

SUMMARY
Immunoediting sculpts immunogenicity and thwarts host anti-tumor responses in tumor cells during tumorigenesis; however, it remains unknown whether metabolic programming of tumor cells can be guided by immunosurveillance. Here, we report that T cell-mediated immunosurveillance in early-stage tumorigenesis instructs c-Myc upregulation and metabolic reprogramming in tumor cells. This previously unexplored tumor-immune interaction is controlled by non-canonical interferon gamma (IFNγ)-STAT3 signaling and supports tumor immune evasion. Our findings uncover that immunoediting instructs deregulated bioenergetic programs in tumor cells to empower them to disarm the T cell-mediated immunosurveillance by imposing metabolic tug-of-war between tumor and infiltrating T cells and forming the suppressive tumor microenvironment.
Graphical Abstract
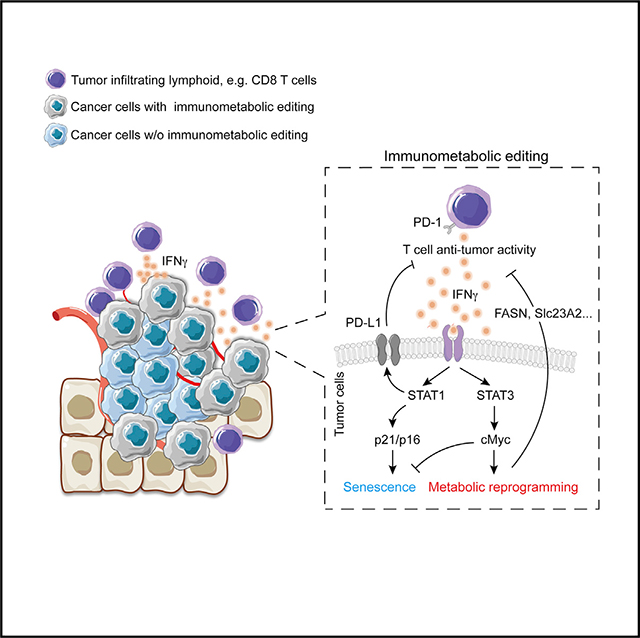
In brief
Metabolic reprogramming, generally believed to be controlled by oncogenic mutations and hypoxia, supports tumor proliferation, anti-apoptosis, and metastasis. Here, we uncover an unexplored action of IFNγ by which T cell-mediated immunosurveillance impacts epigenetic architecture and gene expression in tumor cells that boosts cMyc-dependent metabolic reprogramming and tumor immune evasion.
INTRODUCTION
Oncogenic mutations and hypoxia are believed to be the major drivers in orchestrating metabolic reprogramming in tumors, a cardinal hallmark of most tumor cells that enables the utilization of anabolic metabolism, such as aerobic glycolysis, glutaminolysis, and de novo fatty acid synthesis, for supporting unrestricted growth.1–3 Arguably, hypoxia and mutation-driven metabolic reprogramming does not address the requirements of tumor cells to adjust their metabolic preferences in response to diverse environmental cues and the selective pressure imposed by the host anti-tumor immunity. Metabolic crosstalk and tug-of-war between tumor cells and tumor-infiltrating immune cells have emerged as critical processes for tumor immune evasion by promoting T cell dysfunction and the establishment of an immunosuppressive tumor microenvironment (TME).4–7 In addition, declined dependency of oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) have been reported to minimize immunogenicity and T cell-mediated anti-tumor immunity in melanoma patients.8 These findings highlight that metabolic reprogramming in tumor cells represents an important cellular process for dampening T cell anti-tumor immunity. However, it remains uncharacterized whether metabolic preference and reprogramming in tumor cells can be sculpted as a result of immunosurveillance.
Interferon gamma (IFNγ) is a pleiotropic cytokine driving cellular senescence, blocking proliferation, and modulating multiple layers of immune actions during tumor immunosurveillance.9 Senescence of tumor cells induced by IFNγ represents a central step in achieving tumor immunosurveillance.10,11 However, the beneficial effects of IFNγ on tumor control come with dark sides known as adaptive immune resistance9; this includes stimulation of epithelial-mesenchymal transition and upregulation of programmed death-ligand 1 (PD-L1) and indoleamine 2,3-dioxygenase (IDO) in tumor cells, which promote metastasis, impair T cell anti-tumor responses, and create an immunosuppressive TME, respectively.12 Moreover, IFNγ--STAT1 signal axis is considered as the major driving force for cancer immunoediting, a process whereby the host immune system simultaneously constrains tumor growth and, in doing so, promotes tumor evolution that allows tumor cells with the highest ability to evade immunosurveillance and become the dominant tumor cell populations.13,14 These studies underscore the importance of IFNγ in shaping tumor cells’ capacity for immune evasion; however, it remains unknown whether metabolic reprogramming in tumor cells, as a tumor immune evasion mechanism, can be tailored in response to IFNγ and immunoediting.
Here, we reveal “immunometabolic editing,” controlled by the unexplored action of IFNγ during T cell-mediated immunosurveillance, impacts epigenetic architecture and gene expression in tumor cells for modulating cMyc-dependent metabolic reprogramming. Our results unveil this unexplored format of immunoediting and an unconventional IFNγ signaling cascade on guiding tumor cells to preferentially engage metabolic programs simultaneously supporting proliferation and immune evasion. These findings unveil a new dimension of immunoediting that the metabolic crosstalk occurring between tumor and tumor-infiltrating T cells is an outcome of immunoediting and tumor cells can adjust their metabolic preferences in response to survival pressure imposed by host anti-tumor immunity.
RESULTS
T cell-mediated immunoediting influences metabolic preference in melanoma cells
To investigate whether immunosurveillance could influence the metabolic preference in tumor cells, we took advantage of a genetically engineered melanoma mouse model (referred to as Braf/Pten mice).15,16 We also obtained adaptive immune cell-deficient Braf/Pten mouse strain by crossing Braf/Pten mice with Rag1-deficient (Rag1−/−) mice, which fail to develop mature T and B cells. Then, we generated primary Braf/Pten melanoma cell lines from immunocompetent Braf/Pten mice (referred to as WT cells) and Rag1-deficient Braf/Pten mice (referred to as KO cells) (Figure S1A). Similar to previous reports that tumor cells forming in the absence of adaptive immune cells fail to establish tumors in immunocompetent mice,14 KO cells derived from Braf/Pten Rag1−/− mice formed tumors in Rag1−/− mice but were less capable of growing in the immunocompetent mice (Figure 1A). In contrast, WT cells were more capable of growing in the immunocompetent mice (Figure 1B). Importantly, despite both WT and KO cells having a similar adenosine triphosphate (ATP) level, KO cells exhibited higher oxygen consumption rate to extracellular acidification rate (OCR/ECAR) ratios compared with WT cells (Figures 1C and 1D), suggesting that immunoediting enforces tumors cells to elevate aerobic glycolysis but diminish engagement of OXPHOS to meet their metabolic demands. We also found that primary hepatocellular carcinoma (HCC) cell lines with β-catenin overexpression and Pten deletion derived from Rag1−/− mice (referred to HCC-KO cells) displayed higher OCR/ECAR ratios compared with primary HCC cells from immunocompetent WT mice (Figure S1B). Moreover, melanomas induced from Rag1−/− genetic background displayed distinct metabolic profiles compared with melanomas derived from immunocompetent WT mice determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Figure S1C). Intriguingly, distinct metabolic profiles in tumors growing in the absence of adaptive immune cells were also observed in genetically engineered breast cancer and HCC mouse models (Figures S1D–S1F). By performing enrichment analysis with the differentially expressed metabolites, we observed that metabolites in the tricarboxylic acid (TCA) cycle were highly enriched while metabolites in the glycolytic pathway were downregulated in melanomas from T and B cell-deficient Braf/Pten mice (Figure 1C). Altogether, our results suggest that the presence of adaptive immune cells during tumorigenesis can impact metabolic features and the engagement of aerobic glycolysis in tumors.
Figure 1. Early-stage T cell-mediated immunosurveillance influences tumor metabolism.
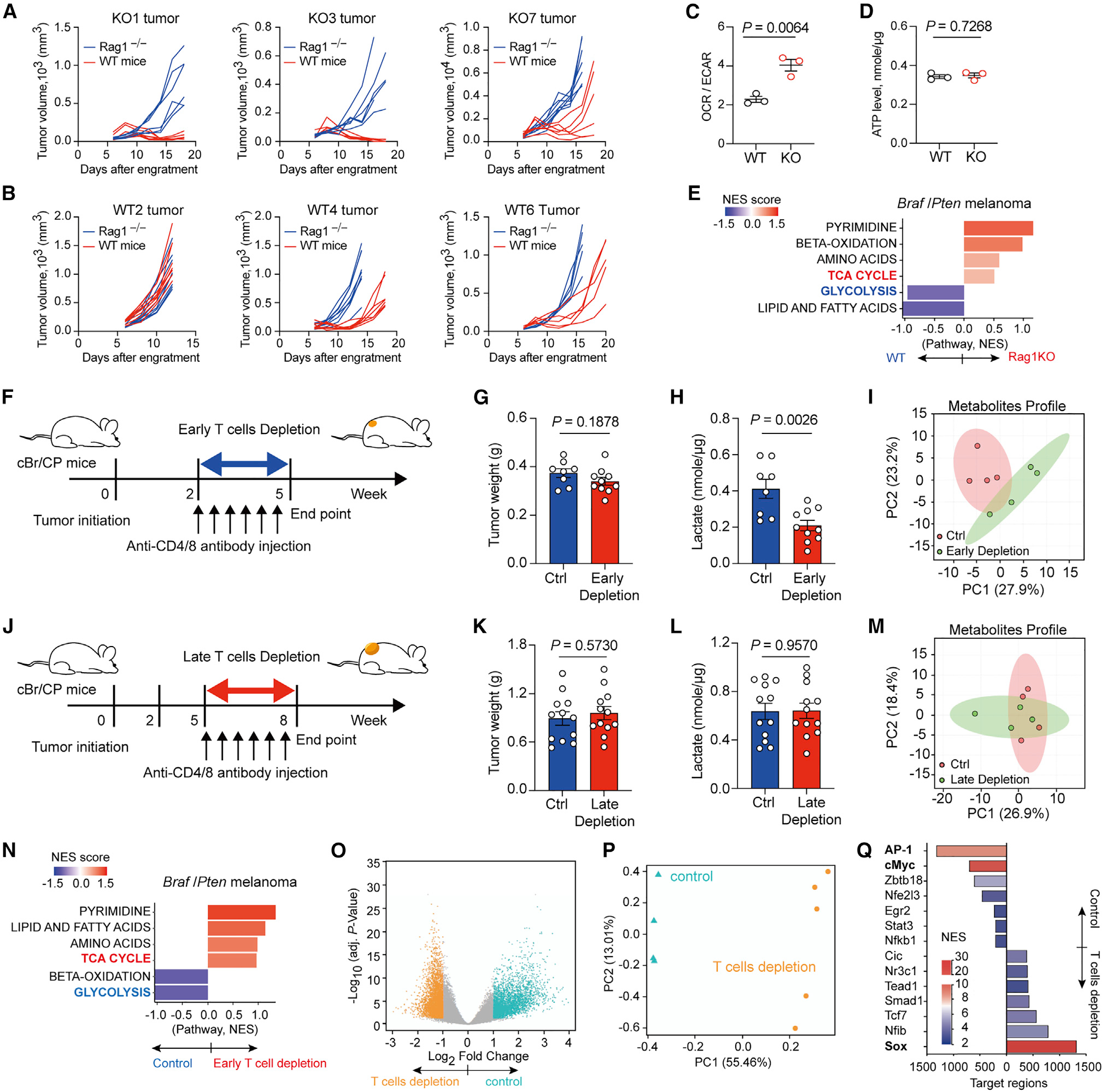
(A) Tumor growth curve for KO1, KO3, and KO7, three Braf/Pten melanoma cell lines derived from adaptive immune cell-deficient Braf/Pten mouse, after subcutaneously engrafting into C57BL/6 wild-type mice and Rag1−/− mice (n = 6–7 per group).
(B) Tumor growth curve for WT2, WT4, and WT6 cell lines derived from immunocompetent Braf/Pten mouse, after subcutaneously engrafting into C57BL/6 wild-type mice and Rag1−/− mice (n = 6–7 per group).
(C and D) Ratios of basal OCR versus ECAR (C) and ATP level (D) of three WT primary melanoma cells and three KO primary melanoma cells.
(E) Metabolite set enrichment analysis of Braf/Pten tumors from immunocompetent wild-type background versus matched Rag1-KO background (bottom panel).
(F) Experimental outline of antibody-based early T cell depletion in murine Braf/Pten melanomas 2 weeks post-tumor induction with 4-hydroxytamoxifen (4-HT).
(G and H) Tumor weight (G) and lactate levels in tumors (H) in Braf/Pten melanomas from control (n = 8–10) and T cell depletion (n = 10) groups.
(I) Principal component analysis (PCA) of metabolites in Braf/Pten tumors from control and early T cell depletion (n = 4 per group).
(J) Experimental scheme of antibody-based late T cell depletion in murine Braf/Pten melanomas 5 weeks post-tumor induction.
(K and L) Tumor weight (K) and lactate levels in tumors (L) in Braf/Pten melanomas from control and late T cell depletion groups (n = 12 per group).
(M) PCA of metabolites in Braf/Pten tumors from control and late T cell depletion (n = 4 per group). (N) Metabolite set enrichment analysis of Braf/Pten tumors from early T cell depletion versus matched control groups.
(O and P) Volcano plot (O) and PCA plot (P) of differentially accessible genomic regions determined by ATAC sequencing in Braf/Pten melanoma cells isolated from Braf/Pten-mTmG mice treated with PBS (control) or early T cell depletion group.
(Q) Enriched transcription factors binding differentially accessible genomic loci in control (Ctrl) or T cell-depleted Braf/Pten melanoma cells.
Data are representative or cumulative results of two independent experiments (A–D, G, H, K, and L). Each symbol represents one individual (G, H, K, and L) or the mean of 5 replicates for each treatment (C and D). Data are mean ± SEM and analyzed by unpaired, two-tailed Student’s t test.
However, the metabolic differences observed between primary tumor cells obtained from immunocompetent WT mice and Rag1−/− mice could result from unknown genetic variations. To rule out this possibility, we applied antibody-based T cell depletion in Braf/Pten mice 2 weeks post-tumor induction for 3 weeks (referred to as early depletion, a starting time point without palpable tumors) (Figure 1F). Similar to findings reported in other genetically engineered murine models,17–20 in which T cell ablation does not affect tumor formation and burden, our results showed that tumor burden in control and T cell-depleted Braf/Pten mice was similar (Figure 1G). Intriguingly, melanomas growing in the absence of T cells displayed a 50% decrease in intratumoral lactate levels (Figure 1H) and a robust difference in the global metabolic profile determined by LC-MS (Figures 1I and S1G) compared with melanomas in Braf/Pten mice receiving PBS treatment. In contrast to early T cell ablation, depleting T cells in Braf/Pten mice that have “established” melanomas for the same duration (late depletion) (Figure 1J) did not affect tumor burden, intratumoral lactate levels, or the global metabolic profile (Figures 1K–1M and S1H). These results suggest that the presence of T cells during early tumorigenesis, but not late-stage tumorigenesis, can impact metabolic features in tumors. We also found that metabolites in the TCA cycle were highly enriched while metabolites in the glycolytic pathway were downregulated in melanomas from T cell-depleted Braf/Pten mice (Figure 1N). Of note, early depletion of regulatory T cells (Tregs) did not impact metabolic profiles in melanomas (Figure S1I). Importantly, neither early nor late T cell depletion altered the hypoxic severity and expression of hypoxia inducible factor 1 alpha (HIF1α) in melanomas (Figures S1J–S1M). Considering that the Braf/Pten mice contain a defined oncogenic driver mutation in B-Raf kinase and deletion of Pten, our results reveal that T cell-mediated immune responses during early-stage tumorigenesis facilitate metabolic switch in tumors from OXPHOS toward aerobic glycolysis via an unexplored mechanism.
