

Abstract
Deficiency of natural killer (NK) cells shows a significant impact on tumor progression and failure of immunotherapy. It is highly desirable to boost NK cell immunity by upregulating active receptors and relieving the immunosuppressive tumor microenvironment. Unfortunately, mobilization of NK cells is hampered by poor accumulation and short retention of drugs in tumors, thus declining antitumor efficiency. Herein, we develop an acid-switchable nanoparticle with self-adaptive aggregation property for co-delivering galunisertib and interleukin 15 (IL-15). The nanoparticles induce morphology switch by a decomposition-metal coordination cascade reaction, which provides a new methodology to trigger aggregation. It shows self-adaptive size-enlargement upon acidity, thus improving drug retention in tumor to over 120 h. The diameter of agglomerates is increased and drug release is effectively promoted following reduced pH values. The nanoparticles activate both NK cell and CD8+ T cell immunity in vivo. It significantly suppresses CT26 tumor in immune-deficient BALB/c mice, and the efficiency is further improved in immunocompetent mice, indicating that the nanoparticles can not only boost innate NK cell immunity but also adaptive T cell immunity. The approach reported here provides an innovative strategy to improve drug retention in tumors, which will enhance cancer immunotherapy by boosting NK cells.
Key words: Nanoparticle, Aggregation, Drug retention, Natural killer cells, Cancer immunotherapy, Acid-switchable, Galunisertib, IL-15
Graphical abstract
An acid-switchable nanoparticle with self-adaptive aggregation property is developed to improve retention of co-delivered galunisertib and IL-15 in tumor for boosting antitumor immunity of natural killer cells.
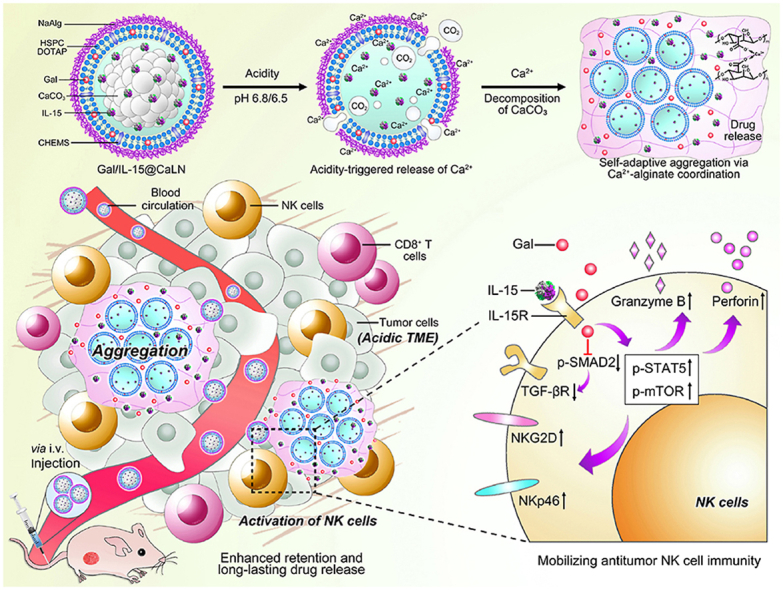
1. Introduction
Innate immunity is vital for combating various diseases including cancer1. Natural killer (NK) cells are critical innate immunity effector cells, which recognize and kill cancer cells in an antigen-independent manner without the need for pre-sensitization2,3. Besides, NK cells bridge innate and adaptive immunity by promoting the secretion of key cytokines and chemokines to enhance antigen presentation and activate T cells4. However, antitumor efficiency of NK cells is hampered by the shortage of activation stimuli in tumor microenvironment (TME) and existing immunosuppressive molecules such as transforming growth factor β (TGF-β), prostaglandin E2 (PGE2) and interleukin 10 (IL-10)5,6. Promoting cellular activation and removing inhibitive factors are equally important to fully mobilize NK cells for immunotherapy7, 8, 9. For example, TGF-β is immunosuppressive and inhibits mTOR pathway in NK cells, which suppresses cell proliferation and activation10. Galunisertib inhibits TGF-β receptor type I kinase to block TGF-β/SMAD pathway and relieve the inhibition of TGF-β on NK cells11. Interleukin 15 (IL-15) is a multi-effect cytokine to activate the JAK/STAT and PI3K/mTOR pathways in NK cells, thus improving proliferation and activation6,12,13. It is highly desirable to improve antitumor efficiency by synergistically activating NK cells and relieving immunosuppressive factors, such as combining the galunisertib and IL-1514.
Although combinational therapies provide attractive tools to boost NK cells, it is hard to ensure efficiency due to poor accumulation and short retention of drugs in tumor11,15. A diversity of nanocarriers has been reported to facilitate targeted delivery of drugs into tumors, but it remains challenging to achieve long-lasting retention due to rapid clearance of nanocarriers via high interstitial fluid and blood washing16, 17, 18. Aggregation strategy is effective to enhance the retention of nanocarriers inside tumor by weakening their ability to pass through blood vessels after accumulation19, 20, 21. The nanocarriers are size-enlargeable by leveraging distinctive features of TME22. Typically, aggregation is triggered through linker form, shape remodel, materials swell and phase transition23, 24, 25. These approaches dominate the aggregation methodology, but most of them require rigorous reaction conditions such as a specific enzyme, high-energy light, elevated temperature and strong redox level26, 27, 28. Convenient and universal methodologies are desirable to weaken external demands for inducing aggregation. In addition, most aggregative strategies perform direct on/off state and lack self-adaptive variations following changes of stimuli, thus strictly limiting the aggregation state as well as retention capability of nanocarriers26,29,30. Therefore, it shows great priority to develop nanocarriers from new aggregative methodology and enable switchable aggregation upon stimuli for further improving drug retention in tumors.
Metal coordination has been widely investigated to fabricate nanocarriers, which enables flexible control of assembly by changing the type and amount of coordinate metal ions31, 32, 33, 34. Herein, we have developed galunisertib- and IL-15-loaded nanoparticles (Gal/IL-15@CaLN) which trigger self-adaptive aggregation through a cascade decomposition–coordination reaction to improve drug retention. The nanoparticle changes input acidic stimulus, a common feature in most solid tumors35,36, to output metal coordination enablement, leading to convenient and universal aggregation in tumors. The nanoparticle is composed by calcium carbonate (CaCO3) core and sodium alginate (NaAlg) outer layer. NaAlg is a biocompatible polymer that reacts with most divalent metal ions including calcium ion (Ca2+) to promote gelation36, 37. Since the release of Ca2+ depends on acid-triggered decomposition of CaCO338, self-adaptive aggregation of nanoparticles is achieved by leveraging controllable release of Ca2+ at different pH values (Fig. 1A). Typically, the nanoparticles keep inert in blood circulation and release Ca2+ in acidic TME. Free Ca2+ facilitates Ca2+-alginate coordination and changes aggregative state of nanoparticles, thus enhancing drug retention in tumors and triggering release of galunisertib and IL-15 (Fig. 1B). The nanoparticles are promising to activate NK cells in tumors and improve their capability to kill tumor cells. Moreover, the promotion of NK cells may further help with T cell immunity to inhibit tumors (Fig. 1C). The immune responses and antitumor effects post treatments are investigated in tumor-bearing immune-deficient mice and immunocompetent mice to display the key role of NK cells. The approach will contribute to NK cell-based cancer immunotherapy through self-adaptive aggregation-enhanced drug retention.
Figure 1.

Schematic illustration of the design of Gal/IL-15@CaLN with self-adaptive aggregation feature and its function to mobilize NK cells (A) Fabrication of Gal/IL-15@CaLN. The nanoparticle was prepared by coating NaAlg on CaCO3-containing lipid nanoparticle. NaAlg was coated via electrostatic adsorption. Gal/IL-15@CaLN was activated in acidic conditions such as pH 6.8 or 6.5 buffers and then release Ca2+. Since the release rate and amount of Ca2+ was associated with the pH values, the nanoparticles enabled self-adaptive aggregation via Ca2+-alginate coordination and induced long-lasting drug release. (B) Gal/IL-15@CaLN accumulated in tumor after intravenous injection. Aggregation was triggered due to the activation of CaCO3 in acidic TME and enhanced the retention of drugs in tumor. The released galunisertib and IL-15 promoted the activation of NK cells. (C) Mechanisms of Gal/IL-15@CaLN for mobilizing antitumor NK cell immunity. IL-15 bound with IL-15R to induce downstream activation signaling pathways. Galunisertib inhibited p-SMAD2 and downregulated the expression of TGF-βR to relieve TGF-β-mediated immunosuppressive effects. The combined therapy enhanced the expression of p-STAT5 and p-mTOR, and promoted the expression of NKG2D and NKp46 and the secretion of granzyme B and perforin.
