

Abstract
With over 2 million new cancer cases and over 600,000 cancer-associated deaths predicted in the U.S. for 2022, this life-debilitating disease continuously impacts the lives of people across the nation every day. Therapeutic treatment options for cancer have historically involved chemotherapies to eradicate tumors with cytotoxic mechanisms which can negatively affect the efficacy versus toxicity ratio of treatment. With a need for more directed and therapeutically active options, targeted small-molecule inhibitors and immunotherapies have since emerged to mitigate treatment-associated toxicities. However, aggressive tumors can employ a wide range of defense mechanisms to evade monotherapy treatment altogether, resulting in the recurrence of therapeutically resistant tumors. Therefore, many clinical routines have included combination therapy in which anti-cancer agents are combined to provide a synergistic attack on tumors. Even with approach, maximizing the efficacy of cancer treatment is contingent upon the dose of drug that reaches the site of the tumor, so often therapy is administered at the site of a tumor via localized delivery platforms. Commonly used platforms for localized drug delivery includes polymeric wafers, nanofibrous scaffolds, and hydrogels where drug combinations can be loaded and delivered synchronously. Attaining synergistic activity from these localized systems is dependent on proper material selection and fabrication methods. Herein, we describe these important considerations for enhancing the efficacy of cancer combination therapy through biodegradable, localized delivery systems.
Graphical Abstract
Introduction
Cancer is a disease that has withstood the span of time. From the medicinal descriptions of Hippocrates and ancient Egyptians to the field of modern medicine, cancer has remained despite the clinical routines that have been developed over this time [1]. However, it was not until the 20th century when treatment strategies began advancing exponentially. Through the mid-1900s, cancer treatment evolved to standardized regimens including surgical resection of tumors, chemotherapy, and radiation therapy. Despite the observable therapeutic benefit seen with this routine, many patients are still hindered with off-target and toxic effects [2]. To help mitigate toxic effects, targeted inhibitors have been developed as the knowledge of cancer mechanisms has progressed.
Small molecule targeted inhibitors have emerged in the field of cancer treatment around the turn of the 21st century. These drugs exhibit an improved efficacy in cancer applications by employing unique mechanisms and specifically target aberrant mutations associated to the disease. Targeted inhibitors have the potential to improve treatment options for a range of cancer patients while reducing the frequency of adverse events [3, 4]. The Food and Drug Association (FDA) approved the first of these drugs, Imatinib (a tyrosine kinase inhibitor), in 2001 for the treatment of chronic myelogenous leukemia [5]. As of 2020, nearly 90 small-molecule targeted inhibitors have been approved by regulating agencies like the FDA and the National Medical Products Administration (China) [6].
Despite the improved outcomes associated with small molecule inhibitors, the long-term efficacy of these therapies is limited because they are mainly for specific genetic aberrations [3]. However, tumors oftentimes display various mechanisms to evade specific treatment which contributes to the development of therapeutically resistant and recurrent tumors. Additionally, to elicit an observable therapeutic benefit, high payloads of drug are needed for those with unfavorable pharmacokinetic properties which requires extensive dosing with dependency on patient compliance. Also, some targeted inhibitors have shown to have affinity for other proteins and receptors resulting in off-target effects upon systemic exposure. These challenges can hinder the success of clinical trials leaving many targeted inhibitors left in preclinical development [7].
More recently, immunotherapies have shifted the landscape of cancer treatment by recruiting and activating an immune response against tumors. Tumors inherently employ a wide range of immunosuppressive mechanisms to evade a host-directed attack; however, immunotherapies, such as checkpoint inhibitors, stimulatory factors, and immune cell therapies, can mitigate these defense mechanisms and sequester an immune response against the tumor. Enhancing an innate and/or adaptive immune response via immunotherapy is a strategy referred to as immunomodulation [8]. The clinical efficacy of these therapies has had limited success since they are often administered systemically and prone to off-target toxicities. Additionally, the high genetic plasticity and immunosuppressive microenvironment of tumors can lead to therapeutically resistant tumors if insufficient treatment is not achieved. To ensure treatment eradicates growing tumors, many FDA approved anti-cancer immunotherapies are administered as an adjuvant or neoadjuvant treatment following chemotherapy treatment [9]. For a more detailed review on the clinical landscape of immunotherapies, refer to Akkin et al. [10].
To improve these aforementioned strategies, steps such as optimization of the drug’s physicochemical properties to improve bioavailability, developing nanocarrier systems to increase drug circulation, or using combination therapy for a more active attack on tumors have been explored [11]. Combination therapies, more specifically, can provide further therapeutic benefits over monotherapies by lowering the amount of drugs needed to achieve tumor regression while also delivering a multi-faceted attack to evade therapeutic resistant and recurrent tumor development [12]. More than 20 of these combination therapies have been approved by the FDA for the treatment of various cancers, and over 10,000 ongoing clinical trials are exploring the therapeutic potential of future combination therapies [13, 14].
Evaluating the improved efficacy of combination therapy is oftentimes laborious. Understanding the mechanisms for metastasis and tumor plasticity is only the first step and it is also important to note that the candidate drugs should not have interfering toxicities or similar therapeutic mechanisms to ensure therapeutic resistance is mitigated [15]. Determining the synergistic ratio and optimum timing of combination treatment is critical. Further, characterizing the pharmacokinetic profile upon administration is necessary for validating effective combination treatment. Even after these crucial factors are determined preclinically, many combinations still face challenges in clinical testing. This, in part, can vary with cancer-associated factors that drive tumor progression and lead to thousands of failed combination therapies [16].
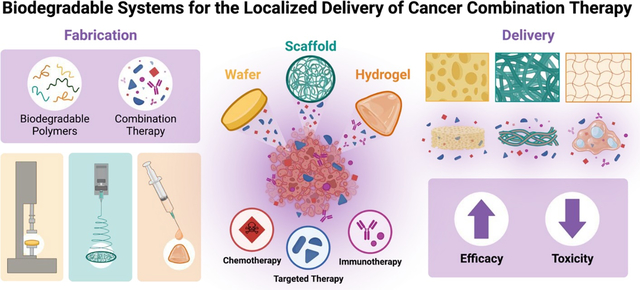
With the increasing advances in cancer screening technology, many tumors, are found in early stages when they are localized in their originating tissues and a strategy that can prevent further disease progression through delivery of combination therapy are localized delivery platforms [17]. Combining drug candidates in a single, localized formulation (Figure 1) can provide added benefits over traditional dosage forms by reducing the chances of off-target toxicities while delivering drug payloads directly to the tumor site. Additionally, many biocompatible polymers with tunable degradation rates can provide opportunities for controlled/sustained release upon administration and relinquish the need for patient compliance throughout treatment. We will review various materials, methods, and design considerations for the development of localized drug delivery platforms using biodegradable and polymeric systems (i.e., wafers, scaffolds, and hydrogels) for combination therapies and review the promising evidence supporting their use in the field of cancer treatment.
Figure 1: Localized Drug Delivery Systems for Cancer Combination Therapy.

Methods for administering localized delivery systems shown on the left (resection cavity implantation, interstitial insertion, and intratumoral injection). Commonly used systems for delivering combination therapies locally with biodegradable polymers include wafers, scaffolds, and hydrogels (schematic of surface morphology shown respectively).
Design Considerations for Localized Drug Delivery Systems
In the context of this review, localized drug delivery systems refer to drug-loaded vehicles that are either implanted or injected at the tumor site. These systems provide therapeutic advantages over systemic or oral administrations by releasing drugs locally to the tumor, significantly reducing systemic exposure. Developing a localized drug delivery systems that can effectively release the loaded drugs requires thorough material consideration regarding the physicochemical compatibility of the polymer with the host as well as sustained release of the active pharmaceutical ingredients (APIs) [18].
To ensure that these therapeutic benefits are achieved, localized delivery strategies should consider the following:
The FDA advises that all materials incorporated into therapeutic implants should be well evaluated for biocompatibility and prevent a foreign body response upon administration [19]. As the immune system is often responsible for triggering these events, the chosen material should not negatively activate the immune system [20]. Similarly, it should be thoroughly tested in vivo for hemocompatibility and thrombogenicity as part of the risk assessment process [19].
The vehicle must have physicochemical compatibility with the therapeutic cargo and ensure the API(s) are stable and not denatured throughout formulation and delivery [21].
The vehicle should provide a controlled release mechanism of drug to the surrounding area. Localized delivery platforms are commonly classified by mechanisms including passive/active diffusion, osmosis driven, or stimuli-activated convection. Furthermore, these systems can be categorized by the site of surgical implantation (e.g., subcutaneous, interstitial, intratumoral) and degradability (i.e., biodegradable, non-biodegradable) [22].
Table 1 summarizes FDA-approved localized drug delivery systems for cancer and cancer-associated therapy with mechanistic, site-specific, and degradation classifications.
Table 1:
FDA-Approved Localized Drug Delivery Systems for Cancer Applications: These systems (implantable medical devices) are distinguished by the loaded therapy, mechanism of drug release, method of implantation, and date of approval.
| Localized Therapy | Drug | Indication | Drug Delivery System | Mechanism of release | Site of Implantation | FDA-approval | Ref. |
|---|---|---|---|---|---|---|---|
| Non-biodegradable | |||||||
| Infusaid Pump | Heparin and Pyrimidine | Hepatic malignancies | Implantable pump | Infusion pump | Intrathecal | 1982 | [23] |
| Intera® 3000 | Floxuridine | Hepatic malignancies | Implantable pump | Infusion pump | Intrathecal | 1996 | [24] |
| Viadur® | Leuprolide | Prostate cancer management | Osmotic pump | Osmosis | Subcutaneous | 2000 | [25] |
| Vantas ™ | Histrelin | Prostate cancer management | Hydrogel Reservoir | Diffusion | Subcutaneous | 2004 | [26] |
| Biodegradable | |||||||
| Gliadel® | Carmustine | High grade gliomas | Wafer | Polymer erosion | Interstitial | 1996 | [27] |
Largely, FDA-approved localized delivery systems for cancer (Table 1) are non-biodegradable systems with pumps and catheter-mediated infusions of a single API. These platforms are unfavorable for localized delivery of combination therapy since their drug release mechanisms do not easily account for the physicochemical properties of multiple APIs (e.g., solubility, hydrophilicity, molecular weight) [21]. Additionally, these non-biodegrading and “permanent” systems may inconvenience patients by requiring routine maintenance and surgical removal once the drug has fully released. For these reasons, this review will focus only on the materials, methods, and considerations for formulating localized combination drug delivery systems that are:
biocompatible and biodegradable;
physicochemically compatible with APIs and provide therapeutic stability throughout formulation and delivery;
characterized with efficient release of each API governed by a controlled mechanism.
