

Significance
Gene regulatory networks (GRNs) expressed in cell-free systems hold great promise for investigating the limits of biological information processing and developing platforms for molecular biosensing and chemical bioproduction. We address the challenge of engineering GRNs that can dynamically activate many targets. The work described here enables classes of deep, wide, and multi-input CRISPR-based genetic circuits. This study represents an important step toward engineered GRNs with complexities approaching those found in nature.
Keywords: activatable promoter engineering, CRISPR activation, gene regulatory networks, genetic circuits, cell-free
Abstract
Dynamic, multi-input gene regulatory networks (GRNs) are ubiquitous in nature. Multilayer CRISPR-based genetic circuits hold great promise for building GRNs akin to those found in naturally occurring biological systems. We develop an approach for creating high-performing activatable promoters that can be assembled into deep, wide, and multi-input CRISPR-activation and -interference (CRISPRa/i) GRNs. By integrating sequence-based design and in vivo screening, we engineer activatable promoters that achieve up to 1,000-fold dynamic range in an Escherichia coli-based cell-free system. These components enable CRISPRa GRNs that are six layers deep and four branches wide. We show the generalizability of the promoter engineering workflow by improving the dynamic range of the light-dependent EL222 optogenetic system from 6-fold to 34-fold. Additionally, high dynamic range promoters enable CRISPRa systems mediated by small molecules and protein–protein interactions. We apply these tools to build input-responsive CRISPRa/i GRNs, including feedback loops, logic gates, multilayer cascades, and dynamic pulse modulators. Our work provides a generalizable approach for the design of high dynamic range activatable promoters and enables classes of gene regulatory functions in cell-free systems.
Natural biological systems employ complex gene regulatory networks (GRNs) to sense diverse environmental cues and respond to them through the coordinated expression of multiple genes (1–3). Cell-free systems (CFS) have emerged as an attractive chassis for building synthetic biological systems as they allow for rapid prototyping of genetic parts and circuits (4–8). To build increasingly complex CFS capable of sensing and responding to diverse inputs, new approaches for increasing the capabilities of synthetic GRNs are needed (9–13). Advances in GRN engineering will accelerate the use of CFS for building multiplexed biosensors (14–16), deploying on-demand bioproduction platforms (17–19), and the construction of synthetic cells (20–23).
CRISPR-Cas transcriptional regulation has proven a promising framework for building sophisticated genetic circuits across a variety of biological systems (24–30). Transcriptional units containing target sequences for CRISPR activation (CRISPRa) and/or CRISPR interference (CRISPRi), termed CRISPRa/i nodes, can be assembled into circuits with network topologies specified by guide RNAs (gRNAs). Experimental and theoretical analyses indicate that the CRISPRa/i system is well suited to design deep and wide control circuits containing internal nodes connected in series or parallel through orthogonal gRNAs (31–33). Large CRISPRi GRNs with up to 7 gRNAs have been constructed in yeast by implementing low leak promoters and high dynamic range repressors (34). In Escherichia coli-based CFS, CRISPRi genetic control is well established (30, 35), and CRISPRa has recently been incorporated (33), greatly expanding the circuit design space. However, CRISPRa circuitry is limited by a lack of promoter-gRNA pairs that can be interconnected with minimal signal degradation (33). Hence, a generalizable approach for engineering activatable promoters with low basal and high activated expression levels would significantly improve CRISPRa and enable the assembly of complex, input-responsive GRNs.
Promoter engineering efforts have traditionally focused on designing constitutive and inducible promoters with predictable expression characteristics (36, 37). Tuning the strength of constitutive promoters involves designing promoter sequences that achieve a desired level of RNA polymerase (RNAP) recruitment to the promoter (38, 39). Inducible promoters contain recognition sites for transcriptional activators or repressors that modulate transcriptional levels upon binding (40, 41). Development of high dynamic range inducible promoters has relied on engineering derepression-based systems (41–45), largely due to the difficulty of rationally designing activatable promoters (46, 47). For effective activation, RNAP recruitment to the promoter should be weak in the absence of an activator; however, transcription initiation should be strong upon activator-mediated RNAP recruitment (40, 46). Hence, tuning RNAP interactions through promoter sequence design could lead to higher dynamic ranges with CRISPRa and other transcriptional activation systems.
We develop an approach integrating sequence-based design and in vivo screening to generate an expandable set of high-performing promoters that exhibit both low basal and high activated expression levels. Through a sequential selection approach, we design activatable promoters with up to 1,000-fold dynamic range, constituting a 33-fold improvement from previous synthetic promoters (33). These promoters enable complex network topologies with performance levels not previously accessible in CFS (33), including a six-layer deep cascade and a four-branch parallel circuit. By engineering activatable promoters, different inputs can be incorporated into CRISPRa/i CFS circuits. We demonstrate that a blue light–responsive transcriptional activator and three different protein–protein interaction (PPI)-dependent CRISPRa systems can function as circuit inputs. We successfully engineer input-responsive CRISPRa/i circuits operating as multilayer activation cascades, positive feedback (PFB) loops, AND-like logic gates, and dynamic two-input pulse modulators. Overall, this work describes a workflow for engineering activatable promoters and provides a toolbox of versatile components with immediate utility for implementing CRISPRa/i circuits. Together, these developments dramatically expand the ability to assemble large, multi-input GRNs in CFS.
Results
We first sought to characterize the impact of sequences affecting RNAP recruitment on both basal and activated expression levels of synthetic activatable promoters. RNAP recruitment is dependent on the affinity of the RNAP sigma subunit (σ) for the −10 and −35 hexamers of the minimal promoter (48, 49) (Fig. 1A). Additionally, recruitment is influenced by the GC content of the intervening sequence between the −10 and −35 sites (48, 49) (Fig. 1A). Promoter recognition is enhanced by the AT-rich UP elements upstream of the minimal promoter, which anchor the α-subunits of RNAP. Collectively, the sequence compositions of these regions influence RNAP recruitment, binding, and initiation at the promoter. We systematically designed libraries of these discrete promoter regions and used a scalable workflow for screening and isolating library variants in E. coli. The libraries were cotransformed with an aTc-inducible CRISPRa plasmid to enable parallel screening of basal and activated expression levels (Fig. 1B, Methods S10) (33). In the absence of aTc, RNAP recruitment is determined by the promoter basal strength. Upon aTc induction, the MCP-SoxS activator is expressed and localized to a CRISPRa complex at the promoter via a modified gRNA, or scaffold RNA (scRNA), containing the MS2 hairpin. MCP-SoxS then recruits RNAP to the promoter through α-CTD interactions, activating transcription (50) (Fig. 1A). This approach allows us to characterize the impact of individual promoter regions on basal and activated expression simultaneously, and combine variants with low basal and high activated expression to construct high-performing activatable promoters.
Fig. 1.
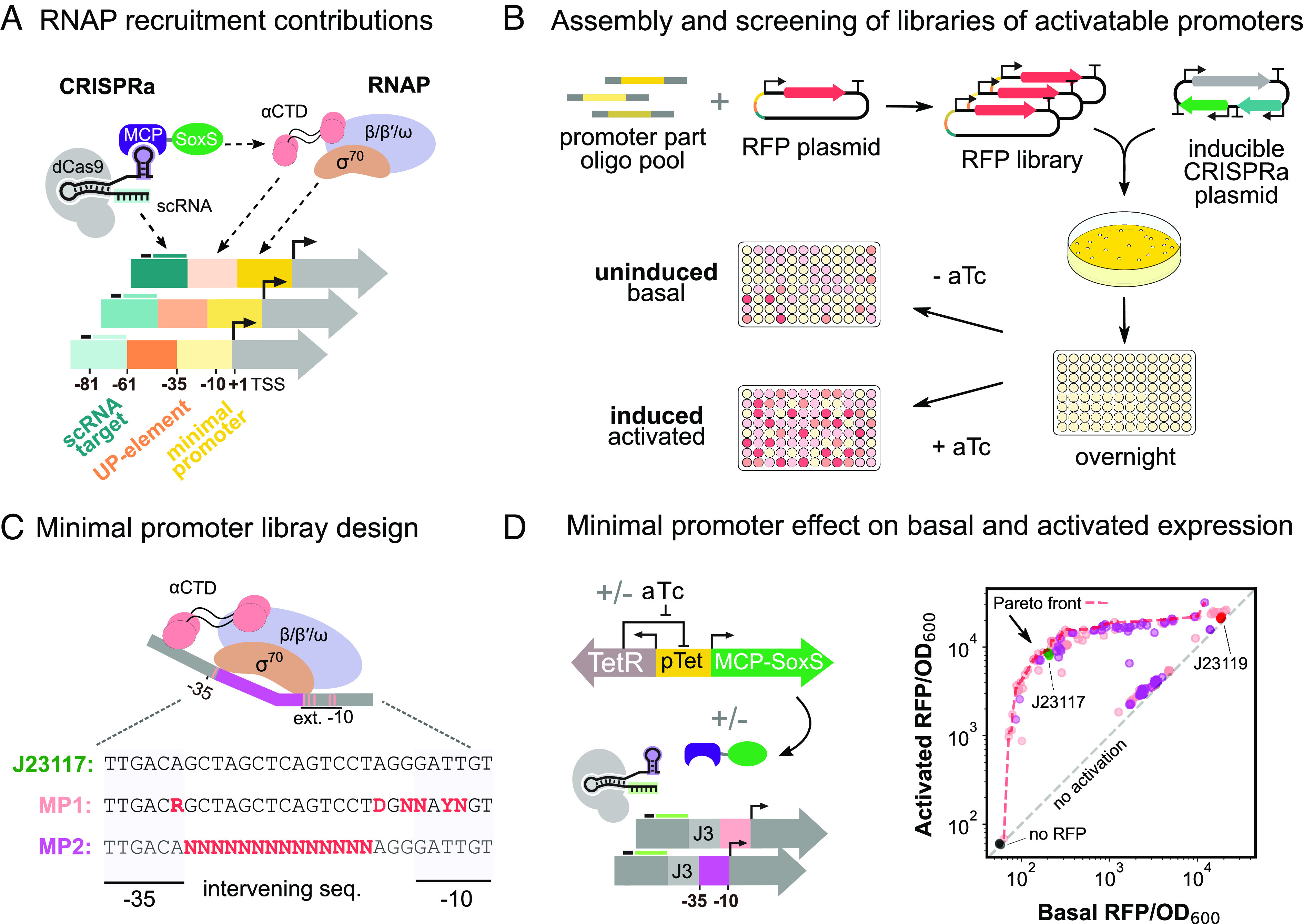
Functional interrogation of promoter regions with CRISPRa. (A) Schematic of RNAP interactions with the CRISPRa complex and target promoter. σ70 affinity for the minimal promoter and α-CTD affinity for the UP element determines RNAP recruitment to a promoter. When CRISPRa is targeted to a promoter with a complementary scRNA target site, the RNAP α-CTD domain is recruited by the SoxS transcriptional activator. RNAP-promoter and CRISPRa-promoter interactions can be modulated by modifying the DNA sequence of the different promoter regions. (B) Workflow for the assembly and characterization of libraries of activatable promoters. A library of RFP genes with varying promoters is generated through PCR (Methods S9). The library is then cotransformed into E. coli with an aTc-inducible CRISPRa plasmid. Colonies are then seeded overnight, and subsequently diluted into media with appropriate concentrations of aTc. For each promoter variant in the libraries, basal and activated RFP levels were measured with 0 nM and 200 nM aTc, respectively (Methods S10. (C) Schematic of RNAP interaction with the minimal promoter and library design. σ70 recognizes specific positions in the extended −10 and −35 regions of the minimal promoter, which informed the design of the library MP1. σ70 binding is also influenced by the GC content, the length, and the −15TGn−1 motif of the intervening sequence, which informed the design of library MP2 (Methods S11). (D) Minimal promoter effect on expression levels. Left: Inducible CRISPRa system and minimal promoter libraries of the J3 synthetic promoter. MCP-SoxS is expressed from the aTc-inducible pTet promoter. dCas9 and J306 scRNA are constitutively expressed. Right: Activated and basal RFP/OD600 for the two minimal promoter libraries (nMP1 = 89, nMP2 = 84). The red dash line defines the Pareto front containing the best-performing promoter variants (SI Appendix, Methods S3), for which no further improvements in basal or activated levels can be achieved without compromising the other. The gray dash line defines promoter variants with equal activated and basal expression levels, indicating they are not activated by CRISPRa. The J23117 minimal promoter (green, triplicates) is included as a standard reference for CRISPRa efficiency. The J23119 minimal promoter (red, triplicates) is an example of a nonactivatable promoter due to high basal expression levels. A plasmid without RFP (black, triplicates) indicates the background fluorescence of the system.