2. Materials and methods
2.1. Materials
Hydrogenated soybean phosphatidylcholine (HSPC) and (2,3-dioleoyloxy-propyl)-trimethylammonium-chloride (DOTAP) were purchased from Advanced Vehicle Technology Pharmaceutical Ltd (Shanghai, China). Cholesteryl hemisuccinate (CHEMS) was purchased from Coupling Pharmaceutical Technology (Shanghai, China). NaAlg (the viscosity of 200–500 mPa s) was purchased from Adamas-beta (Shanghai, China). Calcium colorimetric assay kit was purchased from Beyotime Biotechnology (Shanghai, China). Galunisertib was purchased from CSNpharm (Chicago, IL, USA). IL-15 was purchased from PeproTech (Rocky Hill, NJ, USA). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD) was purchased from Meilun Biotechnology Co., Ltd (Dalian, China). Mouse IL-15 ELISA kit was purchased from Neobioscience Co., Ltd. (Shenzhen, China). The anti-CD3e-PerCP-Cy5.5 (145-2C11) and anti-CD3e-PE (145-2C11) antibodies were purchased from TONBO biosciences (San Diego, CA, USA). The anti-CD49b-FITC (DX5), anti-NKG2D-APC (CX5), anti-NKp46-PE (29A1.4), anti-NKp46-PE-Cy7 (29A1.4), anti-granzyme B-Alexa Fluor 647 (GB11), anti-perforin-PE (S16009B), anti-CD4-FITC (GK1.5), anti-CD8a-PE (53-6.7), anti-CD45-FITC (S18009F), anti-F4/80-PE-Cy7 (BM8), anti-CD80-PE (16-10A1), and anti-CD206-APC (C068C2) antibodies were purchased from Biolegend (San Diego, CA, USA). The anti-TGF-βRI-PE (141231) antibodies were purchased from R&D Systems (Minneapolis, MN, USA). The anti-p-SMAD2 (Ser467) rabbit monoclonal antibody (mAb), anti-p-mTOR (Ser2448) rabbit mAb, anti-CD49b rabbit mAb, and PE-labeled goat anti-rabbit IgG secondary antibodies for flow cytometry examination were purchased from Abcam (Cambridge, MA, USA). The TGF-βRI rabbit polyclonal antibodies (pAb) were purchased from Affinity Biosciences (Changzhou, China). The anti-p-STAT5 (Tyr694) rabbit mAb was purchased from Cell Signaling Technology (Danvers, MA, USA). Alexa Fluor 647-labeled goat anti-rabbit IgG secondary antibodies for cellular CLSM examination and cell counting kit-8 (CCK-8) were purchased from Yeasen biotechnology (Shanghai, China). Anti-CD8a rabbit pAb, Cy3-labeled goat anti-rabbit IgG secondary antibodies, and FITC-labeled goat anti-rabbit IgG secondary antibodies were purchased from Servicebio (Wuhan, China). Other reagents were purchased from Sinopharm Group Chemical Reagent Co., Ltd. (Shanghai, China).
2.2. Cells and animals
The murine colon cancer cell line CT26 was purchased from the cell bank of the Chinese Academy of Sciences (Shanghai, China). CT26 cells were cultured in RPMI 1640 medium containing 10% FBS, 100 U/mL of penicillin, and 100 μg/mL of streptomycin. To isolate NK cells, BALB/c mice spleens were pestled on a 70 μm cell strainer to obtain a single-cell suspension, and then a mouse spleen NK cell separation solution kit was used (NK2011MPK, tbdscience, Tianjin, China). NK cells were cultured in an immune cell culture medium (ANDL-TBD-G, tbdscience, Tianjin, China) containing 10% FBS, 200 U/mL of penicillin, and 200 μg/mL of streptomycin. All cells were incubated at 37 °C with 5% CO2 atmosphere. Female BALB/c nude mice and BALB/c mice (4 weeks, 18–22 g) were provided by Shanghai Experimental Animal Center (Shanghai, China). All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
2.3. Preparation and characterization of Gal/IL-15@CaLN
Gal/IL-15@Ca was initially prepared. Briefly, HSPC (16 mg), CHEMS (4 mg), DOTAP (2 mg), and galunisertib (1.5 mg) in chloroform (10 mL) were mixed with CaCl2 in alcohol (250 μL, 10 mg/mL). Na2CO3 in water (160 μL, 10 mg/mL) was added dropwise to the aforementioned solution under sonication. The emulsion was evaporated using the vacuum rotary evaporation (Heidolph, Schwabach, Germany). The obtained lipid film was hydrated by IL-15 solution (2 mL, 1.5 μg/mL), followed by sonicating under an ice bath, and the unencapsulated IL-15 was removed by ultrafiltration (MWCO, 100 kDa). For the preparation of Gal/IL-15@CaLN, Gal/IL-15@Ca was added dropwise to NaAlg solution (1 mg/mL) with a volume ratio of 1:1 and stirred for 4 h at room temperature. For the preparation of CaCO3-free Gal/IL-15@LN, identical amount of HSPC, CHEMS, DOTAP, and galunisertib was dissolved in chloroform and evaporated to obtain the dry lipid film, and hydrated by IL-15 solution. The release behavior of Ca2+ was examined by using a calcium colorimetric assay kit. Briefly, 1 mL of Gal/IL-15@Ca was dispersed in a dialysis bag with a retained molecular weight of 2000 Da and incubated in 20 mL of PBS of different pH values at 37 °C. One milliliter of release medium was collected at 0.5, 5, 30, 60 and 120 min, respectively. Same volume of fresh release medium was refilled after each collection. The concentration of Ca2+ was detected and the cumulative release rate of Ca2+ was calculated. For preparing DiD-loaded nanoparticles, 1 wt% DiD was added during the process.
The size and zeta potential were determined by dynamic light scanning (DLS) (Malvern, Britain). To detect the encapsulation efficiency (EE) and loading capability (LC), Gal/IL-15@CaLN and Gal/IL-15@LN were dissolved in 25 mmol/L SDS. The concentration of galunisertib was determined by HPLC (C18 reversed-phase column, Thermo Fisher Scientific, MA, USA) and flow rate of 1.0 mL/min. The concentration of IL-15 was determined by an IL-15 ELISA kit. The EE and LC of nanoparticles were calculated by Eqs. (1), (2), respectively:
| (1) |
| (2) |
Gal/IL-15@CaLN and Gal/IL-15@LN were diluted to 0.5 mg/mL by PBS or PBS with 10% FBS and incubated at 37 °C. The size was determined by DLS in PBS for 18 days and in PBS with 10% FBS for 24 h. To investigate the acid-responsiveness, Gal/IL-15@CaLN and Gal/IL-15@LN were incubated in PBS at various pH values (pH 6.5, 6.8, and 7.4) at a concentration of 0.5 mg/mL for 120 min. The size and morphology changes were examined by DLS and TEM, respectively. One milliliter of Gal/IL-15@CaLN was put into a dialysis bag with a retained molecular weight of 2000 Da and performed in 20 mL of PBS with 0.5% tween 80 at various pH values (pH 6.5, 6.8, and 7.4) at 37 °C. Released galunisertib and IL-15 were determined by HPLC and an IL-15 ELISA kit, respectively. And the cumulative release rate of galunisertib and IL-15 were calculated.
