

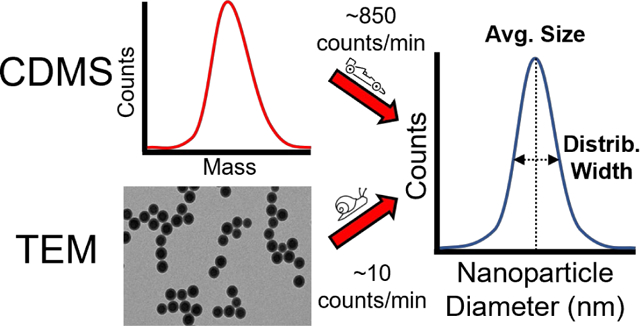
Abstract
The sizes and shapes of nanoparticles play a critical role in their chemical and material properties. Common sizing methods based on light scattering or mobility lack individual particle specificity and microscopy-based methods often require cumbersome sample preparation and image analysis. A promising alternative method for the rapid and accurate characterization of nanoparticle size is charge detection mass spectrometry (CDMS), an emerging technique that measures the masses of individual ions. A recently constructed CDMS instrument designed specifically for high acquisition speed, efficiency, and accuracy is described. This instrument does not rely on an ion energy filter or estimates of ion energy that have been previously required for mass determination, but instead uses direct, in situ measurements. A standardized sample of ~100 nm diameter polystyrene nanoparticles and ~50 nm polystyrene nanoparticles with amine-functionalized surfaces are characterized using CDMS and transmission electron microscopy (TEM). Individual nanoparticle masses measured by CDMS are transformed to diameters and these size distributions are in close agreement with distributions measured by TEM. CDMS analysis also reveals dimerization of ~100 nm nanoparticles in solution that cannot be determined by TEM due to the tendency of nanoparticles to agglomerate when dried onto a surface. Comparing the acquisition and analysis times of CDMS and TEM shows particle sizing rates up to ~80 times faster are possible using CDMS, even when samples ~50x more dilute were used. The combination of both high accuracy individual nanoparticle measurements and fast acquisition rates by CDMS represents an important advance in nanoparticle analysis capabilities.
Keywords: mass, charge, measurement, nanoparticle, sizing, high-throughput, precision
Graphical Abstract
Introduction
Nanoparticles are used in a broad range of chemical and biological applications, including catalysis,1 drug delivery,2 and as materials in electronics and manufacturing.3,4 The size, shape, and composition of nanoparticles are important in determining their functionality in each of these applications.5 There are many different methods to accurately determine nanoparticle size distributions. Methods such dynamic light scattering (DLS) and small-angle x-ray scattering (SAXS), use photon scattering to infer nanoparticle properties. Microscopy-based methods, such as transmission electron microscopy (TEM), scanning electron microscopy (SEM), scanning tunneling electron microscopy (STEM), and atomic force microscopy (AFM), directly image nanoparticles. Scattering-based methods can be performed directly and non-destructively on nanoparticles in solution, which can be an advantageous.6,7 In the case of SAXS, some information about the shape of the nanoparticles can also be determined.7 However, because these techniques rely on an ensemble measurement, they provide only average properties and typically require pure samples for an accurate size determination. This highlights a key advantage of microscopy-based methods for nanoparticle characterization—the ability to resolve and measure individual nanoparticles with high precision and accuracy. Microscopy techniques can also be more readily applied to less “clean” samples containing irregular or multiple distributions of nanoparticles. However, microscopy techniques require significant sample preparation, including drying/affixing the sample onto a measurement substrate. This process can lead to aggregation that is not indicative of solution-phase properties.8 The analysis of individual nanoparticles in microscopy is also resource intensive because 1,000+ individual measurements can be required to produce a statistically meaningful size distribution.9 Automated image analysis methods to size nanoparticles are an active area of research that can help reduce the time cost of compiling the necessary number of individual measurements, but these methods require careful parameterization and monitoring to avoid the inclusion of erroneous measurements or spurious data in the analysis.9
Another method used to size nanoparticle distributions is differential mobility analysis (DMA).10–14 With this method, particles are aerosolized via electrospray ionization (ESI)10–13 or by pneumatic atomization,14 charge reduced to a single charge, and are subsequently analyzed by their relative mobility. Mobilities are related to particle size and shape. Information about these properties is inferred from the measured mobility. Similar to scattering-based methods, DMA experiments are ensemble measurements that are relatively fast, requiring anywhere from a few seconds to a few minutes.15,16 The resolution is limited, with typical experimental mobility errors of ±5%,15,16 although some optimized versions have achieved as low as ±1%.17 DMA experiments are often coupled with secondary analysis techniques, such as inductively coupled plasma mass spectrometry13 and aerosol particle mass analysis,14 which can provide complementary information to the mobilities determined by DMA.
Charge detection mass spectrometry (CDMS) is an alternative method for measuring the size distributions of nanoparticles. In CDMS, molecules or particles are aerosolized and ionized using ESI, as is often done with DMA. However, CDMS provides a direct and accurate measurement of the masses of individual particles from which mass distributions of the sample can be obtained. Conventional mass spectrometry methods for large molecules, which rely on m/z measurements of ensembles of ions, lead to spectral congestion and loss of information when the sample is heterogeneous and high in mass. In contrast, masses of individual ions are determined in CDMS via independent, simultaneous measurements of both m/z and charge, making it possible to eliminate spectral congestion.18–21 Similar to microscopy-based sizing methods, many individual ion measurements are compiled to yield a mass histogram that is representative of the range of molecules, molecular complexes, or particles in the sample. This approach to weighing ions makes it possible to measure mass distributions even when mixtures or other intrinsically heterogenous samples are analyzed.18,22–24 CDMS has been used to characterize a wide variety of large biomolecular assemblies, such as viruses and virus-like particles.22,25–28 CDMS has also been used to analyze polymer-based nanoparticles, but these studies had significant instrumentation-based limitations in either mass accuracy or the speed of data acquisition.18,23
Recent developments in CDMS methodology, including methods to simultaneously analyze multiple ions,29 determine individual ion energies during the measurement process in situ,30 and determine signal amplitudes with greater fidelity,31 has prompted the design of CDMS instrumentation aimed at maximizing acquisition speed while retaining high accuracy mass measurement capabilities. For state-of-the-art CDMS instruments that incorporate electrostatic ion traps to make accurate charge and m/z measurements, ion energies must be known.30,32 Previous CDMS instruments have incorporated energy filtering ion optics to define a narrow band of ion energies to be analyzed, with the centerline of the energy passband used as the assumed energy for calculating the mass of each ion from its oscillation frequency.33,34 However, the use of an energy filter decreases sensitivity because fewer ions are allowed to reach the detection stage. For higher performance mass measurements, the energy bandpass of the filter must be reduced and individual ion trapping times increased,32 meaning that there is a direct trade-off between high performance and high throughput measurements. The recently developed capability to measure the energies of individual ions in situ has made energy filtering optics unnecessary.22,30 Moreover, the simultaneous trapping and mass analysis of multiple ions have greatly improved the acquisition speed for this type of CDMS instrument.22,29
The recently constructed CDMS instrument used in this work does not incorporate an ion energy filter and instead relies on completely in situ ion energy measurements to determine the mass of each analyzed ion. This departure from previous CDMS instruments that incorporate energy filters along with other design optimizations yields increased sensitivity and overall acquisition speed that is demonstrated in this work. Two types of spherical polystyrene nanoparticles with diameters of ~50 nm and ~100 nm were used to evaluate the performance of this instrument. CDMS mass measurements are transformed into diameters via the known density and sphericality of the nanoparticles. These data are compared to the distributions of diameters derived from analysis of TEM images, including analysis of peak values, distribution shapes, and method biases. Because throughput is also an important consideration in the evaluation of a nanoparticle measurement technique, the time required to perform the respective TEM and CDMS experiments and subsequent analyses are also compared.
Results and Discussion
CDMS for Accurate Nanoparticle Size Characterization.