Functional Interrogation of Promoter Regions with CRISPRa.
Impact of minimal promoter region on activatability.
Previous work has demonstrated the importance of the minimal promoter region in determining basal and activated expression levels (40, 46, 50–52). We designed two minimal promoter libraries mutagenizing the −10/−35 hexamers and the intervening sequence of the previously identified best-performing minimal promoter (BBa_J23117) within the J3 synthetic CRISPRa promoter (Fig. 1C, Methods S11) (46). These libraries were cotransformed with the aTc-inducible CRISPRa plasmid expressing the J306 scRNA targeting the cognate J3 promoter. Both libraries yielded high promoter diversity, as measured by RFP expression levels, with basal and activated expression levels ranging from that of a no-reporter control to a strong constitutive promoter (BBa_J23119) (Fig. 1D). The subset of promoters not activated may arise from the generation of sequences resembling tightly regulated native promoters.
The set of minimal promoter variants that maintain both low basal and high activated expression levels can be conceptualized as a Pareto optimal front. In multiobjective optimization, a Pareto front defines the best-performing solutions for which no further improvements in either objective can be achieved without compromising the other (SI Appendix, Methods S3) (53). Three variants from this Pareto front exhibited both lower basal and higher activated expression levels compared to the original BBa_J23117, indicating the original minimal promoter was not a part of the Pareto front. This finding suggests that promoter mutagenesis can yield improved activatable promoters beyond previous screening methods based on promoter basal strengths alone (46). By mutagenizing the minimal promoter of CRISPRa promoters, we generated sequences with >100-fold dynamic range in CRISPRa-mediated gene expression (SI Appendix, Fig. S1).
Impact of UP element region on activatability.
RNAP promoter recognition is enhanced by the AT-rich UP elements upstream of the minimal promoter, which anchor the α-subunits of RNAP (40, 48, 54). For effective CRISPRa, RNAP should only be recruited to the promoter in the presence of an on-target scRNA. Hence, for transcriptional activation with SoxS, improvements in dynamic range could be achieved by minimizing RNAP–UP element interactions and lowering basal expression levels (55). We designed five UP element libraries mutagenizing the AT-rich E. coli consensus sequence with increasing GC content (Fig. 2A, Methods S11). As expected, the consensus UP element and the AT-rich library had the highest basal expression levels (Fig. 2 B, Left). On average, these libraries showed only threefold activation, as compared to 37- to 44-fold activation for the more GC-rich libraries (SI Appendix, Figs. S2 and S3). Qualitatively, we observed a monotonic decrease in basal expression levels and no impact on maximum activated levels with increased GC content (SI Appendix, Fig. S2). We identified the optimal variants from each library and found a shift in the Pareto front toward lower basal and higher activated expression levels with increasing UP element GC content (Fig. 2 B, Right). Specifically, the median basal and activated expression levels of the GC-rich optimal variants were 59.7-fold lower and 1.7-fold higher than that of the AT-rich optimal variants. The original J3 synthetic promoter, with an UP element GC content of 50%, sat in between the Pareto fronts consisting of high and low GC content variants. By mutagenizing the UP element, we generated promoter variants with >350-fold dynamic range in CRISPRa-mediated gene expression.
Fig. 2.
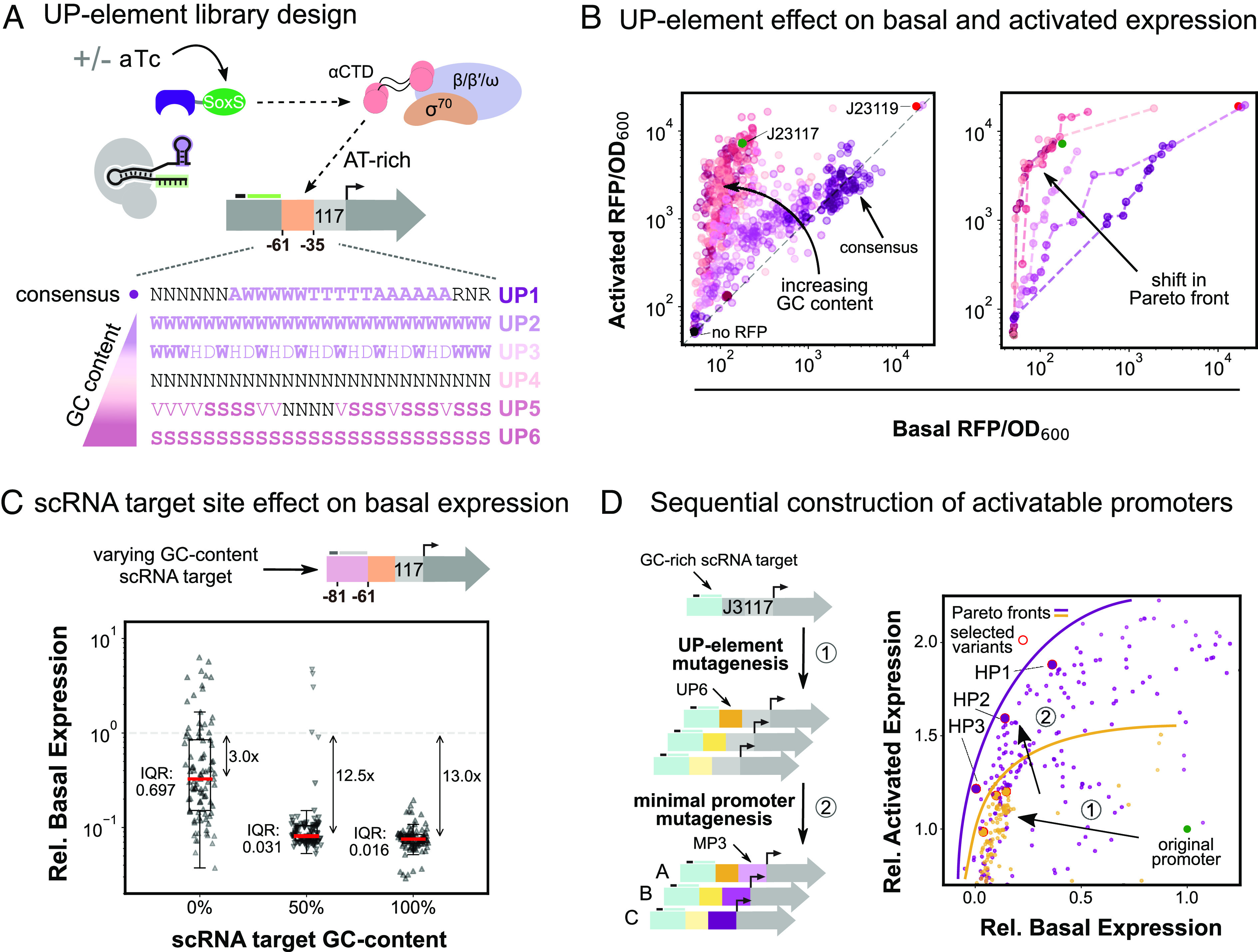
Combining promoter regions to engineer high-performing CRISPRa promoters. (A) Schematic of RNAP interactions with the UP element and library design. α-CTD affinity for AT-rich UP elements upstream of the minimal promoter helps recruit RNAP. Upon targeting with CRISPRa, UP element RNAP recruitment contributions are largely replaced by SoxS-RNAP α-CTD interactions. UP element libraries with increasing GC content were designed to minimize α-CTD interactions (Methods S11). (B) UP element GC content effect on expression levels is shown through activated and basal RFP/OD600 for the six UP element libraries (nUP1 = nUP2 = … = nUP6 = 110). Left: Increasing GC content in the UP element lowers the range of the basal expression level, while maintaining the full range of activated expression levels. The gray dash line defines promoter variants with equal activated and basal expression levels. Right: Colored dash lines define the Pareto front for each UP element library (SI Appendix, Methods S3). Increasing the UP element GC content effectively shifts the Pareto front toward lower basal expression levels. (C) scRNA target site composition effect on basal expression. Comparison of three scRNA libraries with increasing GC content (nS1 = nS2 = nS3 = 93) (Methods S11). Basal expression levels are normalized to the standard J3 promoter basal expression level. Red lines indicate the median expression level of each distribution. The interquartile range is calculated as the difference between the upper and lower quartiles and measures the spread of the distribution. (D) Sequential construction of activatable promoters. Left: Activatable promoters were constructed by sequential library mutagenesis screens starting from the J3 promoter with a GC-rich scRNA target site. Three Pareto optimal UP elements were selected after promoter mutagenesis with a GC-rich UP element library (1). We then mutagenized the minimal promoter of the three previously selected variants (2) and again selected three Pareto optimal variants. Right: Basal and activated expression levels for all mutagenesis variants normalized to the standard J3 promoter expression levels (green). Yellow points represent variants from the UP element mutagenesis (nUP6 = 192) (1), while purple points represent variants from the minimal promoter mutagenesis (nMP3 = 279) (2). Red circles indicate selected variants from each screen, and solid lines depict the Pareto optimal fronts. Each sequential mutagenesis led to variants with both lower basal and higher activated expression levels.
Impact of the scRNA target site region on activatability.
Transcriptional activators bind upstream of the minimal promoter region to recruit RNAP to the transcription start site (TSS) (40). For CRISPRa, the optimal scRNA target site location for SoxS-mediated activation is −81 bp upstream of the TSS (46). Changing the scRNA target sequence enables rapid generation of orthogonal CRISPRa promoters (56, 57). Due to the proximity to the UP element region, we reasoned that the sequence composition of the scRNA target site may have an impact on basal expression levels. We designed three scRNA target site libraries composed of varying GC content (Methods S11) and measured the basal expression of each library. GC-rich libraries had 4.3-fold lower median basal expression compared to AT-rich libraries (Fig. 2C). Additionally, the spread of the basal expression decreased monotonically with increasing GC content of the scRNA target site sequence (Fig. 2C). Together, these results indicate GC-rich scRNA target site sequences lead to low basal expression CRISPRa promoters, perhaps due to reduced interaction with RNAP. To validate the CRISPRa activity at these low basal expression scRNA target sites, we selected 10 GC-rich variants and constructed the corresponding scRNAs. All variants produced a higher fold activation than the original J306 scRNA (SI Appendix, Fig. S4), with 3.5-fold average increase in fold activation.
Combining Promoter Regions to Engineer High-Performing CRISPRa Promoters.
Engineering activatable promoters by combining optimized promoter regions.
We proceeded to test whether the highest-performing variants from the UP element and minimal promoter screens could be combined to yield activatable promoters with improved performance. We selected three high-performing variants from both the UP element and minimal promoter screens, as well as the starting J3 UP element and BBa_J23117 minimal promoter, and constructed a combinatorial set of 16 promoters. Notably, promoter regions that gave the largest improvements in the original context did not necessarily give the largest improvements when tested in different contexts (SI Appendix, Fig. S5). For instance, an UP element that gave 300-fold activation with one minimal promoter only gave 200-fold activation with a different high-performing minimal promoter (P = 0.003), largely due to an increase in basal expression (SI Appendix, Fig. S5, Right). ANOVA results (P = 10−5) support the view that UP element and minimal promoter contributions affecting activation cannot be isolated from one another (SI Appendix, Table S7).
Engineering activatable promoters through sequential promoter region screening.
