

Summary
Although it is known that psoriasis is strongly associated with obesity, the mechanistic connection between diet and skin lesions is not well established. Herein, we showed that only dietary fat, not carbohydrates or proteins, exacerbates psoriatic disease. Enhanced psoriatic skin inflammation was associated with changes in the intestinal mucus layer and microbiota composition by high-fat diet (HFD). Change of intestinal microbiota by vancomycin treatment effectively blocked activation of psoriatic skin inflammation by HFD, inhibited the systemic interleukin-17 (IL-17) response, and led to increased mucophilic bacterial species such as Akkermansia muciniphila. By using IL-17 reporter mice, we could show that HFD facilitates IL-17-mediated γδ T cell response in the spleen. Notably, oral gavage with live or heat-killed A. muciniphila effectively inhibited HFD-induced enhancement of psoriatic disease. In conclusion, HFD exacerbates psoriatic skin inflammation through changing the mucus barrier and the intestine microbial composition, which leads to an enhanced systemic IL-17 response.
Keywords: psoriasis, high-fat diet, γδT cell
Graphical abstract
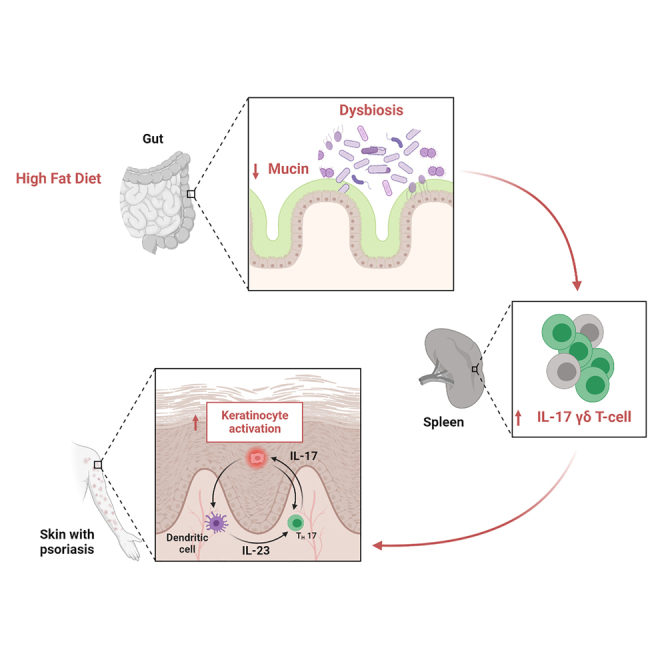
Highlights
-
•
HFD exacerbates psoriasis-like skin inflammation by inducing systemic IL-17 γδT cell response
-
•
HFD altering the intestinal mucus barrier and flora promotes psoriatic skin inflammation
-
•
Vancomycin treatment effectively blocks the activation of psoriatic skin inflammation by HFD
-
•
Akkermansia muciniphila administration inhibits HFD-induced enhancement of psoriatic disease
Sonomoto et al. show that high-fat diet (HFD) facilitates a systemic IL-17-mediated γδ T cell response in an intestine-dependent manner, exacerbating psoriasis-like inflammation. Administration of Akkermansia muciniphila effectively suppressed HFD-induced enhancement of psoriatic disease through intestinal microenvironment improvement.
Introduction
Psoriasis is a systemic immune-mediated inflammatory disease affecting the skin, joints, and cardiovascular system.1 While a substantial part of the susceptibility to psoriasis is based on genetic factors, such as HLA class I alleles and interleukin-23 (IL-23) receptor variants, it is also remarkably associated with metabolic disturbances.1 Thus, (1) obesity, diabetes, and lipid metabolism abnormalities are highly prevalent in patients with psoriasis, who also show an increased cardiovascular risk, despite no or only small elevation of systemic markers of inflammation.1 Furthermore, (2) the level of response to anti-inflammatory treatment depends on the level of metabolic disturbances in patients with psoriasis.2 In addition, (3) dietary measures have been shown to improve the response to anti-inflammatory treatment in patients with psoriasis.3 To date, little is known about the pathogenesis of metabolic disturbances in psoriasis and how they might interact with inflammation.
Diet-induced changes of the intestinal microbiota may explain the link between metabolic changes and inflammation in psoriatic disease. Changes in the intestinal microbiome have been reported in both patients with psoriasis4 and psoriatic arthritis.5 Indeed, intestinal microbiota is known to regulate host metabolism. For example, germ-free mice colonized by intestinal microbiota gain body mass.6 The taxonomic analysis of intestinal microbiomes in mice and humans revealed that in individuals who are obese, the relation between Bacteroidetes and Firmicutes shifts to the latter one, which is referred to as intestinal dysbiosis.7,8 Furthermore, modulation of the intestinal microbiota can influence diseases such as atherosclerosis and diabetes in mice and, to some extent, also in humans.9,10 Some intestinal bacterial species seem to influence the homeostasis in the gut toward a homeostatic anti-inflammatory milieu. Akkermansia (A.) muciniphila, for instance, is a gram-negative, strictly anaerobic bacterium that accounts for 1%–5% of human intestinal microbes.11,12 A. muciniphila can metabolize dietary fibers to produce short-chain fatty acids that will modulate the mucosal gene expression profile of the host. As a consequence, these bacteria directly participate in the maintenance of the gut barrier function, the host immune response, and other homeostatic functions.13 Furthermore, A. muciniphila treatment has been shown to counteract the increased circulating endotoxin level induced by high-fat diet (HFD).14
Of note, bacterial endotoxins are also an important stimulator of IL-23 secretion and sufficient to induce IL-17 secretion by γδ T cells,15 both of which represent the key two inflammatory mediators in psoriatic disease. Therefore, we sought for a mechanistic link between metabolic changes and inflammation in psoriasis and investigated how diet-induced changes of the gut microbiota influence psoriatic skin inflammation by investigating their effects on the key immune effectors in this disease.
Results
HFD exacerbates psoriasis-like skin inflammation
To assess the effects of dietary metabolic stress on psoriatic dermatitis, C57BL/6 male mice were fed diets high in fat, protein, or carbohydrates for 8 weeks. At the end of dietary exposure, mice were challenged to the ear imiquimod (IMQ) model for 3 days since immune cell infiltration in the ear was similar 3, 5, and 7 days post-treatment (Figure S1A). Skin inflammation was determined by measurement of ear thickness and histopathology. Intriguingly, only the fat-enriched diet significantly and dose-dependently exacerbated IMQ-induced skin inflammation (Figure 1A). Hence, ear thickness and histological changes increased with higher dietary fat content (Figure 1A). Significant differences were found between HFD (35.7% fat) and normal diet (ND; 3.3% fat) (Figures S1B and S1C). This phenotype was accompanied by weight gain and impaired glucose tolerance and insulin response in mice fed an HFD (Figures S1D–S1F). In contrast to the HFD, the sugar- or protein-enriched diet did not aggravate psoriatic skin inflammation (Figure 1B). Fluorescence-activated cell sorting (FACS)-based quantification of lesional immune cell infiltration revealed that neutrophils, dendritic cells, monocytes, and αβ and γδ T cells were significantly enriched in the skin lesions after HFD (Figure 1C). Details for gating for the various immune cell subsets are shown in Figures S2A and S2B.
Figure 1.

High-fat diet exacerbates psoriatic skin inflammation
C57BL/6 mice were fed a normal diet (2.9% or 3.3% fat content) or high-fat diet (11.3%, 35.7%, or 79.2% fat content) for 7–10 weeks. Mice were topically treated with imiquimod (IMQ) cream on one ear for the last 3 days of the experiment, while the contralateral ear remained unchallenged (n = 5–10 per group).
(A) Ear thickness measured by caliper and quantification of epidermal thickness in histopathology sections shown. Bars show the mean ± SD. p values were calculated by analysis of variance followed by Jonckheere-Terpstra test. Representative H&E staining of IMQ-treated ears are also shown.
(B) Correlation between fat, sugar, and protein contents of the food and ear thickness. Spearman’s rank correlation coefficient (rS) and p values are shown.
(C) Numbers of immune cells in IMQ-challenged and unchallenged contralateral ears of mice fed with normal diet (ND; 3.3% fat) or high-fat diet (HFD; 35.7% fat) (n = 10). Bars show the mean ± SD. p values were calculated by unpaired Student’s t test.
Quantification of lesional mRNA levels of inflammatory cytokines and chemokines showed that dietary metabolic stress (HFD; 35.7% fat) increased IMQ-induced upregulation of Il17a mRNA levels (Figure S3A). Other inflammatory mediators such as Il23, Il6, tumor necrosis factor α (Tnfa), or Gcsf were not significantly affected (Figure S3A). However, analysis of chemokine expression showed that Cxcl1 and Cxcl2, which attract neutrophils, monocytes, and T cells, were significantly more highly expressed after HFD (Figure S3B). In contrast, no differences in Cxcl3, Cxcl5, Ccl4, and Ccl20 were observed (Figure S3B).
