

Abstract
New Western-style diet 1 (NWD1), a purified diet establishing mouse exposure to key nutrients recapitulating levels that increase human risk for intestinal cancer, reproducibly causes mouse sporadic intestinal and colonic tumors reflecting human etiology, incidence, frequency, and lag with developmental age. Complex NWD1 stem cell and lineage reprogramming was deconvolved by bulk and single-cell RNA sequencing, single-cell Assay for Transposase-Accessible Chromatin using sequencing, functional genomics, and imaging. NWD1 extensively, rapidly, and reversibly, reprogrammed Lgr5hi stem cells, epigenetically downregulating Ppargc1a expression, altering mitochondrial structure and function. This suppressed Lgr5hi stem cell functions and developmental maturation of Lgr5hi cell progeny as cells progressed through progenitor cell compartments, recapitulated by Ppargc1a genetic inactivation in Lgr5hi cells in vivo. Mobilized Bmi1+, Ascl2hi cells adapted lineages to the nutritional environment and elevated antigen processing and presentation pathways, especially in mature enterocytes, causing chronic, protumorigenic low-level inflammation. There were multiple parallels between NWD1 remodeling of stem cells and lineages with pathogenic mechanisms in human inflammatory bowel disease, also protumorigenic. Moreover, the shift to alternate stem cells reflects that the balance between Lgr5-positive and -negative stem cells in supporting human colon tumors is determined by environmental influences. Stem cell and lineage plasticity in response to nutrients supports historic concepts of homeostasis as a continual adaptation to environment, with the human mucosa likely in constant flux in response to changing nutrient exposures.
Implications:
Although oncogenic mutations provide a competitive advantage to intestinal epithelial cells in clonal expansion, the competition is on a playing field dynamically sculpted by the nutritional environment, influencing which cells dominate in mucosal maintenance and tumorigenesis.
Visual Overview
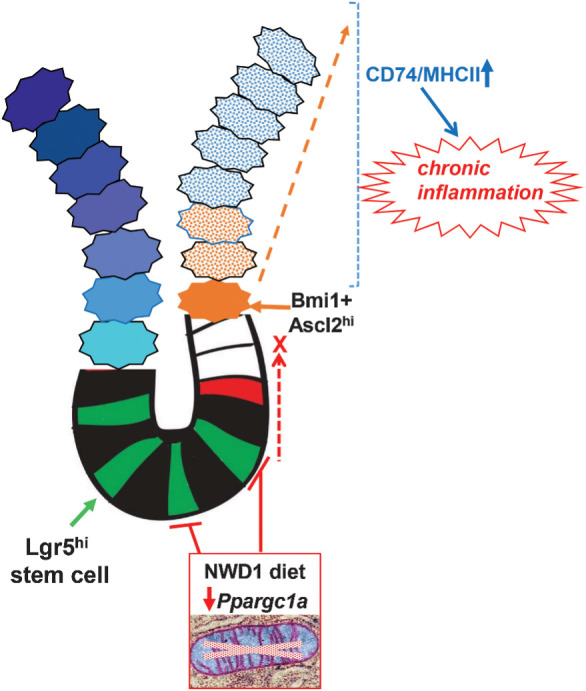
Introduction
Damage or ablation of Lgr5hi intestinal stem cells (ISC) dedifferentiates multiple cell types to function as stem-like cells, restoring homeostasis (1, 2). However, original concepts of Bernard’s “milieu interieur,” and Thompson in coining the term, defined homeostasis as continual tissue adaptation to its environment, not a target state returned to following disruption (3). Indeed, we established that dietary patterns, continually changing in humans, can determine which and how intestinal cells function as stem cells (4–6), and there is rapid intestinal cell adaptation to higher dietary fat by upregulating fat metabolism pathways (7).
Reflecting the complexity and variability of the human diet in the mouse is a major challenge. New Western-style diet 1 (NWD1), a purified diet, causes sporadic intestinal and colon tumors. Sporadic tumors are the majority of human intestinal tumors, broadly defined as tumors with no clear inherited etiology or carcinogen exposure and are the tumors strongly linked to dietary exposures (4, 6, 8, 5). In the NWD1 model, multiple dietary components in control purified diet AIN76A are adjusted to produce mouse exposure to each nutrient at its level linked to elevated incidence of colorectal cancer in high-risk developed countries (9, 10). Fat is at 25%, levels characterizing human high risk, in contrast to 60% in commonly used high-fat mouse diets which cause metabolic alterations differing from those caused by levels better reflecting human exposure (11, 12). NWD1 also is lower in calcium, vitamin D, methyl donors, and fiber, at levels that elevate risk for human colorectal cancer (13). As a result, NWD1 accelerates and amplifies tumor phenotype in multiple genetically initiated models (14–18), and in wild-type (WT) mice, reproducibly causes sporadic small and large intestinal tumors reflecting the etiology, incidence, frequency and lag with developmental age of sporadic human colorectal cancer (19). In the NWD1 model, stem cell reprogramming is caused by interactive nutrient effects (6), important because mouse diet studies are less common than studies of individual nutrients, but it is overall dietary pattern that influences cell physiology, disease risk, and progression (20–22). This is not surprising because even strongly penetrant oncogenes and tumor suppressor genes always depend on underlying cell programming, with very different effects in different tissues.
NWD1 fed mice are not obese, but exhibit reduced energy expenditure without altered food consumption or physical activity, causing glucose intolerance, and increased proinflammatory cytokines (23). Protumorigenic Wnt signaling is elevated throughout the small and large intestine (24–26), and the proliferative compartment expands (27, 28) as in humans at elevated tumor risk (29–32). In NWD1 fed mice, crypt base Lgr5hi ISCs are repressed in supporting mucosal maintenance, show reduced mutation accumulation, and reduced efficiency in producing tumors on introduction of an initiating Apc mutation, but these increase for Bmi1+ cells (4, 6, 8, 5). Our reports anticipate that both Lgr5-positive and -negative stem cells support human colon tumor growth, with balance determined by environmental factors (33), emphasizing significance of NWD1 in determining which cells function as stem cells.
Here single-cell RNA sequencing (scRNA-seq), single-cell Assay for Transposase-Accessible Chromatin using sequencing (scATAC-seq), functional genomics and imaging, with rigorous bioinformatic and statistical approaches, deconvolve complex NWD1 cellular and molecular mucosal remodeling that increases risk for the sporadic tumors. Mechanistically, NWD1 epigenetic downregulation of Ppargc1a in Lgr5hi ISCs altered mitochondrial structure and function that is rapid, which compromised function of these ISCs and developmental maturation of their daughter cells as they proceed along the crypt-villus axis. A cascade of lineage remodeling includes recruitment of alternate Bmi1+, Ascl2hi cells to maintain the mucosa, but with key changes resulting in protumorigenic inflammation. The NWD1 remodeling parallels multiple pathogenic mechanisms in human inflammatory bowel disease (IBD; refs. 34–36), also protumorigenic. Therefore, while oncogenic mutations provide a stem cell with a competitive advantage for clonal expansion (1), the competition is on a playing field sculpted and continually remodeled by the nutritional environment, influencing outcome.
Materials and Methods
Organisms/strains
Mice were obtained from the Jackson Laboratories, Bar Harbor Maine, backcrossed as necessary, and maintained on a C57Bl/6J background (RRID:IMSR_JAX:000664). These are Lgr5tm1(cre/ERT2)Cle/J (Lgr5cre:ER-EGFP) Bmi1tm1(cre/ERT)Mrc/J (Bmicre:ER); Ppargc1a tm2.1Brsp/ (Ppargc1aloxp/loxp); B6.CgGt(ROSA)26Sortm14. Mice were provided food and water ad libitum in a barrier facility at the Albert Einstein College of Medicine (Bronx, NY). For breeding, strains were fed a chow diet (PicoLab 5058, Fisher Feed). Appropriate genotypes were randomized to purified diets (AIN76A, D10011; NWD1, D16378C; Research Diets Inc). Mice of both genders were used (a total of 25 male and 20 female mice). Hydroxy-Tamoxifen (TAM) was a single injection of 1 mg in 100 μL corn oil (Sigma, T5648). Experiments were approved by the Albert Einstein Institutional Animal Care and Use Committee. All authors had access to the study data.
Isolation of intestinal epithelial cells
On sacrifice, excised intestines were opened longitudinally, rinsed with cold saline, crypts isolated, single-cell suspensions prepared, and depending on the experiment, stained with EpCAM-FITC (Miltenyi Biotec, Clone caa7–9G8, catalog no. 130-123-674), and CD45-PerCP (Miltenyi Biotec, Clone 30F11, catalog no. 130-123-879), then Epcam+, CD45-negative cells epithelial cells collected by FACs on a MoFlo instrument as described previously (6).
Bulk RNA-seq analysis
Construction of bulk RNA-seq libraries, sequencing, and data analysis were as described previously (6).
scRNA-seq
scRNA-seq libraries were constructed by the Albert Einstein Genomics Core using the 3′ kit protocols of 10X Genomics with approximately 10,000 single FACS collected cells from each mouse loaded onto a Chromium Chip B microfluidic apparatus. Library quality was verified by size analysis (BioAnalyzer; Agilent) and after passing quality control assays, multiple libraries combined and pair-end sequenced by HiSeq PE150 or NovaSeq PE150 with a readout length of 150 bases (Novogene), and the data assigned to individual cells using the sample indexes.