The results from the previous section highlight the effects that promoter region contexts play in the design of high-performing activatable promoters. Therefore, we tested whether promoters with improved basal and activated expression levels could be achieved by selecting minimal promoters in the context of high-performing UP elements. We first screened the UP element region as these libraries had a larger impact than the minimal promoter libraries on the location of the Pareto front (Figs. 1D and 2B). We mutagenized the UP element of a promoter containing the minimal promoter BBa_J23117 and a GC-rich scRNA target. We selected three Pareto-optimal UP element variants, which had on average 10% of the original basal and 12% higher activated expression than the J3 promoter (Fig. 2D). We then screened minimal promoter libraries in the context of these three selected UP element variants. We again selected three new promoter variants from the Pareto front, which had on average 17% of the basal and 56% higher activated expression than the J3 promoter (Fig. 2D). With this sequential screening approach, we overcame the context effects observed in the previous section and successfully shifted the Pareto front toward lower basal and higher activated expression for both the UP element and minimal promoter screens.
Engineering Deep and Wide Circuits with High-Performing CRISPRa Promoters.
In nature, interconnected, multilayer transcriptional networks coordinate the timing and levels of expression of multiple genes to produce complex responses to environmental stimuli (1, 58, 59). To develop CRISPRa/i GRNs with complexities approaching those found in nature, components enabling construction of circuits with arbitrary interconnections and minimal signal degradation are needed. CRISPRa promoters with high output dynamic ranges are expected to minimize signal degradation by enabling effective level-matching of the input/output dynamic ranges between sequential CRISPRa/i nodes (33, 34). Dynamic range improvements achieved at the promoter level should translate into signal propagation improvements at the circuit level and enable construction of increasingly deep and wide CRISPRa circuits.
Engineering functional CRISPRa/i nodes.
We characterized the three promoter variants selected above (Fig. 2D, HP1-3) in CFS and observed up to ~1,000-fold dynamic range (SI Appendix, Fig. S6) and a high degree of correspondence with in vivo expression levels (R2 = 0.92). We used these promoters to generate a set of orthogonal CRISPRa/i nodes to be assembled into multilayer circuits following previously described methods (33). We combined the highest dynamic range activatable promoter (HP3, SI Appendix, Table S1) with previously screened scRNA target sites to generate orthogonal CRISPRa/i nodes (Fig. 3A). Dose–response curves for each orthogonal node made by titrating the plasmid expressing the cognate scRNA showed that these new nodes gave an average activation of 890-fold (Fig. 3B and SI Appendix, Fig. S7 andTable S1). Given the context dependence of activation, these nodes could be further improved through sequential mutagenesis of promoter elements in the context of the corresponding scRNA target sequence.
Fig. 3.
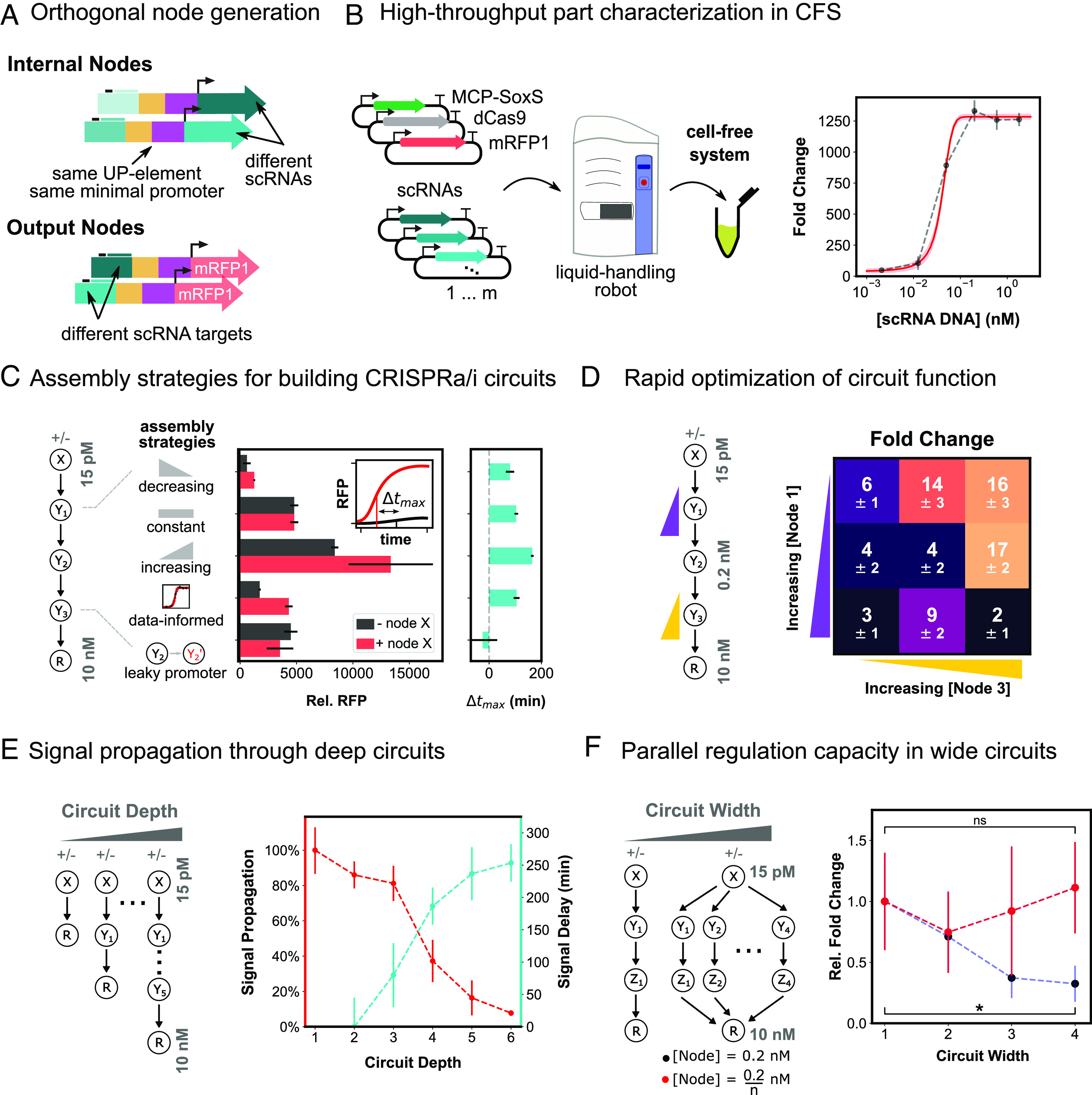
Engineering deep and wide circuits with high-performing CRISPRa promoters. (A) Schematic of orthogonal CRISPRa/i nodes for use in cell-free circuits. Internal nodes contain an orthogonal scRNA target site and express orthogonal scRNAs. Output nodes contain orthogonal scRNA target sites and express RFP. All nodes contain the same UP element and minimal promoter (HP3). (B) High-throughput characterization of scRNA components in CFS. Left: Plasmids encoding each CRISPRa component are mixed using an acoustic liquid handling robot and expressed in CFS. Right: scRNA-dose–response curves for each node are generated by titrating the amount of scRNA plasmid from 0.5 pM to 5 nM. (C) Comparison of assembly strategies for building a four-layer CRISPRa cascade. Left: Internal node concentrations either decreased from 200 pM to 32 pM as depth increased, were held constant at 200 pM, or increased from 200 pM to 1.25 nM as depth increased. A fourth assembly method was tested in which internal node concentrations were 40, 200, and 170 pM, based on individual scRNA-dose response characteristics. A fifth cascade was included in which the high-performing promoter of the second internal node was replaced with the leaky J2 promoter. Input and output node concentrations were held constant across all strategies at 0 or 15 pM and 10 nM, respectively. Center: Cascade output RFP expression for each assembly strategy with scRNA input (red) and without (black), relative to RFP basal expression. Right: Change in time to maximum expression rate (Δtmax) for each assembly strategy (SI Appendix, Methods S3). (D) Rapid fold change optimization of a four-layer CRISPRa cascade. Left: The first and third internal nodes of the cascade were varied between 40 and 160 pM and 85 and 340 pM, respectively. The input node, second internal node, and output node were held constant at 0 or 15 pM, 0.2 nM, and 10 nM, respectively. Right: Fold change between with and without scRNA input for each CRISPRa cascade. (E) Signal propagation through deep CRISPRa/i circuits. Left: CRISPRa cascades with increasing depth. Input and output node concentrations were held constant across all cascades at 0 or 15 pM and 10 nM, respectively. All of the parallel cascade scRNA outputs are connected to the same RFP node. All node concentrations are tabulated in SI Appendix, Table S5. Right: Propagation efficiency and signal delay are shown as a function of circuit depth (SI Appendix, Methods S3). (F) Construction of wide CRISPRa/i circuits. Left: CRISPRa cascades with increasing width. Input and output node concentrations were held constant across all cascades at 0 or 15 pM and 10 nM, respectively. Right: The concentration of each internal node was held at 0.2 nM as circuit width increased (blue), or the internal node concentration was scaled down proportionally to the width of the circuit (red), such that each internal node concentration is 0.2/n nM, where n is the number of parallel cascades. Fold activation is given relative to a single CRISPRa cascade (SI Appendix, Methods S3). For all panels, values represent the mean ± SD of three technical replicates.
Deep CRISPRa circuits.
We investigated whether deep multilayer cascades could be implemented using the improved CRISPRa/i nodes. We first built a two-layer CRISPRa cascade by tuning the expression levels of the input and internal CRISPRa/i nodes and achieved up to 127-fold activation (SI Appendix, Figs. S8 and S9). An excess of either the input or internal node led to decreased performance of the cascade, potentially due to scRNA competition for binding to dCas9 (31, 60). Next, we assembled four-layer activation cascades. To compare circuit performance and dynamics in response to scRNA inputs, we measured RFP expression and time to maximum expression rate (tmax) (Fig. 3C, Inset; SI Appendix, Fig. S10) (SI Appendix, Methods S3). If the input signal propagates faster than the leak from the rest of the nodes, CRISPRa-dependent expression through the network accelerates, reducing tmax. Therefore, a larger tmax between the ± input conditions (Δtmax) corresponds with improved circuit function.
For a cascade with equal node concentrations at each layer, we found no input-dependent change in maximum RFP expression and a Δtmax of 110 min (Fig. 3 C, Middle). We tuned node concentrations by either decreasing (denoted “D”) or increasing (denoted “I”) the concentration of each subsequent node as depth increased (Fig. 3 C, Left). These assemblies D and I had the lowest and highest expression levels, respectively (Fig. 3 C, Middle). These assemblies also had tmax significantly accelerated compared to their no input conditions (Δtmax of 85 min and 165 min, and P = 0.01 and P = 3 × 10−4, respectively) (Fig. 3 C, Right). When scRNA-promoter dose–response curves were used to inform circuit assembly, we obtained threefold higher activated expression than assembly D and 4.5-fold lower basal expression than assembly I, with a similar overall fold activation and Δtmax (Fig. 3C).
We interrogated the impact of high-performing promoters on circuit function by exchanging the high-performing promoter of the second internal node for a previously characterized promoter with leakier basal expression (33). While the leakier promoter was sufficient for constructing functional two-layer cascades (33), the four-layer cascade was no longer input-responsive, as indicated by the fact that expression level and tmax were the same with or without input scRNA. In contrast, all of the assemblies containing high-performing promoters were input-responsive, underscoring the importance of high-performing promoters for building deep transcriptional circuits.
To improve the fold activation of the four-layer cascade, we tuned the concentrations of individual nodes in the cascade. To reduce the experimental search space, we held the second node constant and tuned the concentrations of the first and third internal nodes. With this approach, the tuned four-layer cascade achieved 16-fold activation compared to twofold activation of the initial cascades (Fig. 3 C and D). Qualitatively, higher concentrations of the third node resulted in higher activated states, while lower concentrations of the first node minimized basal expression (SI Appendix, Fig. S11). Notably, these circuits maintained roughly 80% of their maximal activity even when node concentrations varied by nearly 50% (Fig. 3D and SI Appendix, Fig. S11), showing that they are robust to changes in the amounts of gRNAs and targets. Overall, these results indicate that timing and expression level of multilayer, input-responsive circuits can be controlled through node concentration tuning.