To further understand the impact of T cells during tumor progression, we obtained Braf/Pten-mTmG mice by crossing Braf/Pten mice with mTmG mice, a Cre reporter mouse strain that constitutively expresses tandem dimer Tomato fluorescent protein and the expression of Cre recombinase in melanocytes upon tamoxifen treatment will drive the expression of green fluorescent protein (GFP).21 We then depleted T cells as illustrated before (Figure 1F) and isolated GFP+ melanoma cells for examining chromatin accessibility and transcriptome. We identified a profound difference with more than 8,000 differentially accessible chromatin loci between melanoma cells growing in the presence or absence of T cells (Figures 1O and 1P). Moreover, the regions more accessible in melanoma cells growing in the presence of T cells showed enrichment in the binding motifs of AP-1 and cMyc (Figure 1Q), which have been shown to promote invasiveness and metabolic reprogramming in tumors.22–24 Interestingly, the binding motif for SOX, which has been shown to maintain melanocyte lineage, was enriched in melanoma cells growing in the absence of T cells, implying that T cell-mediated immunosurveillance could orchestrate invasiveness and metabolic programming by modulating chromatin architecture in tumor cells. In support of this, RNA sequencing results showed that the presence of T cells during tumorigenesis influenced the transcriptome in melanoma cells (Figure S1N). In addition, melanoma cells growing in the presence of T cells exhibited a higher gene expression associated with IFNγ response and Myc target genes, but lower expression in genes controlling OXPHOS compared with melanoma cells from T cell-depleted Braf/Pten-mTmG mice (Figure S1O). Altogether, these results suggest that T cell-mediated immunosurveillance might impact epigenetic programs empowering tumor cells with the ability to minimize OXPHOS and boost their dependency on aerobic glycolysis.
Immunosurveillance allows melanomas to acquire a metabolic program to disarm T cell anti-tumor immunity
We next investigated whether melanomas arising from T cell-depleted Braf/Pten mice could evade T cell anti-tumor immunity when they displayed distinct metabolic phenotypes. To do so, we took advantage of antibody-based T cell depletion to show that restoration of T cells can be achieved upon discontinuation of mAb treatment. Two weeks post-tumor induction, mice were treated with PBS or mAbs against CD4 and CD8 for 3 weeks. Then, T cell-depleted Braf/Pten mice were divided to receive either anti-CD4 plus anti-CD8 mAbs (continuous depletion) to sustain T cell depletion or PBS (discontinued group) to restore T cell abundance (Figure S2A). Surprisingly, melanomas from the discontinued group, but not the continuous T cell depletion group, displayed reduced growth kinetics and tumor burdens compared with control Braf/Pten melanomas (Figures 2A and 2B). Of note, melanomas from both groups had similar levels of T lymphocytes and myeloid cells in blood and tumors (data not shown; Figures S2B–S2F), highlighting that tumor-infiltrating T lymphocytes in the discontinued group may have higher tumoricidal activities. Moreover, tumors from the discontinued group decreased proliferation as indicated by reduced Ki67 staining but increased apoptosis and senescence as measured by terminal deoxynucleodigyl transferase dUTP nick end labeling (TUNEL) and staining of senescence-associated β-glactosidase (SA-β-gal) and p21 (Figures 2C and 2D). The induction of senescence and apoptosis in melanomas was confirmed by immunoblots against senescence markers, p16 and p21, and the apoptosis marker, cleaved caspase-3 (Figures S2G and S2H). We next investigated the infiltration pattern and functionality of tumor-infiltrating lymphocytes (TILs) and found that CD4+ TILs from control and discontinued groups had similar cytokine-producing ability (Figures S2I and S2J). CD8+ TILs from control and discontinued group displayed a similar diversity in T cell receptor repertoire (Figures S2K–S2M), suggesting that the increased anti-tumor responses in discontinued group were less likely to result from increased expression of more antigens. However, we observed a robust increase of CD8+ TILs in the tumor core of melanomas from the discontinued group (Figure 2E). Interestingly, CD8+ TILs from the discontinued group expressed lower levels of PD-1 and Tim3 (Figure 2F) and exhibited an enhanced ability to produce effector cytokines, including IFNγ and TNF, and granzyme B (Figures 2G–2I). In support of this, melanomas from the discontinued group contained higher levels of IFNγ and IFNγ-induced cytokines (Figure 2J), suggesting that the TME in the discontinued group is less immunosuppressive. Altogether, these results indicate that melanomas forming in the absence of T cells during early-stage tumorigenesis display metabolic features that cannot support their ability to disarm T cell-mediated anti-tumor responses.
Figure 2. Early-stage immunosurveillance supports melanomas to dampen T cell anti-tumor immunity.
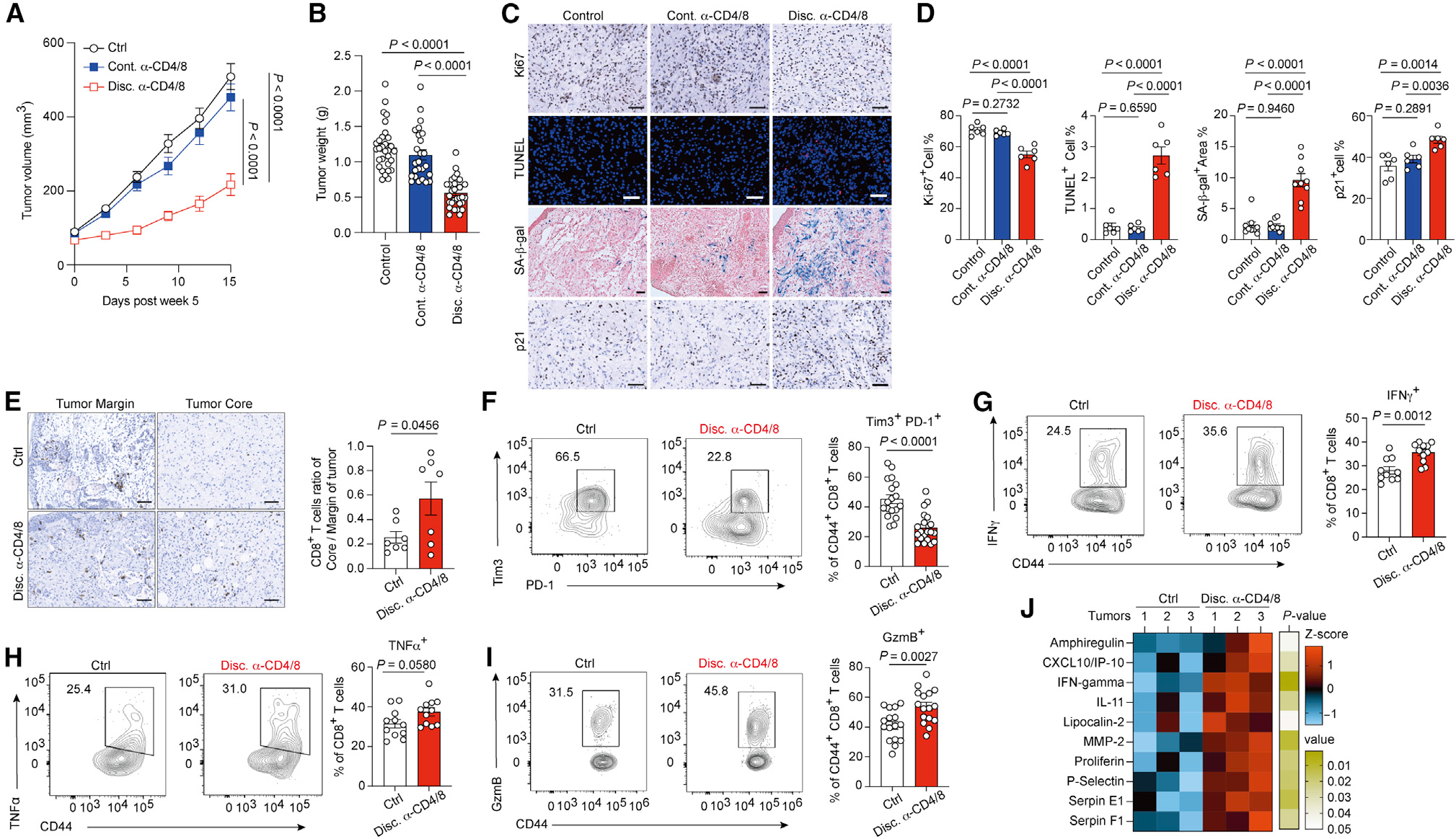
(A and B) Tumor growth (A) and tumor weight (B) of Braf/Pten melanoma tumor from control, continued anti-CD4/8 antibody treatment (Cont. α-CD4/8), or discontinued anti-CD4/8 antibody treatment (Disc. α-CD4/8) groups.
(C) Representative histology images for staining of Ki67, senescence-associated β-galactosidase activity (SA-β-gal), apoptotic cells with TUNEL, and p21 in indicated groups. Slides were counterstained with hematoxylin in chromogenic sections, counterstained with nuclear fast red in β-gal assay, and counterstained with DAPI in immunofluorescence staining. Scale bars, 50 μm.
(D) Quantitative results of IHC staining from indicated markers in indicated groups.
(E) Representative images (left) and quantitative results (right) of CD8+ T cell distribution in the margin region and core region of tumors from indicated Braf/Pten mice.
(F–I) Representative plots and quantitative results of Tim3+ PD-1+ CD8+ tumor-infiltrating T cells (F) and IFNγ-producing (G), TNF-α-producing (H), and granzyme B (GzmB)-producing cells (I) among total tumor-infiltrating CD44+ CD8+ T cells from the indicated mice.
(J) The heatmap of differentially expressed cytokines in tumors from indicated groups (n = 3 per group) determined by cytokine protein array.
Data are the cumulative results from at least two independent experiments (A, B, and F–I). Each symbol represents one individual (B and F–I) or represents the average of positive cell percentage from random 4 field in a section of individual tumor (D and E). Data are means ± SEM and analyzed by two-tailed, unpaired Student’s t test.
The exposure of IFNγ during early-stage tumorigenesis modulates metabolic programming in melanomas
Since we observed melanomas from the discontinued group displayed elevated senescence and apoptosis, which are major cellular processes induced by IFNγ,9,10 we then speculated that the inability to impede IFNγ production in CD8+ TILs could result in declined tumor growth. To investigate this, we temporally depleted T lymphocytes during early-stage tumorigenesis and then discontinued T cell depletion followed with either PBS treatment or neutralizing anti-IFNγ mAb treatment (Figure 3A). Intriguingly, neutralizing anti-IFNγ mAb restored tumor growth upon discontinuation of T cell depletion (Figures 3B and 3C). Given that PD-L1 expression is mainly regulated by IFNγ,9 we also examined PD-L1 expression pattern in tumors to validate the efficacy of the neutralizing strategy (Figure 3D, bottom panel). Most importantly, neutralizing IFNγ was able to prevent the declined proliferation and engagement of senescence and apoptosis occurring upon discontinuation of T cell depletion (Figures 3D and 3E). The action of IFNγ-neutralizing mAb on ameliorating senescence and apoptosis was further confirmed by immunoblots against senescence markers p16 and p21, and the apoptosis marker cleaved caspase-3 (Figures S3A and S3B). Together, these results suggest that T cell-mediated immunosurveillance during the early stage of tumorigenesis may empower tumor cells to restrict IFNγ-elicited anti-tumor responses. In support of this, we found that KO cells from Rag1-deficient Braf/Pten mice were capable of growing in IFNγ-deficient mice (IFNγ−/−) as well as Rag1−/− mice, but not WT mice (Figures 3F–3H). In contrast, WT cells exhibited comparable grow rates in both Rag1−/− and WT mice (Figure S3C). Of note, both WT and KO cells displayed similar IFNγ sensitivity to upregulate major histocompatibility complex I (MHCI) and PD-L1 (Figures S3D and S3E), highlighting that the differences in forming tumor in immunocompetent mice do not result from altered sensitivity to IFNγ. To directly examine whether IFNγ impacts metabolic states in tumors, we next treated Braf/Pten mice with neutralizing anti-IFNγ mAb during early staged tumorigenesis (Figure S3F). Similar to the results obtained by comparing tumors growing in the presence or absence of T cells during early staged tumorigenesis, blocking IFNγ did not affect Braf/Pten melanoma growth (Figure 3I) but led to robust changes in metabolic state in melanomas compared with PBS-treated group. Our result showed that melanomas from anti-IFNγ mAb-treated Braf/Pten mice were characterized by a higher abundance of TCA cycle metabolites but a decreased abundance of glycolytic metabolites (Figure 3J), supporting that IFNγ exposure during early-stage tumorigenesis participates in metabolic reprogramming in tumors. Collectively, these results unveil the importance of IFNγ in orchestrating metabolic preference in tumors and imply that IFNγ-instructed metabolic reprogramming can empower melanoma immune evasion.
Figure 3. IFNγ sculpts metabolic state in tumors during immunoediting.
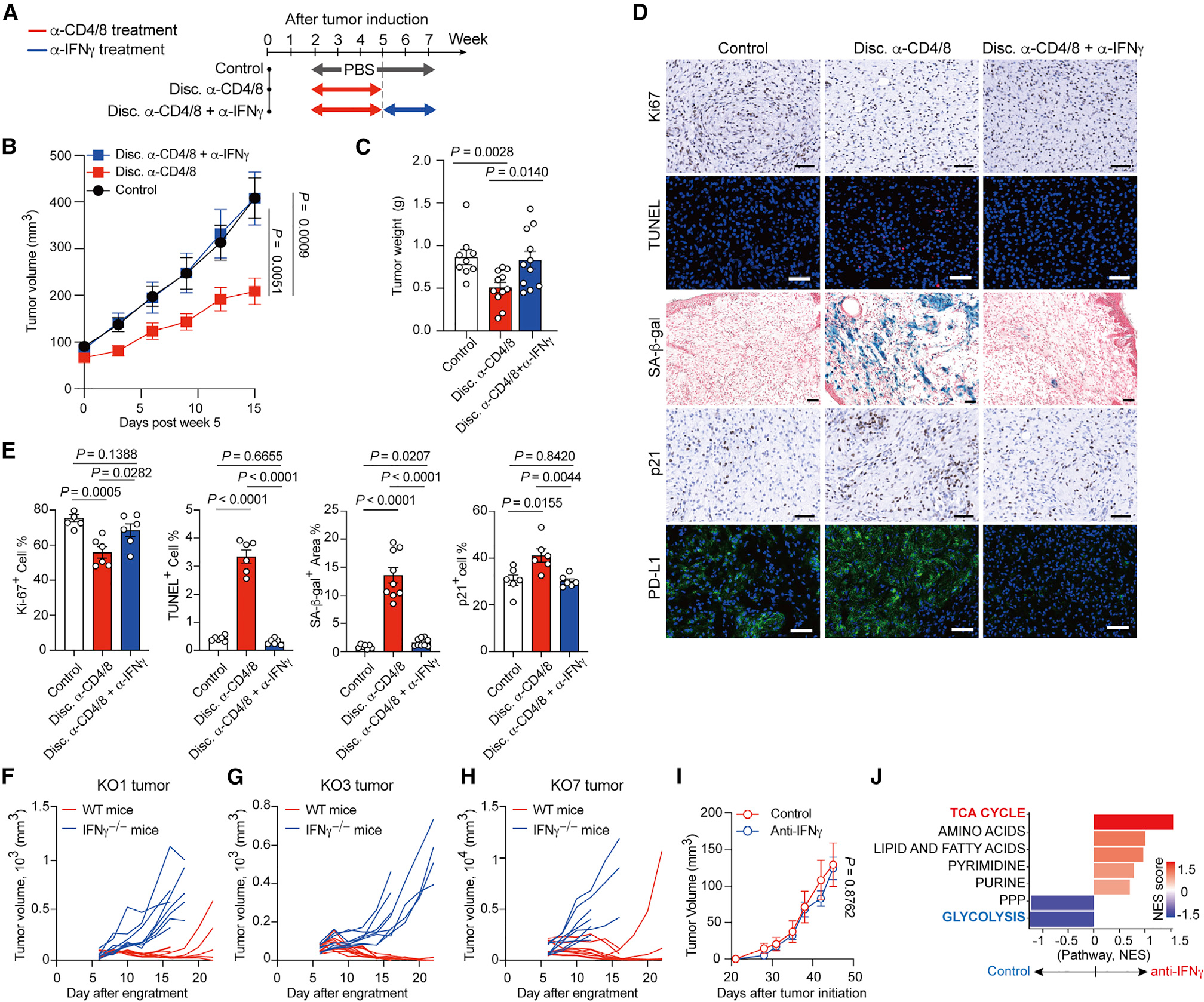
(A) Illustration of experimental design of IFNγ neutralization.