For sequence alignment and subsequent analysis, output Illumina base call files were converted to FASTQ files (bcl2fastq), aligned to the mouse mm10 genome v1.2.0 and converted to count matrices (Cell Ranger software v3.0.2). Force-cell parameter was used, with 5,000–8,000 individual cells identified for each sample, and unique molecular identifiers used to remove PCR duplicates. Quality control and downstream analyses were in R v4.0.2, using Seurat package v3.2.2 (37). To discard doublets or dead cells, cells with gene counts <200 or >5,000, or a mitochondrial gene ratio >20%, were filtered out. Cells from different samples were integrated using Seurat FindIntegrationAnchors and IntegrateData functions and clusters identified from each integrated dataset using Seurat FindClusters. This calculates k-nearest neighbors by principal component analysis, constructs a shared nearest neighbor graph, then optimizes a modularity function (Louvain algorithm) to determine cell clusters. Cell clusters were identified as cell intestinal cell types using established cell-type markers and cluster identification in Seurat FindMarkers function. Differential gene expression compared samples among experimental groups: initial criteria were an expression change of ≥1.5-fold with associated adjusted P value of <0.01 in group comparison (Seurat FindMarkers function). Pathway analysis of differentially expressed genes used clusterProfiler (RRID:SCR_016884; R package v3.16.1) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (RRID:SCR_012773; R package v3.2.4); gene set enrichment analysis (GSEA) used the fgsea R package v1.14.0, the MSigDB (v5.2) hallmark gene set and the KEGG pathway database (RRID:SCR_012773). Pathways at P <0.05 were considered statistically significant. Trajectory analysis used Monocle3 R package v0.2.3.3 (38) with cluster information and cell-type identifications migrated from Seurat, the integrated dataset divided among experimental conditions, and trajectories generated for each experimental group.
RNA-seq data have been deposited in the NCBI Gene Expression Omnibus database (GEO; RRID:SCR_005012): bulk RNA-seq (Fig. 1) - GSE186811; scRNA-seq (Fig. 2, Ppargc1a inactivation) - GSE188339; scRNA-seq (Fig. 3, rapid dietary crossover) - GSE188577; scRNA-seq for Bmi1+ cells (Figs. 4 and 5) - GSE188338.
Figure 1.

Dietary impact on ISCs: A,Lgr5EGFP.cre:ER mice fed AIN76A or NWD1 for 3 or 12 months from weaning, or NWD1 for 3 months then switched to AIN76A for 9 months (cross-over); B–D,Ppargc1a expression by bulk RNA-seq of Lgr5hi cells of two different mouse cohorts fed either NWD1 or AIN76A for 3 months from weaning (B and D), or for NWD1 or AIN76 for 12 months compared with NWD1 for 3 months and then switched to AIN76A for an additional 9 months (C - Arm 3, A). N-3 mice for each group for each cohort. E and F, Pgc1a IHC and quantitation in crypts of mice fed AIN76A or NWD1 for 3 months. N = 3 mice for each group. G,Ppargc1a expression in Lgr5hi and Bmi1+ cells from bulk RNA-seq analysis of mice fed different diets for 3 months. N = 3 mice for each group. H, Examples of mitochondria in WT mice fed AIN76A or NWD1, or mice fed AIN76A and Ppargc1a homozygously inactivated in Lgr5 cells (800X – yellow arrows indicate examples of cristae). I, Quantitation of mitochondrial cristae density in CBC, Paneth, and villus cells, evaluated at 5,000X magnification. N = 3 mice for each group. J, FACs analysis of Lgr5hi cell number. N = 7 mice for each diet group. K, Mitotracker staining of mice fed AIN76A or NWD1 for 3 months. (*, P < 0.05; **, 0.01; ***, 0.001). N = 7 mice for each group. B, C, D, G, and J, Mean ± SD. F and I, Box and whiskers plot. K, Median ± quartiles.
Figure 2.

Genetic inactivation of Ppargc1a in Lgr5 cells: A, Suppressed lineage tracing from Lgr5hi cells by homozygous inactivation of Ppargc1a in Lgr5cre:er-GFP, Ppargc1aF/+ or −/−, Rosa26tom mice fed AIN76A diet, 3 days after a single TAM injection. B, Quantification of lineage tracing. N = 2 mice for each genetic group. C, Cluster map and cell types (nine scRNA-seq libraries). D, Epithelial cell distribution in clusters/lineages in WT mice or with heterozygous or homozygous Ppargc1a inactivation targeted to Lgr5hi cells; shown are stem cells, Replicating cells (R1, R2), Dividing cells (Div), multiple enterocyte populations (EC), Goblet, Enteroendocrine (EE1, EE2), Paneth and Tuft cells and a minor unknown population that could not be aligned with markers (UK). E–G, Trajectory analysis from scRNA-seq of total intestinal epithelial cells from WT mice or 3 days after heterozygous or homozygous Ppargc1a inactivation: yellow arrow, Stem cell cluster; abbreviations as in C and D. H and I, Cell type distribution at branch point “a” and “b”, respectively (red arrows in E–G) of WT, het and hom inactivation of Ppragc1a and GSEA of pathways of Ppargc1a−/− compared with WT mice at those branch points (numbers are P values). For C–I,N = 3 mice for each genetic group.
Figure 3.

Rapid reprogramming of cells by dietary shift: A, Mice fed AIN76A for 3 months (Arm1), switched to NWD1 for 4 days (Arm 2), or then switched back to AIN76A for 4 days (Arm 3), total Epcam+, CD45-negative epithelial cells FACs isolated and analyzed by scRNA-seq. B, Pathways significantly altered by rapid dietary shifts and their negative log P value for significance of pathway change. C, Magnitude of change of each pathway under the different dietary conditions; D, TCA cycle genes repressed by switching mice from AIN76A to NWD1 for 4 days and then elevated when mice switched back to AIN76A control diet for 4 days. E, Altered expression of each gene in the Oxphos pathway by 4-day shift from AIN76A to NWD1, and response of each to subsequent switch back to AIN76A for 4 days. For A–E,N = 3 mice for each arm in A.
Figure 4.

scRNA-seq of Bmi1+ intestinal epithelial cells in response to diet: A, Rosa26tom marked Epcam+, CD45-negative epithelial cells FACs isolated from Bm1cre:er, Rosa26tom mice fed AIN76A or NWD1 for 3 months, then sacrificed at 3 or 66–70 days after TAM injection to activate the Rosatom marker (shorter, longer term, respectively) and analyzed by scRNA-seq (N = 2 for each group). B, Cell trajectory analysis as a function of diet and time after marking by TAM injection: Blue arrows, Ascl2 expressing cells among stem and progenitor cells; or yellow arrows, in goblet and enteroendocrine cells. C and D, Expression of Ascl2 per cell in Stem1, 2, Replicating and Dividing cell clusters: red arrow in D for Stem 2 cells is a population that expressed Ascl2 at a higher level. *** This was highly significant by a likelihood ratio test (P = 0.01), by a negative binomial regression-negative binomial mixed-effect model, with regression and fit using R functions glm.nb and glmer.nb, respectively (84), assuming each cell as independent, and number of Ascl2 reads for each cell as response. Furthermore, Ascl2 expression for NWD1-LT differed significantly from the other three groups (P = 0.002) by post hoc analysis by a similar negative binomial, and the potential that the difference between NWD1-LT and the other groups was random was P = 0.11, tested by a negative binomial mixed-effect model with mice per dietary group treated as random effects, indicating the alternate hypothesis that effects were random is false. E,Ascl2 and Bmi1 expression by ISH in mice fed diets for 3 months: white dotted lines demark the crypt base. For each of the four diet-timepoint groups analyzed in A–D,N = 2 mice per group.
Figure 5.

A, Trajectory analysis of Bmi1+ marked cells at 3 or 66–70 days after Tam activation of the Bmi1 marker, annotated with individual cell types: red arrows/numbers denote branch points analyzed. Cell types identified are stem cells, Replicating cells (R1, R2), Dividing cells (Div), multiple enterocyte populations (EC), Goblet, Enteroendocrine cells (EE1, EE2), Tuft and Paneth cells. B and C, Cell type distribution at branch points “1” and “2” (red arrows in A), D,Ascl2 expression per cell at branch point 2. For each of the four diet-timepoint groups analyzed in A–D,N = 2 mice per group.
In situ analyses
Sections of mouse intestinal Swiss rolls fixed in 10% formalin were dewaxed and rehydrated with reagents from Advanced Cell Diagnostics. Endogenous peroxidases were quenched and heat epitope retrieval done utilizing the RNAscope Multiplex Fluorescent Reagent Kit v2 (catalog no. 323100) and the RNAscope Multiplex Fluorescent Assay v2 protocol followed. RNAscope target probes for Bmi1 and Ascl2 (catalog nos. 466021, 412211, respectively) were hybridized to sections and amplified using the HybEZ Hybridization System (catalog no. 310010). Secondary TSA fluorophores from Akoya Biosciences were Opal 620 (channel 1, catalog no. FP1495001KT) and Opal 570 (channel 2, FP1488001KT), sections counterstained with DAPI and visualized using a Leica SP8 confocal microscope (20X magnification).
scATAC-seq
Total Epcam+, CD45-negative epithelial cells from the small intestine of mice were FACs isolated as for scRNA-seq. Nuclei were isolated following the 10X Genomics protocol, scATAC-seq libraries constructed using the 10X Genomics single-cell ATAC-seq Chromium reagents and protocol, and sequenced as for scRNA-seq. The scATAC-seq data can be accessed at GSE228006.