To evaluate the capabilities of the recently constructed CDMS instrument for accurate quantification of both the mass and diameter distributions of nanoparticles, a sample of Nanosphere™ size standard polystyrene spheres was analyzed using both TEM and with this CDMS instrument. The relatively low polydispersity and spherical shape of these nanoparticles make it an ideal standard for comparing the accuracy of diameters computed from individual ion CDMS mass data to those determined from TEM. With CDMS, the masses of 2,509 individual ions were measured in ~50 min, yielding the mass histogram shown in Figure 1a. There are three main peaks; a low mass peak at ~32 MDa that tails to higher mass, a main peak at ~354 MDa that tails to lower mass, and a minor peak at ~705 MDa that also tails to lower mass. Because the distributions are non-normal, peak values were determined by smoothing the mass histogram using a Savitzsky-Golay filter and picking the local bin maxima (bin width = 0.5 MDa). The lower mass peak likely corresponds to malformed and/or undersized spheres. It is worth noting that because ions are counted individually in CDMS analysis, the relatively high counts for the low mass peak are exaggerated relative to the overall mass of the nanoparticles in a sample. In this case, ions with masses <50 MDa only account for a small portion (0.8%) of the overall mass of the measured ions. The minor peak at 705 MDa is approximately double the mass of the main particle distribution at 354 MDa and is therefore likely due to dimers of nanospheres. Because charge is measured independently of mass in CDMS analysis, a two-dimensional mass vs. charge histogram (Figure 1b) can be generated. In addition to being approximately double the mass of the main peak, the charge states measured for individual ions in the 705 MDa distribution are significantly higher than expected for spherical nanoparticles of similar mass. Figure 1b shows that the main ion distribution centered at ~354 MDa have charge states that are on average 85% of the Rayleigh limit calculated for aqueous spherical droplets (blue dashed line, details provided in the Supporting Information). This extent of charging is consistent with other approximately spherical analytes measured using ESI.28,35 In contrast, the ions corresponding to the 705 MDa peak have higher charge states relative to the Rayleigh charging limit. On average, these ions were charged to 103% of Rayleigh limit, strongly indicating that these ions have a non-spherical geometry, such as that of two conjoined spheres. Charges can be more highly dispersed in this geometry, which lowers Coulomb interactions and leads to higher charging. Although the stochastic nature of particle charging inherently leads to fairly the broad charge distributions, the width of a charge distribution within a particular mass range in CDMS analysis of nanoparticles can also provide useful information about the extent of morphological homogeneity in a sample. A bimodal charging distribution has been observed previously in the CDMS analysis of tobacco mosaic virus disk complexes where two disks linked in an edge-on conformation exhibited a charge state distribution above the Rayleigh limit while a more compact stack of two disks exhibited charge states lower than the Rayleigh limit.24 It is worth noting that measuring both mass and charge directly using CDMS makes identification of nanoparticle dimers straightforward. In contrast, identification of dimers by scattering or mobility-based methods is more challenging because the signals from dimers are indistinguishable from those of larger single particles.
Figure 1.
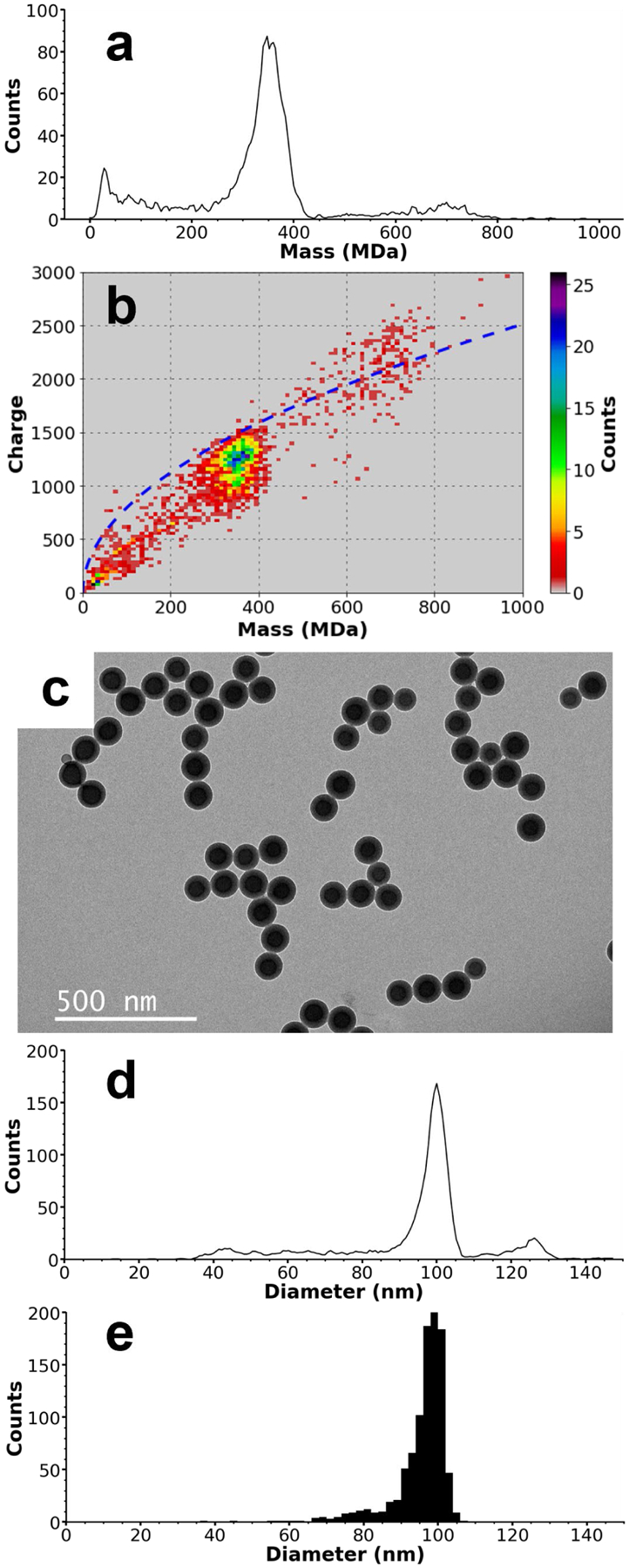
CDMS and TEM data for ~100 nm standardized nanoparticles. A mass histogram composed of 2,509 individual ions is shown in (a), with peaks at ~32 MDa, ~354 MDa, and ~705 MDa, corresponding to contaminants, the ~100 nm standardized nanoparticles, and nanoparticle dimers, respectively. The non-spherical shape of the dimer species is confirmed in (b), a two-dimensional mass v. charge histogram. The blue dashed line is the Rayleigh charging limit of spherical droplets as a function of mass. Spherical molecules, such as the ~100 nm nanoparticles at ~354 MDa, are typically charged to less than ~100% of the Rayleigh limit, but non-spherical molecules, such as the dimers at ~705 MDa, can charge above this value because to their larger relative surface area allows charge to be spread out and reduces Columbic interactions. A representative TEM image of the ~100 nm standard nanoparticles is shown in (c). Diameters derived from the CDMS mass data are shown in (d) and can be compared with the diameters measured using TEM images shown in (e). The main distribution in the CDMS-determined diameters has a peak value of 101.5 nm compared to the 98.5 nm determined from TEM. The CDMS-determined diameters in (d) exhibit a spurious peak at ~127 nm originating from the non-spherical dimer masses, but the similarity in peak shape and peak values between the main distributions of both (d) and (e) demonstrate indicate good agreement between the two methods for sizing nanoparticles.
It is important to consider the origin of the dimer species observed in this sample. Non-specific aggregation of analytes is common in ESI of smaller analytes and can exaggerate the abundances of oligomeric species in solution. However, this only occurs if the initial droplets generated by the ESI emitter are sufficiently large to contain multiple analytes at a given concentration.36–38 The low concentration of sample analyzed here (0.002% w/v, ~60 picomolar (pM) based on the peak mass of 354 MDa), combined with the ESI emitter diameter of ~5 – 6 μm makes it extremely unlikely that two independent nanospheres occupy the same initially formed nanodrop. Previous work by Davidson et al.38 and Susa et al.39 has shown that droplets generated using comparable emitters have an initial diameter of ~1/14 – 1/20 of the diameter of the emitter tip. Using an estimate of 1/17, the 5 – 6 μm emitter tip diameters used in this work should yield initial droplets ~300 – 350 nm in diameter. The known concentration, density, and mean diameter (101 nm) of the nanoparticles analyzed were used along with this initial droplet size estimate to calculate average nanoparticle occupancy of a single droplet at ~0.0005 – 0.0008. Thus, fewer than one in a million droplets should contain more than one independent nanoparticle. Non-specific aggregation inside the evaporating ESI nanodrops is extremely unlikely to occur because the overwhelming majority of droplets contain either one or no analyte particles. However, dimers are ~10% of the total observed ion count. This indicates that the dimers observed here exist in the aqueous sample suspension and enter the initially formed nanodrops as dimers.