(B and C) Tumor growth (B) and tumor weight (C) of Braf/Pten melanomas from indicated groups. (D and E) Representative histology images (D) and quantitative results (E) for staining of Ki67, senescence-associated β-galactosidase activity (SA-β-gal), apoptotic cells with TUNEL, p21, and PD-L1 (labeled with Alexa Fluor-488 conjugated secondary antibody) in indicated groups. Slides were counterstained with hematoxylin in chromogenic sections, counterstained with nuclear fast red in β-gal assay, and counterstained with DAPI in immunofluorescence staining. Scale bars, 50 μm.
(F–H) Tumor growth curve for KO1 (F), KO3 (G), and KO7 (H), three Braf/Pten melanoma cell lines derived from adaptive immune-cell-deficient Braf/Pten mouse, after subcutaneously engrafting into C57BL/6 wild-type mice and IFNγ−/− mice (n = 6–8 per group).
(I) Tumor growth of Braf/Pten melanomas from PBS treatment (Control) and anti-IFNγ mAb treatment group.
(J) Metabolite set enrichment analysis of Braf/Pten tumors from anti-IFNγ antibody treatment group versus matched control group.
Data are the cumulative results from at least two independent experiments (B and C) or are representative images of two independent experiments with similar results (E–I). Each symbol represents one individual (C) or represents the average of positive percentage from random 4 field in a section of individual tumor (E). Data are means ± SEM and analyzed by two-tailed, unpaired Student’s t test.
IFNγ induces metabolic reprogramming in melanoma cells
To further understand whether the different metabolic preference between WT and KO melanoma cell lines results from genetic differences, we performed whole-exome sequencing and found that WT and KO cells contained the same oncogenic events and displayed high similarity in genomic composition and SNPs (Figures S4A and S4B). However, WT and KO cells had robust differences in transcriptome; specifically, gene expression associated with IFNγ response and cMyc target genes was highly enriched in WT cells (Figures 4A and 4B), implying that anabolic metabolism engaged by WT cells may result from cMyc-mediated regulatory circuit. Given that IFNγ has been shown to modulate transcriptome in tumor cells by eliciting epigenetic reprogramming,9,25 we speculated that melanoma cells growing in the presence or absence of T cells could have a distinct transcriptome even though they share similar genomic composition by exhibiting distinct epigenetic programs that can result in persistent differences in the transcriptome. Indeed, WT and KO cells had distinct patterns on chromatin accessibility and DNA methylation (Figures S4C and S4D). Similar to our result obtained from Braf/Pten-mTmG mice, the regions more accessible in melanoma cells growing in the presence of T cells showed enrichment in the binding motifs of AP-1 and cMyc (Figure 4C), supporting that T cell-mediated immunosurveillance imprints an epigenetic program that could influence metabolic preference and invasiveness in melanoma cells.
Figure 4. IFNγ induces metabolic reprogramming in melanoma cells for supporting immune evasion.
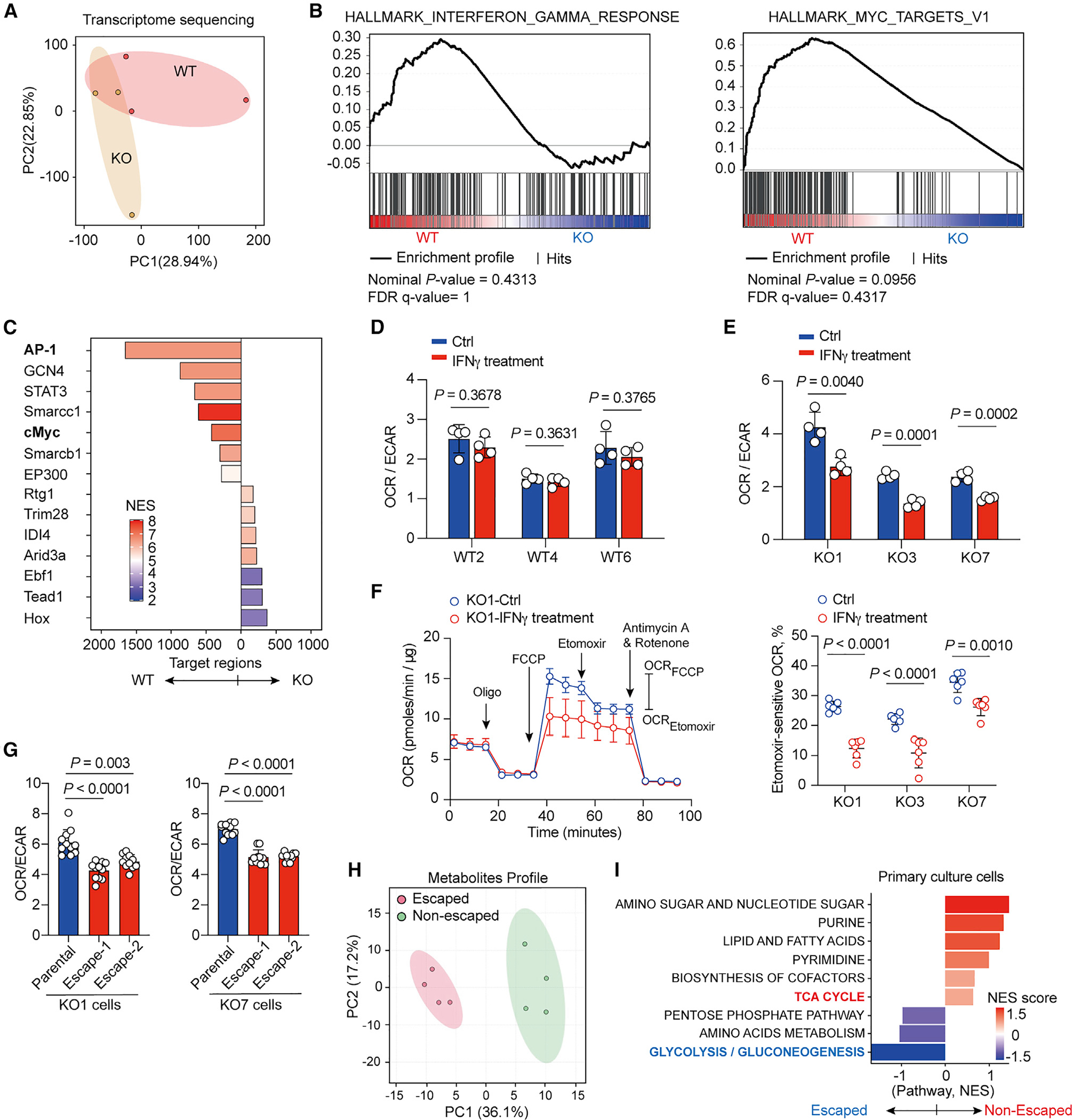
(A) PCA of transcriptome for WT primary melanoma cells and KO primary melanoma cells.
(B) Gene set enrichment analysis showing upregulated expression of genes on IFNγ response and cMyc targets in WT primary melanoma cells compared with KO primary melanoma cells.
(C) Significantly enriched transcription factors binding differentially accessible genomic loci in WT primary melanoma cells compared with KO primary melanoma cells.
(D and E) Ratios of basal OCR versus ECAR in WT primary melanoma cells (D) and KO primary melanoma cells (E) treated with control vehicle (Ctrl) or IFNγ for 48 h prior to assay (n = 4).
(F) Representative kinetic of OCR (left panel) following treatment with oligomycin (2 μM), FCCP (2 μM), etomoxir (40 μM), and antimycin A plus rotenone (0.5 μM each) in KO1 primary melanoma cells. Cells were incubated with control vehicle (Ctrl) or 100 ng mL−1 IFNγ for 48 h prior to assay. Relative etomoxir-sensitive OCR, calculated by measuring the differences of OCR levels between etomoxir and FCCP treatment, in indicated primary melanoma cells treated with control vehicle and IFNγ 48 h prior to assay (right panel) (n ≥ 5 per group).
(G) Ratio of basal OCR versus ECAR in parental KO1 and escaped KO1 cells (left panel) and in parental KO7 and escaped KO7 cells (right panel).
(H) PCA of metabolites in Braf/Pten tumors from non-escaped and escaped inducible Braf/Pten melanomas upon discontinuation of T cells depletion as illustrated in Figure 3A.
(I) Metabolite set enrichment analysis of Braf/Pten melanomas from escaped versus non-escaped tumors.
Data are representative results of two independent experiments with similar results (D–G). Each symbol represents one individual. Data are means ± SEM and analyzed by two-tailed, unpaired Student’s t test.
The above findings imply a model whereby IFNγ directly modulates metabolic machinery in tumor cells and renders tumor cells with the ability to create an immunosuppressive TME, in part via their metabolic programs. We then examined whether IFNγ controls metabolic preference in WT and KO melanoma cell lines. We found that acute IFNγ treatment did not affect OCR/ECAR ratios and intracellular ATP levels in WT cells (Figures 4D and S4E). However, even though IFNγ treatment did not affect ATP levels, IFNγ-treated KO cells reduced OCR/ECAR ratios, indicating a shift toward aerobic glycolysis to meet metabolic demands (Figures 4E and S4E). Moreover, IFNγ treatment abolished lipid utilization in KO cells, but not WT cells (Figures 4F and S4F). Similarly, we also found that etomoxir treatment suppressed OCR in primary HCC cells from Rag1−/− background rather than cells from an immunocompetent background (Figure S4G). These data suggest that non-edited KO cells employ lipid oxidation to meet their metabolic demands and IFNγ exposure could dampen FAO but stimulate aerobic glycolysis in non-edited cells. To further investigate whether the prolonged IFNγ exposure could imprint metabolic changes in melanoma cells, we generated IFNγ-adapted WT and KO cells by culturing parent cell lines in the presence of IFNγ for 4 weeks. We found that prolonged IFNγ exposure induced a persistent shift of metabolic dependency in KO cells, characterized by decreased OCR/ECAR ratios; however, WT cells retained lower OCR/ECA ratios (Figures S4H–S4K). These results suggest that IFNγ exposure could guide metabolic reprogramming in tumor cells to support their immune evasion. To test this, we established four cell lines from mice that formed progressive tumors after initial regression upon engraftment with KO1 or KO7 cells in immunocompetent mice. These escaped KO cells displayed uncontrolled tumor growth upon engraftment into immunocompetent mice (Figure S4L). Importantly, all escaped KO cells increased their metabolic preferences toward aerobic glycolysis but displayed similar sensitivity to IFNγ as measured by PD-L1 upregulation, compared with their parental cell lines (Figures 4G and S4M), highlighting that they acquire the ability to evade T cell-mediated immunosurveillance even though they maintain IFNγ sensitivity. In addition, some inducible Braf/Pten melanoma-bearing mice from the discontinued T cell depletion group (Figure 3A) became progressive melanomas (referred to as escaped melanomas) 5 weeks post-discontinuation of anti-CD4 and anti-CD8 mAb treatment. By examining metabolic profiles of escaped and non-escaped melanomas of inducible Braf/Pten melanoma-bearing mice, we found that escaped Braf/Pten melanomas displayed a distinct metabolic profile and a higher enrichment of metabolites in glycolytic pathway but a lower abundance of metabolites in the TCA cycle (Figures 4H and 4I). Taken together, these results reveal that IFNγ-guided metabolic alterations, potentially via epigenetic reprogramming, are a critical event for tumor immune evasion.
IFNγ stimulates STAT3-dependent cMyc upregulation, allowing tumor immune evasion
Since our results showed that IFNγ is the major factor influencing metabolic states in tumors during early-stage tumorigenesis, and cMyc transcription signature is enriched in tumor cells experiencing T cell-mediated immune responses, we then hypothesized that IFNγ exposure may drive metabolic reprogramming in tumor cells in a cMyc-dependent manner. We found that the acute exposure of IFNγ indeed stimulated cMyc expression in KO cells, but not in WT cells (Figures 5A and 5B). Moreover, IFN-γ-adapted KO cells, but not WT cells, stably expressed higher amounts of cMyc compared with parental cell lines (Figures S5A and S5B), suggesting that prolonged exposure of IFNγ in non-edited tumor cells could induce sustained cMyc expression. Next, we investigated whether cMyc controls metabolic reprogramming induced by IFNγ in KO melanoma cells. By using short hairpin RNAs (shRNAs), we knocked down cMyc in KO cells (Figure S5C). In contrast to KO cells expressing scramble shRNA, cMyc-deficient KO cells sustained OCR/ECAR ratios in response to acute IFNγ treatment (Figure 5C), supporting our postulate that IFNγ orchestrates metabolic reprogramming in a cMyc-dependent manner. In contrast, IFNγ remained ineffective in altering OCR/ECAR ratios in cMyc-deficient WT cells (Figure S5D). We also observed that IFNγ induced senescence in cMyc-deficient KO cells (Figures 5D and S5E). By examining the expression of p16 and p21, molecular regulators controlling senescence, we further confirmed cMyc plays a critical role in suppressing IFNγ-induced cellular senescence (Figures 5E, 5F, and S5F–S5I). Collectively, these results unveil an underexplored action by which IFNγ stimulates c-Myc expression that orchestrates metabolic reprogramming and empowers resistance to cellular senescence in tumor cells.
Figure 5. IFNγ-STAT3 signal axis promotes cMyc-dependent metabolic reprogramming and resistance to senescence.
