveillance in lymphoma. Cancer Discov 2020; 10: 440–59. 10.1158/2159-8290.cd-19-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lue JK, Prabhu SA, Liu Y, Gonzalez Y, Verma A, Mundi PS, et al. Precision targeting with EZH2 and HDAC inhibitors in epigenetically dysregulated lymphomas. Clin Cancer Res 2019; 25: 5271–83. 10.1158/1078-0432.ccr-18-3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adolph LC, Fichaux Q, Strobl CD, Antoniolli M, Passerini V, Keay WD, et al. CHOP but not bendamustine reverses EZH2 Y641 mutation induced MHC-I/II loss in human lymphoma models [abstract]. Blood 2021; 138: 2391-. 10.1182/blood-2021-144803. [DOI] [Google Scholar]
- 65.Pastore A, Jurinovic V, Kridel R, Hoster E, Staiger AM, Szczepanowski M, et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol 2015; 16: 1111–22. 10.1016/s1470-2045(15)00169-2. [DOI] [PubMed] [Google Scholar]
- 66.Jurinovic V, Passerini V, Oestergaard MZ, Knapp A, Mundt K, Araf S, et al. Evaluation of the m7-FLIPI in patients with follicular lymphoma treated within the gallium trial: EZH2 mutation status may be a predictive marker for differential efficacy of chemotherapy [abstract]. Blood 2019; 134: 122-. 10.1182/blood-2019-130208. [DOI] [Google Scholar]
- 67.Dimopoulos K, Søgaard Helbo A, Fibiger Munch-Petersen H, Sjö L, Christensen J, Sommer Kristensen L, et al. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol Oncol 2018; 12: 180–95. 10.1002/1878-0261.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Herviou L, Kassambara A, Boireau S, Robert N, Requirand G, Vincent L, et al. Targeting EZH2 in multiple myeloma could be promising for a subgroup of MM patients in combination with IMiDs [abstract 311]. Blood 2016; 128: 311-. 10.1182/blood.V128.22.311.311. [DOI] [Google Scholar]
- 69.Drew AE, Motwani V, Eichinger L, Smith J, Raimondi A. Mechanism of action of synergistic activity of EZH2 inhibition and IMiDs in preclinical multiple myeloma models [abstract 807]. Cancer Res 2018; 78: 807-. 10.1158/1538-7445.am2018-807. [DOI] [Google Scholar]
- 70.Tong KI, Yoon S, Isaev K, Bakhtiari M, Lackraj T, He MY, et al. Combined EZH2 inhibition and Ikaros degradation leads to enhanced anti-tumor activity in diffuse large B-cell lymphoma. Clin Cancer Res 2021. 10.1158/1078-0432.ccr-20-4027. [DOI] [PubMed] [Google Scholar]
- 71.Batlevi CL, Park SI, Phillips T, Nastoupil L, Amengual J, Andorsky D, et al. Interim analysis of the randomized phase 1b/3 study evaluating the safety and efficacy of tazemetostat plus lenalidomide and rituximab in patients with relapsed/refractory follicular lymphoma [poster 2207]. Presented at: Annual Meeting and Exposition of the American Society of Hematology, Atlanta, GA, December 11, 2021. [Google Scholar]
- 72.Sarkozy C, Morschhauser F, Dubois S, Molina T, Michot JM, Cullières-Dartigues P, et al. A LYSA phase ib study of tazemetostat (EPZ-6438) plus R-CHOP in patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL) with poor prognosis features. Clin Cancer Res 2020; 26: 3145–53. 10.1158/1078-0432.ccr-19-3741. [DOI] [PubMed] [Google Scholar]