Induction of a systemic IL-17 T cell response by HFD
When measuring IL-17 serum levels following IMQ challenge, we found that they increased in mice fed an HFD for 8 weeks compared with mice fed an ND (Figure 2A). To determine the main source of IL-17 in this context, we used IL-17A-IRES-GFP-knockin (KI) reporter mice and analyzed IL-17 production in different organs. IL-17 was produced in a wide range of organs. It was significantly increased in the spleen and, to a lower extent, in the mesenteric lymph nodes (mLNs) of mice exposed to HFD (Figure 2B). The spleen was the main source of IL-17 production in mice exposed to HFD (Figure 2C). This increase was based on IL-17+ γδ T cells but not IL-17+ αβ T cells or IL-17+ non-T cells (Figures 2D–2G and S4A–S4C). To delineate whether HFD alone affects IL-17 production, we analyzed the effect of HFD on IL-17+ cells and IL-17 production in the absence of IMQ. Interestingly, HFD induced IL-17 production, with the spleen being again the main source for IL-17 production (Figures S4D and S4E). At the cellular level, we identified γδ T cells as the key producer of IL-17 in response to HFD (Figures S4F–S4I). The subtyping analyses of γδ T cells by Vγ chains revealed that Vγ4+ and Vγ1−Vγ4− γδ T cells, which are capable of producing IL-17, were increased after HFD (Figure S4I). In contrast, the number of interferon γ (IFN-γ)-producing Vγ1 T cells was not changed (Figures S4I and S4J). In addition, no clonal expansion of γδ T cells was observed in γδ TCR repertoire analysis after HFD (Figures S5A–S5C). Next, we performed an adoptive transfer of splenic T cells isolated from ND- or HFD-fed IL-17A-GFP mice into ND mice (Figures S6A and S6B). However, we noticed that adoptively transferred donor IL-17A+ γδ T cells from HFD into ND mice were recruited to the spleen and the IMQ ear rather than other lymphoid organs such as mLNs (Figures S6C–S6E). Altogether, HFD aggravates systemic IL-17 response by stimulating IL-17 production by γδ T cells and their recruitment to the spleen and the skin-inflamed site.
Figure 2.

Induction of γδ T lymphocytes by HFD
(A) IL-17 serum levels in C57BL/6 mice fed an ND (3.3% fat) or HFD (35.7% fat) after the 3 day challenge with IMQ (n = 60).
(B and C) IL-17 production from different organs (B) and contribution of organs to IL-17 production (C) in IL-17 reporter mice fed an ND or HFD 3 days after being challenged with IMQ (n = 5–9). IL-17 production is defined as FITC-mean fluorescence intensity (MFI) ×IL-17A+ cell number.
(D) Representative dot plot showing IL-17+ γδ+ T lymphocytes pregated on CD3+ T cells in the spleen of mice fed an ND or HFD and challenged with IMQ.
(E and F) Number (E) and level (F) of IL-17 production (MFI) of IL-17+ γδ and αβ T lymphocytes in the spleen (n = 7–9).
(G) Contribution of IL-17-expressing cells to IL-17 production in the spleen. Bars show the mean ± SD.
p values were calculated by Welch’s t test (A) or unpaired Student’s t tests (B, E, and F) and are shown above each group.
HFD impairs the intestinal mucus barrier
Mechanistically, we hypothesized that the systemic production of IL-17 by T cells following HFD was initiated by an alteration of the small intestine since it absorbs fat from the food. To test this idea, we gave 4 kDa fluorescein isothiocyanate (FITC)-labeled dextran to mice to evaluate the intestinal barrier function mediated by tight junctions. However, intestinal permeability was intact after 8 weeks of HFD (Figure S7A). Next, the quality of the mucin barrier was evaluated. Histologic analyses revealed a thick and smooth mucin layer on the edges of the villi in the small intestine of normally fed mice, whereas only a thin, rough, and intermittent mucin layer, accompanied by fewer mucin-producing cells, was observed in the intestine of mice fed with HFD, irrespective of IMQ ear challenge (Figure 3A). In parallel, we observed a marked lipid droplet accumulation in the mucosa of HFD-fed mice (Figure 3A), indicating disruption of the intestinal architecture and possibly inducing inflammation across the mucosa, as reported in previous studies.16 These observations were correlated with decreased levels of Mucin-2 protein (Figure 3B) and decreased expression of Mucin-2, -3, -4, and -13 mRNA in the intestine (Figure 3C), whereas the expression profiles of pro-inflammatory cytokines were unchanged (Figure S7B). Altogether, these data suggest that HFD impaired the mucus barrier in the gut.
Figure 3.

HFD reduces mucin expression in the intestine
C57BL/6 mice were fed an ND (3.3% fat) or HFD (35.7% fat) for 8 weeks and ear challenged with IMQ or without IMQ.
(A) Alcian blue periodic acid-Schiff (AB-PAS) staining of the ileum. Purple layers on the edge of the villus represents the mucin layer (black arrowhead), and mucin-producing cells are depicted by the blue-stained cells.
(A and B) Representative pictures and thickness of PAS layers are shown (A: n = 5; B: n = 10).
(B) Representative pictures of Mucin 2 staining in the ileum. Red: mucin 2; blue: DAPI.
(C) Quantitative real-time PCR for mucin mRNA expression in the first half of ileum (n = 10). Bars in the graphs show the mean ± SD. p values were calculated by unpaired Student’s t test.
Exacerbation of psoriatic skin inflammation by HFD depends on the intestine
Based on our findings on the mucus barrier of the gut, we next investigated whether intestinal microbiota could participate in the exacerbation of psoriatic skin lesions by HFD. Therefore, we treated mice with the antibiotic vancomycin (VCM) in drinking water to specifically target gram-positive bacteria locally in the intestine. HFD-induced weight gain was still present with VCM treatment (Figure S8A). Of note, VCM treatment suppressed HFD-induced skin inflammation following IMQ challenge, as shown by the rescue of ear and epidermal thickening (Figure 4A).
Figure 4.

Intestinal microbiome affects the exacerbation of psoriatic skin inflammation by HFD
C57BL/6 mice were fed an ND (3.3% fat) or HFD (35.7% fat) for 8 weeks and challenged with IMQ. 0.5 mg/dL vancomycin (VCM) was administrated as drinking water for the last 2 weeks.
(A) Ear thickness measured by caliper (n = 5–15). Epidermal thickness in H&E sections (n = 5–10).
(B) Pathway analysis of serum molecules measured by liquid chromatography mass spectrometry. Shown are pathways upregulated in the HFD group but decreased by VCM treatment (n = 10).
(C) Quantitative real-time PCR of Il17a expression in the skin (n = 5).
(D) Circulating IL-17 was measured in the serum by ELISA (HFD; n = 60, HFD + VCM; n = 15).
(E) Immune cell numbers in the total ear measured by flow cytometry (n = 5). Bars show the mean ± SD. p values were calculated by analysis of variance followed by Sidak’s post hoc tests for comparison of multiple groups or by unpaired Student’s t test for comparison of two groups.
As gut microbiota influence host metabolism, we analyzed the possible metabolic perturbations induced by HFD. Therefore, we performed mass spectrometry analyses in the serum of ND mice, HFD mice, and HFD mice receiving VCM. Among all variables, phospholipid biosynthesis pathways related to known γδ T cell activators17 (cardiolipin, phosphatidylethanolamine, phosphatidylcholine, phosphatidylinositol phosphate) were induced by HFD and suppressed after VCM treatment (Figure 4B). Therefore, we hypothesized that fatty acids prompt the translocation of antigen fragments from the intestinal lumen to systemic organs via blood vessels and lymphatic ducts in HFD mice.18 In support of this hypothesis, inhibition of the translocation pathway by lomitapide partially rescued HFD-induced skin inflammation (Figures S8B and S8C). Moreover, induction of IL-17 expression in the affected skin (Figure 4C) and IL-17 level in the serum (Figure 4D) were significantly decreased in VCM-treated mice. Also, infiltration of myeloid cells and T cells, i.e., γδ T lymphocytes, were reduced by VCM treatment (Figures 4E and S8D). These observations suggest that intestinal microbiota mediates enhanced psoriatic skin inflammation in response to HFD.
A. muciniphila plays a key role in intestinal homeostasis
To understand the link between the metabolic changes observed and the gut microbiota composition, we quantified the microbiome in different parts of the intestine by qPCR for 16S rDNA. While HFD strongly diminished the number of bacteria in the ileum, IMQ challenge selectively increased the bacterial load in the caecum of VCM-treated mice (Figure S9). Next, we performed 16S-based microbiome sequencing analysis to characterize the microbiome in intestines of IMQ--challenged mice treated with ND, HFD, and HFD receiving VCM treatment. The genus A., in which A. muciniphila (Verrucomicrobiota phylum) is the only known species, mainly remains after VCM treatment (Figure 5A). Quantitative real-time PCR confirmed that the mice challenged with HFD and treated with VCM had large amounts of A. muciniphila in their ileum and caecum (Figure 5B).