We investigated how input signals are communicated through increasingly deep circuits by measuring signal propagation and signal delay at each layer (Fig. 3 E, Left). We quantify the percent of signal propagated by calculating the fold activation of the full cascade output divided by the activation from the input layer (SI Appendix, Methods S3). We define signal delay as the difference between tmaxFC of the cascade input and output layer, where tmaxFC is the time needed to reach the maximum fold activation (SI Appendix, Methods S3). Signal propagation was sustained above 80% until the 4th layer was added, after which it decreased rapidly (Fig. 3 E, Right). Nevertheless, we observed measurable output differences in circuits of up to six layers. The two-layer cascade gave no significant difference in signal delay compared to a single-layer CRISPRa reaction (P = 0.9). This may suggest there is a slow step in output production, such as fluorophore maturation, that masks the effect of the second layer. Beyond two layers, the signal delay increased with subsequent additions of the third, fourth, and sixth layers (Fig. 3 E, Right), averaging ~50 min/layer, but ranging from ~20 to 100 min.
We investigated whether the signal propagation and signal delay at each layer could be explained by the performance of individual nodes. We used the maximum fold-change of individual nodes from the dose–response curves (SI Appendix, Fig. S7) to predict the delay and signal propagation at the subsequent level (SI Appendix, Methods S4). The model-derived predictions showed high correspondence with the signal propagation and delay at each of the six layers (R2 = 0.92 and R2 = 0.91, respectively) (SI Appendix, Fig. S12). These results are consistent with the ideas that high-performing nodes can quickly activate subsequent layers, propagate signals efficiently, and be assembled into deep circuits.
Wide CRISPRa circuits.
To identify conditions under which orthogonal nodes can compose wide CRISPRa circuits, we constructed one, two, three, and four parallel three-layer cascades operating in the same CFS reaction. We used a single input to activate the downstream nodes, and measured circuit performance by connecting all of the parallel cascade scRNA outputs to the same RFP node (Fig. 3 F, Left). When we maintained the internal node concentrations constant across parallel cascades, we observed up to 66% decrease in output fold activation as the width of the circuit increased from one to four cascades (Fig. 3 F, Right). This decrease came largely from higher output levels in the absence of scRNA input (SI Appendix, Fig. S13), most likely due to higher basal expression of internal scRNAs. We then constructed the same circuits and tuned the node concentrations proportionally to the number of parallel cascades, effectively maintaining the total node concentration constant. When constructed in this manner, we found no statistically significant difference in the fold activation across cascades of different widths (P = 0.61) (Fig. 3 F, Right). Hence, by tuning the concentration of orthogonal CRISPRa/i nodes, we show an arbitrary number of parallel circuits with as many as nine nodes may be regulated.
Developing Activatable Promoters for Blue Light–Responsive CRISPRa/i Circuits.
High-performing blue light–responsive promoters.
Having demonstrated that high-performing CRISPRa promoters can be generated through sequential screening, we tested the same approach for engineering activatable promoters responsive to other transcriptional activators. The EL222 transcriptional activator interactions with the RNAP and the DNA binding site are well-characterized, making it a suitable model system for developing optogenetic inputs for CRISPRa/i circuits (Fig. 4A) (61–64). Briefly, EL222 binds an 18-bp sequence upstream of the −35 region of the luxI promoter and subsequently recruits RNAP through interactions with the α and σ subunits (64). We mutagenized the luxI minimal promoter (Fig. 4A, Methods S11), and screened variants in E. coli in dark and light to select for high dynamic range (Fig. 4B). Starting with a dynamic range of less than twofold, we observed up to fourfold dynamic range in response to blue light. Similar to our CRISPRa promoter screens, minimal promoters with very low (BBa_J23113) or very high (BBa_J23119) basal expressions exhibited low dynamic range in response to blue light.
Fig. 4.
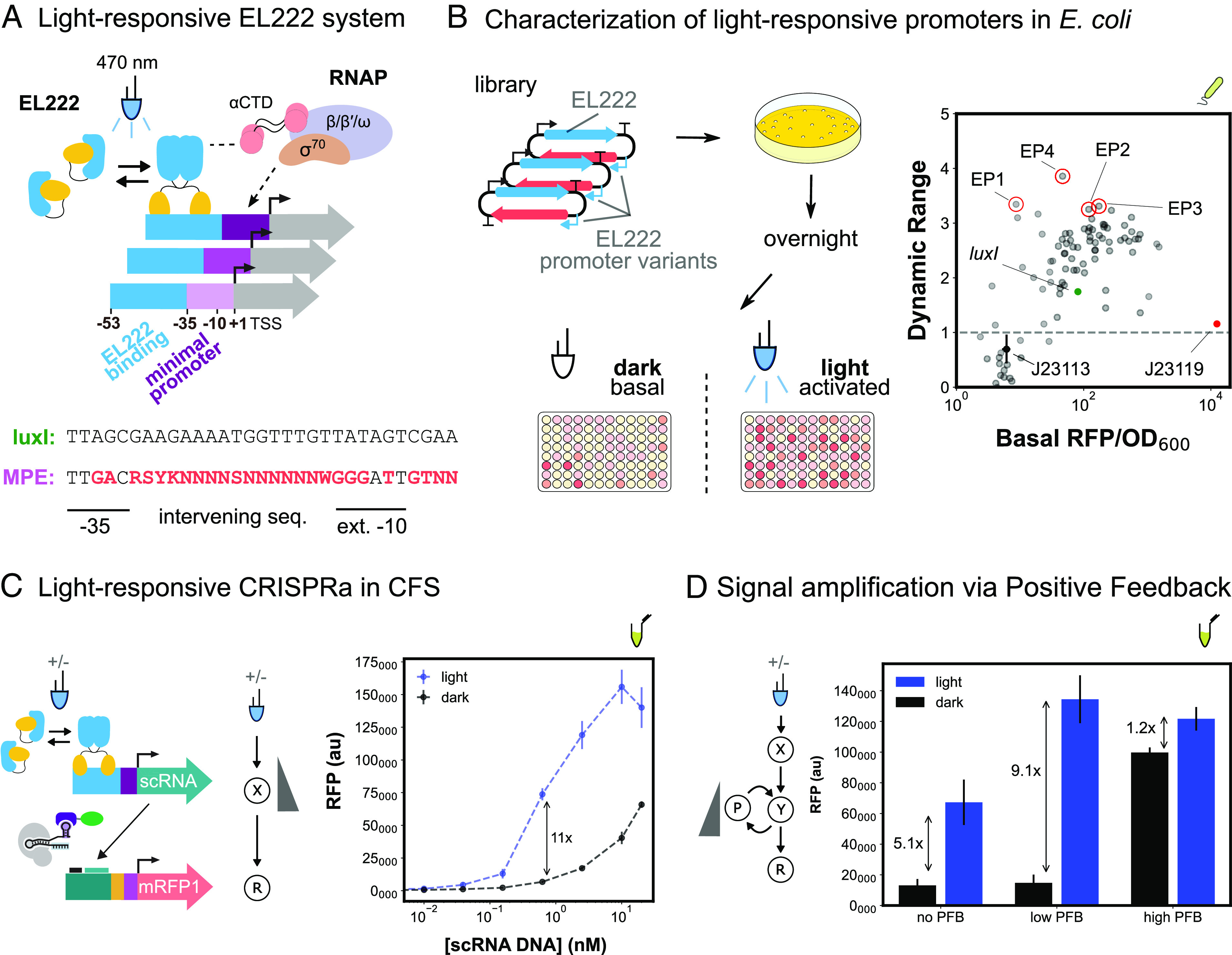
Developing activatable promoters for blue light–responsive CRISPRa/i circuits. (A) Schematic of EL222 light-responsive promoter system and library design. EL222 transcription factor dimerizes in response to 470-nm light and binds a specific sequence upstream of the minimal promoter. EL222 then recruits RNAP through interactions with the α-CTD domain. The minimal promoter library design is based on the original luxI promoter and previous minimal promoter libraries (Methods S11). (B) Characterization of light-responsive promoters in E. coli. Left: Blue-light promoter screening (Methods S13). EL222 protein and promoter library are expressed from a single plasmid. Assembly and screening are carried out as previously described. Basal and activated expression levels are measured from cultures not exposed or continuously exposed to blue light, respectively. Right: Basal expression and dynamic range of blue-light promoter variants (nMP3 = 96). The gray dash line defines promoter variants with equal activated and basal expression levels, indicating they are not activated by EL222. The J23119 minimal promoter (red) and J23113 (black) are examples of nonactivatable promoters. Variants with improved performance (red circles) compared to the original luxI promoter (green) were selected for use in CFS. (C) Light-responsive CRISPRa in CFS. Left: EL222 scRNA expression from an engineered blue-light promoter and downstream CRISPRa. Reactions contain 8 nM and 10 nM of EL222 and RFP plasmids respectively. Right: Titration of blue light–inducible scRNA plasmid concentration to maximize the fold change between blue light–dependent CRISPRa (blue) and CRISPRa due to scRNA leak in the dark (black). (D) Improvement of blue-light CRISPRa dynamic range through the construction of a PFB circuit. Left: Blue light–responsive CRISPRa cascade with PFB. PFB is achieved by including a downstream node that expresses a scRNA targeting an upstream node Reactions contain 15 nM and 10 nM of EL222 and RFP plasmids respectively. Right: Blue light–dependent CRISPRa (blue) and CRISPRa due to scRNA leak in the dark (black). The amount of PFB was tuned by adjusting the concentration of the PFB node. “No”, “Low”, and “High” PFB concentrations correspond to 0, 3 pM, and 2 nM, respectively. For all panels, values represent the mean ± SD of three technical replicates.
We selected 4 variants with >twofold higher dynamic range than the luxI minimal promoter and characterized them in CFS. These variants yielded a 34.1-fold difference in expression between light and dark, compared to just 6.2-fold for the original luxI minimal promoter (SI Appendix, Fig. S14). This improvement comes largely from a reduction in the basal expression from the blue-light promoter, suggesting we successfully minimized the RNAP-minimal promoter interactions without weakening EL222-promoter interactions. More importantly, these results demonstrate this approach for engineering actionable promoters is applicable to other transcriptional activation systems.
Blue light–responsive CRISPRa/i circuits.
We evaluated whether the engineered blue-light promoter transcription levels were suitable for expressing gRNAs for CRISPRa/i circuits. We titrated gRNA-expressing plasmid concentrations and compared RFP expression across dark and light conditions. For CRISPRi, the highest light-dependent change in repression was 50% (SI Appendix, Fig. S15). For CRISPRa, the highest light-dependent fold activation was 11-fold (Fig. 4C), which improved to 14-fold upon increasing EL222 plasmid concentration (SI Appendix, Fig. S16).
We then constructed a light-dependent activation cascade and observed only a fivefold activation in response to blue light (Fig. 4D). To improve the fold activation, we implemented a PFB loop in which a downstream node expresses a scRNA directing CRISPRa to an upstream node. We expected the degree of PFB in the system to be tunable by titrating the PFB node, with high concentrations of this node resulting in activation in the absence of blue light. When optimally tuned, the PFB loop increases the light-dependent CRISPRa output levels almost twofold (Fig. 4D). Excess PFB node led to a 7.6-fold increase in basal expression, decreasing the light-dependent activation to 1.2-fold. These results highlight that rationally designed genetic circuits built from engineered activatable promoters can be used to improve the dynamic range of input-responsive signal processing modules.
Engineered Activatable Promoters Enable Conditional CRISPRa Dependent on PPIs.
The versatility of PPI-mediated genetic regulation for coupling peptide or small-molecule binding to transcriptional outputs has long attracted interest (65–71). In principle, CRISPRa assemblies incorporating dimerizing protein domains could be utilized for PPI-dependent transcriptional activation in bacterial CFS. However, realization of these systems has been limited by strict target site requirements and a low dynamic range of activatable promoters (46, 72). We reasoned that the high dynamic range promoters developed here would allow us to screen for otherwise hard-to-detect functional target sites and component stoichiometries and, if successful, achieve effective conditional, PPI-dependent CRISPRa.
