

Summary
We analyzed 2532 lung adenocarcinomas (LUAD) to identify clinicopathologic and genomic features associated with metastasis, metastatic burden, organotropism, and metastasis-free survival. Patients who develop metastasis are younger and male, with primary tumors enriched in micropapillary/solid histological subtypes and with higher mutational burden, chromosomal instability, and fraction of genome doublings. Inactivation of TP53, SMARCA4, and CDKN2A are correlated with a site-specific shorter time to metastasis. The APOBEC mutational signature is more prevalent among metastases, particularly liver lesions. Analyses of matched specimens show oncogenic and actionable alterations are frequently shared between primary tumors and metastases, whereas copy number alterations of unknown significance are more often private to metastases. Only 4% of metastases harbor therapeutically actionable alterations undetected in their matched primaries. Key clinicopathologic and genomic alterations in our cohort were externally validated. In summary, our analysis highlights the complexity of clinicopathologic features and tumor genomics in LUAD organotropism.
eTOC Blurb
Lengel et al. identify clinicopathologic and genomic features of lung adenocarcinoma associated with the development of metastasis, metastatic burden, organotropism, and metastasis-free survival. The findings illustrate that metastatic tumors have increased chromosomal instability, share the majority of driver mutations with the primary tumor, and rarely develop new clinically targetable alterations.
Graphical Abstract
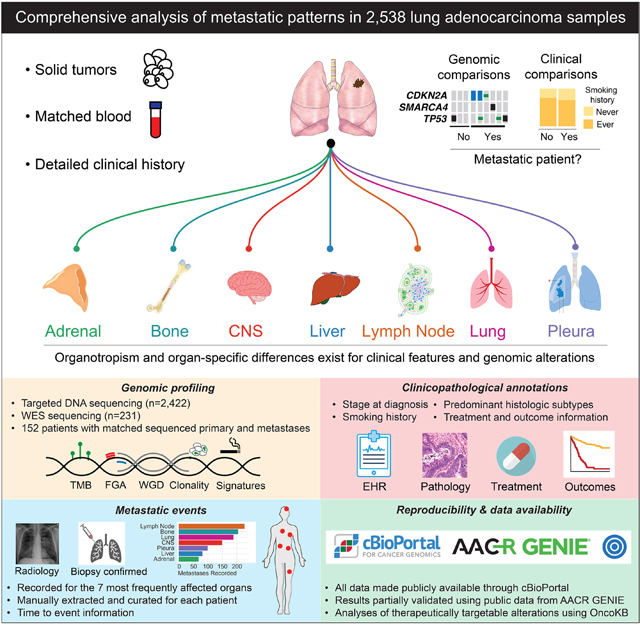
Introduction
Metastasis is the principal determinant of cancer-related mortality, accounting for 90% of all cancer-related deaths.1 Most patients with non-small cell lung cancer (NSCLC) present with stage IV disease, and up to 50% of patients with early-stage disease treated with surgery will develop recurrence and metastasis.2–4 Despite an increasing fractional incidence of early-stage disease, the prognosis for all stages of NSCLC remains poor, with a 5-year overall survival of 26%.5 Lung adenocarcinoma (LUAD) is the most common histologic subtype of NSCLC, and like other solid cancers, it preferentially metastasizes to specific organ sites, a process known as organotropism.6,7 Compared with metastases in most other cancer types, metastases in LUAD typically occur early in the disease course, with 70% to 80% of patients developing recurrence within two years after surgery.3,7
The “seed and soil” hypothesis was first proposed in 1889 by Stephen Paget, who described metastatic organotropism as a nonrandom event.8 In support of the Paget hypothesis, multiple studies have affirmed that certain organs are predisposed to metastatic disease. This predisposition is influenced by intrinsic characteristics of the primary cancer and extrinsic factors, such as vascular and lymphatic anatomy, organ-specific barriers in metastatic organs, and the immune microenvironment.9–11 More recently, the expanded use of next-generation sequencing has allowed investigators to interrogate the contribution of tumor genomic alterations throughout the metastatic cascade.12–14 In a pan-cancer analysis of >25,000 patients, our group recently identified genomic features associated with patterns of metastasis in 50 different types of tumors.15 Pan-cancer analyses provide important insights through the genomic characterization of large numbers of patients and the identification of associations that remain consistent across cancer types. However, they necessarily prioritize breadth over depth and their findings are limited by an inadequacy to investigate cancer-type specific features in detail.12,14–17
In this study, we examined 2532 primary and metastatic LUAD specimens from 2309 patients, using manually reviewed clinicopathologic data, including treatment context, and tumor genomic information obtained from a combination of broad-panel targeted DNA sequencing and whole-exome sequencing (WES). We sought to identify genomic and clinicopathologic features associated with LUAD metastases by analyzing the features of ever-metastatic tumors, as well as features associated with metastatic organotropism. In addition to our characterization of primary tumors, we analyzed metastatic lesions stratified by anatomic site(s) and compared primary and metastatic specimens in a site-specific manner. Finally, we compared molecular profiles of matched primary tumors and metastatic lesions obtained from the same patient to refine our analyses of tumor evolution throughout disease.
Results
We analyzed targeted DNA sequencing data from a total of 2422 LUAD samples classified into subgroups based on the sequenced sample type (primary vs. metastasis), the development of metastases (patient with metastases vs. patient without metastases), and the timing of sequencing relative to the identification of metastasis (Figure 1A–B, Table S1). The first cohort of patients (“Nonmetastatic” or “NM,” n=318) comprised primary LUAD tumors from patients who had at least 2 years of follow-up from the time of sample acquisition and had not developed a metastasis at last follow-up. The second cohort (“Early Stage-Metastatic” or “ES-M,” n=258) consisted of LUAD primary tumor samples from patients diagnosed with clinical or pathologic stage I-III cancer at the time of sample acquisition who developed metastasis. The third cohort (“Late Stage-Metastatic” or “LS-M,” n=190) encompassed primary tumor LUAD samples from patients who presented with stage IV disease. The fourth cohort (“Metastatic Lesions” or “ML,” n=1478) included sequenced metastases from seven distinct organ sites. Table 1 highlights key demographics and details the treatments administered across the cohorts. Finally, we defined a fifth cohort (“Matched Primary-Metastatic” or “MP-M,” n=336) that consisted of 152 primary tumors and 184 matched metastatic samples, where the primary tumor was sequenced before the metastatic sample(s) (Figure 1C). The NM, ES-M, LS-M, and ML cohorts were nonoverlapping, whereas 51 primary tumors from the ES-M and LS-M cohorts and 107 metastatic samples from the ML cohort were also in the MP-M cohort. For the evaluation of ever-metastatic primary tumors, ES-M and LS-M were combined to capture all primary tumors that developed any metastases. Before all ever-metastatic primary tumors were grouped, the ES-M and LS-M groups were compared and were found to be similar in terms of clinicopathologic and genomic features (Figure S1A–E). We also analyzed WES for a set of 231 specimens (120 primary and 111 metastases), including 110 unique LUAD specimens not in our study population (Figure 1D, Table S1). Eight pairs of specimens from patients with matched primary and metastatic lesions were present in the WES cohort.
Figure 1. Cohort overview.

(A) Number of sequenced specimens in unmatched sample analyses, cohorts 1–4. (B) Clinical outcomes of patients with sequenced primary tumors, cohorts 1–3. Top: Kaplan-Meier curve showing overall survival. Bottom: Metastatic burden per patient (left). Number of patients with metastasis to specific anatomic sites (right). (C) Overview of patients with matched primary and metastatic samples, cohort 5. Overlap of samples included in cohorts 2–4 and matched cohort. (D) Primary and metastatic lesions with whole-exome sequencing (WES). Overlap of samples included in the IMPACT sequencing and WES cohorts. CNS, central nervous system.
Table 1.
Demographic and Treatment Characteristics for Unmatched Cohorts
| Characteristic | NM (n=318) | ES-M (n=258) | LS-M (n=190) | ML (n=1478) |
|---|---|---|---|---|
| Age at Date of Tissue Acquisition for IMPACT Sequencing, median (IQR), years | 67.7 (62.7–73.5) | 68.0 (60.1–74.2) | 65.2 (58.0–71.1) | 66.5 (58.8–73.6) |
| Sex | ||||
| Female | 233 (73.3) | 159 (61.6) | 123 (64.7) | 898 (60.8) |
| Male | 85 (26.7) | 99 (38.4) | 67 (35.3) | 580 (39.2) |
| Race | ||||
| Asian | 28 (8.8) | 28 (10.9) | 35 (18.4) | 170 (11.5) |
| Black | 10 (3.1) | 9 (3.5) | 14 (7.4) | 83 (5.6) |
| Other | 14 (4.4) | 15 (5.8) | 12 (6.3) | 80 (5.4) |
| White | 266 (83.6) | 206 (79.8) | 129 (67.9) | 1145 (77.5) |
| Smoking History | ||||
| Ever | 240 (75.5) | 188 (72.9) | 122 (64.2) | - |
| Never | 78 (24.5) | 70 (27.1) | 68 (35.8) | |
| Clinical Stage | ||||
| I | 266 (83.6) | 137 (53.1) | 14 (7.4) | |
| II | 34 (10.7) | 57 (22.1) | 5 (2.6) | |
| III | 17 (5.3) | 58 (22.5) | 2 (1.1) | - |
| IV | 0 (0) | 0 (0) | 169 (88.9) | |
| Unknown | 1 (0.3) | 6 (2.3) | 0 (0) | |
| Pathologic Stage | ||||
| I | 249 (78.3) | 99 (38.4) | 0 (0) | |
| II | 46 (14.5) | 58 (22.5) | 0 (0) | - |
| III | 23 (7.2) | 101 (39.1) | 0 (0) | |
| IV | 0 (0) | 0 (0) | 190 (100) | |
| Surgery | ||||
| Yes | 100 (0) | 241 (93.4) | 32 (16.8) | - |
| No | 0 (0) | 17 (6.6) | 158 (83.2) | |
| Treatment Prior to IMPACT Sequencing | ||||
| Yes | 16 (5.0) | 41 (15.9) | 2 (1.1) | 1116 (75.5) |
| No | 302 (95.0) | 217 (84.1) | 188 (98.9) | 362 (24.5) |
| Type of Treatment Prior to IMPACT Sequencing | ||||
| Chemotherapy | 14 (4.4) | 35 (13.6) | 0 (0) | 513 (34.7) |
| Immunotherapy | 2 (0.6) | 2 (0.8) | 0 (0) | 216 (14.6) |
| Targeted Therapy | 1 (0.3) | 5 (1.9) | 2 (1.1) | 346 (23.4) |
| Radiation | 2 (0.6) | 5 (1.9) | 0 (0) | 845 (57.2) |
| Neoadjuvant Therapy | ||||
| Yes | 16 (5.0) | 40 (15.5) | 0 (0) | - |
| No | 302 (95.0) | 201 (77.9) | 32 (16.8) | |
| NA | 0 (0) | 17 (6.6) | 158 (83.2) | |
| Type of Neoadjuvant Therapy | ||||
| Chemotherapy | 14 (4.4) | 35 (13.6) | ||
| Immunotherapy | 2 (0.6) | 2 (0.8) | - | - |
| Targeted Therapy | 1 (0.3) | 3 (1.2) | ||
| Radiation | 2 (0.6) | 5 (1.9) | ||
| Adjuvant Therapy | ||||
| Yes | 51 (16.0) | 100 (38.8) | 8 (4.2) | |
| No | 267 (84.0) | 126 (48.8) | 14 (7.4) | - |
| NA | 0 (0) | 32 (12.4) | 168 (88.4) | |
| Type of Adjuvant Therapy | ||||
| Chemotherapy | 48 (15.1) | 84 (32.6) | 5 (2.6) | |
| Immunotherapy | 0 (0) | 1 (0.4) | 1 (0.5) | - |
| Targeted Therapy | 0 (0) | 2 (0.8) | 1 (0.5) | |
| Radiation | 11 (3.5) | 37 (14.3) | 1 (0.5) |
NM, nonmetastatic; ES-M, early stage metastatic; LS-M, late stage metastatic; ML, metastatic lesions; IQR, interquartile range; IMPACT, Integrated Mutation Profiling of Actionable Cancer Targets.