2.4. In vitro activation of NK cells
To examine the effects of Gal/IL-15@CaLN on NK cells, CT26 cells were seeded in a 12-well plate at 2 × 105 cells/well overnight, and then NK cells (2 × 105 cells) were added to each well and cultured for 24 h to mimic the adverse effects of TME on NK cells. The cells were subsequently treated with IL-15@CaLN, Gal@CaLN, Gal/IL-15@LN, and Gal/IL-15@CaLN at identical 20 ng/mL IL-15 or/and 8.7 μg/mL galunisertib for 12 h. After that, the cells were stained with anti-NKp46-PE, anti-NKG2D-APC, and anti-TGF-βRI-PE antibodies for 30 min at 4 °C, respectively. For intercellular markers, the cells were treated with fixation-permeabilization buffer and then stained with anti-granzyme B-Alexa Fluor 647, and anti-perforin-PE for 30 min at 4 °C. Additionally, the cells were stained with anti-p-STAT5 or anti-p-SMAD2 rabbit mAb after permeabilization for 1 h at room temperature, followed by incubating with PE-labeled goat anti-rabbit IgG secondary antibodies for 1 h at 4 °C.
For confocal laser scanning microscopy (CLSM) examination, NK cells (2 × 105 cells/well) were co-cultural with CT26 cells (2 × 105 cells/well) in a 12-well plate for 24 h. The cells were fixed with 4% paraformaldehyde and stained with anti-TGF-βRI or anti-p-mTOR rabbit mAb overnight at 4 °C, followed by staining with Alexa Fluor 647-labeled goat anti-rabbit IgG secondary antibodies for 2 h at room temperature. The fluorescence images were examined by CLSM (DAPI: Ex 405 nm, Em 415–485 nm; Alexa Fluor647: Ex 638 nm, Em 649–770 nm).
2.5. In vivo biodistribution of Gal/IL-15@CaLN
BALB/c mice were subcutaneously administered with 2 × 106 CT26 cells/mouse at the right back. Once the tumor volume reached ∼100 mm3, the mice were administered with DiD-labeled Gal/IL-15@CaLN or Gal/IL-15@LN at identical 0.5 mg/kg DiD. The major organs and tumors were harvested at 24 and 120 h after the injection and examined by IVIS (PerkinElmer, Waltham, MA, USA). The tumors were frozen-sectioned into 10 μm pieces, and cell nucleus was stained with DAPI. The fluorescence of DAPI and DiD was evaluated by CLSM.
2.6. Immune-related examination
The immune response of Gal/IL-15@CaLN was initially investigated on CT26 tumor-bearing immune-deficient BALB/c-nu mice, which were subcutaneously administered with 1.5 × 106 CT26 cells/mouse at the right back. Mice were randomly grouped (n = 3) and treated with PBS, Gal/IL-15, IL-15@CaLN, Gal@CaLN, Gal/IL-15@LN, and Gal/IL-15@CaLN with the same IL-15 (30 μg/kg) and galunisertib (13 mg/kg) dosage every three days for four times. Three days after the last treatment, the tumors were harvested and digested into a single-cell suspension. Then the cells were stained with specific biomarkers and examined on flow cytometry. Next, the immune response of Gal/IL-15@CaLN was investigated in the presence of T lymphocytes on CT26 tumor-bearing BALB/c-ic mice, which were subcutaneously administered with 2 × 106 CT26 cells/mouse at the right back. The NK cells and their functions were investigated. Moreover, the macrophages in tumor tissues were evaluated by staining with anti-CD45-FITC and anti-F4/80-PE-Cy7 antibodies. The M1-phenotype macrophages were stained with anti-CD80-PE antibody and the M2-phenotype with anti-CD206-APC antibody after permeabilization. To evaluate the T lymphocyte infiltration in tumors, the lymphocytes were separated by a mouse lymphocyte separation solution (Dakewe Biotech, China). The lymphocytes were extracted, washed, and stained with anti-CD3-PerCP-Cy5.5, anti-CD4-FITC, and anti-CD8-PE antibodies. All the stained cells were examined by flow cytometry (BD, Calibur).
2.7. Tumor growth suppression
To establish CT26 tumor-bearing mice, BALB/c-nu mice and BALB/c-ic mice were subcutaneously administered with 100 μL of 1.5 × 106 or 2 × 106 CT26 cells/mouse at the right back, respectively. The mice were randomly grouped (n = 6) to evaluate tumor growth suppression by Gal/IL-15@CaLN once the tumor volume reached ∼100 mm3. The mice were treated with PBS, Gal/IL-15, IL-15@CaLN, Gal@CaLN, Gal/IL-15@LN, and Gal/IL-15@CaLN every three days for four times, with the same IL-15 (30 μg/kg) and galunisertib (13 mg/kg) dosage. The tumor volume and body weight were monitored every two days. The tumor volume was calculated as Eq. (3):
| (3) |
The major organs and tumors were harvested on Day 14 for BALB/c-nu mice and Day 16 for BALB/c-ic mice. The tumors were weighed and analyzed by hematoxylin and eosin (H&E) staining and the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay. Besides, the infiltration of NK and T cells was examined by fluorescence staining. The injury of major organs was also observed using H&E staining.
2.8. Statistical analysis
Data were expressed as mean ± standard deviation (SD). The statistical significance was displayed by two-sided unpaired Student's t-test. Statistical significance was set as follows: ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
3. Results and discussion
3.1. Preparation and characterization of Gal/IL-15@CaLN
To fabricate Gal/IL-15@CaLN, nanoscale CaCO3 was firstly generated by an emulsion-homogeneity process and formed a thin film with cationic lipids and galunisertib. Then, the galunisertib-loaded lipid film was hydrated in IL-15-containing deionized water. After that, the nanoparticles were incubated with NaAlg solution to obtain Gal/IL-15@CaLN. NaAlg contains a large number of negatively charged carboxyl groups, which enables electrostatic interaction with the positively charged lipid surface. Nanoparticles without NaAlg coating (namely Gal/IL-15@Ca) and nanoparticles without CaCO3 core (namely Gal/IL-15@LN) were prepared by a similar process and set as control groups (Supporting Information Table S1). The diameter and surface charge of Gal/IL-15@CaLN and control nanoparticles were examined by dynamic laser scanning (DLS) examination (Fig. 2A and B). Gal/IL-15@Ca showed a diameter of 98.5 ± 1.5 nm and a ζ-potential of 14.2 ± 2.5 mV. Compared to Gal/IL-15@Ca, Gal/IL-15@CaLN showed an increased diameter to 124.7 ± 6.0 nm and a negative surface charge of −26.8 ± 1.2 mV. The surface charge reversal was due to the coating of NaAlg. The diameter and surface charge of Gal/IL-15@LN were 117.2 ± 2.3 nm and −29.4 ± 1.5 mV, respectively. The LC and EE of galunisertib in Gal/IL-15@CaLN were 4.3 ± 0.01% and 77.9 ± 0.2%, respectively. The LC of galunisertib was comparable to that of most hydrophobic drugs in liposomes. The LC and EE of IL-15 in Gal/IL-15@CaLN were 0.98 ± 0.02 μg/10 mg and 88.0 ± 1.6%, respectively (Supporting Information Table S2). The LC of IL-15 was low, which might be due to the limited inner aqueous core and the protein structure. The EE of both drugs was acceptable for preparing the nanoparticles. Next, the stability of Gal/IL-15@CaLN and Gal/IL-15@LN was studied. The results showed that the nanoparticles performed good stability in PBS and 10% fetal bovine serum (FBS)-containing PBS (Supporting Information Fig. S1).
Figure 2.

Characterization of Gal/IL-15@CaLN (A) Average hydrodynamic size and (B) ζ-Potential of Gal/IL-15@Ca, Gal/IL-15@CaLN, and Gal/IL-15@LN determined by dynamic light scattering (DLS) (n = 3) (C) Cumulative release of Ca2+ from Gal/IL-15@Ca at pH 7.4, 6.8 and 6.5, respectively (n = 3) (D) TEM images of Gal/IL-15@LN (Scale bars, 100 nm) and Gal/IL-15@CaLN at pH 7.4 (Scale bar, 100 nm), pH 6.8 (Scale bar, 500 nm) or pH 6.5 (Scale bar, 1 μm) (E) Hydrodynamic size of Gal/IL-15@LN. (F) Hydrodynamic size of Gal/IL-15@CaLN (n = 3) (G) Cumulative release of galunisertib and (H) IL-15 from Gal/IL-15@CaLN at pH 7.4, 6.8 and 6.5 buffer, respectively (n = 3). Data were shown as mean ± SD. The statistical significance was displayed by two-sided unpaired Student's t-test. (∗∗P < 0.01, ∗∗∗P < 0.001).
