

Abstract
Almost all biological reactions are pH dependent and understanding the origin of pH dependence requires knowledge of the pKa’s of ionizable groups. Here we report a new edition of PKAD, the PKAD-2, which is a database of experimentally measured pKa’s of proteins, both wild type and mutant proteins. The new additions include 117 wild type and 54 mutant pKa values, resulting in total 1742 experimentally measured pKa’s. The new edition of PKAD-2 includes 8 new wild type and 12 new mutant proteins, resulting in total of 220 proteins. This new edition incorporates a visual 3D image of the highlighted residue of interest within the corresponding protein or protein complex. Hydrogen bonds were identified, counted, and implemented as a search feature. Other new search features include the number of neighboring residues <4A from the heaviest atom of the side chain of a given amino acid. Here, we present PKAD-2 with the intention to continuously incorporate novel features and current data with the goal to be used as benchmark for computational methods.
Keywords: electrostatics, computational biophysics, Poisson-Boltzmann equation, hydrogen bonds, surface area
1. Introduction
Ionizable residues play an integral part in the structure, function, and solubility of monomeric proteins and oligomeric protein complexes, and are the key factor contributing to pH-dependence of biological processes (1; 2; 3; 4). Undoubtably salt-bridges, a pair of oppositely charged amino acids, are frequently found to be essential for structural integrity of macromolecules (5; 6; 7), protein-protein (8; 9), protein-RNA (10) and protein-DNA (11) interactions. However, their contribution to the above-mentioned phenomena depends on their ionization state, which requires knowledge of their pKa’s and the corresponding pH of the water phase (12; 13). Thus, the interplay between solution pH and pKa’s of ionizable groups results in pH-dependence of the reaction. The pH at which the process or the reaction are optimal is termed pH-optimum (14; 15; 16), and it was shown that pH-optimum of protein stability and protein activity are correlated (16).
Many pH-dependent processes are documented in literature, both experimentally and computationally. It was suggested that proteins participating in pH regulation can be classified into two categories; positive and negative pH-regulators (17), and such proteins participating in melanosome formation have distinctive different pH-optimum (18). It should be noted that the frequently of protonation states in unbound and bound states are not the same, resulting in proton uptake/release upon macromolecular binding (19; 20; 21). The pH-dependent phenomena are frequently coupled with ligand binding and conformational dynamics (22; 23) . Furthermore, the presence of a lipid membrane affects the proton-coupled conformational dynamics (24), peptide binding (25; 26; 27), and pKa’s of membrane proteins (28; 29; 30; 31). All these examples indicate the importance of pKa’s of ionizable groups in various processes in molecular biology. However, experimental measurements are time-costly and not applicable in genome-scale investigations, and computational methods need large sets of experimentally determined pKa’s for either benchmarking or training.
The most widely used and accurate method for experimentally determining pKa’s of ionizable restudies is the multidimensional nuclear magnetic resonance (NMR) spectroscopy (32; 33). Other methods have been used for measuring pKa’s such as, potentiometric titration, calorimetry, reaction kinetics, spectrelectrophoresis, UV electrophoresis, and high-performance liquid chromatography, but NMR is considered to be superior due to the typical 0.1 pKa unit error (34; 35; 36; 37).
There are two distinctive computational approaches for predicting pKa’s: approaches that do not require training or adjustment, and approaches that require training or optimization of adjustable parameters. For first cases methods, one needs large set of experimentally measured pKa’s to benchmark their accuracy; for the second case of methods, a large set of experimentally measured pKa’s is needed to perform the training or obtaining optimal values of adjustable parameters. Constant-pH Molecular Dynamics Simulation (CpHMD) (38; 39) represent the first class of methods and does not require training. It considers ionization changes simultaneously with conformational changes in MD or Monte Carlo simulations (40; 41). Continuum electrostatics, Poisson-Boltzmann, or Generalized Born based approaches, use static structures which require optimization of the internal dielectric “constant” value (42) or Gaussian-based representation of atomic density (43; 44). Other approaches include Generalized-Born or Debye-Huckel approximation which are based on the chemical properties of residues near ionizable groups (45; 46; 47; 48). Recently a very fast machine learning protocol was developed (49) and database of more than 12M precomputed pKa values were reported (50).
This motivated us to compile experimentally determined pKa’s into a database, the PKAD database (51). The initial version of the PKAD contained 1350 pKa’s of wild type proteins and 232 pKa’s of mutant proteins, so in total 1582 experimentally measured pKa’s. The database also provides 3D structure of the corresponding proteins and several search options. Over the years, the PKAD became a valuable resource for testing and training in the field of pKa’s (52; 53; 54). Due to the continued research interest in ionizable residues pKa’s and the expansion of experimental data, it is imperative to build upon the database. At this time, we present PKAD-2, an upgrade of the pKa database PKAD, with the addition of experimental data points and new features. The PKAD-2 database contains 1456 pKa values for 163 wild-type proteins (117 new data points compared with PKAD) and 286 pKa values for 57 mutant proteins (54 new data points compared with PKAD). The new data was acquired from current literature. In addition to providing experimentally measured pKa’s, the PKAD-2 contains an interactive 3D model of the protein or protein complex, with the ionizable residue of interest visualized for every data point. The new search features include: the number of contacting residues <4A from the side chain of the residue of interest, and the number of hydrogen bonds (HB) associated with the residue of interest, organized by arrangement. Advanced searches for number of hydrogen bonds for each residue are divided into subcategories; #HB Side Chain-Side Chain (sidechain-sidechain), #HB Side Chain(Donor)-Back Bone (Residue of interests side chain acting as donor of hydrogen-Backbone of another residue), #HB Side Chain-Back Bone(Donor) (Residue of interest Backbone is donating hydrogen to another residues side chain), #HB Side Chain(A)-Back Bone (Residue of interest is accepting hydrogen from the Backbone of another residue), #HB Side Chain-Back Bone(A) (Backbone of residue of interest is accepting hydrogen from side chain of another residue), and #HB Back Bone-Back Bone (Number of hydrogen bonds between back of residue of interest and backbone of another residue). The PKAD-2 database is accessible from http://combio.clemson.edu/ lab/software/5/.