The metastatic history of each patient was mapped to the seven most common organ sites: adrenal gland, bone, central nervous system (CNS), liver or biliary tract, lung, lymph node, and pleura. A specific organ site was determined to have a metastatic lesion if the lesion was identified on computed tomography, positron emission tomography, or magnetic resonance imaging reports or confirmed through pathologic analysis of the biopsy or surgical specimen (8th edition AJCC Cancer Staging Manual). For patients who underwent surgery, N1-N3 stage thoracic lymph nodes were not considered to represent metastatic disease. Lung metastases were included from the contralateral lung or separate lobes of the ipsilateral lung. The relative frequencies of organ-specific metastasis in our cohort of primary tumors were consistent with known patterns of metastasis in LUAD.18,19 However, because of differences in sample acquisition, the frequency of these sites was not equivalent among sequenced metastatic lesions (Figure 1B).
Primary LUAD tumors from patients who develop metastases exhibit distinct clinicopathologic and genomic features
Given the impact of metastasis on patient outcomes (Figure 1B), we examined factors associated with the development of metastasis by comparing clinicopathologic and genomic features between nonmetastatic primary tumors (NM, n=318) and ever-metastatic primary tumors (ES-M and LS-M, n=448). Ten genes (TP53, KEAP1, CDKN2A, MDM2, PIK3CA, NKX2–1, RB1, MYC, SMARCA4, FOXA1) were altered more often in ever-metastatic tumors, whereas RBM10 alterations occurred more often in nonmetastatic tumors (16% vs. 7%, q=0.001) (Figure 2A, Table S2). RBM10 alterations are associated with better outcomes and earlier-stage tumors in patients with LUAD.20,21 Overall, 79% of patients with ever-metastatic disease had alterations in 1 or more of these 11 genes. Patients with metastases were younger (median, 67.3 vs. 68.6 years, q=0.032), male (37% vs. 27%, q=0.008), and more often had a micropapillary or solid predominant histologic subtype (18% vs. 8%, q=0.002), which are the most biologically aggressive (Figure 2B).22 Ever-metastatic tumors also had higher tumor mutational burden (TMB) (median, 5.6 vs. 4.9 mut/Mb, p=0.003), higher chromosomal instability as quantified by the fraction of genome altered (FGA) (median, 0.47 vs. 0.26, p<0.001), and higher rate of whole-genome duplication (WGD) (40% vs. 23%, p<0.001) (Figure 2C). Detectable somatic mutational signatures were investigated (Table S2), as previous work has shown that APOBEC mutagenesis is a key driver for many cancers, including NSCLC.23–25 We observed a higher prevalence of APOBEC-related signatures SBS2 and SBS13 in metastatic tumors (18% vs. 8%, p=0.012, and 22% vs. 14%, p=0.069, respectively) (Figure 2D). We then investigated ten canonical oncogenic signaling pathways and found that ever-metastatic tumors had more alterations in the p53 (58% vs. 40%, q<0.001), PI3K (31% vs. 18%, q<0.001), cell cycle (29% vs. 16%, q<0.001), Nrf2 (17% vs. 9%, q=0.010), and TGFβ pathways (7% vs. 3%, q=0.038) (Figure 2E).26,27 Interestingly, the proportions of EGFR and KRAS alterations and RTK/RAS pathway alterations were similar between the nonmetastatic and ever-metastatic groups. To externally validate our results, we used an independent cohort of 318 LUAD samples sequenced at Dana-Farber Cancer Institute (AACR Project GENIE).28,29 Alterations in several genes, including TP53, MYC, and RMB10, as well as demographic trends for age and sex seen in our cohort, were confirmed (Figure S2A–B).
Figure 2. Comparison of nonmetastatic (NM) and ever-metastatic (EM) primary tumors.

(A) Oncoprint displaying clinical attributes and genes altered at significantly different frequencies between the two groups. (B) Comparisons of clinicopathologic features. (C) Tumor mutational burden (TMB), fraction of genome altered (FGA), and whole-genome duplication (WGD). Boxplots display median values, interquartile range (IQR) boxes, and whiskers demonstrating 1.5 x IQR. (D) Percentage of samples with the APOBEC mutational signature present. (E) Percentage of samples with alterations for ten canonical oncogenic pathways. (F) Co-occurrence and mutual exclusivity of genes with significant interaction. (G) Genes with significant associations with metastasis-free survival among patients with clinical stage I-III disease (cohorts 1–2, n=576) on univariable analysis. Statistical analyses: (A, B, D-F) Fisher’s exact test. *p<0.05 unless otherwise indicated; q-values correct for multiple comparisons using false-discovery rate (FDR). (C) Wilcoxon rank-sum test for TMB and FGA. Fisher’s exact test for WGD. (G) Cox proportional hazard model. All p-values as indicated, log-rank test. Squares represent hazard ratio (HR) and whiskers display 95% confidence interval (CI). +, genes that remained significant in multivariable model with clinicopathologic factors. Hx, history; pStage, pathologic stage.
Distinct patterns of gene alteration co-occurrence and mutual exclusivity were observed among ever-metastatic tumors in our cohort. In total, 11 gene pairs were co-altered at a statistically significant frequency, and ten pairs were mutually exclusive (Figure 2F, Table S2). Of these, seven gene co-alterations and five mutually exclusive pairs were unique to ever-metastatic primary tumors (Figure S2C). Of note, co-alteration of TP53 and EGFR was observed only in the ever-metastatic group, suggesting that these alterations cooperate during metastatic progression. Studies have shown that TP53 and EGFR co-alteration is associated with a poor prognosis.30 SMARCA4 alterations co-occurred at a significant frequency with KEAP1 (q<0.001) and STK11 (q<0.001) only in ever-metastatic primary tumors, whereas co-alteration of KEAP1 and STK11 was significant in both groups. Across the ever- and nonmetastatic cohorts, SMARCA4, STK11, KEAP1, and KRAS alterations were mutually exclusive with EGFR alterations.31,32
An exploratory analysis of all surgical patients (NM and ES-M, N=576) was performed to investigate associations between individual gene alterations and time to development of metastatic disease (Figure S2D). After inclusion of clinicopathologic features, MDM2 (p=0.008), MYC (p=0.021), SMARCA4 (p<0.001), and TP53 (p=0.001) were associated with worse metastasis-free survival (MFS), while EGFR (p=0.002) and NF1 (p<0.001) were associated with better MFS (Figure 2G).
Clinicopathologic and genomic changes associated with increased metastatic burden
We explored patterns of metastasis and features associated with metastatic burden among patients with both early (ES-M) and late stage (LS-M) primary tumors (n=448). Overall, 146 patients (33%) had a single site of metastasis, and three patients experienced metastases at all seven sites (median metastatic sites 2 [range, 1–7]) (Figure S3A). Significant co-occurrence of anatomic sites was found among adrenal gland, liver, bone, and lymph node metastases (Figure S3B). In contrast, a single-site metastasis was most likely to occur in the CNS or lung, and neither was found to significantly co-occur with metastases at other sites (Figure 3A, Figure S3B). We next examined the relationships between clinicopathologic features and metastatic burden (Table S3). Younger patients had a higher metastatic burden (p=0.002) (Figure 3B), while smoking history and race were not associated with metastatic burden (Figure S3C). For samples with predominant histologic subtype information (n=249), solid subtype tumors had the highest metastatic burden, although this was not statistically significant. When examining genomic features, higher FGA (p=0.010) and alterations in TP53 (p=0.005) and ERBB2 (p=0.033) were associated with a greater metastatic burden on univariable analysis (Figure 3C–D). Patients with a single site of metastasis had lower TMB and WGD, although the differences were not significant (Figure S3D). Through this analysis, we observed that clinicopathologic and genomic features are associated with higher metastatic burden and variation in metastatic patterns between organ sites, with metastases more likely to occur as a solitary metastasis at certain sites. These findings suggest that features of the primary tumor and characteristics of distant organs may influence LUAD metastasis. Once again, several of these associations were validated with an independent cohort from AACR GENIE (Figure S3E–F).
Figure 3. Clinicopathologic and genomic features of primary tumors associated with site-specific metastasis and metastatic burden.

(A) Metastatic burden stratified by anatomic site. Each bar represents all ever-metastatic patients with metastasis to a given site; colors correspond to proportion of patients with 1, 2, or ≥3 distinct metastatic sites. (B) Age of patients, stratified by metastatic burden. (C) Fraction of genome altered (FGA), stratified by metastatic burden. Boxplots display median values, interquartile range (IQR) boxes, and whiskers demonstrating 1.5 x IQR. (D) Metastatic burden for significantly different genes between altered and wild-type (WT) tumors. (E) Clinicopathologic and genomic features across each organ site. The fill color corresponds to features enriched in tumors ever metastatic to a given site or not metastatic to a given site. (F) Frequency of significant clinicopathologic features and genomic alterations in nonmetastatic and ever-metastatic tumors with metastasis to lymph node and bone on multivariable analysis. (G) Hazard ratio (HR) for clinicopathologic and genomic features in relation to time to metastasis across each organ site. HR >1 indicates shorter time to metastasis; HR <1 indicates longer time to metastasis. (H) Kaplan-Meier curves demonstrating site-specific metastasis-free survival (MFS) for Hippo pathway alterations for central nervous system (CNS) metastases and CDKN2A alterations for bone metastases. Statistical Analyses: (B) Pearson’s correlation. (C-D) Wilcoxon rank-sum test. *p<0.05. (E) Univariate logistic regression. *p<0.05 for clinicopathologic variables on univariable analysis; °q<0.05 for genomic features on univariable analysis. q-values correct for multiple comparisons using the false-discovery rate (FDR). (F) Multivariate logistic regression. Features shown are significant by p<0.05. (G) Cox proportional hazards model. p-values calculated from log-rank test. *p<0.05 for clinicopathologic variables on univariate analysis; °q<0.05 for genomic features on univariate analysis. Met, metastatic; N, no; pStage, pathologic stage; Y, yes.
See also Figures S3, S4, and Table S3.
Inactivation of tumor suppressor genes TP53, SMARCA4, and CDKN2A is associated with site-specific differences in patterns and timing of metastases
To gain insight into factors associated with organotropism, we analyzed clinicopathologic and genomic features associated with site-specific metastasis in ever-metastatic primary tumors (ES-M and LS-M, n=448). On univariable analysis of all ever-metastatic tumors, receipt of neoadjuvant therapy was associated with the development of CNS metastases (odds ratio [OR]=2.59, 95% confidence interval [CI] 1.19–5.97, q=0.016), whereas smoking history was associated with liver (OR=1.77, 95% CI 1.01–3.22, q=0.047) and lymph node metastases (OR=1.85, 95% CI 1.19–2.90, q=0.007) (Figure 3E). In contrast, predominant histologic subtypes were similar across sites (Figure S3G). Inactivating alterations in SMARCA4 and TP53 (including a combination of mutations, deletions, and gene fusions) were associated with specific sites of metastasis in a univariate analysis (Figure 3E). The enrichment of SMARCA4 alterations in patients with bone metastasis (OR=6.47, 95% CI 1.70–42.60, p=0.017) and TP53 alterations in patients with nodal metastasis (OR=2.03, 95% CI 1.28–3.23, p=0.003) remained significant on multivariable analysis (Figure 3F). Multivariate analysis of the association between SMARCA4 alterations and metastasis to the liver was not feasible due to the small number of events, but five of six patients with SMARCA4 alterations developed liver metastasis.