To investigate the acidity-responsive property of Gal/IL-15@CaLN, the nanoparticle and control groups were dispersed in buffers with different pH value. First, the release behavior of Ca2+ was determined. The LC of CaCO3 in Gal/IL-15@Ca was 5.74 ± 0.41%. Gal/IL-15@Ca was used to avoid the influence of NaAlg (Fig. 2C). It was found that only 9.88 ± 1.35% of Ca2+ was released after incubated in pH 7.4 buffer for 120 min. In pH 6.8 buffer, 23.19 ± 1.07% of Ca2+ was released at 0.5 min and 41.75 ± 2.09% was released at 120 min. The release rate and cumulative release amount were further improved in pH 6.5 buffer. 35.14 ± 2.40% of Ca2+ was released at 0.5 min and 61.38 ± 3.22% was detected at 120 min. These results indicated that the release behavior of Ca2+ from Gal/IL-15@Ca was acidity-dependent. More rapid and higher release rate could be achieved at lower pH value. The morphology of Gal/IL-15@LN did not change at pH 7.4, 6.8 or 6.5, respectively (Fig. 2D). Contrastively, aggregation of Gal/IL-15@CaLN was observed at pH 6.8, and more aggressive aggregation was found at pH 6.5 (Fig. 2D). Microscale aggregate complexes were formed at pH 6.5. The diameter of nanoparticles at different pH values was also evaluated by DLS (Fig. 2E and F). There was no significant size change of Gal/IL-15@LN at different pH values. The diameter of Gal/IL-15@CaLN kept unchanged in pH 7.4 buffer, while was size-enlarged to ∼561.2 nm after incubated for 0.5 min at pH 6.8 and further enlarged to ∼935.5 nm after incubated for 30 min. Sharply, the diameter of Gal/IL-15@CaLN was enlarged to ∼983.3 nm after incubated for 0.5 min at pH 6.5 and improved to over 2200 nm at 30 min. The diameter did not further change even with prolonged incubation for 120 min. These results showed that Gal/IL-15@CaLN performed a self-adaptive aggregation property in response to acidity. The aggregation state of Gal/IL-15@CaLN was highly associated with the pH value, and more aggressive aggregation was achieved at pH 6.5 than that at pH 6.8. The aggregation of Gal/IL-15@CaLN was activated via two steps. First, acidity triggered decomposition of CaCO3 and release of Ca2+ from the nanoparticles. Second, Ca2+ reacted with carboxyl group of alginate to promote aggregation by forming Ca2+-alginate complexes. Since the release rate and amount of Ca2+ were strictly controlled by decomposition of CaCO3 in the presence of hydrogen ion (H+), Gal/IL-15@CaLN showed self-adaptive aggregation property in acidic conditions. A previous study has clarified the behavior of Ca2+ release from CaCO3 in different pH buffers39. The coordination in Gal/IL-15@CaLN upon acidity was also confirmed by FITR spectrum (Supporting Information Fig. S2). The asymmetric stretching vibrations of carboxylate salt ion appeared at 1611.44 and 1535.06 cm−1, which shifted to lower wavenumbers. It was owing to the change of the charge density, the radius, and the atomic weight of the cation when Na+ was replaced with Ca2+40.
Next, we investigated the release properties of galunisertib and IL-15 from the nanoparticles in different pH buffers. It was found that 29.50 ± 2.01% of galunisertib was released from the nanoparticles at pH 7.4 within 72 h. The release rate was slightly improved at pH 6.8 and cumulative release amount reached 36.48 ± 1.46% after 72 h incubation. Most importantly, the release rate was further improved at pH 6.5 and 54.26 ± 1.54% of galunisertib was released at 72 h (Fig. 2G). Similar release kinetics of IL-15 from Gal/IL-15@CaLN was detected. The release rate of IL-15 was increased following decreased pH values. The cumulative release amount of IL-15 at 72 h was 33.40 ± 2.29%, 43.65 ± 2.93% and 59.08 ± 1.69% in pH 7.4, 6.8 and 6.5 buffer, respectively (Fig. 2H). These results suggested that Gal/IL-15@CaLN were acid-switchable and triggered controllable release of galunisertib and IL-15 with long-lasting manner.
3.2. Activating NK cells by Gal/IL-15@CaLN in vitro
The killing capability of NK cells to tumor cells is regulated by the surface activating and inhibitory receptors41. However, the immunosuppressive TME and lacking essential nutrients hamper the proliferation and activation of NK cells, which suppresses the antitumor effect of NK cells in vivo5. Immunostimulant IL-15 and TGF-βRI kinase inhibitor galunisertib were encapsulated in Gal/IL-15@CaLN to mobilize the antitumor effect of NK cells. NK cells were firstly prepared from spleen of BALB/c mice and then purified and enriched by flow cytometry. The purity of prepared NK cells was over 90% for in vitro examination (Supporting Information Fig. S3). Firstly, the cytotoxicity of nanoparticles on NK and CT26 cells were evaluated. No significant decrease of cell viability was observed in Gal/IL-15@CaLN treated cells at 43.5 μg/mL of galunisertib and 100 ng/mL of IL-15 (Supporting Information Fig. S4). Then, the NK cells were incubated with CT26 cells for 24 h and treated with various suspensions. The expression of activating receptors including NKG2D and NKp46 on NK cells was investigated (Fig. 3A and B). The percentage of NKG2D+ NK cells was increased from 23.30 ± 1.78% of PBS group to 37.47 ± 3.12% of IL-15@CaLN after the treatments (Fig. 3A and Supporting Information Fig. S5). This could be attributed to IL-15 delivered by IL-15@CaLN. After treated by Gal@CaLN, the percentage of NKG2D+ NK cells was 38.30 ± 4.03%. It was because the released galunisertib inhibited the TGF-βRI on NK cells and relieved the inhibition effect of TGF-β from CT26 cells. Furthermore, 44.17 ± 2.10% and 47.53 ± 4.11% of NKG2D+ NK cells were detected in Gal/IL-15@LN group and Gal/IL-15@CaLN group, respectively (Fig. 3A and Fig. S5). These results indicated a synergistic effect on NK cells by combining IL-15 and galunisertib. Similarly, the expression of NKp46 on NK cells showed a consistent trend as NKG2D (Fig. 3B and Supporting Information Fig. S6). Compared to 26.63 ± 1.42% of NKp46+ NK cells after PBS treatment, the Gal/IL-15@LN and Gal/IL-15@CaLN improved that population to 56.73 ± 3.56% and 56.97 ± 2.68%, respectively. The upregulated expression of NKG2D and NKp46 on NK cells could be attributed to the synergistic effect of IL-15 stimulation and the blockage of TGF-β pathway via galunisertib. Since the expression of activating receptors on NK cells was enhanced after Gal/IL-15@CaLN treatment, we further examined the function of NK cells to secrete granzyme B and perforin. The perforin/granzyme B apoptosis pathway is critical for NK cells to kill tumor cells42. NK cells released perforin to perforate the surface of target cells. Then, granzyme B enters the target cells and triggers apoptosis. Compared with PBS group, the percentage of granzyme B+ NK cells was upregulated to 3.75-, 3.80-, 6.35- and 6.23-fold higher after treated by IL-15@CaLN, Gal@CaLN, Gal/IL-15@LN and Gal/IL-15@CaLN, respectively (Fig. 3C and Supporting Information Fig. S7). Besides, after treated by IL-15@CaLN, Gal@CaLN, Gal/IL-15@LN and Gal/IL-15@CaLN, the percentage of perforin+ NK cells were 2.43-, 2.20-, 4.25- and 4.55-fold as high as that in PBS group, respectively (Fig. 3D and Supporting Information Fig. S8). These results suggested that Gal/IL-15@CaLN enhanced the function of NK cells for killing tumor cells.
Figure 3.

Gal/IL-15@CaLN-promoted activation of NK cells in vitro. The NK cells were pre-cultured with CT26 cells for 24 h and then treated with suspensions at equal 10 μg/mL of galunisertib and 20 ng/mL of IL-15 for 12 h, respectively (A) Percentage of NKG2D+ and (B) NKp46+ NK cells after various treatments (n = 3) (C) Frequency of granzyme B+ and (D) perforin+ expression in NK cells after various treatments (n = 3) (E) p-STAT5 expression in NK cells (n = 3) (F) p-mTOR expression in NK cells. Scale bars, 10 μm. (G) Mean fluorescence intensity (MFI) of p-SMAD in NK cells determined by flow cytometry (n = 3) (H) Quantitation of TGF-βRI + NK cells (n = 3). Data were expressed as mean ± SD. The statistical significance was displayed by two-sided unpaired Student's t-test. (∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001).