Figure 5.

Akkermansia muciniphila regulates the intestinal homeostasis
(A and B) C57BL/6 mice were fed an ND (3.3% fat) or HFD (35.7% fat) for 8 weeks and challenged with IMQ. 0.5 mg/dL VCM was administrated in drinking water for the last 2 weeks.
(A) Microbiome sequencing analysis of the bacterial 16S rDNA in the intestine (n = 5).
(B) Quantitative real-time PCR of 16S rDNA of Akkermansia muciniphila (Akk) in the intestine (n = 5–10).
(C–F) C57BL/6 mice were fed an ND (3.3% fat) or HFD (35.7% fat) for 8 weeks and challenged with IMQ. VCM in drinking water and live or heat-killed Akk oral gavage were given for the last 2 weeks.
(C) Shannon index values indicated gut microbiota diversity.
(D) Microbiome sequencing analysis of the bacterial 16S rDNA in the ileum at the level of phylum and genus (n = 5).
(E) Quantitative real-time PCR of mucins of the first half of ileum (n = 6).
(F) AB-PAS staining of the ileum, and quantification of the PAS layer and the goblet cells/villus (n = 5–12). Bars show the mean ± SD. p values were calculated by analysis of variance followed by Sidak’s post hoc tests for comparison of multiple groups or by unpaired Student’s t test for comparison of two groups.
To determine whether A. muciniphila was responsible for reduction of skin inflammation observed after VCM treatment, HFD mice were gavaged with 2 × 108 colony-forming units (CFUs) of live or heat-killed A. muciniphila during the last 2 weeks of the experiment. First, we analyzed the composition of the gut microbiota following this treatment. According to the Shannon diversity index (Figure 5C) and the phylum and genus quantification (Figure 5D), both live and heat-killed A. muciniphila gavages restored the diversity of microbiome in the ileum that was reduced by HFD. No such changes were observed in the colon (Figures S10A and S10B). A. muciniphila gavages did not rescue the weight gain or the glucose response induced by HFD but ameliorated the insulin tolerance (Figures S10C–S10E). Interestingly, live and heat-killed A. muciniphila restored Muc4 mRNA expression in the ileum (Figure 5E), a transmembrane mucin expressed on the apical surfaces of epithelial cells that forms a glycocalyx layer to protect the gut barrier.19 This was not the case for other mucins like Mucin-2, -3, and -13, which did not respond to the treatment (Figure 5E). In the colon, Muc2 expression was restored after treatment with living A. muciniphila (Figures S10F and S10G). Gavage with live A. muciniphila also increased periodic acid-Schiff (PAS) layer and goblet cell density in the ileum, which was reduced by HFD (Figure 5F). This set of data suggests that administration of A. muciniphila modulates gut microbiota diversity and restores the gut barrier function disturbed by HFD.
A. muciniphila suppresses psoriatic skin inflammation by reducing systemic IL-17 levels
When assessing whether A. muciniphila treatment affects psoriatic disease, we found that both live and heat-killed A. muciniphila gavage attenuated HFD-enhanced ear swelling induced by IMQ (Figures 6A and 6B). Next, we tested if A. muciniphila inhibits systemic IL-17 induction by HFD. First, we checked the IL-17A production by T cells in spleen and MLNs after phorbol myristate acetate (PMA) and ionomycin stimulation in vitro (Figures 6C and 6D). No changes could be seen in MLN IL-17A+, γδ T cells, or IL-17A-producing γδ T cells after in vitro PMA stimulation (Figure 6D). However, HFD-induced upregulation of IL-17A by T cells in the spleen was suppressed by live or heat-killed A. muciniphila (Figures 6E and 6F).
Figure 6.

Akk suppresses psoriatic skin inflammation and systemic IL-17 induced by HFD
C57BL/6 mice were fed an ND (3.3% fat) or HFD (35.7% fat) for 8 weeks and challenged with IMQ. Oral gavages of live or heat-killed Akk were done for the last 2 weeks.
(A) Representative H&E staining of the IMQ-treated ears from HFD and HFD-live Akk-treated mice.
(B) Ear thickness was quantified in these different groups (n = 8–20).
(C and D) Comparison of the proportion of PMA/ionomycin-stimulated IL-17A+ T cells, γδ T cells, and IL-17+ γδ T cells in mLNs from each group (n = 5–6).
(E and F) Flow cytometric analysis of the IL-17+ T lymphocytes in the spleen. The boxed population shows the proportion of CD3 T cells (E). Proportion (%) of IL-17+ T lymphocytes (F) (n = 5). Data are presented as the mean ± SD. p values were calculated by analysis of variance followed by Sidak’s post hoc tests for comparison of groups indicated.
To validate the effect of A. muciniphila on psoriatic inflammation, we applied IMQ treatment on IL-17A-GFP reporter mice fed with ND or HFD. Immune cells were isolated from the IMQ ear, and the IL-17A-producing γδ T cells were detected by flow cytometry. The frequencies of IL-17A-producing γδ T cells in the ear from HFD-fed mice were significantly reduced after A. muciniphila gavage (Figures 7A and 7B). Moreover, mRNA expression of Il17a in the ear was significantly reduced in A. muciniphila treatment groups (Figure 7C) along with a lower IL-17A level in the serum (Figure 7D). Immunohistology revealed alleviated epidermal hyperplasia and less IL-17A+ immune cell infiltration in the IMQ-treated skin from HFD-fed mice treated with live A. muciniphila (Figure 7E). Overall, these data suggest that both heat-killed and live A. muciniphila treatments suppress psoriatic skin inflammation by reducing systemic IL-17 production and by decreasing infiltration of IL-17A-producing γδ T cells in the inflamed skin.
Figure 7.

Akk reduced the increased ear IL-17 levels from HFD
(A) Representative dot plot showing IL-17+ γδ T lymphocytes pregated on CD3+ T cells in the whole IMQ-treated ear of ND- and HFD-fed mice with or without live or heat-killed Akk gavage.
(B) Flow cytometric analysis of the IL-17+ γδ T lymphocytes in the IMQ-treated ear skin. Histogram of IL-17A-GFP population, collective MFI, and number of IL-17+ γδ T lymphocytes in the whole IMQ-treated ear skin (n = 4–6).
(C) Fold induction of Il17a mRNA in the ear skin, as analyzed by qRT-PCR (n = 5).
(D) ELISA quantification of IL-17A concentration (pg/mL) in serum mice from the different groups (n = 5).
(E) Immunofluorescence for IL-17A (red) and DNA (DAPI, blue) and quantification of IL-17 cells/field in the IMQ-treated ear with or without HFD/AKK (n = 4–5). Data are presented as the mean ± SD. p values were calculated by analysis of variance followed by Sidak’s post hoc tests for comparison of groups indicated.
Altogether, our data suggest that both heat-killed and live A. muciniphila treatments suppress HFD-induced, IL-17-associated systemic inflammation via restoration of the gut barrier mucus dysfunction.
Discussion
Herein, we show that HFD exacerbates psoriatic disease via the intestinal microbiome. Mechanistically, the intestinal mucus barrier was impaired by HFD, which triggered a pro-inflammatory milieu and increased production of IL-17 by T cells, highlighting the systemic nature of psoriatic disease.
Exacerbation of psoriatic disease was based on dietary fat, but not sugar or protein, content. As reported previously, fat-based dietary metabolic stress increased the migration of myeloid cells and T cells, in particular IL-17-producing γδ T cells, in the challenged skin and thereby exacerbated disease.20 Likewise, we found that several chemokines were upregulated during HFD, facilitating the migration of immune cells to the skin and supporting the concept of a systemic process that links the intestine with the skin. Of note, HFD cannot act locally on the skin but must have systemic effects to connect the intestine with the skin. Assessment of IL-17 expression upon HFD in several organ systems using an IL-17 reporter construct showed that IL-17-producing γδ T cells are induced in the spleen and thereby contribute to a systemic pro-inflammatory environment. This effect translated to elevated serum levels of IL-17 upon exposure to HFD.
Our data also showed that the effect of dietary metabolic stress on psoriatic skin inflammation is specific, as the expression of several classical inflammatory cytokines, such as TNF-α, IL-6, and G-GCF, remained unchanged, while IL-23, IL-17, and a subset of chemokines were upregulated. This finding supports the concept that microbial changes associated with dietary metabolic stress are triggering a pro-inflammatory, IL-17-based cytokine milieu. This concept is further supported by data showing that IL-17-producing γδ T cells expand upon pathogen challenge.21,22 The observation that intestinal VCM treatment reduced the enhanced IL-17 activation and effectively inhibited psoriatic skin inflammation in metabolically challenged mice strongly supports the concept that the effect of HFD is based on intestinal microbial changes.