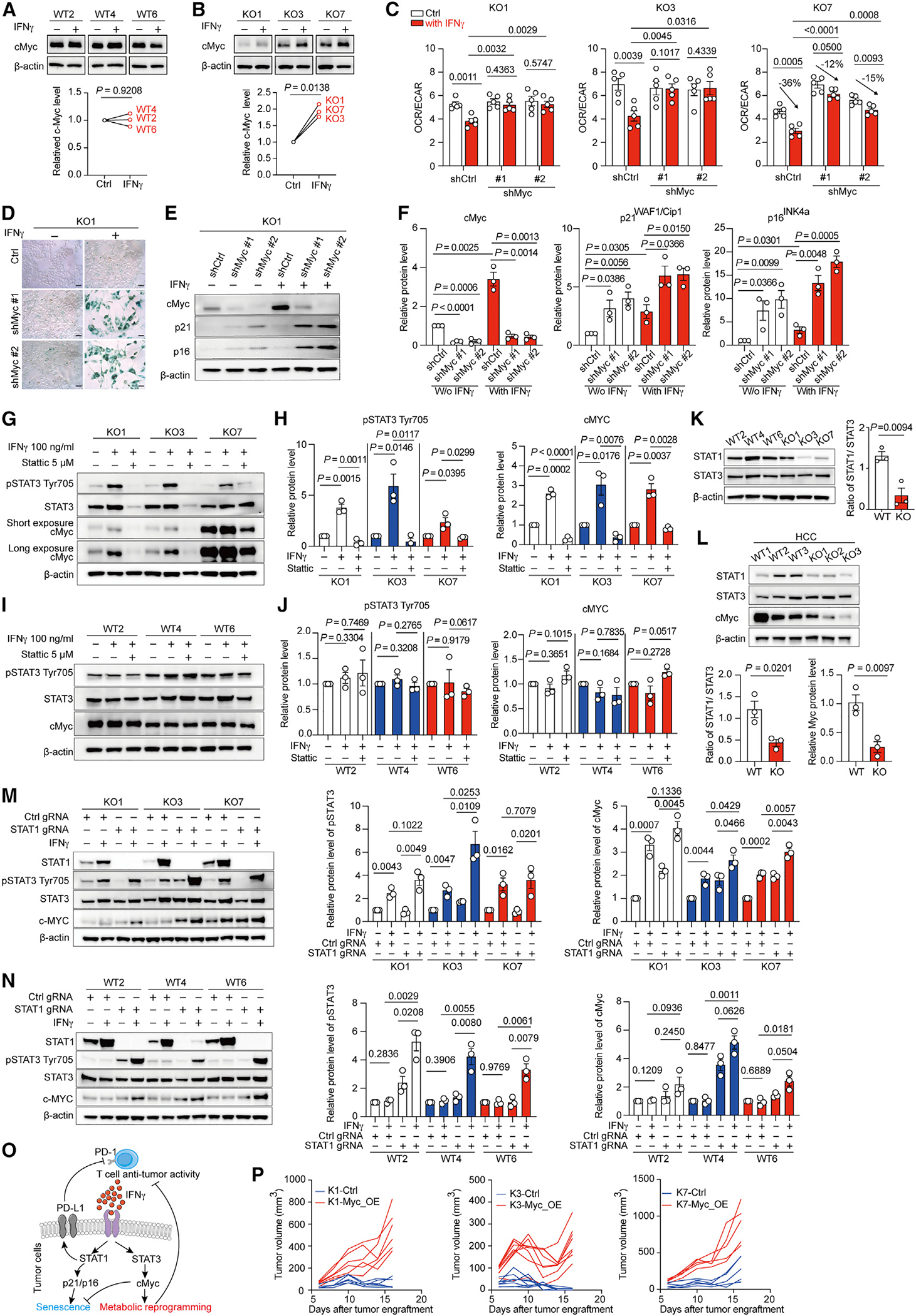
(A and B) Representative immunoblots (upper panel) of cMyc and β-actin and quantitative analysis of cMyc/β-actin ratios in primary WT melanoma cells (A) and KO melanoma cells (B) treated with or without IFNγ for 16 h.
(C) Ratios of basal OCR/EACR in indicated KO cells transfected with short hairpin RNAs targeting scramble sequence (shCtrl) or cMyc sequence (shMyc #1 and shMyc #2). Cells were incubated with or without 100 ng mL−1 IFNγ for 48 h prior to assay.
(D) Represented staining of β-gal activity in KO1 cells transfected with short hairpin RNAs targeting scramble sequence (shCtrl) or cMyc sequence (shMyc #1 and shMyc #2) upon exposure with or without IFNγ for 4 days. Scale bars, 50 μm.
(E and F) Representative immunoblots (E) and quantitative results (F) of indicated proteins in KO1 cells expressing control short hairpin RNA (shCtrl) or cMyc short hairpin RNAs (shMyc #1, #2) upon exposure with or without IFNγ for 16 h.
(G and H) Representative immunoblots (G) and quantitative results (H) of indicated proteins in KO1, KO3, and KO7 cells treated with indicated treatments for 16 h. Stattic: STAT3 inhibitor.
(I and J) Representative immunoblots (I) and quantitative results (J) of indicated proteins in WT primary melanoma cells treated with indicated treatments for 16 h.
(K) Representative immunoblots of indicated proteins in indicated cell lines with quantitative analysis for STAT1/STAT3 ratios (right panel).
(L) Representative immunoblot (upper panel) and quantitative results (bottom panels) for indicated proteins in the primary HCC cells harboring β-catenin overexpression and Pten deletion derived from immunocompetent mice (WT cells) or Rag1−/− mice (KO cells).
(M) Representative immunoblots (left panel) and quantitative results (right panels) of indicated proteins in KO cells expressing control gRNA (Ctrl gRNA) or gRNAs targeting STAT1 (STAT1 gRNA) upon exposure with or without IFNγ for 16 h.
(N) Representative immunoblots (left panel) and quantitative results (right panels) of indicated proteins in WT cells expressing control gRNA (Ctrl gRNA) or gRNAs targeting STAT1 (STAT1 gRNA) upon exposure with or without IFNγ for 16 h.
(O) Illustration of the proposed model.
(P) Tumor growth curve for KO1, KO3, and KO7 expressing either control (Ctrl) or c-Myc overexpression vector (Myc_OE) upon subcutaneously engrafted into C57BL/6 wild-type mice (n = 5–8 per group).
Data are representative or cumulative results of at least two independent experiments with similar results. Each symbol represents one individual. Data are means ± SEM and analyzed by two-tailed, paired (A and B) or unpaired (C, F, H, and J–N) Student’s t test.
Interestingly, IFNγ only promotes metabolic reprogramming in non-edited KO cells (Figures 4D and 4E); however, IFNγ exposure effectively upregulated the expression of PD-L1 and MHCI, STAT1 target genes, in both WT and KO cells (Figures S3C and S3D), suggesting that IFNγ modulates cMyc expression in an unexplored STAT1-independent mechanism to edit metabolic states in tumor cells. Interestingly, IFNγ has been reported to alternatively activate STAT3,26 a molecular hub supporting tumor invasiveness and immune evasion.27 Moreover, melanoma cells growing in the presence of T cells showed more accessible genomic regions with STAT3-binding motifs (Figures 1Q and 4C), implying that STAT3 is active in melanoma cells going through the immunoediting process. Intriguingly, we found that IFNγ simultaneously stimulated STAT3 and STAT1 activation and cMyc expression in KO cells (Figures S5J and S5K). However, IFNγ failed to stimulate STAT3 activation and cMyc expression, but remained effective in activating STAT1, in WT cells (Figures S5L and S5M). We then tested whether IFNγ treatment stimulates cMyc expression in a STAT3-dependent manner. Our results showed that IFNγ stimulated STAT3 activation while treatment with Stattic, a STAT3 inhibitor, hampered IFNγ-induced cMyc upregulation in KO cells (Figures 5G and 5H), but not WT cells (Figures 5I and 5J). Moreover, IFNγ failed to stimulate cMyc upregulation in KO cells with genetic ablation of STAT3 (Figures S5N and S5O), but STAT3 deficiency did not affect the maintenance of high cMyc expression in WT cells (Figures S5P and S5Q). Together, these results suggest that acute exposure of IFNγ to tumor cells triggers STAT3-dependent cMyc upregulation; however, prolonged exposure due to immunosurveillance could lead to fixed increase of cMyc.
IFNγ has been reported to preferentially trigger alternative STAT3 activation in STAT1-null fibroblasts,26 and we indeed found that KO cells displayed lower STAT1/STAT3 ratios compared with WT cells (Figure 5K). In support of our findings, we further found that primary HCC cell lines derived from immunocompetent mice expressed higher STAT1/STAT3 ratios and cMyc compared with cell lines derived from Rag1−/− mice (Figure 5L). To further confirm whether a lower STAT1/STAT3 ratio can facilitate IFNγ-mediated STAT3 activation and cMyc upregulation, we applied CRISPR-based gene ablation and found that IFNγ treatment was more potent to activate STAT3 and cMyc upregulation in KO cells harboring guide RNAs (gRNAs) targeting STAT1 (Figure 5M). Importantly, in STAT1-deficient WT cells, IFNγ exposure was capable of activating STAT3 and slightly stimulating cMyc expression (Figure 5N). Moreover, we found that KO cells stably expressing cMyc acquired the ability to form progression tumors in the immunocompetent mice compared with KO cells expressing control vector (Figures 5P and S5R). Collectively, our data suggest a model whereby the initial exposure of IFNγ in tumor cells during immunosurveillance could simultaneously stimulate STAT1 and STAT3 activation, in which STAT3-mediated cMyc upregulation suppresses senescence elicited by STAT1 activation and synergizes with the STAT1-PD-L1 pathway to hamper T cell immunity via metabolic reprogramming in tumor cells (Figure 5O).
cMyc target genes in melanomas support resistance to T cell anti-tumor responses
Next, to identify metabolic enzymes expressed in tumor cells that support tumor immune evasion, we performed an in vivo CRISPR screening that covered 2,078 metabolic enzymes (Table S1). We engrafted Cas9-expressing YUMM1.7 melanoma cell line (YUMM1.7-Cas9) transduced with gRNAs into immunocompetent WT mice and immunodeficient Rag1−/− mice (Figure 6A). Due to the inability to grow in the immunocompetent WT mice, we expected that tumor cells expressing gRNAs targeting metabolic genes supporting immune evasion will be more abundant in Rag1−/− mice compared with WT mice. In contrast, tumor cells with gRNAs targeting metabolic genes supporting or stimulating immunosurveillance will be more abundant in immunocompetent WT mice. To validate this strategy, we first transduced control gRNA or gRNAs targeting Janus kinase (JAK) to abolish IFNγ signal or PD-L1 into YUMM1.7-Cas9. Our results showed that YUMM1.7-Cas9 cells with gRNAs targeting JAK or PD-L1 expressed low amounts of PD-L1 in response to IFNγ treatment (Figure S6A). Moreover, YUMM1.7-Cas9 cells with gRNAs targeting PD-L1 formed tumors in immunodeficient Rag1−/− mice, but not immunocompetent WT mice (Figure S6B). In contrast, YUMM1.7-Cas9 cells with gRNAs targeting JAK displayed higher tumor growth rates in immunocompetent WT mice, indicating that ablation of genes supporting immunosurveillance in tumor cells indeed facilitates tumor growth in our system. We first confirmed that screening samples and the input cell pool had similar quality (Figure S6C), and our results showed a high mapping rate and distinct clustering of gRNAs between samples from immunocompetent WT mice and Rag1−/− mice (Figures S6D–S6I). A total of 83 and 63 genes were augmented and downregulated in tumors from Rag1−/− mice versus tumors from WT mice, respectively (Figure 6B; Table S2). Of note, deficiency of metabolic enzymes modulating the immunosuppressive features and invasiveness in tumors, including IDO1,28 kyneurenine 3-monoxygenase (KMO),29 proline dehydrogenase (PRODH),30 and phosphoglycerate dehydrogenase (PHGDH),31 increased in tumors growing in Rag1−/− mice, indicating that our in vivo screening strategy captures metabolic enzymes controlling the tumor cell’s ability to evade immunosurveillance. Among the 83 genes supporting tumor immune evasion, cMyc has been reported to directly bind to more than 50% of hits on their genomic loci for modulating gene expression. This result further supports our findings that cMyc-mediated metabolic reprogramming empowers tumor immune evasion.
Figure 6. In vivo CRISPR screening identifies metabolism pathways regulating the anti-tumor immunity.
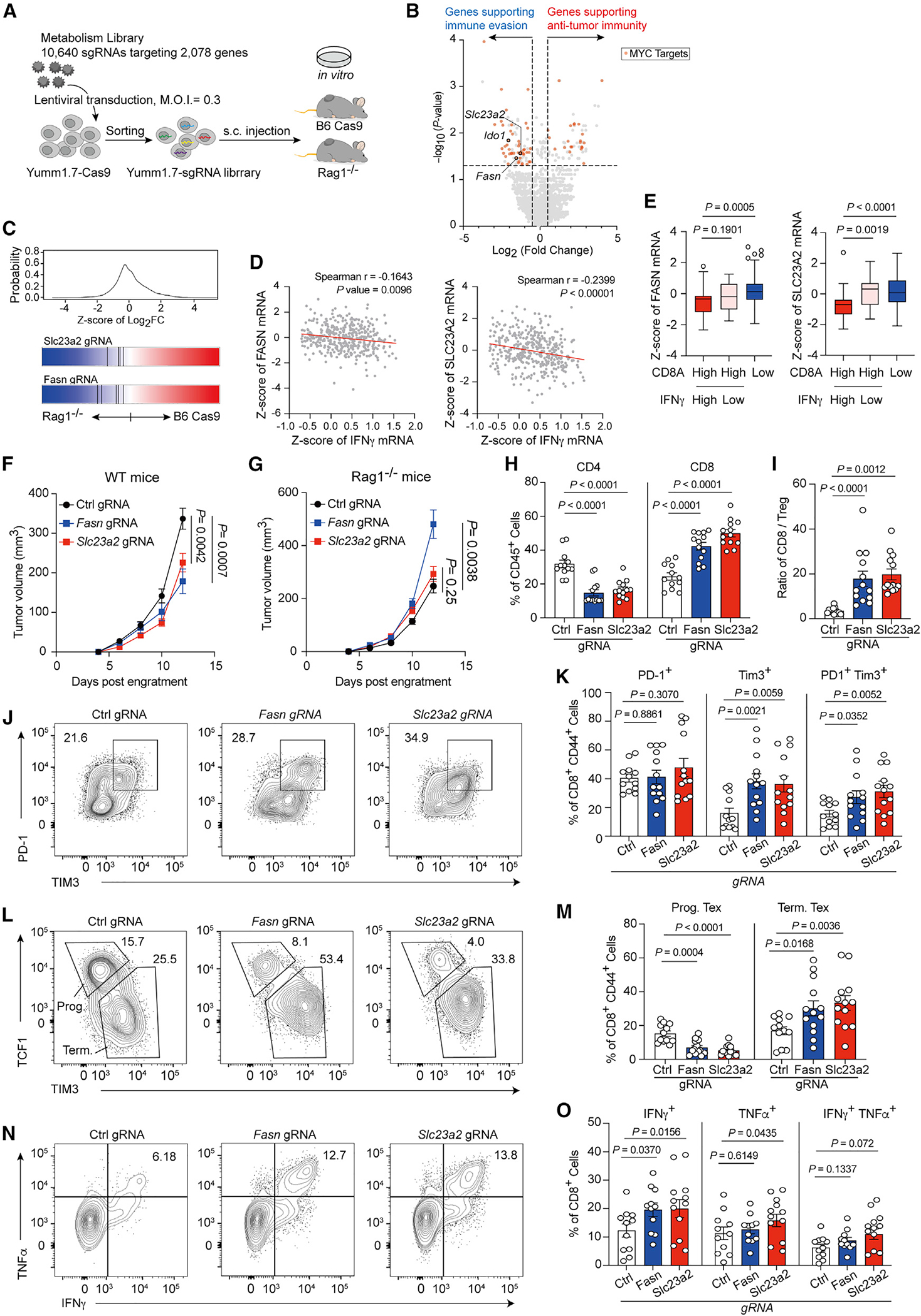
(A) Diagram of in vivo screening system for metabolism-guided immunoediting.