It has been reported that IL-15 regulates JAK/STAT and PI3K/mTOR pathways to facilitate the activation and proliferation of NK cells6. The mean fluorescent intensity of p-STAT5 in NK cells with Gal/IL-15@CaLN treated was 3.63-fold as high as that in PBS group, and the population of NKs with high p-STAT5 expressed in Gal/IL-15@CaLN group was also enhanced (Fig. 3E). The increased expression of p-mTOR which was associated with the activation of NK cells was also confirmed. From CLSM examination, the p-mTOR signal was weak in PBS-treated NK cells. But the signals became stronger in IL-15@CaLN-, Gal/IL-15@LN- and Gal/IL-15@CaLN-treated NK cells than that in PBS group (Fig. 3F). These results suggested that Gal/IL-15@CaLN promoted the activation of STAT-5 and mTOR signals. In addition, galunisertib could block the TGF-β/SMAD pathway in NK cells by inhibiting the TGF-βRI kinase14. We evaluated the expression of p-SMAD2 in NK cells. It was found that the mean fluorescent intensity of p-SMAD2 was decreased after treated by Gal/IL-15@CaLN (Fig. 3G and Supporting Information Fig. S9). The population of TGF-βRI+ NK cells was also decreased with Gal/IL-15@CaLN treatment (Fig. 3H and Supporting Information Figs. S10 and S11). These results indicated that Gal/IL-15@CaLN-mediated activation of NK cells might be contributed to the activation of JAK/STAT and PI3K/mTOR pathways and the inhibition of TGF-β/SMAD pathway.
3.3. Enhanced retention in tumors by Gal/IL-15@CaLN in vivo
The self-adaptive aggregate Gal/IL-15@CaLN was fabricated to improve drug retention in tumors. According to the acidity-responsive properties, the nanoparticles would aggregate in acidic TME and enhance the retention of loaded drugs. To evaluate the capacity of Gal/IL-15@CaLN to improve drug retention in vivo, we treated CT26-tumor bearing BALB/c-ic mice with DiD-loaded Gal/IL-15@CaLN by intravenous administration and examined the fluorescent intensity with an in vivo imaging system (IVIS). Acidity-insensitive nanoparticles Gal/IL-15@LN were set as a control group. Obvious fluorescent signals were detected in tumors of mice in both groups, but the signals in tumors with Gal/IL-15@CaLN treatment were higher than that with Gal/IL-15@LN (Fig. 4A). According to the mean fluorescent intensity kinetics in tumors, the highest accumulation of both nanoparticles was achieved at 24 h after treatments, but the intensity of Gal/IL-15@CaLN was higher than that of Gal/IL-15@LN group. The retention of Gal/IL-15@CaLN in tumor was significantly higher than Gal/IL-15@LN after treated for 120 h (Fig. 4B).
Figure 4.

Biodistribution of Gal/IL-15@CaLN in CT26 tumor-bearing BALB/c-ic mice (A) Fluorescent images of Gal/IL-15@LN and Gal/IL-15@CaLN-treated mice. The mice were intravenously injected at a DiD dose of 0.5 mg/kg. (B) Quantification of average fluorescence intensity at tumors in vivo (n = 3) (C) Ex vivo fluorescence images of major organs and tumors at 24 and 120 h, respectively. (D) Ex vivo quantification of average fluorescence intensity in major organs and tumors at 24 h and (E) at 120 h (n = 3) (F) Examination of Gal/IL-15@LN and Gal/IL-15@CaLN in tumors. Scale bars, 250 μm. Data were expressed as mean ± SD. The statistical significance was displayed by two-sided unpaired Student's t-test. (∗P < 0.05).
3.4. Innate and adaptive immune responses induced by Gal/IL-15@CaLN in vivo
Innate immunity plays a vital role in immune surveillance and killing of tumor cells. Given that NK cells could be activated by Gal/IL-15@CaLN in vitro, we evaluated the immune responses induced by Gal/IL-15@CaLN on CT26 tumor-bearing BALB/c-nu mice. We firstly evaluated the population of NK cells (CD3−CD49b+) and activated NK cells (CD3−CD49b+NKG2D+ or CD3−CD49b+NKp46+) in tumors by flow cytometry (Supporting Information Fig. S12). The percentage of NK cells in tumor tissues was increased after treated by Gal/IL-15@CaLN, which was 3.89- or 3.34-fold as high as that with PBS or Gal/IL-15 treatment, respectively (Fig. 5A and Supporting Information Fig. S13A). Compared with other nanoparticle groups, Gal/IL-15@CaLN improved the infiltration of NK cells most effectively. The NKG2D is an important activating receptor on NK cells, which is responsible for recognizing the NKG2D ligands (e.g., MICA-, MICB-, and UL16-binding proteins) on tumor cells43. It was found that the percentage of NKG2D+ NK cells increased from 12.97 ± 1.26% in PBS group to 35.53 ± 2.48% in Gal/IL-15@LN group, and further increased to 41.43 ± 2.45% in Gal/IL-15@CaLN group (Fig. 5B and Fig. S13B). Similarly, Gal/IL-15@CaLN effectively improved the percentage of NKp46+ NK cells compared to other groups (Fig. 5C and Fig. S13C). These results indicated that Gal/IL-15@CaLN could promote the activation and proliferation of NK cells in tumors. This was attributed to the combinational effects of IL-15 and galunisertib and improved drug retention by the nanoparticles. Inspired by the increase of activating receptors, we next evaluated the function of NK cells to secrete granzyme B and perforin. The population of granzyme B+ and perforin+ NK cells was significantly improved by Gal/IL-15@CaLN (Fig. 5D and E). Although IL-15@CaLN, Gal@CaLN and Gal/IL-15@LN could also enhance the percentage of these NK cells in contrast to PBS or Gal/IL-15 group, higher improvement was achieved by Gal/IL-15@CaLN. These results confirmed that Gal/IL-15@CaLN effectively activated NK cell-mediated innate immunity.
Figure 5.

Antitumor immune responses in vivo (A–E) Immune responses in CT26 tumor-bearing BALB/c-nu mice. (A) Frequency of intratumoral infiltration of NK cells (gating on CD3−CD49b+ cells), (B) NKG2D+ NK cells (gating on CD3−CD49b+NKG2D+ cells), and (C) NKp46+ NK cells (gating on CD3−CD49b+NKp46+ cells) (D) Intratumoral Granzyme B+ NK cells (gating on CD3−CD49b+Granzyme B+ cells), and (E) Perforin+ NK cells (gating on CD3−CD49b+Perforin+ cells) (n = 3) (F–M) Immune responses in CT26 tumor-bearing BALB/c-ic mice. (F) Intratumoral infiltration of NK cells (gating on CD3−CD49b+ cells), (G) NKG2D+ NK cells (gating on CD3−CD49b+NKG2D+ cells), (H) NKp46+ NK cells (gating on CD3−CD49b+ cells), (I) Granzyme B+ NK cells (gating on CD3−CD49b+granzyme B+ cells), (J) Perforin+ NK cells (gating on CD3−CD49b+perforin+ cells), (K) CD8+ T cells (gating on CD3+CD8+ cells), and (L) CD4+ T cells (gating on CD3+CD4+ cells) (n = 3) (M) Ratio of M1 (CD45+F4/80+CD80+)/M2 (CD45+F4/80+CD206+) macrophages in CT26 tumor-bearing BALB/c-ic mice (n = 3). Data were expressed as mean ± SD. The statistical significance was displayed by two-sided unpaired Student's t-test. (∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001).
