

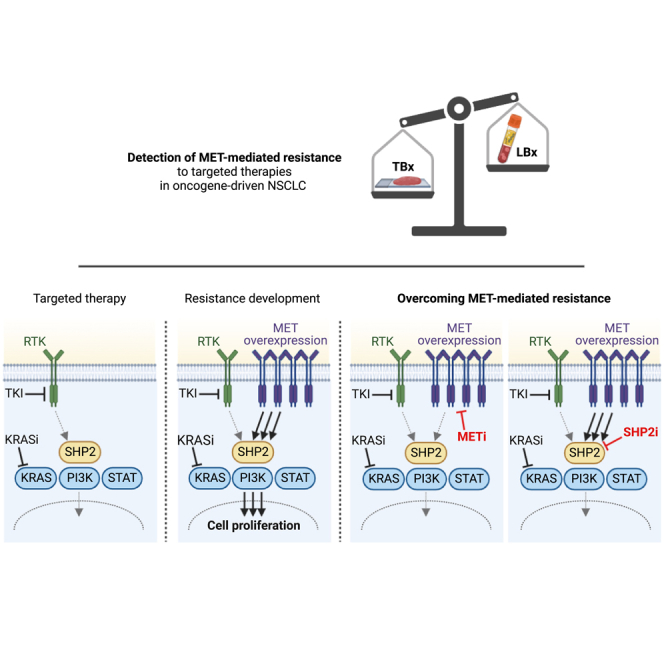
Summary
This study evaluates the efficacy of combining targeted therapies with MET or SHP2 inhibitors to overcome MET-mediated resistance in different NSCLC subtypes. A prevalence study was conducted for MET amplification and overexpression in samples from patients with NSCLC who relapsed on ALK, ROS1, or RET tyrosine kinase inhibitors. MET-mediated resistance was detected in 37.5% of tissue biopsies, which allow the detection of MET overexpression, compared to 7.4% of liquid biopsies. The development of drug resistance by MET overexpression was confirmed in EGFRex19del-, KRASG12C-, HER2ex20ins-, and TPM3-NTRK1-mutant cell lines. The combination of targeted therapy with MET or SHP2 inhibitors was found to overcome MET-mediated resistance in both in vitro and in vivo assays. This study highlights the importance of considering MET overexpression as a resistance driver to NSCLC targeted therapies to better identify patients who could potentially benefit from combination approaches with MET or SHP2 inhibitors.
Subject areas: Health sciences, Molecular physiology, Cancer
Graphical abstract
Highlights
-
•
MET overexpression provokes targeted therapy resistance in NSCLC
-
•
Tissue biopsies surpass liquid ones in detecting MET-mediated resistance
-
•
MET overexpression, a potential resistance driver, warrants deeper study
-
•
MET or SHP2 inhibitors can effectively tackle MET-mediated resistance
Health sciences; Molecular physiology; Cancer
Introduction
Targeted therapies have become a standard treatment for oncogene-driven non-small cell lung cancer (NSCLC),1,2 with epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) rearrangements being the most commonly targeted molecular alterations.3,4,5,6,7 Despite the initial clinical benefit to targeted therapies, most patients have an incomplete response and eventually acquire resistance. Resistance mechanisms include secondary alterations in the respective molecular target or bypass activation of other oncogenic pathways.2
Activation of the MET proto-oncogene, receptor tyrosine kinase (RTK) (MET) pathway by gene amplification constitutes an established and frequent resistance mechanism to EGFR tyrosine kinase inhibitors (TKIs).8 A variety of compounds targeting the MET receptor have been developed, such as the highly selective and potent ATP-competitive TKIs tepotinib9 and capmatinib.10 Prospective evaluation of combination therapies with osimertinib for patients with MET-mediated resistance is ongoing. The savolitinib plus osimertinib combination is being assessed in the phase II SAVANNAH study11 and the phase III SAFFRON study (ClinicalTrials.gov identifier: NCT05261399), while the tepotinib plus osimertinib combination is being evaluated in the INSIGHT2 study.12
Beyond EGFR-mutant tumors,13 MET amplification has also been identified as a resistance driver to other targeted therapies in oncogene-driven NSCLC, including KRASG12C14,15-, HER216-mutant, as well as, ALK17-, ROS118,19-, and RET20-rearranged tumors. Although no studies on resistance mechanisms to neurotrophic tropomyosin receptor kinase (NTRK) inhibitors in patients with NSCLC at progression have been published so far, the acquisition of resistance by MET is highly probable as it has been observed in a patient with NTRK-rearranged cholangiocarcinoma.21
The cytoplasmic tyrosine phosphatase Src homology 2 domain-containing phosphatase 2 (SHP2), encoded by the PTPN11 gene, is a key component of the RAS/RAF/ERK, PI3K/AKT and JAK/STAT pathways and transduces signaling from various RTKs to promote RAS activation and subsequently cell proliferation, differentiation, and survival.22 Several allosteric SHP2 inhibitors, including TNO15523 (ClinicalTrials.gov identifier: NCT03114319), are currently undergoing clinical evaluation and preclinical studies have shown that they can overcome resistance to MET,24 EGFR,25 and MEK inhibitors.26
In the present study, we evaluate the effect of combining targeted therapies with MET or SHP2 inhibitors after occurrence of MET-mediated resistance in different molecular subtypes of NSCLC.
Results
MET amplification and overexpression after relapse on targeted therapies in NSCLC with ALK, ROS1, or RET rearrangements
Although MET amplification has been described as a bypass resistance mechanism to targeted therapies, there are limited data on MET mRNA levels and MET protein expression and phosphorylation in this setting. Consequently, we conducted a prevalence study of MET copy numbers, mRNA levels, as well as MET protein expression and phosphorylation in tumor or cytological biopsies (TBx) and liquid biopsies (LBx) from 28 patients with NSCLC who had disease progression on ALK (n = 15), ROS1 (n = 8) or RET (n = 5) TKIs. For 20 patients only LBx were available, for one patient only a TBx was available and for the remaining seven patients both TBx and at least one LBx were available (blood, cerebrospinal fluid, or pleural effusion) (Table S1). MET amplification was analyzed by next generation sequencing (NGS) and fluorescence in situ hybridization (FISH), MET mRNA levels were determined by nCounter, while MET protein expression and phosphorylation were investigated by immunohistochemistry (IHC). In LBx, MET amplification was detected by NGS in 2/27 (7.4%) patients (Figure 1A and Table S1). Both patients (P6 and P27) had ALK-rearranged tumors (variant 1 and 3) and relapsed on lorlatinib. In TBx, MET alterations were detected in 3/8 (37.5%) patients (Figure 1A and Table S1). Two of them (P24 and P27) had ALK-rearranged NSCLC (variant 1) progressing on lorlatinib and had MET amplification by NGS, with patient P24 also displaying MET mRNA overexpression by nCounter, as well as MET protein overexpression and phosphorylation by IHC. Patient P27 had MET amplification also detected in LBx (pleural effusion), while in patient P24 the LBx (blood) result was negative for MET amplification. The third patient (P26) had a CD74-ROS1-rearranged tumor progressing on crizotinib and had MET mRNA overexpression by nCounter, as well as MET protein overexpression by IHC without MET amplification (Figure 1B).
Figure 1.