Polymeric Systems
Gliadel®, as described in Table 1, is an FDA-approved localized delivery system for the treatment of high-grade gliomas. This therapy releases the chemotherapeutic carmustine (BCNU), a DNA-cross linker, as the drug-loaded polymer wafer gradually degrades in the resected tumor cavity. The creators of Gliadel® used poly [carboxy phenoxy-propane/sebacic acid] anhydride (p[CPP:SA]) to encapsulate 3.85% BCNU (wt./wt.) in the wafer. Initial biocompatibility studies in rodents revealed that this copolymer system would be safe and well-tolerated in human populations. At a molar ratio of 20:80 p[CPP:SA], the wafer promotes an exponential weight loss (in vivo) via hydrolysis and erosion of the wafer. This biphasic destruction of the polymer governs correlating release rates of BCNU with the majority of drug released within the first 10 hours after insertion [28]. Despite the well-characterized properties of localized drug delivery displayed by Gliadel®, the added therapeutic benefit from this system is low for high-grade gliomas. Since BCNU is highly lipophilic, it disperses into the surrounding tissue and rapidly clears from the brain – limiting long-term therapeutic drug release. To achieve sufficient dosing, eight wafers are needed to treat residual tumor cells which might not fit in the tumor cavity due to high wafer rigidity [29]. Moreover, highly malignant tumors can easily become resistant to BCNU treatment if insufficient treatment is achieved [30]. In summary, the limited success of Gliadel® can be attributed to developmental factors such as the chosen polymer system, API, and fabrication method, but also highlights the importance of material selection to the mechanisms of localized drug delivery [31].
Biocompatible polymers are widely used and modified for controlling mechanisms of drug release. These polymers can be further categorized as either biological or synthetic while displaying a wide range of properties that elicit tunable drug releasing mechanisms. Biologically sourced polymers are often biocompatible, but have drawbacks in pharmaceutical development including batch variability, low reproducibility, and risk of immunogenicity. Biosynthetic polymers, however, can be fabricated to display favorable properties similar to biological polymers and overcome these drawbacks through various molecular modifications (e.g., molecular weight adjustments, co-polymerization) [32]. Physicochemical differences between these polymers contributes to various degradation mechanisms including enzymatic reactions, hydrolysis, dissolution, and erosion. Table 2 summarizes commonly used polymers used in drug delivery and correlating physicochemical properties that are critical to their use in pharmaceutical applications.
Table 2: Common Polymers for Drug Delivery Applications.
Polymers indicated by broad classifications including biologically sourced polymers (proteins and saccharides), and biosynthetic polymers. Example(s) of these polymers indicated with associated properties such as molecular weight, soluble solvent systems, degree of hydrophilicity/hydrophobicity, ionic charge, and typical degradation mechanisms. Acronyms: HFP, hexafluoropropylene; TFE, trifluoroethanol; PBS, phosphate buffer saline; EtOH, ethanol; THF, tetrahydrofuran; HFIP, hexafluoro-2-propanol; DMF, dimethylformamide. Example chemical structures taken from Bio Render (Proteins/peptides and Saccharides) and Sigma-Aldrich for the remainder.
| Classification | Examples | Molecular Weight (kDa) | Compatible Solvent System(s) and Solubility | Ionic Charge | Major Degradation Mechanisms | Ref. |
|---|---|---|---|---|---|---|
| Readily Biodegradable Polymers | ||||||
Proteins/Peptides

|
Collagen (I) | 300 | HFP, TFE, PBS/EtOH | Neutral | Enzymatic | [33, 34] |
| Elastin | 60–70 | Water/EtOH (40% alcohol content solubility) | Anionic | Enzymatic | [35, 36] | |
| Resilin | 28.1 | Alkaline (pH 8.2) water/PBS | Neutral | Enzymatic | [37–39] | |
Saccharides

(glucose) |
Chitosan | 50–2000 | Insoluble in aqueous systems, increases in water with decreasing pH (< 6.5) | Cationic | Enzymatic | [33, 40, 41] |
| Alginate | 32–400 | Water and organic solvent insoluble, Increases at lower pH | Anionic | Dissolution | [33, 42, 43] | |
| Cellulose | 30–200 | Insoluble, increases in acidic and alkaline environments | Anionic | Enzymatic, Hydrolysis | [33, 44–46] | |
Polyamino acids
 (PLL)
(PLL)
|
Poly-lysine (PLL) | 70–150 | Polar solvents (water, dimethyl sulfoxide, DMF) | Cationic | Enzymatic, Hydrolysis | [47] |
|
Polyanhydrides
|
Poly-sebacic anhydride (PSA) | 0.1–137 | Organic solvents (dichloromethane), slightly water soluble | Neutral | Surface Erosion | [48, 49] |
Polyesters
 (PLA)
(PLA)
|
Poly-lactic acid (PLA) | 128–152 | Dioxane, acetonitrile, chloroform (water insoluble) | Neutral | Hydrolysis | [50, 51] |
| Polycaprolactone (PCL) | 14 | Methylene chloride, anisole (lower MW aqueous solubility) | Neutral | Enzymatic, Hydrolysis | [52, 53] | |
| Poly-glycolic acid (PGA) | 20–140 | HFIP (low water and organic solvent solubility) | Neutral | Enzymatic, Hydrolysis | [54, 55] | |
| Poly-lactide-co-glycolide (PLGA) | 10–100 | THF, ethyl acetate, dichloromethane (poor water solubility) | Neutral | Hydrolysis | [56, 57] | |
| Polymers with Very Limited Biodegradation | ||||||
Polyethers
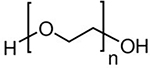 (PEG)
(PEG)
|
Polyethylene glycol (PEG) | 20 | Polar solvents (water, acetone, EtOH) | Neutral | Enzymatic (oxidative) | [58] |
| Polypropylene glycol (PPG) | 0.5–3.5 | Water, acetone, diethyl ether | Neutral | Enzymatic (oxidative) | [58] | |
Polymethacrylates
 (HEMA)
(HEMA)
|
2-Hydroxyethyl Methacrylate p(HEMA) | 2–50 | Organic polar solvents (alcohols), relatively water soluble with increasing MW | Neutral | Dissolution | [59, 60] |
Polyvinyls
 (PVA)
(PVA)
|
Poly-vinyl alcohol (PVA) | 26.3–30 | Polar solvents (water, dimethyl sulfoxide) | Neutral | Enzymatic (oxidation), Hydrolysis | [61, 62] |
Various polymeric properties, such as those listed in Table 2, and the physiological conditions of tumors can contribute to the mechanisms of polymer degradation following surgical insertion. These factors include polymer morphology, molecular weight, melting point, pH/temperature sensitivity, surface charge, and hydrophilic moieties. Additionally, the presence of microorganisms/bacteria, oxidative species, enzymes, and water surrounding tumors can affect the degradation rates [63]. Of note, the polymers listed above differ by the extent to which they can biodegrade under physiological conditions. For example, polyethylene glycol (PEG) is a very common polymer used in drug delivery formulations due to low protein adherence and aqueous solubility [64]. However, biodegradation of PEG alone can only be accomplished in highly oxidative or bacteria-rich environments which causes many mono-PEG formulations to have a high residence time in the body (with relatively non-toxic effects) [65]. Co-polymerization of PEG or other less-physiologically degradable polymers is oftentimes used to, instead, alter degradation rates. A common application of this technique is the chemical modification of poly-lactic acid (PLA) polymers with inserted PEG constituents. This shortens the degradation rate of hydrolysable PLA by increasing its overall hydrophilicity [66]. Copolymerization opportunities are numerous to generate biodegradable systems and provides an opportunity to enhance drug encapsulation and prolong release [67]. For a more detailed review on mechanisms of polymeric biodegradation under in vivo conditions, refer to Murthy et al. [68].
The following sections will discuss localized platforms that apply the favorable properties of polymers to the mechanisms which govern localized release of combination therapies. This includes considerations involved in polymer selection, system fabrication, and tuning drug release mechanisms to optimize the therapeutic efficacy of synergistic drug combinations. Specifically, we will discuss the use of polymeric wafers, scaffolds, and hydrogels (Figure 1), which are widely utilized as localized drug delivery platforms, and can be used to enhance the synergistic effects of loaded drug combinations.
Polymeric Wafers:
Polymeric wafers are common localized drug delivery systems using biodegradable and biocompatible polymers. Fabricating these systems can be relatively simple; drugs are emulsified with polymers in aqueous solutions (Figure 2A), or in solid dispersions under mortar-and-pestle mixing (Figure 2B), to form polymer-drug microparticles. Another commonly used fabrication method is electrospraying (Figure 2C) which generates polymeric particles with size and morphological tunability [69]. Dried polymeric particles as solid dispersions can then then be compressed under pressure to form compact polymer systems in the form of a wafer. This system can be tuned via changing the surface-to-volume ratio and adjusting the compression force (Figure 2D). With a higher surface to volume ratio, drug release rates are altered by changing the surface area available for drug release. Additionally, polymeric wafers can exhibit different structural and surface morphologies for unique drug-releasing profiles in vivo. Polymeric formulations, including wafers, can display different degradation mechanisms in aqueous environments including bulk degradation and surface erosion which are dependent upon scaffold hydrophilicity, water solubility, and enzymatic activity (Figure 2F). As internal and surface structural integrity weakens with water exposure, drug-release rates are influenced by these degradation mechanisms as well as the physicochemical characteristics of the loaded cargo. These systems are highly scalable and compatible with most small-molecule drugs, but often lack feasibility when using biological drugs (e.g., antibodies) or other APIs that are sensitive to mechanical stress [70].