Immune responses induced by Gal/IL-15@CaLN were also evaluated on CT26 tumor-bearing BALB/c-ic mice. The infiltration of NK cells increased from 5.40 ± 1.07% in PBS group to 24.17 ± 1.36% in Gal/IL-15@CaLN group (Fig. 5F and Supporting Information Fig. S14). Then, to determine the activation of NK cells in mice, we evaluated the NKG2D and NKp46 expression on NK cells. The percentage of NKG2D+ NK cells treated with Gal/IL-15@CaLN was increased to 40.67 ± 2.10%, compared with 7.83 ± 1.28% in PBS group (Fig. 5G and Supporting Information Fig. S15). The percentage of NKp46+ NK cells treated with Gal/IL-15@CaLN was increased to 22.23 ± 2.20% while that in PBS group was only 3.14 ± 0.45% (Fig. 5H and Supporting Information Fig. S16). Then, we evaluated the granzyme B+ and perforin+ NK cells in tumor tissues after different treatments. The proportion of granzyme B+ NK cells was increased from 5.41 ± 0.85% in PBS group to 23.77 ± 0.55% in Gal/IL-15@CaLN group (Fig. 5I and Supporting Information Fig. S17). The proportion of perforin+ NK cells was increased from 2.94 ± 0.63% in PBS group to 16.70 ± 1.77% in Gal/IL-15@CaLN group (Fig. 5J and Supporting Information Fig. S18). These results indicated that Gal/IL-15@CaLN improved the amount and function of NK cells in tumor tissues, thus enhancing the secretion of granzyme B and perforin to kill cancer cells. IL-15 could also induce the activation of T cells44,45. Besides, since TGF-β directly inhibited the proliferation and activation of T cells and the antigen-presenting capacity of antigen-presenting cells (APCs), galunisertib might relieve immunosuppressive effects of TGF-β on T cells46. Moreover, the activation of NK cells also showed a positive effect on T cells and antigen presentation2,47. So we evaluated the immune responses of T cells after treated by different suspensions. The proportion of CD8+ T cells (CD3+CD8+ cells) was increased after treated by drug-loaded nanoparticles comparted to PBS and Gal/IL-15 groups. The population of CD8+ T cells in PBS group was only 1.61 ± 1.26%, which was 18.80 ± 1.90% in Gal/IL-15@CaLN group (Fig. 5K and Supporting Information Fig. S19). For CD4+ T cells (CD3+CD4+ cells), IL-15@CaLN, Gal@CaLN, Gal/IL-15@LN and Gal/IL-15@CaLN induced elevation of CD4+ T cells to over 50% percentage (Fig. 5L and Fig. S19). It indicated that the activation of T cells was attributed to both the activation of NK cells and Gal/IL-15@CaLN. We also determined the polarization of macrophages in tumor tissues (Supporting Information Figs. S20 and S21). The proportion of M1-phenotype macrophages (CD80+CD45+F4/80+) was increased from 6.79 ± 0.94% in PBS group to 18.10 ± 1.31% in Gal/IL-15@CaLN group (Fig. S20 and Supporting Information Fig. S22A). In contrast, the proportion of M2-phenotype macrophages (CD206+CD45+F4/80+) was decreased from 28.83 ± 1.66% in PBS group to 14.17 ± 1.00% in Gal/IL-15@CaLN group (Figs. S21 and S22B). Among the macrophage population, the CD80+ to CD206+ cells ratio was 0.24 ± 0.04 in PBS group, and the ratio was 1.29 ± 0.18 after treated by Gal/IL-15@CaLN (Fig. 5M). These results indicated that Gal/IL-15@CaLN could not only activate NK cells but also boost adaptive T cell immunity for killing cancer cells.
3.5. Inhibition of CT26 tumor growth in BALB/c-nu and BALB/c-ic mice
Cancer immunotherapy has performed great advances in suppressing tumor growth48,49. An IL-15 superagonist ALT-803 has been combined with nivolumab to treat advanced non-small cell lung cancer in a clinical study50. Galunisertib or galunisertib with chemotherapeutics or immune checkpoint inhibitor has been studied in several clinical studies for the treatment of solid tumors51. However, the combination of IL-15 with galunisertib has not been applied in clinic yet. Inspired by the antitumor immune responses induced by Gal/IL-15@CaLN, we first investigated the antitumor effects of Gal/IL-15@CaLN on CT26 tumor-bearing BALB/c-nu mice. The mice were treated with different suspensions for 4 times at a time interval of 2 days (Fig. 6A). The tumor volume and body weight were recorded every two days after the first administration (Fig. 6B and C). Compared to PBS group, Gal/IL-15 showed no significant delay of tumor growth within 14 days. The mice treated with IL-15@CaLN or Gal@CaLN showed moderate therapeutic effects. Improved therapeutic effects were observed in Gal/IL-15@LN and Gal/IL-15@CaLN group, which was attributed to the combination of IL-15 and galunisertib. More significant inhibition of tumor growth was achieved by Gal/IL-15@CaLN than that with Gal/IL-15@LN, which suggested that acidity-mediated self-adaptive aggregation could further improve antitumor efficiency. On Day 14, the tumors were harvested and weighed in all groups (Fig. 6D and Supporting Information Fig. S23). Compared to PBS group, the tumor weight was significantly reduced in Gal/IL-15@CaLN group. The result confirmed that Gal/IL-15@CaLN could significantly suppress tumor growth. The antitumor mechanisms of Gal/IL-15@CaLN were investigated by H&E and TUNEL staining of tumor sections (Supporting Information Figs. S24A and S24B). The antitumor effects of Gal/IL-15@CaLN were attributed to the innate immune response triggered by IL-15 and galunisertib, which could enhance the proliferation and activation of NK cells. After Gal/IL-15@CaLN therapy, the signal of NK cells (CD49b+ cells) was increased in tumor tissues (Fig. S24C). Moreover, H&E staining showed that no significant injury or inflammation was observed in major organs of mice in all groups (Supporting Information Fig. S25). There was no significant difference of body weight change in control and treated groups (Supporting Information Fig. S26). Above results indicated that Gal/IL-15@CaLN induced effective innate immune responses to inhibit tumor even in the absence of T cell immunity.
Figure 6.

Antitumor effects of Gal/IL-15@CaLN in CT26 tumor-bearing BALB/c-nu mice and CT26 tumor-bearing BALB/c-ic mice. The CT26 tumor-bearing BALB/c-nu mice and BALB/c-ic mice were treated with PBS, Gal/IL-15, IL-15@CaLN, Gal@CaLN, Gal/IL-15@LN and Gal/IL-15@CaLN every 3 days for 4 times. The dosage of IL-15 and galunisertib was 30 μg/kg and 15 mg/kg, respectively (A) Schematic illustration of the treatments in CT26 tumor-bearing mice. (B) Tumor growth curves and (C) individual tumor growth curves in CT26 tumor-bearing BALB/c-nu mice with treatments (n = 6) (D) Tumor weight in all groups on Day 14 of antitumor study (n = 6) (E) Tumor growth curves and (F) individual tumor growth curves in CT26 tumor-bearing BALB/c-ic mice with treatments (n = 6) (G) Tumor weight (n = 6) (H) Photographs of tumors on Day 16 of antitumor study in CT26 tumor-bearing BALB/c-ic mice (1# PBS, 2# Gal/IL-15, 3# IL-15@CaLN, 4# Gal@CaLN, 5# Gal/IL-15@LN, 6# Gal/IL-15@CaLN). Scale bars, 1 cm (I) H&E staining. Scale bars, 200 μm. (J) TUNEL staining. Scale bars, 200 μm. Data were expressed as mean ± SD. The statistical significance was displayed by two-sided unpaired Student's t-test. (∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001).
To evaluate the antitumor effects of both innate and adaptive immune responses induced by Gal/IL-15@CaLN, CT26 tumor-bearing BALB/c-ic mice were established for antitumor studies. The tumor growth was significantly inhibited by Gal/IL-15@CaLN in contrast to other control groups (Fig. 6E and F). The tumors were collected and weighed on Day 16. It was found that tumor weight in Gal/IL-15@CaLN group was much lighter than other treatments (Fig. 6G and H). Obvious lysis and apoptosis were observed in tumor tissues with Gal/IL-15@CaLN treatment by H&E and TUNEL staining (Fig. 6I and J). Increased CD49b+ and CD8+ signals were observed in tumor sections after Gal/IL-15@CaLN therapy, indicating the increased infiltration of NK cells and CD8+ T cells in tumors (Supporting Information Fig. S27). Gal/IL-15@CaLN inhibited 82.4% of the tumor growth in CT26 tumor-bearing BALB/c-ic mice, compared with 70.7% of the tumor growth in CT26 tumor-bearing BALB/c-nu mice. These results indicated that Gal/IL-15@CaLN showed better tumor growth inhibition in the presence of adaptive T cell immune responses. Similarly, no significant injury or inflammation was observed in major organs of mice in all groups by H&E staining (Supporting Information Fig. S28). And the body weight of mice was not significantly changed in all groups (Supporting Information Fig. S29).