Consistent with PKAD database, PKAD-2, lists the associated Protein Data Bank (PDB) (55) ID, the error (uncertainly) in pKa’s measurements, salt concentration, pH range and temperature used for pKa’s measurement along with computed relative solvent accessibility surface area (%SASAs). The database contains pKa values for buried and exposed resides. The %SASA provided in the database can aid the user in filtering residues by the degree of exposure, while the new 3D image display will contribute to the visualization of a desired residue within the structure. Number of hydrogen bonds is an advanced search feature and provides integral information about the residue within the structure, as well as the proteins secondary and tertiary structure. If a user is interested in the pKa’s of residues within a particular secondary structure, the search provides the subcategory (#HB Back Bone- Back Bone) to filter all residues with this interaction. Neighboring residues contribute greatly to pKa’s and thus PKAD-2 provides a list of neighboring residues within 4A of the side chain, as well as grants the user the ability to sift through data points by number of neighboring residues, within this distance. This calculated data provides the user with information pertaining to the microenvironment of a residue and what closely located neighbors may interact or affect its pKa. Our goal is to provide an up to date compilation of pKa values, number of hydrogen bonds, and number of contacting residues for corresponding 3D structures to assist in the development and benchmarking of computational approaches to predict pKa’s.
2. Database Development
The PKAD-2 database contains 1456 pKa values for 163 wild-type proteins and 286 pKa values for 57 mutant proteins. All the entries from the previous development, the PKAD database, were retained and new entries were added. The new data was acquired from published literature (52–54; 56–72). This resulted in the following distribution: among the experimental pKa’s listed in the database 31% are Glu, 28% Asp, 12% Lys and 21% His residues. Sparce data points for Arg, Tyr , Cys, N and C -termini were available. The pKa’s of Ser and Arg resides are high and therefore difficult to determine by titration due the denaturing of protein at such an alkaline pH, leading to few, if any experimental datapoints (73).
The number of hydrogen bonds and neighboring residues, for a specific amino acid, within its subunit was calculated using ChimeraX-1.4 (74). To be counted, the bond was required to classified as a strong hydrogen bond. This was defined by the distance between the proton acceptor and donor to be between 1.6A to 2.4A or the distance from heavy atoms to be <=3A apart. Data analyzed was broken up into categories based on the roles and structural level of the hydrogen bond. For instance, a hydrogen bond between two side chains and one between the backbones of two residues were listed in separate categories. Neighboring residues were defined as residues of the same subunit that could be found less than 4A from the heavy atom in the side chain of the residue of interest.
The application comprises two layers: a backend layer and a frontend layer. The backend layer is built on top of MongoDB (www.mongodb.com), a non-relational database management system that accommodates archiving a large amount of structured pKa data. A Node.js (nodejs.org/en/) server communicates with the database server to generate a cached version of data to be used by the frontend application, reducing the overall server load in the production environment. The frontend layer is built with React (reactjs.org/), Typescript (www.typescripting.org/) & iCn3D, which loads the cached JSON file, renders it on a table, and provides an interface for users to sort and query the data (75). It also implements the iCn3d library for an in-house interactive 3D viewer, which allows rendering of the 3D structure of protein and its residue.
The application presents two distinct tabs allowing users to navigate between the pKa’s of ionizable residues in wild-type and mutant proteins. The search field allows for querying table data based on the PDB ID, residue name, or residue ID. Users can also filter the table data with the pKa range, %SASA range, number of contacting residues, and types of hydrogen bonds. Each PDB ID links to an interactive 3D structure viewer allowing users to view, zoom, and highlight the residue and the protein structure within the website.
3. Results and Discussions
Wild type proteins
Summarized in Table 1 is the average pKa’s for all wild type protein residues and the number of values for each residue type. Out of the 1456 wild type resides, the most experimental pKa values came from Asp, His, Glu, and Lys (Table 1). The ranges of measured pKa’s for each amino acid type varied significantly, with Asp and Cys showing the largest range. Furthermore, the Table 1 provides the intrinsic pKa’s as well which allows for estimation of the effect of protein microenvironment on the corresponding pKa’s. It was found that residues with the greatest % difference away from their intrinsic values are Glu (44.6%), Cys (34.0%), and His (42.3%). Figure 1 displays the distribution of pKa values by amino acid type for the four residues with a substantial amount of experimental data.
Table 1.
Summary of experimentally measured 1456 residue-specific wild-type pKa values collected from literature
| Residue ID | No. of measurements | Average pKa | Lowest pKa | Highest pKa | Intrinsic pKa |
|---|---|---|---|---|---|
|
| |||||
| ASP | 450 | 3.56 | 0.5 | 9.9 | 3.9 |
| GLU | 455 | 4.13 | 2.1 | 7.2 | 6.5 |
| HIS | 264 | 6.61 | 2.5 | 9.19 | 4.3 |
| LYS | 168 | 10.69 | 6.5 | 12.12 | 9.8 |
| TYR | 50 | 10.50 | 9.14 | >12.5 | 10.4 |
| CYS | 22 | 6.10 | 2.88 | >14.9 | 8.6 |
| N-term | 22 | 7.64 | 6.91 | 9.14 | 8.0 |
| C-term | 25 | 3.15 | 2.4 | 4.03 | 3.7 |
footnoteA.
footnote B.