We next performed a temporal analysis in primary tumors from patients without metastasis at the time of initial sequencing (ES-M, n=258) to elucidate features associated with site-specific patterns of metastasis. Genomic variables associated with organotropism in the previous site-specific analysis of ever-metastatic tumors (Figure 3E) remained significant in the time-to-metastasis analysis: SMARCA4 inactivation was associated with shorter time to bone metastasis (OR=3.37, 95% CI 1.47–7.73, p=0.004), while SMARCA4 (OR=2.13, 95% CI 1.03–4.39, p=0.041), TP53 gene (OR=2.21, 95% CI 1.47–3.33, p<0.001), and p53 pathway (OR=2.29, 95% CI 1.43–3.67, p<0.001) alterations were associated with shorter time to lymph node metastasis (Figure 3G). In addition, CDKN2A alterations were associated with shorter time to bone metastasis (OR=2.79, 95% CI 1.71–4.56, p<0.001), Hippo pathway alterations were associated with shorter time to CNS metastasis (OR=5.21, 95% CI 2.21–12.30, p<0.001), and Nrf2 pathway alterations were associated with shorter time to liver (OR=3.85, 95% CI 1.70–8.74, p=0.001) and nodal metastases (OR=2.29, 95% CI 1.43–3.67, p<0.001). Of note, SMARCA4 alterations were associated with a shorter time to metastasis in a site-agnostic manner, whereas Hippo pathway and CDKN2A alterations were site-specific and involved shorter time to metastasis to the CNS and bone, respectively (Figure 3H, Figure S4A–C).
Increased mutational burden and chromosomal instability in sequenced metastases do not translate to a higher fraction of clinically targetable alterations
We explored the genomic differences among metastatic lesions from each organ site. Across sites, TMB and FGA varied, as did the proportion of samples with EGFR, KRAS, CDKN2A, TERT, ALK, PIK3CA, RBM10, BRAF, and Notch, Cell Cycle, and NRF2 pathway alterations (Figure S5A–C, Table S4). Pleural metastases had a lower mutational burden (median, 4.4 vs. 6.6 mut/Mb, q<0.001), less chromosomal instability (median FGA, 0.5 vs. 0.7, q<0.001), and a greater proportion of EGFR alterations (48% [96/212] vs. 32% [410/1275], q=0.002), whereas other sites like CNS were associated with factors of poor prognosis, such as STK11 alterations.33
We next investigated differences between primary tumors and metastases, first in aggregate and then stratified by site. Overall, metastatic samples had a significantly higher TMB and chromosomal instability (FGA and WGD) (all q<0.001) (Figure 4A, Table S4). Nine genes (TP53, EGFR, CDKN2A, NKX2–1, FOXA1, MET, NF1, ARID1A, and MGA) were altered more often in metastases, and four (KRAS, MDM2, PIK3CA, and ERBB2) were altered more often in primary tumors. These differences in TMB, chromosomal instability, and gene alternations were replicated in an independent validation cohort of LUAD samples from AACR GENIE (Figure S6A–B). Six pathways (RTK/RAS, cell cycle, p53, MYC, Wnt, HIPPO) were altered more often in metastases (Figure 4A). These pathway differences were often driven by alterations in specific genes such as CDKN2A in a site-specific manner. For example, in CNS and liver, CDKN2A inactivation was more frequent in metastases than in primary tumors with metastasis to these sites (q=0.018 for both).
Figure 4. Genomic comparisons between primary tumors and metastases.

(A) Left to right: distributions of tumor mutational burden (TMB), fraction of genome altered (FGA), whole-genome duplication (WGD), gene alteration frequencies, pathway alteration frequencies, and frequency of samples presenting APOBEC signatures across groups. Boxplots display median values, interquartile range (IQR) boxes, and whiskers demonstrating 1.5 x IQR. Heat map lists alteration frequencies for genes and pathways as percentages. Darker colors correspond to higher percentage of altered samples. Statistical analysis: Wilcoxon rank-sum test for TMB and FGA; Fisher’s exact test for WGD, gene, pathway, and APOBEC signature frequencies. Significance indicated with a red or green * for q<0.05, false-discovery rate (FDR) adjusted. (B) Top to bottom: distributions of oncogenic alteration types, number of actionable alterations (levels 1 to 3A), and highest level of actionability across grouped sites. (C) Percentage of samples with actionable alterations. Color in the left column corresponds to level of actionability. (D) Mutational signature profiles of primary and metastatic lesions in WES cohort, with metastases stratified by anatomic site. (E) Frequency of samples with signature present for three mutational signatures of interest (SBS2, SBS4, SBS13). Statistical analysis: Fisher’s exact test. ^ and * indicate q<0.05, FDR adjusted, for comparisons of primary vs. lesion site and metastatic lesion site vs. all other metastatic lesions, respectively. CNS, central nervous system; M, metastasis; P, primary tumor; VUS, variant of unknown significance.
See also Figures S5–S7 and Tables S4–S6.
In our analysis, >50% of all primary tumors and metastases contained at least one actionable alteration, except for adrenal gland metastases. Adrenal gland metastases had the lowest proportion of level 1 actionable alterations (26/63 [41%]), whereas pleural metastases had the highest proportion (133/202 [66%]), mirroring the rate of EGFR activating mutations in these sites (Figure 4B). Comparison of site-specific alterations highlighted important differences between primary tumors and metastases. For example, CDKN2A deletions are currently not actionable (level 4 alteration); however, these were observed more often in metastatic samples across sites (24% vs. 14%, p<0.001). In primary tumors that metastasized to the liver, KRAS G12C mutations were particularly high compared to metastatic lesions at the liver (17/81 [21%] and 12/188 [6%], respectively, p<0.001), while in pleural metastases, the rate of KRAS G12C mutations was similar between primary tumors and metastases (12/99 [12%] and 24/202 [12%], respectively, p=1) (Figure 4C). Overall, metastases were associated with greater mutational burden and chromosomal instability compared to primary tumors, yet there was not a corresponding increased number of actionable targets.
Since the number of mutations identified by MSK-IMPACT is limited, we performed WES on 231 LUAD specimens to better investigate differences in mutational signatures across primary tumors (n=120) and metastases (n=111) (Figure 4D, Tables S5 and S6). The most frequently detected signatures were SBS4 (smoking signature), SBS2/SBS13 (APOBEC signatures), and SBS1 (a mitotic clock signature associated with aging). Consistent with our results using MSK-IMPACT (Figure S6C), the APOBEC-related signatures were more prevalent in sequenced metastases than in primary tumors. In the WES cohort, liver metastases were more often positive for SBS13 (10/15 [67%] vs. 21/78 [27%], q=0.034) and less often positive for SBS4 (4/15 [27%] vs. 51/78 [65%], q=0.034) than metastases sequenced at other organs (Figure 4E). Further comparison of cases sequenced with MSK-IMPACT and WES shows good concordance for estimates of genomic alterations (Figure S7A–C).
Oncogenic and actionable alterations are enriched among somatic changes shared between primary tumors and matched metastases
Comparisons using unmatched cohorts of primary tumors and metastases can be informative, but the intrinsic interpatient heterogeneity is a limitation. To address this, we compared paired LUAD specimens from primary tumors (n=152) and metastatic lesions (n=184) from the same patients. Every patient had at least one metastatic specimen, and some had multiple sequenced metastases. In all pairs, the primary lesion was sequenced before the metastatic lesion. FGA was higher in metastatic samples (median FGA, 0.085, p<0.001), while WGD was most often shared between the primary and metastatic samples (59/125 patients [47.2%]) than private to the metastatic sample (40/125 [32.0%]) or private to the primary (26/125 [20.8%]) (Figure 5A). The alteration types private to the primary or metastatic samples or shared between them varied. Mutations were the most common shared alteration compared to copy number alterations (940/1826 [51.5%] vs. 205/827 [24.8%], p<0.001) and fusions (940/1826 [51.5%] vs. 33/114 [28.9%], p<0.001) (Figure 5B). Shared mutations and copy number alterations included a larger fraction of oncogenic variants than private (mutations: 379/940 [40.3%] vs. 229/886 [25.8%], p<0.001; CNA: 125/205 [60.1%] vs. 289/622 [46.5%], p<0.001). Copy number alterations unique to the metastatic lesions (489/827 [59.1%]) were most frequently observed compared to those private to the primary (133/827 [16.1%], p<0.001) and shared between the two (205/827 [24.8%], p<0.001). These findings are consistent with the results from our previous unmatched analyses, where we observed more fusions and copy number alterations in metastatic samples.15,20 Actionable alterations were enriched among alterations shared between the primary and metastatic samples (141/282 [50.0%] vs. 41/159 [25.8%], p<0.001); private metastatic alterations were rarely actionable. Only 4% (7/184) of metastatic lesions developed a level 1 targetable mutation not observed in the primary tumor (Figure 5C, Figure S8A).
Figure 5. Genomic comparisons between patient-matched primary tumors and metastases.

(A) Median tumor mutational burden (TMB) and fraction of genome altered (FGA) between matched primary tumors and metastases. Statistical analysis: Wilcoxon rank-sum test. p-values as indicated. Right: Proportion of samples with whole-genome duplication (WGD) private or shared between primary and metastatic samples. (B) Distribution of shared and private alterations stratified by alteration types across sites. (C) Actionability of alterations private or shared between primary and metastatic samples across sites. (D) Cancer cell fraction (CCF) of shared mutations between primary and metastatic samples. CCF <0.8 is considered subclonal. (E) Proportion of shared and private mutations, stratified by clonality status for each gene (left) and specific amino acid changes (right). Clonality status nomenclature describes mutation clonality first in metastasis and then primary sample. CNA, copy number alteration; CNS, central nervous system; Fus, fusion; LN, lymph node; Met, metastasis; Mut, mutation; VUS, variant of unknown significance.
See also Figure S8.
We next assessed differences in clonality between matched primary tumors and metastases. Clonal mutations accounted for 73.8% of all mutations and were more often shared between primary and metastasis than subclonal mutations (314/472 [66.5%] vs. 74/168 [44.0%], p<0.001) (Figure 5D). At an individual gene level, this same pattern was present, with a few exceptions: RBM10, PIK3CA, and ARID1A had the lowest proportion of shared clonal mutations (Figure 5E). We also analyzed common mutations in TP53, KRAS and EGFR, of which only EGFR T790 mutations were not primarily clonal in both primary tumors and metastases, consistent with the expected behavior of a resistance mutation.34 We compared cancer cell fraction for mutations detected in matched primary and metastatic WES samples for eight patients, and identified clonal oncogenic mutations shared between primary and metastasis in six of them (Figure S8B). Paired WES tumors also resembled each other in terms of detected mutational signatures, although statistical significance was difficult to assess given the small sample size (Figure S8C–D).