4. Conclusions
In this study, we have developed self-adaptive aggregate nanoparticle Gal/IL-15@CaLN to co-deliver galunisertib and IL-15 and enhance their retention for boosting NK cells. Since the decomposition of inner CaCO3 nanoparticles was acidity-dependent, the release rate and amount of Ca2+ was highly associated with pH values. The aggregation of Gal/IL-15@CaLN was activated by Ca2+–alginate coordination, hence its aggregation state was also affected by pH values and performed a self-adaptive property. This property could facilitate its aggregation state in normal tissues, TME or even endosomes, leading to varied retention behaviors and drug release features. Compared with nanoparticles without aggregative capacity, Gal/IL-15@CaLN showed higher retention at tumor sites in duration of over 120 h. Synergistic delivery of galunisertib and IL-15 to tumors enhanced the infiltration, activation and functions of NK cells both in vitro and in vivo. On the one hand, IL-15 activated NK cells by binding IL-15 receptor on cell membrane, which improved the expression of p-STAT5 and p-mTOR in cells. On the other hand, galunisertib inhibited p-SMAD2 and suppressed the expression of TGF-βRI on cell membrane, thus blocking the suppressive effects of TGF-β. These changes regulated NK cells by promoting expression of activation receptors including NKG2D and NKp46 and secretion of granzyme B and perforin. Moreover, the mobilization of NK cells also facilitated the antitumor immunity of CD8+ T cells. In vivo antitumor studies showed that NK cell immunity could inhibit CT26 tumor growth without the help of T cells, while more effective antitumor benefits could be achieved by leveraging the synergistic effects between NK cells and T cells. Our approach reported here might provide a clinically transformable strategy to enhance cancer immunotherapy by boosting antitumor NK cell immunity.
Acknowledgments
We thank the National Center for Protein Science for the platform of flow cytometer. Financial supports from the National Natural Science Foundation of China (32170935, 81903548, and 31930066), the Youth Innovation Promotion Association of CAS (2019283, China) and Shandong Provincial Natural Science Foundation (ZR2019PH013, China) are gratefully acknowledged.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2023.02.002.
Contributor Information
Xia Chen, Email: chenxjluedu@163.com.
Dangge Wang, Email: dgwang@simm.ac.cn.
Yaping Li, Email: ypli@simm.ac.cn.
Author contributions
Dangge Wang and Xiangshi Sun designed the project. Xiangshi Sun did the experiment and data analysis. Xiaoxuan Xu, Jue Wang, Xinyue Zhang, Zitong Zhao, Xiaochen Liu and Guanru Wang provided technical helps for the experiment. Lesheng Teng, Xia Chen, Dangge Wang and Yaping Li supervised, wrote and reviewed the final manuscript. All of the authors have read and approved the final manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Appendix A. Supplementary data
The following is the Supplementary data to this article.
References
- 1.Demaria O., Cornen S., Daeron M., Morel Y., Medzhitov R., Vivier E. Harnessing innate immunity in cancer therapy. Nature. 2019;574:45–56. doi: 10.1038/s41586-019-1593-5. [DOI] [PubMed] [Google Scholar]
- 2.Bald T., Krummel M.F., Smyth M.J., Barry K.C. The NK cell-cancer cycle: advances and new challenges in NK cell-based immunotherapies. Nat Immunol. 2020;21:835–847. doi: 10.1038/s41590-020-0728-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y., Wang L.L., Song Q.Q., Ali M., Crowe W.N., Kucera G.L., et al. Intrapleural nano-immunotherapy promotes innate and adaptive immune responses to enhance anti-PD-L1 therapy for malignant pleural effusion. Nat Nanotechnol. 2022;17:206–216. doi: 10.1038/s41565-021-01032-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiossone L., Dumas P.Y., Vienne M., Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. 2018;18:671–688. doi: 10.1038/s41577-018-0061-z. [DOI] [PubMed] [Google Scholar]
- 5.Guillerey C., Huntington N.D., Smyth M.J. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17:1025–1036. doi: 10.1038/ni.3518. [DOI] [PubMed] [Google Scholar]
- 6.Myers J.A., Miller J.S. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18:85–100. doi: 10.1038/s41571-020-0426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim H.S., Kim J.Y., Seol B., Song C.L., Jeong J.E., Cho Y.S. Directly reprogrammed natural killer cells for cancer immunotherapy. Nat Biomed Eng. 2021;5:1360–1376. doi: 10.1038/s41551-021-00768-z. [DOI] [PubMed] [Google Scholar]
- 8.Maskalenko N.A., Zhigarev D., Campbell K.S. Harnessing natural killer cells for cancer immunotherapy: dispatching the first responders. Nat Rev Drug Discov. 2022;21:559–577. doi: 10.1038/s41573-022-00413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J., Lin Y.D., Lin Z., Wei Q., Qian J.Q., Ruan R.J., et al. Stimuli-responsive nanoparticles for controlled drug delivery in synergistic cancer immunotherapy. Adv Sci. 2022;9 doi: 10.1002/advs.202103444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Viel S., Marcais A., Guimaraes F.S., Loftus R., Rabilloud J., Grau M., et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. 2016;9:ra19. doi: 10.1126/scisignal.aad1884. [DOI] [PubMed] [Google Scholar]
- 11.Derynck R., Turley S.J., Akhurst R.J. TGFβ biology in cancer progression and immunotherapy. Nat Rev Clin Oncol. 2021;18:9–34. doi: 10.1038/s41571-020-0403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delconte R.B., Kolesnik T.B., Dagley L.F., Rautela J., Shi W., Putz E.M., et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol. 2016;17:816–824. doi: 10.1038/ni.3470. [DOI] [PubMed] [Google Scholar]
- 13.Marcais A., Cherfils-Vicini J., Viant C., Degouve S., Viel S., Fenis A., et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. 2014;15:749–757. doi: 10.1038/ni.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu B., Zhu X., Kong L., Wang M., Spanoudis C., Chaturvedi P., et al. Bifunctional TGF-β trap/IL-15 protein complex elicits potent NK cell and CD8+ T cell immunity against solid tumors. Mol Ther. 2021;29:2949–2962. doi: 10.1016/j.ymthe.2021.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo J.Y., Liang Y., Xue D.Y., Shen J., Cai Y.Q., Zhu J.K., et al. Tumor-conditional IL-15 pro-cytokine reactivates anti-tumor immunity with limited toxicity. Cell Res. 2021;31:1190–1198. doi: 10.1038/s41422-021-00543-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z., Meng F.H., Zhong Z.Y. Emerging targeted drug delivery strategies toward ovarian cancer. Adv Drug Deliv Rev. 2021;178 doi: 10.1016/j.addr.2021.113969. [DOI] [PubMed] [Google Scholar]
- 17.Dane E.L., Belessiotis-Richards A., Backlund C., Wang J.N., Hidaka K., Milling L.E., et al. STING agonist delivery by tumour-penetrating PEG-lipid nanodiscs primes robust anticancer immunity. Nat Mater. 2022;21:710–720. doi: 10.1038/s41563-022-01251-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren H., Yong J.H., Yang Q.Q., Yang Z., Liu Z.Y., Xu Y., et al. Self-assembled FeS-based cascade bioreactor with enhanced tumor penetration and synergistic treatments to trigger robust cancer immunotherapy. Acta Pharm Sin B. 2021;11:3244–3261. doi: 10.1016/j.apsb.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X.S., Chen Y.J., Li H., Huang N., Jin Q., Ren K.F., et al. Enhanced retention and cellular uptake of nanoparticles in tumors by controlling their aggregation behavior. ACS Nano. 2013;7:6244–6257. doi: 10.1021/nn402201w. [DOI] [PubMed] [Google Scholar]