Because mass is directly proportional to volume, a sample that is normally distributed in the diameter domain should yield a non-normal distribution in the mass domain. Such a distribution should tail to high mass in a manner similar to a Boltzmann distribution. However, the opposite distribution shape is observed in the mass data of Figure 1a, with tailing to lower masses. To find the distribution of diameters expected from the data of Figure 1a, masses were transformed into the diameter domain assuming sphericality and using the bulk density of polystyrene (1.05 g/mL).40 These data are shown in Figure 1d. The main distribution has a peak value of 101.7 nm and full width at half maximum (FWHM) of ~6 nm, in excellent agreement with manufacturer-certified mean value of 101 nm ± 3 nm and reported standard deviation of 6.2 nm. The apparent peak at ~127 nm corresponding to the dimer distribution can be disregarded due to the non-spherical shape of the dimers.
The diameter distribution measured using CDMS was compared to diameters obtained from TEM images of this sample. A representative image is shown in Figure 1c. Relatively wide-field images (2 – 3 μm) were obtained in order to increase counts for statistical purposes. Images were analyzed using a FIJI/ImageJ plugin41 utilizing the Hough circle transform42 to identify and determine the diameters of 976 nanoparticles. These results were compiled into the histogram shown in Figure 1e. Due to the natural tendency of nanoparticles to agglomerate when dried on a TEM grid, it is difficult to differentiate aggregates from actual suspended dimers using TEM images without additional sample preparation steps.8 However, nanoparticles are analyzed directly from their working suspensions in the CDMS analysis, making it possible to quantify the extent of oligomerization that exists in the original solution. The capability of CDMS to provide this information can be useful in gauging the quality of nanoparticle samples and their chemical functionality.
The distribution of diameters determined from the TEM images in Figure 1e has a peak at 98.5 nm, ~3 nm lower than measured by CDMS and the manufacturer certified value. However, the TEM peak width (~6 nm FWHM) and overall peak shape match well with the CDMS data, with significant tailing of particle diameters toward lower values. The TEM distribution does not tail to as low particle diameters as CDMS distribution. It is possible that the lower masses detected by CDMS do not correspond to well-formed spherical particles and therefore are excluded by the automated image analysis method used in the TEM analysis. Several different factors could contribute to the ~3 nm difference in the peak values determined by the two methods. The mass uncertainty for an individual ion with the average charge of a non-dimerized particle (1190 e) is ~1.5%, which, when propagated into the diameter domain, is an uncertainty of only ~0.5%. Thus, the mass uncertainty in the CDMS measurements cannot account for all the observed difference between the two techniques. Solvent, surfactants, or salts can adduct onto ions, increasing their mass and hence apparent diameter. Nonspecific adduction during electrospray has been reported to add as much as ~1% to the nominal mass of analytes near 1 MDa in size43 and it is feasible that the extent of adduction could be higher for the ~350 MDa nanoparticles measured here. However, even if 3% of the measured mass is due to nonspecific adduction, the reported diameter would only be too high by 1%. Thus, adduction is unlikely to fully account for the small difference in diameters obtained from TEM and CDMS measurements.
The relatively large number of measurements made using CDMS (2,509) and TEM (976) makes statistical variance an unlikely explanation for the slightly different diameters obtained with these methods, but the non-normal distribution of diameters makes particle counting statistics more difficult to quantify. A potentially significant contributor to the observed difference in peak diameters is the TEM instrument calibration and image resolution (0.51 nm/pixel), as well as the automated particle sizing method used here.41,42 It is worth noting that the intended use of the Nanosphere™ size standards measured here (with a certified mean diameter of 101 nm ± 3 nm) is to calibrate electron microscopy instruments.
To test the capabilities of the recently constructed CDMS instrument for the analysis of less homogeneous nanoparticles, CDMS and TEM measurements of a highly polydisperse sample of amine-functionalized polystyrene spheres with ~50 nm nominal diameters were performed. In the CDMS analysis, the masses of 14,455 individual ions were measured over a period of ~17 min and compiled into the mass histogram shown in Figure 2a. For these nanoparticles, there is a single broad distribution spanning 10 – 70 MDa centered at ~37.0 MDa. The distribution in the mass domain is non-normal and tails toward higher masses. Ion masses were transformed into the diameter domain using the same process described above for the ~100 nm nanoparticles, and the resultant diameter histogram is shown in Figure 2b. The mass-based diameter distribution ranges from ~30 – 60 nm with a peak at 47.5 nm and tails toward lower diameters. TEM images of this nanoparticle sample were also analyzed, and a representative image is shown in Figure 2d. Diameters were measured for 1,291 individual particles in the TEM images and compiled into the histogram of Figure 2c. The diameters measured with TEM have a similar distribution shape to those measured by CDMS (Figure 2b) but have a slightly lower range (25 – 55 nm) and peak value (42 nm).
Figure 2.
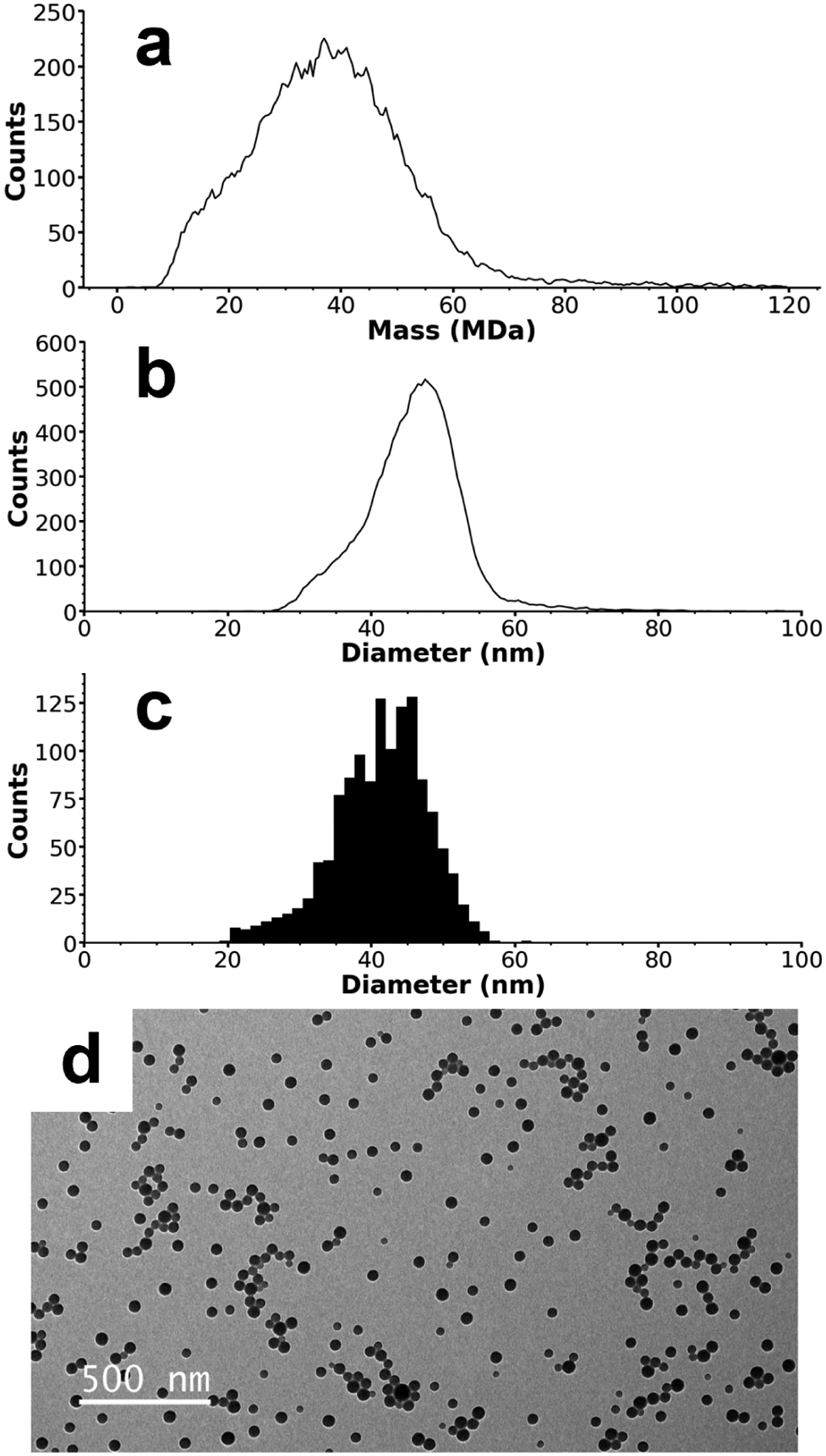
CDMS and TEM data for ~50 nm amine-functionalized nanoparticles. A mass histogram composed of 14,455 individual ions is shown in (a), with a single broad peak centered at ~37 MDa. Diameters derived from the CDMS mass data are shown in (b) and can be compared with the diameters measured using TEM shown in (c). The CDMS diameters have a peak centered at 47.5 nm compared to 42 nm for the TEM data. Despite this difference, both size distributions have approximately the same shape, indicating a relative systematic error between the two methods that may originate from the amine-functionalized surface of these nanoparticles. A representative TEM image of the nanoparticles is shown in (d).
