

Abstract
In addition to preventing crosstalk among related signaling pathways, scaffold proteins might facilitate signal transduction by preforming multimolecular complexes that can be rapidly activated by incoming signal. In many cases, such as mitogen-activated protein kinase (MAPK) cascades, scaffold proteins are necessary for full activation of a signaling pathway. To date, however, no detailed biochemical model of scaffold action has been suggested. Here we describe a quantitative computer model of MAPK cascade with a generic scaffold protein. Analysis of this model reveals that formation of scaffold–kinase complexes can be used effectively to regulate the specificity, efficiency, and amplitude of signal propagation. In particular, for any generic scaffold there exists a concentration value optimal for signal amplitude. The location of the optimum is determined by the concentrations of the kinases rather than their binding constants and in this way is scaffold independent. This effect and the alteration of threshold properties of the signal propagation at high scaffold concentrations might alter local signaling properties at different subcellular compartments. Different scaffold levels and types might then confer specialized properties to tune evolutionarily conserved signaling modules to specific cellular contexts.
The mitogen-activated protein kinase (MAPK) cascades are an evolutionary conserved feature of a variety of receptor-mediated signal transduction schemes (1–3). A MAPK cascade consists of three sequentially acting kinases. The last member of the cascade, MAPK, is activated by dual phosphorylation at tyrosine and threonine residues by the second member of the cascade: MAPKK. MAPKK is activated by phosphorylation at threonine and serine by the first member of the cascade: MAPKKK. Activation of MAPKKK apparently proceeds through different mechanisms in different systems. The cascade reactions occur in the cytosol with the activated MAPK phosphorylating various targets in the cytosol and nucleus.
At least one property of a MAPK cascade that permits its consideration as a separate module in the signal transduction pathway is the existence of scaffold proteins (4–6). These proteins have been proposed to serve as organizing centers for signal transduction because they can bind several members of a signaling cascade to form a multimolecular complex. Scaffolds were first identified in yeast, with a member of the mating pathway Ste5 serving as the prototype (3, 7–10). Functionally analogous proteins in mammalian cells include MP1 specifically binding MAPKK MAPK kinase/ERK kinase (MEK) and MAPK extracellular signal-regulated kinase (ERK)-1 (11), JNK-interacting protein (JIP)-1 binding MAPKKK MLK, MAPKK MKK7, and MAPK c-Jun kinase (JNK) (12), and KSR-1, interacting with MEK and ERK (13–15). In addition, MEKK1 can serve both as a scaffold and as MAPKKK, interacting specifically with MAPKK MKK4/7 and MAPK JNK (1, 6, 16). Scaffolds are also found in signaling pathways other than MAPK cascade, implying that their function is general (17, 18).
One function of scaffold proteins might be to reduce the extent of crosstalk between different pathways sharing molecular components (1, 6). For example, Ste5 specifically binds the MAPKKK Ste11, the MAPKK Ste7, and the MAPK Fus3. Because Ste11 participates in at least two other pathways and Ste7 in one other pathway, formation of a preexisting signaling complex by Ste5 specifically connects Ste11 and Ste7 to Fus3 and facilitates activation of this but not other MAP kinases. Another putative yeast scaffold protein Pbs2, itself a MAPKK, specifically mediates activation of the MAPK Hog1 by Ste11 in response to osmotic stress (10). However, the presence of scaffolds, although potentially necessary, is not a sufficient condition for absence of Ste11-mediated crosstalk in yeast. Indeed, elimination of the negative feedback from Hog1 to the elements upstream in the pathway resulted in a high-degree mating pathway crossactivation despite continued presence of the scaffold proteins (19).
Although it is likely that scaffold proteins increase the specificity of signaling by physically separating various signaling complexes, their function may not be limited to this. Indeed, it is invariably observed that the rate of kinase activation within a scaffold complex is higher than in solution. At the same time, having a scaffold-type linker molecule in excess may lead to separation of kinases into nonfunctional complexes and inhibit signaling. How then can addition of a scaffold molecule be predicted to modify the rate and amplitude of signal transduction in a multikinase pathway?
Here we attempt to study numerically the role of a generic scaffold protein in a MAPK pathway. We show that the presence of the optimal scaffold concentration can substantially increase the signaling output. If the scaffold concentration is greater than optimal, a significant decrease in signaling can occur. The value of the optimal concentration and the effect of the scaffold on threshold properties of the signaling are also examined.