IHC
Swiss roll sections were dewaxed, rehydrated, endogenous peroxidases blocked (3% hydrogen peroxide in methanol), and heat induced epitope retrieval done with 10 mmol/L sodium citrate buffer (pH 6.0) for all antibodies. Tissues were incubated overnight at 4°C with primary CD3 antibody (Novus catalog no. NB600-1441, RRID:AB780192, 1:10 dilution). Protein block and secondary antibody for CD3 employed ImmPRESS- Horse Anti-Rabbit IgG Polymer Kit-Alkaline Phosphatase (Vector Laboratories, catalog no. MP-5401). F4/80 antibody (1:200, Cell Signaling Technology, catalog no. 70076) was incubated on tissue overnight at 4°C. Secondary antibody was SignalStain Boost IHC Detection Reagent (horseradish peroxidase, Rabbit, Cell Signaling Technology, catalog no. 8114). For Pgc1a staining, tissues were incubated overnight at 4°C with rabbit anti-Pgc1a Ab (diluted 1:500, NBP1-04676, Novusbio) followed by biotinylated goat anti-rabbit IgG (1:100, BA-1000; Vector Lab) for 30 minutes. Visualization used the Vectastain Elite ABC Kit (Vector Lab) and DAB Quanto kit (Thermo Fisher Scientific). Counterstaining was with hematoxylin and visualization by bright field microscopy, or a CY5 filter for CD3.
Electron microscopy
Samples were fixed with 2.5% glutaraldehyde, 2% paraformaldehyde in 0.1 mol/L sodium cacodylate buffer, post-fixed with 1% osmium tetroxide followed by 2% uranyl acetate, dehydrated through a graded ethanol series and embedded in LX112 resin (LADD Research Industries). Ultrathin sections were cut on a Leica Ultracut UC7, stained with uranyl acetate followed by lead citrate and viewed on a JEOL 1400 Plus transmission electron microscope at 120 kV. Images were captured at 800X magnification, and at 5,000X for quantitative measurements of mitochondrial size and cristae density.
Mitochondrial membrane potential
Crypts were dispersed into single-cell suspensions and incubated with MitoTracker Red FM (Invitrogen, catalog no. M22425, 25 nmol/L) for 20 minutes at 37°C. Zombie NIR (Invitrogen, catalog no. L10119) was then added to mark dead cells. Flow cytometry used a Cytek Aurora Instrument (Cytek Biosciences) with analysis using Flowjo 10 software (RRID:SCR_008520; Treestar Inc.).
Lineage tracing of Ppargc1a knockout mice
Induction of creERT2 activity was by a 100 μL intraperitoneal injection of 1 mg freshly prepared TAM in sterile corn oil. Mice were sacrificed 3 days later, intestinal tissues fixed with 4% paraformaldehyde for 2 hours, hydrated with 15% sucrose for 3 hours, and 30% sucrose overnight. Fixed tissues were embedded in optimal cutting temperature compound (Sakura), frozen on dry ice, cryostat sectioned, and stained with DAPI for identification of nuclei before imaging.
Image analysis
Quantification of anti-Pgc1a Ab staining was at 60× magnification. A threshold was applied for all slides, and the area above threshold calculated and divided by total area. The crypt-villus axis was divided by 20 μm increments from the crypt base and measured up to 80 μm. For analysis of mitochondrial morphometry, crypt base columnar cells, Paneth cells, and villus cells imaged at 5,000× magnification were selected. Cristae density was calculated by the sum of individual cristae divided by mitochondrial size. Mitochondrial size (μm2) and cristae area (μm2) were manually traced using Fiji (RRID:SCR_002285). For lineage tracing quantification, the crypt-villus axis was manually traced and the threshold automatically adjusted for each crypt. Red fluorescent positive area (μm2) was measured and divided by total crypt-villus area (μm2).
Data availability
All bulk and scRNA-seq data and scATAC-seq data are available in the NCBI GEO database, accession numbers GSE186811, GSE188339, GSE188577, GSE188338, and GSE228006.
Results
NWD1 impact on Lgr5hi ISCs
Bulk RNA-seq analyzed Lgr5hi cells from Lgr5EGFP-creER mice fed NWD1 or AIN76A control diet for 3 or 12 months from weaning, or 3 months NWD1 then switched to AIN76A until 12 months (Fig. 1A). NWD1 increased expression of 164 genes, and decreased 63 at 3 months (≥50% and P ≤ 0.01; Supplementary Fig. S1A), expanded >3-fold at 12 months to 551 and 229 genes, but reverted to more limited perturbation when 3 months NWD1 fed mice were switched to control AIN76A until 12 months (Supplementary Fig. S1A). By GSEA, NWD1 significantly upregulated and downregulated 25 and 30 pathways, respectively, in Lgr5hi cells at 3 months, most persisting to 12 months, and reversed by shift to control AIN76A diet (Supplementary Figs. S1B and S2). Thus, NWD1 had a major effect on transcriptional profile of Lgr5hi cells.
The Oxphos and tricarboxylic acid (TCA) cycle pathways were downregulated by NWD1 at 3 months, persisted until 12 months, and reversed after switching to control diet at 3 months (Supplementary Figs. S1B and S2). Oxphos is necessary for intestinal Lgr5hi cells, hematopoietic, and embryonic cells to function as stem cells (39–43). NWD1 downregulated 58 of 132 genes in the Oxphos pathway at 3 months, confirmed in an independent mouse cohort also fed NWD1 or AIN76A for 3 months (6). Mean expression of these genes decreased by 14% and 11% in the datasets (Supplementary Fig. S3A), but the overall altered profile was highly significant (P = 10−26, Fisher exact test in comparison with the ∼8,000 genes expressed). The downregulated gene expression and pathway profiles persisted at 12 months (Supplementary Figs. S1B, S2, and S3B), encompassing multiple genes encoding subunits of each mitochondrial electron transport chain complex (Supplementary Fig. S3C). Greatest downregulation for both cohorts were Cox6a2 and Cox7a1 of cytochrome oxidase, the terminal respiratory chain enzyme, and Sdhb, and Sdhd of complex II, succinic dehydrogenase (Supplementary Fig. S3A), downregulated in colorectal cancer and other cancers causing succinate accumulation and tumor development (44, 45). In shift to control diet (Arm 3), 52 (82%) of downregulated Oxphos genes increased toward control levels (Supplementary Fig. S3B; P = 10−18), reflecting GSEA reversal (Supplementary Figs. S1B and S2).
Ppargc1a encodes Pgc1a, a master regulator of metabolism and mitochondrial biogenesis (46, 47) and Ppargc1a overexpression elevates mitochondrial biogenesis and function of colonic epithelial cells, altering their fate (48). NWD1 for 3 months reduced Lgr5hi ISC Ppargc1a expression by ≥60% in both mouse cohorts (Fig. 1B and D, P = 0.008 and 0.030), similarly decreased after 12 months of NWD1 feeding (P = 0.06), and switch to control AIN76A produced levels indistinguishable from mice continuously fed AIN76A (Fig. 1C). Pgc1a, the encoded protein, was highly expressed in crypt cells of control mice, but much lower in crypts of NWD1 mice, paralleling decreased expression of the gene in Lgr5hi ISCs (Fig. 1E). The decrease was significant in NWD1 mouse crypts (P < 0.01), but there was much lower expression, and no difference with diet, at any position above 20 μm from the crypt base (Fig. 1F), confirmed by decreased Ppargc1a expression in Lgr5hi cells, but not in Bmi1+ cells most abundant in cells further up the villi (Fig. 1G). Cristae density in crypt base epithelial cells decreased in NWD1 mice (P < 0.01), with no change in Paneth cells at the base, or in cells higher up (Fig. 1H and I). GFPhi cells of Lgr5EGFP mice fed NWD1 decreased by 60% (Fig. 1J, P = 0.03) and their retention of mitotracker dye decreased (Fig. 1K, P < 0.0001), an index of mitochondrial function. Therefore, NWD1 suppressed Ppargc1a expression, the encoded Pgc1a protein, and expression of many genes encoding proteins of the electron transport chain, which altered mitochondrial structure and membrane potential in stem cells at the crypt base.
Mechanistic role of Ppargc1a downregulation
Ppargc1a was inactivated heterozygously or homozygously in Lgr5hi cells of mice fed control diet (Lgr5EGFP-cre:ER, Ppargc1aloxp/+ or loxp/loxp, Rosa26tom mice) by a single TAM injection 3 days before sacrifice. FACs isolated intestinal epithelial cells (Epcam+, CD45-negative) were analyzed by scRNA-seq.