Development of conditional CRISPRa systems.
As experimental testbeds, we incorporated three previously characterized protein–protein heterodimerization domains into our CRISPRa system: the synthetic coiled-coil SYNZIP 5/6 pairs (73), the abscisic acid (ABA) responsive ABI–PYL1 (65, 74, 75), and the gibberellic acid (GA) responsive GID1–GAI (65, 76). We fused these heterodimerization domains to SoxS and MCP to enable conditional recruitment of SoxS to the CRISPRa complex. We generated the MCP-SZP6 and SoxS-SZP5 domains for SYNZIP-CRISPRa, the MCP-ABI and SoxS-PYL1 domains for ABA-CRISPRa, and MCP-GAI and SoxS-GID1 for GA-CRISPRa (Fig. 5A). In the original J3 promoter context, we observed 5.7-fold activation when cognate SYNZIPs were fused to the C termini of MCP (MCP-SYNZIP) and SoxS (SoxS-SYNZIP) (SI Appendix, Fig. S17). This combination of orientations gave the best activation, compared to 2.8-fold when MCP was fused in the opposite orientation (SYNZIP-MCP & SoxS-SYNZIP), and 1.4-fold when SoxS was fused in the opposite orientation (MCP-SYNZIP & SYNZIP-SoxS) (SI Appendix, Fig. S17). The fold activation of ABA-CRISPRa was also maximized when SoxS was at the N terminus (SI Appendix, Fig. S17, Right), therefore we moved forward with all SoxS N-terminal fusions. For ABA- and GA-CRISPRa, only C-terminal MCP fusions were tested due to the MCP-SYNZIP result and the strong precedent for using C-terminal MCP fusions in CRISPRa systems (50, 77).
Fig. 5.
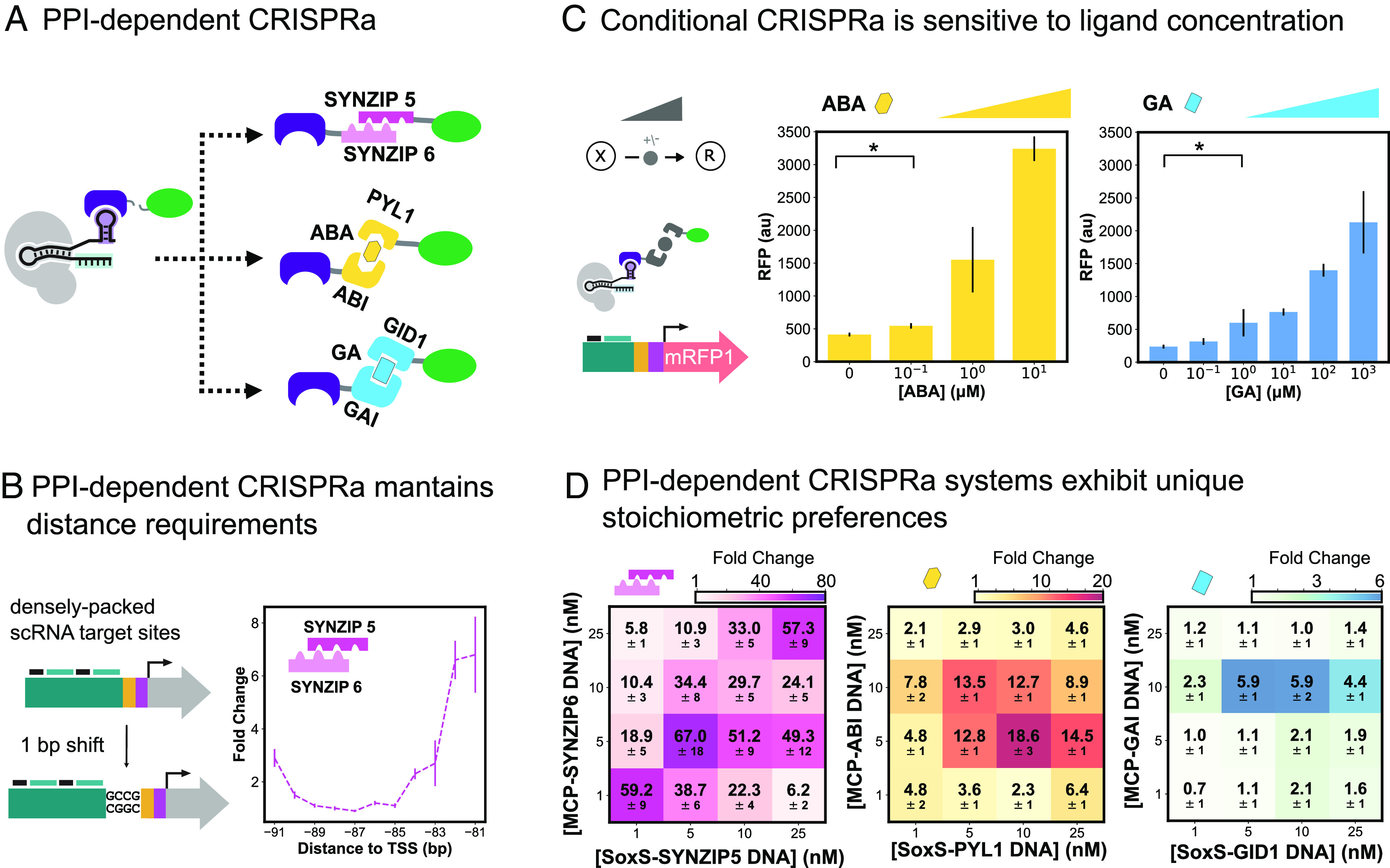
Engineered activatable promoters enable PPI-dependent conditional CRISPRa. (A) Schematic of different PPI-dependent CRISPRa systems. MCP-SoxS fusion is split and the two proteins are instead fused to one end of a heterodimerization domain. The heterodimerization domains used to build PPI-dependent CRISPRa systems are the SYNZIP5/SYNZIP6 pair, the ABA-responsive ABI/PYL1 domain, and the gibberellic acid (GA)-responsive GAI/GID1 domain. (B) Distance requirements of PPI-dependent CRISPRa. Left: Engineered promoter containing densely packed scRNA target sites and single base pair 5′ additions allows for CRISPRa targeting between −81 and −111 bp from the TSS. Right: Testing SYNZIP-CRISPRa between −81 and −91 bp from the TSS. SYNZIP-CRISPRa components are expressed at 5 nM. Fold change is calculated relative to an off-target scRNA for each promoter variant. (C) Tuning conditional CRISPRa response through titration of small molecule concentration. For ABA- and GA- CRISPRa, the corresponding small molecule was titrated between 0 and 10 or 0 and 103 μM, respectively to find the optimal concentration. ABA- and GA-CRISPRa components are expressed at 10 nM. (D) Improving PPI-dependent and conditional CRISPRa response by optimizing component stoichiometries. The concentration of the plasmids expressing the MCP and SoxS components for each dimerization system was varied 1 to 25 nM and tested combinatorially to find the best ratio of the two heterodimers. ABA is added at 10 μM and GA is added at 103 μM. Fold change is given relative to a reaction with no MCP and SoxS plasmids added.
CRISPRa operates narrowly within -101:-71 bp from the TSS in a phase-dependent manner (46). We tested whether the introduction of an additional protein linkage into MCP-SoxS affects the relative scRNA target site requirements (72). We designed a CRISPRa promoter with densely packed scRNA target sites every 10 bp (50), as well as variants with 1-bp frameshifts (46, 72) to allow screening with single base pair resolution between -111:-81 bp from the TSS (Fig. 5B and SI Appendix, Table S1). Surprisingly, SYNZIP-CRISPRa maintains the same preference for the targeting site −81 bp from the TSS and the same stringent 10- to 11-bp phase dependency seen in conventional CRISPRa (Fig. 5B and SI Appendix, Fig. S18). We then tested SYNZIP-CRISPRa using a high-performing promoter with an optimal target site (HP3, SI Appendix, Table S1) and found a 5.4-fold improvement compared to the original J3 promoter (SI Appendix, Fig. S19).
To create functional systems for coupling peptide or small-molecule binding to CRISPRa-directed transcriptional outputs, we identified permissible small-molecule input and component expression levels. For ABA- and GA-CRISPRa, small-molecule titrations showed that ABA-CRISPRa is responsive between 0.1 and 10 μM with up to 7.9 fold activation, and GA-CRISPRa is responsive between 1 and 1,000 μM with up to 9.0 fold activation (Fig. 5C). We screened dimer stoichiometries and expression levels by surveying a range of concentrations for the MCP- and SoxS-fused components. SYNZIP-CRISPRa performs the best of the three systems, giving a maximal activation of 67-fold compared to a control without MCP or SoxS. Even at low concentrations of MCP- and SoxS-fused components, SYNZIP-CRISPRa still achieves 59-fold activation (Fig. 5 D, Left). ABA-CRISPRa gave a maximum activation of 18.6-fold (Fig. 5 D, Middle). For GA-CRISPRa, the maximum activation of 5.9-fold was accessible in a relatively narrow range of component concentrations (Fig. 5 D, Right). For all three PPI-dependent CRISPRa systems, higher expression of the MCP- and SoxS-fused heterodimer components did not necessarily improve activation. In line with the behaviors of natural scaffolds (78, 79), we observed a unique optimal concentration for each PPI-dependent CRISPRa system. The differences between systems may be due to the different affinities of each PPI.
Engineering Multi-Input CRISPRa/i Circuits.
Multilayer and multi-input circuits with conditional CRISPRa.
We built two types of input-responsive circuits to explore the use of conditional CRISPRa for multi-input and multilayer input processing: an AND-like logic gate and a CRISPRa cascade. We began by characterizing the scRNA dose–response curve of the CRISPRa systems. For both conditional CRISPRa systems, the amount of scRNA needed to saturate the CRISPRa response was similar to that of direct CRISPRa (SI Appendix, Figs. S7 and S20). We tested the orthogonality of the small molecule systems to evaluate whether they could be used together for independent gene regulation (Fig. 6A). ABA-CRISPRa is highly specific to its target ligand, showing no significant activation in the presence of GA (P = 0.21). GA-CRISPRa showed 3.1-fold cross-activation from ABA, in line with reports in eukarya (65). Nonetheless, GA-CRISPRa maintained a threefold higher specificity for its cognate ligand, giving 10.5-fold activation from GA. These results suggested that the ABA- and GA-CRISPRa systems could be used for orthogonal gene regulation.
Fig. 6.