The Mathematical and Computational Model
The model is based on the mathematical description of MAPK cascade as a series of phosphorylation reactions as proposed previously (20–22). The core of the model consists of a system of ordinary differential equations (ODEs) describing sequential phosphorylation of the kinases of the MAPK cascade and their dephosphorylation by the corresponding phosphatases. Only dually phosphorylated MAPKK and MAPK are presumed to be active. The ODEs are derived from the general description of enzymatic reactions based on Michaelis–Menten kinetics. Each reaction scheme is of the following form:
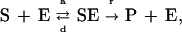 |
where S is the substrate, E the enzyme, and P the product of the reaction proceeding with corresponding association (a), dissociation (d), and reaction (r) constants. The assumptions underlying this model can be found in the studies referred to above. This treatment has been adequate for prediction of many key properties of MAPK cascade in various experimental systems.
We extended this model to include explicitly various molecular scaffold species corresponding to all possible scaffold–kinase complexes, as illustrated for a two-member scaffold in Fig. 1. Transitions between different scaffold complex species can be caused by binding of various kinases to the scaffold, their dissociation from it, and phosphorylation reactions taking place between kinases bound to a scaffold molecule. Dephosphorylation of kinases in scaffolded complexes was assumed to be precluded because of sterical obstruction of the phosphate groups (relaxation of this assumption does not change the results appreciably with the parameters chosen).
Figure 1.

Various kinase–scaffold combinations (scaffold species) Ci postulated by the model and transitions among them. Transitions by kinase association or dissociation (magenta arrows; only transitions for C1 are shown for clarity) and intrascaffold reactions (red arrows) are allowed in the model. P designates phosphorylation of a kinase.
We made additional assumptions based on experimental studies of a two-member scaffold MP1 (11) and the putative scaffold MEF (23). In particular, Schaffer et al. found that “MP1 was without effect on MEK-1 previously activated by phosphorylation in vitro by B-Raf, suggesting that MP1 interacts preferentially with inactive or partially active MEK-1.” Although this finding is related specifically to active MAPKK, we assumed that the same is true for MAPK–scaffold interaction. Hence our first assumption: (i) there is no binding of partially or fully activated MAPKK or MAPK to the scaffold. The same study finds that “MP1 increases the ability of MEK-1 to be activated by B-Raf and also enhances the ability of mutationally activated MEK-1 to activate ERK in vitro.” In addition, the data on a putative scaffold MEF revealed that, in the absence of MEF, both phosphorylations of MAPK necessary for activation are simultaneous, whereas in the absence of MEF, the second phosphorylation is delayed by about 20 min (23). We interpreted these findings as indicating that activation of MAPK within a scaffold is processive, that is, both phosphorylation reactions occur at the same time. We therefore assumed that: (ii) the activation of MEK and MAPK when bound to a scaffold is processive rather than distributive with the reaction rate equal to the rate of a single phosphorylation reaction. The remaining assumptions are: (iii) kinases bind to the scaffold independently of one another, i.e., there is no cooperativity in the binding; (iv) scaffold molecules do not possess catalytic properties, so that the reaction rates within a scaffold complex and in solution are equal. Thus, according to assumption iv, any increase in MAPK activation because of the scaffold is caused by making the corresponding reactions less diffusion limited and hence more reaction limited. We also assumed that the reactions take place in a homogenous environment with no additional mechanism for compartmentalization of molecules; thus we ignored MAPK translocation to the nucleus. Because assumptions ii and iii may be invalid for some systems, we examined the sensitivity of our model to these assumptions with results described below.
In choosing reaction parameters, we referred whenever possible to estimates available in the literature (22, 24, 25). The nomenclature and values of various coefficients used are given in Table 1 of the supplementary materials (www.pnas.org), unless stated otherwise. The full system of equations describing MAPK cascade with a generic two-member scaffold can also be found in the supplementary materials. Because our major goal was to elucidate possible differences between scaffolded and nonscaffolded signal transduction, we considered sensitivity of the observed results to variations in parameters related to the scaffold proteins, rather than the parameters of the phosphorylation cascade [these studies have been reported by others (22)].
We next developed a mathematica 3.0 (Wolfram Research, Champaign, IL) package (available on request) allowing simulation of solutions to these equations. The accuracy of the numerical simulations was verified by numerically checking the law of conservation of mass and by running the model with simplified assumptions allowing analytical solutions. In all simulations, we first allowed the system to equilibrate in the absence of signaling, which resulted in simulation of preformation of scaffold–kinase complexes. The concentrations of these complexes and molecular species were then used as the initial conditions for simulations of the response to a constant nonzero signal. The signal was interpreted as the concentration of a molecule capable of MAPKKK activation. We assumed that the level of signaling input is unaffected by the signaling output within the time frame considered. As signaling read-outs, we used either the concentration of free dually phosphorylated MAPK (MAPK-PP), not bound to the scaffold, or its time integral over the first 100 sec of signaling.