Sequence analysis of the single-cell libraries showed transcripts proceeding into exons 3 to 5, the region targeted for deletion, were reduced by 80% (P = 0.016) in Lgr5hi cells of the homozygous knockout mice (Supplementary Fig. S4A). In contrast, there was a nonsignificant reduction of approximately half that (30%) in Lgr5hi cells of the heterozygotes, and no differences in the total cell population of WT, homozygous or heterozygous mice (Supplementary Fig. S4A, and figure legend). These data confirm the expected inactivation of Ppargc1a in relation to both genotype and Lgr5hi cell targeting. Homozygous inactivation phenocopied effects of NWD1: lineage tracing from Lgr5-positive cells was suppressed (Fig. 2A and B, P = 0.02), and crypt base columnar (CBC) cell cristae density decreased (P < 0.05) with no alteration in Paneth or in villus cells (Fig. 1H and I).
scRNA-seq identified 20 cell clusters that aligned with intestinal cell types and lineages and a minor cluster not aligned (UK; <1% of cells in any single-cell library; Fig. 2C and D). Cell representation across clusters was similar among genotypes, but homozygous inactivation significantly increased early enterocyte populations, EC1 and EC2 (Fig. 2D). By developmental trajectory analysis (Monocle), EC1 was proximal to dividing cells and then EC2 emerged, with other EC cells and lineages appearing further downstream (Fig. 2E–G), consistent with markers identifying cell position along the crypt-villus axis (Supplementary Fig. S4B; ref. 49). Therefore, increased EC1 and EC2 cell number suggests that Ppargc1a inactivation in Lgr5hi cells reprogrammed dynamics of cell maturation as cells undergo differentiation.
Progressive maturation of progenitors begins at the stem cell cluster, characterized by branch points. Mitochondrial function suppresses ability of Lgr5hi cells to function as stem cells (39–43), determines the balance between stem cell self-renewal or differentiation (50), and mitochondrial fusion and hence activation is necessary for ISC progeny to differentiate (51). Therefore, cell-type representation was determined at early developmental branch points. At branch point “a,” developmental trajectory from the Replicating 1 cell compartment (R1) bifurcates to secretory goblet or R2 cells (red arrow, Fig. 2E–G). For both WT and Ppargc1a het mice, R1 cells were the major cell type at branch point “a” with few or no stem cells (Fig. 2H) indicating transition from stem to R1. However, with homozygous Ppargc1a inactivation, 100% of cells at “a” still identified as stem cells, a highly significant repression of reprogramming with maturation from stem to R1 cells (Fig. 2H; P = 10−5, by a weighted linear regression based on cell number at each cluster for each mouse). This altered cell distribution at branch “a” with homozygous inactivation was characterized by significant suppression of Oxphos, TCA cycle, cell cycle, and glycolysis pathways (Fig. 2H), reflecting the altered mitochondrial structure and function (Fig. 1H and I).
With further cell developmental progression, branch point “b” was transition from R1 to R2 cells that differentiate into enteroendocrine and Tuft cells (Fig. 2E–G). At “b,” most cells in WT or het mice had progressed to the R2 compartment, but with homozygous Ppargc1a inactivation, 90% remained in R1 (P = 0.016), with significant repression of the Oxphos pathway persisting (Fig. 2I). Therefore, Ppargc1a inactivation altered stem cell maturation to R1 cells, and repressed transition out of R1 with downregulation of the Oxphos pathway, and a parallel enrichment in the glycolytic pathway.
Rapidity and reversibility of dietary alterations
scRNA-seq analyzed intestinal epithelial cells of mice fed AIN76A for 3 months (Fig. 3A, Arm 1), switched to NWD1 for 4 days (Arm 2), or 4dNWD1 mice switched back again to AIN76A for 4 days (Arm 3). Stem and progenitor cells, and all lineages were identified, with no significant changes in cell distribution for clusters in these short time periods (Supplementary Fig. S5A and S5B), and cell trajectory confirmed by expression of genes that characterize different positions (Supplementary Fig. S4C). For the stem cell cluster, 4 days of NWD1 altered 30 pathways with high significance (P = 10−3 to 10−43) including repression (negative Z-score) of the TCA cycle and Oxphos (P = 10−6 and 10−43, respectively; Fig. 3B and C). A total of 19 of the 30 pathways reverted to control values in 4dNWD1 mice switched back to AIN76A for 4 days, including the TCA cycle pathway, downregulated by 4 days of NWD1 (P = 10−6), then upregulated when switched back to AIN76A ((Fig. 3B and C). Expression of eight genes encoding TCA cycle enzymes were downregulated by 4dNWD1 and reversed by 4 days switch-back to AIN76A (Fig. 3D), accounting for the significant GSEA for dietary shifts (Fig. 3B and C). Four days of NWD1 decreased expression of 51 Oxphos pathway genes (Fig. 3E), consistent with the significant negative GSEA (P = 10−43; Fig. 3B and C), but only 15 reversed on AIN76A switch back (Fig. 3E), reflecting the absence of reversal by GSEA (Fig. 3C). Therefore, by 4 days after shift to NWD1, stem cells at the crypt base underwent significant mitochondrial reprogramming, in large part, but not entirely, reversed after 4 days back to control diet.
Using stringent criteria (P ≤ 0.001), pathway changes caused by the 4-day switch to NWD1 were compared with changes at 3 days after homozygous Ppargc1a inactivation targeted to Lgr5hi cells (Supplementary Fig. S5C). Focus was on the Stem 1 and Stem 2 clusters, since the targeted Lgr5hi cells are in these clusters at early timepoints. For Stem 1, 17 of 25 pathways (76%) altered by NWD1 were also altered by Ppargc1a genetic inactivation, and for Stem 2, 15 of 24 (64%; Supplementary Fig. S5C). For pathways altered by both diet and genetic inactivation, extent of increase or decrease was similar for both datasets in both Stem 1 and 2 (Supplementary Fig. S5C), with considerable overlap for the two clusters. Mitochondrial dysfunction was the most highly enriched for both Stem 1 and 2, also reflected in Oxphos as the most negatively enriched pathway for both interventions in both stem cell compartments (green arrows, Supplementary Fig. S5C). The next most enriched pathway for both clusters was Granzyme A signaling that drives cell death and induces inflammation (52), potentially linked to commonly enriched pathways for immunogenic cell death signaling (red arrows), The third most commonly enriched pathway was Sirtuin signaling (orange arrows), linked to histone modification and growth control in cancers (53), and regulated at multiple levels by miRNAs (54), also a commonly altered pathway (black arrows). Finally, Bag2 signaling, a mediator of Myc, Erk, p53, and other signaling pathways, and linked to apoptosis and cell survival in cancer (55), was also commonly enriched (blue arrows).
Subsets of pathways are also altered in Stem1 or 2 by NWD1 but not altered at this stringency by Ppargc1a inactivation (Supplementary Fig. S5C). Therefore, there was extensive overlap for the most dominant effects, with the combination of nutrient changes in NWD1 also having a broader impact on both stem cell compartments. For this subset in Stem 2, the cell cycle:G1–S checkpoint pathway is altered only by NWD1, of potential significance since this compartment harbors the Bmi1+, Ascl2 cells mobilized as alternate stem cells by NWD1 (below, Fig. 4).
A cascade of cell remodeling by diet
NWD1 expands the mouse intestinal proliferative cell compartment (27, 28), as in humans at elevated colorectal cancer risk (29–32). Consistent with this, NWD1 induced lineage tracing from Bmi1+ cells above the crypt base that persists for as long as mice are fed NWD1 (6, 8), and targeting Apc inactivation to Bmi1+ cells increased tumor development when mice were fed NWD1, demonstrating that plasticity for alternate cells to function as stem cells takes place in cell adaptation to their nutritional environment (4–6).
scRNA-seq data were generated for Bmi1+ cells and their marked epithelial cell progeny isolated from Bmi1cre-ERT2, Rosa26tom mice fed diets for 3 months from weaning (Tom+, Epcam+, CD45-negative cells). The Bmi1 marker was activated at 3 months by a single TAM injection, and each mouse continued on its diet for 3 or 66–70 days before sacrifice to analyze cells soon after marking, or in cells accumulated over 2 months (Fig. 4A; Supplementary Fig. S6A and S6B). There was similar cell distribution for the stem, replicating, dividing cells and all lineages regardless of diet (Supplementary Fig. S6B and S6C), and developmental trajectory was confirmed by expression of markers that identify cell position along the crypt-luminal axis (ref. 49; Supplementary Fig. S4D). Therefore, although NWD1 repressed Lgr5hi ISC functions, the mucosa was maintained.
The stem cell transcription factor Ascl2 drives dedifferentiation of Bmi1+ cells to replace damaged Lgr5hi cells (1, 2). Regardless of diet or time after Bmi1+ cell marking, Ascl2 was expressed in the Bmi1+ Stem1 and 2, Replicating and Dividing cells of the mice (Fig. 4B, blue arrows, quantified in Supplementary Fig. S6D). A small number of Tuft and goblet cells also expressed Ascl2 where goblet and enteroendocrine lineages diverged (Fig. 4B, orange arrows; Supplementary Fig. S6D).
Mean Ascl2 expression per cell among the four progenitor cell compartments was highest in Stem2 (Fig. 4C). Ascl2 expression across individual cells was low per cell for Stem 1, Replicating and Dividing cells, with no difference by diet or length of time after cell marking (Fig. 4D). In contrast, for Stem2, expression per cell was heterogeneous, and a subset of Stem2 cells expressed elevated Ascl2 that accumulated longer term [red arrow, Fig. 4D: highly significant by linked statistical tests (Fig. 4 legend)]. ISH identified Bmi+, Ascl2hi expressing cells at the crypt base in AIN76A mice, but in NWD1 mice, elevated Ascl2 was in Bmi1+ cells above the crypt base (Fig. 4E), as in mice after acute Lgr5hi cell damage (1, 2), although in NWD1 mice, these cells were further above the crypt base (Discussion).