Frequency of MET amplification and MET overexpression in 28 patients with NSCLC who had disease progression on ALK (n = 15), ROS1 (n = 8) or RET (n = 5) tyrosine kinase inhibitors (TKIs) (refer to Table S1 for details)
(A) Left: Pie chart visualizing the frequency of MET amplification or on-target ALK/ROS1 mutations as potential resistance mechanisms to ALK, ROS1 or RET TKIs in 27 patients with liquid (blood, pleural effusions, cerebrospinal fluids) biopsies (LBx) available. Patients P6 and P27 tested positive for MET amplification. Right: Pie chart visualizing the frequency of MET amplification, MET overexpression or on-target ALK/ROS1 mutations as potential resistance mechanisms to ALK, ROS1 or RET TKIs in 8 patients with tumor or cytological biopsies (TBx) available. Patients P24 and P27 tested positive for MET amplification (P24 also showed MET overexpression) and patient P26 tested positive for MET overexpression without MET amplification.
(B) FISH and IHC analyses of patients P24 and P26 progressing on TKIs presumably by MET alterations. Left: patient P24 harbored an ALKv1-rearranged NSCLC relapsing on lorlatinib and had MET amplification. Right: patient P26 harbored a CD74-ROS1-rearranged NSCLC relapsing on crizotinib and had MET overexpression without MET amplification. As indicated in the images, scale bars refer to 5, 100 and 200 µm, respectively.
Our prevalence study demonstrates a higher percentage of MET-mediated resistance to targeted therapies detected in TBx (37.5%) than in LBx (7.4%). Nonetheless, the ease of LBx in clinical practice at the time of resistance needs to be acknowledged as seen by the fact that 27/28 patients had LBx to analyze. However, the detection of MET amplification is frequently missed and can be detected in TBx. Moreover, the use of TBx also allows the analysis of MET overexpression that can help further identify patients with a MET-mediated mechanism of TKI resistance who may benefit from MET TKI treatment.
Generation of cell line models with MET-mediated resistance to targeted therapies
To explore the functional role of MET overexpression in a defined genetic background of oncogene-driven NSCLC, we generated stable cell lines using lentiviral transduction in which the wild-type MET protein can be dynamically overexpressed in a doxycycline-inducible manner (tetON-MET cells). Five NSCLC cell lines (EGFRex19del-mutant: HCC827, KRASG12C-mutant: NCI-H358, HER2ex20ins-mutant: NCI-H1781; EML4-ALK-mutant: NCI-H2228, SLC34A2-ROS1-mutant: HCC-78) and one colorectal cancer cell line (TPM3-NTRK1-mutant: KM12) were selected as model systems. Overexpression of the 170 kDa immature pro-MET protein could be confirmed in all cell lines by Western blot (Figure 2A). It has been reported that cleavage of the pro-MET protein into the 50 kDa α-subunit and the 145 kDa β-subunit may not be required for receptor activity, as the uncleaved precursor protein is still constitutively active.27 Likewise, increased phosphorylation at Y1234/1235 of the pro-MET protein could be detected in all MET-overexpressing cells, suggesting that MET overexpression is sufficient for MET-receptor self-activation. To exclude fetal calf serum (FCS)-dependent activation in the MET-overexpressing cells, KM12 cells were analyzed in the absence of FCS (Figure S1A). Doxycycline treatment led to a strong enhancement of the activating phosphorylation site Y1234/1235 of the pro-MET protein independently of FCS supplementation, further suggesting self-activation of the MET receptor by MET overexpression.
Figure 2.

MET overexpression causes resistance to osimertinib, sotorasib, poziotinib, and entrectinib
(A) The indicated tetON-MET cells were induced with doxycycline for 48 h before lysis. Western blot analysis was performed with the indicated antibodies.
(B) Cells were cultured with doxycycline for 24 h before being treated with the respective targeted therapies. Cellular viability was measured six days from treatment onset. Data are represented as mean of n = 3 ± SD.
Next, we examined the effect of MET overexpression on resistance development by treating all cells with their corresponding targeted therapies (Figure 2B). The EGFR inhibitor osimertinib, the KRASG12C inhibitor sotorasib, the HER2 inhibitor poziotinib, the ALK inhibitor alectinib and the NTRK-/ROS1 inhibitor entrectinib were used. While the cells without MET overexpression were sensitive to their corresponding targeted therapy, doxycycline-induced MET overexpression caused resistance to osimertinib and sotorasib in HCC827 and NCI-H358 cells, respectively. In NCI-H1781 and KM12 cells, a partial resistance development to poziotinib and entrectinib, respectively, could be observed. In contrast, MET overexpression did not alter the sensitivity of NCI-H2228 and HCC-78 to alectinib and entrectinib, respectively. We also tested whether a higher MET overexpression, achieved by an increased lentiviral multiplicity of infection (MOI), would confer alectinib resistance in NCI-H2228 cells. However, although the cells displayed increased levels of MET protein and phosphorylation, they did not become resistant (Figure S1B). Changes in drug sensitivity by the lentiviral transduction could be excluded by comparing the cell growth behavior of transfected cells in the absence of doxycycline to their parental cell lines (Figure S2A).
In line with the viability assay results, only HCC827, NCI-H358, NCI-H1781, and KM12 cells demonstrated morphological changes upon MET overexpression (Figure S2B). While their counterparts without MET overexpression grew in defined epithelial clusters, MET-overexpressing cells exhibited a mesenchymal spindle-like cell shape with single cell movement.
MET or SHP2 inhibitors revert MET-mediated resistance in vitro
Next, we explored whether direct targeting of MET or the simultaneous blockade of various RTKs by inhibition of SHP2 can overcome the MET-mediated resistance. We analyzed the response of the four cell lines recapitulating MET-driven resistance (HCC827, NCI-H358, NCI-H1781, KM12) upon treatment with their respective targeted therapies combined with the MET inhibitor tepotinib or the SHP2 inhibitor TNO155 by Western blot. Upon single-treatment with osimertinib or sotorasib stronger phosphorylation of AKT, ERK, and STAT3 could be observed in the MET overexpressing HCC827 and NCI-H358 cells, respectively, compared to the cells with endogenous MET expression (Figure 3A). These results indicate remaining MAPK/PI3K downstream signaling upon treatment with the targeted therapies in the MET-overexpressing cells and support the observation of resistance development. In both MET-overexpressing cell lines, the combination of the targeted agent with either tepotinib or TNO155 decreased AKT and ERK phosphorylation more potently than either of the agents alone, suggesting reversion of the MET-driven resistance. The phosphorylation of STAT3 increased upon SHP2 inhibition, since SHP2 is known to negatively regulate STAT3 activity.28 MET overexpressing NCI-H1781 and KM12 cells also displayed stronger phosphorylation of AKT, ERK and STAT3 upon single-treatment with poziotinib and entrectinib, respectively, than the cells without MET overexpression (Figure 3B). However, the effect was not as pronounced as in HCC827 and NCI-H358 cells, in which the targeted therapy nearly abolished ERK phosphorylation in the cells without MET overexpression but only resulted in a minor decrease of ERK phosphorylation in the MET-overexpressing cells (Figure 3A). This is in line with the partial development of MET-driven resistance in NCI-H1781 and KM12 compared to HCC827 and NCI-H358 cells. Yet, combination treatment of poziotinib and entrectinib with tepotinib or TNO155 in MET overexpressing NCI-H1781 and KM12 cells decreased the phosphorylation of ERK stronger than either of the inhibitors alone. Thus, the Western blot data suggest that the combination of targeted therapies with MET or SHP2 inhibitors could be efficacious in overcoming MET-mediated resistance.