Results
1. Kinetics of MAPK Activation.
To model the effect of adding a scaffold protein on kinetics of MAPK activation, we investigated the time dependence of MAPK-PP production in the presence or absence of a two-member scaffold (Fig. 2A). For the parameter values chosen, addition of the scaffold up to approximately 0.3 μM resulted in an earlier onset and increased rate of MAPK-PP production. As a result, the plateau levels of MAPK-PP increased. However, as the scaffold concentration increased above 0.3 μM, MAPK activation slowed down, with plateau levels decreasing. Thus addition of a scaffold results in progressive facilitation of MAPK activation only within a limited range of scaffold concentration. We will discuss below the reasons for this nonmonotonic behavior.
Figure 2.

The role of scaffold proteins in kinetics and input–output sensitivity of MAPK activation. (A) Kinetics of MAPK activation as a function of a two-member scaffold concentration. The scaffold concentrations considered are indicated in the figure. The dashed line represents the kinetics of unscaffolded reaction. (B) Dependence of MAPK activation on the level of signaling. For each scaffold concentration, the kinetics of MAPK activation was computed as in A, and integral over the first 100 sec was plotted as a function of the signal. The level of the signal corresponds to the concentration in micromolars of a MAPKKK activator. The dashed curve represents unscaffolded reaction. (C) Input–output relationships for various scenarios of reactions in the scaffold. The processive (all values are divided by four), intermediate, and distributive (multiplied by 108) reactions are considered (see details in the text). MAPK activation with no scaffold is presented by the dashed curve for comparison.
2. Threshold Properties of Scaffolded Signaling.
Presence of thresholds is a common feature of signal transduction systems. As discussed by Ferrell et al. (24, 26, 27), one of the ways to produce a threshold or switch-like response is to have a distributive rather than processive mechanism of dual phosphorylation of MAPKK and MAPK. The distributive mechanism presupposes that the two phosphorylation reactions necessary for MAPKK and MAPK activation are separated by full dissociation of the intermediary kinase–substrate complexes, resulting in a sigmoidal signaling input–output curve. Attachment of the kinase and substrate to a scaffolding molecule might prevent this dissociation and thus result in the interaction becoming processive, with signaling losing its threshold properties. In our model, we assumed processive kinase activation within a scaffold complex and explored how the presence of various amounts of the scaffold affected the overall response to the signaling input. As is clear from Fig. 2B, an increase in the scaffold concentration from 0 μM to 2 μM resulted in a gradual disappearance of the sigmoidal character of the response curves, signifying reduction of threshold character of activation. This effect represents a potential qualitative difference between the properties of response between scaffolded and nonscaffolded signaling systems.
It may be argued that the threshold reduction observed in Fig. 2B is “built in” to our model by assumption ii (processive rather than distributive kinase activation within a scaffold). Although, as described above, there is some experimental support for this assumption, we considered the effect of its relaxation. First we note that purely distributive activation would make scaffolds completely ineffectual. Indeed, in distributive activation of MAPK by MAPKK, two separate interactions are required for two phosphorylations. Therefore, that MAPKK needs to dissociate from the scaffold complex after its activation and then reassociate with the complex twice for MAPK activation, a situation equivalent to (or, according to our assumption i, less effective than) activation in solution.