A detailed evaluation of 22 patients with one sequenced primary tumor and two or more sequenced metastases showed that most detected driver alterations (75/91, 82.4%) were shared across at least two matched samples from the same patient. Oncogenic alterations detected in only one matched sample from a given patient were often gene amplifications or deletions (13/16, 81.3%) (Figure 6A). The time course and overall genomic landscape varied between patients. For some patients, there was little change between the primary tumor and metastases despite many months between samples (e.g., Patient 17); in contrast, others had metastatic disease identified shortly after initial diagnosis and the metastases had different TMB, WGD, and FGA profiles compared to their primary tumor (e.g., Patient 3).
Figure 6. Individual patient comparisons of matched primary and metastatic samples.

(A) Summary of genomic and clinical characteristics of patients with one primary and ≥2 matched sequenced metastases. Each numbered cluster represents a patient. One column represents a sequenced sample with the first column representing the primary sample and subsequent columns representing metastatic samples. For example, Patient 1 has one primary and five metastatic samples. (B) Phylogenetic trees for two patients with multiple matched samples, including timeline of sample acquisition and treatment course. CNS, central nervous system; FGA, fraction of genome altered; LN, lymph node; TMB, tumor mutational burden; Tx, treatment; WGD, whole-genome duplication; XRT, radiation.
Our analysis of matched samples also illustrates the relationship between treatment and genomic changes. Of the metastatic lesions, 79% (145/184) had treatment before tissue acquisition (Table 2). Six of the seven metastatic lesions that developed a level 1 alteration not observed in the primary tumor had treatment between primary and metastatic tissue acquisition. All six patients received TKI therapy and developed level 1 alterations that are known mechanisms of resistance to TKI therapy (5 had EGFR T790M mutations after erlotinib, 1 developed an ALK I1171T mutation after loratinib and alectinib). The last patient who was treatment-naïve developed a new KRAS G12C mutation. At an individual level, Patient 2 demonstrated genomic variation across five samples despite being acquired within 30 months of each other. Though all metastatic samples shared a CDKN2A deletion, three different MET alterations were present. All samples shared a MET amplification, and the primary tumor and liver metastasis shared a MET fusion. However, following CNS progression on crizotinib, two of the CNS metastases developed a MET:Y1230N mutation, which is related to crizotinib-resistance (Figure 6B). In contrast, Patient 5 had remarkably similar genomic profiles between samples despite almost four years between resection of the primary tumor and identification and biopsy of a lung metastasis. One can hypothesize that the similarity in samples despite the longer time interval may be related to the lack of systemic treatment. Overall, our analysis of matched primary and metastatic samples confirms the higher mutational burden and increased FGA observed in the unmatched analysis, while demonstrating that metastases often acquire new alterations through increased chromosomal instability and following systemic treatment.
Table 2.
Demographic and Treatment Characteristics for Matched Cohort
| Characteristic | Primary Samples (n = 152) | Metastatic Samples (n = 184) |
|---|---|---|
| Age at Time of Primary Sample, median (IQR), y | 65.8 (57.0–72.0) | - |
| Sex | ||
| Female | 100 (65.8) | - |
| Male | 52 (34.2) | |
| Race Asian |
18 (11.8) |
|
| Black | 11 (7.2) | - |
| Other | 11 (7.2) | |
| White | 112 (73.7) | |
| Smoking History | ||
| Ever | 100 (65.8) | - |
| Never | 52 (34.2) | |
| Clinical Stage I |
44 (28.9) | |
| II | 11 (7.2) | |
| III | 26 (17.1) | - |
| IV | 68 (44.7) | |
| Unknown | 3 (2.0) | |
| Treatment Prior to IMPACT Sequencing Yes |
47 (30.9) | 145 (78.8) |
| No | 105 (69.1) | 39 (21.2) |
| Type of Treatment Prior to IMPACT Sequencing Chemotherapy |
25 (16.4) | 79 (42.9) |
| Immunotherapy | 4 (2.6) | 32 (17.4) |
| Targeted Therapy | 28 (18.4) | 93 (50.5) |
| Radiation | 3 (2.0) | 37 (20.1) |
| Definitive Local Therapy for Primary | ||
| Surgery | 71 (46.7) | - |
| Radiation | 14 (9.2) |
IQR, interquartile range; IMPACT, Integrated Mutation Profiling of Actionable Cancer Targets.
Discussion
Previous work from our group has identified clinicopathologic and genomic factors related to recurrence and disease-free survival following surgical resection in LUAD.20,24,26,35–37 In this study, we investigated LUAD-specific organotropism, with a focus on the incorporation of clinicopathologic features across analyses, temporal relationships between genomic and clinicopathologic features, and time to metastatic dissemination within the context of site-specific metastasis. As part of a larger institutional effort, we recently performed a pan-cancer analysis that found significant associations between metastatic patterns and genomic features across 50 different cancer types, including a cohort of patients with LUAD.15 While there are similarities between these two studies, there are critical methodologic differences. First, the previous pan-cancer study relied exclusively on tumor registry information and billing codes for the annotation of metastatic sites in individual patients, whereas this study adopted a more comprehensive process of manual curation aimed at reducing false-negatives and inter-physician variability through individualized review of imaging, pathology, and clinical reports. A head-to-head comparison of our extraction methods for a group of shared patients revealed that we identified 407 additional metastatic diagnoses, including a nearly 4-fold increase in lymph node metastases (28% vs. 7.5%). Second, we also substantially increased the breadth and depth of our clinical annotations to include detailed treatment information and variables of translational relevance for LUAD patients, such as smoking history, predominant histologic subtypes, and detailed pathological staging, which allowed us to investigate additional questions such as the role of mutational signatures and intra-patient tumor heterogeneity and tumor evolution throughout the metastatic cascade. Third, we performed a comprehensive time-to-event analysis of matched primary and metastatic samples to better understand tumor clonality, selected therapeutic pressures on the tumor genome, and spatiotemporal patterns of organotropism. Collectively, this study offers a dedicated analysis aimed at understanding how clinicopathological and genomic features correlate with metastatic organotropism in LUAD.
The methodological improvements in our study have led to previously unreported, clinically relevant findings. For example, we observed that patients with primary tumors that eventually metastasize are younger, male, and have tumors with a micropapillary or solid predominant histological subtype. In contrast, no significant differences in metastatic burden (number of metastatic sites) or specific organotropism were observed based on predominant histologic subtype.
SMARCA4 and TP53 alterations were enriched in ever-metastatic tumors, associated with shorter MFS, and exhibited organotropism (SMARCA4 was enriched in bone metastases and TP53 in lymph node metastases). Previous studies have shown that alterations in SMARCA4 and TP53 were independently associated with recurrence-free survival in early-stage LUAD and that mutations in both genes were enriched in ever-metastatic tumors,15,20 but there is limited information regarding their role in organotropism.20,31,36,38 Others have shown a relationship between TP53 mutations and nodal metastases in gastric and head and neck cancers.39,40 In LUAD, mutations in both genes have been associated with adrenal gland metastasis, whereas TP53 mutations have also been associated with CNS metastasis.15 In contrast to previous site-specific analyses, primary tumors in our study were predominantly untreated (only 10% of ES-M and LS-M patients had prior treatment before sequencing).14,15 This is relevant, as studies have revealed that systemic therapies can lead to private alterations, particularly in metastases.14,41 Our data suggests that alterations in SMARCA4 and TP53 are related to metastasis in primary LUAD and that the higher risk associated with these alterations is unequally represented among organ sites.
Clinicopathologic and genomic features, including younger age, higher chromosomal instability, TP53 alteration, and ERBB2 alteration, were associated with higher metastatic burden. One can speculate that behavioral differences may account for some of the observed associations—younger individuals may be more likely to ignore early symptoms of lung cancer. However, prior work suggests possible alternative explanations, such as a more aggressive biology of LUAD in younger patients.42 Various patterns of metastatic development also exist among patients with adrenal gland and liver metastases, which rarely occurred as solitary metastases. We found that CDKN2A alterations were associated with shorter time to bone metastasis and Hippo pathway alterations were associated with shorter time to CNS metastasis. Recently, Shih et al. showed that genes in the Hippo pathway were amplified at a higher frequency in LUAD brain metastases.43 In contrast to CDKN2A and the Hippo pathway, SMARCA4 alterations were associated with worse MFS across all sites. While both SMARCA4 and CDKN2A are known to be associated with poor outcomes in LUAD, our study describes potential relationships with organotropisms.31,32,38,43,44 We show that SMARCA4 alterations were associated with higher probability of bone-specific metastasis, whereas CDKN2A alterations result in shorter time to bone metastasis. Collectively, these findings highlight the complexity of metastatic organotropism, as various factors may affect the site, timing, and number of metastases.
Tumor evolution over time in response to external factors such as treatments and during the metastatic process is well established.14 A pan-cancer study showed that brain metastases possess unique genomic alterations compared with matched primary tumors, and despite temporospatial differences, brain metastases were more alike compared to the primary tumor or other organ site metastases.45 Similarly, we identified multiple genomic differences—including alterations in specific genes and mutational signatures—among metastases from different sites and between metastases and primary tumors, with important treatment implications. For example, some of the most frequently targeted alterations, including KRAS and other alterations in the RTK/RAS pathway, were significantly less frequent among metastatic samples. Across metastatic sites, the difference in actionable alterations was even more pronounced. Despite previous studies showing that both primary tumors and metastases possess level 1 actionable alterations in >50% of samples, there was robust variation among sites, with adrenal metastases possessing few actionable alterations and pleural metastases possessing many.46
The potential clinical implications of tumor evolution are best illustrated through comparisons of paired primary and metastatic samples. Metastases often arise from major clones within the primary tumor, consistent with an evolutionary bottleneck, and many driver mutations occur early in tumor evolution, whereas alterations private to the metastasis are often caused by treatment effect.12,14,47 In concordance with these observations, we found that driver and actionable alterations were often clonal and shared, while alterations private to metastases were frequently copy number alterations of unknown significance, rarely actionable, and, in some cases, related to treatment resistance. Collectively, these differences illustrate the considerable complexity of tumor evolution, and that higher chromosomal instability leads to distinct cell populations in metastases, with potentially important clinical consequences.
The primary strength of our study is its use of a richly annotated clinicopathologic and genomic data set curated from our large cohorts of primary tumors and metastatic samples. Additionally, we were able to externally validate several of our key findings using the AACR GENIE data set. Limitations include the sampling of a single site of the primary tumor or metastatic lesion, rather than multiple regions, which may fail to capture the intra-tumoral heterogeneity described by Swanton et al.48 The use of a targeted sequencing panel for most of our cohort is another limitation. This was partially addressed by performing WES on a subset of patients, which allowed us to investigate mutational signatures in more detail. Consistent with previous findings,49,50 a comparison of cases sequenced with MSK-IMPACT and WES shows good concordance for estimates of TMB, FGA, and WGD, suggesting that our results would likely remain unchanged if we had performed WES on the entire cohort. Further, the number of patients in our study with matched primary and metastatic samples was relatively small. The unmatched analyses allowed us to highlight general trends of organotropism; however, the statistical power of our matched cohort was insufficient to draw definitive conclusions about tumor evolution after stratification for specific metastatic sites. The sample size challenges for our matched cohort are a direct reflection of current practice for clinically indicated tissue sampling. Confirmation and treatment of metastatic disease with the acquisition of tissue is an inherently biased process. Biopsy may be avoided altogether for suspected diffusely metastatic disease, or biopsy may be performed on the most accessible site of disease. Additionally, patient factors may play a role in tissue sampling. However, the results of this study are still generalizable, as they are real-world data based on accepted clinical practice.