Mucosal remodeling by mobilized Bmi1+ cells
There were two early branch points among progenitor cell populations in the developmental trajectory of Bmi1+ derived cells (Fig. 5A, red arrows 1 and 2). At branch point “1”, Stem1 cells give rise to secretory lineages (enteroendocrine, goblet, and Paneth cells). At this branch point, there were no significant changes in cell type representation by diet, shorter or longer after Bmi1+ cell marking (Fig. 5B). In contrast, at branch point 2 leading to absorptive cell differentiation, in Bmi1+ cells shorter term after marking (Fig. 5C, top), cells from AIN76A control mice were equally divided between R and Div cells, but in NWD1 mice, all cells remained in R with none in the Div cell compartment (P = 0.001), with identical patterns from independent mice. This became more pronounced in Bmi1+ cells that accumulated longer term (Fig. 5C, bottom): nearly all cells at branch point 2 of AIN76A mice are Div cells, but for NWD1 mice, 100% of the cells retained a transcriptional profile of R cells (P = 0.001, independent mouse replicates identical). Notably, R cluster cells at branch point 2 longer term after marking express much higher Ascl2 levels (Fig. 5D, P = 10−8) similar to the Bmi1+, Ascl2hi cells that express higher Ascl2 in Bmi1+ cells that accumulate in Stem2 of NWD1 mice (Fig. 4D) and the cells above the crypt base in NWD1 mice (Fig. 4E). This dietary suppression of cell developmental progression was also reported in mice in which genetic manipulation of mitochondrial structure downregulated stem cell function (51) and in human patients with IBD (36), and was also identified by us in aging mice, reversed by geroprotectors (56).
NWD1 cell reprogramming is proinflammatory and adaptive
Although Bmi1+ marked cells give rise to all lineages at equivalent levels in NWD1 and AIN76A mice, NWD1 altered gene expression profiles in all Bmi1+ cell populations both long- and short-term after marking (≥50% and P ≤ 0.01). Transcriptome alteration was most pronounced in enterocytes, greatest in the most mature EC7 enterocytes accumulating longer term (Fig. 6A).
Figure 6.

NWD1 reprogramming and adaptation of cells: A, number of differentially expressed genes (>50% and P = 0.01) in Bmi1 cell clusters of Supplementary Fig. S6B. B, Heatmap of genes differentially expressed by diet in EC7 cells and cell pathways enriched (GSEA) as a function of diet in Bmi1+ EC7 cells longer term after marking of Bmi1+ cells (statistical analysis in text). C, Fraction of cells in each cluster expressing CD74. D, CD74 expression per cell, in each cell cluster as a function of diet and time after marking of Bmi1+ cells. E, GSEA for each cell cluster for the fatty acid metabolism pathway in the rapid dietary cross-over experiment (Fig. 3A)—red bars, pathway change in mice fed AIN76A for 3 months, then switched to NWD1 for 4 days before sacrifice; blue bars, the mice then switched back to AIN76A for 4 more days. F, scATAC-seq data for Cyp4a10 in enterocytes. G, scRNA-seq data for Trpv6 Bmi1+ derived cells from AIN and NWD1 fed mice 3 days or 66–70 days after cells were marked (Fig. 4A). H, scATAC-seq data for Trpv6 in enterocytes for mice fed either AIN76A or NWD1 for 4 months from weaning. For each of the diet-time point groups analyzed in A–D, F, and H,N = 2 mice for each diet-time point group; for E,N = 3 mice per group.
The pathway most enriched in Bmi1+ marked EC7 cells was antigen processing and presentation (Fig. 6B; NES 1.75, P = 0.009). The CD74 gene characterizing this pathway is expressed in epithelial cells (57–66), mediating interaction of Lgr5hi ISCs and their immune environment (66). The fraction of cells expressing CD74 was highly enriched across all cell clusters derived from Bmi1+ cells that accumulated with time (Fig. 6C), paralleled by elevated expression per cell, with much higher expression in EC7 mature enterocytes compared with all other clusters (Fig. 6D, P = 10−25, by negative binomial regression analysis). CD74 interacts with proteins of the MHCII complex to process and present cell surface antigens (61), and MHCII complex genes also exhibited greatest upregulation in EC7 cells (Supplementary Fig. S7A). These expression patterns are consistent with CD74 and MHCII expression predominantly in cells in the upper third of human villi (67), suggesting a unique role in these differentiated cells.
Cell surface expression of CD74 characterizes dysplastic cells of the intestinal epithelium, and cells of human Crohn’s, Ulcerative, and Amebic colitis (63, 68, 69), chronic inflammation in each elevating colorectal cancer risk. NWD1 is also proinflammatory, elevating serum levels of inflammatory cytokines (23), and there was a 2- to 3-fold significant increase in cells expressing CD3, a pan T-cell marker, in 3-month NWD1 compared with AIN76A mice (Supplementary Fig. S7B–S7D).
The second most highly NWD1-enriched pathway in EC7 cells is “Intestinal Immune Network/IgA production” (Fig. 6B; NES = 1.50, P = 0.027). IgA is abundant in the intestinal and colonic mucosa, generating a SigA complex interacting with the proinflammatory myeloid cell FcαRI receptor. NWD1 also elevated F/480+ myeloid cells (P = 0.01; Supplementary Fig. S7E and S7F), also characterizing protumorigenic IBD (70).
Multiple metabolic pathways were also altered in EC7 cells, including Ppar signaling, cholesterol metabolism, and fat digestion/absorption (Fig. 6B), adaptive responses to increased fat (25%) in NWD1. In the independent scRNA-seq data from rapid dietary cross-over (Fig. 3A), fatty acid metabolism was elevated in all cell clusters by the 4-day switch to NWD1 from AIN76A, and except for Goblet2 and EE2, reverted for the clusters within 4 days of switch back to AIN76A (Fig. 6E), consistent with rapid shift of metabolic pathways (Fig. 3B).
Another adaptive response to NWD1 was increased enterocytes expressing Trpv6 in Bmi1+ marked cells, especially longer term (Fig. 6G). Trpv6 encodes the enterocyte brush border channel mediating calcium uptake under low dietary calcium conditions (71). Therefore, it is likely elevated Trpv6 expression contributes to NWD1 mice maintaining serum calcium levels (23), despite lower calcium and vitamin D3 in NWD1.
scATAC-seq data of epithelial cells of replicate mice fed AIN76A control diet for 4 months showed no areas of open chromatin structure for either Cp4a10 in enterocytes, a gene common to multiple pathways of fat metabolism, nor for the Trpv6 gene, but substantial accessibility of chromatin structure at and upstream of the promoter of each for replicate NWD1 fed mice (Fig. 6F and H), demonstrating classic epigenetic transcriptional activation of these genes underlie the adaptive dietary response.
scRNA-seq established that Stem 1 harbored the Lgr5hi ISCs at the crypt base, consistent with trajectory analysis identifying Stem1 at initiation of maturation of all cell types/lineages (Fig. 4B). The Signace “Reference Mapping” function (NY Genome Center) aligned 1 of the 13 scATAC-seq clusters with Stem 1 from the scRNA-seq data (Fig. 7A). The number of cells in this stem cell cluster identified by scATAC-seq decreased 50% in NWD1 mice compared with AIN76A (Fig. 7B, P = 0.009), independently confirming NWD1 epigenetic suppression of Lgr5hi cells. For Ppargc1a, downregulated in Lgr5hi cells by NWD1, the position and magnitude of accessible chromatin regions at the promotor (“P”) in the Stem 1 cluster is similar regardless of diet (Fig. 7C). However, in the boxed region of Ppargc1a, expanded in Fig. 7D, “a” and “b” are areas harboring open chromatin regions in AIN76A mice absent or substantially reduced in NWD1 mice, quantified and shown to be highly significant (Fig. 7E). These regions of reduced accessibility in NWD1 mice correspond to location of a strong enhancer in the gene that regulates gene expression in intestinal epithelial cells (Fig. 7C and D; Enhanceratlas 2.0). The scATAC-seq data confirm NWD1 epigenetic downregulation of Ppargc1a expression, but likely by a more complex mechanism than altering accessibility of sites at promoter.
Figure 7.

scATAC-seq data, stem cells: A, Clusters from the scATAC-seq data. B, Per cent Stem 1 cells in mice fed AIN76A or NWD1 based on the scATAC-seq data. C, scATAC-seq data for the Ppargc1a gene in the Stem 1 cluster. Box delineates region of diminished peaks in NWD1 compared with AIN76A fed mice, and P denotes the promoter region. D, Boxed region in C expanded, with “a” and “b” denoting areas within this region where peaks in AIN fed mice are substantially diminished in NWD1 fed mice. E, Quantification of reads in regions a and b relative to reads at the promotor for mice fed the different diets; statistical analysis by a Poisson regression on aggregated read counts over all cells per mouse, normalized by coverage per sample in the promoter region of Ppargc1a. E, Mean ± SD. For A–E,N = 2 mice for each dietary group.