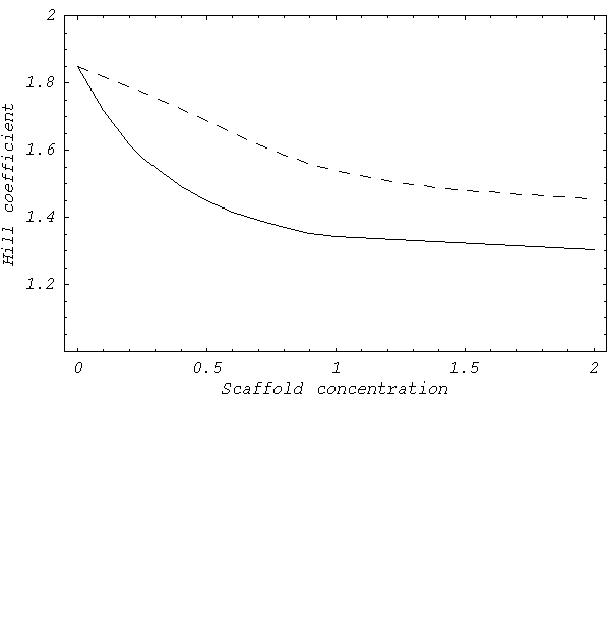
The intrascaffold activation scheme that approaches purely distributive activation most closely is completely distributive activation of MAPKK in the scaffold, followed by single phosphorylation of MAPK in the scaffold with the second MAPK phosphorylation to occur in solution, the scenario assumed in our control studies (Fig. 2C). Another scheme presented in Fig. 2C is an intermediate case of processive MAPKK activation but with only single MAPK phosphorylation within the scaffold. A reduction in the sigmoidness of the input–output dependence in the presence of the scaffold is evident only in the intermediate distributive activation scenario. The estimated Hill coefficients at 2 μM of scaffold were 1.3 in the fully processive scheme, 1.45 in the partially distributive, and 1.8 in the maximally distributive case. Unscaffolded MAPK activation (Hill coefficient 1.85) is shown in Fig. 2C for comparison. A more detailed dependence of the Hill coefficient on the scaffold concentration can be found in supplementary materials (www.pnas.org). As activation is made more distributive, the absolute value of activity decreases. In fact, for the parameter values chosen, the maximally distributive activation within the scaffold leads to a dramatic inhibition of activation regardless of the scaffold concentration applied. Thus, although the threshold properties of MAPK activation can be reconstituted if the reactions occur in a more distributive manner, the scaffold efficiency in signaling activation decreases. We predict that various combinations of distributive and processive activation suitable for the required signal activation and threshold reduction properties may occur in particular biochemical scaffolds.
3. Existence of a Scaffold Concentration Optimal for Signaling.
MAPK activation, as plotted in Fig. 2, displays nonmonotonic dependence of MAPK activation on the variation of scaffold concentration. To explore this behavior further, we computed the time integral of the activated MAPK as a function of the scaffold concentration. This dependence has a clear maximum at approximately 0.3 μM for the chosen set of parameters (Fig. 3A). Clearly, the presence of a scaffold can have two opposing effects on the level of response. Because we do not assume that scaffolds can confer any additional catalytic properties, both these effects must depend on the efficiency of colocalization of the MAPKK and MAPK components in the signaling complex.
Figure 3.

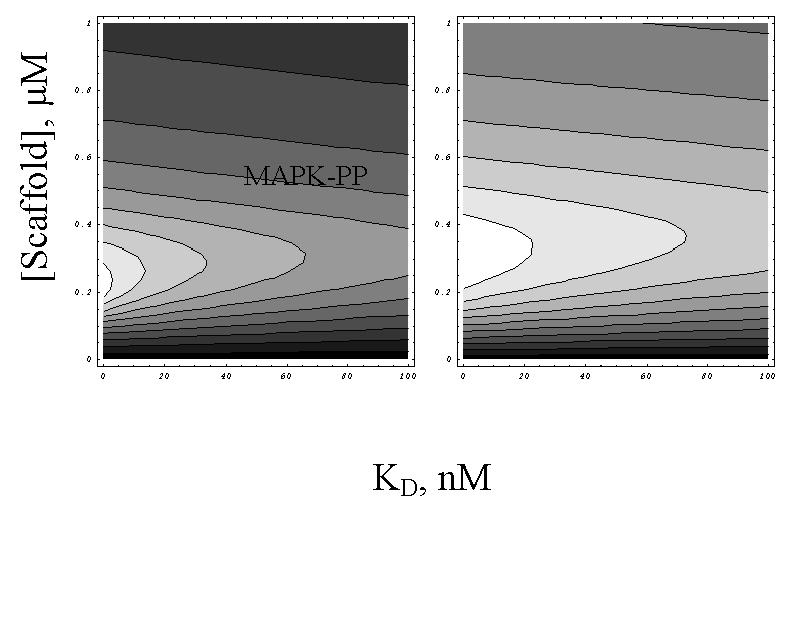
Existence of an optimal scaffold concentration. (A) Dependence of MAPK activation (red) and functional scaffold–kinase complexes C6 (blue) and C8 (dashed magenta) on the two-member scaffold concentration. MAPK activation at 100-fold lower MAPKK phosphatase concentration is shown in light blue. C6 is normalized by dividing by 40 and C8 by multiplication by 17 for better comparison and illustration purposes. The results are compared with the plot of 0.15/[Scaffold] (green). (B) The optimum of MAPK activation or C6 formation is not sensitive to the dissociation constant KD of MAPK–scaffold interaction. The lighter areas in the contour plots correspond to higher MAPK or C6 concentrations. (C and D) The optimal scaffold concentration (two-member scaffold) is a function of MAPKK (C) and MAPK (D). Both three-dimensional and contour plots of MAPK activation are shown.