In conclusion, we have performed an integrative analysis of clinicopathologic and genomic variables, including manually curated annotations across multiple sites and timing of metastasis, for a large cohort of patients with LUAD. Our results highlight features associated with the development of metastasis, metastatic burden, organotropisms, and MFS. By using a subset of patients with matched primary and metastatic samples, we have obtained additional insights into the spatiotemporal evolution of metastases. While chromosomal instability clearly plays an important role in metastatic dissemination, unfortunately the higher frequency of copy number changes observed in metastatic specimens does not translate to an increase in therapeutically targetable alterations.
STAR Methods
Resource Availability
Lead contact
Further information and requests for resources or data should be directed to and will be fulfilled by David R. Jones (jonesd2@mskcc.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The cBioPortal link for all the study data and information to access the account are listed below: https://www.cbioportal.org/study/summary?id=luad_mskcc_2023_met_organotropism.
All the data from the AACR GENIE BPC NSCLC v2.0 public cohort and the AACR GENIE v13.0 cohort that were used for validation purposes are publicly available and can be accessed using this link: https://genie.cbioportal.org/
The MSK-IMPACT data analysis pipeline can be found at https://github.com/rhshah/IMPACT-Pipeline. The mutational signature decomposition code can be found at https://github.com/mskcc/tempoSig. Genomic alterations were annotated with information from OncoKB using the OncoKB annotator tool (https://github.com/oncokb/oncokb-annotator). Additional custom written tools and programs used in our analyses are available from https://github.com/mskcc.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study population
This study was approved by the institutional review board at Memorial Sloan Kettering Cancer Center (IRB#18–391). All patients included in the study received a pathologic diagnosis of LUAD using either a tissue biopsy specimen or a surgical specimen from October 2006 to July 2021 and provided written consent to participate in the protocol. Exclusion criteria included mucinous or minimally invasive LUAD, no available matched-normal sample, and low tumor purity. NM and ES-M patients were excluded if they had microscopic (R1) or macroscopic (R2) residual disease after surgery. LS-M patients who received consolidative, maintenance, or palliative chemoradiotherapy before sample acquisition were excluded because of the potential for therapy-related changes to the tumor genomic profile.51,52 Patients from all other groups were included regardless of treatment status. The receipt of induction therapy among NM and ES-M patients was not an exclusion criterion, since it remains unknown whether short-course chemotherapy or radiotherapy induces meaningful tumor genomic alterations.
Clinical annotation and data curation
Clinical characteristics, imaging findings (including preoperative computed tomography, positron emission tomography, and/or magnetic resonance imaging), and pathologic reports from biopsy or surgical specimens (8th edition AJCC Cancer Staging Manual) were manually reviewed and annotated. A subset of samples included in the study underwent annotation for use in the NSCLC cohort of the American Association for Cancer Research Project Genomics Evidence Neoplasia Information Exchange biopharma consortium.53 Follow-up was performed in accordance with National Comprehensive Cancer Network guidelines.54 For primary tumors, median follow-up was 3.1 years, which was calculated from the date of sample acquisition to the date of last follow-up. Recurrences were distinguished from new primary tumors using the Martini and Melamed criteria, with differentiation according to pathologic and genomic relatedness when available, as previously reported by our group.55,56
Organ site and date of metastasis were obtained for the first lesion at a given organ site. Any additional lesions at a preexisting metastatic site were not included. Therefore, each site was only accounted for once at the time of the initial diagnosis of metastasis. Metastatic burden was defined as the number of distinct organ sites affected in a patient’s clinical course, consistent with the definition used in previous studies.15 Any lesions deemed suspicious on a radiology report were considered metastatic if the treating clinician reported the lesion as metastatic in clinic notes and/or tailored therapy to a diagnosis of metastatic disease.
METHOD DETAILS
DNA sequencing
LUAD specimens were profiled by use of hybridization capture-based next-generation sequencing (MSK-IMPACT) at Memorial Sloan Kettering Cancer Center.26,57 There were 4 versions of the MSK-IMPACT sequencing platform used during the study, as the panel of sequenced genes increased during the study: 341 (n=127, 5.2% of samples), 410 (n=606, 25.0% of samples), 468 (n=1356, 55.9% of samples), or 505 (n=333, 13.7% of samples) (Table S7). All reported alteration frequencies were adjusted to account for the specific genes included in each version of the MSK-IMPACT panel by dividing by the number of samples for which a given gene was sequenced. Similarly, estimates of TMB were adjusted to account for differences in genomic coverage across panels. The MSK-IMPACT assay achieves a high depth of sequencing (800x) and is performed in a Clinical Laboratory Improvement Amendments (CLIA) certified molecular laboratory, as previously described.13,57 For WES, DNA sequence libraries for 231tumor and matched blood normal specimens that had previously been sequenced with MSK-IMPACT were recaptured using the Agilent SureSelect Exome Kit (v3) and sequenced on an Illumina HiSeq 2500. One hundred twenty one of these 231 tumors and blood pairs were a subset of the core cohort of 2422 cases shown in Figure 1. The remaining 110 pairs were additional LUAD cases added to increase the size of the WES cohort. WES was performed at a depth of 150x for tumor DNA and 70x for blood DNA.
Genomic analyses
TMB was defined as the total number of nonsynonymous single-nucleotide variants per megabase (mut/Mb) and was normalized by panel size. FGA was defined as the number of bases with log2 copy number variation (gain or loss) >0.2 divided by the total number of genome bases profiled. Allele-specific analyses of copy number deletions, amplifications, and WGD were computed using the Fraction and Allele-specific Copy number Estimates from Tumor Sequencing (FACETS) algorithm.58 FACETS corrects for purity and ploidy and quantifies chromosomal instability by estimating integer DNA copy number calls from the sequencing samples. Tumors were considered to have undergone WGD if greater than 50% of their autosomal genome had a major copy number (the more frequent allele in a given segment) greater than or equal to two.50 Mutations were considered subclonal when the cancer cell fraction was less than 0.8, as estimated by FACETS. The clonal fraction was calculated by dividing the total number of clonal mutations by the sum of all clonal and subclonal mutations.
Ten canonical signaling pathways (cell cycle, Hippo, Myc, Notch, Nrf2, PI3K, RTK/RAS, TGFβ, p53, and Wnt) and all genes mutated in ≥3% of the overall cohort were investigated for associations with site-specific metastases (Table S7).26,27 The oncogenic effects and treatment implications of individual variants were annotated using the OncoKB precision oncology knowledgebase, an FDA-recognized human genetic variant database curated by experts at Memorial Sloan Kettering.59 The term “driver” refers to functionally relevant alterations, including mutations and copy number changes known to activate oncogenes or inactivate tumor suppressor genes. These are labeled as oncogenic, likely oncogenic, or predicted oncogenic in the OncoKB knowledge base. A “passenger” alteration has no known oncogenic effect according to OncoKB. Information on therapeutic actionability was also annotated using OncoKB, and each genomic alteration was stratified into 1 of 4 levels of clinical actionability, including FDA-recognized biomarkers of response (level 1), standard of care biomarkers (level 2), investigational biomarkers (level 3), and biomarkers of hypothetical relevance based on preclinical evidence (level 4). Similar to the definitions used in other studies, levels 1 to 3A were considered “actionable” or “targetable” alterations.15,46,59 Mutational signatures were computed for MSK-IMPACT samples with at least 13.8 mut/Mb, as justified in previous publications (Table S7).13,24,37 We used our own publicly available code for signature decomposition (https://github.com/mskcc/tempoSig) and the single-base substitution signatures defined in the Catalogue of Somatic Mutations in Cancer (COSMIC) database.60 Samples were considered to have a signature presence if the mean signature value was >0.1. All genomic and clinical data for patients in this study are publicly available through the cBioPortal for Cancer Genomics.61,62
In the matched analysis, alterations were labeled as private if they occurred only in the primary sample or only in the metastatic sample(s) for an individual patient. Shared alterations were observed in both primary and metastatic sample(s) from the same patient. In the event a patient had multiple metastases, alterations were considered shared if they were found in the primary and at least one metastatic sample. Alterations observed in multiple metastases from the same patient were not considered shared in these analyses. Phylogenetic trees were manually curated and drawn based on co-occurrence of oncogenic somatic alterations across samples. The lengths of the lines indicate time between sample collection and are not necessarily an estimate of sample similarity. The location of the alteration labels distinguishes alterations that are shared across samples from those unique to a particular sample. For example, in Figure 6B, amplifications of CDK4 and MDM2 are shared across all samples, whereas CDKN2A deletion was present in all metastatic lesions but missing in the primary tumor.
QUANTIFICATION AND STATISTICAL ANALYSIS
The cohorts included for each individual analysis are outlined in Table S8. For comparisons between ever-metastatic tumors (ES-M and LS-M) and nonmetastatic tumors (NM) and between primary tumors (ES-M and LS-M) and metastatic lesions (ML), Fisher’s exact test was used to compare categorical variables, and the Wilcoxon rank-sum test was used to compare continuous variables. Fisher’s exact test was used to compare the alteration frequencies of genes (altered in ≥3% of the entire cohort) and pathways and the presence of APOBEC signatures between groups.
Mutual exclusivity and co-occurrence of genes and pathways were assessed using Fisher’s exact test. P values were adjusted to correct for multiple comparisons using the false-discovery rate. The same methodology was applied to analysis of the ML samples, where one lesion site was compared to the other lesion sites.
Univariable logistic regression analysis was performed to quantify the relationships between preoperative clinicopathologic features and site-specific metastatic outcomes in ever-metastatic (ES-M and LS-M) samples. For the analysis of site-specific metastasis in primary tumors, any patient who developed a metastasis at the site of interest was compared with patients who did not have metastasis to that site. The nonmetastatic to site group included patients with no recorded metastasis at the site of interest and at least 2 years of follow-up from the time of surgery or biopsy. Two separate univariable logistic regression analyses were performed to quantify the relationships between genomic features and site-specific metastatic outcomes—one inclusive of gene alterations and one inclusive of pathway alterations. The regressions, including genomic features, were corrected for multiple testing with the false-discovery rate. A multivariable logistic regression model was then constructed with features with q<0.1 in univariable analyses by use of a backward-selection method. Again, pathways and genes were included in two distinct multivariable models. All analyses were two-sided, and p<0.05 was considered statistically significant.
Time to recurrence was defined as the time from biopsy or surgery to the first report of site-specific metastasis in the patient record or, if no recurrence was observed, from biopsy or surgery to last follow-up. For patients without metastasis at the time of diagnosis, MFS was defined from the time of surgery to the development of the first metastasis and was censored at last follow-up or death. An exploratory analysis of the effects of gene alterations and MFS was performed using multivariable Cox proportional hazards regression on the NM and ES-M patients.
Site-specific MFS was defined from the time of surgery to the development of the first metastasis at the site of interest and was censored at last follow-up or death. In an analysis restricted to ES-M patients, univariable cause-specific Cox proportional hazards regression was performed to quantify the relationships between preoperative clinicopathologic features and time to site-specific metastasis. The time to first metastasis (other than the metastatic site of interest) was incorporated into the analyses as a time-varying variable for each patient. Univariable Cox proportional hazards models were then constructed to quantify the relationships between clinicopathologic and genomic factors and the hazard of site-specific metastasis, incorporating the time-varying variable of time to first metastasis. For this analysis, two distinct sets of univariable models were constructed—one inclusive of gene alterations and one inclusive of pathway alterations. The regressions, including genomic features, were corrected for multiple testing correction with the false-discovery rate. Two-sided q<0.1 indicated statistical significance; features with q<0.1 were used to generate a clinicopathologic-adjusted multivariable model constructed using the backward-selection method. If alterations in a pathway and in a gene within that pathway were both statistically significant, the pathway was preferentially selected. All analyses were two-sided, and p<0.05 was considered to indicate statistical significance. All analyses were performed using Stata 15.0 (StataCorp, College Station, TX) and R 4.1.1 (R Core Team, Vienna, Austria).