Figure 3.

The combination of targeted therapies with tepotinib or TNO155 decreases MAPK/PI3K downstream signaling more potently than either of the agents alone
(A) HCC827 tetON-MET and NCI-H358 tetON-MET, as well as (B) NCI-H1781 tetON-MET and KM12 tetON-MET cells were induced with doxycycline for 48 h before they were treated with the respective inhibitors and combinations for 6 h. Western blot analysis was performed with the indicated antibodies.
Next, we analyzed the viability of the cell lines recapitulating MET-driven resistance upon combination treatment of the respective targeted therapy with the MET inhibitors tepotinib and capmatinib or with the SHP2 inhibitors M674824 and TNO155 (Figures 4A and S3). As already suggested by the pathway phosphorylation analyses, the combination of osimertinib, sotorasib, poziotinib, and entrectinib with either MET or SHP2 inhibitors were efficacious in reverting the MET-mediated resistance. Moreover, synergism between the targeted therapies and MET or SHP2 inhibitors could be confirmed by combination dose matrices in MET overexpressing HCC827, NCI-H358, and KM12 cells using the Loewe combination method29 (Figure S4). Similar results were obtained by colony formation assays (Figure 4B). While the single-treatment with the targeted therapies prevented most colony growth of the cells with endogenous MET, increased colony forming ability was detected in the MET overexpressing HCC827 and NCI-H358 cells. In NCI-H1781 and KM12 cells, MET overexpression only conferred increased colony formation ability to a small number of cells. Yet, in all cell lines the combination of osimertinib, sotorasib, poziotinib, and entrectinib with MET or SHP2 inhibitors reduced the colony count of the MET-overexpressing cells stronger than either of the inhibitors alone.
Figure 4.

MET or SHP2 inhibitors revert MET-mediated resistance in vitro
The indicated tetON-MET cells were cultured with doxycycline for 24 h before they were treated with the indicated targeted therapies and combinations.
(A) The cell lines were analyzed in 6-day viability assays. Data are represented as mean of n = 3 ± SD.
(B) The cell lines were analyzed in two week colony formation assays.
Tepotinib overcomes MET-mediated resistance in vivo
Finally, we explored whether adding tepotinib to a targeted therapy in vivo can revert MET-mediated resistance. For this, we selected the KRASG12C-mutant model NCI-H358, in which doxycycline-induced MET overexpression caused resistance to sotorasib. Mice were subcutaneously injected with NCI-H358 tetON-MET cells and fed with a doxycycline-containing diet to establish MET overexpressing KRASG12C-mutant tumors. MET overexpression and induction of MET autophosphorylation upon doxycycline was confirmed in the established xenograft tumors (Figure S5A). Tumor bearing mice were kept on doxycycline-containing diet during the experiment and were randomized (n = 10/group) to receive once daily oral treatments of either vehicle control, sotorasib, or tepotinib monotherapy or the combination of the two compounds for 75 days. Sotorasib and tepotinib monotherapy delayed tumor growth compared to the vehicle control, but MET overexpression conferred sotorasib resistance with tumor volumes reaching the humane endpoint after 75 days of treatment (Figure 5A). In contrast, the combination of tepotinib with sotorasib significantly inhibited tumor growth and resulted in tumor regression in all treated animals (Figures 5A and 5B). This result is supported by the decrease of ERK and AKT phosphorylation upon tepotinib plus sotorasib combination treatment detected in tumor lysates at the end of the study (Figure S5B). The combination therapy was well tolerated based on body weight change (Figure 5C) and clinical symptoms.
Figure 5.

Tepotinib overcomes MET-mediated sotorasib resistance in vivo
Antitumor activity and tolerability of sotorasib and tepotinib as mono- and combination therapy was investigated in mice bearing subcutaneous NCI-H358 tetON-MET xenografts. Ten mice were randomized to each treatment. Mice were fed with a doxycycline-containing diet and compounds were applied daily via oral gavage.
(A) Graph displaying the tumor volumes. Data are represented as mean ± SEM.
(B) Waterfall plot showing the relative tumor volume change from baseline in each individual mouse at day of best overall response in combination group (d42).
(C) Graph displaying the relative body weight change. Data are represented as mean ± SEM.
Discussion
To date, the best predictive biomarkers for sensitivity to MET inhibitors are the METex14 skipping mutation and MET gene amplification.9,30 While METex14 is mostly a primary oncogenic driver, gene amplification constitutes an established and frequent mechanism of resistance in EGFR-mutant NSCLC.13 Increasing evidence also suggests dysregulated MET signaling as resistance driver to other targeted therapies, such as ALK,17 ROS1,18,19 and RET20 TKIs. The observation that acquired resistance to targeted therapies is oncogene or drug agnostic indicates that MET-driven resistance may potentially occur in patients with NSCLC who relapse on various targeted therapies.31
Most studies have focused on the detection of MET amplification through FISH or NGS as a resistance mechanism to TKIs. MET protein overexpression through IHC has also been evaluated, particularly in earlier resistance studies with mixed results in terms of efficacy,32 but the use of MET mRNA levels is still quite exploratory.33 The underlying complex machinery of transcription, translation, and protein degradation determining protein abundance34 warrants an extensive evaluation of the different determinants of MET-altered resistance in an effort to be able to identify the largest population that may benefit from a targeted treatment at the time of TKI progression. In our prevalence study, the detection of MET amplification in 7.4% of patients tested in LBx is in line with previous studies reporting MET amplification as a resistance driver in approximately 10% of NSCLC with ALK, ROS1, or RET rearrangements.35 Our TBx analysis showed a higher prevalence (37.5%) of MET-mediated resistance than what can be detected by LBx. NGS testing in either TBx or LBx is a valuable method for the detection of the mechanism of resistance but still has limitations for the detection of MET amplification.12,36 This may be due to transcriptional or translational enhancement that cannot be detected by NGS. Additionally, low fractions of circulating tumor DNA (ctDNA) can limit the sensitivity of NGS in LBx, leading to poor concordance with genomic profiling of TBx. Therefore, negative LBx results should be confirmed with TBx testing.37 Previous reports in the EGFR mutant setting have shown that low ctDNA content limits NGS sensitivity for detecting MET amplification in LBx.36
High MET protein levels are not only associated with higher pathological tumor stage and worse prognosis,38 but some evidence suggests that high MET expression and phosphorylation also correlate with drug sensitivity.39,40 Therefore, our analyses highlight the need to consider MET overexpression, besides MET amplification, as a resistance driver to better identify patients potentially benefiting from combination treatments and prioritize tissue biopsies compared to plasma. The SAVANNAH study has shown clinically relevant efficacy with the combination of the MET inhibitor savolitinib with osimertinib for patients with EGFR-mutant NSCLC who have developed resistance to osimertinib and have high level MET amplification and/or MET overexpression as detected on TBx with FISH and IHC, respectively.11
Nonetheless, future studies with larger sample size from patients with NSCLC who relapsed on ALK, ROS1 or RET TKIs and, at best, paired TBx and LBx are needed for further investigation. Moreover, baseline biopsies to rule out MET alterations are already present before treatment, and the use of other measurements, such as ELISA for the detection of shed MET41 or whole transcriptome analysis, would provide detailed insights on the MET status.