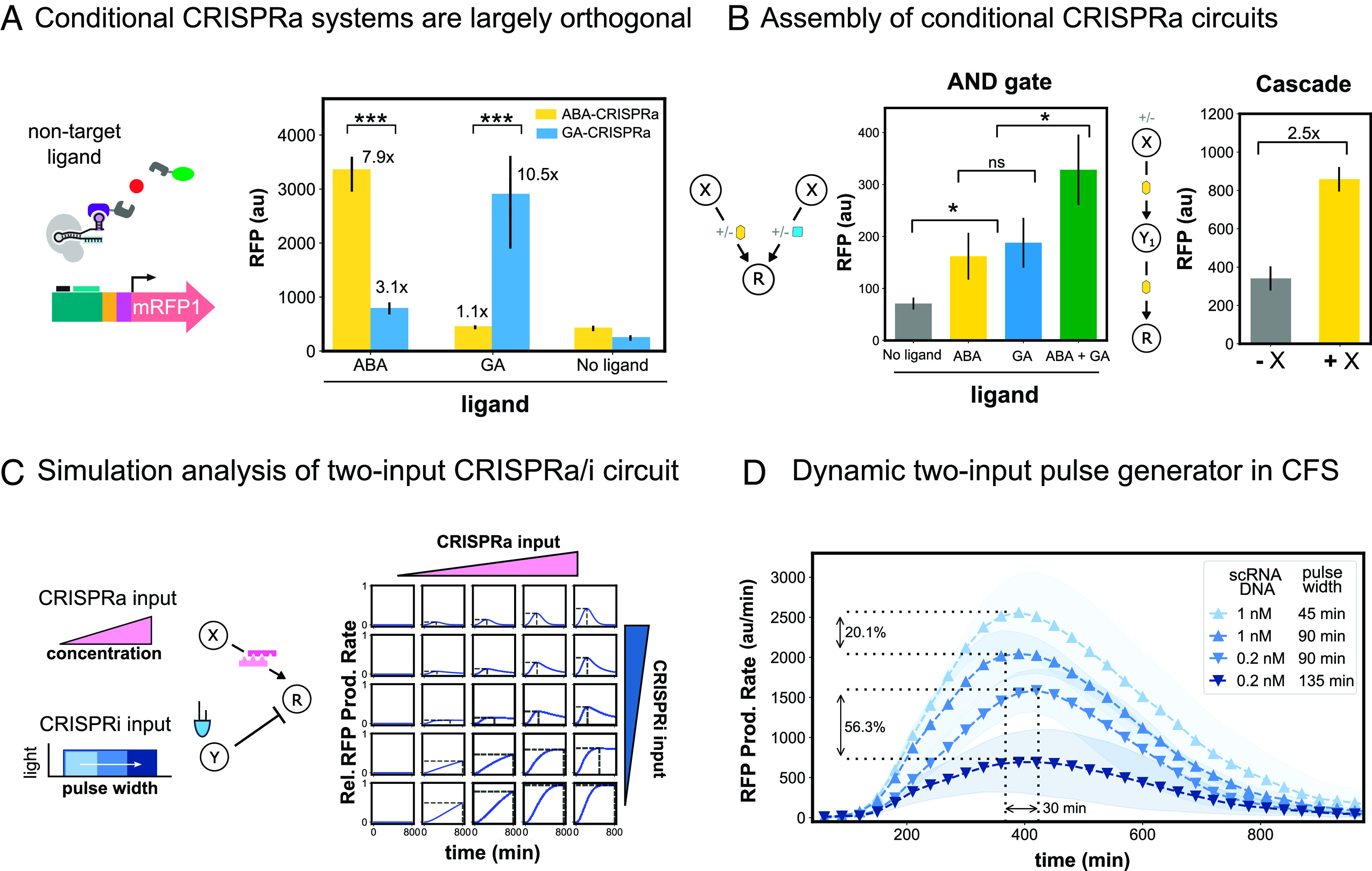
Engineering multi-input CRISPRa/i circuits. (A) Conditional CRISPRa response to noncognate ligands. The orthogonality of the small molecule-responsive conditional CRISPRa systems was tested by adding either the corresponding or noncorresponding small molecule to cell-free reactions containing the components for ABA- or GA-CRISPRa. All components are added at their respective optimal screened concentrations. ABA is added at 10 μM and GA is added at 103 μM. (B) Assembly of conditional CRISPRa circuits. For both circuits, all components are added at their respective optimal screened concentrations. ABA is added at 10 μM and GA is added at 103 μM. Left: AND-like behavior was constructed by adding the components for both ABA- and GA- CRISPRa in a cell-free reaction. Right: The CRISPRa cascade was assembled by using ABA-CRISPRa to activate expression of both the first and second nodes in an activation cascade. The first node was added at either 0.05 or 0 nM, and the internal and output nodes were added at 10 nM. (C) Simulation analysis of a two-input CRISPRa/i circuit using SYNZIP5/SYNZIP6 heterodimerization mediated-CRISPRa and blue-light CRISPRi (CFS Blue-light CRISPRa/imodeling). (D) SYNZIP-CRISPRa and blue-light CRISPRi were integrated to construct a tunable pulse generator. The amount of CRISPRa was tuned by adding either 0.2 nM or 1 nM of constitutively expressed scRNA plasmid to the CFS reaction. The sgRNA targeting RFP for CRISPRi was driven from the blue light–responsive engineered EL222 promoter. The amount of CRISPRi was tuned by adjusting the time of blue-light exposure between 45 and 135 min. RFP production rates (SI Appendix, Methods S3) are plotted as a function of CRISPRa and CRISPRi inputs. For all panels, values represent the mean ± SD of three technical replicates.
We constructed a multi-input circuit for AND logic by coexpressing components for ABA- and GA-CRISPRa (Fig. 6 B, Left). The addition of either ABA or GA resulted in twofold activation compared to the no-ligand condition (P = 0.03). Therefore, we specified that the average of the technical replicates must be above or below twofold activation for the circuit to be considered ON or OFF. Consistent with AND-like logic, the circuit generated 4.5-fold activation in the presence of both ligands, a level of activation that was clearly distinct from either of the one-input states (P = 0.03). We built a multilayer conditional CRISPRa cascade by having both internal layers dependent on ABA. The cascade gave 2.5-fold activation upon addition of ABA, showing that conditional CRISPRa can also support multilayer information processing (Fig. 6 B, Right).
Two-input dynamic pulse generator.
Synthetic biologists aim to recreate complex, dynamic signaling networks with multiple input-responsive regulators to tightly program the expression timing and magnitude of downstream targets (2, 3, 80). As a proof of concept, we aimed to engineer a GRN that integrates blue-light CRISPRi with PPI-dependent CRISPRa. The result would be a tunable pulse generator with two-input control over the level and timing of gene expression (Fig. 6 C, Left). Because SYNZIP-CRISPRa has high-fold activation (SI Appendix, Fig. S20), we expected that it could be well suited for integration with blue-light CRISPRi.
We first employed simulation analysis to identify system designs for combining two inputs and dynamically regulating reporter gene expression. We built upon a coarse-grained mechanistic model of CRISPRa/i regulation [32] by introducing blue-light pulses regulating gRNA expression (Methods S14). We simulated changes in the pulse width as CRISPRi inputs and scRNA concentration as CRISPRa inputs. By evaluating changes in reporter production rates, our analysis suggested that there are broad ranges of CRISPRi and CRISPRa input parameter values compatible with multi-input-responsive regulatory control (Fig. 6 C, Left).
We proceeded to experimentally validate the results of the simulation analysis. To dynamically tune CRISPRi, we changed the blue-light exposure time. To tune SYNZIP-CRISPRa, we changed the scRNA-expressing plasmid concentration. In all cases, we first kept the CFS in the dark for one hour to allow for EL222 expression. As predicted by the model, the RFP production rate pulse was tunable by the scRNA plasmid concentration and the blue-light exposure time (Fig. 6D). When compared across conditions with the same CRISPRa input, higher CRISPRi input led to 20 to 56% lower maximum production rates. In conditions with the same CRISPRi input, higher CRISPRa input increased maximum production rates by 20 to 40 min. Collectively, these results highlight the potential of input-responsive CRISPRa/i GRNs for complex signal processing applications.
Discussion
Natural biological systems have evolved GRNs containing wide ranges of activatable promoters that enable dynamic responses to changing environmental conditions. Engineering activatable promoters has traditionally been thought to involve a trade-off between basal and activated expression levels (36, 42, 46). In this work, we show that this trade-off can be relaxed to generate activatable promoters with both lower basal and higher activated expression levels than previously possible (Figs. 1B and 2B). Sequential screening of promoter regions allowed us to overcome context effects and identify high-performing activatable promoters (Fig. 2D). With this approach, we successfully engineered a suite of orthogonal CRISPRa promoters that match the basal and activated expression levels of the canonical Tet–inducible system (SI Appendix, Fig. S21) and exceed those of the IPTG-inducible system (81).
The E. coli transcriptional network is governed by a hierarchical structure containing nine layers of regulation (1). Engineered activatable promoters allowed us to build multilayer CRISPRa/i GRNs in E. coli-based CFS with depths and widths significantly larger than the state of the art (33, 82), approaching the complexity of natural GRNs. Specifically, a 33-fold improvement in promoter dynamic range resulted in 80% lower signal degradation in two-layer cascades (33), and enabled deep GRNs with up to six layers of regulation (Fig. 3E). Additionally, we demonstrated wide GRNs regulating up to four parallel cascades (Fig. 3F), indicating that the CRISPRa/i framework is well suited for the design of wide control circuits for parallel computing and multigene regulation (26). We also showed that these circuits can be implemented in E. coli by constructing three-layer activation cascades with the internal nodes expressed from either high or low copy number plasmids (SI Appendix, Fig. S22). The E. coli cascades maintained more than threefold activation regardless of the copy numbers of the internal node plasmids (SI Appendix, Fig. S22), demonstrating the high correspondence between CFS and in vivo component function (6, 83). To our knowledge, these represent the deepest CRISPRa cascades in CFS and in vivo. Further improvements in GRN complexity may be limited by resource constraints, including upstream gRNAs outcompeting downstream gRNAs for dCas9 binding (SI Appendix, Figs. S8 and S22). Strategies to dynamically regulate upstream gRNA expression, such as reversing CRISPRa complex binding or implementing negative autoregulation motifs (84), could enable even larger GRNs.
Biological systems continuously monitor and process environmental signals by using signal transduction modules as inputs to complex GRNs (85, 86). Our work provides a general framework for optimizing transcriptional activation systems at the promoter level and integrating them into CRISPRa/i GRNs. Promoter engineering of the optogenetic EL222 system enabled high light-dependent dynamic ranges (Fig. 4B and SI Appendix, Fig. S14), with relevant expression levels for downstream applications (Fig. 4C and SI Appendix, Fig. S15). Through inducible gRNA expression, we demonstrated input signal modulation with various GRN topologies, including PFB loops and CRISPRa/i cascades (Fig. 4D), as well as integration of different signal transduction modules into the same CRISPRa/i GRN (Fig. 6B). Overall, our work highlights the potential for achieving more complex biocomputing functions, including multi-input AND and NOT gates for targeted therapeutics and next-generation biosensors (87), through multi-input CRISPRa/i GRNs.
PPIs have been used widely to execute complex, input-responsive functions in eukaryotes (88–92) Developing similar systems in prokaryotes has been difficult, and the development of high dynamic range promoters allowed us to successfully prototype and optimize conditional CRISPRa systems in E. coli-based CFS. Implementation of novel conditional CRISPRa systems may be streamlined by the fact that all systems tested here are effective when targeted −81 bp from the TSS (Fig. 5B and SI Appendix, Fig. S18), despite the presence of additional PPIs up to 500 amino acids in length. Additionally, conditional CRISPRa fold activation is proportional to the strength of the PPI (SI Appendix, Table S2) (73, 76, 93–97). informing the a priori selection of heterodimers for use in conditional CRISPRa. Collectively, our work suggests that other heterodimerization domains could be implemented, with minimal prototyping, as signal transduction modules for CRISPRa/i GRNs for multiplexed biosensing or screening of PPIs in CFS.
Our workflow for activatable promoter engineering enables the dynamic specification of expression levels for large networks of orthogonal gene targets. The classes of deep, wide, and input-responsive CRISPRa/i GRNs developed here have immediate application in CFS for investigating the rules of genetic circuit design (10, 13) and biological information processing (20–23), as well as for building dynamic, multienzyme expression programs for self-assembling bioproduction platforms (17–19, 98, 99). Moreover, CRISPRa/i GRNs could be integrated with existing field-deployable medical diagnostics and environmental monitors to enable complex, multi-input signal processing (4, 15, 16, 100). Moving forward, this work could serve as a stepping stone for building entirely synthetic cells and engineered living materials with GRNs that match or go beyond the complexity of natural systems.
Methods
Please see SI Appendix, Methods for a full description of the methods used.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank members of the J.M.C. and J.G.Z. groups for advice, materials, and comments on the manuscript. We also thank the Thachuk and Baker labs for access to their Echo instruments. This work was supported by US NSF Award MCB 2032794 (J.M.C. and J.G.Z.), NSF Award CBET 1844152 (J.M.C.), NSF Award EF-1935087 (J.M.C.), US Department of Energy (DOE) BETO DE-EE0008927 (J.M.C. and J.G.Z.) and DOE ARPA-E DE-AR00002387-1567 (J.M.C). This material is based upon work supported by the NSF Graduate Research Fellowship Program under Grant No. DGE-2140004 (D.A.B.). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the NSF.