(B) Hits from screen for regulating tumor immunity. The x axis shows the log2 (fold change) comparing gRNA levels of tumors from WT (B6 cas9) with those from Rag1−/− mice, and the y axis denotes the −log10 of p value for differential expression obtained by MAGeCK. (Red symbol represents c-Myc target genes; Ido1, Fasn, and Slc23a2 are highlighted as open circle.)
(C) Frequency histograms of enrichment (Z score) for sgRNAs targeting indicated genes.
(D) Spearman correlation of Fasn (left) or Slc23a2 (right) expression (Z score) with IFNγ (n = 401 biologically independent melanoma tumor samples from TCGA cohort).
(E) Boxplot with Tukey whiskers showing varied Fasn (left) or Slc23a2 (right) expression (Z score) in CD8Ahigh and CD8Alow melanoma patients from TCGA cohort (n = 272).
(F and G) Tumor volume for Yumm1.7 tumors with control gRNA or gene deletions as indicated in WT (F) and Rag1−/− mice (G).
(H) Population of CD4+ (right) and CD8+ (left) T cells in Fasn-null, Slc23a2-null, or control tumors.
(I) Ratio of CD8+ T cells/FoxP3+ Treg in Fasn-null, slc23a2-null, or control tumors.
(J–M) Representative plots and percentages of the PD-1+ Tim3+ T cells (J and K) and the TCF1+ Tim3+ T cells (L and M) among total tumor-infiltrating CD44+ CD8+ T cells from the indicated mice.
(N and O) Representative plots (N) and percentages (O) of the IFNγ and TNF-α-producing tumor-infiltrating T cells from the indicated tumor samples after ex vivo restimulation with CD3/CD28.
Data are the cumulative results from at least two independent experiments (F–O). Each symbol represents one individual. Data are means ± SEM and analyzed by two-tailed, unpaired Student’s t test.
We next focused on two genes, FASN and Slc23a2, due to their involvement in supporting YUMM1.7 melanoma immune evasion (Figure 6C), and the expression of both genes negatively associated with IFNγ mRNA levels in melanoma patients of The Cancer Genome Atlas (TCGA) cohort (Figure 6D). Moreover, the expression levels of FASN and Slc23a2 associated with poor CD8+ TIL infiltration, and in patients with a high CD8A expression, IFNγ mRNA expression was downregulated in patients expressing high levels of Slc23a2 (Figure 6E). Of note, in contrast to Slc23a2, the other two members of the solute carrier family 23 that control uptake of vitamin C,32 Slc23a1 and Slc23a3, did not show negative association with IFNγ mRNA levels in melanoma patients of TCGA cohort (Figure S6J), suggesting that Slc23a2 supports melanoma immune evasion via an undefined and vitamin-C-uptake-independent mechanism. To validate whether FASN and Slc23a2 support melanoma immune evasion, we generated knockout YUMM1.7 cell lines and confirmed the knockout efficiency with immunoblots (Figure S6K). The loss of FASN and Slc23a2 hampered tumor growth in the immunocompetent WT mice compared with YUMM1.7 melanoma cells expressing control gRNAs (Figure 6F). However, both Fasn-deficient and Slc23a2-deficient YUMM1.7 melanoma cell displayed similar and even higher tumor growth rates in Rag1−/− mice compared with control YUMM1.7 cells (Figure 6G). We also found that Fasn- and Slc23a2-deficient B16 melanoma cells failed to form tumor cells in the immunocompetent mice (Figure S6L). Furthermore, the reduction of tumor growth of Fasn-deficient and Slc23a2-deficient YUMM1.7 melanoma in immunocompetent mice was accompanied by increased CD8+ TILs but reduced intratumoral CD4+ T cell abundance, which resulted in higher ratios of CD8+ TILs to Tregs (Figures 6H and 6I). Intriguingly, Fasn-deficient and Slc23a2-deficient melanomas contained more PD-1/TIM3 double-positive population (Figures 6J and 6K) but reduced progenitor exhausted T cells (defined by TCF1+ TIM3lo activated CD8+ T cells) (Figures 6L and 6M). By examining the cytokine-producing ability, we found that more CD8+ TILs from Fasn-deficient and Slc23a2-deficient melanomas were capable of producing IFNγ and TNF-α (Figures 6N and 6O), suggesting that loss of Fasn and Slc23a2 in melanoma cells could impair their ability to restrict infiltration of CD8+ T lymphocytes and to drive T cell dysfunction. To rule out the possibility that these differences resulted from different tumor burdens among groups, we collected TILs from control, Fasn-deficient, and Slc23a2-deficient melanomas with similar tumor weights. Our results confirmed that the above changes remained (Figures S6M–S6P). Taken together, our results further support the importance of cMyc-mediated metabolic program in facilitating tumor immune evasion and highlight the therapeutic potential of targeting the metabolic program acquired by tumor cells in response to immunosurveillance to revert the immunosuppressive features in tumors to an immunostimulatory context.
DISCUSSION
The astonishing metabolic flexibility of tumor cells underscores the concept that multiple metabolic programs can be used by tumor cells to support their growth and evokes the fundamental question of which factors occurring during tumorigenesis orchestrate metabolic preferences in tumor cells. Here, we uncover that the exposure of IFNγ during early-stage tumorigenesis facilitates engagement of aerobic glycolysis and declined FAO in tumor cells by driving cMyc-dependent metabolic reprogramming. As a result of this, upregulation of cMyc empowers tumor cells to restrict IFNγ-induced senescence and metabolic reprogramming allows tumor cells to impede tumor infiltration of CD8+ TILs and drive dysfunction in CD8+ TILs. Together, our findings unveil the existence of immunometabolic editing, an unexplored format and signaling cascade of immunoediting, that guides tumor cells to preferentially engage metabolic programs simultaneously supporting proliferation and immune evasion.
cMyc overexpression is a common feature in many cancers that synergizes with oncogenic mutations to promote tumor growth and metabolic reprogramming.23,24 Furthermore, cMyc overexpression has been shown to dampen adaptive and innate immune responses by promoting PD-L1 and CD47 expression in tumor cells.33 In support of this, cMyc upregulation in melanoma and neuroblastoma has been shown to be associated with poor immune infiltration in tumors,34 and melanoma patients with the elevated cMyc signature fail to respond to PD-1 blockade treatment in clinical trials.35,36 However, amplification of cMyc gene is a rare genomic change in cancers.37,38 In this study, we reveal that a non-canonical IFNγ-STAT3 signaling cascade is involved in the immunoediting process to stimulate cMyc-mediated metabolic reprogramming in tumors and support tumor immune evasion. Moreover, by applying in vivo CRISPR screening, we identified more than 40 metabolic enzymes controlled by cMyc participating in tumor immune evasion, which points out the diversity of metabolic programs that can be utilized by tumor cells to impede immunosurveillance. Altogether, these findings highlight that cMyc overexpression can coordinate metabolic reprogramming and immune-related pathways, including PD-L1 and CD47 expression, to maximize tumor immune evasion. Future studies are needed to precisely elucidate how those metabolic enzymes in tumor cells could support tumor immune evasion and their impacts on T cell dysfunction, which may lay a new foundation for developing treatments to reprogram immune states in the TME.
Our findings support the rationale for targeting STAT3 for cancer treatment, and we also speculate that targeting STAT3 in tumor cells may partially impair their metabolic reprogramming and stimulate sensitivity to IFNγ-induced senescence. In support of this, genetic ablation of STAT3 in tumor cells has been reported to promote immune infiltration into the TME, stimulate tumor regression, and induce apoptosis and senescence in the genetically engineered breast mouse model,27 B16 melanoma engraftment model,39 and carcinogen-induced skin cancer and HCC.40,41 Thus, the understanding of the contributions of non-canonical IFNγ-STAT3 signaling cascade during immunoediting may reveal a new dimension of tumor evolution. Interestingly, IFNγ treatment has been shown to induce epigenetic reprogramming in human melanoma cells without detailed mechanisms.25 In fact, STAT3 activation can reshape epigenetic landscape in somatic and melanoma cells,42,43 and the upregulation of cMyc has recently been shown to alter chromatin interactions at super-enhancer regions.44 Thus, it is likely that prolonged exposure of IFNγ during immunosurveillance could result in STAT3- and cMyc-dependent chromatin remodeling to modulate the transcriptome of tumor cells as we observed.
Of note, several studies did not observe differences in tumor incidence and growth kinetics between immunocompetent and Rag1- or Rag2-deficient mice by using genetic engineered tumor models, which led to the argument that immunoediting and T cell anti-tumor immunity did not exist.17–20 Here, our findings unveil that tumor cells could engage OXPHOS, which supports slow proliferation rates, to meet their metabolic demands when T cell-mediated immune responses are diminished. Thus, our work implies a possible scenario that the net growth outcome due to the metabolic program and diminished immunosurveilance in tumors growing in the absence of adaptive immunity are similar to tumors forming in immunocompetent mice, in which tumors use metabolic program supporting high proliferation rates to survive in response to anti-tumor immunity.
Overall, our findings describe a new dimension of tumor-immune interaction by which IFNγ produced by T cells instructs metabolic reprogramming in tumor cells for supporting tumor immune evasion. Of note, other immune cells such as NK cells also secrete IFNγ and participate in immunosurveillance. Thus, it is possible that immunosurveillance elicited by other immune cells can also tailor metabolic preference of tumor cells depending on which immune cell types are the major providers of immune pressure in the TME. Moreover, we also expect that how this tumor-immune interaction guides metabolic reprogramming in tumor cells can be influenced by the metabolic nature in the tissue context. The investigations of these questions would provide important information for elucidating the evolvement of the TME and development of metabolic targeting approaches that could unleash host anti-tumor immunity by alleviating metabolic stress imposed by tumors.
Limitations of study
Although we unveil that T cell-mediated immunosurveillance could impact the metabolic program and cMyc expression in murine tumor models, it remains challenging to explore whether this can be observed in human tumor settings. One major limitation is that human tumor cells exhibit high genomic instability that plays a major role in tumor evolution and immune evasion; however, our murine studies cannot fully recapitulate this. Furthermore, the metabolic heterogeneity of tumor microclusters within the same tumor is not explored in this study due to technical limitations. Altogether, this study uncovers a new dimension of immunoediting, immunometabolic editing, in guiding metabolic preferences in tumor cells, but the role of immunometabolic editing in human tumors remains to be investigated in future studies.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ping-Chih Ho (ping-chih.ho@unil.ch).
Materials availability
All the materials generated and used in this study will be available upon request
Data and code availability
The RNA-seq, ATAC–seq, RRBS data, Exome-seq and CRISPR screening are available in the Gene Expression Omnibus database under accession codes GSE217480 for RNA-seq, GSE190993 and GSE217475 for ATAC-seq, GSE217481 for RRBS data, GSE217479 for Exome-seq, GSE217861 for TCR-seq, GSE217862 for Smart-seq2, and GSE217859 for CRISPR screening. Data S1 contains uncropped immunoblots and raw data of all graphs. Uncropped blots and values of graphs are included in the zip file, Data S1.
No original code was generated in this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J, Rag1−/− (B6.129S7-Rag1tm1Mom/J), mTmG (B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J), and IFNγ−/− (B6.129S7-Ifngtm1Ts/J) mice were purchased from Jackson Laboratory. BRafCA; Tyr::CreER; Ptenlox4-5 (Braf/Pten) mice were obtained from M. Bonsenburg at Yale University. Mice were bred and crossed in-house to generate Tyr::CreERT2/+; BRafCA/+; Ptenl/l; ROSA26RLSL-tdTomato/+ and Tyr::CreERT2/+; BRafCA/+; Ptenl/l; Rag1tm1Mom. All mice were housed in specific pathogen-free facilities and allowed to acclimate to the animal facility prior to experimental use. All techniques and procedures conducted on the mice were in accordance with and approved by the Institutional Animal Care and Use Committee. For tumor induction, 3-week-old Braf/Pten mice were treated with 4-hydroxytamoxifen (4-HT) on the skin surface to induce tumor formation. For tumor engraftment, 1 × 105 cells tumor cells in 50 μl phosphate-buffered saline (PBS) were injected subcutaneously in 6-week-old mice, and tumors were measured every 2–3 d after tumor engraftment. Tumor volume was calculated using volume = (length × width2)/2. For antibody treatment, tumor-bearing mice were treated with anti-CD8 antibody (100 μg per injection, BioXcell, clone 2.43) plus anti-CD4 antibody (100 μg per injection, BioXcell, clone GK1.5), anti-CD8/4 antibody plus anti-IFNγ antibody (200 μg per injection; BioXcell, clone XMG1.2) or anti-CD25 antibody (350 μg per injection, BioXcell, clone PC61.5.3) according to the indicated combination by intraperitoneal injection. To allow T cell recovery, tumor-bearing Braf/PTEN mice, two weeks after tumor induction, were treated with a gradually decreased dosage of anti-CD4 and anti-CD8 antibodies of 100/100, 50/50, 50/50, and 50/25 μg, respectively. To confirm the T cell depletion and recovery, CD4 and CD8 cell population in blood of treated mice were analyzed in flow cytometry. For detection of hypoxia, tumors were harvested 2 hours after mice received 60 mg/kg of pimonidazole HCl. Animals were observed on a daily basis and sick mice were euthanized humanely in accordance with the Guidelines for Humane End Points for Animals used in biomedical research. Animals were housed in specific-pathogen-free facilities at the University of Lausanne and all experimental studies were approved and performed in accordance with guidelines and regulations implemented by the Swiss Animal Welfare Ordinance.
HCC Tumor generation
A sterile 0.9% NaCl mixture was prepared containing 10 ug of pT3-N90-CTNNB1 (CTNNB1; Addgene plasmid #31785), 5 ug of px330-sgPTEN1 and 5 ug of px330-sgPTEN2, and a 4:1 ratio of transposon to SB13 transposase-encoding plasmid dissolved in 2 ml of 0.9% NaCl solution and injected 10% of the weight of each mouse in volume into the lateral tail vein within 5–7 seconds. The px330-sgPTEN1, px330-sgPTEN2, and SB13 transposase-encoding plasmid were kindly provided by Dr. Amaia Lujambio and designed as previously described.45 Vectors for hydrodynamic delivery were produced using the QIAGEN plasmid PlusMega kit (QIAGEN). Equivalent DNA concentration between different batches of DNA was confirmed to ensure reproducibility among experiments.