Cell line models harboring NSCLC driver mutations and acquired MET-mediated resistance to targeted therapies are rare, probably because their generation by long-term exposure with targeted agents is time consuming and random genetic mutations are acquired. For this reason, we generated stable cell lines in which the wild-type MET protein was artificially overexpressed in a doxycycline-inducible manner in the defined genetic background of oncogene-driven human cell line models. Resistance development to the respective targeted therapies by MET overexpression was observed in the EGFRex19del-, and KRASG12C-mutant cell lines and partially in the HER2ex20ins- and TPM3-NTRK1-mutant cell lines. In contrast to the findings from Dagogo-Jack et al.,17 we did not observe resistance development by MET overexpression in the ALK-mutant cell line. One possible explanation could be the choice of different cell line models. While the NCI-H3122 cells used by Dagogo-Jack and colleagues express EML4-ALK variant 1, NCI-H2228 cells harbor variant 3a/b.42 It has been shown that the EML4-ALK variant 1 displays less protein stability and therewith greater sensitivity toward ALK inhibitors.43 Moreover, NCI-H3122 cells exhibit stronger epithelial characteristics than NCI-H2228 cells,44 rendering the latter potentially less susceptible to MET overexpression. Similarly, the SLC34A2-ROS1-mutant HCC-78 cell line, in which no resistance development by MET overexpression could be observed, already possesses high MET expression and particularly high MET phosphorylation in the parental state. In this cell line, MET phosphorylation was not considerably increased after doxycycline treatment, suggesting saturated MET receptor activity.
Together, these findings demonstrate that MET overexpression causes resistance to targeted therapies in four out of six tested human cell line models, indicating that the downstream effects of MET overexpression can vary depending on the oncogenic driver in the cancer cell. Therefore, the observation that MET overexpression causes resistance to targeted therapies in some oncogene-driven models but not in others suggests that there is heterogeneity in the underlying mechanisms of resistance, which is dependent on the specific oncogenic driver.
Since strategies combining targeted therapies with inhibitors of components of the ERK/MAPK pathway could restore drug sensitivity and potentiate therapeutic benefit,2,24 we tested the concept of combining them with selective MET or SHP2 inhibitors. In addition to supporting the combination efficacy of EGFR with MET inhibitors already being tested in clinical studies,12,45,46 our data reveal that combinations not only with specific MET inhibitors but also with SHP2 inhibitors can overcome MET-mediated resistance, and these findings extend beyond EGFR-mutant models25 to other molecular NSCLC subtypes. The efficacy of crizotinib and capmatinib in acquired resistance models of sotorasib has been shown previously.47 However, our sotorasib-resistant KRASG12C-cell line model demonstrates combination benefit with both MET and SHP2 inhibitors and, for the first time, the reversion of MET-driven resistance by tepotinib with sotorasib could also be shown in vivo. This is of great importance, since there is a high medical need for patients progressing on KRASG12C inhibitors by MET alterations.14,15 The mechanisms of acquired resistance to poziotinib and entrectinib for patients with NSCLC with HER2ex20ins mutations and patients with solid tumors harboring an NTRK gene fusion, respectively, remain unclear. Molecular profiling of afatinib-resistant HER2-mutant NSCLC cell lines suggests MET amplification as probable resistance driver.16 The publication by Elamin et al.48 reports that MET amplification can also serve as a mechanism of resistance to poziotinib in patients with EGFR exon 20-mutant NSCLC. Likewise, Cocco et al. identified MET amplification as resistance mechanism to entrectinib in a patient with NTRK-rearranged cholangiocarcinoma.21 Therefore, to the best of our knowledge, our study reveals for the first time that MET overexpression causes resistance to poziotinib and entrectinib in HER2ex20ins- and NTRK1-mutant cell line models, respectively, and demonstrates that combination with MET or SHP2 inhibitors may restore sensitivity to the targeted therapies.
In conclusion, our preclinical models of resistance to osimertinib, sotorasib, poziotinib, and entrectinib recapitulate MET overexpression as a resistance mechanism to targeted therapies and provide powerful tools to explore reversion of resistance development. With the use of these model systems, we show the broad clinical application of MET and SHP2 inhibitors as combination partners to overcome MET-mediated resistance. Together with the results of the prevalence study, our data highlights the importance to consider MET overexpression as a potential resistance driver in oncogene-driven NSCLC to identify the largest population that may benefit from combination approaches with MET or SHP2 inhibitors after relapse on targeted therapy.
Limitations of the study
This study has limitations that should be noted. Firstly, the small sample size in our prevalence study of patients with NSCLC who relapsed on ALK, ROS1 or RET TKIs restricts our ability to perform robust statistical analysis and draw definitive conclusions. Secondly, paired TBx and LBx would be necessary to directly compare the detection of MET alterations within patients, and the analysis of baseline biopsies would be beneficial to distinguish between intrinsic and acquired MET-mediated resistance. Thirdly, our preclinical cell line models are an artificial system with variable MET overexpression levels and thus limited comparison to MET levels in patient samples. However, the focus on MET overexpression as a resistance driver and the prevention of random mutations from long-term exposures with targeted therapies justified the use of this approach. Finally, while the choice of a single cell line to represent a specific oncogene-driven tumor subtype is a limitation in drawing definitive conclusions regarding the role of MET-mediated resistance in that subtype, our study using six cell lines underscores the importance of considering the context specificity of MET overexpression as a resistance driver. These findings highlight the need for further investigation of the mechanisms underlying resistance to targeted therapies across various oncogene-driven tumor subtypes.