Unexpectedly, intestinal barrier function was not altered after HFD. This observation may be related to the time of exposure to dietary metabolic stress, as longer feeding of HFD has been shown to increase intestinal permeability,23,24 while shorter exposure had no effect.25 However, profound changes in the mucus layer, mucus-producing cells, and mucin expression in the small intestine were found after HFD. Thus, exacerbation of skin inflammation and induction of IL-17-producing T cells by dietary metabolic stress may cause changes of the intestinal mucus barrier. Further experiments remain necessary to delineate the mucus barrier link to spleen IL-17 expression.
Our study point to a role of A. muciniphila in mitigating intestine-triggered inflammatory responses that exacerbate psoriatic disease. This bacterium is an intestinal commensal that resides in the mucus and is shown to be relevant for intact mucus layers. Thus, A. muciniphila could prevent age-related decline in thickness of the mucus layer and thereby attenuates inflammation.26 Furthermore, A. muciniphila was shown to mediate the anti-inflammatory effect of fish oil27 and inhibit the development of obesity and inflammation.28 A. muciniphila has also been described as an intestinal bacterial species found in patients with psoriasis.4,29 Treatment with VCM spared A. muciniphila, which becomes a dominant species in the gut, as previously reported,30 and may therefore explain the immunomodulatory role of VCM in psoriasis. Hence, dysbiosis, rather than absence of certain microbes in the gut, may be the key driver of alterations in the hosts’ immune response.31 A. muciniphila encodes a wide repertoire of enzymes32 that can degrade the mucin protein backbone and release sugars from the glycan chains. On the other hand, A. muciniphila was also found to stimulate mucin production, preserving the mucus thickness and the integrity of the gut barrier.33,34 Supplementation with A. muciniphila reversed the HFD-induced exacerbation of psoriasis, indicating that A. muciniphila may block the detrimental effect of metabolic factors on the development of psoriasis. It has been reported that heat-killed A. muciniphila had even stronger effects on body weight gain and glucose intolerance in HFD-fed mice compared with live A. muciniphila because the effective outer membrane proteins can remain stable at high temperature and pasteurization increases the accessibility of the effector proteins to the host.28 Nonetheless, in our study, A. muciniphila gavages did not rescue weight gain or glucose response but ameliorated insulin tolerance and psoriatic disease. Both heat-killed and live A. muciniphila had comparable effects on psoriatic skin inflammation.
In conclusion, these data indicate that HFD enhances psoriatic disease by changing gut microbiota and enhancing IL-17 production by γδ T cells in the spleen.
Limitations of the study
We elucidated the key role of intestinal flora on the exacerbation of psoriasis caused by HFD. Probiotics such as A. muciniphila could help to maintain or restore the intestinal barrier. However, improving the intestinal microenvironment with probiotics still requires further investigations, notably in the analysis of the metabolic links downstream of lipid metabolism. For instance, metabolic intermediates or metabolic molecular modifications are specifically altered by probiotics, which would need to be better characterized to optimize future clinical treatments. In addition, although our study showed that HFD alters IL-17 production by γδ T cells in the spleen, the detailed mechanisms need to be further studied.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse Ly6G-APC (clone 1A8) | BD Biosciences | Cat#560599; RRID:AB_1727560 |
| Anti-mouse CD45-eFluor780 (clone 30-F11) | eBioscience | Cat#47-0451-82; RRID:AB_1548781 |
| Anti-mouse Ly6C-BV421 (clone AL-21) | BD Biosciences | Cat#562727; RRID:AB_2737748 |
| Anti-mouse CD11c-PE (clone N418) | Biolegend | Cat#117308; RRID:AB_313777 |
| Anti-mouse CD11b-PE/Cy7 (clone M1/70) | eBioscience | Cat#25-0112-81; RRID:AB_469588 |
| Anti-mouse CD4-PerCP (clone GK1.5) | Biolegend | Cat#100431; RRID:AB_893329 |
| Anti-mouse CD3ε-APC (clone 145-2C11) | Biolegend | Cat#100312; RRID:AB_312677 |
| Anti-mouse CD45-alexafluor488 (clone 30-F11) | Biolegend | Cat#103128; RRID:AB_493715 |
| Anti-mouse CD8a-APC/Cy7 (clone 53–6.7) | BD Biosciences | Cat#557654; RRID:AB_396769 |
| Anti-mouse TCRγ/δ-BV421 (clone GL3) | Biolegend | Cat#118119; RRID:AB_10896753 |
| Anti-mouse TCRβ-BV510 (clone H57-597) | Biolegend | Cat#109233; RRID:AB_2562349 |
| Anti-mouse CD19-PE (clone 1D3) | BD Biosciences | Cat#553786; RRID:AB_395050 |
| Anti-mouse TCR Vγ1.1/Cr4-PerCP/Cy5.5 (clone 2.11) | Biolegend | Cat#141111; RRID:AB_2750516 |
| Anti-mouse CD11b-APC/Cy7 (clone M1/70) | Biolegend | Cat#101225; RRID:AB_830641 |
| Anti-mouse TCR Vγ2-PE/Cy7 (clone UC3-10A6) | ThermoFisher | Cat#25-5828-82; RRID:AB_2573474 |
| Anti-mouse TCR Vγ1.1/Cr4-APC (clone 2.11) | Biolegend | Cat#141107; RRID:AB_10897806 |
| Anti-mouse CD19-APC/Cy7 (clone 1D3) | BD Biosciences | Cat#557655; RRID:AB_396770 |
| Anti-mouse CD3-PE (clone 17A2) | Biolegend | Cat#100205; RRID:AB_312662 |
| TruStain FcX™ (anti-mouse CD16/32) Antibody | Biolegend | Cat#101320; RRID:AB_1574975 |
| Anti-CD28 (clone CD28.2) | eBioscience | Cat#16-0281-82 |
| Anti-MUC2 | Novus | Cat#NBP1-31231 |
| Anti-IL-17A | abcam | Cat#ab79056 |
| Bacterial and virus strains | ||
| Akkermansia muciniphila | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase II | Merck | Cat#C 2-22 |
| HBSS | Gibco | Cat#14025050 |
| DNase I | Sigma-Aldrich | Cat#10104159001 |
| HEPES | Merck | Cat#L1613 |
| DTT | Roth | Cat#6908.1 |
| PEQGOLD RNAPURE | VWR | Cat#732-3312 |
| AccuGENE 0.5 M EDTA Solution | Lonza | Cat#51201 |
| Fetal calf serum | Gibco | Cat#26400044 |
| VECTASHIELD with DAPI | Vector labs | Cat#H-1200 |
| DPBS | Gibco | Cat#14040141 |
| Imiquimod cream | MEDA | N/A |
| vancomycin | Merck | Cat#1404-93-9 |
| Critical commercial assays | ||
| TCRγ/δ+ T cell Isolation Kit, mouse | Miltenyi | Cat#130-092-125 |
| QIAamp Fast DNA Stool Mini Kit | Qiagen | Cat# 51604 |
| IL-17A (homodimer) Mouse Uncoated ELISA Kit | Invitrogen | Cat#88-7371-22 |
| High-Capacity cDNA Reverse Transcription Kit | LifeTech | Cat#4368813 |
| SYBR qPCR Master Mix Kit | Applied biosystems | Cat#4472920 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| Deposited data | ||
| The Liquid Chromatography - Mass Spectrometry metabolomics datasets | This paper | https://www.ebi.ac.uk/metabolights/MTBLS7964 |
| 16S rRNA sequence datasets | This paper | BioPoject: PRJNA979156; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA979156 |
| Experimental models: Organisms/strains | ||
| C57BL/6NRj | Janvier labs | N/A |
| C57BL/6-Il17atm1Bcgen/J | The Jackson Laboratory | RRID:IMSR_JAX:018472 |
| Oligonucleotides | ||
| Primers for qPCR are listed in Table S3 | N/A | N/A |
| Software and algorithms | ||
| Graphpad Prism 9 | GraphPad Software | https://www.graphpad.com/ |
| ZEN 2012 SP1 v8.1.0484 | Zeiss | https://www.zeiss.com.cn/microscopy/products/microscopesoftware/zen.html |
| Biorender | Biorender | https://www.biorender.com |
| Flowjo | Flowjo | https://www.flowjo.com/ |
| QuantStudio Software v1.3 | Thermo Fisher | https://www.thermofisher.cn/cn/zh/home/global/forms/life-science/quantstudio-6-7-flex-software.html |
| METAGENassist | METAGENassist | http://www.metagenassist.ca/METAGENassist/faces/Home.jsp |
| mzVault | Thermo Fisher | N/A |
| mzcloud™ | Thermo Fisher | https://www.mzcloud.org |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Aline Bozec (aline.bozec@uk-erlangen.de).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
Animal studies
Mice and HFD-IMQ mouse model
This work employed male C57BL/6NRj (Janvier labs) and both male and female C57BL/6-Il17atm1Bcgen/J (IL17A-IRES-GFP-KI) mice (Jackson laboratory, Stock number #018472). The mice were housed with ad libitum access to food and drinking water in the local specific pathogen-free barrier facility with 20–24°C, 55 ± 10% and 12h light/dark cycle. For feeding experiments, 6–7 weeks old mice were maintained on diets with different fat content (ssniff Spezialdiaeten, Table S4) for 7 to 10 weeks. Treatment with vancomycin (VCM, DEMO Pharmaceuticals) was performed by supplementing the drinking water with 0.5 g/L of vancomycin for the last 2 weeks of the experiment. Live and heat killed A. muciniphila was suspended in 200 μL of anaerobic Dulbecco’s phosphate buffered saline (DPBS, ThermoFisher Scientific) with indicated concentrations, and gavaged orally once daily for the last two weeks. Control group also received 200 μL of DPBS in the same manner. For heat killed A. muciniphila, the bacteria were inactivated by pasteurization for 30 min at 70°C. In chylomicron inhibition experiment, mice were gavaged indicated doses of lomitapide (Sigma-Aldrich) was suspended in 5 μL/g body weight of DPBS, and gavaged orally once daily for the last two weeks. Control group also received oral gavage of 5 μL/g body weight of DPBS. IMQ treatment was performed by administering 25mg/mouse of Aldara cream (Meda Pharmaceuticals) once daily. The cream was applied topically to the right ear of the mouse while the left ear was left untreated as a control. The treatment was applied for the last 3 days of the experiment. Total ear thickness was measured by caliper. Mice were sacrificed by CO2 asphyxiation at the end of the experiment at an age of 14–17 weeks.