Discussion
NWD1 adjusts multiple nutrient exposures to levels elevating risk for human sporadic colon tumors causing rapid, reversible and hence dynamic adaptation of stem cells and lineages through epigenetic mechanisms that also alter pathways establishing a protumorigenic environment. The data herein establish that increasing risk by NWD1 parallels pathogenic mechanisms of human inflammatory bowel disease and human tumorigenesis in multiple ways: (i) low-level chronic inflammation that is protumorigenic; (ii) NWD1 epigenetically reduced Ppargc1a expression in Lgr5hi ISCs and lower expression also represses Lgr5hi cell stem cell functions in IBD (72–74). Furthermore, downregulation of the encoded protein, Pgc1a, exacerbates human IBD (35), with both diet and IBD altering mitochondrial structure, metabolically reprogramming the cells. This establishes a link between Ppargc1a and mitochondrial function to IBD (discussed in refs. 73, 74), and Ppargc1a role in chronic protumorigenic inflammation; (iii) NWD1 or genetic inactivation of Ppargc1a in Lgr5hi cells retarded developmental maturation of cells migrating along the crypt-lumen axis, similar to retarded cell differentiation in human IBD along this axis determined by scRNA-seq (36); (iv) NWD1 altered expression profile most extensively in mature villus-tip enterocytes, with the most prominent change elevated CD74/MHCII pathways of antigen presentation and processing. Highest expression of these pathways is in upper villi of the human small intestine (67) and upper crypt colonocytes (34), and the elevation by NWD1 in the mouse parallels elevation of the pathways in human Crohn's, ulcerative, and amebic colitis (63, 68, 69), confirmed by single-cell analysis of human IBD tissue (34), and also in Helicobacter pylori–infected gastric tissue (60, 75), chronic inflammatory states increasing risk for human tumor development. In NWD1 mice, as in IBD, this is associated with increased proinflammatory cytokines (23), and CD3 and myeloid cell mucosal infiltration (Supplementary Fig. S7); (v) NWD1 induces high ectopic expression of Lyz and other Paneth cell markers in mouse colon (26), similar to Lyz elevation in human IBD (34). scRNA-seq documented expression of these markers in cells of the human mucosa that are not Paneth cells (76) and there is rapid alteration of Paneth cell markers in mice fed a 60% fat diet (7). (vi) NWD1 induced extensive remodeling of stem cells and lineages parallels cell reprogramming in both the involved and uninvolved mucosa in human IBD (34, 36), and in the earliest benign human tumors (77), demonstrating remodeling is initiated early in the mucosa at dietary risk; (vii) Establishment that the nutritional environment determines which stem cells function in mucosal maintenance anticipates the recent report that both Lgr5-positive and -negative stem cells support human tumor growth, with the balance determined by the mutational signature and environmental signals (33).
The substantially increased CD74/MHCII pathways by NWD1, similar to that in involved and uninvolved mucosa in human IBD, contrasts with the repression of these pathways in Lgr5 ISCs in mice fed a 60% fat diet, suggested to be protumorigenic by dampening immune surveillance (78). This raises a major issue: while modeling human dietary exposure in the mouse is complex and not readily achieved, better attention to this is similar to the importance of more closely recapitulating human genetic influences and in pharmacologic studies, compound exposure levels relevant to humans. Reported data establish this is fundamental. For example, commonly used 60% dietary fat to cause mouse obesity exceeds fat intake in even obese humans, causing metabolic alterations differing from those induced by dietary fat levels more common in humans (11, 12). Furthermore, common rodent diets, including high-fat diets, expose mice to levels of vitamin D exceeding the mean level in humans by 200%–300%, and well above even the 1% of the population at very highest levels (4, 6, 8, 5). This is fundamental since robust expression of the vitamin D receptor is a core component of the Lgr5hi ISC signature (79), and we established an essential role of vitamin D in Lgr5hi stem cell function, as in hair follicle and other stem cells (5, 8). As discussed in detail (4–6, 8), the much lower vitamin D in humans may contribute to the fact that it takes 50-fold longer for CBC stem cells in humans to reach crypt clonality than in the mouse (1, 80, 81). Thus, commonly used mouse diets establish a nutritional environment that strongly supports Lgr5hi ISC functions, but this environment necessary for robust Lgr5hi ISC function is simply not present in nearly all humans, potentially complicating how well data translate to human physiology and disease.
The significance of metabolic reprogramming of Lgr5hi stem cells by NWD1 is emphasized by necessity of Oxphos for stem cell functions of mouse Lgr5hi and Drosophila ISCs (39–41) and hematopoietic stem cells (42), for embryonic stem cell pluripotency (43), and a major role of mitochondrial function in determining whether stem cells self-renew or differentiate (50). The repression of Lgr5hi cells as ISCs recruits alternate Bmi1+, Ascl2hi cells above the crypt base for mucosal maintenance. Ascl2 is regulated by Wnt signaling (82), elevated by NWD1 throughout mouse intestinal villi and colonic cells (24–26). Ascl2 encodes a stem cell transcription factor essential for Bmi1+ cell dedifferentiation in response to Lgr5hi cell damage (1, 2). However, the roles of Bmi1+, Ascl2hi cells may differ in NWD1 versus damage induced plasticity. Acute damage purges Lgr5hi ISCs, Ascl2hi cells mobilizing to migrate into the crypt restoring the stem cell population. However, limited crypt space establishes competition among crypt Lgr5hi cells (1), and single-cell laser ablation of crypt base cells triggers reorganization of remaining stem cells, confirming importance of physical space in regulating stem cell dynamics (83). It is not clear whether crypt space is available in NWD1 fed mice. More important, cells migrating into the niche would still be repressed by NWD1, and reduced Lgr5hi cell lineage tracing from the crypt base, and Oxphos and TCA pathway repression, persist to at least 1 year of feeding NWD1 (ref. 8; Supplementary Figs. S1 and S2), with Bmi1+ cells above the crypt base lineage tracing for months in mice continuously fed NWD1 (6).
Tumors reflect growth advantage of transformed cells (1), but tumorigenesis is rare: lower-risk individuals develop no sporadic tumors, and those at higher risk only one to two tumors despite billions of mucosal cell divisions over decades, suggesting mechanisms establishing risk are subtle and complex. Therefore, it is fundamental that the nutritional environment, through metabolic reprogramming of stem cells, dynamically and continually sculpts the playing field on which ISC competition takes place, significantly influencing outcome.
Study limitations
Nutrients are interactive in programming stem cells and lineages, emphasizing the importance of dietary patterns as drivers of tumor risk and development rather than individual nutrients. However, human dietary complexity and variability would require understanding large matrices of interactions for a clearer understanding of the impact of dietary variables. Furthermore, exacerbation of IBD by downregulation of Pgc1a that alters mitochondrial function in stem cells establishes the link to protumorigenic chronic inflammation, but the mechanistic determinants in this cascade can include multiple roles of mitochondria and their interaction with other cellular compartments not yet clear. Finally, understanding coordination of extensive dietary induced changes of gene expression in stem cells and lineages requires investigation of higher-order interactions among genes and regulatory elements throughout the genome, which is now technically possible.
Supplementary Material
The western diet extensively reprograms Lgr5hi stem cells that expands with time and is reversible. This includes the TCA cycle and Oxphos pathways
The western diet elevates and suppresses multiple pathways by 3 months, most persisting to 12 months, and reversible by switching to control diet.
The western diet suppresses expression of a large number of genes encoding subunits of each of the 5 complexes of mitochondrial electron transport.
Targeted knockout of Ppargc1a in Lgr5hi cells was confirmed, and trajectory analyses of dietary and genetic alterations confirmed by marker expression.
Single cell RNAsequencing following 4 days of dietary switch, or then switch back and comparison to pathways altered in stem cells by Ppargc1a genetic inactivation.
Single cell analyses of cell progeny of the alternate stem cells mobilized by the western diet to maintain the mucosa.
The western diet causes chronic pro-tumorigenic inflammation.
S1. The western diet extensively reprograms Lgr5hi stem cells that expands with time and is reversible. This includes the TCA cycle and Oxphos pathways.S2. The western diet elevates and suppresses multiple pathways by 3 months, most persisting to 12 months, and reversible by switching to control diet.S3. The western diet suppresses expression of a large number of genes encoding subunits of each of the 5 complexes of mitochondrial electron transport.S4. Mouse genotyping identified presence of the wild-type, or heterozygous or homozygous conditionally targeted Ppargc1a allele.S5. Rapid dietary cross-over experiment of Fig 3.S6. scRNAseq analysis of Bmi1+ derived cells.S7. Inflammatory response to NWD1.
Acknowledgments
This study was supported in part by R01CA229216, R01CA214625, R01CA222358 to L. Augenlicht, and P30CA013330 from the NCI, NIH and P30 AG038072 and 5T32AG023475 to J. Choi from the NIA, NIH.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
This article is featured in Highlights of This Issue, p. 753
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Authors' Disclosures
J. Choi reports grants from National Institute on Aging during the conduct of the study. L. Augenlicht reports grants from NCI during the conduct of the study. No disclosures were reported by the other authors.