Supplementary Material
Table S1. Clinical and genomic features for patients in the MSK-IMPACT and WES cohorts, Related to Figure 1
Table S2. Comparison of clinical and genomic features and mutational signatures in nonmetastatic (NM) and ever-metastatic (EM) cohorts, Related to Figure 2
Table S3. Analysis of clinical and genomic associations with metastatic burden, site-specific metastasis, and time to metastasis in ever-metastatic (EM) patients, Related to Figure 3
Table S4. Genomic and mutational signature analysis for primary samples with history of metastasis to specific site and metastatic lesions at these sites, Related to Figure 4
Table S5. Mutational signature decomposition for samples from the MSK-IMPACT and WES cohorts, Related to Figure 4
Table S6. Analyses of mutational signatures for primary versus metastatic lesions and by metastatic site in WES cohort, Related to Figure 4
Table S7. Genes in MSK-IMPACT panels, pathway alterations, and MAF for the MSK-IMPACT and WES cohorts, Related to STAR Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Human tumor and matched normal samples (blood) | This paper | This paper |
| Deposited Data | ||
| Clinical data, including metastatic events at the patient level is deposited for visualization, and download in the cBioPortal for Cancer Genomics. |
This paper | https://www.cbioportal.org/study/summary?id=luad_mskcc_2023_met_organotropism. |
| Genomic alterations data (mutations, copy number alterations, fusions) is deposited for visualization, and download in the cBioPortal for Cancer Genomics. | This paper |
https://www.cbioportal.org/study/summary?id=luad_mskcc_2023_met_organotropism. |
| GENIE-BPC NSCLCv2.0 | Pugh et al, 2022 | https://www.aacr.org/professionals/research/aacr-project-genie/bpc/nsclc/ https://genie.cbioportal.org/ |
| Software and Algorithms | ||
| cBioPortal | Cerami et al., 2012 | https://www.cbioportal.org/ |
| FACETS | Shen and Seshan, 2016 |
https://github.com/mskcc/facets-suite |
| IMPACT Pipeline and Analysis tools | Cheng et al., 2015 | https://github.com/rhshah/IMPACT-Pipeline https://github.com/mskcc |
| OncoKB | Chakravarty et al., 2017 | https://github.com/oncokb/oncokb |
| tempoSig | Alexandrov et al., 2013 | https://github.com/mskcc/tempoSig |
| R (v4.1.1) | R CRAN | https://cran.r-project.org/ |
| Stata(v1.5) | StataCorp | https://www.stata.com/ |
Highlights.
Primary tumors that metastasize have distinct genomic and clinical profiles
TP53, SMARCA4, and CDKN2A alterations influence time to and site of metastasis
Driver mutations are frequently clonal and are shared by primaries and metastases
Alterations private to metastases are rarely actionable and relate to prior treatment
Acknowledgments
We thank David Sewell for his superb editing and handling of the manuscript submission.
Funding
National Institutes of Health (NIH) grant R01CA217169 and R01CA240472 (both to D.R.J.), NIH grant R01CA236615 (to P.S.A.), NIH grant R01CA192399 (to M.W.M.), Hamilton Family Foundation (to D.R.J.), Al-Asmakh Foundation (to D.R.J.), US Department of Defense grant LC160212 (to P.S.A.), NIH grant T32CA009501 (to J.G.C.), NIH grant P30CA008748 (to Memorial Sloan Kettering Cancer Center).
C.M.R. has consulted regarding oncology drug development with AbbVie, Amgen, Astra Zeneca, Epizyme, Genentech/Roche, Ipsen, Jazz, Lilly, and Syros and serves on the scientific advisory boards of Bridge Medicines, Earli, and Harpoon Therapeutics. G.J.R. has institutional research funding from Mirait, Takeda, Merck, Roche, Novartis, and Pfizer. D.B.S. has consulted for and received honoraria from Pfizer, Lilly/Loxo Oncology, Vividion Therapeutics, Scorpion Therapeutics and BridgeBio. M.F.B. has consulted for Eli Lilly and PetDx and has received research funding from Grail not related to the work presented. D.R.J. is a member of the Advisory Council for Astra Zeneca and member of the Clinical Trial Steering Committee for Merck.
Footnotes
Declaration of Interests
All other authors have no relevant competing interests to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lambert AW, Pattabiraman DR, Weinberg RA, (2017), Emerging biological principles of metastasis, Cell, 168, 670–691, 10.1016/j.cell.2016.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lou F, Huang J, Sima CS, Dycoco J, Rusch V, Bach PB, (2013), Patterns of recurrence and second primary lung cancer in early-stage lung cancer survivors followed with routine computed tomography surveillance, J Thorac Cardiovasc Surg, 145, 75–81; discussion 81–72, 10.1016/j.jtcvs.2012.09.030 [DOI] [PubMed] [Google Scholar]
- 3.Watanabe K, Tsuboi M, Sakamaki K, Nishii T, Yamamoto T, Nagashima T, Ando K, Ishikawa Y, Woo T, Adachi H, Kumakiri Y, Maehara T, Nakayama H, Masuda M, (2016), Postoperative follow-up strategy based on recurrence dynamics for non-small-cell lung cancer, Eur J Cardiothorac Surg, 49, 1624–1631, 10.1093/ejcts/ezv462 [DOI] [PubMed] [Google Scholar]
- 4.Brandt WS, Yan W, Zhou J, Tan KS, Montecalvo J, Park BJ, Adusumilli PS, Huang J, Bott MJ, Rusch VW, Molena D, Travis WD, Kris MG, Chaft JE, Jones DR, (2019), Outcomes after neoadjuvant or adjuvant chemotherapy for cT2–4N0–1 non-small cell lung cancer: A propensity-matched analysis, J Thorac Cardiovasc Surg, 157, 743–753 e743, 10.1016/j.jtcvs.2018.09.098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ganti AK, Klein AB, Cotarla I, Seal B, Chou E, (2021), Update of incidence, prevalence, survival, and initial treatment in patients with non-small cell lung cancer in the US, JAMA Oncol, 7, 1824–1832, 10.1001/jamaoncol.2021.4932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tamura T, Kurishima K, Nakazawa K, Kagohashi K, Ishikawa H, Satoh H, Hizawa N, (2015), Specific organ metastases and survival in metastatic non-small-cell lung cancer, Mol Clin Oncol, 3, 217–221, 10.3892/mco.2014.410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen DX, Bos PD, Massagué J, (2009), Metastasis: from dissemination to organ-specific colonization, Nat Rev Cancer, 9, 274–284, 10.1038/nrc2622 [DOI] [PubMed] [Google Scholar]
- 8.Paget S, (1989), The distribution of secondary growths in cancer of the breast. 1889, Cancer Metastasis Rev, 8, 98–101. [PubMed] [Google Scholar]
- 9.Fidler IJ, (2003), The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited, Nat Rev Cancer, 3, 453–458, 10.1038/nrc1098 [DOI] [PubMed] [Google Scholar]
- 10.Massagué J, Ganesh K, (2021), Metastasis-initiating cells and ecosystems, Cancer Discov, 11, 971–994, 10.1158/2159-8290.Cd-21-0010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, Singh S, Williams C, Soplop N, Uryu K, Pharmer L, King T, Bojmar L, Davies AE, Ararso Y, Zhang T, Zhang H, Hernandez J, Weiss JM, Dumont-Cole VD, Kramer K, Wexler LH, Narendran A, Schwartz GK, Healey JH, Sandstrom P, Labori KJ, Kure EH, Grandgenett PM, Hollingsworth MA, de Sousa M, Kaur S, Jain M, Mallya K, Batra SK, Jarnagin WR, Brady MS, Fodstad O, Muller V, Pantel K, Minn AJ, Bissell MJ, Garcia BA, Kang Y, Rajasekhar VK, Ghajar CM, Matei I, Peinado H, Bromberg J, Lyden D, (2015), Tumour exosome integrins determine organotropic metastasis, Nature, 527, 329–335, 10.1038/nature15756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R, Salm M, Horswell S, Escudero M, Matthews N, Rowan A, Chambers T, Moore DA, Turajlic S, Xu H, Lee SM, Forster MD, Ahmad T, Hiley CT, Abbosh C, Falzon M, Borg E, Marafioti T, Lawrence D, Hayward M, Kolvekar S, Panagiotopoulos N, Janes SM, Thakrar R, Ahmed A, Blackhall F, Summers Y, Shah R, Joseph L, Quinn AM, Crosbie PA, Naidu B, Middleton G, Langman G, Trotter S, Nicolson M, Remmen H, Kerr K, Chetty M, Gomersall L, Fennell DA, Nakas A, Rathinam S, Anand G, Khan S, Russell P, Ezhil V, Ismail B, Irvin-Sellers M, Prakash V, Lester JF, Kornaszewska M, Attanoos R, Adams H, Davies H, Dentro S, Taniere P, O’Sullivan B, Lowe HL, Hartley JA, Iles N, Bell H, Ngai Y, Shaw JA, Herrero J, Szallasi Z, Schwarz RF, Stewart A, Quezada SA, Le Quesne J, Van Loo P, Dive C, Hackshaw A, Swanton C, Consortium TR, (2017), Tracking the evolution of non-small-cell lung cancer, N Engl J Med, 376, 2109–2121, 10.1056/NEJMoa1616288 [DOI] [PubMed] [Google Scholar]
- 13.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, Hellmann MD, Barron DA, Schram AM, Hameed M, Dogan S, Ross DS, Hechtman JF, DeLair DF, Yao J, Mandelker DL, Cheng DT, Chandramohan R, Mohanty AS, Ptashkin RN, Jayakumaran G, Prasad M, Syed MH, Rema AB, Liu ZY, Nafa K, Borsu L, Sadowska J, Casanova J, Bacares R, Kiecka IJ, Razumova A, Son JB, Stewart L, Baldi T, Mullaney KA, Al-Ahmadie H, Vakiani E, Abeshouse AA, Penson AV, Jonsson P, Camacho N, Chang MT, Won HH, Gross BE, Kundra R, Heins ZJ, Chen HW, Phillips S, Zhang H, Wang J, Ochoa A, Wills J, Eubank M, Thomas SB, Gardos SM, Reales DN, Galle J, Durany R, Cambria R, Abida W, Cercek A, Feldman DR, Gounder MM, Hakimi AA, Harding JJ, Iyer G, Janjigian YY, Jordan EJ, Kelly CM, Lowery MA, Morris LGT, Omuro AM, Raj N, Razavi P, Shoushtari AN, Shukla N, Soumerai TE, Varghese AM, Yaeger R, Coleman J, Bochner B, Riely GJ, Saltz LB, Scher HI, Sabbatini PJ, Robson ME, Klimstra DS, Taylor BS, Baselga J, Schultz N, Hyman DM, Arcila ME, Solit DB, Ladanyi M, Berger MF, (2017), Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients, Nat Med, 23, 703–713, 10.1038/nm.4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu Z, Li Z, Ma Z, Curtis C, (2020), Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases, Nat Genet, 52, 701–708, 10.1038/s41588-020-0628-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen B, Fong C, Luthra A, Smith SA, DiNatale RG, Nandakumar S, Walch H, Chatila WK, Madupuri R, Kundra R, Bielski CM, Mastrogiacomo B, Donoghue MTA, Boire A, Chandarlapaty S, Ganesh K, Harding JJ, Iacobuzio-Donahue CA, Razavi P, Reznik E, Rudin CM, Zamarin D, Abida W, Abou-Alfa GK, Aghajanian C, Cercek A, Chi P, Feldman D, Ho AL, Iyer G, Janjigian YY, Morris M, Motzer RJ, O’Reilly EM, Postow MA, Raj NP, Riely GJ, Robson ME, Rosenberg JE, Safonov A, Shoushtari AN, Tap W, Teo MY, Varghese AM, Voss M, Yaeger R, Zauderer MG, Abu-Rustum N, Garcia-Aguilar J, Bochner B, Hakimi A, Jarnagin WR, Jones DR, Molena D, Morris L, Rios-Doria E, Russo P, Singer S, Strong VE, Chakravarty D, Ellenson LH, Gopalan A, Reis-Filho JS, Weigelt B, Ladanyi M, Gonen M, Shah SP, Massague J, Gao J, Zehir A, Berger MF, Solit DB, Bakhoum SF, Sanchez-Vega F, Schultz N, (2022), Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients, Cell, 185, 563–575.e511, 10.1016/j.cell.2022.