Fig. 1.
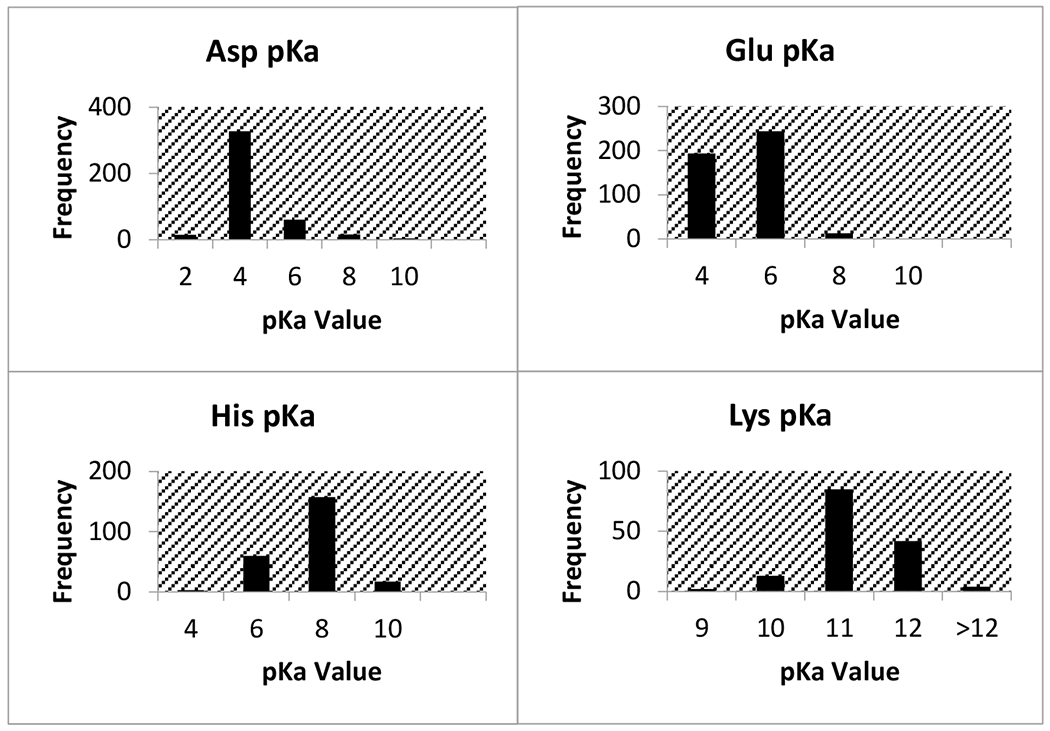
Distribution of pKa values for wild type residues with the most experimental data points
The lowest pKa found was 0.5 for Asp76 in Ribonuclease T1 (RNase T1) and for Asp70 in T4 lysozyme. The side chain of Asp76 in RNase T1 is 99% buried, and forms four hydrogen bonds with (i) a buried, conserved water molecule, (ii) Thr91, (iii) Tyr11 and (iv) Asn9. These hydrogen bonds stabilize the aspartate and therefore drastically lower the pKa. In T4 lysozyme, the side chain of Asp70 makes a salt bridge with His31, resulting in the low pKa of 0.5. The highest marked pKa is 12.1 from multiple Lys residues in the P43G mutant form of calbindin D9K. The residue Cys40 in rhh protein (UniProtKB Q54323), was determined to have a pKa above 14.9. This cystine is found in a hydrophobic dimer interface, thus experiencing large de-solvation penalty.
Figure 2 shows %SASA plotted against the wild type pKa’s. Despite a lack of an appropriate sample size for comparison, acidic residues show a negative correlation between %SASA and pKa, indicating the more buried the residue is the higher the pKa and the more energy required to remove its proton (Figure 2a.). A positive correlation was found for basic residues; the more buried the residue, the smaller is the pKa (Figure 2b). One might expect that buried residues, if not supported by favorable interactions, should have their pKa’s shifted toward an uncharged state (higher for acidic and lower pKa’s for basic groups). However, in wild type proteins, the de-solvation penalty is frequently compensated by favorable interactions.
Fig. 2.