Author contributions
D.A.B., R.A.L.C., B.I.T., C.K., C.J.M., J.G.Z., and J.M.C. designed research; D.A.B., R.A.L.C., B.I.T., C.K., and C.J.M. performed research; D.A.B., R.A.L.C., B.I.T., C.K., and C.J.M. analyzed data; and D.A.B., R.A.L.C., J.G.Z., and J.M.C. wrote the paper.
Competing interests
D.A.B., R.C., J.G.Z., and J.M.C are inventors on patents and/or patent applications filed by the University of Washington that describe promoter engineering, conditional CRISPRa technologies, and CRISPRa/i circuits in prokaryotic systems. J.G.Z. and J.M.C are members of the Wayfinder Biosciences scientific advisory board.
Footnotes
This article is a PNAS Direct Submission. C.D.S. is a guest editor invited by the Editorial Board.
Contributor Information
Jesse G. Zalatan, Email: zalatan@uw.edu.
James M. Carothers, Email: jcaroth@uw.edu.
Data, Materials, and Software Availability
Supporting Information
References
- 1.Ma H.-W., et al. , An extended transcriptional regulatory network of Escherichia coli and analysis of its hierarchical structure and network motifs. Nucleic Acids Res. 32, 6643–6649 (2004), 10.1093/nar/gkh1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cai L., Dalal C. K., Elowitz M. B., Frequency-modulated nuclear localization bursts coordinate gene regulation. Nature 455, 7212 (2008), 10.1038/nature07292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benzinger D., Ovinnikov S., Khammash M., Synthetic gene networks recapitulate dynamic signal decoding and differential gene expression. Cell Syst. 13, 353–364.e6 (2022), 10.1016/j.cels.2022.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Jung J. K., et al. , Cell-free biosensors for rapid detection of water contaminants. Nat. Biotechnol. 38, 12 (2020), 10.1038/s41587-020-0571-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karim A. S., et al. , In vitro prototyping and rapid optimization of biosynthetic enzymes for cell design. Nat. Chem. Biol. 16, 912–919 (2020), 10.1038/s41589-020-0559-0. [DOI] [PubMed] [Google Scholar]
- 6.Lehr F.-X., et al. , Cell-free prototyping of AND-logic gates based on heterogeneous RNA activators. ACS Synth. Biol. 8, 2163–2173 (2019), 10.1021/acssynbio.9b00238. [DOI] [PubMed] [Google Scholar]
- 7.Moore S. J., et al. , Rapid acquisition and model-based analysis of cell-free transcription–translation reactions from nonmodel bacteria. Proc. Natl. Acad. Sci. U.S.A. 115, E4340–E4349 (2018), 10.1073/pnas.1715806115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swank Z., Maerkl S. J., CFPU: A cell-free processing unit for high-throughput, automated in vitro circuit characterization in steady-state conditions. BioDesign Res. 2021, 2968181 (2021), 10.34133/2021/2968181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bassalo M. C., Liu R., Gill R. T., Directed evolution and synthetic biology applications to microbial systems. Curr. Opin. Biotechnol. 39, 126–133 (2016), 10.1016/j.copbio.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 10.Brophy J. A. N., Voigt C. A., Principles of genetic circuit design. Nat. Methods 11, 5 (2014), 10.1038/nmeth.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swank Z., Laohakunakorn N., Maerkl S. J., Cell-free gene-regulatory network engineering with synthetic transcription factors. Proc. Natl. Acad. Sci. U.S.A. 116, 5892–5901 (2019), 10.1073/pnas.1816591116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xia P.-F., Ling H., Foo J. L., Chang M. W., Synthetic genetic circuits for programmable biological functionalities. Biotechnol. Adv. 37, 107393 (2019), 10.1016/j.biotechadv.2019.04.015. [DOI] [PubMed] [Google Scholar]
- 13.Alon U., Network motifs: Theory and experimental approaches. Nat. Rev. Genet. 8, 450–461 (2007), 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 14.Jung J. K., et al. , Cell-free biosensors for rapid detection of water contaminants. Nat. Biotechnol. 38, 1451–1459 (2020), 10.1038/s41587-020-0571-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung J. K., Archuleta C. M., Alam K. K., Lucks J. B., Programming cell-free biosensors with DNA strand displacement circuits. Nat. Chem. Biol. 18, 385–393 (2022), 10.1038/s41589-021-00962-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen P. Q., et al. , Wearable materials with embedded synthetic biology sensors for biomolecule detection. Nat. Biotechnol. 39, 1366–1374 (2021), 10.1038/s41587-021-00950-3. [DOI] [PubMed] [Google Scholar]
- 17.Pardee K., et al. , Portable, on-demand biomolecular manufacturing. Cell 167, 248–259.e12 (2016), 10.1016/j.cell.2016.09.013. [DOI] [PubMed] [Google Scholar]
- 18.Stark J. C., et al. , On-demand biomanufacturing of protective conjugate vaccines. Sci. Adv. 7, eabe9444 (2021), 10.1126/sciadv.abe9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou C., Lin X., Lu Y., Zhang J., Flexible on-demand cell-free protein synthesis platform based on a tube-in-tube reactor. React. Chem. Eng. 5, 270–277 (2020), 10.1039/C9RE00394K. [DOI] [Google Scholar]
- 20.Adamala K. P., Martin-Alarcon D. A., Guthrie-Honea K. R., Boyden E. S., Engineering genetic circuit interactions within and between synthetic minimal cells. Nat. Chem. 9, 431–439 (2017), 10.1038/nchem.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aufinger L., Brenner J., Simmel F. C., Complex dynamics in a synchronized cell-free genetic clock. Nat. Commun. 13, 1 (2022), 10.1038/s41467-022-30478-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garamella J., Garenne D., Noireaux V., TXTL-based approach to synthetic cells. Methods Enzymol. 617, 217–239 (2019), 10.1016/bs.mie.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 23.Karzbrun E., Tayar A. M., Noireaux V., Bar-Ziv R. H., Synthetic biology. Programmable on-chip DNA compartments as artificial cells. Science 345, 829–832 (2014), 10.1126/science.1255550. [DOI] [PubMed] [Google Scholar]
- 24.Qi L. S., et al. , Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013), 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banerjee D., et al. , Genome-scale metabolic rewiring improves titers rates and yields of the non-native product indigoidine at scale. Nat. Commun. 11, 5385 (2020), 10.1038/s41467-020-19171-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim H., Bojar D., Fussenegger M., A CRISPR/Cas9-based central processing unit to program complex logic computation in human cells. Proc. Natl. Acad. Sci. U.S.A. 116, 7214–7219 (2019), 10.1073/pnas.1821740116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landberg J., Wright N. R., Wulff T., Herrgård M. J., Nielsen A. T., CRISPR interference of nucleotide biosynthesis improves production of a single-domain antibody in Escherichia coli. Biotechnol. Bioeng. 117, 3835–3848 (2020), 10.1002/bit.27536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reis A. C., et al. , Simultaneous repression of multiple bacterial genes using nonrepetitive extra-long sgRNA arrays. Nat. Biotechnol. 37, 1294–1301 (2019), 10.1038/s41587-019-0286-9. [DOI] [PubMed] [Google Scholar]
- 29.Santos-Moreno J., Schaerli Y., CRISPR-based gene expression control for synthetic gene circuits. Biochem. Soc. Trans. 48, 1979–1993 (2020), 10.1042/BST20200020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Westbrook A., et al. , Distinct timescales of RNA regulators enable the construction of a genetic pulse generator. Biotechnol. Bioeng. 116, 1139–1151 (2019), 10.1002/bit.26918. [DOI] [PubMed] [Google Scholar]
- 31.Clamons S., Murray R., Modeling predicts that CRISPR-based activators, unlike CRISPR-based repressors, scale well with increasing gRNA competition and dCas9 bottlenecking. bioXriv [Preprint] (2019), 10.1101/719278. Accessed 30 October 2022. [DOI]
- 32.Nielsen J., Keasling J. D., Engineering cellular metabolism. Cell 164, 1185–1197 (2016), 10.1016/j.cell.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 33.Tickman B. I., et al. , Multi-layer CRISPRa/i circuits for dynamic genetic programs in cell-free and bacterial systems. Cell Syst. 13, 215–229.e8 (2021), 10.1016/j.cels.2021.10.008. [DOI] [PubMed] [Google Scholar]
- 34.Gander M. W., Vrana J. D., Voje W. E., Carothers J. M., Klavins E., Digital logic circuits in yeast with CRISPR-dCas9 NOR gates. Nat. Commun. 8, 1 (2017), 10.1038/ncomms15459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marshall R., Noireaux V., Quantitative modeling of transcription and translation of an all- E. coli cell-free system. Sci. Rep. 9, 1 (2019), 10.1038/s41598-019-48468-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alper H., Fischer C., Nevoigt E., Stephanopoulos G., Tuning genetic control through promoter engineering. Proc. Natl. Acad. Sci. U.S.A. 102, 12678–12683 (2005), 10.1073/pnas.0504604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelly J. R., et al. , Measuring the activity of BioBrick promoters using an in vivo reference standard. J. Biol. Eng. 3, 4 (2009), 10.1186/1754-1611-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brewster R. C., Jones D. L., Phillips R., Tuning promoter strength through RNA polymerase binding site design in Escherichia coli. PLoS Comput. Biol. 8, e1002811 (2012), 10.1371/journal.pcbi.1002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fleur T. L., Hossain A., Salis H. M., Automated model-predictive design of synthetic promoters to control transcriptional profiles in bacteria. bioXriv [Preprint] (2021), 10.1101/2021.09.01.458561. Accessed 30 October 2022. [DOI] [PMC free article] [PubMed]
- 40.Lee D. J., Minchin S. D., Busby S. J. W., Activating transcription in bacteria. Annu. Rev. Microbiol. 66, 125–152 (2012), 10.1146/annurev-micro-092611-150012. [DOI] [PubMed] [Google Scholar]
- 41.Yu T. C., et al. , Multiplexed characterization of rationally designed promoter architectures deconstructs combinatorial logic for IPTG-inducible systems. Nat. Commun. 12, 325 (2021), 10.1038/s41467-020-20094-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cress B. F., et al. , Rapid generation of CRISPR/dCas9-regulated, orthogonally repressible hybrid T7-lac promoters for modular, tuneable control of metabolic pathway fluxes in Escherichia coli. Nucleic Acids Res. 44, 4472–4485 (2016), 10.1093/nar/gkw231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Groseclose T. M., Rondon R. E., Herde Z. D., Aldrete C. A., Wilson C. J., Engineered systems of inducible anti-repressors for the next generation of biological programming. Nat. Commun. 11, 1 (2020), 10.1038/s41467-020-18302-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu X., et al. , De novo design of programmable inducible promoters. Nucleic Acids Res. 47, 10452–10463 (2019), 10.1093/nar/gkz772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roney I. J., Rudner A. D., Couture J.-F., Kærn M., Improvement of the reverse tetracycline transactivator by single amino acid substitutions that reduce leaky target gene expression to undetectable levels. Sci. Rep. 6, 27697 (2016), 10.1038/srep27697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fontana J., et al. , Effective CRISPRa-mediated control of gene expression in bacteria must overcome strict target site requirements. Nat. Commun. 11, 1618 (2020), 10.1038/s41467-020-15454-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y., et al. , Tuning the dynamic range of bacterial promoters regulated by ligand-inducible transcription factors. Nat. Commun. 9, 64 (2018), 10.1038/s41467-017-02473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ross W., Aiyar S. E., Salomon J., Gourse R. L., Escherichia coli promoters with UP elements of different strengths: Modular structure of bacterial promoters. J. Bacteriol. 180, 5375–5383 (1998), 10.1128/JB.180.20.5375-5383.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shultzaberger R. K., Chen Z., Lewis K. A., Schneider T. D., Anatomy of Escherichia coli σ 70 promoters. Nucleic Acids Res. 35, 771–788 (2007), 10.1093/nar/gkl956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dong C., Fontana J., Patel A., Carothers J. M., Zalatan J. G., Synthetic CRISPR-Cas gene activators for transcriptional reprogramming in bacteria. Nat. Commun. 9, 1 (2018), 10.1038/s41467-018-04901-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hook-Barnard I. G., Hinton D. M., Transcription initiation by mix and match elements: Flexibility for polymerase binding to bacterial promoters. Gene Regul. Syst. Biol. 1, 275–293 (2007). [PMC free article] [PubMed] [Google Scholar]