Despite these limitations, our study provides important information for future clinical trials on MET protein expression as diagnostic biomarker to identify patients with NSCLC who progressed on targeted therapy and who may benefit from combination strategies with MET or SHP2 inhibitors.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-phospho-MET, 1:1000 | Cell Signaling | #3077; RRID:AB_2143884 |
| mouse anti-Met, 1:1000 | Cell Signaling | #3127; RRID:AB_331361 |
| mouse anti-AKT, 1:1000 | Cell Signaling | #2920; RRID:AB_1147620 |
| rabbit anti-phospho-Akt (Ser473), 1:1000 | Cell Signaling | #4058; RRID:AB_331168 |
| mouse anti-STAT3, 1:1000 | Cell Signaling | #9139; RRID:AB_331757 |
| rabbit anti- phospho-Stat3 (Tyr705) (D3A7) XP, 1:2000 | Cell Signaling | #9145; RRID:AB_2491009 |
| rabbit anti-phospho p44/42 MAP Kinase (Erk1/2), 1:1000 | Cell Signaling | #9101; RRID:AB_331646 |
| mouse anti-ERK, 1:5000 | BD | #610123; RRID:AB_397529 |
| mouse anti-GAPDH, 1:50000 | Santa Cruz | #sc-32233; RRID:AB_627679 |
| goat anti-rabbit Alexa Fluor 680, 1:25000 | Thermo Fisher | #A21076; RRID:AB_2535736 |
| donkey anti-mouse IRDye 800CW, 1:25000 | LI-COR | #926-32212; RRID:AB_621847 |
| MET SP44 clone | Roche, Mannheim, Germany | NA |
| Chemicals, peptides, and recombinant proteins | ||
| alectinib | BOC Sciences | NA |
| capmatinib | ChemShuttle | NA |
| entrectinib | Medchemexpress | NA |
| M6748 | The healthcare business of Merck KGaA, Darmstadt, Germany International Patent Application Publication No.: WO2020/033828 A1, Example #10b) |
NA |
| osimertinib | VWR | NA |
| poziotinib | AChemBlock | NA |
| sotorasib | Medchemexpress/MedKoo Biosciences | NA |
| tepotinib | The healthcare business of Merck KGaA, Darmstadt, Germany | NA |
| TNO155 | Synthesized at the healthcare business of Merck KGaA, Darmstadt, Germany | NA |
| Deposited data | ||
| Patient 1 | This paper, deposited in NCBI SRA: PRJNA975367 | SAMN35325464 |
| Patient 2 | This paper, deposited in NCBI SRA: PRJNA975367 | SAMN35325465 SAMN35325466 |
| Patient 3 | This paper, deposited in NCBI SRA: PRJNA975367 | SAMN35325467 |
| Patient 4 | This paper, deposited in NCBI SRA: PRJNA975431 | SAMN35326201 |
| Patient 5 | This paper, deposited in NCBI SRA: PRJNA975431 | SAMN35326202 |
| Patient 6 | This paper, deposited in NCBI SRA: PRJNA975431 | SAMN35326203 SAMN35326204 |
| Patient 7 | This paper, deposited in NCBI SRA: PRJNA975431 | SAMN35326205 |
| Patient 8 | This paper, deposited in NCBI SRA: PRJNA975431 | SAMN35326206 SAMN35326207 |
| Patient 9 | This paper, deposited in NCBI SRA: PRJNA975431 | SAMN35326208 |
| Patient 10 | This paper, deposited in NCBI SRA: PRJNA975431 | SAMN35326209 |
| Patient 11 | This paper, deposited in NCBI SRA: PRJNA975431 | SAMN35326210 |
| Patient 12 | This paper, deposited in NCBI SRA: PRJNA975444 | SAMN35326335 |
| Patient 13 | This paper, deposited in NCBI SRA: PRJNA975444 | SAMN35326336 |
| Patient 14 | This paper, deposited in NCBI SRA: PRJNA975444 | SAMN35326337 |
| Patient 15 | This paper, deposited in NCBI SRA: PRJNA975444 | SAMN35326338 |
| Patient 16 | This paper, deposited in NCBI SRA: PRJNA975444 | SAMN35326339 |
| Patient 17 | This paper, deposited in NCBI SRA: PRJNA975444 | SAMN35326340 |
| Patient 18 | This paper, deposited in NCBI SRA: PRJNA975444 | SAMN35326341 |
| Patient 19 | This paper, deposited in NCBI SRA: PRJNA975444 | SAMN35326342 |
| Patient 20 | This paper, deposited in NCBI SRA: PRJNA975444 | SAMN35326343 |
| Patient 22 | This paper, deposited in NCBI SRA: PRJNA975361 | SAMN35325436 SAMN35325437 |
| Patient 23 | This paper, deposited in NCBI SRA: PRJNA975361, PRJNA975444 | SAMN35325438 SAMN35326344 |
| Patient 24 | This paper, deposited in NCBI SRA: PRJNA975456 | SAMN35326492 |
| Patient 25 | This paper, deposited in NCBI SRA: PRJNA975361, PRJNA975456 | SAMN35325439 SAMN35326493 SAMN35326494 |
| Patient 26 | This paper, deposited in NCBI SRA: PRJNA975456 | SAMN35326495 SAMN35326496 |
| Patient 27 | This paper, deposited in NCBI SRA: PRJNA975456 | SAMN35326497 SAMN35326498 SAMN35326499 SAMN35326500 SAMN35326501 |
| Patient 28 | This paper, deposited in NCBI SRA: PRJNA975456 | SAMN35326502 SAMN35326503 |
| Experimental models: Cell lines | ||
| HCC827 | ATCC | ATCC CRL-2868 |
| NCI-H358 | ATCC | ATCC CRL-5807 |
| NCI-H1781 | ATCC | ATCC CRL-5894 |
| KM12 | M.D. Anderson Cancer Center | NA |
| NCI-H2228 | ATCC | ATCC CRL-5935 |
| HCC-78 | DSMZ | ACC 563 |
| Oligonucleotides | ||
| Probe set for nCounter hybridization, refer to Table S2 | NanoString Technologies, customized nCounter panel | NA |
| Recombinant DNA | ||
| pcLVi(3G)-MET-EF1a-TetON-T2A-Puro-WPRE | SIRION Biotech GmbH | NA |
| Software and algorithms | ||
| Prism (version 8.2.0) | GraphPad | NA |
| GeneData Screener® Software (version 16.0.5) | Genedata | NA |
| nSolver™ (version 2.6) | NanoString Technologies | NA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Niki Karachaliou (niki.karachaliou@merckgroup.com).
Materials availability
This study did not generate new unique reagents. The used drugs were synthesized at the healthcare business of Merck KGaA, Darmstadt, Germany or purchased from commercial vendors as stated in the key resources table.
Experimental model and subject details
Patients
Lung cancer patients from three institutions were included in the study; Dexeus University Hospital, Sagrat Cor Hospital and General de Catalunya Hospital. Previous informed patient consent was obtained in all cases. The study had been approved by the ethical committees of each hospital (approval number 04/2020, 25 February 2020) and was conducted according to the Declaration of Helsinki. Refer to Table S1 for details.
Cell lines
All cell lines used were obtained from commercial vendors (refer to key resources table) and cultured according to provider’s recommendations. The identity of the cell lines was confirmed by their respective STR profiles and the absence of mycoplasma and bacterial contamination were tested.
Mice
Xenograft tumors were established in eight-to ten-week-old female H2d Rag2 [C; 129P2-H2d-TgH(II2rg)tm1Brn-TgH(Rag2)tm1AltN4] mice (Taconic Biosciences, Denmark). The study design and animal usage were approved by local animal welfare authorities (Regierungspräsidium Darmstadt, Germany, protocol registration number DA4/Anz.1040).
Method details
FISH and IHC
FISH (Fluorescent in situ hybridization) for MET was performed with the ZytoLight SPEC MET/centromere 7 (MET/CEP7) Dual Color Probe (ZytoVision, Bremerhaven, Germany) according to manufacturer’s instructions. A sample was considered positive for MET amplification if (i) a MET/CEP7 ratio ≥2 and (ii) MET gene copy number (GCN) per cell ≥6 were observed. IHC (immunohistochemistry) staining for total and phospho-MET protein was performed with MET SP44 clone (Roche, Mannheim, Germany) and phospho-MET (Tyr1234/1235) D26 XP Rabbit mAb (Cell Signaling, Danvers, MA), respectively, on a BenchMark ULTRA automated tissue staining system (Ventana Medical Systems, Tucson, Arizona, USA). A sample was considered positive if intense membrane staining (3+) was observed in ≥50% of tumor cells.