Figure 2: Fabricating and Characterizing Drug-Loaded Polymeric Wafers.

Polymeric wafers for localized drug delivery are highly compatible with small molecule therapies. (A-C) Fabrication methods for generating drug-loaded particles for wafer development including (A) emulsification, (B) mortar-and-pestle mixing, and (C) electrospraying. (D) High-force compression to form polymeric wafer (E) with an example graphic for the surface morphology. (F) Biodegradation mechanisms of wafers can follow surface erosion (red dashed arrow, wafer volume decreases as density remains constant) and bulk degradation (solid blue arrow, wafer volume remains constant but polymer density decreases). (H) Commonly associated drug-release rates (bulk degradation shown in gradual solid blue line and surface erosion as the dashed red line depicting rapid burst release).
As previously described, Gliadel® wafers lack significant efficacy over the standard of care for treating malignant brain tumors with the mono-therapeutic delivery of BCNU. As the polymer degrades, BCNU is quickly washed away from the delivery site, limiting its therapeutic activity in the tumor cavity. To combat rapid BCNU release and clearance, Shapira-Furman et al. described a method of improving the therapy by locally delivering drug payloads in combination with temozolomide (TMZ) in a single, drug-loaded wafer [71]. TMZ is a DNA alkylating agent (chemotherapy) and is given orally as the standard of care for patients with glioblastoma multiforme. Although this standardized treatment has clinical benefits, such as relatively high permeability through the blood brain barrier (BBB), there are various disadvantages of using this drug as a monotherapy including a short half-life of less than 2 hours and risk of off-target toxicities [72]. These authors describe an approach using a biodegradable wafer to improve the efficacy of BCNU Gliadel® wafers and TMZ in a uniform and localized delivery platform.
To optimize the release rates of two therapeutic agents upon localized delivery, the researchers carefully selected an alternative polymer as the bulk material for the polymeric wafer. Poly-lactic-co-glycolic acid (PLGA) was chosen to overcome the hydrophilicity of p[CPP:SA] and subsequent burst release of BCNU as seen with Gliadel®. In contrast, PLGA is much more hydrophobic than p[CPP:SA] and it was hypothesized to have higher structural integrity upon delivery in vivo. Hydrophobic polymers typically display longer degradation rates as the biodegradation and drug release relies on bulk erosion of the polymer more so than surface erosion [73]. Using a layer-by-layer technique in dichloromethane (DCM) TMZ was coated with PLGA in a continuously stirred solution. Once the solvent evaporated, the process was repeated (below the solubility limit of PLGA in DCM) with BCNU to obtain a second polymer/drug layer over the coated TMZ. This double coating process left TMZ and BCNU completely covered in PLGA as a white, solid powder. The product was compressed by 1.5 tons of pressure into a 3 mm by 1 mm wafer with a final loading of 50% TMZ and 3.8% BCNU by weight. Although limited by in vivo methods to assess total BCNU release rates, TMZ was found to release uniformly over 35 days and had no observable difference in release rates when compared to TMZ-only wafers. Additionally, they found in a rodent 9L gliosarcoma model, intertumoral insertion of the combined drug-loaded wafers provided significantly improved survival following treatment relative to the controls with 25% of this treatment group identified as long-term survivors (> 120 days) [71].
Despite the therapeutic advantages seen with this chemotherapeutic combination in PLGA wafers, these methods described are limited by the poor reproducibility and variable drug dispersion upon localized delivery. Thorough characterization is required to ensure that consistent drug-loading is achieved during drug-polymer conjugation and wafer compression. To improve drug loading accuracy, freeze-dried casting and lyophilization has been used to formulate similar drug-loaded polymeric wafers [74]. Additionally, drugs with extreme differences in physicochemical properties may not release synergistically upon wafer bulk degradation therefore increasing the wafer’s porosity by using CO2 sponging may provide for higher uniformity in biodegradation mechanisms and subsequent drug release [75].
Polymeric Scaffolds
Fibrous polymer systems, or scaffolds, are commonly used for biomedical applications since their structure can be made to closely mimic that of the extracellular matrix. These systems can also be applied to localized drug-delivery systems by encapsulating a broad range of therapeutic agents into drug-loaded fibers. Throughout fabrication, polymeric scaffolds can display a variety of drug release rates with a high surface area-to-volume ratio - characteristic of the nano-dispersed fibers. The kinetics of drug-release can be high tunable based on the chosen solvent systems, fabrication parameters, and various environmental factors [76]. These systems can be formulated using a method known as electrospinning, as shown in Figure 3A where a high-voltage field is applied to a continuously expelled spinneret and collection plate. As a polymer/drug solution protrudes from the tip of the spinneret, the high-voltage field allows the forming droplet to overcome surface-tension while forming a conical liquid projection (Taylor Cone). The electrostatic force causes the polymer-drug solution to jet from the spinneret to towards the oppositely charged collection plate as the solution is continuously expelled. As the liquid mixture travels towards the collection plate, the solvent evaporates quickly before drugs can crystallize and results in a solid, drug-loaded polymeric fiber.
Figure 3: Electrospinning Polymeric Nanofibrous Scaffolds for Localized Drug Delivery.

(A) Electrospinning apparatus containing a continuously pumped syringe with a drug/polymer solution. A high-voltage field (V) is applied to the spinneret and collection plate distributing the opposite charges across a specified working distance (WD). Zoomed images depicts the formed Taylor Cone from the tip of the spinneret. (B) Coaxial electrospinning uses a similar set-up (voltage field, collection plate, and working distance not shown) with an additional continuously pumped syringe to form an outer shell and inner core regions of a fiber. The zoomed image shows the convergence of shell and core solutions as it reaches the spinneret tip. (C) These coaxial fibers can also be formed from emulsion electrospinning where dispersed phase converges towards the tip of the spinneret forming the core (zoomed image).
Gurysh et al. used electrospinning methods to fabricate drug-loaded nanofibrous scaffolds using the polymer acetalated dextran (Ace-DEX; Figure 4A). This polysaccharide derivative provides many added benefits in localized delivery systems for cancer treatment including (1) tunable degradation rates (ranging from hours to months) can be achieved by modulating the reaction time of the polymer; (2) acid sensitivity; (3) pH neutral non-toxic degradation products. By using electrospinning methods, the authors individually encapsulated paclitaxel, a microtubule inhibitor (chemotherapy), and everolimus, an mTOR inhibitor (targeted inhibitor), in different Ace-DEX formulations to achieve similar release kinetics of ~3% drug by weight per day (Figure 4E–F). The efficacy of this treatment was assessed in vivo using an orthotopic glioblastoma model (U87-MG) in nude mice. Upon localized co-delivery of the paclitaxel and everolimus Ace-DEX scaffolds in tumor resection cavities, 100% of mice receiving this treatment displayed progression-free survival following treatment. Comparatively, mice receiving the vehicle Ace-DEX control scaffold or individual drug-loaded scaffolds had recurrent tumors (Fig. 4G) [77]. This work demonstrates how fabricating polymeric scaffolds can be applied with combination therapy for treating cancer in a localized delivery system.
Figure 4: Ace-DEX Nanofibrous Scaffolds as a Localized Delivery Platform for Glioblastoma with Synergistic Drug Combination Therapy.

(A) The reaction scheme used to generate Ace-DEX from dextran. Increasing the reaction time increases the polymer backbone coverage with cyclic acetal groups (%CAC). The higher the %CAC, the longer the degradation rate of Ace-DEX. (B-D) Scanning electron micrographs of electrospun Ace-DEX scaffolds containing (B) no drug (blank), (C) 20% wt. paclitaxel (Ace-PTX), and (D) 5% wt. everolimus (Ace-EVR). (E-F) In vitro release studies (in PBS at 37°C) demonstrated zero-order release rates with ~3% per day of drug released from (E) Ace-PTX and (F) Ace-EVR. Data points are mean values with ± standard deviation. (G) Results from in vivo studies using a U87-MG tumor model in nude mice showed 100% of mice with progression free survival following combination treatment with Ace-PTX + Ace-EVR. Redacted with permission from author.
Fabricating nanofibrous polymer systems using coaxial or emulsion electrospinning allows for the encapsulation of multiple APIs into a single polymeric scaffold (Figure 3 B–C). Coaxial electrospinning allows for drug-specific formulations to be combined during fiber formation so that drugs are dispersed within polymer systems with desired release kinetics (Figure 3B). The general process of fabricating these scaffolds is similar to the electrospinning methods previously described; however, coaxial capillary tubes converge at the spinneret tip comprising of distinct polymer solutions forming a core-shell fiber. A general rule to follow regarding drug-polymer solvent selection for each layer is that immiscible solvents are more likely to result in stabilized and distinguishable fiber layers. Although much more difficult to obtain, miscible solvents can also be used to form distinguished layers so long as the transit time of the liquid jet from the spinneret tip to the collector and solvent evaporation time is quicker than the diffusion of the two solvents (determined by the solvent diffusion rate constant) [78]. Adjusting polymer concentrations or viscosity, voltage strength, flow rate, or transient distance can additionally influence fiber layer structure and integrity [79].