It has been proposed before that a molecular crosslinker can promote formation of a macromolecular complex at low concentrations and inhibit it at high concentrations. A detailed study of this “pro-zone” effect has been reported recently (28). The pro-zone effect (“combinatorial inhibition” can be proposed as a more descriptive term) in terms of the scaffold-mediated MAPK activation means that at low scaffold concentration, when both kinases are in excess, formation of a functional complex containing both kinases is likely, whereas in excess of the scaffold, nonfunctional complexes containing none or only one of the kinases become relatively more abundant. This situation clearly should be true in the absence of signaling, when there are only four possible kinase–scaffold combinations (C1, C2, C4, and C6 in Fig. 1). In the presence of a signal, however, more combinations are possible, and the functional read-out is proportional to the concentration of free MAPK-PP, rather than C6. We studied this issue by plotting the time integral of MAPK-PP and C6 concentrations (Fig. 3A). Remarkably, it can be seen that the maxima of the time integrals of C6 and MAPK-PP coincide at [Scaffold] = 0.3 μM, although the overall shapes of the curves differ. In fact, there is a much better correlation between the combination of the MAPK-PP containing scaffold complexes (of which the most abundant is C8) and the free MAPK-PP than between C6 and MAPK-PP. All of the curves in Fig. 3A display excellent correlation in the tail portion and vary as 1/[Scaffold]. It is shown in the supplementary materials (www.pnas.org) that, in general, at high scaffold concentrations (the tails of the curves), the concentration of C6 varies as: 1/[Scaffold]m−1, where m is the number of molecular species a scaffold needs to bind to assemble a functional complex.
Another important feature of MAPK-PP dependence on the scaffold concentration illustrated in Fig. 3A is the relative change in MAPK-PP compared with unscaffolded reaction. Indeed, we found that, depending on the concentration or KM of MAPKK phosphatase, there may be a substantial relative increase or decrease in MAPK activation. The absolute activation levels as well as the position of the optimum of the scaffold dependency do not change appreciably. This observation can be explained if one recalls that the efficiency of MAPK activation in solution is in direct relationship with the availability of activated MAPKK. Thus at low MAPKK phosphatase activity, MAPK activation in solution becomes comparable to that in the scaffold. Therefore, MAPKK phosphatase activity might determine whether a particular scaffold is efficient in enhancing or inhibiting MAPK activation.
The coincidence of the maxima of the activation curves in Fig. 3A can be explained only if the factors determining the positions of the maxima are identified. Because the positions of maxima for complexes such as C6 have not been studied before (it can be shown that analytically, this problem is intractable), we thus investigated the factors determining the optimal scaffold concentration for C6 concentration and MAPK activation (Fig. 3B). We found that variation of MAPKK and MAPK concentrations, but not binding parameters of the kinases to the scaffold, affected the position of the peaks. Therefore, substantial variations of kinase-binding affinities between the complexes C6 through C9 affect neither the positions of the optima for formation of these complexes nor, by extension, the optimum for MAPK activation. We illustrate below that this argument is consistent with the shift in the optima positions when cooperation in kinase binding is assumed. A more detailed analysis (Fig. 3 C and D) revealed that as the concentrations of both kinase and scaffold were varied, the MAPK reached a plateau along the “line” corresponding to a certain ratio of scaffold-to-kinase concentrations (approximately 2:1 for MAPK and 3:1 for MAPKK for the parameters chosen).
4. Two-Member vs. Three-Member Scaffolds.
The combinatorial nature of kinase–scaffold complex formation leads to the prediction that MAPK activation is sensitive to the number of kinases a particular scaffold can bind (“scaffold membership”). In particular, we hypothesized that a higher membership scaffold can form more nonfunctional complexes, so that the inhibition of signaling at high scaffold concentrations is more pronounced. To confirm this, we simulated a three-member scaffold capable of binding all members of MAPK module, with the same binding parameters as the two-member scaffold. This model is based on the same principles as those illustrated in Fig. 1 for the two-member scaffold. As shown in Fig. 4A, we indeed observed higher sensitivity of MAPK activation to variation of the scaffold concentration. In particular, although the peak level of MAPK activation is somewhat greater for the three-member scaffold-mediated signaling, the inhibition of activity to the level of 0.1 μM⋅sec requires approximately twice as much two-member scaffold as three-member scaffold.
Figure 4.