Cell lines and in vitro cultures
YUMM1.7 melanoma cell line was kindly provided by Marcus Bosenberg.46 YUMM1.7 were cultured in DMEM with 10% fetal bovine serum and 1% penicillin-streptomycin and used for experiments when in exponential growth phase. Braf/Pten melanoma and HCC primary cells were established from non-necrotic, endpoint murine tumors in C57BL/6 and Rag1−/− mice. During necropsy, individual tumors were extracted and transferred into sterile PBS. Under sterile conditions, the tumors were finely chopped mechanically then digested with collagenase type IV (1mg/mL; Sigma Aldrich) and Dispase II (2 U/ml; Gibco) prepared in 5mL of DMEM for 30 minutes at 37 °C) with agitation. The digestion was then neutralized and washed twice with 5mL DMEM media supplemented with 10% (v/v) heat-inactivated FBS (Gibco), and 1% penicillin-streptomycin. The resulting pellet was disaggregated with a solution of 0.25% Trypsin and incubated for 5 min at 37 °C. For immune-escaped KO1 and KO7 cells, 1 × 106 cells were subcutaneously injected in immune-competent mice, and tumors with continued growth two weeks following engraftment were harvested. The cells were maintained in culture for a minimum of 8 passages prior to experimental use to select tumor cells and reduce contamination from other cell types. The primary cells with a passage number of 9–15 were used in the experiment. To derive IFNγ-resistance, cells were incubated in IFNγ-containing medium using gradually increased dosage from 10 to 100 ng mL−1 of recombinant murine IFNγ (PEPROTECH) in 2 weeks. All cells were maintained in DMEM with 10% FBS and 1% penicillin-streptomycin and used for experiments when in the exponential growth phase.
METHOD DETAILS
Flow cytometric analysis
Tumors were minced into small pieces in RPMI containing 2% FBS, 1% penicillin-streptomycin, DNase I (1 μg ml−1; Sigma-Aldrich) and collagenase (1 mg ml−1; Sigma-Aldrich) and kept for digestion for 40 min at 37 °C, followed by the filtration with a 70-μm cell strainer. Filtered cells were incubated with ACK Lysing Buffer (Invitrogen) to lyse red blood cells and then washed with FACS buffer (phosphate-buffered saline with heat-inactivated 2% FBS and 0.1% sodium azide). Tumor-infiltrating leukocytes were further enriched by Percoll density gradient centrifugation (800g for 30 min) at room temperature. Single-cell suspensions were incubated with anti-CD16/32 Fc receptor-blocker on ice for 10 min before staining. Viable cells were first stained using a LIVE/DEAD Fixable Dead Cell Stain Kit (Thermo Fisher Scientific) at room temperature for 15 min. After washing, surface proteins were stained for 30 min at 4 °C. To detect cytokine production, cell suspensions were resuspended in RPMI 1640 with 10% FBS and incubated with Dynabeads covalently coupled to anti-CD3 and anti-CD28 antibodies (Invitrogen) for another 5 h at 37 °C in the presence of 2.5 μg ml−1 Brefeldin A Solution (BioLegend). Cells were processed for surface marker staining, as described above, and then intracellular cytokine staining. Samples were analyzed on LSRII flow cytometers (BD Biosciences), and data were analyzed with FlowJo v10. Cells were sorted on a FACSAria III sorter (BD Biosciences). The following antibodies were used for flow cytometry: anti-CD3ε (17A2), anti-CD4 (RM4–5), anti-CD8a (53.6.7), anti-CD11b (M1/70), anti-CD11c (N418), anti-CD19 (6D5), anti-CD45 (30-F11), anti-CD103 (2E7), anti-Gr-1 (RB6-8C5), anti-MHC class II I-Ab/I-E (M5/114.15.2), anti-MHC class I (28-8-6), anti-FoxP3 (MF-14), anti-NK1.1 (PK136), anti-IFN-γ (XMG1.2), anti-TNF-α (MP6-XT22), anti-CD274 (10F.9G2), anti-Tim3 (RMT3–23), and anti-PD-1 (RMP1–30). All antibodies were purchased from Biolegend, BD Biosciences or Invitrogen (eBioscience).
Immunoblot analysis
Cell pellets were lysed in the presence of complete EDTA-free Protease Inhibitor Cocktail (Sigma-Aldrich, no. 11873580001). Protein lysates were mixed with SDS–PAGE loading dye and then subjected to SDS-PAGE for immunoblotting. The following antibodies were used for immunoblots: anti-cMyc (Y69) rabbit monoclonal Ab (Abcam, no. ab32072), anti-p21 (EPR18021) rabbit monoclonal Ab (Abcam, no. ab188224), anti-CDKN2A/p16INK4a (EPR20418) rabbit monoclonal Ab (Abcam, no. ab211542), anti-rabbit IgG HRP-linked antibody (Cell Signaling, no. 7074), anti-mouse IgG HRP-linked antibody (Cell Signaling, no. 7076), anti-HIF-1α (D1S7W) rabbit monoclonal Ab (Cell Signaling, no.36169), anti-STAT1 ((D1K9Y) rabbit monoclonal Ab (Cell Signaling, no. 14994), anti-STAT3 (D1B2J) rabbit monoclonal Ab (Cell Signaling, no. 30835), anti-pSTAT1 Tyr701 (58D6) rabbit monoclonal Ab (Cell Signaling, no. 9167), pSTAT3 Tyr705 (D3A7) rabbit monoclonal Ab (Cell Signaling, no. 9145), anti-Fasn rabbit polyclonal Ab (Cell Signaling, no. 3189), anti-Slc23a2 rabbit polyclonal Ab (Invitrogen, no. PA5-111911) and anti-β-Actin mouse monoclonal antibody (Sigma-Aldrich, no. A2228). For proteome cytokine array, tumor samples were frozen on dry ice immediately following resection from tumor-bearing mice. Tumor samples were homogenized in Tris-buffered saline containing protease inhibitors and 1% Triton X-100 through 30 sec-high-speed shaking in TissueLyser (Qiagen). From each sample, 200 μg of protein lysate was applied to each membrane of the Proteome Profiler Mouse XL Cytokine Array (R&D, ARY028). Staining and exposure were performed according to the manufacturer’s instructions. The signal intensities of indicated cytokines and target proteins were calculated by ImageJ.
Seahorse extracellular flux analysis
Extracellular flux analysis was performed with a Seahorse XF96 Extracellular Flux Analyzer. After equilibrium in a non-CO2 37°C incubator for 45 minutes, Cells were treated with oligomycin (2 μM, Sigma-Aldrich), FCCP (2 μM, Sigma-Aldrich), etomoxir (40 μM, Sigma-Aldrich), rotenone (0.5 μM, Sigma-Aldrich), and antimycin A (0.5 μM, Sigma-Aldrich) in the DMEM containing glucose (10 mM), sodium pyruvate (10 mM), and L-glutamine (20 mM). For response to IFNγ, cancer cells were treated with 100 ng mL−1 of recombinant murine IFNγ (PEPROTECH) for 48 hours prior to assay. Each condition was analyzed, with 4–6 replicates in each single experiment.
Immunohistochemistry analysis and β-galactosidase staining
Paraffin sections at a thickness of 4–5 μm were deparaffinized and stained with antibodies against CD8α (Cell Signaling, no. 98941), p21 (Abcam no. ab188224), and Ki67 (Abcam no. ab16667) followed by STAT-Q IHC chromogenic staining system (Innovex Biosciences). Chromogenic staining was determined in the Zeiss Axioscan Z1 slide scanner. For frozen samples, tumor tissues were put in buffered formalin containing 10 % sucrose for 1 hour at 4 °C, transferred into PBS with 20% sucrose for 16 hours, and embedded in Tissue-Tek O.C.T. compound (Sakura). Frozen tumors were sectioned at a thickness of 8 μm and incubated with anti-PD-L1 antibody (MIH5, Invitrogen, no. 14-5982-82), detected by Alex flour 488-conjugated anti-Rat (H+L) secondary antibody (Invitrogen, no. A-11006). Fluorescent images were acquired and exported on a Zeiss Axio Imager Z1microscope. Hypoxia level, apoptosis, and senescence in tumor tissues were determined according to the manufacturer’s instructions for Hypoxyprobe Plus Kit (Hypoxyprobe), ApopTag Fluorescein In Situ Apoptosis Detection Kit (Merck), and Senescence β-Galactosidase Staining Kit (Cell signaling), respectively. For response to IFNγ, indicated cells were incubated with 100 ng mL−1 for 4 days. Four random regions were selected to quantify positive levels for each tumor tissue section, and the value for each image was quantified using ImageJ. To determine the distribution of CD8+ T cells, tumor margin was defined as a 250-μm-wide region from their distance to the boundary and a region of 250–750 μm inside the tumor boundary was defined as the tumor core region.
Broad-scale targeted metabolomics
Tissue samples were immersed in extraction solvent (methanol/water, 80/20, v/v, pre-cooled in −80°C freezer) using a homogenizer with ceramic beads. The extracts were centrifuged for 10 min at16,000g at 4 °C and the resulting supernatant was collected and evaporated to dryness in a vacuum concentrator (LabConco, Missouri, US). Dried sample extracts were resuspended in MeOH:H2O (4:1, v/v) according to the total protein content (evaluated using BCA protein assay kit) and analyzed by hydrophilic interaction liquid chromatography coupled to tandem mass spectrometry (HILIC–MS/MS) in both positive and negative ionization modes using a TQ-S triple quadrupole mass spectrometer (6495 Ion Funnel, Agilent Technologies). Optimized compound-dependent parameters were used for data acquisition in scheduled multiple reaction monitoring mode.47 Pooled quality control samples (representative of the entire sample set) were analyzed periodically (every four to five samples) throughout the overall analytical run, to assess the quality of the data, correct the signal intensity drift and remove peaks with poor reproducibility (coefficient of variation > 30%). Hepatocellular carcinoma metabolite extracts were analyzed using Ultimate 3000 Dionex UHPLC (Thermo Fisher Scientific) coupled to Q Exactive Plus mass spectrometer (Thermo Fisher Scientific). A hydrophilic interaction chromatography method (HILIC) with an Xbridge amide column (100 × 2.1 mm i.d., 3.5 μm; Waters) was used for compound separation at 25 °C. Mobile phase A: water with 5 mM ammonium acetate (pH 6.8), and mobile phase B: 100 % acetonitrile. Linear gradient is: 0 min, 85% B; 1.5 min, 85% B; 5.5 min, 35% B; 6.9 min, 35% B; 10.5 min, 35% B; 10.6 min, 10% B; 12.5 min, 10% B; 13.5 min, 85% B; 17.9 min, 85% B; 18 min, 85% B; 20 min, 85% B. The flow rate is: 0–5.5 min, 0.15 ml/min; 6.9–10.5 min, 0.17 ml/min; 10.6–17.9 min, 0.3 ml/min; 18–20 min, 0.15 ml/min. Q Exactive Plusmass spectrometer was equipped with a HESI probe and operated in the positive/negative switching mode. The relevant parameters are as listed: 120 °C; sheath gas, 30; auxiliary gas, 10; sweep gas, 3; spray voltage, 3.6 kV for positive mode and 2.5 kV for negative mode; capillary temperature, 320°C; S-lens, 55. The resolution was set at 70,000 (at m/z 200). Maximum injection time (max IT) was set at 200 ms and automated gain control (AGC) was set at 3 × 106.
Metabolite identification was based on exact mass to charge ratio and retention time, which was determined using in house library. Integrated peak area of each metabolite was first normalized to the corresponding tissue weight and was then used to calculate the relative changes of metabolites in different samples. The obtained tables (containing peak areas of detected metabolites across all samples) were exported to “R” software http://cran.r-project.org/ where signal intensity drift was corrected in the LOWESS/Spline normalization program followed by noise filtering (coefficient of variation (QC features) > 30%) and visual inspection of the linear response. Unsupervised principal component analysis (PCA) was performed using the MetaboAnalyst (version 4.0).48 Metabolite intensities with auto scaling were used for the analysis. To investigate the potential molecular biological pathway of indicated treatments, the Gene Set Enrichment Analysis (GSEA; version 4.1.0) was performed to explore the enriched pathway based on differentially expressed metabolites.49,50 The log2-transformed ratio of indicated groups were calculated and used as pre-ranked list for GSEA analysis. Normalized enrichment score (NES) was calculated by GSEA using self-defined metabolic pathway.
RNA-seq data processing and computational analysis
500 viable GFP+ cancer cells from Braf/Pten-mTmG mice with indicated treatment were isolated by FACS cell sorters (with at least 99% purity) and directly into 4 μl lysis buffer consisting of 0.2% (v/v) Triton X-100 solution and RNase inhibitor (Clontech). Plates containing samples were sealed, flash-frozen and kept at −80 °C before further processing following a version of the Smart-seq2 protocol described before.51 The sequencing library was prepared by illumine Truseq Stranded RNA kit and sequenced by single-end 150bp on illumina HiSeq 4000. Raw reads were trimmed using TrimGalore (v.0.6.6) according to quality from FastQC and adaptor. The trimmed reads were mapped to the mm10 using STAR (v.2.7.9a). The summarizeOverlaps function from the GenomicAlignments package (v.1.30.0) in R/Bioconductor (v.3.13) was used to define gene expression level by counting the alignments to exons. The differential expression analysis was performed using DESeq2 (v.1.34.0). The Gene Sets Enrichment Analysis (GSEA) was performed by GSEA software and represented the results by R. The Principal Component Analysis (PCA) was performed by R.
Chromatin accessibility data processing and analysis
Sorted tumor cells or primary culture tumor cells from the indicated conditions were collected and chromatin accessibility mapping performed by ATAC–seq, as described previously.51 Base calling was performed by Illumina Real Time Analysis (v2.7.7) software and the base calls were converted to short reads using the IlluminaBasecallsToSam tool from the Picard toolkit (v2.19.2). Sequencing adapters were removed, and the low-quality reads were filtered using the fastp software (v 0.20.1). The short reads were aligned to the mm10/GRCm38 assembly of the mouse reference genome using Bowtie2 (v2.4.1) with the “-very-sensitive” parameter. PCR duplicates were marked using samblaster (v0.1.24), and the reads mapped to the ENCODE black-listed regions were discarded prior to peak calling. BigWig files were generated using the computeMatrix from the deepTools (v3.5.0), using the following parameters: -p max –binSize 10 –normalizeUsing RPGC –effectiveGenomeSize 2407883318 –extendReads 175. To detect the open chromatin regions, we performed peak calling using the MACS2 (v2.2.7.1) software using the function callpeak with the following options: –nomodel –keep-dup auto –g 2407883318 –p 1e-09 –extsize 147. Peaks overlapping with blacklisted regions were discarded. Finally, the consensus peak list was created by merging the ATAC-seq peaks from all samples using the BEDTools (v2.29.2) merge command. The presences of open chromatin were used to performed PCA analysis by R. The differential accessibility was calculated using DESeq2. Differentially accessible regions between populations were defined using P value ≤ 0.05 and |log2(fold change)| ≥ 1 for following transcription factor enrichment analysis. The transcription factor enrichment analysis was performed by i-cisTarget.52
TF-binding motif enrichment analysis
To identify TF-binding motifs enriched in the sets of differentially accessible regions, the i-cisTarget tool was used (online version v6.0, input bed files, genome mm9, 24453 PWMs database).53 Genome coordinates of the analyzed regions were converted from mm10 to mm9 using the UCSC liftover tool (using the -minMatch=0.95 parameter). The motifs from the same cluster were merged and TFs annotated for motifs specifically enriched in C or TD that also have at least one ATAC peak assigned were selected.