Kumar et al. demonstrated the drug-releasing tunability of coaxial nanofiber scaffolds for the co-delivery of synergistic drug combinations against various cancer cells in vitro [80]. 5-fluorouracil (5-FU), an inhibitor of DNA replication (chemotherapy), and curcumin, an anti-inflammatory compound, were coaxially electrospun into polymeric fibers within layers of polyethylene oxide (PEO) and polyethylenimine (PEI). This copolymer system is inherently hydrophilic and susceptible to rapid hydrolytic swelling and erosion. Dissolution of PEO-PEI crosslinks causes a burst release of drug when exposed to aqueous environments. However, chemical cross-linking of PEI molecules with glutaraldehyde vapor decreases the hydrophilicity of PEO-PEI copolymer systems by making the copolymer cross-links less susceptible to hydrolysis and forming a protective diffusion layer around encapsulated drugs [81]. By increasing the chemical cross-linking time of PEI, the coaxial fiber systems displayed proportionally longer degradation rates in vitro. The authors leveraged this physicochemical property of cross-linked copolymer systems to coaxially electrospin 5-FU in a highly concentrated PEO core and curcumin in the chemically crosslinked PEO-PEI shell. These polymer solutions allowed for distinct layering of core and shell regions with amorphous and stable drugs found in each respective layer. In vitro, curcumin and 5-FU released gradually as fibers absorbed surrounding water. Due to the hydrophilicity of the inner core (PEO) layer, the fiber began to expand while driving the initial release of curcumin by diffusion. Subsequently, the aqueous environment dissolves 5-FU (3.85% wt/wt. PEO) from the core and releases it through the expanding cross-linked shell. This dual-release significantly reduced viability of both non-small cell lung cancer (A549) cells and glioblastoma (U87-MG) cells. Additionally, this controlled release allowed for increased apoptosis and necrosis events following treatment of curcumin and 5-FU in tumor cells, highlighting the significance of sustained synergistic activity throughout combinational treatment. Although this treatment was assessed for only 96 hours in vitro, these strategies can be employed to other polymer systems to prolong the delivery and treatment of localized combination therapies against tumors.
A similar approach to generating multi-phase polymeric fibers for drug encapsulation and delivery is through emulsion electrospinning (Figure 3C). This emerging strategy increases the amount of drug options that can be incorporated into polymeric scaffolds such as highly lipophilic drugs, biological compounds, and genetic vectors which also expands the number of therapeutic combinations that can be administered locally via electrospun fibers. Methods for generating these fibers first requires forming an emulsion of two immiscible solvent solutions. By using oil in water or water in oil emulsions with surfactants at various ratios, different sized droplets can be suspended in the bulk, or continuous, phase (5–500nm). Increasing surfactant concentrations can stabilize the droplets while encapsulating various APIs in a dispersed phase [82]. Emulsified polymer-drug solutions can be electrospun using the previously described equipment set-up with slightly different mechanisms for fiber formation. As the single solution flows through the high-voltage field at the spinneret, the bulk phase quickly begins to evaporate, increasing the radial distribution of polymeric viscosity. This forces the dispersed droplets to consolidate at the polymer core while forming a biphasic fiber with the bulk phase polymer as the shell. Adjusting solvent and surfactant ratios, flow rates, voltage intensity, and transient distance towards the collector can all impact fiber stability and core formation during electrospinning emulsions. It is critical to ensure that APIs remain intact and bioactively stable throughout formulating these polymeric scaffold systems to achieve controlled delivery following emulsification [83].
Emulsion electrospinning techniques were adopted by He et al. for the combined delivery of doxorubicin, a DNA intercalator (chemotherapy), and apatinib, a P-glycoprotein inhibitor (targeted inhibitor) [84]. Doxorubicin micelles were self-assembled with copolymerized polycaprolactone (PCL)-PEG and 3-aminophenyl boronic acid in a tumor-targeting copolymer system and stabilized in a water suspension with a glycerin surfactant. This was emulsified in a bulk oil phase containing PLA and free apatinib. Electrospinning the emulsified water-in-oil solution gave rise to coaxial polymeric fibers existing of a hydrophilic core, loaded with doxorubicin micelles, and a hydrophobic PLA shell containing apatinib. Assessing the drug release of the degrading scaffold in vitro revealed that fiber expansion and hydrolysis of the core rapidly released micelles to the surrounding area. With the copolymer conjugation to doxorubicin, the terminal 3-aminophenyl boronic acid could traffic these drug-loaded particles to solid tumors which express sialic acid receptors. Concurrently, gradual expansion of the fiber shell allowed apatinib to be diffuse slowly from PLA and to the tumor. Results from locally delivering these developed scaffolds in mice with multi-drug resistant adenocarcinoma tumors revealed rapid tumor suppression after 21 days and improved survival when compared to control mice. Additionally, less toxic events were observed upon this localized platform when compared to mice receiving free doxorubicin or apatinib injections [85]. These results provide evidence of the therapeutic potential existing in localized drug combination therapy for treating cancer. By formulating nanofibrous polymeric scaffolds that display optimum drug releasing connects in a controlled and sustained manner, tumors can be eradicated efficiently - with lower adverse toxicities - upon localized delivery.
In summary, fabricating these electsopun polymeric scaffolds reveal many opportunities to create drug-releasing fibers with tunable kinetics. These systems offer other added benefits to developing drug carriers such as an increased drug loading, encapsulation efficiency, surface area-to-volume ratio, and scalability throughout formulation. However, finding the optimum fabricating parameters (e.g., solvent system, flow rate, voltage strength, working distance, polymer concentration) to generate stable fibers can be very cumbersome [86]. Furthermore, these parameters can easily hinder drug stability and escape from fibers once delivered locally. Immunotherapeutic antibodies, for example, are highly susceptible to degradation by environmental factors and can easily degrade under electrospinning conditions. Recent efforts of conjugating these therapeutic components to the outer surface of fibers have, instead, been explored to combine immunotherapy with the localized delivery of anti-cancer polymeric scaffolds [87]. Understanding these physicochemical interactions between polymer systems and APIs which govern scaffold degradation and drug release is foundational to achieving effective combination treatment from localized delivery to tumors.
Hydrogels
Hydrogels are another localized delivery platform that can provide a sustained release of loaded drug combinations (Figure 5). These carrier systems consist of a three-dimensional crosslinked networks of polymers (typically hydrophilic) which form gelatinous structures once injected at the tumor site. In situ forming hydrogels can be used to overcome the solubility limits of many drugs (e.g., chemotherapies, targeted molecules) and maintain the stability of bioactive compounds as polymer crosslinks form protective borders around the loaded cargo [88]. Additionally, hydrogels can form dense structures under specific environmental conditions, ensuring that the localized drug delivery is precisely at the tumor. With a low rigidity and physiological compatibility, hydrogels have promising therapeutic potential in cancer treatment while offering reduced off-target toxicities [89].
Figure 5: Fabricating Drug Loaded Hydrogels for Localized Delivery.

(B) Forming the polymeric network can happen spontaneously or in situ via physical or chemical cross-linking. (C) Following exposure to physiological environments hydrogels releases loaded therapeutic cargo in a variety of mechanisms including passive diffusion through pores, mechanical contraction/swelling upon water absorption, hydrolysis of cross-links, polymer dissolution/erosion, and enzymatic cleavage.
Hydrogels, much like that of other polymer systems, can be classified by their physicochemical properties. These classifications include the bases of polymer composition (e.g., monopolymer, copolymer), physical structure (e.g., amorphous, crystalline, mixed), cross-linking (e.g., chemical, physical), physiological response (e.g., thermoresponsive, pH responsive, stimuli-responsive), and overall ionic charge (e.g., cation, anion, nonionic). These properties and hydrogel behavior can be tuned or modified throughout formulation to generate hydrogels with unique degradation rates and drug-releasing kinetics [90]. Due to their high stability and water content, many different classes of drugs can be incorporated within these polymeric systems such as anti-cancer immunotherapies (e.g., antibodies, cell-based therapies) as well as other bioactive agents. It is critical to determine the physicochemical properties of the localized drug-delivery system that will be suitable for effective loading and delivery of cargo, which is in part governed by the synthesis methods.
To synthesize drug-loaded hydrogels, four critical components are needed: polymeric or copolymeric monomers, an initiator, a cross-linker, and loaded APIs. Drug can be added at the time of polymerization or the drug molecules can be later passively added through submerging the hydrogel in a solution of drug at a high concentration (Figure 5A). Table 3 describes the various physical and chemical cross-linking methods used in hydrogel formation along with examples of compatible polymers that are commonly used. It is important to ensure the crosslinked components are mixed at specified ratios to attain the ideal properties of hydrogels [91].
Table 3: Methods for Fabricating Hydrogels.
Various methods used for fabricating chemically and physically cross-linked hydrogels. Each method is described with the mechanism of cross-linking three-dimensional polymeric frameworks. Commonly used polymers that are physiochemically compatible with these processes listed respectively.
| Hydrogel Type | Methods | Description | Examples of Compatible Polymers | Ref(s) |
|---|---|---|---|---|
| Chemically Cross-Linked | Addition Reactions | Bis- and functional groups link via Michael type reactions | Chitosan, Dextran, PEG | [92, 93] |
| Aldehyde Reactions | Hydroxyl or amine groups targeted and become cross-linked via aldehyde | Albumin, Chitosan, Gelatin, PVA | [94] | |
| Condensation Reactions | Amide, hydroxyl, or carboxyl groups are deprotonated to become cross-linking site | Polyamides, Polyesters | [95] | |
| Enzymatic | Substrate-specific activity of enzymes initiates polymeric cross-linking | Albumin, Fibrin, PEG | [96] | |
| Free Radical Polymerization | Photoinitiated vinyl groups become radicalized for polymerized linking | PVA | [97] | |
| Radiation | Electron beam/gamma radiation initiates polymerization with chemical cross-linker | Acrylic acid-co-vinyl acetate, Gelatin, PVA | [94] | |
| Physically Cross-Linked | Heating/Cooling | Rigid helices self-assemble as temperature fluctuates from hot to cold | Carrageenan, Gelatin | [98] |
| Hydrogen Bonding | Lowering the pH of polymer-salt solutions causes H-bonding | Cellulose, Chitosan | [94, 99] | |
| Ionic Interactions | In the presence of free ions, oppositely charged monomers form polyplexes | Alginate, Chitosan, Dextran | [100] | |
| Protein Binding | Polymer and Peptides fused during protein generation | Albumin, Elastin, Polyacrylamide | [94] |
The size, strength, and distribution of polymeric cross-links can influence drug-release rates and mechanisms out of the polymeric system (Figure 5C). Of these mechanisms, passive diffusion can occur with respect to the size and water solubility of an encapsulated drug. When the size of the drug is less than the hydrogel pores and soluble in water, drugs can passively diffuse into the surrounding aqueous environment. As the drug size increases or pore size decreases below the size of the drug, diffusion is limited. When drug is large and pore size small, hydrogels must undergo physical or chemical biodegradation mechanisms to elicit drug-release. Hydrolysis or enzymatic degradation of cross-linked polymers is common to facilitate drug release from the gels. Additionally, cleavable drug-polymer conjugations can be added to these networks to immobilize drugs within the mesh and prolong their release from degrading hydrogels. These drug-polymer modifications, along with others, have been considered throughout hydrogel development allowing for optimized drug-release from these localized delivery platforms [101].