The scaffold membership and cooperativity of MAPKK and MAPK binding to scaffold affect MAPK activation. (A) Comparison of the effects of two- (red) and three- (blue) member scaffolds on the signaling properties. Assumptions of the three-member scaffold model are the same as the two-member model considered above with the additional assumption of MAPKKK binding to the scaffold. The dashed line illustrates that it takes considerably more two-member than three-member scaffolds to inhibit activity to the same level at high scaffold concentrations. (B) Cooperativity does not affect the property of existence of an optimal scaffold concentration but results in a shift of the optimum. No cooperativity (red), partial positive (green), and partial negative (blue) cooperativity are shown. In partial cooperativity, only inactive kinases interact. The dashed lines of the corresponding color represent C6 normalized as in Fig. 3. (C) Full (red) and partial (green) positive cooperation is compared with noncooperative binding (blue). Inset illustrates the concomitant dependence of C3 (see Fig. 1). In all figures, cooperativity is defined as an increase or decrease in the second kinase association constant by the factor of 10 when the first kinase is bound. See more information in the text.
5. Sensitivity of the Model to Cooperativity of MAPKK and MAPK Binding.
We have already discussed the sensitivity of the model to the assumption of the processive character of intrascaffold reactions. To determine the sensitivity to the assumption of independent MAPKK and MAPK binding to a scaffold molecule (assumption iii), we modified the equations to introduce partial positive and negative cooperativity by allowing the on-rates for the scaffold binding of MAPKK and MAPK for transitions C4 → C6 and C2→ C6 (see Fig. 1) to be respectively either increased or decreased by a factor of 10. Thus the fact of an inactive kinase presence in the scaffold complex increases or decreases the chance of the second kinase binding. We also examined the case of full cooperation between both active and inactive kinases, with the additional changes in the rates of transition: C3 → C7 and C5 → C9. We found that the overall dependence of MAPK activation on the scaffold concentration remains the same even if positive or negative cooperation exists in binding of the kinases (Fig. 4B). Significantly, however, there is a shift in the optimal position to the left in partial positive and to the right in partial negative cooperation. No such shift is observed for C6 or for the case of full kinase-binding cooperation (data not shown). This can be explained by the following reasoning. As mentioned before, the concentration of MAPK-PP is directly related to the concentration of C8. The main route of C8 formation is through C7, which, in turn, can be formed either through C4 → C6→ C7 (a) or C2 → C6→ C7 (b) or C2 → C3→ C7 (c). Routes a and b are affected by cooperation assumption, whereas route c may or may not be affected. It can be shown that route c overwhelmingly prevails at high but not low scaffold concentrations. As illustrated in Fig. 4C, this means that the MAPK activation curve for partial cooperation is close to the curve for full cooperation at low scaffold concentrations, while approaching the curve for no cooperation as the scaffold concentration increases and then following intermediate values according to the behavior of C3. It can be shown graphically that, although the limiting curves have the same maxima, the maximum of the partial cooperation curve is necessarily shifted.
Discussion
Numerical studies of MAPK cascade activation presented here revealed that molecular scaffolds may both facilitate and inhibit signal propagation depending on their concentration. Indeed, the rate of MAPK activation, the steady-state value of the activated MAPK during signaling, and the time integral of the concentration of the activated MAPK during onset of the signaling all displayed nonmonotonic dependence on the scaffold concentration. At a particular scaffold concentration, all these read-outs were found to have a maximal value, which decreased as the scaffold concentration was either increased or reduced.
The mechanism responsible for this behavior was addressed by considering the concentration of functional scaffold–kinase complexes before and during signaling. Because of the pro-zone (combinatorial inhibition) effect, the concentration of functional complexes before signaling can be shown to have a maximum (28). However, the existence of a maximum for MAPK activation and its position cannot be inferred directly from this effect, because phosphorylation reactions increase the number of possible complexes, and free rather than bound MAPK-PP represents the signaling output. Our results suggest that not only is there an optimal scaffold concentration for MAPK activation, but the position of the optimum does not depend on the binding constants of the kinase–scaffold interactions. The only determinants of the optimal scaffold concentration are the concentrations of the kinases and the character of their mutual interactions. For example, positive cooperation in binding of inactive kinases shifts the position of the optimum to scaffold lower concentrations.
Our model also predicts that sensitivity of pathway activation to scaffold concentration increases with membership of the scaffold. In particular, at high scaffold concentrations, signaling is inhibited as 1/[Scaffold]m−1. This prediction can be tested by experiments in which the binding sites of kinases in a scaffold are mutated to change or abolish affinity of the kinases to the scaffold.