Authors' Contributions
J. Choi: Conceptualization, data curation, formal analysis, investigation, methodology, writing–original draft, writing–review and editing. X. Zhang: Data curation, software, formal analysis, investigation, methodology, writing–original draft, writing–review and editing. W. Li: Data curation, formal analysis, validation, investigation, methodology, writing–original draft, writing–review and editing. M. Houston: Formal analysis, investigation, writing–review and editing. K. Peregrina: Formal analysis, investigation, writing–review and editing. R. Dubin: Data curation, software, formal analysis, supervision, writing–review and editing. K. Ye: Data curation, software, formal analysis, investigation, methodology, writing–review and editing. L. Augenlicht: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, validation, investigation, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Shivdasani RA. Race to the bottom: darwinian competition in early intestinal tumorigenesis. Cell Stem Cell 2021;28:1340–2. [DOI] [PubMed] [Google Scholar]
- 2. Murata K, Jadhav U, Madha S, van Es J, Dean J, Cavazza A, et al. Ascl2-dependent cell dedifferentiation drives regeneration of ablated intestinal stem cells. Cell Stem Cell 2020;26:377–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Davies KJ. Adaptive homeostasis. Mol Aspects Med 2016;49:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li W, Peregrina K, Houston M, Augenlicht LH. Vitamin D and the nutritional environment in functions of intestinal stem cells: implications for tumorigenesis and prevention. J Steroid Biochem Mol Biol 2019;198:105556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Augenlicht LH. Environmental impact on intestinal stem cell functions in mucosal homeostasis and tumorigenesis. J Cell Biochem 2017;118:943–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li W, Zimmerman S, Peregrina K, Houston M, Mayoral J, Zhang J, et al. The nutritional environment determines which and how intestinal stem cells contribute to homeostasis and tumorigenesis. Carcinogenesis 2019;40:937–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Enriquez JR, McCauley HA, Zhang KX, Sanchez JG, Kalin GT, Lang RA, et al. A dietary change to a high-fat diet initiates a rapid adaptation of the intestine. Cell Rep 2022;41:111641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peregrina K, Houston M, Daroqui C, Dhima E, Sellers RS, Augenlicht LH. Vitamin D is a determinant of mouse intestinal Lgr5 stem cell function. Carcinogenesis 2015;36:25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Newmark HL. Nutrient density: an important and useful tool for laboratory animal studies. Carcinogenesis 1987;8:871–3. [DOI] [PubMed] [Google Scholar]
- 10. Newmark HL, Yang K, Kurihara N, Fan K, Augenlicht LH, Lipkin M. Western-style diet-induced colonic tumors and their modulation by calcium and vitamin D in C57Bl/6 mice: a preclinical model for human sporadic colon cancer. Carcinogenesis 2009;30:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Speakman JR. Use of high-fat diets to study rodent obesity as a model of human obesity. Int J Obes 2019;43:1491–2. [DOI] [PubMed] [Google Scholar]
- 12. Bastias-Perez M, Serra D, Herrero L. Dietary options for rodents in the study of obesity. Nutrients 2020;12:3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Newmark HL, Yang K, Lipkin M, Kopelovich L, Liu Y, Fan K, et al. A Western-style diet induces benign and malignant neoplasms in the colon of normal C57Bl/6 mice. Carcinogenesis 2001;22:1871–5. [DOI] [PubMed] [Google Scholar]
- 14. Yang W, Bancroft L, Liang J, Zhuang M, Augenlicht LH. p27kip1 in intestinal tumorigenesis and chemoprevention in the mouse. Cancer Res 2005;65:9363–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang K, Lipkin M, Newmark H, Rigas B, Daroqui C, Maier S, et al. Molecular targets of calcium and vitamin D in mouse genetic models of intestinal cancer. Nutr Rev 2007;65:S134–7. [DOI] [PubMed] [Google Scholar]
- 16. Bi X, Tong C, Dockendorff A, Bancroft L, Gallagher L, Guzman-Hartman G, et al. Genetic deficiency of decorin causes intestinal tumor formation through disruption of intestinal cell maturation. Carcinogenesis 2008;29:1435–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Augenlicht LH, Yang W, Mariadason J, Velcich A, Klampfer L, Lipkin M, et al. Interaction of genetic and dietary factors in mouse intestinal tumorigenesis. J Nutr 2006;136:2695S–6S. [DOI] [PubMed] [Google Scholar]
- 18. Yang K, Edelmann W, Fan K, Lau K, Leung D, Newmark H, et al. Dietary modulation of carcinoma development in a mouse model for human familial polyposis. Cancer Res 1998;58:5713–7. [PubMed] [Google Scholar]
- 19. Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol 2019;16:713–32. [DOI] [PubMed] [Google Scholar]
- 20. Simpson SJ, Le Couteur DG, Raubenheimer D. Putting the balance back in diet. Cell 2015;161:18–23. [DOI] [PubMed] [Google Scholar]
- 21. Warden CH, Fisler JS. Comparisons of diets used in animal models of high-fat feeding. Cell Metab 2008;7:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lien EC, Vander Heiden MG. A framework for examining how diet impacts tumour metabolism. Nat Rev Cancer 2019;19:651–61. [DOI] [PubMed] [Google Scholar]
- 23. Bastie C, Gaffney-Stomberg E, Ting-Wen L, Dhima E, Pessin J, Augenlicht L. Dietary cholecalcifero and calcium levels in a Western-style defined rodent diet alter energy metabolism and inflammatory response in mice. J Nutrition 2012;142:859–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang K, Kurihara N, Fan K, Newmark H, Rigas B, Bancroft L, et al. Dietary induction of colonic tumors in a mouse model of sporadic colon cancer. Cancer Res 2008;68:7803–10. [DOI] [PubMed] [Google Scholar]
- 25. Augenlicht LH, Taylor J, Anderson L, Lipkin M. Patterns of gene expression that characterize the colonic mucosa in patients at genetic risk for colonic cancer. Proc Natl Acad Sci U S A 1991;88:3286–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang D, Peregrina K, Dhima E, Lin EY, Mariadason JM, Augenlicht LH. Paneth cell marker expression in intestinal villi and colon crypts characterizes dietary induced risk for mouse sporadic intestinal cancer. Proc Natl Acad Sci U S A 2011;108:10272–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Newmark HL, Lipkin M, Maheshwari N. Colonic hyperplasia and hyperproliferation induced by a nutritional stress diet with four components of Western-style diet. J Natl Cancer Inst 1990;82:491–6. [DOI] [PubMed] [Google Scholar]
- 28. Newmark HL, Lipkin M, Maheshwari N. Colonic hyperproliferation induced in rats and mice by nutritional-stress diets containing four components of a human Western-style diet (series 2). Am J Clin Nutr 1991;54:209S–14S. [DOI] [PubMed] [Google Scholar]
- 29. Lipkin M, Winawer SJ, Sherlock P. Early identification of individuals at increased risk for cancer of the large intestine. Part I: definition of high risk populations. Clin Bull 1981;11:13–21. [PubMed] [Google Scholar]
- 30. Lipkin M, Blattner WE, Fraumeni JF, Lynch HT, Deschner E, Winawer S. Tritiated thymidine (0p,0h)labeling distribution as a marker for hereditary predisposition to colon cancer. Cancer Res 1983;43:1899–904. [PubMed] [Google Scholar]
- 31. Lipkin M, Newmark H. Effect of added dietary calcium on colonic epithelial cell proliferation in subjects at high-risk for familial colon cancer. N Engl J Med 1985;313:1381–4. [DOI] [PubMed] [Google Scholar]
- 32. Lipkin M, Friedman E, Winawer SJ, Newmark H. Colonic epithelial cell proliferation in responders and nonresponders to supplemental dietary calcium. Cancer Res 1989;49:248–54. [PubMed] [Google Scholar]
- 33. Vasquez EG, Nasreddin N, Valbuena GN, Mulholland EJ, Belnoue-Davis HL, Eggington HR, et al. Dynamic and adaptive cancer stem cell population admixture in colorectal neoplasia. Cell Stem Cell 2022;29:1213–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parikh K, Antanaviciute A, Fawkner-Corbett D, Jagielowicz M, Aulicino A, Lagerholm C, et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 2019;567:49–55. [DOI] [PubMed] [Google Scholar]
- 35. Hou Y, Sun X, Gheinani PT, Guan X, Sharma S, Zhou Y, et al. Epithelial SMYD5 exaggerates IBD by down-regulating mitochondrial functions via post-translational control of PGC-1alpha stability. Cell Mol Gastroenterol Hepatol 2022;14:375–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 2019;178:714–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive integration of single-cell data. Cell 2019;177:1888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014;32:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rodriguez-Colman MJ, Schewe M, Meerlo M, Stigter E, Gerrits J, Pras-Raves M, et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 2017;543:424–7. [DOI] [PubMed] [Google Scholar]
- 40. Chen L, Vasoya RP, Toke NH, Parthasarathy A, Luo S, Chiles E, et al. HNF4 regulates fatty acid oxidation and is required for renewal of intestinal stem cells in mice. Gastroenterology 2020;158:985–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koehler CL, Perkins GA, Ellisman MH, Jones DL. Pink1 and Parkin regulate Drosophila intestinal stem cell proliferation during stress and aging. J Cell Biol 2017;216:2315–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, et al. A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med 2012;18:1350–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Khoa LTP, Tsan YC, Mao F, Kremer DM, Sajjakulnukit P, Zhang L, et al. Histone acetyltransferase MOF blocks acquisition of quiescence in ground-state ESCs through activating fatty acid oxidation. Cell Stem Cell 2020;27:441–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta 2011;1807:1432–43. [DOI] [PubMed] [Google Scholar]
- 45. Zhang D, Wang W, Xiang B, Li N, Huang S, Zhou W, et al. Reduced succinate dehydrogenase B expression is associated with growth and de-differentiation of colorectal cancer cells. Tumour Biol 2013;34:2337–47. [DOI] [PubMed] [Google Scholar]