DNA purification and NGS sequencing analysis
DNA was purified from liquid biopsies (blood, cerebrospinal fluid and pleural effusion), cytological samples and FFPE biopsies using the DNeasy Blood &Tissue Kit and the GeneRead DNA FFPE Kit, respectively (QIAGEN, Hilden, Germany), following the manufacturer’s instructions. DNA concentration was measured by Qubit and those samples with DNA ≥2.5 ng/μL were diluted to achieve this concentration. DNA-based next generation sequencing (NGS) was performed using a GeneRead QIAact Custom extended Panel (Qiagen, Hilden, Germany) according to manufacturer’s instructions. The panel targets mutations and copy number variations (CNVs) in 30 genes frequently altered in lung cancer (EGFR, BRAF, MET, ERBB2, ALK, ROS1, RET, PIK3CA, KRAS, NRAS, KIT, PDGFRA, TP53, STK11, KEAP1, ARID1A, FAT1, NFE2L2, SETD2, POLE, POLD1, IDH1, IDH2, ERBB4, FGFR1, FGFR2, FGFR3, MYC, CDK4, CDK6). Up to 40 ng of purified DNA were used as a template. Clonal amplification was performed on pooled libraries (625 pg) and, following bead enrichment, GeneReader instrument was used for sequencing. Qiagen Clinical Insight Analyze (QCI-A) software was employed to align the read data and call sequence variants, which were imported into the Qiagen Clinical Insight Interpret (QCI-I) web interface for data interpretation and generation of final custom report.
CNVs were identified using an in-house algorithm, of which the validation was extensively described elsewhere.40,49 First, for each sample, the sum of median UMI read coverage for the 30 genes in the panel was calculated. Then, the UMI read coverages for each gene were normalized using this sum, generating the so-called Ngx. Next, the geometric mean (geomean) and standard deviation (SD) of the normalized coverages were calculated for each gene across all samples analyzed in the laboratory. A gene was considered amplified if Ngx ≥ geomean +2 SD.
RNA extraction and NanoString nCounter assay
In the case of tissue samples (TBx), the high Pure RNA isolation Kit (Roche Diagnostics) was employed for RNA isolation, purity and concentration of the extracts were determined by NanoDrop 8000 (Thermo Scientific). Total RNA was hybridized with a custom-designed mixture of biotinylated capture tags and fluorescently labeled reporter probes (Elements Chemistry).40,50 All processes of hybridization, capture, clean-up and digital data acquisition were performed with nCounter Prep Station and Digital Analyzer (NanoString Technologies) according to the manufacturer’s instructions. The hybridization mixture included specific probes for fusions, MET-wt and METΔex14 target sequences (see Table S2). The analytical validation of the nCounter methodology to identify relevant gene rearrangements in advanced NSCLC (including result analysis, establishment of cut-off values and orthogonal validation with FISH and IHC) has been extensively described elsewhere.40,50,51 Reporter counts were uploaded into the nSolver analysis software version 2.6 and converted into an Excel Sheet. Samples were considered not evaluable if the geometrical mean (geomean) of counts corresponding to the housekeeping genes was lower than 100. Counts from MET probes were normalized in two steps and subjected to a logarithmic transformation to obtain the log-MET mRNA expression values.40
In the case of liquid biopsies (LBx), blood samples were collected in a 10-mL sterile Vacutainer tubes (BD) while pleural effusions and cerebrospinal fluids were collected in sterile tubes or containers with no additives or anticoagulants. Samples were centrifuged twice for 10 min at 500 xg and the circulating-free RNA (cfRNA) was isolated from 4 mL of supernatant using the QIAsymphony DSP Virus/Pathogen Midi Kit in a QIAsymphony robot (Qiagen), following the manufacturer’s instructions. Final elution volume was 50 μL in all cases. RNA concentration was estimated by Qubit and 5 μL were retrotranscribed to cDNA with the nCounter Low RNA Input Amplification Kit (NanoString Technologies). The cDNAs obtained were pre-amplified using the nCounter Low RNA Input Amplification Kit (NanoString Technologies). The 30-cycle pre-amplification step was performed in a Verity thermal cycler (Applied Biosystems) using a custom primer pool. Amplified cDNA was denatured at 95°C for 5 min and hybridized at 67°C for 18–24 h with a customized nCounter panel (see Table S2) including probes for housekeeping genes, common ALK, ROS1 or RET fusion transcripts and METΔex14 transcripts. Capture, clean-up and digital data acquisitions were performed as described for tissue samples. Samples were considered evaluable if the geometric mean of housekeeping gene counts was higher than 30. A sample was considered positive for a specific fusion if the counts of the corresponding transcript were higher than the geomean plus six times the standard deviation (6xSD) of the counts in the negative samples. Finally, liquid biopsy samples need a pre-amplification step (see above) and were considered not adequate to estimate tumor mRNA levels. This technique was validated using 84 LBx from healthy donors and patients with NSCLC, breast, prostate and pancreatic cancer as well as eight cell lines (NCI-H2228, NCI-H3122, HCC78, LC-2/ad, Hs746.T, NCI-H23, A-549, PC9).
Generation of MET-overexpressing cell lines
To generate the MET-overexpressing cell lines, lentiviral particles were produced by SIRION Biotech GmbH using the pcLVi(3G)-MET-EF1a-TetON-T2A-Puro-WPRE transfer plasmid and third generation lentiviral packaging plasmids. To construct the vector, the wild-type MET sequence (NM_001127500.3) was used. Cells were transduced by lentiviral infection and selected with puromycin for at least 2 weeks. To induce the MET overexpression, cells were treated with 0.5 μg/mL (0.1 μg/mL for NCI-H358 tetON-MET cells) doxycycline (Sigma-Aldrich #D3072) for at least 24 h.
Cell viability assays
Cells were seeded into clear 96-well plates at densities between 2000 and 2500 cells per well (9000/well for NCI-H1781 cells). The next day, cells were treated with serial dilutions of compounds with a constant DMSO concentration using a Tecan D300e Digital Dispenser (Tecan, Männedorf, Switzerland). After six days, cell viability was assessed using the Resazurin assay (R&D Systems, Minneapolis, MN) according to manufacturer’s instructions. Data was analyzed by the Loewe combination method29 using GeneData Screener Software (Version 16.0.5).
Colony formation assays
Cells were seeded into clear 6-well plates at densities between 2200 and 6000 cells per well and compound exposure initiated on the following day. Cells were cultured for two weeks, while medium and compounds were changed every 2–3 days. Colonies were fixed and stained with neutral red (in 70% Ethanol) for 15 min at room temperature (RT). Following incubation, the plates were rinsed thoroughly under running water and dried.
Western blot analysis
Cell or mouse tumor samples were lysed using RIPA lysis buffer: 0.5% Sodium deoxycholate, 50 mM Tris-HCl pH 7.4, 1% Triton, 137 mM NaCl, 1% Glycerin, 0.5 mM EDTA pH 8.0, 0.1% SDS. Following protein concentrations determination using the BCA Protein Assay Kit (Pierce, Appleton, WI), cell lysates were mixed with 4x NuPAGE LDS Sample Buffer (Invitrogen) and 10x NuPAGE Reducing Agent (Invitrogen) and heated at 98°C for 10 min. Lysates were separated using NuPAGE 4–12% BisTris Midi Protein gels (Invitrogen) and NuPAGE MOPS SDS Running Buffer (Invitrogen). Proteins were subsequently transferred onto Immun-Blot Low Fluorescence PVDF membranes (Bio-Rad) using the Bio-Rad Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were blocked using the Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE) on a rocking platform for 1 h. Primary antibodies (refer to key resources table) were incubated at 4°C overnight. Following washing steps, membranes were incubated with secondary antibodies (refer to key resources table) for 1 h at RT. Signals were detected using the Odyssey Imager (LI-COR Biosciences, Model 9120).