Combinatorial inhibition offers a resolution to the apparent conflict existing in the literature regarding whether scaffolds facilitate or suppress signaling. For example, JIP-1 has been shown to inhibit JNK-mediated signaling by retaining JNK in the cytoplasm. Based on our model, we suggest that the concentration at which this molecule is present in the cells studied is in the range of combinatorial inhibition, and reducing this concentration to an optimal level would lead to a response higher than that of JNK in the absence of JIP-1. Thus JIP-1 might serve as both an inhibitor and a facilitator of signaling. This suggestion is corroborated by data indicating that JNK activation within the JIP-1-mediated complex is increased (6). The discrepancies observed in the effect of KSR-1 on MAPK-mediated signaling can be explained similarly. Indeed, although identified as a positive regulator, KSR-1 can suppress signaling when overexpressed, as reported by several groups. One study reports both positive and negative regulation by KSR-1, depending on the level of the protein expression (29), in direct agreement with the model proposed here. Qualitative reasoning similar to our model has been offered for KSR-1 action (14, 15).
Our results indicate that scaffold proteins can also affect such qualitative characteristics as the presence of thresholds. Indeed, increasing scaffold concentrations gradually diminished the sigmoid character of MAPK activation as a function of signal strength. This threshold attenuation resulted from the assumption that activation of MAPK by MAPKK is processive rather than distributive when both molecules are bound to a scaffold. This assumption has some experimental support and is corroborated by our control simulations, indicating that purely distributive activation would severely limit the effectiveness of MAPKK-MAPK colocalization.
Reduction of the cascade activation threshold coupled with augmented signal response could increase sensitivity of the signal-transduction pathway to low levels of signaling. This might be undesirable physiologically because the ability of the signaling system to filter out low nonspecific signals in a generally very noisy environment might decrease. Thus, the way scaffold concentration is regulated in the absence of and during signaling is extremely important.
Scaffold concentration potentially could be modulated globally, through variation of gene expression and stability of the scaffold protein, and locally, by concentrating scaffolds in specific subcellular cellular compartments at the expense of the other parts of the cell. Apparently, both mechanisms are used in scaffold regulation in vivo. Indeed, the yeast scaffold Ste5 is substantially more stable when bound to MAPKKK Ste11 (30). Ste5 concentration is also regulated locally. Receptor activation results in translocation of Ste5 from the cytosol and nucleus to the portions of the cell membrane, where the concentration of the activated receptor is the highest. Therefore, in a relatively small volume, concentration of the scaffold may transiently achieve levels significantly higher than the average concentration in the cytosol. Concentration of Ste5 in other subcellular compartments is concomitantly reduced.
The differential spatial distribution of scaffold proteins hints at their potential role in gradient sensing. Indeed, as reviewed by Parent and Devreotes (31), in eukaryotic cells heterotrimeric G protein-linked signaling pathways mediate directional sensing. At least two scaffold proteins, Ste5 and KSR-1(13), have been shown to translocate to the cell membrane in response to G-protein activation. If before signaling Ste5 concentration is suboptimal, specific scaffold translocation to the area of the cell membrane facing the highest concentration of chemoattractant can increase the signaling in a small portion of the cell membrane dramatically because of the loss of threshold properties and increased rate of MAPK activation as described above. Signaling in the rest of membrane would be decreased. Thus scaffolding signaling reactions can amplify the spatial signaling inhomogeneity thought to be essential for gradient sensing.
Finally, our data suggest that scaffolds can lead to channeling cascade reactions not by sequestering various molecular components from the cytosol, but by specific enhancement or inhibition of the reaction that would otherwise occur differently in solution.
Supplementary Material
Acknowledgments
We thank B. J. Wold and three anonymous reviewers for discussion and critical review. This work was supported by Office of Naval Research Grant N00014–97-1–0293 and by a Jet Propulsion Laboratory grant to J.B. and P.W.S. (an investigator with the Howard Hughes Medical Institute), by a Sloan Research Fellowship to J.B., and by Burroughs–Wellcome Fund Computational Molecular Biology Postdoctoral Fellowship to A.L.