Animal experiments were approved by local ethics committee of Regierung von Mittelfranken (No. 55.2-2532-2-855).
Metabolic cage
Food consumption was measured using a metabolic cage system (OxyletPro System – Physiocage, Panlab). Briefly, 5 mice in the same cage were transferred to a metabolic cage 4 weeks after the start of the experiment, along with their food. They were monitored for food intake every 2 min for 48 h under free access to food and water in the animal house conditions described above. Data was analyzed and summarized using METABOLISM V3.0 software (Panlab).
Method details
Flow cytometry
Flow cytometry was performed using the Cytoflex S (Beckman Coulter). Organs were taken from the mice and prepared as outlined below for the different organs.
Ear sample preparation
The ears were cut into small pieces and digested with 10 mL of HBSS with Ca2+, Mg2++ 10% fetal bovine serum (FBS) + 1000 U/ml collagenase II (Merck Millipore) + 0.1 mg/mL DNase I (Sigma-Aldrich) at 37°C with shaking at 300 rpm for 30 min. The buffer was replaced with flow cytometry buffer (DPBS +2% FBS +20 mM EDTA) and the resulting suspension was then transferred into gentleMACS C tubes (Miltenyi Biotec). Tissue dissociation was performed by using program B_01 3 times in the gentleMACS tissue dissociator (Miltenyi Biotec). This was followed by washing step and straining through a 40 μm cell strainer, and then suspended into flow cytometry buffer for staining.
Blood sample preparation
Blood was harvested from the mice using syringe and needles by cardiac puncture after euthanizing. To prevent clogging the blood was placed in a heparin tube (Micro tube 1.3mL LH, Sarstedt). The blood was then diluted with erythrocyte lysis buffer (150 mM NH4CL, 20 mM HEPES and 0.1 mM EDTA, pH 7.2) and incubated for 5 min at room temperature. The samples were washed and strained through a 40 μm cell strainer, re-suspended into flow cytometry buffer for staining.
Spleen, lymph nodes and Peyer’s patches sample preparation
The tissue was homogenized by a syringe plunger on a 40 μm strainer containing flow cytometry buffer. The resulting suspension was transferred through a new 40 μm strainer, washed and re-suspended in flow cytometry buffer for staining.
Intestinal epithelial fraction sample preparation
Surrounding adipose tissue was removed from the small intestine, and the intestine was flushed with HBSS without Ca2+, Mg2+ + 10 mM HEPES to remove intestinal content. Next, Peyer’s patches were removed, and the intestine was longitudinally cut open followed by vigorous washing in fresh HBSS without Ca2+, Mg2++ 10 mM HEPES. This was repeated with fresh buffer until no visible increase in buffer turbidity was observed. The intestine was then cut into 1cm pieces and transferred into 20 mL of HBSS without Ca2+, Mg2+ + 10 mM HEPES +1% FBS +1 mM dithiothreitol (DTT). The suspension was incubated at 37°C with 150 rpm shaking for 20 min. Follow incubation, the tissue was filtered on a 100 μm strainer, the tissue pieces retained on the strainer were then transferred to 20 mL of pre-digestion buffer (HBSS without Ca2+, Mg2+ + 10 mM HEPES +1% FBS +1 mM DTT +2 mM EDTA) and incubated with shaking at 300 rpm at 37°C for 20 min. After the incubation, the tissues were vortexed for 20 s and again filtered on a 100 μm strainer. This filtrate contained the intestinal epithelial fraction. The retentate was then re-incubated in pre-digestion buffer and the steps repeated to obtain two intestinal epithelial fractions. The two intestinal epithelial fractions were pooled and washed by flow cytometry buffer followed by filtering through a 40μm strainer. The filtrate was then again washed and re-suspended in flow cytometry buffer for staining.
Lamina propria fraction sample preparation
Lamina propria was prepared by taking the leftover retained tissue-pieces from filtering to produce the intestinal epithelial fractions and washing it with 20 mL of HBSS without Ca2+, Mg2+ + 10 mM HEPES +1% FBS by incubating at 37°C with 150 rpm shaking for 20 min. The resulting suspension was filtered on a 100 μm strainer and the retentate transferred into 10 mL of digestion buffer (HBSS with Ca2+, Mg2+ + 10% FBS +250 U/ml collagenase II + 0.1 mg/mL DNase I). The tissue was then digested by incubating at 37°C with shaking at 300 rpm for 30 min. Following digestion, the tissues were vortexed for 20 s and again filtered on a 100 μm strainer. The flow-through was the lamina propria fraction. The retentate was once again transferred into 10 mL of digestion buffer, digested, vortexed, and filtered to obtain two lamina propria fractions. The fractions were pooled and washed by flow cytometry buffer for two times, and then re-suspended into flow cytometry buffer for staining.
Colon sample preparation
Colon was flushed with DPBS, opened longitudinally, and washed in a dish with flow cytometry buffer followed by cutting it into pieces. The pieces were homogenized in a gentleMACS C tube containing 3mL of flow cytometry buffer using the gentleMACS tissue dissociator, program m_intestine twice. Following homogenization, the sample was filtered through a 40μm strainer. This step was repeated if necessary, when the disuse was deemed as to viscous for the flow cytometer. The filtrate was washed and re-suspended in flow cytometry buffer for staining.
Sample staining for analysis with flow cytometer
Staining was carried out by adding antibodies to a final dilution of 1:1000 in flow cytometry buffer to an aliquot of the final suspension obtained for each organ sample. The resulting mixture was incubated at 4°C protected from light for 20 min. To obtain the IL-17 production for each organ, the amount of IL-17+ cells were multiplied with the mean fluorescence intensity (MFI) of the IL-17+ cell population to obtain a normalized value of total contribution of IL-17 for each organ. The various staining panels employed are shown in Table S1. The Heilig and Tonegawa’s nomenclature is employed in this manuscript to show γδ TCR.35 TruStain FcX (anti-mouse CD16/32) Antibody was used as Fc block.
Adoptive transfer model of IMQ dermatitis mice
γδ T cell were purified by MACS Kit (Miltenyi Biotec) from IL-17A-GFP mice after 8-week HFD. 2 X 106 γδ T cell per mouse were injected intravenously into C57BL/6 recipient d1 post challenge with IMQ model as described in experimental model. The IMQ treatment was applied for 3 days once daily. Control mice received PBS +0.2% BSA alone. IL-17-GFP+ γδ T cell infiltration of target organs in recipients were analyzed 2 days post transfer by flow cytometry. Spleen, IMQ ear and mLN were collected and prepared as described in the section: Ear sample preparation and Spleen, lymph nodes and Peyer’s patches sample preparation.
RNA extraction and cDNA synthesis
After mice were euthanized, organ samples for real-time PCR analysis were collected and snap frozen in Precellys lysing kit CK14 (Bertin instruments) immediately. The samples were kept in −20°C until further procedure. Total tissue was homogenized using a Precellys 24 homogenizer (Bertin instruments) after adding 1mL of peqGOLD RNA PureFL (VWR). Following homogenization, the RNA was extracted according to the manufacturer’s specifications. cDNA was synthesized by first pre-treating RNA with DNase I (ThermoFisher Scientific) followed by heat and EDTA inactivation of the DNase I according to manufacturer’s specifications. cDNA was then synthesized using the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific) according to the manufacturer’s specifications. The synthesized cDNA was then used for real-time PCR.