The biocompatible materials and methods used in formulating hydrogels offers vast opportunities to incorporate various biologically active therapies. Additionally, the physiochemical properties of hydrogels allow for a complex organization of drug combinations within the cross-linked framework while displaying drug-specific release mechanisms [102]. Wang et al. demonstrated this concept by first generating a prodrug form of cisplatin, a DNA crosslinker (chemotherapy), with enzymatically cleavable polypeptide moieties. In aqueous environments, the cisplatin prodrug rapidly forms organized hydrogen bonds, entrapping the hydrophobic drug in the core of nanotubes (~10 μm in length). The aqueous solution of the cisplatin (150 μg) loaded nanotubes was mixed with a therapeutic payload of anti-PD-1 antibodies (50 μg), a checkpoint blockade inhibitor (immunotherapy), and injected into the tumors of mice that bore GL-261 glioma or CT-26 colon tumors. Upon exposure to ionically charged species in physiological environments, the nanotubes self-assemble into a hydrogel. The in situ gelation encapsulated anti-PD-1 and cisplatin within the cross-linked nanotube framework with a subsequent, zero-ordered degradation rate (~1.73% per day). In both tumor models, cisplatin and anti-PD-1 were predominately contained within the tumor following release. Cisplatin demonstrated a steady release rate governed by enzymatic cleavage of the prodrug nanotube fibers. Anti-PD-1 slowly diffused out of the degrading hydrogel eliciting a prominent immune response to the local tumor (e.g., increased immune cell infiltration). In both models, this hydrogel treatment provided complete tumor regression and survival in tumor-baring mice, and significantly improved an effective immune response following a rechallenge in surviving mice [103]. These results indicate that the therapeutic efficacy of immunotherapies can be enhanced when administered as combination therapies and delivered locally via hydrogels.
Hydrogels can also be fabricated to release drugs in response to the environmental conditions surrounding tumors. For example, tumors inherently maintain an acidic (pH 6.3–7) microenvironment that can be toxic to healthy tissues. This further protects tumors from infiltrating immune cells and aids in suppressing a cytotoxic immune response [104]. Through the incorporation of pH-sensitive polymers, peptides, and cross-linkers, hydrogels can trigger drug release mechanisms within these acidic environments. This design was employed by Liu et al., where an acid-sensitive polypeptide was used to form drug loaded hydrogels at the site of the tumor. The polypeptide was first synthesized with repeating polar and non-polar amino acids so that hydrogen bonded cross-links can form in acidic conditions. In a concentration and pH dependent manner, peptides spontaneously formed stable hydrogels in neutral environments with physically cross-linked peptides. With a 1:4 mass ratio of paclitaxel to gemcitabine, a DNA intercalator (chemotherapy [105]), these two drugs were encapsulated in a stabilized gel at a physiological pH of 7.4. In vitro release studies revealed that the hydrogel matrix loses structural integrity more rapidly at a pH of 5.8 and ~97% of the hydrogel degraded under these conditions after 1 week. This pH-driven degradation mechanism promoted different release rates of the encapsulated drugs. Gemcitabine rapidly diffused out of the hydrogel due to its hydrophilicity, with nearly 100% of drug released in the first 3 days. Paclitaxel, a much more hydrophobic drug, released more gradually over 1 week in acidic conditions. In vivo studies used mice with orthotopic breast cancer tumors (4T1) that received subcutaneous injection of the combined gemcitabine and paclitaxel hydrogel. This treatment stunted tumor growth much more rapidly than mice receiving free gemcitabine and paclitaxel. Additionally, the released drugs were retained much more efficiently from degraded hydrogels than other delivery forms, reducing systemic exposure to toxic chemotherapies [106]. Using these strategies to make delivery systems responsive to the physiological environment of tumors allows for a more controlled mechanism of combination treatment and drug delivery in cancer.
Hydrogels have also been useful in controlling the delivery and reducing the toxicity of cell-based therapies for cancer. Immunomodulatory cells such as activated dendritic cells and T-cells have shown promising potential as anti-cancer immunotherapy platforms [102, 107]. However, the efficacy of these bioactive therapies is limited due to the extreme sensitivity of cells to exogenous environments. Throughout formulation of anti-cancer vaccines, for example, incorporated immune cells can lose their therapeutic activity before being administered. Additionally, activated immune cells that are introduced systemically can cause a toxic inflammatory response [108]. To mitigate these issues, Yang et al. generated a dendritic cell loaded hydrogel. Additionally, doxorubicin and CpG, an immunostimulatory agent, were made into nanoparticles and co-loaded into the hydrogel to further provide anti-cancer effects [109]. These doxorubicin/CpG nanoparticles were self-assembled following the conjugation of doxorubicin to the PEI linker and electrostatic interactions with CpG. The highly biocompatible polymer cyclodextrin was added in solution to the nanoparticles and dendritic cells to serve as the foundation of the hydrogel matrix. At physiological conditions, the hydrogel forms via physical cross-linking of adjacent hydrogen bonds and forms a three-dimensional framework around the loaded agents which maintains the integrity and viability of cell-based components [110]. Due to the pH-sensitivity of these cross-linked matrices, nanoparticles and cells are released from the swollen hydrogel into the tumor environment. The nanoparticles were found to quickly release and degrade in acidic conditions, exposing tumor cells to the cytotoxic effects of doxorubicin. Subsequently, released CpG was able to interact with loaded dendritic cells and immune cells around the tumor to initiate the immune activation. Once activated, a cascade of events led to evidence of a stronger immune response against tumors. The authors injected this drug and dendritic cell loaded hydrogel intratumorally in mice with colon (C-26) tumors. This treatment led to inhibited tumor growth with an enhanced anti-tumor immune response. Immunosuppressive agents (e.g., regulatory T cells, IL-10) were significantly reduced as the activated dendritic cells infiltrated into the tumor microenvironment. Moreover, systemic toxicities were marginal since doxorubicin, CpG, and activated dendritic cells were retained at the site of the tumor. These results promote hydrogels as an effective localized delivery platform for tumors while highlighting the feasibility of using them to deliver active and therapeutic stable APIs [111].
Although the use of hydrogels as localized drug delivery vehicles has seen success therapeutically, the long-term implications following injection or implantation can be variable [112]. In situ cross-linking hydrogels, for example, can vary in volume and distribution following injection. It is critical to optimize and characterize the cross-linking time so that hydrogels can quickly form at the injection site without syringe clogging or off-target systemic distribution [113]. Additionally, cross-linking mechanisms should not interfere with the stability and therapeutic activity of the loaded APIs. Many catalyst driven chemical cross-linking methods can hinder the stability of live biotherapies, limiting the materials and methods that can be used in fabricating these drug-delivery systems [114]. Moreover, hydrogels inherently have a high-water content which can impair scale-up processes and hydrogel characterization [115]. It is critical to characterize, in vitro and in vivo, the drug encapsulation efficiency, long-term stability, cross-linking mechanisms, biodistribution, degradation, and release-kinetics of hydrogel systems to maximize the efficacy and limit systemic toxicity of this drug-loaded, combination therapy platform.
Emerging Concepts in Combination Local Drug Delivery Systems
To maximize the controllability of localized delivery platforms, external stimuli-responsive materials have gained traction in localized drug-delivery development. Fabricating these “smart” devices can be done using many of the methods previously described for wafers, scaffolds, or hydrogels. Similarly, they can be physically delivered or formed at the site of the tumor; but, they can also be made to mechanistically respond to external stimuli to aid in the formulation and drug release. External stimuli can include photothermal radiation, ultrasound, magnetism, or a combination thereof which physically modifies the carrier’s structure or behavior. Accomplishing stimuli-responsive drug delivery typically involves synthesizing unique polymer systems with structural moieties that react to an external stimulus resulting in cross-linking initiation (as in smart hydrogels) or drug release. For example, adding iron, cobalt, or nickel containing nanoparticles to hydrogel solutions can spatiotemporally cross-link hydrogel injections at quicker rates (as demonstrated by Dai et al.) [116]. Additionally, near-infrared (NIR) absorbing hydrogels containing biocompatible photothermal agents (e.g., dopamine nanoparticles) can increase the rate of drug release upon light absorption [117].
Generating external stimuli-responsive vehicles for combination treatment can oftentimes impair drug stability [118]. Exposing sensitive therapies to highly reactive agents needed for fabricating responsive materials poses the risk of depleting the therapeutic activity of the loaded cargo [119]. When choosing to deliver multiple drugs locally to tumors, it is imperative to first consider the release profiles needed to exhibit a synergistic effect. The platform (i.e., wafer, scaffold, or hydrogel) and various release mechanisms in the tumor environment should then be explored to ensure high compatibility with the chosen APIs. Optimizing the release profiles with various stimuli-responsive moieties should be considered as a final-stage effort to increase the efficacy of localized delivery.