The difference in peak diameter values obtained from TEM and CDMS measurements may be due to a combination of adduction in the ESI process, calibration and measurement errors, or statistical variance, but the larger difference between the results of the two methods for the ~50 nm sample than the ~100 nm standard nanoparticles indicates that another factor may play a role. For these nanoparticles, their smaller overall size and potential fluidity and flexibility in the amine-functionalized surfaces may cause the particle diameters to be underestimated in the TEM image analysis. It is interesting to note that no dimers are observed in the CDMS analysis of the amine-functionalized ~50 nm nanoparticles compared to the clear dimer peak observed for the unfunctionalized ~100 nm nanoparticles. Dimers of lower mass nanoparticles could be concealed by the width of the mass distribution, but the mass vs. charge plot of the ~50 nm particles (Figure S1), shows charge states that are normally distributed across the entire mass range. A bimodal distribution would be expected if a non-spherical population existed. Thus, we conclude that no significant dimerization occurs for these particles. These results indicate that the amine functionalization is effective at eliminating particle aggregation.
Disparities of a few nanometers in the measured diameters for nanoparticles are common when comparing different sizing techniques such as TEM, SEM, STEM, AFM, DLS, and SAXS and these differences have been well documented elsewhere.44 The overall qualitative and very close quantitative matches of the distribution shapes and size between the two methods used in this work for both the ~50 nm particles (Figure 2) and ~100 nm size standards (Figure 1) indicate that CDMS is a viable nanoparticle sizing method capable of high-precision mass and diameter measurements.
Rapid Nanoparticle Characterization by CDMS.
Rapid analysis of nanoparticles would be useful to both the development of automated nanoparticle synthesis workflows and quality assurance of large-scale production. However, due to the polydispersity inherent to most nanoparticle samples, hundreds or even thousands of individual particle size measurements are required to attain a meaningful representation of the size distribution. Sizing individual nanoparticles using microscopy image-based methods can be a time-consuming process.9 Choosing the proper concentration to avoid oversaturating the measurement substrate while still depositing a single layer of nanoparticles with sufficient area coverage for good counting statistics can require multiple imaging iterations. For the ~50 nm and ~100 nm nanoparticle samples analyzed by TEM in this work, dilution factors were optimized at 100 and 40, respectively. Preparing the sample and acquiring multiple images with independent fields of view required ~1 hr for each type of nanoparticle. Methods to process images and extract size information from each individual particle can also be time consuming. The automated Hough circle transform-based method used here41,42 required ~3 – 4 s per particle using a desktop computer. Analysis of the 1,291 and 976 particles for the ~50 nm and ~100 nm nanoparticle samples, respectively, required ~1 hr each. Thus, the total combined experiment and analysis time required for each type of nanoparticle in these TEM analyses was ~2 hr. Software-assisted manual diameter measurement, a method still commonly used to size nanoparticles in microscopy images, can add even more to the time and effort required.44
The recently constructed CDMS instrument presented in this work is designed to maximize sensitivity and reduce the total acquisition time required to meaningfully quantify MDa-sized analytes on an individual particle basis. In contrast to the TEM analyses, CDMS experiments measuring 14,455 and 2,509 ions took ~17 min and ~50 min respectively, from the ~50 nm and ~100 nm nanoparticle samples. The difference in count rates for the two samples is partially due to their differing dilution factors and average molecular masses. The initial 2.5% w/v ~50 nm sample, with an average mass of 37.0 MDa, was diluted by a factor of 5000, yielding a concentration of ~140 pM, whereas the ~100 nm sample (1% w/v, average mass of 354 MDa) has a concentration of ~60 pM. The difference in counting rate between the ~50 nm and ~100 nm samples may be also result of the different surfaces of the two nanoparticle types. The ~50 nm particles are functionalized with amine groups whereas the ~100 nm spheres are unfunctionalized polystyrene. The presence of the amine groups may promote a higher ionization efficiency in the ESI process, leading to the observed difference. Electrophoretic enrichment45 of the amine-functionalized particles in the electrospray could contribute to this difference. However, there was no significant variation in ion counts measured over time, even when nearly all the sample solution had been exhausted from the emitter capillary. This indicates that any electrophoretic enrichment that may occur does not lead to a measurable change in signal. It is also possible the larger, much more massive ~100 nm particles are not as efficiently transmitted and confined in the early stages of the CDMS instrument due to their large masses and aerodynamic acceleration effects that have been explored in other work.18 However, regardless of the number of particles, recent advances in CDMS data analysis methods31 have made it possible to measure and plot the masses of individual ions in real-time using a desktop computer (the same computer used for the automated TEM image analysis method) such that no significant time cost outside of the experiment duration was incurred for data processing.
To compare the time necessary for TEM and CDMS measurements, the time needed to obtain and analyze data was normalized to take into account the different particle counts, experiment times, and processing times to obtain a particle per minute (particles/min) acquisition rate for each method. The CDMS analysis method has rates of 850 and 50 particles/min for the ~50 nm and ~100 nm samples, respectively. The TEM method yielded rates of 11 and 8 particles/min for the ~50 nm and ~100 nm samples, respectively. Thus, CDMS analysis acquisition rates are ~6 – 80x faster than the TEM imaging method used in this work. It is worth noting that while the electrospray emitter tips used in this work were filled manually, sizing nanoparticles by CDMS is a process highly amenable to automation using auto-sampling techniques that are commonly used in other types of ESI-MS-based workflows.46
The greater degree of dilution (CDMS dilution factors of 5000 and 500, compared to TEM dilution factors of 100 and 40 for the ~50 nm and ~100 nm samples, respectively) used for the CDMS analysis is an additional advantage of the technique. Nanoparticles with poor solubilities or that are available only in small quantities that would preclude analysis by less sensitive techniques can still be analyzed by CDMS, rendering the main advantage of non-destructive analysis methods moot. The high degree of sample dilution also demonstrates the high sensitivity and detection efficiency enabled by the CDMS instrument design detailed here. Because the average mass, total ion count, and volume of expended sample were measured, and the sample concentration is known, it is possible to calculate the fraction of total particles expended in the analysis that were counted, i.e., a detection efficiency. For the ~50 nm and ~100 nm particles, the detection efficiencies were 2.22 × 10−4 (~1/4500) and 3.65 × 10−5 (~1/27400), respectively. The detection efficiency for the ~100 nm spheres is comparable to the best detection efficiency (4.4 × 10−5 for ~4 MDa hepatitis B virus capsids) previously demonstrated by CDMS,47 despite the nanoparticles having ~100x higher masses. The detection efficiency for the ~50 nm nanoparticles is ~5 times higher than the previous best demonstrated by CDMS,47 despite the relatively high and broad mass range spanned by the sample. The higher detection efficiency of the current instrument may be due to several factors, but it is likely that elimination of an ion energy filter that inherently reduces transmission plays an important role. The high acquisition speed and detection efficiency demonstrated here are important advances in the analysis of nanoparticles (and other analytes), both for quality control and research purposes.
Conclusions
Accurate size characterization of nanoparticles has primarily been done using methods capable of imaging individual particles at these small sizes, such as TEM, which report the diameters of individual particles. The number of particles necessary to characterize a size distribution varies but increases substantially with sample heterogeneity, leading to relative long analysis times that can involve both optimization of the sample preparation and the actual analysis of the resulting data. CDMS is an alternative method for determining particle size and has the advantage of high accuracy and speed for measuring the masses of individual particles. Total measurement and analysis time can be up to 80x less per particle compared to TEM, making it practical to acquire many more individual ion measurements and more accurately determine particle size distributions. Equally important is the ability to deduce the extent of aggregation that exists in solution from the preservation of aggregates though the measurement process. A mass accuracy of ~2% or better is readily achievable, which corresponds to a diameter accuracy of ~0.7% for spherical particles.