Real-time PCR
Real time PCR was performed using the Applied Biosystems SYBR Select Master Mix (ThermoFisher scientific) according to the manufacturer’s specifications using CFX96 Touch Real-Time PCR Detection System (BioRad) or QuantStudioTM 6 Flex system (ThermoFisher scientific). The cycling parameters of the PCR program are 50°C for 2 min, 95°C for 10 min as the initial denaturation step followed by 40 cycles of 95°C for 15 s and 60°C for 1 min annealing/extension step. This is followed by a melting curve analysis; 95°C for 15 s, 60°C for 1 min and 95°C for 15 s. Data was analyzed using the ΔΔCt method using Actb as a housekeeping gene.
Enzyme-linked immunosorbent assay (ELISA)
Serum was isolated using BD Microtainer Tubes (BD) according to manufacturer’s specifications. ELISA was performed using commercially available kit according to the manufacturer’s specifications. TMB Solution (ThermoFisher scientific) was used as enzyme substrate. Optical density was measured by SPECTRA MAX 190 (Molecular Devices).
Histology
Tissues for histology were fixed in ROTI Histofix 4% (Carl Roth) overnight immediately after collection. Intestinal samples were embedded in 0.5% w/v of agarose low melt dissolved in DPBS prior to fixation with the agarose allowed to harden on ice before the tissue was fixed. Paraffin sections with the thickness of 6-μm were prepared and stained with hematoxylin and eosin (H&E) and Alcian Blue-Periodic acid-Schiff (AB-PAS) stainings. Semi quantitative measurements for epidermal thickness and average PAS layer thickness were performed using Adobe Photoshop (Adobe). Epidermal thickness was calculated by manually measuring the area of the epidermis of whole ear and dividing by its length. Average PAS layer thickness was calculated by manually measuring the PAS positive area surrounding the villi and the villi surface length. The length of the villi surfaces then divided the positive area to obtain the average PAS layer thickness. Unless otherwise mentioned, displayed small intestine histology is from tissue collected at ∼25% of the length of the small intestine.
Immunofluorescence
Immunofluorescence was performed after antigen retrieval as follows. 2 min sequential boiling steps in a microwave with TE (10 mM Tris +1 mM EDTA +0.05% Tween, pH 9.0) buffer and Citrate (10 mM Citrate, pH 6.0) buffer in order, Citrate, TE, Citrate, TE, Citrate. The slides were cooled at room temperature for 30 min following antigen retrieval, washed once with DPBS and blocked with 5% horse serum for 1h at room temperature. The blocking solution was then removed and staining with primary antibodies: rabbit anti-MUC2 (1:100, Novus, NBP1-31231) or rabbit anti-IL-17A (1:300, abcam, ab79056) were carried out in 5% horse serum in DPBS overnight at 4°C. The next day, the slides were washed in DPBS and the secondary antibody was applied and slides were incubated for 2h at room temperature followed by an additional two washes in DPBS. Washed slides were mounted using VECTASHIELD Antifade Mounting Medium with DAPI (Vector labs) prior to fluorescence microscopy.
Microscopy
Bright field and Fluorescence microscopy was performed using the Keyence BZ-X710 microscope.
Intestinal permeability assay
Mice were fed by oral-gavage with 0.44 mg/g body weight of FITC-labelled 4 kDa dextran that has a diameter of approx. 6nm (Tdb Labs). Dextran was dissolved at 100 mg/mL in DPBS. Following gavage, the mice were fasted for 4 h, serum isolated and the fluorescence of the serum was measured using an Infinite F200 Pro plate reader (Tecan). Filter setup was as follows: excitation wavelength filters of 485 nm and Emission Wavelength filters of 535 nm. Excitation bandwidth was 20 nm and emission bandwidth 25 nm.
Stool DNA isolation
Stool from the small intestine and caecum was collected in Precellys lysing MK28 (Bertin instruments) separately, followed by storage at −20°C until DNA extraction. DNA was extracted from frozen stool by using the QIAamp Fast DNA Stool Mini Kit (Qiagen) according to the manufacturer’s specifications with modifications as follows. A homogenization step was inserted between step 2 and step 3 of the protocol supplied with the kit (following the addition of “inhibitEX” buffer and prior to sample heating). Briefly, 100–150 mg stool samples are lysed in InhibitEX Buffer effectively by incubating the stool homogenate at 95°C to separate DNA-degrading substances and PCR inhibitors. The sample matrix is pelleted by centrifugation and the DNA in the supernatant is purified on QIAamp Mini spin columns. The QIAamp DNA purification procedure involves digestion of proteins, binding DNA to the QIAamp silica membrane. Proteins are digested and degraded under denaturing conditions during a 70°C incubation with proteinase K. Optimized buffering conditions (Buffer AL + fresh ethanol) in the lysate ensure that the remains of digested proteins and other impurities, which can inhibit PCR and other downstream enzymatic reactions, are not retained on the QIAamp membrane and allow optimal binding of DNA to the QIAamp membrane. DNA is adsorbed onto the QIAamp silica membrane during a brief centrifugation step. DNA bound to the QIAamp membrane is washed by Buffer AW1 and Buffer AW2 to ensure complete removal of any residual impurities. At last, purified, concentrated DNA is eluted from the QIAamp Mini spin column in low-salt buffer (Buffer ATE) equilibrated to 37°C.
16s ribosomal RNA sequence analysis
The V4 regions of the 16S rRNA genes were amplified using 10 ng of bacterial template DNA with region-specific primers (515F: 5′-GTGYCAGCMGCCGCGGTAA-3′; 806R: 5′-GACTACNVGGGTWTCTAAT-3′) containing barcodes and Illumina flow cell adaptor sequences (PMID: 26271760) in a reaction consisting of 25 PCR cycles (95°C 45 s, 50°C 60 s, 72°C 90 s) using the NEBNext Ultra II Q5 Master Mix (New England Biolabs, Ipswich, MA). Amplicons were purified with Agencourt AMPure XP Beads (Beckmann Coulter, Brea, CA), normalized and pooled before sequencing on an Illumina MiSeq device using the Illumina V2 chemistry. The DADA2 (PMID: 27214047) plugin within Qiime2 (PMID: 31341288) was used to establish amplicon sequencing variants (ASVs). QIIME2 q2-feature-classifier using the SILVA database (v138) was used to assign taxonomy to ASVs OTUs. Data was analyzed by METAGENassist platform (http://www.metagenassist.ca/METAGENassist/faces/Home.jsp).
γδ T cell sorting
γδ T cells were sorted from spleen of C57BL/6 male mice fed ND or HFD for 8 weeks. Spleen was cut into pieces and digested in HBSS with Ca2+, Mg2++ 10% FBS +400 U/ml collagenase II + 0.1 mg/mL DNase I at 37°C for 20 min. The resulting suspension was filtered on a 40 μm strainer and was washed by sorting buffer (DPBS +2% FBS +2 mM AccuGENE EDTA), and then diluted with erythrocyte lysis buffer and incubated for 10 min at room temperature. Following ery-lysis, cell suspension was washed and re-suspended in sorting buffer containing staining antibodies shown in Table S2. Cells were stained for 30 min in the dark at 4°C, and then washed and re-suspended in the sorting buffer at the concentration of 5 x 107/mL. Vγ4+TCRδ+CD3+ lymphocytes and Vγ1-Vγ4-TCRδ+CD3+ lymphocytes were sorted using MoFlo Astrios EQ (Beckman Coulter), and then the RNA was extracted using RNeasy Mini Kit (Qiagen) according to the manufacturer’s specifications.
TCR repertoire analysis
A molecular amplification fingerprinting (MAF) strategy was incorporated during library preparation. Extracted RNA was transcribed into cDNA using primers specific for TCRD and TCRG constant regions including a Reverse Identifier (RID) and Illumina Adapter sequences. cDNA synthesis was followed by clean-up with SPRI beads (Agencourt AMPure XP, Beckman Coulter). cDNA was used in a multiplex PCR for various TCRG and TCRD V genes. V-gene specific primers included Forward Identifiers (FID) and Illumina Adapter sequences. After clean-up with Ampure Beads, individual sample indices were added to the PCR amplicons using Illumina Nextera i5 and i7 primers (Nextera XT Index Kit, Illumina). Sequencing was performed on the MiSeq platform (Illumina) with a 300 bp x 300 bp configuration. Paired-end reads were merged with the IgRepertoireConstructor (IgReC). Read Quality was ensured by FASTQC quality analysis. PCR bias or sequencing errors were corrected by considering sequences containing both RIDs and FIDs only. Read counts were normalized to unique RID counts. VDJ annotation was conducted with the Change-O wrapper for IgBLAST using IMGT references. Normalized reads were summarized in clonotype tables according to their CDR3 amino acid sequence using dplyr, tidyr and visualized with ggplot2. Diversity estimates based on clonotype distributions were calculated with the immunarch package.