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, Jamal-Hanjani M, Shafi S, Murugaesu N, Rowan AJ, Grönroos E, Muhammad MA, Horswell S, Gerlinger M, Varela I, Jones D, Marshall J, Voet T, Van Loo P, Rassl DM, Rintoul RC, Janes SM, Lee SM, Forster M, Ahmad T, Lawrence D, Falzon M, Capitanio A, Harkins TT, Lee CC, Tom W, Teefe E, Chen SC, Begum S, Rabinowitz A, Phillimore B, Spencer-Dene B, Stamp G, Szallasi Z, Matthews N, Stewart A, Campbell P, Swanton C, (2014), Spatial and temporal diversity in genomic instability processes defines lung cancer evolution, Science, 346, 251–256, 10.1126/science.1253462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turajlic S, Xu H, Litchfield K, Rowan A, Chambers T, Lopez JI, Nicol D, O’Brien T, Larkin J, Horswell S, Stares M, Au L, Jamal-Hanjani M, Challacombe B, Chandra A, Hazell S, Eichler-Jonsson C, Soultati A, Chowdhury S, Rudman S, Lynch J, Fernando A, Stamp G, Nye E, Jabbar F, Spain L, Lall S, Guarch R, Falzon M, Proctor I, Pickering L, Gore M, Watkins TBK, Ward S, Stewart A, DiNatale R, Becerra MF, Reznik E, Hsieh JJ, Richmond TA, Mayhew GF, Hill SM, McNally CD, Jones C, Rosenbaum H, Stanislaw S, Burgess DL, Alexander NR, Swanton C, (2018), Tracking cancer evolution reveals constrained routes to metastases: TRACERx Renal, Cell, 173, 581–594.e512, 10.1016/j.cell.2018.03.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Budczies J, von Winterfeld M, Klauschen F, Bockmayr M, Lennerz JK, Denkert C, Wolf T, Warth A, Dietel M, Anagnostopoulos I, Weichert W, Wittschieber D, Stenzinger A, (2015), The landscape of metastatic progression patterns across major human cancers, Oncotarget, 6, 570–583, 10.18632/oncotarget.2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao Y, Bado I, Wang H, Zhang W, Rosen JM, Zhang XH, (2019), Metastasis organotropism: redefining the congenial soil, Dev Cell, 49, 375–391, 10.1016/j.devcel.2019.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones GD, Brandt WS, Shen R, Sanchez-Vega F, Tan KS, Martin A, Zhou J, Berger M, Solit DB, Schultz N, Rizvi H, Liu Y, Adamski A, Chaft JE, Riely GJ, Rocco G, Bott MJ, Molena D, Ladanyi M, Travis WD, Rekhtman N, Park BJ, Adusumilli PS, Lyden D, Imielinski M, Mayo MW, Li BT, Jones DR, (2021), A genomic-pathologic annotated risk model to predict recurrence in early-stage lung adenocarcinoma, JAMA Surg, 156, e205601, 10.1001/jamasurg.2020.5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen H, Carrot-Zhang J, Zhao Y, Hu H, Freeman SS, Yu S, Ha G, Taylor AM, Berger AC, Westlake L, Zheng Y, Zhang J, Ramachandran A, Zheng Q, Pan Y, Zheng D, Zheng S, Cheng C, Kuang M, Zhou X, Zhang Y, Li H, Ye T, Ma Y, Gao Z, Tao X, Han H, Shang J, Yu Y, Bao D, Huang Y, Li X, Zhang Y, Xiang J, Sun Y, Li Y, Cherniack AD, Campbell JD, Shi L, Meyerson M, (2019), Genomic and immune profiling of pre-invasive lung adenocarcinoma, Nat Commun, 10, 5472, 10.1038/s41467-019-13460-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, Beer DG, Powell CA, Riely GJ, Van Schil PE, Garg K, Austin JHM, Asamura H, Rusch VW, Hirsch FR, Scagliotti G, Mitsudomi T, Huber RM, Ishikawa Y, Jett J, Sanchez-Cespedes M, Sculier JP, Takahashi T, Tsuboi M, Vansteenkiste J, Wistuba I, Yang PC, Aberle D, Brambilla C, Flieder D, Franklin W, Gazdar A, Gould M, Hasleton P, Henderson D, Johnson B, Johnson D, Kerr K, Kuriyama K, Lee JS, Miller VA, Petersen I, Roggli V, Rosell R, Saijo N, Thunnissen E, Tsao M, Yankelewitz D, (2011), International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society International Multidisciplinary Classification of Lung Adenocarcinoma, J Throac Oncol, 6, 244–285, 10.1097/JTO.0b013e318206a221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langenbucher A, Bowen D, Sakhtemani R, Bournique E, Wise JF, Zou L, Bhagwat AS, Buisson R, Lawrence MS, (2021), An extended APOBEC3A mutation signature in cancer, Nat Commun, 12, 1602, 10.1038/s41467-021-21891-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caso R, Sanchez-Vega F, Tan KS, Mastrogiacomo B, Zhou J, Jones GD, Nguyen B, Schultz N, Connolly JG, Brandt WS, (2020), The underlying tumor genomics of predominant histologic subtypes in lung adenocarcinoma, J Throac Oncol, 15, 1844–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swanton C, McGranahan N, Starrett GJ, Harris RS, (2015), APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity, Cancer Discov, 5, 704–712, 10.1158/2159-8290.CD-15-0344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou J, Sanchez-Vega F, Caso R, Tan KS, Brandt WS, Jones GD, Yan S, Adusumilli PS, Bott M, Huang J, Isbell JM, Sihag S, Molena D, Rusch VW, Chatila WK, Rekhtman N, Yang F, Ladanyi M, Solit DB, Berger MF, Schultz N, Jones DR, (2019), Analysis of tumor genomic pathway alterations using broad-panel next-generation sequencing in surgically resected lung adenocarcinoma, Clin Cancer Res, 25, 7475–7484, 10.1158/1078-0432.Ccr-19-1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, Chakravarty D, Daian F, Gao Q, Bailey MH, Liang WW, Foltz SM, Shmulevich I, Ding L, Heins Z, Ochoa A, Gross B, Gao J, Zhang H, Kundra R, Kandoth C, Bahceci I, Dervishi L, Dogrusoz U, Zhou W, Shen H, Laird PW, Way GP, Greene CS, Liang H, Xiao Y, Wang C, Iavarone A, Berger AH, Bivona TG, Lazar AJ, Hammer GD, Giordano T, Kwong LN, McArthur G, Huang C, Tward AD, Frederick MJ, McCormick F, Meyerson M, Van Allen EM, Cherniack AD, Ciriello G, Sander C, Schultz N, (2018), Oncogenic signaling pathways in The Cancer Genome Atlas, Cell, 173, 321–337.e310, 10.1016/j.cell.2018.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Consortium APG, (2017), AACR Project GENIE: powering precision medicine through an international consortium, Cancer Discov, 7, 818–831, 10.1158/2159-8290.CD-17-0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pugh TJ, Bell JL, Bruce JP, Doherty GJ, Galvin M, Green MF, Hunter-Zinck H, Kumari P, Lenoue-Newton ML, Li MM, Lindsay J, Mazor T, Ovalle A, Sammut SJ, Schultz N, Yu TV, Sweeney SM, Bernard B, AACR Project Genie Consortium G, Analysis Working G, (2022), AACR Project GENIE: 100,000 cases and beyond, Cancer Discov, 12, 2044–2057, 10.1158/2159-8290.CD-21-1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skoulidis F, Heymach JV, (2019), Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy, Nat Rev Cancer, 19, 495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schoenfeld AJ, Bandlamudi C, Lavery JA, Montecalvo J, Namakydoust A, Rizvi H, Egger J, Concepcion CP, Paul S, Arcila ME, Daneshbod Y, Chang J, Sauter JL, Beras A, Ladanyi M, Jacks T, Rudin CM, Taylor BS, Donoghue MTA, Heller G, Hellmann MD, Rekhtman N, Riely GJ, (2020), The genomic landscape of SMARCA4 alterations and associations with outcomes in patients with lung cancer, Clin Cancer Res, 26, 5701–5708, 10.1158/1078-0432.Ccr-20-1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lehtiö J, Arslan T, Siavelis I, Pan Y, Socciarelli F, Berkovska O, Umer HM, Mermelekas G, Pirmoradian M, Jönsson M, Brunnström H, Brustugun OT, Purohit KP, Cunningham R, Asl HF, Isaksson S, Arbajian E, Aine M, Karlsson A, Kotevska M, Hansen CG, Haakensen VD, Helland Å, Tamborero D, Johansson HJ, Branca RM, Planck M, Staaf J, Orre LM, (2021), Proteogenomics of non-small cell lung cancer reveals molecular subtypes associated with specific therapeutic targets and immune evasion mechanisms, Nat Cancer, 2, 1224–1242, 10.1038/s43018-021-00259-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.La Fleur L, Falk-Sörqvist E, Smeds P, Berglund A, Sundström M, Mattsson JS, Brandén E, Koyi H, Isaksson J, Brunnström H, Nilsson M, Micke P, Moens L, Botling J, (2019), Mutation patterns in a population-based non-small cell lung cancer cohort and prognostic impact of concomitant mutations in KRAS and TP53 or STK11, Lung Cancer, 130, 50–58, 10.1016/j.lungcan.2019.01.003 [DOI] [PubMed] [Google Scholar]
- 34.Jia Y, Yun C-H, Park E, Ercan D, Manuia M, Juarez J, Xu C, Rhee K, Chen T, Zhang H, Palakurthi S, Jang J, Lelais G, DiDonato M, Bursulaya B, Michellys P-Y, Epple R, Marsilje TH, McNeill M, Lu W, Harris J, Bender S, Wong K-K, Jänne PA, Eck MJ, (2016), Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors, Nature, 534, 129–132, 10.1038/nature17960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lengel HB, Connolly JG, Jones GD, Caso R, Zhou J, Sanchez-Vega F, Mastrogiacomo B, Isbell JM, Li BT, Liu Y, Rekhtman N, Jones DR, (2021), The emerging importance of tumor genomics in operable non-small cell lung cancer, Cancers (Basel), 13, 10.3390/cancers13153656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caso R, Connolly JG, Zhou J, Tan KS, Choi JJ, Jones GD, Mastrogiacomo B, Sanchez-Vega F, Nguyen B, Rocco G, Molena D, Sihag S, Adusumilli PS, Bott MJ, Jones DR, (2021), Preoperative clinical and tumor genomic features associated with pathologic lymph node metastasis in clinical stage I and II lung adenocarcinoma, NPJ Precis Oncol, 5, 70, 10.1038/s41698-021-00210-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones GD, Caso R, Tan KS, Mastrogiacomo B, Sanchez-Vega F, Liu Y, Connolly JG, Murciano-Goroff YR, Bott MJ, Adusumilli PS, (2021), KRASG12C mutation is associated with increased risk of recurrence in surgically resected lung adenocarcinoma, Clin Cancer Res, 27, 2604–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Concepcion CP, Ma S, LaFave LM, Bhutkar A, Liu M, DeAngelo LP, Kim JY, Del Priore I, Schoenfeld AJ, Miller M, Kartha VK, Westcott PMK, Sánchez-Rivera FJ, Meli K, Gupta M, Bronson RT, Riely GJ, Rekhtman N, Rudin CM, Kim CF, Regev A, Buenrostro JD, Jacks T, (2022), Smarca4 Inactivation promotes lineage-specific transformation and early metastatic features in the lung, Cancer Discov, 12, 562–585, 10.1158/2159-8290.Cd-21-0248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi JH, Kim YB, Ahn JM, Kim MJ, Bae WJ, Han SU, Woo HG, Lee D, (2018), Identification of genomic aberrations associated with lymph node metastasis in diffuse-type gastric cancer, Exp Mol Med, 50, 1–11, 10.1038/s12276-017-0009-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biswas NK, Das C, Das S, Maitra A, Nair S, Gupta T, D’Cruz AK, Sarin R, Majumder PP, (2019), Lymph node metastasis in oral cancer is strongly associated with chromosomal instability and DNA repair defects, Int J Cancer, 145, 2568–2579, 10.1002/ijc.32305 [DOI] [PubMed] [Google Scholar]