- 46. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999;98:115–24. [DOI] [PubMed] [Google Scholar]
- 47. Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev 2003;24:78–90. [DOI] [PubMed] [Google Scholar]
- 48. D'Errico I, Salvatore L, Murzilli S, Lo Sasso G, Latorre D, Martelli N, et al. Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1alpha) is a metabolic regulator of intestinal epithelial cell fate. Proc Natl Acad Sci U S A 2011;108:6603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moor AE, Harnik Y, Ben-Moshe S, Massasa EE, Rozenberg M, Eilam R, et al. Spatial reconstruction of single enterocytes uncovers broad zonation along the intestinal villus axis. Cell 2018;175:1156–67. [DOI] [PubMed] [Google Scholar]
- 50. Iwata R, Casimir P, Vanderhaeghen P. Mitochondrial dynamics in postmitotic cells regulate neurogenesis. Science 2020;369:858–62. [DOI] [PubMed] [Google Scholar]
- 51. Ludikhuize MC, Meerlo M, Gallego MP, Xanthakis D, Burgaya Julia M, Nguyen NTB, et al. Mitochondria define intestinal stem cell differentiation downstream of a FOXO/notch axis. Cell Metab 2020;32:889–900. [DOI] [PubMed] [Google Scholar]
- 52. van Daalen KR, Reijneveld JF, Bovenschen N. Modulation of inflammation by extracellular granzyme A. Front Immunol 2020;11:931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao E, Hou J, Ke X, Abbas MN, Kausar S, Zhang L, et al. The roles of sirtuin family proteins in cancer progression. Cancers 2019;11:1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Karbasforooshan H, Roohbakhsh A, Karimi G. SIRT1 and microRNAs: the role in breast, lung and prostate cancers. Exp Cell Res 2018;367:1–6. [DOI] [PubMed] [Google Scholar]
- 55. Qin L, Guo J, Zheng Q, Zhang H. BAG2 structure, function and involvement in disease. Cell Mol Biol Lett 2016;21:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Choi J, Houston M, Wang R, Ye K, Li W, Zhang X, et al. Intestinal stem cell aging at single cell resolution: transcriptional perturbations alter cell developmental trajectory reversed by gerotherapeutics. Aging Cell 2023Mar 2. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Volc-Platzer B, Majdic O, Knapp W, Wolff K, Hinterberger W, Lechner K, et al. Evidence of HLA-DR antigen biosynthesis by human keratinocytes in disease. J Exp Med 1984;159:1784–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Collins T, Korman AJ, Wake CT, Boss JM, Kappes DJ, Fiers W, et al. Immune interferon activates multiple class II major histocompatibility complex genes and the associated invariant chain gene in human endothelial cells and dermal fibroblasts. Proc Natl Acad Sci U S A 1984;81:4917–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Momburg F, Koch N, Moller P, Moldenhauer G, Butcher GW, Hammerling GJ. Differential expression of Ia and Ia-associated invariant chain in mouse tissues after in vivo treatment with IFN-gamma. J Immunol 1986;136:940–8. [PubMed] [Google Scholar]
- 60. Beswick EJ, Reyes VE. CD74 in antigen presentation, inflammation, and cancers of the gastrointestinal tract. World J Gastroenterol 2009;15:2855–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schroder B. The multifaceted roles of the invariant chain CD74–more than just a chaperone. Biochim Biophys Acta 2016;1863:1269–81. [DOI] [PubMed] [Google Scholar]
- 62. Jiang Z, Xu M, Savas L, LeClair P, Banner BF. Invariant chain expression in colon neoplasms. Virchows Arch 1999;435:32–6. [DOI] [PubMed] [Google Scholar]
- 63. Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Iwashige H, Aridome K, et al. Invariant chain expression in gastric cancer. Cancer Lett 2001;168:87–91. [DOI] [PubMed] [Google Scholar]
- 64. Gold DV, Stein R, Burton J, Goldenberg DM. Enhanced expression of CD74 in gastrointestinal cancers and benign tissues. Int J Clin Exp Pathol 2010;4:1–12. [PMC free article] [PubMed] [Google Scholar]
- 65. Tamori Y, Tan X, Nakagawa K, Takai E, Akagi J, Kageshita T, et al. Clinical significance of MHC class II-associated invariant chain expression in human gastric carcinoma. Oncol Rep 2005;14:873–7. [PubMed] [Google Scholar]
- 66. Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, Schnell A, et al. T helper cell cytokines modulate intestinal stem cell renewal and differentiation. Cell 2018;175:1307–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wosen JE, Mukhopadhyay D, Macaubas C, Mellins ED. Epithelial MHC class II expression and its role in antigen presentation in the gastrointestinal and respiratory tracts. Front Immunol 2018;9:2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. de Jong YP, Abadia-Molina AC, Satoskar AR, Clarke K, Rietdijk ST, Faubion WA, et al. Development of chronic colitis is dependent on the cytokine MIF. Nat Immunol 2001;2:1061–6. [DOI] [PubMed] [Google Scholar]
- 69. Lawrance IC, Fiocchi C, Chakravarti S. Ulcerative colitis and Crohn's disease: distinctive gene expression profiles and novel susceptibility candidate genes. Hum Mol Genet 2001;10:445–56. [DOI] [PubMed] [Google Scholar]
- 70. Breedveld A, van Egmond M. IgA and FcalphaRI: pathological roles and therapeutic opportunities. Front Immunol 2019;10:553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Suzuki Y, Landowski CP, Hediger MA. Mechanisms and regulation of epithelial Ca2+ absorption in health and disease. Annu Rev Physiol 2008;70:257–71. [DOI] [PubMed] [Google Scholar]
- 72. Davidson LA, Goldsby JS, Callaway ES, Shah MS, Barker N, Chapkin RS. Alteration of colonic stem cell gene signatures during the regenerative response to injury. Biochim Biophys Acta 2012;1822:1600–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Denson LA. Mitochondrial networks: a new therapeutic target in colitis. Cell Mol Gastroenterol Hepatol 2020;10:426–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cunningham KE, Vincent G, Sodhi CP, Novak EA, Ranganathan S, Egan CE, et al. Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1alpha) protects against experimental murine colitis. J Biol Chem 2016;291:10184–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xia HH, Lam SK, Chan AO, Lin MC, Kung HF, Ogura K, et al. Macrophage migration inhibitory factor stimulated by Helicobacter pylori increases proliferation of gastric epithelial cells. World J Gastroenterol 2005;11:1946–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Burclaff J, Bliton RJ, Breau KA, Ok MT, Gomez-Martinez I, Ranek JS, et al. A proximal-to-distal survey of healthy adult human small intestine and colon epithelium by single-cell transcriptomics. Cell Mol Gastroenterol Hepatol 2022;13:1554–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chen B, Scurrah CR, McKinley ET, Simmons AJ, Ramirez-Solano MA, Zhu X, et al. Differential pre-malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell 2021;184:6262–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Beyaz S, Chung C, Mou H, Bauer-Rowe KE, Xifaras ME, Ergin I, et al. Dietary suppression of MHC class II expression in intestinal epithelial cells enhances intestinal tumorigenesis. Cell Stem Cell 2021;28:1922–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Munoz J, Stange DE, Schepers AG, van de Wetering M, Koo BK, Itzkovitz S, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. EMBO J 2012;31:3079–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nicholson AM, Olpe C, Hoyle A, Thorsen AS, Rus T, Colombe M, et al. Fixation and spread of somatic mutations in adult human colonic epithelium. Cell Stem Cell 2018;22:909–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hodder MC, Flanagan DJ, Sansom OJ. Intestinal stem cell dynamics: a story of mice and humans. Cell Stem Cell 2018;22:785–7. [DOI] [PubMed] [Google Scholar]
- 82. Schuijers J, Junker JP, Mokry M, Hatzis P, Koo BK, Sasselli V, et al. Ascl2 acts as an R-spondin/Wnt-responsive switch to control stemness in intestinal crypts. Cell Stem Cell 2015;16:158–70. [DOI] [PubMed] [Google Scholar]
- 83. Choi J, Rakhilin N, Gadamsetty P, Joe DJ, Tabrizian T, Lipkin SM, et al. Intestinal crypts recover rapidly from focal damage with coordinated motion of stem cells that is impaired by aging. Sci Rep 2018;8:10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw 2015;67:1–48. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The western diet extensively reprograms Lgr5hi stem cells that expands with time and is reversible. This includes the TCA cycle and Oxphos pathways
The western diet elevates and suppresses multiple pathways by 3 months, most persisting to 12 months, and reversible by switching to control diet.
The western diet suppresses expression of a large number of genes encoding subunits of each of the 5 complexes of mitochondrial electron transport.
Targeted knockout of Ppargc1a in Lgr5hi cells was confirmed, and trajectory analyses of dietary and genetic alterations confirmed by marker expression.
Single cell RNAsequencing following 4 days of dietary switch, or then switch back and comparison to pathways altered in stem cells by Ppargc1a genetic inactivation.
Single cell analyses of cell progeny of the alternate stem cells mobilized by the western diet to maintain the mucosa.
The western diet causes chronic pro-tumorigenic inflammation.
S1. The western diet extensively reprograms Lgr5hi stem cells that expands with time and is reversible. This includes the TCA cycle and Oxphos pathways.S2. The western diet elevates and suppresses multiple pathways by 3 months, most persisting to 12 months, and reversible by switching to control diet.S3. The western diet suppresses expression of a large number of genes encoding subunits of each of the 5 complexes of mitochondrial electron transport.S4. Mouse genotyping identified presence of the wild-type, or heterozygous or homozygous conditionally targeted Ppargc1a allele.S5. Rapid dietary cross-over experiment of Fig 3.S6. scRNAseq analysis of Bmi1+ derived cells.S7. Inflammatory response to NWD1.
Data Availability Statement
All bulk and scRNA-seq data and scATAC-seq data are available in the NCBI GEO database, accession numbers GSE186811, GSE188339, GSE188577, GSE188338, and GSE228006.