Liquid chromatography - Mass spectrometry (LC/MS)
Cells were lysed with 1 mL of ice-cold methanol/warer (80/20 v/v) containing four recovery standards in order to monitor sample preparation. The standards included tridecanoic acid (25 μg/mL), DL-2-fluorophenylglycine (2.5 μg/mL), DL-4-chlorophenylalanine (10 μg/mL) (all three from Merck, Darmstadt, Germany) and [2H6]-cholesterol (25 μg/mL; Avanti Polar Lipids, Alabaster, AL, USA). After centrifugation at 16000 rpm and 4°C for 5 min using an Eppendorf 5427 R centrifuge (Eppendorf, Hamburg, Germany), 2 x 350 μL of the supernatant of each sample were pipetted into two separate LC vials (VWR, Darmstadt, Germany). A pool sample was prepared by mixing 230 μL of the supernatant of each sample. From this mixture, 350 μL were taken into LC vials for each QC sample. Reference samples were prepared by pipetting 350 μL of the lysis solution. All samples were dried under a gentle stream of nitrogen at 30°C. Subsequently, the dried samples were reconstituted in 200 μL of hydrophilic interaction chromatography (HILIC) eluent (acetonitrile 95%, 5% water, 0.1% formic acid (all three from VWR chemicals, Darmstadt, Germany), 10 mM ammonium formate (Merck, Darmstadt, Germany) or 200 μL reverse phase (RP) eluent (99.5% water, 0.5% methanol (VWR chemicals, Darmstadt, Germany), both 0.1% formic acid). The eluents contained nine internal standard in order to monitor instrument performance: [2H28]-hexadecanedioic acid (10 ng/mL), [2H24]-tetradecanedioic acid (5 ng/mL), [2H3]-carnitine (1 ng/mL) (all three EQ Laboratories, Augsburg, Germany), imipramine (0.5 ng/mL; Merck, Darmstadt, Germany), [2H3]-creatinine (2 ng/mL; CDN Isotopes, Pointe-Claire, QC, Canada), [2H9]-trimethylamine-N-oxide (0.86 ng/mL; Cambridge Isotope Laboratories, Tewksbury, MA, USA), [2H5]-tryptophan (40 ng/mL; Santa Cruz Biotechnology, Heidelberg, Germany), [2H7]-arginine (100 ng/mL; Cambridge Isotope Laboratories, Tewksbury, MA, USA) and [2H7]-glucose (1 ng/mL; Merck, Darmstadt, Germany).
All analyses were performed on a Dionex Ultimate 3000 chromatographic system hyphenated to a Q Exactive Focus mass spectrometer (both from Thermo Fisher Scientific, Dreieich, Germany) with a heated electrospray source. The instrument was controlled over TraceFinder 4.1 (Thermo Fisher Scientific, Dreieich, Germany). Each sample was analyzed via both, HILIC and RP chromatographic separation. Furthermore, each chromatographic separation was analyzed in positive and negative mode resulting in four runs per sample. HILIC separation was achieved on an Acquity UPLC BEH Amide, 1.7 μm, 2.1 × 100 mm column, whereas RP separation was performed on an Acquity UPLC BEH C18, 1.7 μm, 2.1 × 100 mm columns. For both columns, 2.1 × 5 mm guard columns were installed in order to prolong column lifetime (all columns from Waters, Eschborn, Germany). For both chromatographic separations, the column temperature was 40°C, the flow rate 0.35 mL/min and the injection volume 2 μL. The HILIC eluent was A: water, 0.1% formic acid, 10 mM ammonium formate and B: acetonitrile 95%, 5% water, 0.1% formic acid, 10 mM ammonium formate. The RP eluent was A: water, 0.1% formic acid and B: methanol, 0.1% formic acid. For HILIC and RP a gradient elution program was used with details shown in Table S2. The Q Exactive Focus mass spectrometer was operated with a capillary voltage of −3 kV in negative mode and 3 kV in positive mode. The capillary temperature was 380°C and the auxiliary gas temperature was 400°C. The sheath gas pressure, auxiliary gas pressure and sweep gas flow rate were set to 60, 20, and 0 arbitrary units, respectively. Nitrogen 5.0 was used for these gases. The scanning range was 66.7–1000 m/z in both ionization modes. Furthermore, the mass spectrometer was operated in discovery acquisition mode, whereby the instrument switches between full scan and ddMS2 mode allowing for a better annotation of the most abundant features. Resolution of the analyser in full scan was set to 70000 and the maximum inject time set at auto with an AGC target of 1e6. In ddMS2, resolution was set to 35000 with the maximum injection time set to auto and an AGC target of 5e4. Prior to each run, the mass spectrometer performance was evaluated and calibrated according to the manufacturer’s recommendations.
For each of the four LC-MS modes, i.e. HILIC positive, HILIC negative, RP positive and RP negative, a sequence was established including samples from the present study, blanks, QC standards (pooled samples) and reference standards, whereby the samples from this study were randomly distributed over the sequence. Retention times and peak areas of the QC sample compounds were monitored in all samples (tolerance for retention time variations = ±0.1 min; tolerance for peak area variations = ±15%).
For metabolomics data analysis, Compound Discoverer 3.1 was used (Thermo Fisher Scientific, Dreieich, Germany). Minimum peak intensity for detection was set to 1e5. Features were grouped and identified with a mass tolerance of 5 ppm and an RT tolerance of 0.2 min. Normalization factors for each sample were based on the cell number of the respective sample from the present study. Differential analysis was performed using the log-transformed values for the peak area of a feature. Adjusted p values were calculated using Benjamini-Hochberg correction, whereby adjusted p values of ≤0.05 were considered as statistically significant. Compound annotations were made by the software using mzcloud, mzVault and ChemSpider databases. Annotations were defined in four levels based on which software was used and the match quality (for mzcloud and mzVault).
Metabolic pathway enrichment analysis was performed by MetaboAnalyst platform (https://www.metaboanalyst.ca/).
Quantification and statistical analysis
Statistical analysis was carried out using GraphPad Prism software v9.1.1(GraphPad software) with indicated methods in the figure legends. All values were conducted as the mean ± SD. p values less than 0.05 were regard as statistically significant.
Acknowledgments
We thank Christine Zech and Barbara Happich for great technical assistance. The authors thank Dr. Wolfgang Baum for expertise in mice experiments. We thank Uwe Appelt and Markus Mroz of the Core Unit Cell Sorting and Immunomonitoring facility for their technical support in flow cytometry and for cell sorting. This study was supported by the German Research Foundation (BO-3811/5-1, BO-3811/6-1, and INST 90/1048-1 FUGG), the Collaborative Research Center (CRC) 1181 project A01; the SPP μBone; FOR 2886 Pandora TPO A2 and A1; FOR2799 to T.H.W. (project number 395236335); the Fritz Thyssen Foundation; the Interdisciplinary Center for Clinical Research (IZKF) grant A77; and the European Research Council (ERC) Synergy Grant 4D Nanoscope.
Author contributions
K.S., R.S., D.E., and A.B. designed the study, performed the experiments, interpreted the results, and wrote the manuscript. A.M.H. designed and performed the experiments and interpreted the data. N.L. and R.V.T. performed experiments and corrected data. K.S., R.S., D.E., and R.V.T. performed bioinformatics analysis and interpreted the data. S.W. and D.S. provided expertise in microbiome and bacteria analyses. Y.T., G.S., and A.B. acquired funding, designed the study and experiments, and wrote the manuscript. All authors read and commented on the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: July 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112713.