CDMS mass measurements can be applied to nanoparticles of nearly any type. The mass of a particle is a fundamental property that does not depend on shape. The accurate measurement of mass distributions of nanoparticles enabled by CDMS has many potential advantages in the characterization of nanoparticles. For nanoparticles of known shape, such as the spherical nanoparticles analyzed here, masses can be transformed directly into particle dimensions. For nanoparticles of unknown or mixed morphologies, some information about particle shape can be determined from the extent of particle charging. Alternatively, limited TEM measurements could provide sufficient information about shape and enable subsequent fast, high accuracy CDMS determination of the distribution of nanoparticle dimensions.
Although only aqueous nanoparticle suspensions were analyzed in this work, ESI is regularly performed using many different solvents and solvent mixtures. Thus, this technique is applicable to a broad range of nanoparticle types, including nanoparticles prone to aggregation in oxidative environments, such as some types of quantum dots.48,49 The minimal sample preparation (typically dilution) and introduction through electrospray ionization that can be readily automated makes this a potentially powerful method for automated particle size characterization. CDMS analysis could be combined with high-throughput chemical synthetic methods to analyze data in real time and provide feedback about particle sizes in automated synthetic workflows. Further improvements to this instrument to increase data acquisition rates and mass measuring accuracy are ongoing.
Methods
CDMS Instrument Description.
A CDMS instrument that does not incorporate ion optics for energy filtration was designed and constructed. Ion energies are measured directly from the duty cycle of ion oscillation within an electrostatic ion trap making it possible to introduce and measure the masses of ions with a wide range of energies. This method for ion energy measurements has been demonstrated on previous instrumentation and has been shown to be in good agreement with energy filter-based determinations of ion energies.30 In addition to simplifying the design of CDMS instruments, a key advantage of this configuration is that no ions are lost to an energy filter, resulting in improved sensitivity at low sample concentrations. Energy based mass biases stemming from the differing extents of aerodynamic acceleration for nanoparticles with different cross-sections entering a vacuum system18 are mitigated by the removal of energy filter. This recently constructed instrument also includes several other design optimizations aimed at maximizing the sensitivity, analyte mass range, and speed of individual ion analyses. The entire assembly, including a mounting cart and all power supplies and electronics needed for operation, has dimensions of 160 × 56 × 122 cm (L × W × H), excluding mechanical backing pumps. Figure 3 shows a schematic of the instrument divided into several main regions that are described in more detail below. Approximate DC potentials that ions experience as they transit the various ion optics is also provided in Figure 3 below the corresponding regions of the instrument.
Figure 3.
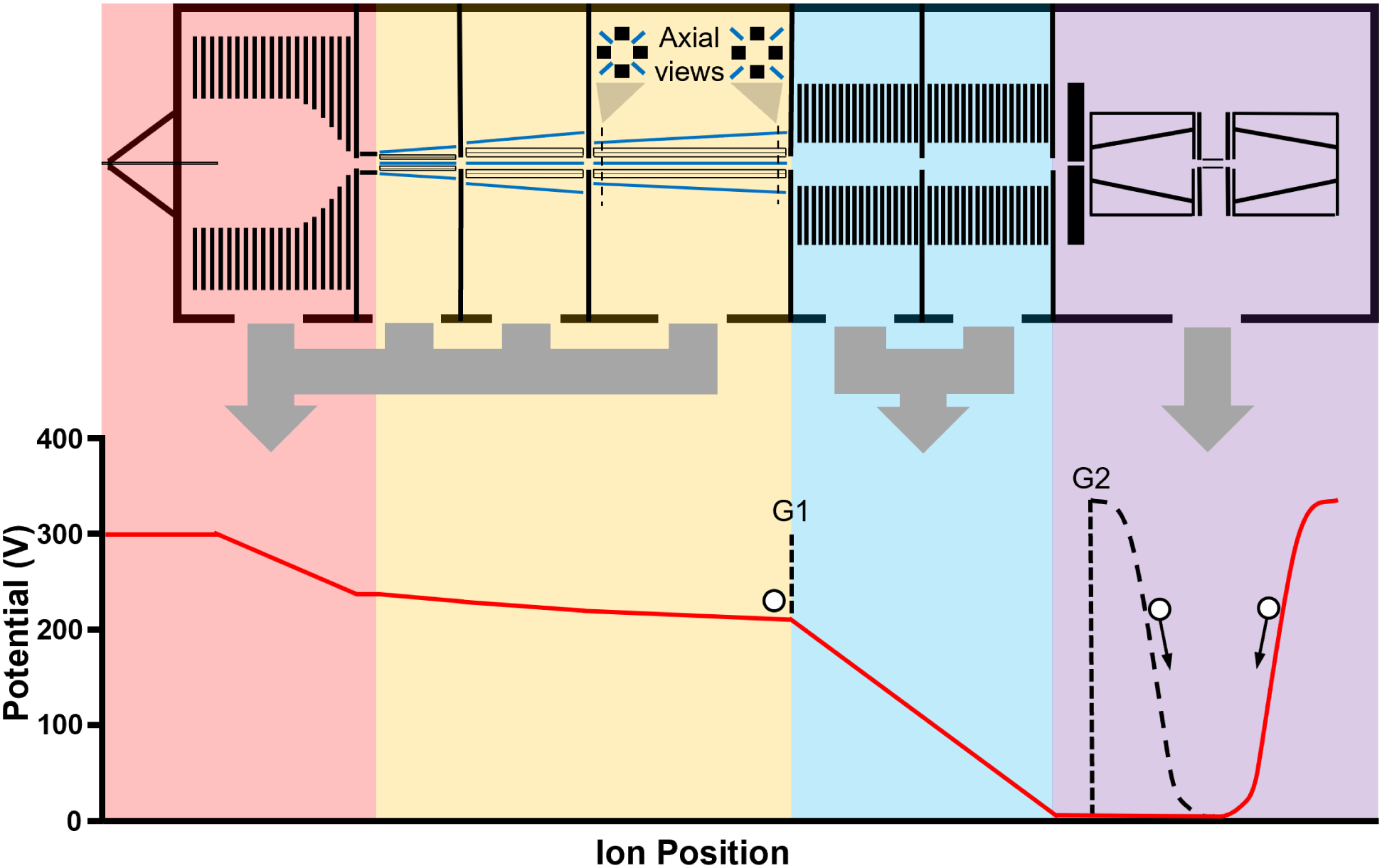
A schematic of the recently constructed CDMS instrument used in this work and the DC ion optics potentials experienced by an ion as it transits the instrument. The instrument is divided into four main regions: an initial drying and funnel stage (red), a quadrupole thermalization and accumulation stage (yellow), an ion acceleration stage (blue), and the electrostatic ion trap stage (purple) where the induced current of ions oscillating within the trap is used to measure ion masses. The black dashed lines at points G1 and G2 indicate potentials experienced by ions during the “closed” accumulation and trapping state while the red line indicates potentials during the “open” state.
An ESI source, drying tube, ion funnel, and gate valve are contained in the first region highlighted in red in Figure 3. A nanoelectrospray source is used in all experiments performed in this work. Briefly, an electrospray emitter is mounted to a block and is connected to a ±4 kV power supply (Ardara Technologies) through a platinum wire that is in contact with the sample solution. ESI voltages between +1.3–1.7 kV relative to the inlet capillary were used and currents of ~50–100 nA were typically observed. These voltages and currents correspond to the onset of electrospray and were not increased further during the course of the experiments. Borosilicate capillaries pulled to tip diameters of 5 – 6 μm are placed ~5 mm from the instrument inlet.50 ESI-generated droplets enter the instrument via a heated metal capillary that is 11.4 cm long with an inner diameter of 0.5 mm. The heated capillary serves to evaporate solution from the nanodrops in order to produce “bare” or unsolvated ions. This drying capillary is equipped with an isolated cartridge heater and PID temperature controller and can be heated to temperatures between 25 and 200 °C. A drying capillary temperature of 140 °C was used in this work. The drying capillary is maintained at a voltage of ~250 – 350 V.