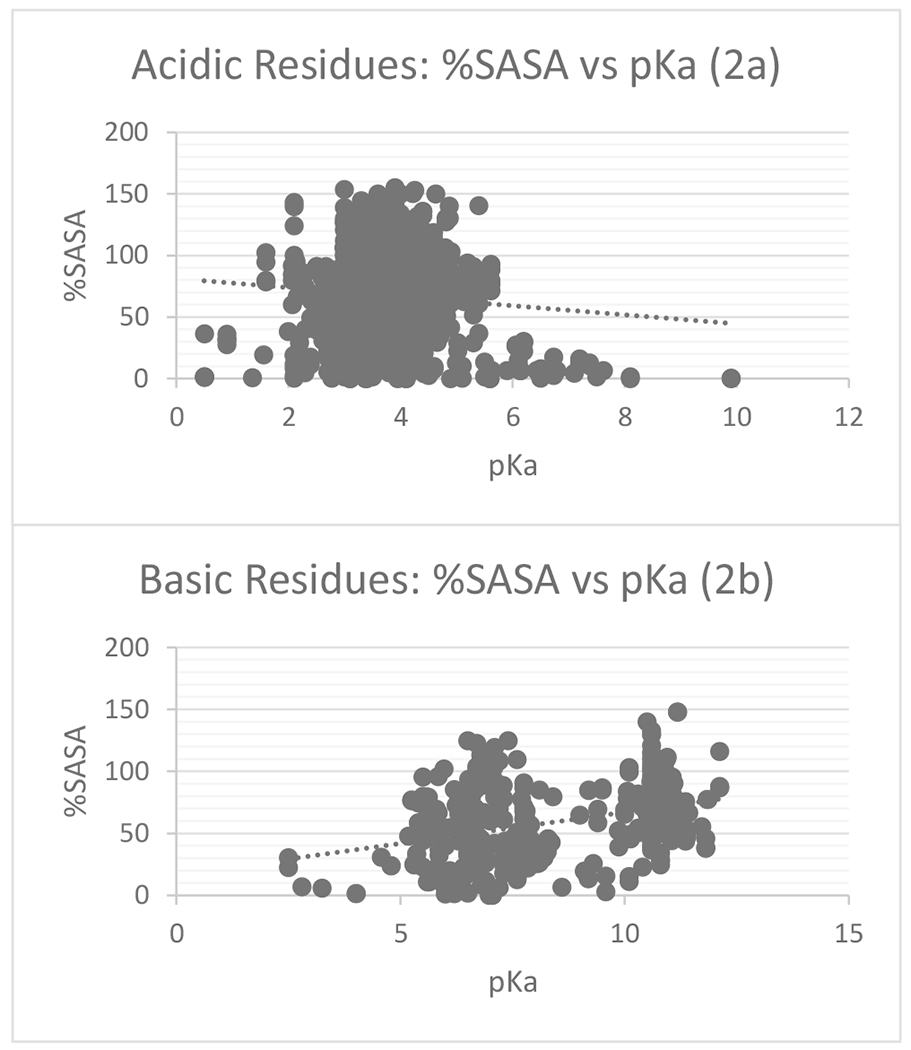
Scatterplots of %SASA vs. pKa for acid wild type residues Glu and Asp (2a) and basic wild type residues His and Lys (2b)
Figure 3 shows the relationship between number of hydrogen bonds and measured pKa’s for wild type proteins. The wild type data, together, represents a correlation between the greater number of hydrogen bonds and the ease of ionizability (lower pKa). It is expected that hydrogen bonds stabilize a residue granting an increased ability to hold the ionization charge, lowering the pKa value for acidic groups. For basic residues, it is expected the opposite trend. The hydrogen bonds that basic donates should make their pKa’s higher. The data points also indicate that other factors beyond or in addition to hydrogen bond interactions influence a specific residues pKa’s.
Fig. 3.
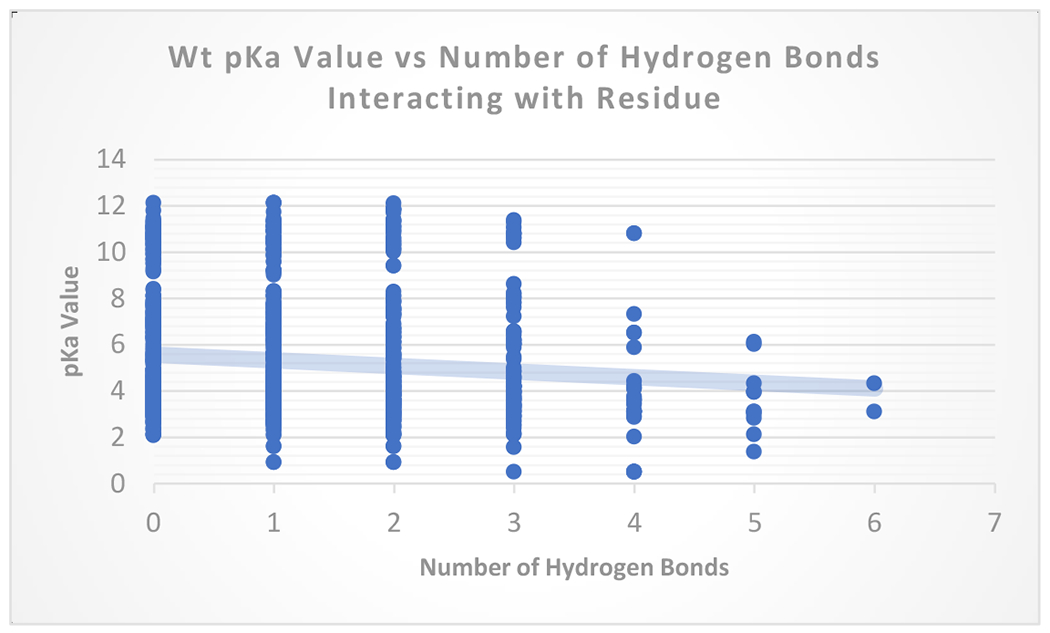
pKa vs number of hydrogen bonds for wildtype residues
Mutant proteins
Experimentally measured pKa values were collected for 286 residues in mutant proteins, and out of them 37% His residues, 30% Glu, 15.2% Lys. Less frequently found residues included Arg, Tyr, Cys, and the C-terminus. Mutant protein pKa values carried a range of 10.9. We consider two types of mutant proteins: (a) mutant proteins (type I) that the mutation is not at the site where pKa’s is measured, and (b) mutant protein (type II) such that the mutated residue pKa value is reported. Furthermore, in some cases, pKa values from proteins with multiple replacements are listed. Data points classified as mutant type I compose 62% of the values from mutant proteins and there are 21 pKa values from proteins with multiple mutations (Table 2). Table 3 provides the number of pKa values for each amino acid residue.