Conclusions and Future Considerations
Localized drug delivery platforms provide an effective solution to the age-long challenge of supplying efficacious cancer treatments to tumors. These polymeric systems can be fabricated into a variety of complex structures that are biocompatible and biodegradable while offering unique mechanisms to release therapies at the site of tumors. The mechanisms of localized drug delivery from polymeric wafers, scaffolds, and hydrogels can mitigate systemic exposure and toxic side effects while maximizing the therapeutic activity in the tumor microenvironment. However, localized delivery platforms still face challenges in reaching clinical translation - many of which are central to poor reproducibility and ineffective drug release [120]. Considerable efforts are needed to optimize the formulation and characterization of these systems to strengthen the clinical feasibility of localized therapies.
A powerful tool that has gained traction in recent years of pharmaceutical development is that of mathematical modeling and machine learning [121, 122]. Researchers can use these strategies to correlate trends, optimize experimental parameters, and predict future outcomes using this computational approach to interpret their results. With an organized data set containing an array of input and output variables, traditional statistic methods such as principal component analysis (PCA) and orthogonal partial least squares (OPLS) can be applied with a computational software to identify statistically significant trends and intervariable relationships [123]. Highly significant trends can be extrapolated as a predictive model for approximating (with immense accuracy) the output from interpreting the known inputs. Stiepel et al., for example, used these methods with a diffusion-erosion model to adequately predict the diffusion coefficients and in vitro release rates of dexamethasone and 3’3’-cyclic guanosine monophosphate-adenosine monophosphate from Ace-DEX nanoparticles; the results of which highlight the utility of machine learning for optimizing development of drug delivery systems [124].
Machine learning has also been incorporated to applications of other localized drug delivery systems with the goal of better understanding drug-polymer interactions. Bannigan et al. used this technique to predict fractional drug release from various biodegradable long-acting injectable formulations. With 43 unique drug-polymer systems (containing PLGA, PLA, and PCL), and nearly 4,000 observed fractional releases, these authors generated a supervised data set to identify a model for predicting drug release from polymeric injectables. Additionally, the authors also used various physicochemical properties of loaded drugs and polymers that could potentially influence release rates as input parameters to distinguish the most influential properties existing within the fitted model. Following a series of performance and validation tests within the supervised data set, the most accurate model was identified as the light gradient boosting model. This model also identified polymer molecular weight and drug molecular weight as the two most influential factors to the observed release rate where higher molecular weights of both drug and polymer correlate to “slow” release rates whereas low molecular weights reveal “fast” release rates [125]. Although these results are limited to in vitro release studies, it demonstrates a useful method to identify general release rates that correlate to various polymeric formulations for localized delivery platforms.
To the best of our knowledge, multi-drug release kinetics from localized drug delivery systems have not been extensively characterized with machine learning strategies. With the infinite number of possible formulations based on drug combinations and polymers, future development strategies may benefit from employing this useful tool to aid in identifying the best polymeric formulations to sustain synergistic activity. Nevertheless, machine learning has been useful for identifying and successfully predicting synergistic drug combinations for a variety of cancers [126, 127] – which is a requisite to developing effective localized combination drug delivery platforms. Many of these strategies, which interpret extensive data sets with countless variables and observations, are limited to making in vitro predictions. Translating future localized therapies to the clinic also requires extensive characterization of release and efficacy from in vivo data. A more thorough approach to assess the physiological relevance and characterization of drug release from these systems is through pharmacokinetic/pharmacodynamic modeling. As demonstrated by Al-Zu’bi et al., this technique was used to assess the efficacy and quantify the drug release of a doxorubicin-loaded implant administered in combination with photothermal therapy [128].
In summary, this review has described various materials and methods used to successfully encapsulate and co-deliver synergistic drug combinations via biodegradable polymer systems and discusses important considerations for optimizing drug release mechanisms. Although opportunities for fine-tuning these mechanisms are numerous, various strategies may compromise the stability and integrity of therapeutic payloads. Improving the future development of localized delivery systems for combination therapy requires extensive drug release characterization that is reproducible and sufficient for achieving synergistic activity against tumors. With the number of FDA-approved combination treatments growing for cancer treatment, localized drug delivery systems have significant therapeutic potential as novel drug delivery platforms for enhancing cancer treatment.
Acknowledgements.
Images made in BioRender.
References
- 1.Anand U, et al. , Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes & Diseases, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeVita VT Jr. and Chu E, A History of Cancer Chemotherapy. Cancer Research, 2008. 68(21): p. 8643–8653. [DOI] [PubMed] [Google Scholar]
- 3.Sun G, et al. , Role of Small Molecule Targeted Compounds in Cancer: Progress, Opportunities, and Challenges. Frontiers in Cell and Developmental Biology, 2021. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Megino-Luque C, et al. , Small-Molecule Inhibitors (SMIs) as an Effective Therapeutic Strategy for Endometrial Cancer. Cancers (Basel), 2020. 12(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen MH, et al. , Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin Cancer Res, 2002. 8(5): p. 935–42. [PubMed] [Google Scholar]
- 6.Zhong L, et al. , Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal Transduction and Targeted Therapy, 2021. 6(1): p. 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sochacka-Ćwikła A, Mączyński M, and Regiec A, FDA-Approved Small Molecule Compounds as Drugs for Solid Cancers from Early 2011 to the End of 2021. Molecules, 2022. 27(7): p. 2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hegmans JPJJ and Aerts JGJV, Immunomodulation in cancer. Current Opinion in Pharmacology, 2014. 17: p. 17–21. [DOI] [PubMed] [Google Scholar]
- 9.Kruger S, et al. , Advances in cancer immunotherapy 2019 – latest trends. Journal of Experimental & Clinical Cancer Research, 2019. 38(1): p. 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akkin S, Varan G, and Bilensoy E, A Review on Cancer Immunotherapy and Applications of Nanotechnology to Chemoimmunotherapy of Different Cancers. Molecules, 2021. 26(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang RX, et al. , Nanomedicine of synergistic drug combinations for cancer therapy – Strategies and perspectives. Journal of Controlled Release, 2016. 240: p. 489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sochacka-Ćwikła A, Mączyński M, and Regiec A, FDA-Approved Small Molecule Compounds as Drugs for Solid Cancers from Early 2011 to the End of 2021. Molecules, 2022. 27(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Q, et al. , Small-molecule inhibitors, immune checkpoint inhibitors, and more: FDA-approved novel therapeutic drugs for solid tumors from 1991 to 2021. Journal of Hematology & Oncology, 2022. 15(1): p. 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu S, et al. , Combination strategies to maximize the benefits of cancer immunotherapy. Journal of Hematology & Oncology, 2021. 14(1): p. 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilad Y, et al. , Drug Combination in Cancer Treatment-From Cocktails to Conjugated Combinations. Cancers (Basel), 2021. 13(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bayat Mokhtari R, et al. , Combination therapy in combating cancer. Oncotarget, 2017. 8(23): p. 38022–38043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolinsky JB, Colson YL, and Grinstaff MW, Local drug delivery strategies for cancer treatment: gels, nanoparticles, polymeric films, rods, and wafers. J Control Release, 2012. 159(1): p. 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pons-Faudoa FP, et al. , Advanced implantable drug delivery technologies: transforming the clinical landscape of therapeutics for chronic diseases. Biomedical Microdevices, 2019. 21(2): p. 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Association Fa.D., Biological evaluation of medical devices - Part 1: Evaluation and testing within a risk management process, Health C.f.D.a.R., Editor. 2020: Dockets Management. [Google Scholar]
- 20.Fayzullin A, et al. , Implantable Drug Delivery Systems and Foreign Body Reaction: Traversing the Current Clinical Landscape. Bioengineering (Basel), 2021. 8(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stewart SA, et al. , Implantable Polymeric Drug Delivery Devices: Classification, Manufacture, Materials, and Clinical Applications. Polymers (Basel), 2018. 10(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quarterman JC, Geary SM, and Salem AK, Evolution of drug-eluting biomedical implants for sustained drug delivery. Eur J Pharm Biopharm, 2021. 159: p. 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bryant DD 3rd, DeWitty RL Jr., and Dennis GC, The Infusaid pump in the management of intractable cancer pain. J Natl Med Assoc, 1987. 79(3): p. 305–11. [PMC free article] [PubMed] [Google Scholar]