Ions exiting the drying capillary enter a vacuum pumped region containing an ion funnel at ~1.7 Torr. The ion funnel is 11.4 cm long and has 57 discrete (0.6 mm thick) lens elements. These elements are spaced by 2.54 mm between centers in the upstream portion of the funnel; this spacing decreases to 1.27 mm between centers in the last ~23 mm of the funnel where the element orifices have their smallest diameters. The funnel elements are resistively connected to form a DC gradient across the length of the funnel between setpoints supplied to the furthest upstream and downstream elements. In typical operation, a “downhill” gradient of 20 – 80 V across the length of the ion funnel is used, starting at the voltage of drying capillary. 180° out of phase RF potentials of up to ~300 Vpp are also applied on alternating lens elements of the funnel to confine and assist in thermalization of the ions. The RF potential is supplied by a Broad-Head broadband RF power supply (Ardara Technologies) that can produce the required 300 Vpp output voltages at frequencies between 10 and 500 kHz. A frequency of 200 kHz for the ion funnel RF was used in this work. At the downstream exit of the ion funnel, ions pass through a conductance limiting orifice (2.0 mm) and a thin gate valve region (Ardara Technologies) with a single tube lens with potentials a few volts below that of the downstream end of the ion funnel.
After transiting the gate valve region, ions enter a region (highlighted in yellow) containing three sets of rectilinear quadrupole ion guides (Ardara Technologies) with RF potentials provided by Broad-Head RF supplies (Ardara Technologies) with typical operating values of 600–1000 Vpp and adjustable DC offset voltages. The frequencies and amplitudes of the applied RF potentials can be selected to provide optimal containment of ions in a desired m/z range. Typical operating frequencies were between 70–200 kHz.
The chamber design and associated split-flow turbomolecular pump results in three differentially pumped regions with operating pressures of ~100 mTorr, ~3 mTorr, and ~0.1 mTorr (pumping stages represented by gray arrows). The first rectilinear quadrupole has an inscribed diameter of 5.0 mm; the second and third quadrupoles have inscribed diameters of 8.0 mm. All three quadrupoles are equipped with asymptotic field shaping electrodes (Ardara Technologies), which provide gentle z-axis acceleration to prevent stalling of ions. An axial view of these electrodes relative to the quadrupole ion guides is shown inset in Figure 3. The electrodes are closer to the center of the quadrupole on the upstream side and further away on the downstream side. When the potential of these electrodes is set to a value greater than the DC offset of the quadrupole rods, this geometry generates a potential gradient along the centerlines of the guides. A gentle gradient of ~5 V along the length of the quadrupole rods helps to move ions through this region and, when operating any of these guides as an ion accumulation region, concentrates ions near the downstream ‘exit’ of the quadrupole rods to facilitate more efficient extraction. The downstream quadrupole is capped with an exit lens with 5.0 mm aperture.
To facilitate accumulation and gated extraction of ions prior to the final analyzer stage, a custom MOSFET-based (2SK1835) switch can change the DC offset potential of the appropriate quadrupole section or downstream exit lens with <1 μs rise times. In this work, this switching is applied exclusively to the downstream exit lens and the approximate extent potential change is shown in Figure 3 by a black dotted line at point G1. The timings of this MOSFET switch and an additional identical switch used to control the DC potential of the upstream cone electrode (G2 in Figure 3) in the analyzer stage are controlled by computer via a NI PCIe-6612 counter/timer device and SCB-68A connector block that provide the 5 V switching signals to the gates of the MOSFETs with ~10 ns precision. This precise control of voltage switching and timing is necessary to ensure that a gated pulse of ions exiting from the last rectilinear quadrupole ion guide (at point G1) reaches the electrostatic trap containing the detection electrode at times when the trap is in an “open” voltage state (red solid line) immediately prior to entering the “closed” state (black dotted line) when ions are confined and measured. In this work, this ion time-of-flight delay was varied in even steps across a narrow range of delay times (~1 ms) centered around the delay time of optimal ion current for the sample being analyzed.
After extraction from the quadrupole region, ion pass through two sets of custom-built ion accelerators in two differentially pumped regions (highlighted in blue in Figure 3) separated by a 5 mm aperture. Each region is pumped by an Edwards EXT250 turbomolecular pump (Edwards Vacuum), yielding pressures of ~2 × 10−6 Torr and ~1 × 10−7 Torr in the upstream and downstream regions, respectively. The ion accelerators are composed of 31 and 43 discrete lens elements (19 mm i.d.) for the upstream and downstream units, respectively. Potentials are supplied to the first upstream lens and the final downstream lens in each set. The internal lenses are connected resistively to form a non-focusing linear potential gradient along the length of the lens set. At the end of the second ion acceleration region, there is a conductance-limiting aperture (5.0 mm diameter) into the final vacuum chamber.
The final region of the instrument contains the electrostatic ion trap and detection cylinder (highlighted in purple in Figure 3). Upon entering the final vacuum chamber, ions must pass through a ‘collimator’, a 2.54 cm thick plate with a 0.5 mm aperture that is aligned with the center of the electrostatic ion trap. This effectively produces a thin beam of collimated ions because ions that are successful in transiting this aperture must have a relatively narrow angular and spatial spread. While this restriction results in ion losses, it ensures that ions entering the electrostatic trap have initial trajectories that are conducive to long-term, stable trapping.
The electrostatic ion trap is modeled after the original ‘conetrap’ design of Schmidt et al.51 and has the same dimensions as a trap used for CDMS that was reported previously for a different instrument,33 with one key difference. The dimensions of the detection cylinder (14 mm length, 3.5 mm O. D. in this work) are half that of the previous CDMS instrument33 and the positions of the cone electrodes and shielding plates were adjusted accordingly to maintain the same spacing as in the previous design. SIMION modeling of ion trajectories shows that decreasing the length of the detection cylinder increases the width of the ion energy per charge distribution that can be stably trapped at a given trapping voltage. At the 330 V trapping potential used both in this work and previous CDMS work, the range of stably trappable ion energies is ~184–273 eV/z compared to 193–253 eV/z reported for previous instrumentation.33 The shorter length of the detection cylinder also yields higher ion oscillation frequencies that increase S/N by reducing effects of 1/f noise and increasing the number of passes through the detector electrode. The trap is operated in a similar way to previous CDMS instruments,33 with the upstream cone potential reduced to 0 V (red line at point G2 in Figure 3) for a brief period of <1 ms to allow ions to enter the trap followed by a fast (<1 μs rise time) return to the full trapping potential (330 V in this work, shown by the black dotted line at point G2 in Figure 3). Ions in the process of transiting the detector tube or turning around in the downstream cone electrode region when the upstream cone returns to the full trapping potential are trapped. After trapping, ions can be retained for a user-selected length of time. A 100 ms trapping period was used for all experiments carried out in this work and only ions trapped for the full 100 ms period (>95% of all trapped ions) were included in the data analysis.
Data Measurement and Analysis.
Trapped ions oscillate between the cone electrodes and induce a current on the detection cylinder on each pass. This induced current is passed into the input of a charge-sensitive pre-amplifier (Amptek A250 CoolFET) with a Peltier-cooled JFET inside a shielded box within the vacuum chamber. The induced current is converted into a voltage signal with pulse amplitudes proportional to the charge of the ion. The signal is then routed outside of the vacuum chamber and passes through a custom bandpass filter with a flat (<0.11% ripple) passband from ~1 kHz to ~300 kHz with an overall gain of ~10. The signal is subsequently digitized at 1 MHz (AlazarTech ATS9120) and analyzed using a Python program implementing short-time Fourier transform (STFT)-based methods.29 Peaks corresponding to individual ion signals within the STFT are traced and fit with their characteristic functions to avoid error due to scalloping and sampling artifacts in the FT and ensure amplitudes (and therefore charge) and frequency are measured with high accuracy. This method, which also uses custom-built Python software, has been described in detail elsewhere.31 The individually traced ion frequencies and amplitudes are then converted to measurements of m/z, charge, and ion energy.29,30,33,52,53 Calibration values used to convert frequency to m/z and convert harmonic amplitude ratios (HARs) to ion energies were determined from SIMION simulations of the instrument trap geometry.30 Calibration values used to convert fundamental amplitude to charge were determined using standards of known mass and a trap-specific SIMION simulation-based correction of raw amplitudes based on ion energies.53
Materials.
A sample of Nanosphere™ size standard polystyrene spheres (1% w/v in aqueous suspension) with a calibrated mean diameter of 101 nm ± 3 nm and a standard deviation of 6.2 nm was obtained from the manufacturer (Thermo Scientific, catalog #: 3100/3100A) and diluted by a factor of 500 into 0.5% aqueous acetic acid. The calibrated mean diameter (certified batch #: 3100–009) is certified by the manufacturer via transfer by TEM from National Institute of Standards and Technology certified microspheres. A ~50 nm surface amine-functionalized polystyrene sphere sample (≥2.5% w/v aqueous suspension) was obtained from the manufacturer (Sigma Aldrich, product #: L0780) and diluted by a factor of 5,000 into 0.5% acetic acid. The manufacturer reported mean diameter range for this sample is 45–55 nm. The density of polystyrene is 1.0502 g/mL;40 this value is used in all calculations of diameter determined from measured masses.