Table 2.
Summary of mutation type of proteins from collected literature
| Mutation | No. of measurements |
|---|---|
|
| |
| Mut type I | 176 |
| Mut type II | 91 |
| Multiple Mut. | 19 |
Table 3.
Number of measurements of Mutant residues by amino acid type
| Residue ID | No. of measurements |
|---|---|
|
| |
| Asp | 39 |
| Cys | 13 |
| Glu | 81 |
| His | 103 |
| Lys | 41 |
| Arg | 3 |
| Tyr | 3 |
| C-Term | 3 |
His pKa values in this set of data are typically larger than its intrinsic value, which is similar to the wild type protein data for this residue. Despite large clusters of similar pKa values, the range was widespread. His was typically not the residue in the sequence that incurred the mutation, thus 95% of the values were mutant type I.
Glu pKa composes almost a third of the mutant data and was the replacement residue (mutation II) about half of the time. Due to Glu’s acidic and polar structure it is an excellent candidate for knocking out the function of nonpolar and basic amino acids in knock out trials, due to the contrasting properties. With most of Glu pKa between 3 and 5, this factor may also play a role in the choice of Glu to replace another residue with a different pKa, perhaps to study its enzymatic function. This residue also has a wide range of exhibited pKa values in various proteins, from 2.9 to 9.4. The mean pKa of Glu in the mutant protein data was also higher than its intrinsic value, indicating a general destabilization within protein structure.
In mutant protein data, Lys showed the largest range of pKa values. Its average pKa value was lower than its intrinsic value. The reason is that many Lys pKa values come from mutant type II proteins, where Lys was introduced in the hydrophobic core of the protein.
4. Conclusion
PKAD-2 database holds experimentally measured pKa values and many autonomous residue specific informational features. It provides a user-friendly interface and visualization of a residues location. As new experimental values are measured the size of the database will increase to include these data points. The database is accessible via http://compbio.clemson.edu/lab/software/5/.
Acknowledgments
National Institutes of Health (R01GM093937).
Footnotes
Conflict of interest. None declared.
Contributor Information
Nicolas Ancona, Department of Biological Sciences, College of Science, Clemson University, 105 Sikes Hall, Address, Clemson, SC 29634, United States of America.
Ananta Bastola, School of Computing, College of Engineering, Computing and Applied Sciences, Clemson University, 105 Sikes Hall, SC 29634, United States of America.
Emil Alexov, Department of Physics, College of Science, Clemson University, 105 Sikes Hall, Address, Clemson, SC 29634, United States of America.
References
- 1.Talley K, & Alexov E (2010). On the pH-optimum of activity and stability of proteins. Proteins, 78(12), 2699–2706. 10.1002/prot.22786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Z, Witham S, & Alexov E (2011). On the role of electrostatics in protein-protein interactions. Physical biology, 8(3), 035001. 10.1088/1478-3975/8/3/035001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matthew JB, Gurd FR, Garcia-Moreno B, Flanagan MA, March KL, & Shire SJ (1985). pH-dependent processes in proteins. CRC critical reviews in biochemistry, 18(2), 91–197. 10.3109/10409238509085133 [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Moreno B (2009). Adaptations of proteins to cellular and subcellular pH. Journal of biology, 8(11), 98. 10.1186/jbiol199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hendsch ZS, & Tidor B (1994). Do salt bridges stabilize proteins? A continuum electrostatic analysis. Protein science : a publication of the Protein Society, 3(2), 211–226. 10.1002/pro.5560030206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bosshard HR, Marti DN, & Jelesarov I (2004). Protein stabilization by salt bridges: concepts, experimental approaches and clarification of some misunderstandings. Journal of molecular recognition : JMR, 17(1), 1–16. 10.1002/jmr.657 [DOI] [PubMed] [Google Scholar]
- 7.Peng Y, Alexov E, & Basu S (2019). Structural Perspective on Revealing and Altering Molecular Functions of Genetic Variants Linked with Diseases. International journal of molecular sciences, 20(3), 548. 10.3390/ijms20030548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheinerman FB, Norel R, & Honig B (2000). Electrostatic aspects of protein-protein interactions. Current opinion in structural biology, 10(2), 153–159. 10.1016/s0959-440x(00)00065-8 [DOI] [PubMed] [Google Scholar]
- 9.Bertonati C, Honig B, & Alexov E (2007). Poisson-Boltzmann calculations of nonspecific salt effects on protein-protein binding free energies. Biophysical journal, 92(6), 1891–1899. 10.1529/biophysj.106.092122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng Y, & Alexov E (2017). Computational investigation of proton transfer, pKa shifts and pH-optimum of protein-DNA and protein-RNA complexes. Proteins, 85(2), 282–295. 10.1002/prot.25221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Y, Kucukkal TG, Li J, Alexov E, & Cao W (2016). Binding Analysis of Methyl-CpG Binding Domain of MeCP2 and Rett Syndrome Mutations. ACS chemical biology, 11(10), 2706–2715. 10.1021/acschembio.6b00450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Onufriev AV, & Alexov E (2013). Protonation and pK changes in protein-ligand binding. Quarterly reviews of biophysics, 46(2), 181–209. 10.1017/S0033583513000024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isom DG, Castañeda CA, Cannon BR, & García-Moreno B (2011). Large shifts in pKa values of lysine residues buried inside a protein. Proceedings of the National Academy of Sciences of the United States of America, 108(13), 5260–5265. 10.1073/pnas.1010750108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitra RC, Zhang Z, & Alexov E (2011). In silico modeling of pH-optimum of protein-protein binding. Proteins, 79(3), 925–936. 10.1002/prot.22931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexov E (2004). Numerical calculations of the pH of maximal protein stability. The effect of the sequence composition and three-dimensional structure. European journal of biochemistry, 271(1), 173–185. 10.1046/j.1432-1033.2003.03917.x [DOI] [PubMed] [Google Scholar]