Mouse xenograft experiments
Xenograft tumors were established by subcutaneous injection of 5 million NCI-H358 tetON-MET cells suspended in 100 μL medium/Matrigel (1:1) into the right flanks of eight-to ten-week-old female H2d Rag2 [C; 129P2-H2d-TgH(II2rg)tm1Brn-TgH(Rag2)tm1AltN4] mice (Taconic Biosciences, Denmark). Mice were fed with a doxycycline-containing diet starting from the day of cell injection. When tumor xenografts reached a volume of 120–250 mm3, mice (N = 10 per treatment arm, randomized from 15 mice per arm to obtain a similar mean and median within the treatment groups) received the respective treatment. Tepotinib was formulated in 0.5% HPMC (Hydroxy propyl methyl cellulose) and 0.25% Tween 20 in water, sotorasib was administered in 2% HPMC and 1% Tween80 in water. Both drugs were administered by oral gavage once daily.
Quantification and statistical analysis
If not stated otherwise, results are presented as mean ± standard deviation (SD).
Dose-response curves were generated using GraphPad Prism (version 8.2.0) and synergy analysis was performed using GeneData Screener Software (Version 16.0.5).
Additional resources
This study has not generated or contributed to a new website/forum and is not part of a clinical trial.
Acknowledgments
We thank Jacqueline Buttler and Catherine Eichhorn for their technical guidance and support with cell culture activities.
Author contributions
Conceptualization: N.R. and N.K.; methodology: N.R., C.S., MA.M-V., J.A., C.E., N.K.; formal analysis: N.R., MA.M-V., J.A., N.K.; investigation: N.R., C.S., MA.M-V., N.J-A., D.K., S.G-R.; writing—original draft: N.R., N.K.; writing—review & editing: N.R., C.S., MA.M-V., N.J-A., D.K., S.G-R., MA.S., D.M., C.E., J.A., N.K.; visualization: N.R., MA.M-V., J.A., N.K.; supervision: J.A., N.K.
Declaration of interests
N.R., C.S., D.K., MA.S., D.M., C.E., J.A., and N.K. are employees of the healthcare business of Merck KGaA, Darmstadt, Germany. MA.M-V., N.J-A., and S.G-R., are employees of Pangaea Oncology, Hospital Universitario Quiron-Dexeus, Barcelona, Spain.
No other potential conflict of interest relevant to this article was reported.
Published: May 29, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107006.
Supplemental information
Data and code availability
-
•
Raw NGS and nCounter data related to Figure 1 and Tables S1 and S2 are deposited in the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI), under the following project codes and accession numbers (the single accession numbers are listed in the key resources table):
SRA: PRJNA975361 (https://www.ncbi.nlm.nih.gov/bioproject/975361): SAMN35325436 - SAMN35325439.
SRA: PRJNA975367 (https://www.ncbi.nlm.nih.gov/bioproject/975367): SAMN35325464 - SAMN35325467.
SRA: PRJNA975431 (https://www.ncbi.nlm.nih.gov/bioproject/975431): SAMN35326201 - SAMN35326210.
SRA: PRJNA975444 (https://www.ncbi.nlm.nih.gov/bioproject/975444): SAMN35326335 - SAMN35326344.
SRA: PRJNA975456 (https://www.ncbi.nlm.nih.gov/bioproject/975456): SAMN35326492 - SAMN35326503.
Microscopy images and original western blot images reported in this paper will be shared by the lead contact upon request.
-
•
This study did not analyze codes.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Rosell R., Karachaliou N. Large-scale screening for somatic mutations in lung cancer. Lancet. 2016;387:1354–1356. doi: 10.1016/S0140-6736(15)01125-3. [DOI] [PubMed] [Google Scholar]
- 2.Lamberti G., Andrini E., Sisi M., Rizzo A., Parisi C., Di Federico A., Gelsomino F., Ardizzoni A. Beyond EGFR, ALK and ROS1: current evidence and future perspectives on newly targetable oncogenic drivers in lung adenocarcinoma. Crit. Rev. Oncol. Hematol. 2020;156:103119. doi: 10.1016/j.critrevonc.2020.103119. [DOI] [PubMed] [Google Scholar]
- 3.Shaw A.T., Kim D.W., Nakagawa K., Seto T., Crinó L., Ahn M.J., De Pas T., Besse B., Solomon B.J., Blackhall F., et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 4.Ou S.H.I., Ahn J.S., De Petris L., Govindan R., Yang J.C.H., Hughes B., Lena H., Moro-Sibilot D., Bearz A., Ramirez S.V., et al. Alectinib in crizotinib-refractory ALK-rearranged non-small-cell lung cancer: a phase II global study. J. Clin. Oncol. 2016;34:661–668. doi: 10.1200/jco.2015.63.9443. [DOI] [PubMed] [Google Scholar]
- 5.Kim D.W., Mehra R., Tan D.S.W., Felip E., Chow L.Q.M., Camidge D.R., Vansteenkiste J., Sharma S., De Pas T., Riely G.J., et al. Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol. 2016;17:452–463. doi: 10.1016/S1470-2045(15)00614-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lynch T.J., Bell D.W., Sordella R., Gurubhagavatula S., Okimoto R.A., Brannigan B.W., Harris P.L., Haserlat S.M., Supko J.G., Haluska F.G., et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 7.Paez J.G., Jänne P.A., Lee J.C., Tracy S., Greulich H., Gabriel S., Herman P., Kaye F.J., Lindeman N., Boggon T.J., et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 8.Chmielecki J., Gray J.E., Cheng Y., Ohe Y., Imamura F., Cho B.C., Lin M.C., Majem M., Shah R., Rukazenkov Y., et al. Candidate mechanisms of acquired resistance to first-line osimertinib in EGFR-mutated advanced non-small cell lung cancer. Nat. Commun. 2023;14:1070. doi: 10.1038/s41467-023-35961-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paik P.K., Felip E., Veillon R., Sakai H., Cortot A.B., Garassino M.C., Mazieres J., Viteri S., Senellart H., Van Meerbeeck J., et al. Tepotinib in non-small-cell lung cancer with MET exon 14 skipping mutations. N. Engl. J. Med. 2020;383:931–943. doi: 10.1056/NEJMoa2004407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolf J., Seto T., Han J.Y., Reguart N., Garon E.B., Groen H.J.M., Tan D.S.W., Hida T., de Jonge M., Orlov S.V., et al. Capmatinib in MET exon 14-mutated or MET-amplified non-small-cell lung cancer. N. Engl. J. Med. 2020;383:944–957. doi: 10.1056/NEJMoa2002787. [DOI] [PubMed] [Google Scholar]
- 11.Ahn M.j., De Marinis F., Bonanno L., Cho B.C., Kim T.M., Cheng S., Novello S., Proto C., Kim S.W., Lee J.S., et al. EP08.02-140 MET biomarker-based preliminary efficacy analysis in SAVANNAH: savolitinib+osimertinib in EGFRm NSCLC post-osimertinib. J. Thorac. Oncol. 2022;17:S469–S470. doi: 10.1016/j.jtho.2022.07.823. [DOI] [Google Scholar]