Transmission Electron Microscopy (TEM).
Prior to TEM analysis, both the ~50 nm and ~100 nm polystyrene nanoparticle samples were diluted in pure water by factors of ~100 and ~40, respectively. Formvar/carbon coated 300 mesh copper grids (Ted Pella Inc.) were made hydrophilic with an easiGlow (Pelco) benchtop glow discharge unit. 5 μL of sample were placed on each grid for 5 min before being manually dried using clean filter paper. Sample coated grids were imaged using a Tecnai 12 120 kV transmission electron microscope (FEI). Images were recorded using a Rio 16 CMOS with DigitalMicrograph software (Gatan Inc.). The resulting TEM images were analyzed using FIJI/ImageJ version 1.53v.54 Tiff-formatted files were converted to binary and contrast was enhanced using the Outline and Dilate features. Noise was removed using Despeckle. Circle radii were obtained using the Hough Circle Transform (HCT) plugin by University of California, Berkeley Vision Sciences with a radius search increment of 1 pixel and a Hough score threshold of 0.5 (more details on this plugin can be found here: https://imagej.net/plugins/hough-circle-transform).41,42 TEM images with a resolution of ~0.5 nm/pixel were analyzed to generate a diameter and mass distribution for each sample. In total, 976 and 1291 particles were analyzed to generate the data shown for the ~50 nm and ~100 nm diameter nanosphere samples, respectively.
Supplementary Material
Acknowledgements
The authors are grateful for financial support from the National Institutes of Health (5R01GM139338), the Arnold and Mabel Beckman Foundation Postdoctoral Fellowship in Chemical Instrumentation (C.C.H.), and the Merck DBL SEEDS program (J.S.J.). The authors thank Reena Zalpuri at the University of California, Berkeley Electron Microscope Laboratory for advice and assistance in TEM data collection.
Footnotes
Supporting Information
Description of Rayleigh limit calculations and 2D mass vs. charge histogram for ~50 nm nanoparticles.
References
- (1).Astruc D Introduction: Nanoparticles in Catalysis. Chem. Rev 2020, 120 (2), 461–463. [DOI] [PubMed] [Google Scholar]
- (2).Couvreur P Nanoparticles in Drug Delivery: Past, Present and Future. Adv. Drug Deliv. Rev 2013, 65 (1), 21–23. [DOI] [PubMed] [Google Scholar]
- (3).Tan HW; An J; Chua CK; Tran T Metallic Nanoparticle Inks for 3D Printing of Electronics. Adv. Electron. Mater 2019, 5 (5), 1800831. [Google Scholar]
- (4).Mohajerani A; Burnett L; Smith JV; Kurmus H; Milas J; Arulrajah A; Horpibulsuk S; Kadir AA Nanoparticles in Construction Materials and Other Applications, and Implications of Nanoparticle Use. Materials (Basel). 2019, 12 (19), 3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Modena MM; Rühle B; Burg TP; Wuttke S Nanoparticle Characterization: What to Measure? Adv. Mater 2019, 31 (32), 1901556. [DOI] [PubMed] [Google Scholar]
- (6).Ramos AP Dynamic Light Scattering Applied to Nanoparticle Characterization. In Nanocharacterization Techniques; William Andrew Publishing, Norwich, New York, 2017; pp 99–110. [Google Scholar]
- (7).Li T; Senesi AJ; Lee B Small Angle X-Ray Scattering for Nanoparticle Research. Chem. Rev 2016, 116 (18), 11128–11180. [DOI] [PubMed] [Google Scholar]
- (8).Michen B; Geers C; Vanhecke D; Endes C; Rothen-Rutishauser B; Balog S; Petri-Fink A Avoiding Drying-Artifacts in Transmission Electron Microscopy: Characterizing the Size and Colloidal State of Nanoparticles. Sci. Rep 2015, 5 (1), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lee B; Yoon S; Lee JW; Kim Y; Chang J; Yun J; Ro JC; Lee JS; Lee JH Statistical Characterization of the Morphologies of Nanoparticles through Machine Learning Based Electron Microscopy Image Analysis. ACS Nano 2020, 14 (12), 17125–17133. [DOI] [PubMed] [Google Scholar]
- (10).De Juan L; Fernández de la Mora J On-Line Sizing of Colloidal Nanoparticles via Electrospray and Aerosol Techniques. In ACS Symposium Series; UTC, 1996; Vol. 622, pp 19–41. [Google Scholar]
- (11).Kaufman SL Analysis of Biomolecules Using Electrospray and Nanoparticle Methods: The Gas-Phase Electrophoretic Mobility Molecular Analyzer (GEMMA). J. Aerosol Sci 1998, 29 (5–6), 537–552. [Google Scholar]
- (12).Song DK; Lenggoro IW; Hayashi Y; Okuyama K; Kim SS Changes in the Shape and Mobility of Colloidal Gold Nanorods with Electrospray and Differential Mobility Analyzer Methods. Langmuir 2005, 21 (23), 10375–10382. [DOI] [PubMed] [Google Scholar]
- (13).Tan J; Liu J; Li M; El Hadri H; Hackley VA; Zachariah MR Electrospray-Differential Mobility Hyphenated with Single Particle Inductively Coupled Plasma Mass Spectrometry for Characterization of Nanoparticles and Their Aggregates. Anal. Chem 2016, 88 (17), 8548–8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Li C; Lee AL; Chen X; Pomerantz WCK; Haynes CL; Hogan CJ Multidimensional Nanoparticle Characterization through Ion Mobility-Mass Spectrometry. Anal. Chem 2020, 92 (3), 2503–2510. [DOI] [PubMed] [Google Scholar]
- (15).Flagan RC Continuous-Flow Differential Mobility Analysis of Nanoparticles and Biomolecules. Annu. Rev. Chem. Biomol. Eng 2014, 5, 255–279. [DOI] [PubMed] [Google Scholar]
- (16).Guha S; Li M; Tarlov MJ; Zachariah MR Electrospray-Differential Mobility Analysis of Bionanoparticles. Trends Biotechnol. 2012, 30 (5), 291–300. [DOI] [PubMed] [Google Scholar]
- (17).Fernandez de la Mora J A Singularly Narrow 29 Nm Aerosol Size Standard Based on the ΦX174 Bacteriophage. J. Aerosol Sci 2022, 161, 105949. [Google Scholar]
- (18).Elliott AG; Merenbloom SI; Chakrabarty S; Williams ER Single Particle Analyzer of Mass: A Charge Detection Mass Spectrometer with a Multi-Detector Electrostatic Ion Trap. Int. J. Mass Spectrom 2017, 414, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Contino NC; Jarrold MF Charge Detection Mass Spectrometry for Single Ions with a Limit of Detection of 30 Charges. Int. J. Mass Spectrom 2013, 345–347, 153–159. [Google Scholar]
- (20).Doussineau T; Kerleroux M; Dagany X; Clavier C; Barbaire M; Maurelli J; Antoine R; Dugourd P Charging Megadalton Poly(Ethylene Oxide)s by Electrospray Ionization. A Charge Detection Mass Spectrometry Study. Rapid Commun. Mass Spectrom 2011, 25 (5), 617–623. [DOI] [PubMed] [Google Scholar]
- (21).Benner WH A Gated Electrostatic Ion Trap to Repetitiously Measure the Charge and m/z of Large Electrospray Ions. Anal. Chem 1997, 69 (20), 4162–4168. [Google Scholar]
- (22).Harper CC; Miller ZM; Lee H; Bischoff AJ; Francis MB; Schaffer DV; Williams ER Effects of Molecular Size on Resolution in Charge Detection Mass Spectrometry. Anal. Chem 2022, 94 (33), 11703–11712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Doussineau T; Bao CY; Antoine R; Dugourd P; Zhang W; D’Agosto F; Charleux B Direct Molar Mass Determination of Self-Assembled Amphiphilic Block Copolymer Nanoobjects Using Electrospray-Charge Detection Mass Spectrometry. ACS Macro Lett. 2012, 1 (3), 414–417. [DOI] [PubMed] [Google Scholar]