- 52.Paget M. S., Helmann J. D., The σ70family of sigma factors. Genome Biol. 4, 203 (2003), 10.1186/gb-2003-4-1-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Censor Y., Pareto optimality in multiobjective problems. Appl. Math. Optim. 4, 41–59 (1977), 10.1007/BF01442131. [DOI] [Google Scholar]
- 54.Estrem S. T., Gaal T., Ross W., Gourse R. L., Identification of an UP element consensus sequence for bacterial promoters. Proc. Natl. Acad. Sci. U.S.A. 95, 9761–9766 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaal T., et al. , DNA-binding determinants of the alpha subunit of RNA polymerase: Novel DNA-binding domain architecture. Genes Dev. 10, 16–26 (1996), 10.1101/gad.10.1.16. [DOI] [PubMed] [Google Scholar]
- 56.Fontana J., Sparkman-Yager D., Zalatan J. G., Carothers J. M., Challenges and opportunities with CRISPR activation in bacteria for data-driven metabolic engineering. Curr. Opin. Biotechnol. 64, 190–198 (2020), 10.1016/j.copbio.2020.04.005. [DOI] [PubMed] [Google Scholar]
- 57.Lian J., HamediRad M., Hu S., Zhao H., Combinatorial metabolic engineering using an orthogonal tri-functional CRISPR system. Nat. Commun. 8, 1688 (2017), 10.1038/s41467-017-01695-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bolouri H., Davidson E. H., Transcriptional regulatory cascades in development: Initial rates, not steady state, determine network kinetics. Proc. Natl. Acad. Sci. U.S.A. 100, 9371–9376 (2003), 10.1073/pnas.1533293100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Banuett F., Signalling in the yeasts: An informational cascade with links to the filamentous fungi. Microbiol. Mol. Biol. Rev. 62, 249–274 (1998), 10.1128/MMBR.62.2.249-274.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen P.-Y., Qian Y., Del Vecchio D., “A model for resource competition in CRISPR-mediated gene repression” in 2018 IEEE Conference on Decision and Control (CDC) (IEEE, Miami, FL, 2018), pp. 4333–4338, 10.1109/CDC.2018.8619016. [DOI] [Google Scholar]
- 61.Camsund D., Jaramillo A., Lindblad P., Engineering of a promoter repressed by a light-regulated transcription factor in Escherichia coli. BioDesign Res. 2021, 9857418 (2021), 10.34133/2021/9857418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jayaraman P., et al. , Blue light-mediated transcriptional activation and repression of gene expression in bacteria. Nucleic Acids Res. 44, 6994–7005 (2016), 10.1093/nar/gkw548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Motta-Mena L. B., et al. , An optogenetic gene expression system with rapid activation and deactivation kinetics. Nat. Chem. Biol. 10, 196–202 (2014), 10.1038/nchembio.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zoltowski B. D., Motta-Mena L. B., Gardner K. H., Blue light-induced dimerization of a bacterial LOV-HTH DNA-binding protein. Biochemistry 52, 6653–6661 (2013), 10.1021/bi401040m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao Y., et al. , Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nat. Methods 13, 1043–1049 (2016), 10.1038/nmeth.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jain P. K., et al. , Development of light-activated CRISPR using guide RNAs with photocleavable protectors. Angew. Chem. Int. Ed. Engl. 55, 12440–12444 (2016), 10.1002/anie.201606123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kundert K., et al. , Controlling CRISPR-Cas9 with ligand-activated and ligand-deactivated sgRNAs. Nat. Commun. 10, 1 (2019), 10.1038/s41467-019-09985-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nihongaki Y., Kawano F., Nakajima T., Sato M., Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 33, 755–760 (2015), 10.1038/nbt.3245. [DOI] [PubMed] [Google Scholar]
- 69.Polstein L. R., Gersbach C. A., A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat. Chem. Biol. 11, 198–200 (2015), 10.1038/nchembio.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ying Z.-M., Wang F., Chu X., Yu R.-Q., Jiang J.-H., Activatable CRISPR transcriptional circuits generate functional RNA for mRNA sensing and silencing. Angew. Chem. Int. Ed. Engl. 59, 18599–18604 (2020), 10.1002/anie.202004751. [DOI] [PubMed] [Google Scholar]
- 71.Yu Y., et al. , Engineering a far-red light–activated split-Cas9 system for remote-controlled genome editing of internal organs and tumors. Sci. Adv. 6, eabb1777 (2020), 10.1126/sciadv.abb1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Villegas Kcam M. C., Tsong A. J., Chappell J., Rational engineering of a modular bacterial CRISPR–Cas activation platform with expanded target range. Nucleic Acids Res. 49, 4793–4802 (2021), 10.1093/nar/gkab211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thompson K. E., Bashor C. J., Lim W. A., Keating A. E., SYNZIP protein interaction toolbox: In vitro and in vivo specifications of heterospecific coiled-coil interaction domains. ACS Synth. Biol. 1, 118–129 (2012), 10.1021/sb200015u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cunningham-Bryant D., Sun J., Fernandez B., Zalatan J. G., CRISPR–Cas-mediated chemical control of transcriptional dynamics in yeast. ChemBioChem 20, 1519–1523 (2019), 10.1002/cbic.201800823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liang F.-S., Ho W. Q., Crabtree G. R., Engineering the ABA plant stress pathway for regulation of induced proximity. Sci. Signal. 4, rs2 (2011), 10.1126/scisignal.2001449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miyamoto T., et al. , Rapid and orthogonal logic gating with a Gibberellin-induced dimerization system. Nat. Chem. Biol. 8, 465–470 (2012), 10.1038/nchembio.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zalatan J. G., et al. , Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 160, 339–350 (2015), 10.1016/j.cell.2014.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Douglass E. F. Jr., Miller C. J., Sparer G., Shapiro H., Spiegel D. A., A comprehensive mathematical model for three-body binding equilibria. J. Am. Chem. Soc. 135, 6092–6099 (2013), 10.1021/ja311795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Levchenko A., Bruck J., Sternberg P. W., Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc. Natl. Acad. Sci. U.S.A. 97, 5818–5823 (2000), 10.1073/pnas.97.11.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang B., Barahona M., Buck M., Engineering modular and tunable genetic amplifiers for scaling transcriptional signals in cascaded gene networks. Nucleic Acids Res. 42, 9484–9492 (2014), 10.1093/nar/gku593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee T. S., et al. , BglBrick vectors and datasheets: A synthetic biology platform for gene expression. J. Biol. Eng. 5, 12 (2011), 10.1186/1754-1611-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garamella J., Marshall R., Rustad M., Noireaux V., The all E. coli TX-TL toolbox 2.0: A platform for cell-free synthetic biology. ACS Synth. Biol. 5, 344–355 (2016), 10.1021/acssynbio.5b00296. [DOI] [PubMed] [Google Scholar]
- 83.Borkowski O., et al. , Cell-free prediction of protein expression costs for growing cells. Nat. Commun. 9, 1457 (2018), 10.1038/s41467-018-03970-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Specht D. A., Cortes L. B., Lambert G., Overcoming leak sensitivity in CRISPRi circuits using antisense RNA sequestration and regulatory feedback. ACS Synth. Biol. 11, 2927–2937 (2022), 10.1021/acssynbio.2c00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kaplan S., Bren A., Zaslaver A., Dekel E., Alon U., Diverse two-dimensional input functions control bacterial sugar genes. Mol. Cell 29, 786–792 (2008), 10.1016/j.molcel.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Krishna S., Orosz L., Sneppen K., Adhya S., Semsey S., Relation of intracellular signal levels and promoter activities in the gal regulon of Escherichia coli. J. Mol. Biol. 391, 671–678 (2009), 10.1016/j.jmb.2009.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kempton H. R., Goudy L. E., Love K. S., Qi L. S., Multiple input sensing and signal integration using a split Cas12a system. Mol. Cell. 78, 184–191.e3 (2020), 10.1016/j.molcel.2020.01.016. [DOI] [PubMed] [Google Scholar]
- 88.Beltrán J., et al. , Rapid biosensor development using plant hormone receptors as reprogrammable scaffolds. Nat. Biotechnol. 40, 1855–1861 (2022), 10.1038/s41587-022-01364-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Foight G. W., et al. , Multi-input chemical control of protein dimerization for programming graded cellular responses. Nat. Biotechnol. 37, 10 (2019), 10.1038/s41587-019-0242-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guntas G., et al. , Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc. Natl. Acad. Sci. U.S.A. 112, 112–117 (2015), 10.1073/pnas.1417910112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huang Z., et al. , Creating red light-switchable protein dimerization systems as genetically encoded actuators with high specificity. ACS Synth. Biol. 9, 3322–3333 (2020), 10.1021/acssynbio.0c00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Piraner D. I., Abedi M. H., Moser B. A., Lee-Gosselin A., Shapiro M. G., Tunable thermal bioswitches for in vivo control of microbial therapeutics. Nat. Chem. Biol. 13, 75–80 (2017), 10.1038/nchembio.2233. [DOI] [PubMed] [Google Scholar]
- 93.Carey J., Cameron V., De Haseth P. L., Uhlenbeck O. C., Sequence-specific interaction of R17 coat protein with its ribonucleic acid binding site. Biochemistry 22, 2601–2610 (1983), 10.1021/bi00280a002. [DOI] [PubMed] [Google Scholar]
- 94.Dupeux F., et al. , A thermodynamic switch modulates abscisic acid receptor sensitivity. EMBO J. 30, 4171–4184 (2011), 10.1038/emboj.2011.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Miyazono K., et al. , Structural basis of abscisic acid signalling. Nature 462, 7273 (2009), 10.1038/nature08583. [DOI] [PubMed] [Google Scholar]
- 96.Ueguchi-Tanaka M., et al. , GIBBERELLIN INSENSITIVE DWARF1 encodes a soluble receptor for gibberellin. Nature 437, 7059 (2005), 10.1038/nature04028. [DOI] [PubMed] [Google Scholar]
- 97.Yoshida H., et al. , Evolution and diversification of the plant gibberellin receptor GID1. Proc. Natl. Acad. Sci. U.S.A. 115, E7844–E7853 (2018), 10.1073/pnas.1806040115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Grubbe W. S., Rasor B. J., Krüger A., Jewett M. C., Karim A. S., Cell-free styrene biosynthesis at high titers. Metab. Eng. 61, 89–95 (2020), 10.1016/j.ymben.2020.05.009. [DOI] [PubMed] [Google Scholar]
- 99.Kruyer N. S., et al. , Membrane augmented cell-free systems: A new frontier in biotechnology. ACS Synth. Biol. 10, 670–681 (2021), 10.1021/acssynbio.0c00625. [DOI] [PubMed] [Google Scholar]
- 100.Peruzzi J. A., Galvez N. R., Kamat N. P., Engineering transmembrane signal transduction in synthetic membranes using two-component systems. bioXriv [Preprint] (2022), 10.1101/2022.10.30.514420. Accessed 30 October 2022. [DOI] [PMC free article] [PubMed]
- 101.Alba D., Cardiff R., Data for Engineering activatable promoters for scalable and multi-input CRISPRa/i circuits. Zenodo. https://zenodo.org/record/7384227. Accessed 30 November 2022.
- 102.Alba D., Cardiff R., Data for Engineering activatable promoters for scalable and multi-input CRISPRa/i circuits. Github. https://github.com/carothersresearch/CRISPRai_Circuits_2022. Accessed 7 April 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)