- 41.Hu Z, Curtis C, (2020), Looking backward in time to define the chronology of metastasis, Nat Commun, 11, 3213, 10.1038/s41467-020-16995-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sacher AG, Dahlberg SE, Heng J, Mach S, Janne PA, Oxnard GR, (2016), Association between younger age and targetable genomic alterations and prognosis in non-small-cell lung cancer, JAMA Oncol, 2, 313–320, 10.1001/jamaoncol.2015.4482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shih DJH, Nayyar N, Bihun I, Dagogo-Jack I, Gill CM, Aquilanti E, Bertalan M, Kaplan A, D’Andrea MR, Chukwueke U, Ippen FM, Alvarez-Breckenridge C, Camarda ND, Lastrapes M, McCabe D, Kuter B, Kaufman B, Strickland MR, Martinez-Gutierrez JC, Nagabhushan D, De Sauvage M, White MD, Castro BA, Hoang K, Kaneb A, Batchelor ED, Paek SH, Park SH, Martinez-Lage M, Berghoff AS, Merrill P, Gerstner ER, Batchelor TT, Frosch MP, Frazier RP, Borger DR, Iafrate AJ, Johnson BE, Santagata S, Preusser M, Cahill DP, Carter SL, Brastianos PK, (2020), Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma, Nat Genet, 52, 371–377, 10.1038/s41588-020-0592-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fernando TM, Piskol R, Bainer R, Sokol ES, Trabucco SE, Zhang Q, Trinh H, Maund S, Kschonsak M, Chaudhuri S, Modrusan Z, Januario T, Yauch RL, (2020), Functional characterization of SMARCA4 variants identified by targeted exome-sequencing of 131,668 cancer patients, Nat Commun, 11, 5551, 10.1038/s41467-020-19402-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, Van Allen EM, Lawrence MS, Horowitz PM, Cibulskis K, Ligon KL, Tabernero J, Seoane J, Martinez-Saez E, Curry WT, Dunn IF, Paek SH, Park SH, McKenna A, Chevalier A, Rosenberg M, Barker FG 2nd, Gill CM, Van Hummelen P, Thorner AR, Johnson BE, Hoang MP, Choueiri TK, Signoretti S, Sougnez C, Rabin MS, Lin NU, Winer EP, Stemmer-Rachamimov A, Meyerson M, Garraway L, Gabriel S, Lander ES, Beroukhim R, Batchelor TT, Baselga J, Louis DN, Getz G, Hahn WC, (2015), Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets, Cancer Discov, 5, 1164–1177, 10.1158/2159-8290.Cd-15-0369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chakravarty D, Solit DB, (2021), Clinical cancer genomic profiling, Nat Rev Genet, 22, 483–501, 10.1038/s41576-021-00338-8 [DOI] [PubMed] [Google Scholar]
- 47.Birkbak NJ, McGranahan N, (2020), Cancer genome evolutionary trajectories in metastasis, Cancer Cell, 37, 8–19, 10.1016/j.ccell.2019.12.004 [DOI] [PubMed] [Google Scholar]
- 48.Swanton C, Govindan R, (2016), Clinical implications of genomic discoveries in lung cancer, N Engl J Med, 374, 1864–1873, 10.1056/NEJMra1504688 [DOI] [PubMed] [Google Scholar]
- 49.Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, Plodkowski A, Long N, Sauter JL, Rekhtman N, Hollmann T, Schalper KA, Gainor JF, Shen R, Ni A, Arbour KC, Merghoub T, Wolchok J, Snyder A, Chaft JE, Kris MG, Rudin CM, Socci ND, Berger MF, Taylor BS, Zehir A, Solit DB, Arcila ME, Ladanyi M, Riely GJ, Schultz N, Hellmann MD, (2018), Molecular determinants of response to anti-programmed cell death (PD)-1 and Anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing, J Clin Oncol, 36, 633–641, 10.1200/JCO.2017.75.3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bielski CM, Zehir A, Penson AV, Donoghue MTA, Chatila W, Armenia J, Chang MT, Schram AM, Jonsson P, Bandlamudi C, Razavi P, Iyer G, Robson ME, Stadler ZK, Schultz N, Baselga J, Solit DB, Hyman DM, Berger MF, Taylor BS, (2018), Genome doubling shapes the evolution and prognosis of advanced cancers, Nat Genet, 50, 1189–1195, 10.1038/s41588-018-0165-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mouw KW, Cleary JM, Reardon B, Pike J, Braunstein LZ, Kim J, Amin-Mansour A, Miao D, Damish A, Chin J, Ott PA, Fuchs CS, Martin NE, Getz G, Carter S, Mamon HJ, Hornick JL, Van Allen EM, D’Andrea AD, (2017), Genomic evolution after chemoradiotherapy in anal squamous cell carcinoma, Clin Cancer Res, 23, 3214–3222, 10.1158/1078-0432.Ccr-16-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murugaesu N, Wilson GA, Birkbak NJ, Watkins T, McGranahan N, Kumar S, Abbassi-Ghadi N, Salm M, Mitter R, Horswell S, Rowan A, Phillimore B, Biggs J, Begum S, Matthews N, Hochhauser D, Hanna GB, Swanton C, (2015), Tracking the genomic evolution of esophageal adenocarcinoma through neoadjuvant chemotherapy, Cancer Discov, 5, 821–831, 10.1158/2159-8290.Cd-15-0412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lavery JA, Brown S, Riely GJ, Bedard PL, Park BH, Warner JL, Kehl KL, Lepisto EM, Rizvi H, LeNoue-Newton M. Pan-cancer evaluation of homologous repair deficiency somatic mutations and response to first-line anti-neoplastic therapy. In: Wolters Kluwer Health; 2021. [Google Scholar]
- 54.Ettinger DS, Wood DE, Aggarwal C, Aisner DL, Akerley W, Bauman JR, Bharat A, Bruno DS, Chang JY, Chirieac LR, D’Amico TA, Dilling TJ, Dobelbower M, Gettinger S, Govindan R, Gubens MA, Hennon M, Horn L, Lackner RP, Lanuti M, Leal TA, Lin J, Loo BW Jr., Martins RG, Otterson GA, Patel SP, Reckamp KL, Riely GJ, Schild SE, Shapiro TA, Stevenson J, Swanson SJ, Tauer KW, Yang SC, Gregory K, Hughes M, (2019), NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 1.2020, J Natl Compr Canc Netw, 17, 1464–1472, 10.6004/jnccn.2019.0059 [DOI] [PubMed] [Google Scholar]
- 55.Chang JC, Alex D, Bott M, Tan KS, Seshan V, Golden A, Sauter JL, Buonocore DJ, Vanderbilt CM, Gupta S, (2019), Comprehensive NGS unambiguously distinguishes separate primary lung carcinomas from intra-pulmonary metastases: Comparison with standard histopathologic approach, Clin Cancer Res, 25, 7113–7125, 10.1158/1078-0432.CCR-19-1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martini N, Melamed MR, (1975), Multiple primary lung cancers, J Thorac Cardiovasc Surg, 70, 606–612. https://www.ncbi.nlm.nih.gov/pubmed/170482. Published 1975/10/01. [PubMed] [Google Scholar]
- 57.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, Brannon AR, O’Reilly C, Sadowska J, Casanova J, Yannes A, Hechtman JF, Yao J, Song W, Ross DS, Oultache A, Dogan S, Borsu L, Hameed M, Nafa K, Arcila ME, Ladanyi M, Berger MF, (2015), Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology, J Mol Diagn, 17, 251–264, 10.1016/j.jmoldx.2014.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen R, Seshan VE, (2016), FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing, Nucleic Acids Res, 44, e131, 10.1093/nar/gkw520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH, (2017), OncoKB: a precision oncology knowledge base, JCO Prec Oncol, 1, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, (2020), The repertoire of mutational signatures in human cancer, Nature, 578, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. In. Vol 2: AACR; 2012:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, (2013), Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal, Sci Sig, 6, pl1-pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical and genomic features for patients in the MSK-IMPACT and WES cohorts, Related to Figure 1
Table S2. Comparison of clinical and genomic features and mutational signatures in nonmetastatic (NM) and ever-metastatic (EM) cohorts, Related to Figure 2
Table S3. Analysis of clinical and genomic associations with metastatic burden, site-specific metastasis, and time to metastasis in ever-metastatic (EM) patients, Related to Figure 3
Table S4. Genomic and mutational signature analysis for primary samples with history of metastasis to specific site and metastatic lesions at these sites, Related to Figure 4
Table S5. Mutational signature decomposition for samples from the MSK-IMPACT and WES cohorts, Related to Figure 4
Table S6. Analyses of mutational signatures for primary versus metastatic lesions and by metastatic site in WES cohort, Related to Figure 4
Table S7. Genes in MSK-IMPACT panels, pathway alterations, and MAF for the MSK-IMPACT and WES cohorts, Related to STAR Methods
Data Availability Statement
The cBioPortal link for all the study data and information to access the account are listed below: https://www.cbioportal.org/study/summary?id=luad_mskcc_2023_met_organotropism.
All the data from the AACR GENIE BPC NSCLC v2.0 public cohort and the AACR GENIE v13.0 cohort that were used for validation purposes are publicly available and can be accessed using this link: https://genie.cbioportal.org/
The MSK-IMPACT data analysis pipeline can be found at https://github.com/rhshah/IMPACT-Pipeline. The mutational signature decomposition code can be found at https://github.com/mskcc/tempoSig. Genomic alterations were annotated with information from OncoKB using the OncoKB annotator tool (https://github.com/oncokb/oncokb-annotator). Additional custom written tools and programs used in our analyses are available from https://github.com/mskcc.