- (24).Bischoff AJ; Harper CC; Williams ER; Francis MB Characterizing Heterogeneous Mixtures of Assembled States of the Tobacco Mosaic Virus Using Charge Detection Mass Spectrometry. J. Am. Chem. Soc 2022, 144 (51), 23368–23378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Keifer DZ; Motwani T; Teschke CM; Jarrold MF Acquiring Structural Information on Virus Particles with Charge Detection Mass Spectrometry. J. Am. Soc. Mass Spectrom 2016, 27 (6), 1028–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Dunbar CA; Callaway HM; Parrish CR; Jarrold MF Probing Antibody Binding to Canine Parvovirus with Charge Detection Mass Spectrometry. J. Am. Chem. Soc 2018, 140 (46), 15701–15711. [DOI] [PubMed] [Google Scholar]
- (27).Jarrold MF Applications of Charge Detection Mass Spectrometry in Molecular Biology and Biotechnology. Chem. Rev 2022, 122 (8), 7415–7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Harper CC; Brauer DD; Francis MB; Williams ER Direct Observation of Ion Emission from Charged Aqueous Nanodrops: Effects on Gaseous Macromolecular Charging. Chem. Sci 2021, 12 (14), 5185–5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Harper CC; Elliott AG; Oltrogge LM; Savage DF; Williams ER Multiplexed Charge Detection Mass Spectrometry for High-Throughput Single Ion Analysis of Large Molecules. Anal. Chem 2019, 91 (11), 7458–7468. [DOI] [PubMed] [Google Scholar]
- (30).Harper CC; Elliott AG; Lin HW; Williams ER Determining Energies and Cross Sections of Individual Ions Using Higher-Order Harmonics in Fourier Transform Charge Detection Mass Spectrometry (FT-CDMS). J. Am. Soc. Mass Spectrom 2018, 29 (9), 1861–1869. [DOI] [PubMed] [Google Scholar]
- (31).Miller ZM; Harper CC; Lee H; Bischoff AJ; Francis MB; Schaffer DV; Williams ER Apodization Specific Fitting for Improved Resolution, Charge Measurement, and Data Analysis Speed in Charge Detection Mass Spectrometry. J. Am. Soc. Mass Spectrom 2022, 33 (11), 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Todd AR; Barnes LF; Young K; Zlotnick A; Jarrold MF Higher Resolution Charge Detection Mass Spectrometry. Anal. Chem 2020, 92 (16), 11357–11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Elliott AG; Harper CC; Lin HW; Williams ER Mass, Mobility and MSN Measurements of Single Ions Using Charge Detection Mass Spectrometry. Analyst 2017, 142 (15), 2760–2769. [DOI] [PubMed] [Google Scholar]
- (34).Contino NC; Pierson EE; Keifer DZ; Jarrold MF Charge Detection Mass Spectrometry with Resolved Charge States. J. Am. Soc. Mass Spectrom 2013, 24 (1), 101–108. [DOI] [PubMed] [Google Scholar]
- (35).Fernandez De La Mora J Electrospray Ionization of Large Multiply Charged Species Proceeds via Dole’s Charged Residue Mechanism. Anal. Chim. Acta 2000, 406 (1), 93–104. [Google Scholar]
- (36).Jordan JS; Williams ER Effects of Electrospray Droplet Size on Analyte Aggregation: Evidence for Serine Octamer in Solution. Anal. Chem 2021, 93 (3), 1725–1731. [DOI] [PubMed] [Google Scholar]
- (37).Xia Z; Williams ER Effect of Droplet Lifetime on Where Ions Are Formed in Electrospray Ionization. Analyst 2019, 144 (1), 237–248. [DOI] [PubMed] [Google Scholar]
- (38).Davidson KL; Oberreit DR; Hogan CJ; Bush MF Nonspecific Aggregation in Native Electrokinetic Nanoelectrospray Ionization. Int. J. Mass Spectrom 2017, 420, 35–42. [Google Scholar]
- (39).Susa AC; Xia Z; Williams ER Small Emitter Tips for Native Mass Spectrometry of Proteins and Protein Complexes from Nonvolatile Buffers That Mimic the Intracellular Environment. Anal. Chem 2017, 89 (5), 3116–3122. [DOI] [PubMed] [Google Scholar]
- (40).Godin M; Bryan AK; Burg TP; Babcock K; Manalis SR Measuring the Mass, Density, and Size of Particles and Cells Using a Suspended Microchannel Resonator. Appl. Phys. Lett 2007, 91 (12), 123121. [Google Scholar]
- (41).Caval-Holme FS; Aranda ML; Chen AQ; Tiriac A; Zhang Y; Smith B; Birnbaumer L; Schmidt TM; Feller MB The Retinal Basis of Light Aversion in Neonatal Mice. J. Neurosci 2022, 42 (20), 4101–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Mirzaei M; Rafsanjani HK An Automatic Algorithm for Determination of the Nanoparticles from TEM Images Using Circular Hough Transform. Micron 2017, 96, 86–95. [DOI] [PubMed] [Google Scholar]
- (43).McKay AR; Ruotolo BT; Ilag LL; Robinson CV Mass Measurements of Increased Accuracy Resolve Heterogeneous Populations of Intact Ribosomes. J. Am. Chem. Soc 2006, 128 (35), 11433–11442. [DOI] [PubMed] [Google Scholar]
- (44).Teulon JM; Godon C; Chantalat L; Moriscot C; Cambedouzou J; Odorico M; Ravaux J; Podor R; Gerdil A; Habert A; Herlin-Boime N; Chen SWW; Pellequer JL On the Operational Aspects of Measuring Nanoparticle Sizes. Nanomaterials 2019, 9 (1), 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhang Z; Pulliam CJ; Flick T; Cooks RG Electrophoretic Desalting to Improve Performance in Electrospray Ionization Mass Spectrometry. Anal. Chem 2018, 90 (6), 3856–3862. [DOI] [PubMed] [Google Scholar]
- (46).Wolters DA; Washburn MP; Yates JR An Automated Multidimensional Protein Identification Technology for Shotgun Proteomics. Anal. Chem 2001, 73 (23), 5683–5690. [DOI] [PubMed] [Google Scholar]
- (47).Todd AR; Jarrold MF Dramatic Improvement in Sensitivity with Pulsed Mode Charge Detection Mass Spectrometry. Anal. Chem 2019, 91 (21), 14002–14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Yeh CW; Chen GH; Ho SJ; Chen HS Inhibiting the Surface Oxidation of Low-Cadmim-Content ZnS:(Cd,Se) Quantum Dots for Enhancing Application Reliability. ACS Appl. Nano Mater 2019, 2 (8), 5290–5301. [Google Scholar]
- (49).Calvin JJ; O’Brien EA; Sedlak AB; Balan AD; Alivisatos AP Thermodynamics of Composition Dependent Ligand Exchange on the Surfaces of Colloidal Indium Phosphide Quantum Dots. ACS Nano 2021, 15 (1), 1407–1420. [DOI] [PubMed] [Google Scholar]
- (50).Jordan JS; Xia Z; Williams ER Tips on Making Tiny Tips: Secrets to Submicron Nanoelectrospray Emitters. J. Am. Soc. Mass Spectrom 2022, 33 (3), 607–611. [DOI] [PubMed] [Google Scholar]
- (51).Schmidt HT; Cederquist H; Jensen J; Fardi A Conetrap: A Compact Electrostatic Ion Trap. Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. with Mater. Atoms 2001, 173 (4), 523–527. [Google Scholar]
- (52).Elliott AG; Harper CC; Lin HW; Susa AC; Xia Z; Williams ER Simultaneous Measurements of Mass and Collisional Cross-Section of Single Ions with Charge Detection Mass Spectrometry. Anal. Chem 2017, 89 (14), 7701–7708. [DOI] [PubMed] [Google Scholar]
- (53).Elliott AG; Harper CC; Lin HW; Williams ER Effects of Individual Ion Energies on Charge Measurements in Fourier Transform Charge Detection Mass Spectrometry (FT-CDMS). J. Am. Soc. Mass Spectrom 2019, 30 (6), 946–955. [DOI] [PubMed] [Google Scholar]
- (54).Schindelin J; Arganda-Carreras I; Frise E; Kaynig V; Longair M; Pietzsch T; Preibisch S; Rueden C; Saalfeld S; Schmid B; Tinevez JY; White DJ; Hartenstein V; Eliceiri K; Tomancak P; Cardona A Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9 (7), 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.