Abbreviations
- MAPK
mitogen-activated protein kinase
- ERK
extracellular signal-regulated kinase
- MEK
MAPK kinase/ERK kinase
- JIP
JNK-interacting protein
- JNK
c-Jun kinase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Garrington T P, Johnson G L. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 2.Widmann C, Gibson S, Jarpe M B, Johnson G L. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 3.Gustin M C, Albertyn J, Alexander M, Davenport K. Microbiol Mol Biol Rev. 1998;62:1264–1300. doi: 10.1128/mmbr.62.4.1264-1300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elion E A. Science. 1998;281:1625–1626. doi: 10.1126/science.281.5383.1625. [DOI] [PubMed] [Google Scholar]
- 5.Faux M C, Scott J D. Cell. 1996;85:9–12. doi: 10.1016/s0092-8674(00)81075-2. [DOI] [PubMed] [Google Scholar]
- 6.Whitmarsh A J, Davis R J. Trends Biochem Sci. 1998;23:481–485. doi: 10.1016/s0968-0004(98)01309-7. [DOI] [PubMed] [Google Scholar]
- 7.Marcus S, Polverino A, Barr M, Wigler M. Proc Natl Acad Sci USA. 1994;91:7762–7766. doi: 10.1073/pnas.91.16.7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi K Y, Satterberg B, Lyons D M, Elion E A. Cell. 1994;78:499–512. doi: 10.1016/0092-8674(94)90427-8. [DOI] [PubMed] [Google Scholar]
- 9.Printen J A, Sprague G F J. Genetics. 1994;138:609–619. doi: 10.1093/genetics/138.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Posas F, Saito H. Science. 1997;276:1702–1705. doi: 10.1126/science.276.5319.1702. [DOI] [PubMed] [Google Scholar]
- 11.Schaeffer H J, Catling A D, Eblen S T, Collier L S, Krauss A, Weber M J. Science. 1998;281:1668–1671. doi: 10.1126/science.281.5383.1668. [DOI] [PubMed] [Google Scholar]
- 12.Whitmarsh A J, Cavanagh J, Tournier C, Yasuda J, Davis R J. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- 13.Bell B, Xing H, Yan K, Gautam N, Muslin A J. J Biol Chem. 1999;274:7982–7986. doi: 10.1074/jbc.274.12.7982. [DOI] [PubMed] [Google Scholar]
- 14.Denouel-Galy A, Douville E M, Warne P H, Papin C, Laugier D, Calothy G, Downward J, Eychene A. Curr Biol. 1998;8:46–55. doi: 10.1016/s0960-9822(98)70019-3. [DOI] [PubMed] [Google Scholar]
- 15.Yu W, Fantl W J, Harrowe G, Williams L T. Curr Biol. 1998;8:56–64. doi: 10.1016/s0960-9822(98)70020-x. [DOI] [PubMed] [Google Scholar]
- 16.Xu S, Cobb M H. J Biol Chem. 1997;272:32056–32060. doi: 10.1074/jbc.272.51.32056. [DOI] [PubMed] [Google Scholar]
- 17.Cohen L, Henzel W J, Baeuerle P A. Nature (London) 1998;395:292–296. doi: 10.1038/26254. [DOI] [PubMed] [Google Scholar]
- 18.Rothwarf D M, Zandi E, Natoli G, Karin M. Nature (London) 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 19.O'Rourke S M, Herskowitz I. Genes Dev. 1998;12:2874–2886. doi: 10.1101/gad.12.18.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldbeter A, Koshland D E J. J Biol Chem. 1984;259:14441–14447. [PubMed] [Google Scholar]
- 21.Goldbeter A, Koshland D E J. Proc Natl Acad Sci USA. 1981;78:6840–6844. doi: 10.1073/pnas.78.11.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang C Y, Ferrell J E J. Proc Natl Acad Sci U S A. 1996;93:10078–10083. doi: 10.1073/pnas.93.19.10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott A, Haystead C M, Haystead T A. J Biol Chem. 1995;270:24540–24547. doi: 10.1074/jbc.270.41.24540. [DOI] [PubMed] [Google Scholar]
- 24.Ferrell J E J, Bhatt R R. J Biol Chem. 1997;272:19008–19016. doi: 10.1074/jbc.272.30.19008. [DOI] [PubMed] [Google Scholar]
- 25.Bhalla U S, Iyengar R. Science. 1999;283:381–387. doi: 10.1126/science.283.5400.381. [DOI] [PubMed] [Google Scholar]
- 26.Ferrell J E J. Trends Biochem Sci. 1996;21:460–466. doi: 10.1016/s0968-0004(96)20026-x. [DOI] [PubMed] [Google Scholar]
- 27.Ferrell J E J. Trends Biochem Sci. 1997;22:288–289. doi: 10.1016/s0968-0004(97)82217-7. [DOI] [PubMed] [Google Scholar]
- 28.Bray D, Lay S. Proc Natl Acad Sci USA. 1997;94:13493–13498. doi: 10.1073/pnas.94.25.13493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cacace A M, Michaud N R, Therrien M, Mathes K, Copeland T, Rubin G M, Morrison D K. Mol Cell Biol. 1999;19:229–240. doi: 10.1128/mcb.19.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim S H, Lee S K, Choi K Y. Mol Cells. 1998;8:130–137. [PubMed] [Google Scholar]
- 31.Parent C A, Devreotes P N. Science. 1999;284:765–770. doi: 10.1126/science.284.5415.765. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}