- 16.Talley K, & Alexov E (2010). On the pH-optimum of activity and stability of proteins. Proteins, 78(12), 2699–2706. 10.1002/prot.22786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiriyasermkul P, Moriyama S, & Nagamori S (2020). Membrane transport proteins in melanosomes: Regulation of ions for pigmentation. Biochimica et biophysica acta. Biomembranes, 1862(12), 183318. 10.1016/j.bbamem.2020.183318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koirala M, Shashikala HBM, Jeffries J, Wu B, Loftus SK, Zippin JH, & Alexov E (2021). Computational Investigation of the pH Dependence of Stability of Melanosome Proteins: Implication for Melanosome formation and Disease. International journal of molecular sciences, 22(15), 8273. 10.3390/ijms22158273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petukh M, Stefl S, & Alexov E (2013). The role of protonation states in ligand-receptor recognition and binding. Current pharmaceutical design, 19(23), 4182–4190. 10.2174/1381612811319230004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onufriev AV, & Alexov E (2013). Protonation and pK changes in protein-ligand binding. Quarterly reviews of biophysics, 46(2), 181–209. 10.1017/S0033583513000024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng Y, & Alexov E (2017). Computational investigation of proton transfer, pKa shifts and pH-optimum of protein-DNA and protein-RNA complexes. Proteins, 85(2), 282–295. 10.1002/prot.25221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henderson JA, & Shen J (2022). Exploring the pH- and Ligand-Dependent Flap Dynamics of Malarial Plasmepsin II. Journal of chemical information and modeling, 62(1), 150–158. 10.1021/acs.jcim.1c01180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma S, Henderson JA, & Shen J (2021). Exploring the pH-Dependent Structure-Dynamics-Function Relationship of Human Renin. Journal of chemical information and modeling, 61(1), 400–407. 10.1021/acs.jcim.0c01201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y, Henderson JA, & Shen J (2021). Continuous Constant pH Molecular Dynamics Simulations of Transmembrane Proteins. Methods in molecular biology (Clifton, N.J.), 2302, 275–287. 10.1007/978-1-0716-1394-8_15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lousa D, Pinto ART, Campos SRR, Baptista AM, Veiga AS, Castanho MARB, & Soares CM (2020). Effect of pH on the influenza fusion peptide properties unveiled by constant-pH molecular dynamics simulations combined with experiment. Scientific reports, 10(1), 20082. 10.1038/s41598-020-77040-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silva TFD, Vila-Viçosa D, & Machuqueiro M (2022). Increasing the Realism of in Silico pHLIP Peptide Models with a Novel pH Gradient CpHMD Method. Journal of chemical theory and computation, 18(11), 6472–6481. 10.1021/acs.jctc.2c00880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silva TFD, Vila-Viçosa D, & Machuqueiro M (2021). Improved Protocol to Tackle the pH Effects on Membrane-Inserting Peptides. Journal of chemical theory and computation, 17(7), 3830–3840. 10.1021/acs.jctc.1c00020 [DOI] [PubMed] [Google Scholar]
- 28.Oliveira NFB, Silva TFD, Reis PBPS, & Machuqueiro M (2021). pK a Calculations in Membrane Proteins from Molecular Dynamics Simulations. Methods in molecular biology (Clifton, N.J.), 2315, 185–195. 10.1007/978-1-0716-1468-6_11 [DOI] [PubMed] [Google Scholar]
- 29.Gupta C, Khaniya U, Vant JW, Shekhar M, Mao J, Gunner MR, & Singharoy A (2021). Poor Person’s pH Simulation of Membrane Proteins. Methods in molecular biology (Clifton, N.J.), 2315, 197–217. 10.1007/978-1-0716-1468-6_12 [DOI] [PubMed] [Google Scholar]
- 30.Kaur D, Zhang Y, Reiss KM, Mandal M, Brudvig GW, Batista VS, & Gunner MR (2021). Proton exit pathways surrounding the oxygen evolving complex of photosystem II. Biochimica et biophysica acta. Bioenergetics, 1862(8), 148446. 10.1016/j.bbabio.2021.148446 [DOI] [PubMed] [Google Scholar]
- 31.Wei RJ, Khaniya U, Mao J, Liu J, Batista VS, & Gunner MR (2022). Tools for analyzing protonation states and for tracing proton transfer pathways with examples from the Rb. sphaeroides photosynthetic reaction centers. Photosynthesis research, 10.1007/s11120–022-00973–0. Advance online publication. 10.1007/s11120-022-00973-0 [DOI] [PubMed] [Google Scholar]
- 32.Szakács Z, Kraszni M, & Noszál B (2004). Determination of microscopic acid-base parameters from NMR-pH titrations. Analytical and bioanalytical chemistry, 378(6), 1428–1448. 10.1007/s00216-003-2390-3 [DOI] [PubMed] [Google Scholar]
- 33.van Vlijmen HW, Schaefer M, & Karplus M (1998). Improving the accuracy of protein pKa calculations: conformational averaging versus the average structure. Proteins, 33(2), 145–158. [DOI] [PubMed] [Google Scholar]