Supplemental information
Data and code availability
-
•
The 16S rRNA sequence datasets of this article have been deposited at the NCBI Sequence Read Archive (SRA) database under accession number PRJNA979156, (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA979156) and the metabolomics datasets have been deposited at the EMBL-EBI MetaboLights database with the identifier MTBLS7964 (https://www.ebi.ac.uk/metabolights/MTBLS7964). These data are publicly available as of the date of publication. Accession numbers of these data are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Boehncke W.H., Schön M.P. Psoriasis. Lancet. 2015;386:983–994. doi: 10.1016/S0140-6736(14)61909-7. [DOI] [PubMed] [Google Scholar]
- 2.Jensen P., Skov L. Psoriasis and Obesity. Dermatology. 2016;232:633–639. doi: 10.1159/000455840. [DOI] [PubMed] [Google Scholar]
- 3.Alotaibi H.A. Effects of Weight Loss on Psoriasis: A Review of Clinical Trials. Cureus. 2018;10:e3491. doi: 10.7759/cureus.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Codoñer F.M., Ramírez-Bosca A., Climent E., Carrión-Gutierrez M., Guerrero M., Pérez-Orquín J.M., Horga de la Parte J., Genovés S., Ramón D., Navarro-López V., Chenoll E. Gut microbial composition in patients with psoriasis. Sci. Rep. 2018;8:3812. doi: 10.1038/s41598-018-22125-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scher J.U., Ubeda C., Artacho A., Attur M., Isaac S., Reddy S.M., Marmon S., Neimann A., Brusca S., Patel T., et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol. 2015;67:128–139. doi: 10.1002/art.38892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bäckhed F., Ding H., Wang T., Hooper L.V., Koh G.Y., Nagy A., Semenkovich C.F., Gordon J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turnbaugh P.J., Ley R.E., Mahowald M.A., Magrini V., Mardis E.R., Gordon J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 8.Ley R.E., Turnbaugh P.J., Klein S., Gordon J.I. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 9.Gregory J.C., Buffa J.A., Org E., Wang Z., Levison B.S., Zhu W., Wagner M.A., Bennett B.J., Li L., DiDonato J.A., et al. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J. Biol. Chem. 2015;290:5647–5660. doi: 10.1074/jbc.M114.618249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karlsson F.H., Tremaroli V., Nookaew I., Bergström G., Behre C.J., Fagerberg B., Nielsen J., Bäckhed F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498:99–103. doi: 10.1038/nature12198. [DOI] [PubMed] [Google Scholar]
- 11.Derrien M., Vaughan E.E., Plugge C.M., de Vos W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004;54:1469–1476. doi: 10.1099/ijs.0.02873-0. [DOI] [PubMed] [Google Scholar]
- 12.Derrien M., Collado M.C., Ben-Amor K., Salminen S., de Vos W.M. The Mucin degrader Akkermansia muciniphila is an abundant resident of the human intestinal tract. Appl. Environ. Microbiol. 2008;74:1646–1648. doi: 10.1128/AEM.01226-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Derrien M., Van Baarlen P., Hooiveld G., Norin E., Müller M., de Vos W.M. Modulation of mucosal immune response, tolerance, and proliferation in mice colonized by the mucin-degrader Akkermansia muciniphila. Front. Microbiol. 2011;2:166. doi: 10.3389/fmicb.2011.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J., Lin S., Vanhoutte P.M., Woo C.W., Xu A. Akkermansia muciniphila protects against atherosclerosis by preventing metabolic endotoxemia-induced inflammation in Apoe−/− mice. Circulation. 2016;133:2434–2446. doi: 10.1161/CIRCULATIONAHA.115.019645. [DOI] [PubMed] [Google Scholar]
- 15.Shibata K., Yamada H., Hara H., Kishihara K., Yoshikai Y. Resident Vδ1+ γδ T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J. Immunol. 2007;178:4466–4472. doi: 10.4049/jimmunol.178.7.4466. [DOI] [PubMed] [Google Scholar]
- 16.Sferra R., Pompili S., Cappariello A., Gaudio E., Latella G., Vetuschi A. Prolonged chronic consumption of a high fat with sucrose diet alters the morphology of the small intestine. Int. J. Mol. Sci. 2021;22:7280. doi: 10.3390/ijms22147280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reijneveld J.F., Ocampo T.A., Shahine A., Gully B.S., Vantourout P., Hayday A.C., Rossjohn J., Moody D.B., Van Rhijn I. Human γδ T cells recognize CD1b by two distinct mechanisms. Proc. Natl. Acad. Sci. USA. 2020;117:22944–22952. doi: 10.1073/pnas.2010545117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y., Ghoshal S., Ward M., de Villiers W., Woodward J., Eckhardt E. Chylomicrons promote intestinal absorption and systemic dissemination of dietary antigen (ovalbumin) in mice. PLoS One. 2009;4:e8442. doi: 10.1371/journal.pone.0008442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grondin J.A., Kwon Y.H., Far P.M., Haq S., Khan W.I. Mucins in intestinal mucosal defense and inflammation: learning from clinical and experimental studies. Front. Immunol. 2020;11:2054. doi: 10.3389/fimmu.2020.02054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakamizo S., Honda T., Adachi A., Nagatake T., Kunisawa J., Kitoh A., Otsuka A., Dainichi T., Nomura T., Ginhoux F., et al. High fat diet exacerbates murine psoriatic dermatitis by increasing the number of IL-17-producing γδ T cells. Sci. Rep. 2017;7:14076. doi: 10.1038/s41598-017-14292-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin B., Hirota K., Cua D.J., Stockinger B., Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity. 2009;31:321–330. doi: 10.1016/j.immuni.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 22.Zákostelská Z., Málková J., Klimešová K., Rossmann P., Hornová M., Novosádová I., Stehlíková Z., Kostovčík M., Hudcovic T., Štepánková R., et al. Intestinal Microbiota Promotes Psoriasis-Like Skin Inflammation by Enhancing Th17 Response. PLoS One. 2016;11:e0159539. doi: 10.1371/journal.pone.0159539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stenman L.K., Holma R., Korpela R. High-fat-induced intestinal permeability dysfunction associated with altered fecal bile acids. World J. Gastroenterol. 2012;18:923–929. doi: 10.3748/wjg.v18.i9.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cani P.D., Bibiloni R., Knauf C., Waget A., Neyrinck A.M., Delzenne N.M., Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 25.Kless C., Müller V.M., Schüppel V.L., Lichtenegger M., Rychlik M., Daniel H., Klingenspor M., Haller D. Diet-induced obesity causes metabolic impairment independent of alterations in gut barrier integrity. Mol. Nutr. Food Res. 2015;59:968–978. doi: 10.1002/mnfr.201400840. [DOI] [PubMed] [Google Scholar]
- 26.van der Lugt B., van Beek A.A., Aalvink S., Meijer B., Sovran B., Vermeij W.P., Brandt R.M.C., de Vos W.M., Savelkoul H.F.J., Steegenga W.T., Belzer C. Akkermansia muciniphila ameliorates the age-related decline in colonic mucus thickness and attenuates immune activation in accelerated aging Ercc1 (-/Delta7) mice. Immun. Ageing. 2019;16:6. doi: 10.1186/s12979-019-0145-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caesar R., Tremaroli V., Kovatcheva-Datchary P., Cani P.D., Bäckhed F. Crosstalk between Gut Microbiota and Dietary Lipids Aggravates WAT Inflammation through TLR Signaling. Cell Metabol. 2015;22:658–668. doi: 10.1016/j.cmet.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plovier H., Everard A., Druart C., Depommier C., Van Hul M., Geurts L., Chilloux J., Ottman N., Duparc T., Lichtenstein L., et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat. Med. 2017;23:107–113. doi: 10.1038/nm.4236. [DOI] [PubMed] [Google Scholar]
- 29.Tan L., Zhao S., Zhu W., Wu L., Li J., Shen M., Lei L., Chen X., Peng C. The Akkermansia muciniphila is a gut microbiota signature in psoriasis. Exp. Dermatol. 2018;27:144–149. doi: 10.1111/exd.13463. [DOI] [PubMed] [Google Scholar]
- 30.Hansen C.H.F., Krych L., Nielsen D.S., Vogensen F.K., Hansen L.H., Sørensen S.J., Buschard K., Hansen A.K. Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia. 2012;55:2285–2294. doi: 10.1007/s00125-012-2564-7. [DOI] [PubMed] [Google Scholar]
- 31.Reading N.C., Kasper D.L. The starting lineup: key microbial players in intestinal immunity and homeostasis. Front. Microbiol. 2011;2:148. doi: 10.3389/fmicb.2011.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puertollano E., Kolida S., Yaqoob P. Biological significance of short-chain fatty acid metabolism by the intestinal microbiome. Curr. Opin. Clin. Nutr. Metab. Care. 2014;17:139–144. doi: 10.1097/MCO.0000000000000025. [DOI] [PubMed] [Google Scholar]
- 33.Everard A., Belzer C., Geurts L., Ouwerkerk J.P., Druart C., Bindels L.B., Guiot Y., Derrien M., Muccioli G.G., Delzenne N.M., et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA. 2013;110:9066–9071. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shin N.-R., Lee J.-C., Lee H.-Y., Kim M.-S., Whon T.W., Lee M.-S., Bae J.-W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut. 2014;63:727–735. doi: 10.1136/gutjnl-2012-303839. [DOI] [PubMed] [Google Scholar]
- 35.Heilig J.S., Tonegawa S. Diversity of murine gamma genes and expression in fetal and adult T lymphocytes. Nature. 1986;322:836–840. doi: 10.1038/322836a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The 16S rRNA sequence datasets of this article have been deposited at the NCBI Sequence Read Archive (SRA) database under accession number PRJNA979156, (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA979156) and the metabolomics datasets have been deposited at the EMBL-EBI MetaboLights database with the identifier MTBLS7964 (https://www.ebi.ac.uk/metabolights/MTBLS7964). These data are publicly available as of the date of publication. Accession numbers of these data are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.