- 24.Cavnar M, et al. , Considerations and barriers to starting a new HAI pump program: an international survey of the HAI Consortium Research Network. HPB, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moul JW and Civitelli K, Managing advanced prostate cancer with Viadur (leuprolide acetate implant). Urologic nursing, 2001. 21(6): p. 385–8, 393–4; quiz 395–6. [PubMed] [Google Scholar]
- 26.Shore N, Introducing Vantas: The First Once-Yearly Luteinising Hormone-Releasing Hormone Agonist. European Urology Supplements, 2010. 9(8): p. 701–705. [Google Scholar]
- 27.Dang W, Daviau T, and Brem H, Morphological Characterization of Polyanhydride Biodegradable Implant Gliadel® During in Vitro and in Vivo Erosion Using Scanning Electron Microscopy. Pharmaceutical Research, 1996. 13(5): p. 683–691. [DOI] [PubMed] [Google Scholar]
- 28.Fleming AB and Saltzman WM, Pharmacokinetics of the Carmustine Implant. Clinical Pharmacokinetics, 2002. 41(6): p. 403–419. [DOI] [PubMed] [Google Scholar]
- 29.Panigrahi M, Das PK, and Parikh PM, Brain tumor and Gliadel wafer treatment. Indian J Cancer, 2011. 48(1): p. 11–7. [DOI] [PubMed] [Google Scholar]
- 30.Khair-ul-Bariyah S, Carmustine (gliadel wafers) for fight against brain tumor: A review. International Journal of Research, 2015. 24. [Google Scholar]
- 31.Pena ES, et al. , Design of Biopolymer-Based Interstitial Therapies for the Treatment of Glioblastoma. Int J Mol Sci, 2021. 22(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marin E, Briceño MI, and Caballero-George C, Critical evaluation of biodegradable polymers used in nanodrugs. Int J Nanomedicine, 2013. 8: p. 3071–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baranwal J, et al. , Biopolymer: A Sustainable Material for Food and Medical Applications. Polymers (Basel), 2022. 14(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Law JX, et al. , Electrospun Collagen Nanofibers and Their Applications in Skin Tissue Engineering. Tissue Eng Regen Med, 2017. 14(6): p. 699–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mills CE, Michaud Z, and Olsen BD, Elastin-like Polypeptide (ELP) Charge Influences Self-Assembly of ELP–mCherry Fusion Proteins. Biomacromolecules, 2018. 19(7): p. 2517–2525. [DOI] [PubMed] [Google Scholar]
- 36.Mills CE, Ding E, and Olsen BD, Cononsolvency of Elastin-like Polypeptides in Water/Alcohol Solutions. Biomacromolecules, 2019. 20(6): p. 2167–2173. [DOI] [PubMed] [Google Scholar]
- 37.Li L, et al. , Tunable Mechanical Stability and Deformation Response of a Resilin-Based Elastomer. Biomacromolecules, 2011. 12(6): p. 2302–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McGann CL, Akins RE, and Kiick KL, Resilin-PEG Hybrid Hydrogels Yield Degradable Elastomeric Scaffolds with Heterogeneous Microstructure. Biomacromolecules, 2016. 17(1): p. 128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGann CL, Akins RE, and Kiick KL, Resilin-PEG Hybrid Hydrogels Yield Degradable Elastomeric Scaffolds with Heterogeneous Microstructure. Biomacromolecules, 2016. 17(1): p. 128–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aranaz I, et al. , Chitosan: An Overview of Its Properties and Applications. Polymers (Basel), 2021. 13(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qin C, et al. , Water-solubility of chitosan and its antimicrobial activity. Carbohydrate Polymers, 2006. 63(3): p. 367–374. [Google Scholar]
- 42.Lee KY and Mooney DJ, Alginate: properties and biomedical applications. Prog Polym Sci, 2012. 37(1): p. 106–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tønnesen HH and Karlsen J, Alginate in Drug Delivery Systems. Drug Development and Industrial Pharmacy, 2002. 28(6): p. 621–630. [DOI] [PubMed] [Google Scholar]
- 44.Kabir SMF, et al. , Cellulose-based hydrogel materials: chemistry, properties and their prospective applications. Progress in Biomaterials, 2018. 7(3): p. 153–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou Y, et al. , Molecular weight characterization of cellulose using ionic liquids. Polymer Testing, 2021. 93: p. 106985. [Google Scholar]
- 46.Minnick DL, et al. , Cellulose Solubility in Ionic Liquid Mixtures: Temperature, Cosolvent, and Antisolvent Effects. The Journal of Physical Chemistry B, 2016. 120(32): p. 7906–7919. [DOI] [PubMed] [Google Scholar]
- 47.Kodama Y, et al. , Application of biodegradable dendrigraft poly-l-lysine to a small interfering RNA delivery system. J Drug Target, 2017. 25(1): p. 49–57. [DOI] [PubMed] [Google Scholar]
- 48.Santos CA, et al. , Poly(fumaric–co-sebacic anhydride): A degradation study as evaluated by FTIR, DSC, GPC and X-ray diffraction. Journal of Controlled Release, 1999. 60(1): p. 11–22. [DOI] [PubMed] [Google Scholar]
- 49.Bagherifam S, et al. , Poly(sebacic anhydride) nanocapsules as carriers: effects of preparation parameters on properties and release of doxorubicin. Journal of Microencapsulation, 2015. 32(2): p. 166–174. [DOI] [PubMed] [Google Scholar]
- 50.da Silva D, et al. , Biocompatibility, biodegradation and excretion of polylactic acid (PLA) in medical implants and theranostic systems. Chem Eng J, 2018. 340: p. 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Casalini T, et al. , A Perspective on Polylactic Acid-Based Polymers Use for Nanoparticles Synthesis and Applications. Frontiers in Bioengineering and Biotechnology, 2019. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abdelfatah J, et al. , Experimental Analysis of the Enzymatic Degradation of Polycaprolactone: Microcrystalline Cellulose Composites and Numerical Method for the Prediction of the Degraded Geometry. Materials (Basel), 2021. 14(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bordes C, et al. , Determination of poly(ɛ-caprolactone) solubility parameters: Application to solvent substitution in a microencapsulation process. International Journal of Pharmaceutics, 2010. 383(1): p. 236–243. [DOI] [PubMed] [Google Scholar]
- 54.Budak K, Sogut O, and Aydemir Sezer U, A review on synthesis and biomedical applications of polyglycolic acid. Journal of Polymer Research, 2020. 27(8): p. 208. [Google Scholar]
- 55.Sanko V, et al. , A versatile method for the synthesis of poly(glycolic acid): high solubility and tunable molecular weights. Polymer Journal, 2019. 51(7): p. 637–647. [Google Scholar]
- 56.Gentile P, et al. , An overview of poly(lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. Int J Mol Sci, 2014. 15(3): p. 3640–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Makadia HK and Siegel SJ, Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers (Basel), 2011. 3(3): p. 1377–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kawai F, Biodegradation of Polyethers (Polyethylene Glycol, Polypropylene Glycol, Polytetramethylene glycol, and Others), in Biopolymers Online. [Google Scholar]
- 59.Moghadam MN and Pioletti DP, Biodegradable HEMA-based hydrogels with enhanced mechanical properties. Journal of Biomedical Materials Research Part B: Applied Biomaterials, 2016. 104(6): p. 1161–1169. [DOI] [PubMed] [Google Scholar]
- 60.Weaver JVM, et al. , Stimulus-Responsive Water-Soluble Polymers Based on 2-Hydroxyethyl Methacrylate. Macromolecules, 2004. 37(7): p. 2395–2403. [Google Scholar]
- 61.Chiellini E, et al. , Biodegradation of poly (vinyl alcohol) based materials. Progress in Polymer Science, 2003. 28(6): p. 963–1014. [Google Scholar]
- 62.Gupta D, Jassal M, and Agrawal AK, The electrospinning behavior of poly(vinyl alcohol) in DMSO–water binary solvent mixtures. RSC Advances, 2016. 6(105): p. 102947–102955. [Google Scholar]
- 63.Sung YK and Kim SW, Recent advances in polymeric drug delivery systems. Biomaterials Research, 2020. 24(1): p. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen J, et al. , Biocompatibility studies of poly(ethylene glycol)–modified titanium for cardiovascular devices. Journal of Bioactive and Compatible Polymers, 2012. 27(6): p. 565–584. [Google Scholar]
- 65.Ulbricht J, Jordan R, and Luxenhofer R, On the biodegradability of polyethylene glycol, polypeptoids and poly(2-oxazoline)s. Biomaterials, 2014. 35(17): p. 4848–61. [DOI] [PubMed] [Google Scholar]
- 66.Tanaka M, et al. , Design of biocompatible and biodegradable polymers based on intermediate water concept. Polymer Journal, 2015. 47(2): p. 114–121. [Google Scholar]
- 67.Soni V, et al. , Chapter 11 - Biodegradable Block Copolymers and Their Applications for Drug Delivery, in Basic Fundamentals of Drug Delivery, Tekade RK, Editor. 2019, Academic Press. p. 401–447. [Google Scholar]
- 68.Murthy N, Wilson S, and Sy JC, 9.28 - Biodegradation of Polymers, in Polymer Science: A Comprehensive Reference, Matyjaszewski K and Möller M, Editors. 2012, Elsevier: Amsterdam. p. 547–560. [Google Scholar]
- 69.Steipel RT, et al. , Electrospray for generation of drug delivery and vaccine particles applied in vitro and in vivo. Materials Science and Engineering: C, 2019. 105: p. 110070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kamaly N, et al. , Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem Rev, 2016. 116(4): p. 2602–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shapira-Furman T, et al. , Biodegradable wafers releasing Temozolomide and Carmustine for the treatment of brain cancer. Journal of Controlled Release, 2019. 295: p. 93–101. [DOI] [PubMed] [Google Scholar]
- 72.Tosoni A, et al. , Relapsed Glioblastoma: Treatment Strategies for Initial and Subsequent Recurrences. Curr Treat Options Oncol, 2016. 17(9): p. 49. [DOI] [PubMed] [Google Scholar]
- 73.Gopferich A and Langer R, Modeling of polymer erosion. Macromolecules, 1993. 26(16): p. 4105–4112. [Google Scholar]
- 74.Boateng JS, et al. , Characterisation of freeze-dried wafers and solvent evaporated films as potential drug delivery systems to mucosal surfaces. Int J Pharm, 2010. 389(1–2): p. 24–31. [DOI] [PubMed] [Google Scholar]
- 75.Jacob J, et al. , Biopolymer based nanomaterials in drug delivery systems: A review. Materials Today Chemistry, 2018. 9: p. 43–55. [Google Scholar]
- 76.Prabaharan M, Jayakumar R, and Nair SV, Electrospun Nanofibrous Scaffolds-Current Status and Prospects in Drug Delivery, in Biomedical Applications of Polymeric Nanofibers, Jayakumar R and Nair S, Editors. 2012, Springer Berlin Heidelberg: Berlin, Heidelberg. p. 241–262. [Google Scholar]