- 34.Tan YJ, Oliveberg M, Davis B, & Fersht AR (1995). Perturbed pKA-values in the denatured states of proteins. Journal of molecular biology, 254(5), 980–992. 10.1006/jmbi.1995.0670 [DOI] [PubMed] [Google Scholar]
- 35.Fitch CA, Platzer G, Okon M, Garcia-Moreno BE, & McIntosh LP (2015). Arginine: Its pKa value revisited. Protein science : a publication of the Protein Society, 24(5), 752–761. 10.1002/pro.2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Griffiths SW, King J, & Cooney CL (2002). The reactivity and oxidation pathway of cysteine 232 in recombinant human alpha 1-antitrypsin. The Journal of biological chemistry, 277(28), 25486–25492. 10.1074/jbc.M203089200 [DOI] [PubMed] [Google Scholar]
- 37.Webb H, Tynan-Connolly BM, Lee GM, Farrell D, O’Meara F, Søndergaard CR, Teilum K, Hewage C, McIntosh LP, & Nielsen JE (2011). Remeasuring HEWL pK(a) values by NMR spectroscopy: methods, analysis, accuracy, and implications for theoretical pK(a) calculations. Proteins, 79(3), 685–702. 10.1002/prot.22886 [DOI] [PubMed] [Google Scholar]
- 38.Martins de Oliveira V, Liu R, & Shen J (2022). Constant pH molecular dynamics simulations: Current status and recent applications. Current opinion in structural biology, 77, 102498. Advance online publication. 10.1016/j.sbi.2022.102498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oliveira NFB, & Machuqueiro M (2022). Novel US-CpHMD Protocol to Study the Protonation-Dependent Mechanism of the ATP/ADP Carrier. Journal of chemical information and modeling, 62(10), 2550–2560. 10.1021/acs.jcim.2c00233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radak BK, Chipot C, Suh D, Jo S, Jiang W, Phillips JC, Schulten K, & Roux B (2017). Constant-pH Molecular Dynamics Simulations for Large Biomolecular Systems. Journal of chemical theory and computation, 13(12), 5933–5944. 10.1021/acs.jctc.7b00875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song Y, Mao J, & Gunner MR (2009). MCCE2: improving protein pKa calculations with extensive side chain rotamer sampling. Journal of computational chemistry, 30(14), 2231–2247. 10.1002/jcc.21222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anandakrishnan R, Aguilar B, & Onufriev AV (2012). H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic acids research, 40(Web Server issue), W537–W541. 10.1093/nar/gks375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pahari S, Sun L, Basu S, & Alexov E (2018). DelPhiPKa: Including salt in the calculations and enabling polar residues to titrate. Proteins, 86(12), 1277–1283. 10.1002/prot.25608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang L, Zhang M, & Alexov E (2016). DelPhiPKa web server: predicting pKa of proteins, RNAs and DNAs. Bioinformatics (Oxford, England), 32(4), 614–615. 10.1093/bioinformatics/btv607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Warwicker J (2004). Improved pKa calculations through flexibility based sampling of a water-dominated interaction scheme. Protein science : a publication of the Protein Society, 13(10), 2793–2805. 10.1110/ps.04785604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kieseritzky G, & Knapp EW (2008). Optimizing pKa computation in proteins with pH adapted conformations. Proteins, 71(3), 1335–1348. 10.1002/prot.21820 [DOI] [PubMed] [Google Scholar]
- 47.Li H, Robertson AD, & Jensen JH (2005). Very fast empirical prediction and rationalization of protein pKa values. Proteins, 61(4), 704–721. 10.1002/prot.20660 [DOI] [PubMed] [Google Scholar]
- 48.Cvitkovic JP, Pauplis CD, Carney PC, & Kaminski GA (2021). Expansion and Additional Validation of PKA17: A Fast Real-Time and Web-Based pKa Predictor. Journal of Computational Biophysics and Chemistry, 20(2), 141–152. [Google Scholar]
- 49.Reis PBPS, Bertolini M, Montanari F, Rocchia W, Machuqueiro M, & Clevert DA (2022). A Fast and Interpretable Deep Learning Approach for Accurate Electrostatics-Driven pKa Predictions in Proteins. Journal of chemical theory and computation, 18(8), 5068–5078. 10.1021/acs.jctc.2c00308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reis PBPS, Clevert DA, & Machuqueiro M (2021). pKPDB: a Protein Data Bank extension database of pKa and pI theoretical values. Bioinformatics (Oxford, England), btab518. Advance online publication. 10.1093/bioinformatics/btab518 [DOI] [PubMed] [Google Scholar]
- 51.Pahari S, Sun L, & Alexov E (2019). PKAD: a database of experimentally measured pKa values of ionizable groups in proteins. Database : the journal of biological databases and curation, 2019, baz024. 10.1093/database/baz024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reis PBPS, Vila-Viçosa D, Rocchia W, & Machuqueiro M (2020). PypKa: A Flexible Python Module for Poisson-Boltzmann-Based pKa Calculations. Journal of chemical information and modeling, 60(10), 4442–4448. 10.1021/acs.jcim.0c00718 [DOI] [PubMed] [Google Scholar]
- 53.Kozlowski LP (2021). IPC 2.0: prediction of isoelectric point and pKa dissociation constants. Nucleic acids research, 49(W1), W285–W292. 10.1093/nar/gkab295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang H, Eerland J, Horn V, Schellevis R, & van Ingen H (2021). Mapping the electrostatic potential of the nucleosome acidic patch. Scientific reports, 11(1), 23013. 10.1038/s41598-021-02436-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burley SK, Bhikadiya C, Bi C, Bittrich S, Chao H, Chen L, Craig PA, Crichlow GV, Dalenberg K, Duarte JM, Dutta S, Fayazi M, Feng Z, Flatt JW, Ganesan S, Ghosh S, Goodsell DS, Green RK, Guranovic V, Henry J, … Zardecki C (2022). RCSB Protein Data Bank (RCSB.org): delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic acids research, gkac1077. Advance online publication. 10.1093/nar/gkac1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang H (2020). Computational, biochemical, and NMR-driven structural studies on histone variant H2A. B (Doctoral dissertation, Leiden University; ). [Google Scholar]