Exome-seq data processing and computational analysis
High-molecular-weight gDNA was extracted by Blood & Cell Culture DNA Kit (Qiagen, Cat. 13323) according to manufacture protocol. The sequencing library was prepared by mouse exome panel (Twist Bioscicences, Cat. 102035) and sequenced by paired-end 150bp on illumina HiSeq 4000. For SNV/Indel calling, raw reads were converted to unmapped bam by using samtools, and the unmapped bam files were marked duplicates with MarkDuplicatesSpark function from GATK4 (v.4.2.3.0). The processed bam files were mapped to the mouse reference genome (mm10) by using BWA (v.0.7.17), and called variation by Mutect2 individually. For calling copy number variations, the flat reference was first generated according to targets from Twist mouse exome panel. The aligned sam files were sorted and converted to bam files by using samtools. The copy number variations were called by CNVkit with amplicon method and adjusted to β-allele with SNV/indel results from GATK4. The results of SNV/Indel and CNV were present by R with the UpSetR, CopyNumbePlots, and KaryoploteR packages.
RRBS data processing and computational analysis
Full length gDNA was extracted by Blood & Cell Culture DNA Kit (Qiagen, Cat. 13323) according to manufacture protocol. The RRBS samples were sequenced by single-end 50bp on Illumina HiSeq3000 platform. Base calling was performed by Illumina Real Time Analysis (v2.7.7) software and the base calls were converted to short reads using Illumina2bam (http://gq1.github.io/illumina2bam/) tool. Trimmomatic (v0.32) (Bioinformatics 30.15 (2014): 2114–2120) was used for trimming the adapter sequences. Trimmed short read sequences were aligned onto the mm10/GRCm38 mouse reference genome with BSMAP(v2.90)54 aligner in RRBS mode which was optimized for aligning the RRBS data while being aware of the restriction sites. Methylation levels at the CpG sites were detected by using an in-house software called biseqMethCalling (https://github.com/epigen/biseqMethCalling), which was developed to better support RRBS data analysis. Finally, RnBeads(v1.6.1) package55 was used for further quality control, exploratory and differential methylation analysis. The Principle Component Analysis (PCA) was performed by R.
TCR clonality in Braf/Pten melanomas
Tumors tissues from with indicated group were minced into small pieces in RPMI containing 2% FBS, 1% penicillin-streptomycin, DNase I (1 μg/ml) and collagenase (2 mg/ml) and dissociated by gentleMACS followed by digestion for 30 min at 37 °C. Tumor-infiltrating leukocytes were further enriched by Percoll density gradient centrifugation (800g for 30 min) at room temperature and alive tumor-infiltrated T cells were sorted according to positive expression of CD45, CD3, CD8, and CD44. Briefly, 3000–5000 of TILs were lysis by Lysis/Binding Buffer (Invitrogen) for further mRNA purification. Murine TCR beta-chain specific primer was used for amplification and followed by amplicon-sequencing. TCR repertoire analysis and clonotype identification was done by MiXCR in Linux platform, and the diversity estimation was quantified by “immunarch” package in R.56 The diversity of TCR was quantify by D50 index, chao1, and Gini-simpson index.
Integration of RNA-seq, ATAC-seq, and RRBS
The differential express genes obtained from DEseq2 were first filtered according to log2 foldchange and p-value. The top 100 differentially expressed genes, which are dominant in WT cells or KO cells were selected for further analysis. The gene loci of top differential express genes were obtained from the UCSC genome browser, and 2,000 bp around TSS (+- 1,000 bp) were defined as promoter regions in this study. The integration of ATAC-seq and RRBS was processed by Easeq software. Overall, the top differential express genes were first ranked according to log2 foldchange, and ATAC-seq & RRBS data were extracted according to the promoter region. To maximize the differences of RRBS data which is single-base resolution, a 200 bp window was used to calculate accumulated methylation percentage. The scale of ATAC-seq signal was normalized according to the number of mapped reads.
In vivo CRISPR screening
For screening the Yumm1.7-Cas9 cell line, we used a library of 10,640 optimized sgRNAs targeting 2,078 metabolism genes, 5 gRNAs per genes and 250 non-targeting control sequences, which designed and provided by Massimiliano Mazzone. The metabolism gRNAs pools were delivered to Yumm1.7-Cas9 cells via lentiviral infection at an infection rate of 30%. Transduced cells were purified using an Thy1.1 reporter by FACS and then expanded in vitro before being implanted into animals. For in vivo CRISPR screening, Yumm1.7 cells-gRNA library were treated with doxycycline (10μg/mL) for 3 days and refreshed the medium without doxycycline for another 3 days before 1×106 cells were engrafted into Rag1−/− mice and Cas9 mice. Mice were euthanized 15–18 days after tumor implantation and tumor genomic DNA was prepared from whole tumor tissue by salt precipitation method (https://doi.org/10.1016/j.cell.2015.02.038). Briefly, tumor tissues were homogenized in lysis Buffer (50mM Tris, 50mM EDTA, 1% SDS, pH8) containing 0.1 mg/mL proteinase K at 55°C overnight, and genomic DNA were extracted by isopropanol following removal of RNA and protein by RNase A and ammonium acetate, respectively. PCR was used to amplify the sgRNA region according to the adaptor forward primer (5’ TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG ATC TTG TGG AAA GGA CGA AAC 3’) and reverse primer (5’ GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GCA AGT TGA TAA CGG ACT AGC C 3’) and the sequencing library was sequenced by single-end 150bp on illumina HiSeq 4000. The analysis pipeline and quality control were performed according to MAGeCK-VISPR algorithm. The MYC targets were got from Harmonizome.57
Generation and engraftment of CRISPR-edited cell lines
For the cell line generation, all the CRISPR-edited primary, Yumm1.7, and B16-F10 melanoma cell lines were generated with SCAR system as described previously.58 Firstly, pSCAR_Cas9-hygro lentivirus were generated, and then cancer cell lines were infected in media containing polybrene. After 48 h, cells were selected with hygromycin B at least 2 passages (more than 5–7 days). Cas9-expressing cancer cells were further infected with lentivirus for pSCAR_sgRNAs as listed below for 48 h, followed by the selection with puromycin for at least 2 passages (more than 5–7 days). The expression of GFP and mKate2 markers for the pSCAR_Cas9-hygro and pSCAR_sgRNA vectors, respectively, were examined by flow cytometry. GFP+ mKate2+ double-positive cells were sorted and proceeded to the infection with IDLV-Cre lentivirus, followed by being monitored for the loss of GFP and mKate2. After 5–7 days, GFP-mKate2-double-negative cancer cells were sorted and expanded for the indicated experiments, including western blotting and tumor engraftment. sgRNAs: Control, GCGAGGTATTCGGCTCCGCG; Fasn-1, TTGATGTGAGGGGAGATGAG; Fasn-2, TGTCTCCGAAAAGAGCCGGG; Slc23a2-1, TGTTTCAGTGGCACGATCG; Slc23a2-2, CAGCGGAGAGCAGGACAACG; Cdkn2a-1, CGCTGCGTCGTGCACCGGG; Cdkn2a-2, GTCTGGGCGACGTTCCCAG. For the tumor engraftment, tumor cell lines were injected (Yumm1.7 and B16-F10 cell lines: 1 × 105 cells; primary cancer cell lines: 1 × 106 cells) subcutaneously into B6 or Rag1−/− mice, followed by the tumor monitoring started from Day 4 every other day and analysis on Day 14.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using the two-tailed, unpaired Student’s t-test. Each point represented a biological replicate, and all data are presented as means ± s.d. or means ± s.e.m., as indicated. Comparison of survival curve was analyzed by Log-rank test. Correlation between groups was determined by computing Pearson correlation coefficient r and the associated P value.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | DENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| In vivo anti-CD8, clone 2.43 | BioXcell | Cat# BE0061; RRID: AB_1125541 |
| In vivo anti-CD4, clone GK1.5 | BioXcell | Cat# BE0003–1; RRID: AB_1107636 |
| In vivo anti-IFNg antibody, clone XMG1.2 | BioXcell | Cat# BE0055; RRID: AB_1107694 |
| In vivo anti-CD25 antibody, clone PC-61.5.3 | BioXcell | Cat# BE0012; RRID: AB_1107619 |
| TruStain Fc (anti-mouse CD16/32) Antibody | BioLegend | Cat# 101320; RRID: AB_1574975 |
| APC/Fire 750 anti-CD3ε, clone 145–2C11 | BioLegend | Cat# 100362; RRID: AB_2629687 |
| BUV737 anti-CD4, clone RM4–5 | BD Biosciences | Cat# 612843; RRID: AB_2870165 |
| BV605 anti-CD8a, clone 53.6.7 | BioLegend | Cat# 100743; RRID: AB_2561352 |
| PE/Cyanine7 anti-CD11b, clone M1/70 | BioLegend | Cat# 101216; RRID: AB_312799 |
| BV650 anti-CD11c, clone N418 | BioLegend | Cat# 117339; RRID: AB_2562414 |
| Alexa Fluor 700 anti-CD19, clone 6D5 | BioLegend | Cat# 115528; RRID: AB_493735 |
| Alexa Fluor 488 anti-mouse CD45 Antibody, clone 30-F11 |
BioLegend | Cat# 103122; RRID: AB_493531 |
| BUV563 anti-CD103, clone M290 | BD Biosciences | Cat# 741261; RRID: AB_2870808 |
| BV711 anti-Gr-1, clone RB6–8C5 | BioLegend | Cat# 108443; RRID: AB_2562549 |
| BV786 Rat anti-mouse I-Ab/I-E, clone M5/114.15.2 | BD Biosciences | Cat# 742894; RRID: AB_2734759 |
| APC anti-mouse H-2Kb/H-2Db Antibody, clone 28–8-6 |
BioLegend | Cat# 114614; RRID: AB_2750194 |
| BV421 anti-FoxP3, clone MF-14 | BioLegend | Cat# 126419; RRID: AB_2565933 |
| PerCP/Cy5.5 anti-NK1.1, clone PK136 | BioLegend | Cat# 108728; RRID: AB_2132705 |
| PE anti-IFN-γ, clone XMG1.2 | Thermo Fisher | Cat# 12–7311-82; RRID: AB_466193 |
| PerCP-eFluor 710 anti-TNF-α, clone MP6-XT22 | Thermo Fisher | Cat# 46–7321-82; RRID: AB_1834445 |
| PE anti-CD274 (PD-L1) Antibody, clone 10F-9G2 | biolegend | Cat# 124308; RRID: AB_2073556 |
| PE anti-mouse CD366 (Tim3) antibody, clone RMT3–23 | BioLegend | Cat# 119704; RRID: AB_345378 |
| BV711 anti-mouse CD279 (PD-1) antibody, clone RMP1–30 | BioLegend | Cat# 135231; RRID: AB_2566158 |
| PE/Dazzle594 F4/80, clone BM8 | BioLegend | Cat# 123146; RRID: AB_2564133 |
| Monoclonal Anti-β-Actin antibody produced in mouse | Sigma-Aldrich, | Cat# A2228; RRID: AB_476697 |
| Stat1 (D1K9Y) Rabbit mAb | Cell Signaling | Cat# 14994; RRID: AB_2737027 |
| Stat3 (D1B2J) Rabbit mAb | Cell Signaling | Cat# 30835; RRID: AB_2798995 |
| Phospho-Stat1 (Tyr701) Rabbit, clone 58D6 | Cell Signaling | Cat# 9167; RRID: AB_561284 |
| Phospho-Stat3 (Tyr705) Antibody, clone D3A7 | Cell Signaling | Cat# 9145; RRID: AB_2491009 |
| anti-rabbit IgG HRP-linked antibody | Cell Signaling | Cat# 7074; RRID: AB_2099233 |
| anti-mouse IgG HRP-linked antibody | Cell Signaling | Cat# 7076;RRID: AB_330924 |
| Fatty Acid Synthase Antibody | Cell Signaling | Cat# 3189; RRID: AB_2100798 |
| Cleaved Caspase-3 (Asp175)Rabbit mAb, clone 5A1E | Cell Signaling | Cat# 9664; RRID: AB_2070042 |
| Recombinant Anti-p21 antibody, clone EPR18021 | Abcam | Cat# ab188224; RRID: AB_2734729 |
| Anti-CDKN2A/p16INK4a antibody | Abcam | Cat# ab211542; RRID: AB_2891084 |
| Recombinant Anti-Ki67 antibody, clone SP6 | Abcam | Cat# ab16667; RRID: AB_302459 |
| Recombinant Anti-c-Myc antibody, clone Y69 | Abcam | Cat# ab32072; RRID: AB_731658 |
| SLC23A2 (extracellular) Polyclonal Antibody | Invitrogen | Cat# PA5–111911; RRID: AB_2857319 |
| HIF-1 alpha (D1S7W) rabbit mAb | Cell Signaling | Cat# 36169; RRID: 2799095 |
| CD274 (PD-L1) Monoclonal Antibody, clone MIH5 | Invitrogen | Cat# 14–5982-82; RRID: AB_467781 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Rotenone | Sigma-Aldrich | Cat#SI-R8875–1G |
| Antimycin A | Sigma-Aldrich | Cat#FL-75351–5MG |
| FCCP | Sigma-Aldrich | Cat#SI-C2920–10MG |
| Oligomycin A | Sigma-Aldrich | Cat#FL-75351–5MG |
| Collagenase | Sigma-Aldrich | Cat#C5138 |
| 4-Hydroxytamoxifen | Sigma-Aldrich | Cat# H7904–25MG |
| DMEM | GIBCO | Cat# 11965118 |
| RPMI 1640 Medium | GIBCO | Cat# 21875034 |
| 2-Mercaptoethanol | GIBCO | Cat# 31350010 |
| Murine IFN-g | Peprotech | Cat# 315–05 |
| ACK Buffer | Invitrogen | Cat# A10492–01 |
| Percoll | GE Healthca | Cat# 17–0891-01 |
| Dispase II | Roche (Sigma) | Cat#4942078001 |
| DnaseI | Sigma | Cat# D5025–150KU |
| True-Nuclear Transcription Factor Buffer Set | Biolegend | Cat#424401 |
| Permeabilization buffer 10X | eBioscience | Cat# 00–8333-56 |
| Stattic | Cayman | Cat# 14590 |
| Dynabeads Mouse T-Activator CD3/CD28 for T-Cell Expansion and Activation |
Invitrogen | Cat# 11452D |
|
| ||
| Critical commercial assays | ||
|
| ||
| Mouse XL Cytokine Array | R&D, | Cat#ARY028 |
| Senescence β-Galactosidase Staining Kit | Cell Signaling | Cat# 9860S |
| ApopTag Red In Situ Apoptosis Detection kit | Merck | Cat# S7165 |
| Hypoxyprobe Plus Kit | Hypoxyprobe | Cat# HP6–200Kit |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq data of cell lines | This paper | GEO: GSE217480 |
| ATAC-seq data of cell lines and primary tumor cells | This paper | GEO: GSE190993 and GSE217475 |
| RRBS data of cell lines | This paper | GEO: GSE21748 |
| Exome-seq data of cell lines | This paper | GEO: GSE217479 |
| TCR-seq of primary tumor cells | This paper | GEO: GSE217861 |
| RNA-seq data of primary tumor cells | This paper | GEO: GSE217862 |
| CRISPR screening | This paper | GEO: GSE217859 |
| Data and code availability | This paper | Data S1 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| mouse: Yumm1.7 | ATCC | CRL-3362 |
| mouse: B16 | ATCC | CRL-6322 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | Jackson Laboratory | #:000664; RRID: IMSR_JAX:000664 |
| Mouse: Rag1−/− (B6.129S7-Rag1tm1Mom/J) | Jackson Laboratory | #:002216; RRID: IMSR_JAX:002216 |
| Mouse: mTmG (B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato-EGFP)Luo/J) |
Jackson Laboratory | #:007676; RRID: IMSR_JAX:007676 |
| Mouse: IFNγ−/− (B6.129S7-Ifngtm1Ts/J) | Jackson Laboratory | #:002287; RRID: IMSR_JAX:002287 |
| Mouse: BRafCA; Tyr::CreER; Ptenlox4–5 (Braf/Pten) | Kindly provided by Dr. Marcus Bosenberg | N/A |
| Mouse: Kcre;Ecdf;P53f;RAG1 | Provided by Dr. Karin de Visser | N/A |
| Mouse: Neut;RAG1 | Provided by Dr. Karin de Visser | N/A |
| Mouse: Tyr::CreERT2/+; BRafCA/+; PtenVI; ROSA26RLSL-tdTomato/+ |
This paper | N/A |
| Mouse: Tyr::CreERT2/+; BRafCA/+; PtenVI; Rag1tm1Mom |
This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pSCAR_sgRNA_puro-mKate-lox2272 | Addgene | Cat# 162076 |
| pSCAR_Cas9-hygro_GFP | Addgene | Cat# 162075 |
| pLX_EFS-Cre_ppt-del | Addgene | Cat# 162073 |
| psPAX2-D64V | Addgene | Cat# 63586 |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism 9 | GraphPad | N/A |
| Flow Jo 10.1 | FlowJo | N/A |
| ImageJ | NIH | https://imagej.nih.gov |
|
| ||
| Other | ||
|
| ||
| LSRFortessa Flow Cytometer | BD Biosciences | N/A |
| LSRII Flow Cytometer | BD Biosciences | N/A |
| Seahorse XFe96 FluxPak | Agilent | Cat#102416–100 |
| Seahorse XFe96 Analyzer | Agilent | N/A |
Highlights.