- 77.Graham-Gurysh EG, et al. , Synergistic drug combinations for a precision medicine approach to interstitial glioblastoma therapy. J Control Release, 2020. 323: p. 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Han D and Steckl AJ, Coaxial Electrospinning Formation of Complex Polymer Fibers and their Applications. ChemPlusChem, 2019. 84(10): p. 1453–1497. [DOI] [PubMed] [Google Scholar]
- 79.Lu Y, et al. , Coaxial electrospun fibers: applications in drug delivery and tissue engineering. WIREs Nanomedicine and Nanobiotechnology, 2016. 8(5): p. 654–677. [DOI] [PubMed] [Google Scholar]
- 80.Uday Kumar S, et al. , Differentially cross-linkable core–shell nanofibers for tunable delivery of anticancer drugs: synthesis, characterization and their anticancer efficacy. RSC Advances, 2014. 4(72): p. 38263–38272. [Google Scholar]
- 81.Gölander C-G and Eriksson JC, ESCA studies of the adsorption of polyethyleneimine and glutaraldehyde-reacted polyethyleneimine on polyethylene and mica surfaces. Journal of Colloid and Interface Science, 1987. 119(1): p. 38–48. [Google Scholar]
- 82.Ye C and Chi H, A review of recent progress in drug and protein encapsulation: Approaches, applications and challenges. Materials Science and Engineering: C, 2018. 83: p. 233–246. [DOI] [PubMed] [Google Scholar]
- 83.Nikmaram N, et al. , Emulsion-based systems for fabrication of electrospun nanofibers: food, pharmaceutical and biomedical applications. RSC Advances, 2017. 7(46): p. 28951–28964. [Google Scholar]
- 84.Luraghi A, Peri F, and Moroni L, Electrospinning for drug delivery applications: A review. Journal of Controlled Release, 2021. 334: p. 463–484. [DOI] [PubMed] [Google Scholar]
- 85.He Y, et al. , Programmable Codelivery of Doxorubicin and Apatinib Using an Implantable Hierarchical-Structured Fiber Device for Overcoming Cancer Multidrug Resistance. Small, 2019. 15(8): p. 1804397. [DOI] [PubMed] [Google Scholar]
- 86.Cavo M, et al. , Electrospun nanofibers in cancer research: from engineering of in vitro 3D cancer models to therapy. Biomaterials Science, 2020. 8(18): p. 4887–4905. [DOI] [PubMed] [Google Scholar]
- 87.Li L, et al. , Electrospun Fibers Control Drug Delivery for Tissue Regeneration and Cancer Therapy. Advanced Fiber Materials, 2022. [Google Scholar]
- 88.Norouzi M, Nazari B, and Miller DW, Injectable hydrogel-based drug delivery systems for local cancer therapy. Drug Discovery Today, 2016. 21(11): p. 1835–1849. [DOI] [PubMed] [Google Scholar]
- 89.Fan D. y., Tian Y, and Liu Z.-j., Injectable Hydrogels for Localized Cancer Therapy. Frontiers in Chemistry, 2019. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ma J, et al. , Hydrogels for localized chemotherapy of liver cancer: a possible strategy for improved and safe liver cancer treatment. Drug Deliv, 2022. 29(1): p. 1457–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mohamed M, Hydrogel Preparation Technologies: Relevance Kinetics, Thermodynamics and Scaling up Aspects. Journal of Polymers and the Environment, 2019. 27. [Google Scholar]
- 92.FitzSimons TM, Anslyn EV, and Rosales AM, Effect of pH on the Properties of Hydrogels Cross-Linked via Dynamic Thia-Michael Addition Bonds. ACS Polymers Au, 2022. 2(2): p. 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Quadrado RFN, et al. , Chitosan-based hydrogel crosslinked through an aza-Michael addition catalyzed by boric acid. International Journal of Biological Macromolecules, 2021. 193: p. 1032–1042. [DOI] [PubMed] [Google Scholar]
- 94.Ali F, et al. , Emerging Fabrication Strategies of Hydrogels and Its Applications. Gels, 2022. 8(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ray D, et al. , Comparative delivery of Diltiazem hydrochloride through synthesized polymer: Hydrogel and hydrogel microspheres. Journal of Applied Polymer Science, 2010. 116(2): p. 959–968. [Google Scholar]
- 96.Teixeira LS, et al. , Enzyme-catalyzed crosslinkable hydrogels: emerging strategies for tissue engineering. Biomaterials, 2012. 33(5): p. 1281–90. [DOI] [PubMed] [Google Scholar]
- 97.Matsumoto A, Free-radical crosslinking polymerization and copolymerization of multivinyl compounds, in Synthesis and Photosynthesis. 1995, Springer Berlin Heidelberg: Berlin, Heidelberg. p. 41–80. [Google Scholar]
- 98.Varghese JS, Chellappa N, and Fathima NN, Gelatin-carrageenan hydrogels: role of pore size distribution on drug delivery process. Colloids Surf B Biointerfaces, 2014. 113: p. 346–51. [DOI] [PubMed] [Google Scholar]
- 99.Yu H, et al. , A Hydrogen Bonds-Crosslinked Hydrogels With Self-Healing and Adhesive Properties for Hemostatic. Frontiers in Bioengineering and Biotechnology, 2022. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lu L, et al. , The Formation Mechanism of Hydrogels. Curr Stem Cell Res Ther, 2018. 13(7): p. 490–496. [DOI] [PubMed] [Google Scholar]
- 101.Li J and Mooney DJ, Designing hydrogels for controlled drug delivery. Nat Rev Mater, 2016. 1(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu Y, et al. , Injectable Hydrogel as a Unique Platform for Antitumor Therapy Targeting Immunosuppressive Tumor Microenvironment. Front Immunol, 2021. 12: p. 832942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang F, et al. , Supramolecular prodrug hydrogelator as an immune booster for checkpoint blocker-based immunotherapy. Sci Adv, 2020. 6(18): p. eaaz8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lee SH and Griffiths JR, How and Why Are Cancers Acidic? Carbonic Anhydrase IX and the Homeostatic Control of Tumour Extracellular pH. Cancers (Basel), 2020. 12(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Toschi L, et al. , Role of gemcitabine in cancer therapy. Future Oncology, 2005. 1(1): p. 7–17. [DOI] [PubMed] [Google Scholar]
- 106.Liu Y, et al. , pH-Sensitive Peptide Hydrogels as a Combination Drug Delivery System for Cancer Treatment. Pharmaceutics, 2022. 14(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xie Z, et al. , Polymer-based hydrogels with local drug release for cancer immunotherapy. Biomedicine & Pharmacotherapy, 2021. 137: p. 111333. [DOI] [PubMed] [Google Scholar]
- 108.Ventola CL, Cancer Immunotherapy, Part 2: Efficacy, Safety, and Other Clinical Considerations. P t, 2017. 42(7): p. 452–463. [PMC free article] [PubMed] [Google Scholar]
- 109.Yang A, et al. , Doxorubicin/CpG self-assembled nanoparticles prodrug and dendritic cells co-laden hydrogel for cancer chemo-assisted immunotherapy. Chemical Engineering Journal, 2021. 416: p. 129192. [Google Scholar]
- 110.Liu J, et al. , Cyclodextrin-Containing Hydrogels: A Review of Preparation Method, Drug Delivery, and Degradation Behavior. Int J Mol Sci, 2021. 22(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim J, et al. , Injectable Hydrogel-Based Combination Cancer Immunotherapy for Overcoming Localized Therapeutic Efficacy. Pharmaceutics, 2022. 14(9): p. 1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Narayanaswamy R and Torchilin VP, Hydrogels and Their Applications in Targeted Drug Delivery. Molecules, 2019. 24(3): p. 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Parhi R, Cross-Linked Hydrogel for Pharmaceutical Applications: A Review. Adv Pharm Bull, 2017. 7(4): p. 515–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hu W, et al. , Advances in crosslinking strategies of biomedical hydrogels. Biomaterials Science, 2019. 7(3): p. 843–855. [DOI] [PubMed] [Google Scholar]
- 115.Almawash S, et al., Current and Future Prospective of Injectable Hydrogels—Design Challenges and Limitations. Pharmaceuticals, 2022. 15(3): p. 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dai G, et al. , Catechol-metal coordination-mediated nanocomposite hydrogels for on-demand drug delivery and efficacious combination therapy. Acta Biomater, 2021. 129: p. 84–95. [DOI] [PubMed] [Google Scholar]
- 117.GhavamiNejad A, et al. , pH/NIR Light-Controlled Multidrug Release via a Mussel-Inspired Nanocomposite Hydrogel for Chemo-Photothermal Cancer Therapy. Sci Rep, 2016. 6: p. 33594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yang Y, et al. , Smart materials for drug delivery and cancer therapy. VIEW, 2021. 2(2): p. 20200042. [Google Scholar]
- 119.Lu Q, et al. , Magnetic Polymer Composite Particles: Design and Magnetorheology. Polymers (Basel), 2021. 13(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Abdelkader H, et al. , Polymeric long-acting drug delivery systems (LADDS) for treatment of chronic diseases: Inserts, patches, wafers, and implants. Advanced Drug Delivery Reviews, 2021. 177: p. 113957. [DOI] [PubMed] [Google Scholar]
- 121.Staszak M, et al. , Machine learning in drug design: Use of artificial intelligence to explore the chemical structure–biological activity relationship. WIREs Computational Molecular Science, 2022. 12(2): p. e1568. [Google Scholar]
- 122.Boso DP, et al. , Drug delivery: Experiments, mathematical modelling and machine learning. Computers in Biology and Medicine, 2020. 123: p. 103820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jiang T, Gradus JL, and Rosellini AJ, Supervised Machine Learning: A Brief Primer. Behav Ther, 2020. 51(5): p. 675–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Stiepel RT, et al. , A predictive mechanistic model of drug release from surface eroding polymeric nanoparticles. Journal of Controlled Release, 2022. 351: p. 883–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bannigan P BZ, Hickman R, Aldeghi M, Häse F, Aspuru-Guzik A, et al. , Machine Learning Models to Accelerate the Design of Polymeric Long-Acting Injectables. ChemRxiv., 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kuenzi BM, et al. , Predicting Drug Response and Synergy Using a Deep Learning Model of Human Cancer Cells. Cancer Cell, 2020. 38(5): p. 672–684.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Anderson E, et al. , Synergistic drug combinations and machine learning for drug repurposing in chordoma. Sci Rep, 2020. 10(1): p. 12982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Al-Zu’bi M and Mohan A, Modelling of combination therapy using implantable anticancer drug delivery with thermal ablation in solid tumor. Sci Rep, 2020. 10(1): p. 19366. [DOI] [PMC free article] [PubMed] [Google Scholar]