- 57.Hiebler K, Lengyel Z, Castañeda CA, & Makhlynets OV (2017). Functional tuning of the catalytic residue pKa in a de novo designed esterase. Proteins: Structure, Function, and Bioinformatics, 85(9), 1656–1665. [DOI] [PubMed] [Google Scholar]
- 58.Dhembla C, Arya R, Kumar A, Kundu S, & Sundd M (2021). L. major apo-acyl carrier protein forms ordered aggregates due to an exposed phenylalanine, while phosphopantetheine inhibits aggregation in the holo-form. International Journal of Biological Macromolecules, 179, 144–153 [DOI] [PubMed] [Google Scholar]
- 59.Thomas NE, Wu C, Morrison EA, Robinson AE, Werner JP, & Henzler-Wildman KA (2018). The C terminus of the bacterial multidrug transporter EmrE couples drug binding to proton release. Journal of Biological Chemistry, 293(49), 19137–19147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zou J, Xiao S, Simmerling C, & Raleigh DP (2021). Quantitative Analysis of Protein Unfolded State Energetics: Experimental and Computational Studies Demonstrate That Non-Native Side-Chain Interactions Stabilize Local Native Backbone Structure. The Journal of Physical Chemistry B, 125(13), 3269–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raum HN, & Weininger U (2019). Experimental pKa value determination of all ionizable groups of a hyperstable protein. ChemBioChem, 20(7), 922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han CT, Song J, Chan T, Pruett C, & Han S (2020). Electrostatic environment of proteorhodopsin affects the pka of its buried primary proton acceptor. Biophysical journal, 118(8), 1838–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang H, Eerland J, Horn V, Schellevis R, & van Ingen H (2021). Mapping the electrostatic potential of the nucleosome acidic patch. Scientific reports, 11(1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kot EF, Wang Y, Goncharuk SA, Zhang B, Arseniev AS, Wang X, & Mineev KS (2020). Oligomerization analysis as a tool to elucidate the mechanism of EBV latent membrane protein 1 inhibition by pentamidine. Biochimica et Biophysica Acta (BBA)-Biomembranes, 1862(10), 183380. [DOI] [PubMed] [Google Scholar]
- 65.Hofer F, Dietrich V, Kamenik AS, Tollinger M, & Liedl KR (2019). pH-Dependent protonation of the Phl p 6 pollen allergen studied by NMR and cpH-aMD. Journal of chemical theory and computation, 15(10), 5716–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerland L, Friedrich D, Hopf L, Donovan EJ, Wallmann A, Erdmann N, ... & Oschkinat H (2020). pH‐Dependent Protonation of Surface Carboxylate Groups in PsbO Enables Local Buffering and Triggers Structural Changes. ChemBioChem, 21(11), 1597–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barnes CA, Shen Y, Ying J, & Bax A Modulating the Stiffness of the Myosin VI Single a-Helical Domain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kougentakis CM, Grasso EM, Robinson AC, Caro JA, Schlessman JL, Majumdar A, & García-Moreno E,B (2018). Anomalous properties of Lys residues buried in the hydrophobic interior of a protein revealed with 15N-detect NMR spectroscopy. The Journal of Physical Chemistry Letters, 9(2), 383–387. [DOI] [PubMed] [Google Scholar]
- 69.Li J, Sae Her A, & Traaseth NJ (2020). Site-specific resolution of anionic residues in proteins using solid-state NMR spectroscopy. Journal of biomolecular NMR, 74(6), 355–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brockerman JA, Okon M, Withers SG, & McIntosh LP (2019). The pKa values of the catalytic residues in the retaining glycoside hydrolase T26H mutant of T4 lysozyme. Protein Science, 28(3), 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Payliss BJ, Vogel J, & Mittermaier AK (2019). Side chain electrostatic interactions and pH-dependent expansion of the intrinsically disordered, highly acidic carboxyl-terminus of γ-tubulin. Protein science : a publication of the Protein Society, 28(6), 1095–1105. 10.1002/pro.3618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mishra P, Patni D, & Jha SK (2021). A pH-dependent protein stability switch coupled to the perturbed pKa of a single ionizable residue. Biophysical chemistry, 274, 106591. 10.1016/j.bpc.2021.106591 [DOI] [PubMed] [Google Scholar]
- 73.Fink AL, Calciano LJ, Goto Y, Kurotsu T, & Palleros DR (1994). Classification of acid denaturation of proteins: intermediates and unfolded states. Biochemistry, 33(41), 12504–12511. [DOI] [PubMed] [Google Scholar]
- 74.Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, & Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein science : a publication of the Protein Society, 27(1), 14–25. 10.1002/pro.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang J, Youkharibache P, Zhang D, Lanczycki CJ, Geer RC, Madej T, Phan L, Ward M, Lu S, Marchler GH, Wang Y, Bryant SH, Geer LY, & Marchler-Bauer A (2020). iCn3D, a web-based 3D viewer for sharing 1D/2D/3D representations of biomolecular structures. Bioinformatics (Oxford, England), 36(1), 131–135. 10.1093/bioinformatics/btz502 [DOI] [PMC free article] [PubMed] [Google Scholar]