Immunometabolic editing orchestrates metabolic preferences in tumor cells
Metabolic program acquired by edited tumor cells supports tumor immune evasion
IFNγ-driven editing modulates cMyc expression in a non-canonical STAT3 pathway
Immunosurveillance impacts tumor epigenetic program for boosting aggressiveness
ACKNOWLEDGMENTS
We thank Li-Fan Lu and Stanley Ching-Cheng Huang for providing valuable comments. P.-C. Hsueh is funded by the European Research Council starting grant (802773-MitoGuide), SNSF grants (31003A_182470, CRSII5_205930, and IZLCZ0_206083), the Cancer Research Institute (CLIP and Lloyd J. Old STAR award), the Melanoma Research Alliance Young and Established Investigator Awards, EMBO Young Investigator Award, and Ludwig Cancer Research. C.-H.T. is supported in part by Ministry of Science and Technology (MOST 111-2314-B-016-004), Instrument Center of National Defense Medical Center, and Agricultural Biotechnology Research Center. Y.-M.C. and P.-C.H. are supported in part by Ministry of Science and Technology Taiwan, and P.-W.H. is supported by Academia Sinica. J.I. is supported by SNSF R’Equip 316030_183377. X. Li and X. Liu are supported by North Carolina State University startup fund. A.L. was supported by a Damon Runyon-Rachleff Innovation Award (DR52-18) and R37 Merit Award (R37CA230636). S.Y.L. acknowledges funding from the METAvivor Early Career Investigator Grant and the American Association for Cancer Research-Incyte NextGen Grant for Transformative Cancer Research. S.M.K. is supported by the NIH (R01 CA206483), the Melanoma Research Alliance, and a Salk innovation grant.
Footnotes
DECLARATION OF INTERESTS
P.-C.H. is a member of the scientific advisory board for Elixiron Immunotherapeutics and received research grants from Elixiron Immunotherapeutics. P.-C.H. is also a founder of Pilatus Biosciences.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cmet.2022.12.003.
REFERENCES
- 1.Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Kim J, and DeBerardinis RJ (2019). Mechanisms and implications of metabolic heterogeneity in cancer. Cell Metab. 30, 434–446. 10.1016/j.cmet.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fendt SM, Frezza C, and Erez A (2020). Targeting metabolic plasticity and flexibility dynamics for cancer therapy. Cancer Discov. 10, 1797–1807. 10.1158/2159-8290.CD-20-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, et al. (2015). Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162, 1217–1228. 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. (2015). Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241. 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Wenes M, Romero P, Huang SC, Fendt SM, and Ho PC (2019). Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 16, 425–441. 10.1038/s41571-019-0203-7. [DOI] [PubMed] [Google Scholar]
- 7.Lim AR, Rathmell WK, and Rathmell JC (2020). The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. eLife 9, e55185. 10.7554/eLife.55185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harel M, Ortenberg R, Varanasi SK, Mangalhara KC, Mardamshina M, Markovits E, Baruch EN, Tripple V, Arama-Chayoth M, Greenberg E, et al. (2019). Proteomics of melanoma response to immunotherapy reveals mitochondrial dependence. Cell 179, 236–250.e18. 10.1016/j.cell.2019.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castro F, Cardoso AP, Gonc alves, R.M., Serre K, and Oliveira MJ (2018). Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 9, 847. 10.3389/fimmu.2018.00847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braumüller H, Wieder T, Brenner E, Aßmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C, et al. (2013). T-helper-1-cell cytokines drive cancer into senescence. Nature 494, 361–365. 10.1038/nature11824. [DOI] [PubMed] [Google Scholar]
- 11.Brenner E, Schörg BF, Ahmetlić F, Wieder T, Hilke FJ, Simon N, Schroeder C, Demidov G, Riedel T, Fehrenbacher B, et al. (2020). Cancer immune control needs senescence induction by interferon-dependent cell cycle regulator pathways in tumours. Nat. Commun. 11, 1335. 10.1038/s41467-020-14987-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jorgovanovic D, Song M, Wang L, and Zhang Y (2020). Roles of IFN-gamma in tumor progression and regression: a review. Biomark. Res. 8, 49. 10.1186/s40364-020-00228-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schreiber RD, Old LJ, and Smyth MJ (2011). Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570. 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 14.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, and Schreiber RD (2001). IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410, 1107–1111. 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 15.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr., You MJ, DePinho RA, McMahon M, and Bosenberg M (2009). Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 41, 544–552. 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho PC, Meeth KM, Tsui YC, Srivastava B, Bosenberg MW, and Kaech SM (2014). Immune-based antitumor effects of BRAF inhibitors rely on signaling by CD40L and IFNgamma. Cancer Res. 74, 3205–3217. 10.1158/0008-5472.CAN-13-3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJM, Ciampricotti M, Hawinkels LJAC, Jonkers J, et al. (2015). IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522, 345–348. 10.1038/nature14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans RA, Diamond MS, Rech AJ, Chao T, Richardson MW, Lin JH, Bajor DL, Byrne KT, Stanger BZ, Riley JL, et al. (2016). Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight 1, e88328. 10.1172/jci.insight.88328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kubick BJ, Fan X, Crouch A, McCarthy R, and Roop DR (2020). Tracing the equilibrium phase of cancer immunoediting in epidermal neoplasms via longitudinal intravital imaging. J. Invest. Dermatol. 140, 891–900.e10. 10.1016/j.jid.2019.08.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rogers LM, Olivier AK, Meyerholz DK, and Dupuy AJ (2013). Adaptive immunity does not strongly suppress spontaneous tumors in a Sleeping Beauty model of cancer. J. Immunol. 190, 4393–4399. 10.4049/jimmunol.1203227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muzumdar MD, Tasic B, Miyamichi K, Li L, and Luo L (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605. 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 22.Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G, Christiaens V, Svetlichnyy D, Luciani F, Van den Mooter L, et al. (2015). Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 6, 6683. 10.1038/ncomms7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pelengaris S, Khan M, and Evan G (2002). c-MYC: more than just a matter of life and death. Nat. Rev. Cancer 2, 764–776. 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- 25.Kim YJ, Sheu KM, Tsoi J, Abril-Rodriguez G, Medina E, Grasso CS, Torrejon DY, Champhekar AS, Litchfield K, Swanton C, et al. (2021). Melanoma dedifferentiation induced by IFN-gamma epigenetic remodeling in response to anti-PD-1 therapy. J. Clin. Invest. 131, e145859. 10.1172/JCI145859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qing Y, and Stark GR (2004). Alternative activation of STAT1 and STAT3 in response to interferon-gamma. J. Biol. Chem. 279, 41679–41685. 10.1074/jbc.M406413200. [DOI] [PubMed] [Google Scholar]
- 27.Jones LM, Broz ML, Ranger JJ, Ozcelik J, Ahn R, Zuo D, Ursini-Siegel J, Hallett MT, Krummel M, and Muller WJ (2016). STAT3 establishes an immunosuppressive microenvironment during the early stages of breast carcinogenesis to promote tumor growth and metastasis. Cancer Res. 76, 1416–1428. 10.1158/0008-5472.CAN-15-2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhai L, Bell A, Ladomersky E, Lauing KL, Bollu L, Sosman JA, Zhang B, Wu JD, Miller SD, Meeks JJ, et al. (2020). Immunosuppressive IDO in cancer: mechanisms of action, animal models, and targeting strategies. Front. Immunol. 11, 1185. 10.3389/fimmu.2020.01185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park SY, and Nam JS (2020). Kynurenine pathway enzyme KMO in cancer progression: a tip of the iceberg. EBiomedicine 55, 102762. 10.1016/j.ebiom.2020.102762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Y, Mao C, Wang M, Liu N, Ouyang L, Liu S, Tang H, Cao Y, Liu S, Wang X, et al. (2020). Cancer progression is mediated by proline catabolism in non-small cell lung cancer. Oncogene 39, 2358–2376. 10.1038/s41388-019-1151-5. [DOI] [PubMed] [Google Scholar]
- 31.Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. (2011). Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43, 869–874. 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.May JM (2011). The SLC23 family of ascorbate transporters: ensuring that you get and keep your daily dose of vitamin C. Br. J. Pharmacol. 164, 1793–1801. 10.1111/j.1476-5381.2011.01350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gütgemann I, Eilers M, and Felsher DW (2016). MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231. 10.1126/science.aac9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu X, Nelson M, Basu M, Srinivasan P, Lazarski C, Zhang P, Zheng P, and Sandler AD (2021). MYC oncogene is associated with suppression of tumor immunity and targeting Myc induces tumor cell immunogenicity for therapeutic whole cell vaccination. J. Immunother. Cancer 9, e001388. 10.1136/jitc-2020-001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson DB, Bao R, Ancell KK, Daniels AB, Wallace D, Sosman JA, and Luke JJ (2019). Response to anti-PD-1 in uveal melanoma without high-volume liver metastasis. J. Natl. Compr. Canc. Netw. 17, 114–117. 10.6004/jnccn.2018.7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, et al. (2018). A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984–997.e24. 10.1016/j.cell.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB, et al. (2015). Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell 162, 1271–1285. 10.1016/j.cell.2015.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, Koya RC, Samatar AA, Khanlou N, Braun J, et al. (2015). Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 27, 240–256. 10.1016/j.ccell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, et al. (2004). Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 10, 48–54. 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 40.Chan KS, Sano S, Kiguchi K, Anders J, Komazawa N, Takeda J, and DiGiovanni J (2004). Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J. Clin. Invest. 114, 720–728. 10.1172/JCI21032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abe M, Yoshida T, Akiba J, Ikezono Y, Wada F, Masuda A, Sakaue T, Tanaka T, Iwamoto H, Nakamura T, et al. (2017). STAT3 deficiency prevents hepatocarcinogenesis and promotes biliary proliferation in thioacetamide-induced liver injury. World J. Gastroenterol. 23, 6833–6844. 10.3748/wjg.v23.i37.6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang Y, Luo Y, Jiang Z, Ma Y, Lin CJ, Kim C, Carter MG, Amano T, Park J, Kish S, and Tian XC (2012). Jak/Stat3 signaling promotes somatic cell reprogramming by epigenetic regulation. Stem Cells 30, 2645–2656. 10.1002/stem.1225. [DOI] [PubMed] [Google Scholar]
- 43.Swoboda A, Soukup R, Eckel O, Kinslechner K, Wingelhofer B, Schörghofer D, Sternberg C, Pham HTT, Vallianou M, Horvath J, et al. (2021). STAT3 promotes melanoma metastasis by CEBP-induced repression of the MITF pathway. Oncogene 40, 1091–1105. 10.1038/s41388-020-01584-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.See YX, Chen K, and Fullwood MJ (2022). MYC overexpression leads to increased chromatin interactions at super-enhancers and MYC binding sites. Genome Res. 32, 629–642. 10.1101/gr.276313.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Molina-Sánchez P, Ruiz de Galarreta M, Yao MA, Lindblad KE, Bresnahan E, Bitterman E, Martin TC, Rubenstein T, Nie K, Golas J, et al. (2020). Cooperation between distinct cancer driver genes underlies intertumor heterogeneity in hepatocellular carcinoma. Gastroenterology 159, 2203–2220.e14. 10.1053/j.gastro.2020.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meeth K, Wang JX, Micevic G, Damsky W, and Bosenberg MW (2016). The YUMM lines: a series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment Cell Melanoma Res. 29, 590–597. 10.1111/pcmr.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Medina J, van der Velpen V, Teav T, Guitton Y, Gallart-Ayala H, and Ivanisevic J (2020). Single-step extraction coupled with targeted HILIC-MS/MS approach for comprehensive analysis of human plasma lipidome and polar metabolome. Metabolites 10, 495. 10.3390/metabo10120495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chong J, Wishart DS, and Xia J (2019). Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinformatics 68, e86. 10.1002/cpbi.86. [DOI] [PubMed] [Google Scholar]
- 49.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273. 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 50.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu YR, Imrichova H, Wang H, Chao T, Xiao Z, Gao M, Rincon-Restrepo M, Franco F, Genolet R, Cheng WC, et al. (2020). Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat. Immunol. 21, 1540–1551. 10.1038/s41590-020-0793-3. [DOI] [PubMed] [Google Scholar]
- 52.Herrmann C, Van de Sande B, Potier D, and Aerts S (2012). i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Res. 40, e114. 10.1093/nar/gks543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Imrichová H, Hulselmans G, Atak ZK, Potier D, and Aerts S (2015). i-cisTarget 2015 update: generalized cis-regulatory enrichment analysis in human, mouse and fly. Nucleic Acids Res. 43, W57–W64. 10.1093/nar/gkv395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xi Y, and Li W (2009). BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 10, 232. 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Assenov Y, Müller F, Lutsik P, Walter J, Lengauer T, and Bock C (2014). Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 11, 1138–1140. 10.1038/nmeth.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, and Chudakov DM (2015). MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods 12, 380–381. 10.1038/nmeth.3364. [DOI] [PubMed] [Google Scholar]
- 57.Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, and Ma’ayan A (2016). The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford) 2016, baw100. 10.1093/database/baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dubrot J, Lane-Reticker SK, Kessler EA, Ayer A, Mishra G, Wolfe CH, Zimmer MD, Du PP, Mahapatra A, Ockerman KM, et al. (2021). In vivo screens using a selective CRISPR antigen removal lentiviral vector system reveal immune dependencies in renal cell carcinoma. Immunity 54, 571–585.e6. 10.1016/j.immuni.2021.01.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq, ATAC–seq, RRBS data, Exome-seq and CRISPR screening are available in the Gene Expression Omnibus database under accession codes GSE217480 for RNA-seq, GSE190993 and GSE217475 for ATAC-seq, GSE217481 for RRBS data, GSE217479 for Exome-seq, GSE217861 for TCR-seq, GSE217862 for Smart-seq2, and GSE217859 for CRISPR screening. Data S1 contains uncropped immunoblots and raw data of all graphs. Uncropped blots and values of graphs are included in the zip file, Data S1.
No original code was generated in this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.