- 20.Li J., Wang H., Wang Y.Q., Gong X., Xu X.X., Sha X.Y., et al. Tumor-activated size-enlargeable bioinspired lipoproteins access cancer cells in tumor to elicit anti-tumor immune responses. Adv Mater. 2020;32 doi: 10.1002/adma.202002380. [DOI] [PubMed] [Google Scholar]
- 21.Liu R., Hu C., Yang Y.Y., Zhang J.Q., Gao H.L. Theranostic nanoparticles with tumor-specific enzyme-triggered size reduction and drug release to perform photothermal therapy for breast cancer treatment. Acta Pharm Sin B. 2019;9:410–420. doi: 10.1016/j.apsb.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X.D., Chen X.K., Song J., Zhang J.M., Ren X.Z., Zhao Y.L. Size-transformable nanostructures: from design to biomedical applications. Adv Mater. 2020;32 doi: 10.1002/adma.202003752. [DOI] [PubMed] [Google Scholar]
- 23.Yu W.Q., Liu R., Zhou Y., Gao H.L. Size-tunable strategies for a tumor targeted drug delivery system. ACS Cent Sci. 2020;6:100–116. doi: 10.1021/acscentsci.9b01139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang L., Jing D., Jiang N., Rojalin T., Baehr C.M., Zhang D.L., et al. Transformable peptide nanoparticles arrest HER2 signalling and cause cancer cell death. in vivo. Nat Nanotechnol. 2020;15:145–153. doi: 10.1038/s41565-019-0626-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peng J.R., Xiao Y., Yang Q., Liu Q.Y., Chen Y., Shi K., et al. Intracellular aggregation of peptide-reprogrammed small molecule nanoassemblies enhances cancer chemotherapy and combinatorial immunotherapy. Acta Pharm Sin B. 2021;11:1069–1082. doi: 10.1016/j.apsb.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ai X.Z., Ho C.J.H., Aw J.X., Attia A.B.E., Mu J., Wang Y., et al. In vivo covalent cross-linking of photon-converted rare-earth nanostructures for tumour localization and theranostics. Nat Commun. 2016;7 doi: 10.1038/ncomms10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu Q.Y., Sun W.J., Lu Y., Bomba H.N., Ye Y.Q., Jiang T.Y., et al. Tumor microenvironment-mediated construction and deconstruction of extracellular drug-delivery depots. Nano Lett. 2016;16:1118–1126. doi: 10.1021/acs.nanolett.5b04343. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen A., Bottger R., Li S.D. Recent trends in bioresponsive linker technologies of prodrug-based self-assembling nanomaterials. Biomaterials. 2021;275 doi: 10.1016/j.biomaterials.2021.120955. [DOI] [PubMed] [Google Scholar]
- 29.Mura S., Nicolas J., Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mater. 2013;12:991–1003. doi: 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]
- 30.Li Y.N., Zhang C.G., Li G., Deng G.W., Zhang H., Sun Y.B., et al. Protease-triggered bioresponsive drug delivery for the targeted theranostics of malignancy. Acta Pharm Sin B. 2021;11:2220–2242. doi: 10.1016/j.apsb.2021.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Z.S., Wang Y.X., Yao D., Wu J.H., Hu Y.Q., Yuan A.H. Nanoscale coordination polymers induce immunogenic cell death by amplifying radiation therapy mediated oxidative stress. Nat Commun. 2021;12:145. doi: 10.1038/s41467-020-20243-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie L.S., Li J., Wang G.H., Sang W., Xu M.Z., Li W.X., et al. Phototheranostic metal-phenolic networks with antiexosomal PD-L1 enhanced ferroptosis for synergistic immunotherapy. J Am Chem Soc. 2022;144:787–797. doi: 10.1021/jacs.1c09753. [DOI] [PubMed] [Google Scholar]
- 33.Yan J., Wang G.H., Xie L.S., Tian H., Li J., Li B., et al. Engineering radiosensitizer-based metal-phenolic networks potentiate STING pathway activation for advanced radiotherapy. Adv Mater. 2022;34 doi: 10.1002/adma.202105783. [DOI] [PubMed] [Google Scholar]
- 34.He S.Y., Wu L., Li X., Sun H.Y., Xiong T., Liu J., et al. Metal-organic frameworks for advanced drug delivery. Acta Pharm Sin B. 2021;11:2362–2395. doi: 10.1016/j.apsb.2021.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu M.D., Huang L.Q., Zhang W.N., Wang X.C., Geng Y.Y., Zhang Y.H., et al. A transistor-like pH-sensitive nanodetergent for selective cancer therapy. Nat Nanotechnol. 2022;17:541–551. doi: 10.1038/s41565-022-01085-5. [DOI] [PubMed] [Google Scholar]
- 36.Zhu D., Lu Y., Gui L., Wang W.J., Hu X., Chen S., et al. Self-assembling, pH-responsive nanoflowers for inhibiting PAD4 and neutrophil extracellular trap formation and improving the tumor immune microenvironment. Acta Pharm Sin B. 2022;12:2592–2608. doi: 10.1016/j.apsb.2021.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dalheim M.O., Omtvedt L.A., Bjorge I.M., Akbarzadeh A., Mano J.F., Aachmann F.L., et al. Mechanical properties of Ca-saturated hydrogels with functionalized alginate. Gels. 2019;5:23. doi: 10.3390/gels5020023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang C.J., Dong Z.L., Hao Y., Zhu Y.J., Ni J., Li Q.G., et al. Coordination polymer-coated CaCO3 reinforces radiotherapy by reprogramming the immunosuppressive metabolic microenvironment. Adv Mater. 2022;34 doi: 10.1002/adma.202106520. [DOI] [PubMed] [Google Scholar]
- 39.Zheng P., Ding B.B., Shi R., Jiang Z.Y., Xu W.G., Li G., et al. A multichannel Ca2+ nanomodulator for multilevel mitochondrial destruction-mediated cancer therapy. Adv Mater. 2021;33 doi: 10.1002/adma.202007426. [DOI] [PubMed] [Google Scholar]
- 40.Daemi H., Barikani M. Synthesis and characterization of calcium alginate nanoparticles, sodium homopolymannuronate salt and its calcium nanoparticles. Sci Iran. 2012;19:2023–2028. [Google Scholar]
- 41.Morvan M.G., Lanier L.L. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16:7–19. doi: 10.1038/nrc.2015.5. [DOI] [PubMed] [Google Scholar]
- 42.Trapani J.A., Smyth M.J. Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol. 2002;2:735–747. doi: 10.1038/nri911. [DOI] [PubMed] [Google Scholar]
- 43.Huntington N.D., Cursons J., Rautela J. The cancer-natural killer cell immunity cycle. Nat Rev Cancer. 2020;20:437–454. doi: 10.1038/s41568-020-0272-z. [DOI] [PubMed] [Google Scholar]
- 44.Waldmann T.A. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. 2006;6:595–601. doi: 10.1038/nri1901. [DOI] [PubMed] [Google Scholar]
- 45.Mishra A., Sullivan L., Caligiuri M.A. Molecular pathways: interleukin-15 signaling in health and in cancer. Clin Cancer Res. 2014;20:2044–2050. doi: 10.1158/1078-0432.CCR-12-3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gorelik L., Flavell R.A. Transforming growth factor-beta in T-cell biology. Nat Rev Immunol. 2002;2:46–53. doi: 10.1038/nri704. [DOI] [PubMed] [Google Scholar]
- 47.Vivier E., Ugolini S., Blaise D., Chabannon C., Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012;12:239–252. doi: 10.1038/nri3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng P., Ding B.B., Jiang Z.Y., Xu W.G., Li G., Ding J.X., et al. Ultrasound-augmented mitochondrial calcium ion overload by calcium nanomodulator to induce immunogenic cell death. Nano Lett. 2021;21:2088–2093. doi: 10.1021/acs.nanolett.0c04778. [DOI] [PubMed] [Google Scholar]
- 49.Feng X.R., Xu W.G., Li Z.M., Song W.T., Ding J.X., Chen X.S. Immunomodulatory nanosystems. Adv Sci. 2019;6 doi: 10.1002/advs.201900101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wrangle J.M., Velcheti V., Patel M.R., Garrett-Mayer E., Hill E.G., Ravenel J.G., et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018;19:694–704. doi: 10.1016/S1470-2045(18)30148-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamazaki T., Gunderson A.J., Gilchrist M., Whiteford M., Kiely M.X., Hayman A., et al. Galunisertib plus neoadjuvant chemoradiotherapy in patients with locally advanced rectal cancer: a single-arm, phase 2 trial. Lancet Oncol. 2022;23:1189–1200. doi: 10.1016/S1470-2045(22)00446-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.