- 12.Mazieres J., Kim T.M., Lim B.K., Wislez M., Dooms C., Finocchiaro G., Hayashi H., Liam C.K., Raskin J., Tho L.M., et al. LBA52 Tepotinib + osimertinib for EGFRm NSCLC with MET amplification (METamp) after progression on first-line (1L) osimertinib: initial results from the INSIGHT 2 study. Ann. Oncol. 2022;33:S1419–S1420. doi: 10.1016/j.annonc.2022.08.054. [DOI] [Google Scholar]
- 13.Camidge D.R., Davies K.D. MET copy number as a secondary driver of epidermal growth factor receptor tyrosine kinase inhibitor resistance in EGFR-mutant non-small-cell lung cancer. J. Clin. Oncol. 2019;37:855–857. doi: 10.1200/JCO.19.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Awad M.M., Liu S., Rybkin I.I., Arbour K.C., Dilly J., Zhu V.W., Johnson M.L., Heist R.S., Patil T., Riely G.J., et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N. Engl. J. Med. 2021;384:2382–2393. doi: 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Y., Murciano-Goroff Y.R., Xue J.Y., Ang A., Lucas J., Mai T.T., Da Cruz Paula A.F., Saiki A.Y., Mohn D., Achanta P., et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature. 2021;599:679–683. doi: 10.1038/s41586-021-04065-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torigoe H., Shien K., Takeda T., Yoshioka T., Namba K., Sato H., Suzawa K., Yamamoto H., Soh J., Sakaguchi M., et al. Therapeutic strategies for afatinib-resistant lung cancer harboring HER2 alterations. Cancer Sci. 2018;109:1493–1502. doi: 10.1111/cas.13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dagogo-Jack I., Yoda S., Lennerz J.K., Langenbucher A., Lin J.J., Rooney M.M., Prutisto-Chang K., Oh A., Adams N.A., Yeap B.Y., et al. MET alterations are a recurring and actionable resistance mechanism in ALK-positive lung cancer. Clin. Cancer Res. 2020;26:2535–2545. doi: 10.1158/1078-0432.CCR-19-3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J., Zhou P., Yu M., Zhang Y. Case report: high-level MET amplification as a resistance mechanism of ROS1-tyrosine kinase inhibitors in ROS1-rearranged non-small cell lung cancer. Front. Oncol. 2021;11:645224. doi: 10.3389/fonc.2021.645224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin J.J., Johnson T., Lennerz J.K., Lee C., Hubbeling H.G., Yeap B.Y., Dagogo-Jack I., Gainor J.F., Shaw A.T. Resistance to lorlatinib in ROS1 fusion-positive non-small cell lung cancer. J. Clin. Oncol. 2020;38:9611. doi: 10.1200/JCO.2020.38.15_suppl.9611. [DOI] [Google Scholar]
- 20.Lin J.J., Liu S.V., McCoach C.E., Zhu V.W., Tan A.C., Yoda S., Peterson J., Do A., Prutisto-Chang K., Dagogo-Jack I., et al. Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Ann. Oncol. 2020;31:1725–1733. doi: 10.1016/j.annonc.2020.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cocco E., Schram A.M., Kulick A., Misale S., Won H.H., Yaeger R., Razavi P., Ptashkin R., Hechtman J.F., Toska E., et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat. Med. 2019;25:1422–1427. doi: 10.1038/s41591-019-0542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karachaliou N., Cardona A.F., Bracht J.W.P., Aldeguer E., Drozdowskyj A., Fernandez-Bruno M., Chaib I., Berenguer J., Santarpia M., Ito M., et al. Integrin-linked kinase (ILK) and src homology 2 domain-containing phosphatase 2 (SHP2): novel targets in EGFR-mutation positive non-small cell lung cancer (NSCLC) EBioMedicine. 2019;39:207–214. doi: 10.1016/j.ebiom.2018.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LaMarche M.J., Acker M., Argintaru A., Bauer D., Boisclair J., Chan H., Chen C.H.T., Chen Y.N., Chen Z., Deng Z., et al. Identification of TNO155, an allosteric SHP2 inhibitor for the treatment of cancer. J. Med. Chem. 2020;63:13578–13594. doi: 10.1021/acs.jmedchem.0c01170. [DOI] [PubMed] [Google Scholar]
- 24.Pudelko L., Jaehrling F., Reusch C., Vitri S., Stroh C., Linde N., Sanderson M.P., Musch D., Lebrun C.J., Keil M., et al. SHP2 inhibition influences therapeutic response to tepotinib in tumors with MET alterations. iScience. 2020;23:101832. doi: 10.1016/j.isci.2020.101832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu C., Lu H., Wang H., Loo A., Zhang X., Yang G., Kowal C., Delach S., Wang Y., Goldoni S., et al. Combinations with allosteric SHP2 inhibitor TNO155 to block receptor tyrosine kinase signaling. Clin. Cancer Res. 2021;27:342–354. doi: 10.1158/1078-0432.CCR-20-2718. [DOI] [PubMed] [Google Scholar]
- 26.Fedele C., Ran H., Diskin B., Wei W., Jen J., Geer M.J., Araki K., Ozerdem U., Simeone D.M., Miller G., et al. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov. 2018;8:1237–1249. doi: 10.1158/2159-8290.CD-18-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J., Babic A. Regulation of the MET oncogene: molecular mechanisms. Carcinogenesis. 2016;37:345–355. doi: 10.1093/carcin/bgw015. [DOI] [PubMed] [Google Scholar]
- 28.Mohrherr J., Uras I.Z., Moll H.P., Casanova E. STAT3: versatile functions in non-small cell lung cancer. Cancers. 2020;12:1107. doi: 10.3390/cancers12051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loewe S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung. 1953;3:285–290. [PubMed] [Google Scholar]
- 30.Le X., Paz-Ares L.G., Van Meerbeeck J., Viteri S., Cabrera Galvez C., Vicente Baz D., Kim Y.-C., Kang J.-H., Schumacher K.-M., Karachaliou N., et al. Tepotinib in patients (pts) with advanced non-small cell lung cancer (NSCLC) with MET amplification (METamp) J. Clin. Oncol. 2021;39:9021. doi: 10.1200/JCO.2021.39.15_suppl.9021. [DOI] [Google Scholar]
- 31.Doebele R.C. Acquired resistance is oncogene and drug agnostic. Cancer Cell. 2019;36:347–349. doi: 10.1016/j.ccell.2019.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirsch F.R., Govindan R., Zvirbule Z., Braiteh F., Rittmeyer A., Belda-Iniesta C., Isla D., Cosgriff T., Boyer M., Ueda M., et al. Efficacy and safety results from a phase II, placebo-controlled study of onartuzumab plus first-line platinum-doublet chemotherapy for advanced squamous cell non-small-cell lung cancer. Clin. Lung Cancer. 2017;18:43–49. doi: 10.1016/j.cllc.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 33.Ruiz de Garibay G., Roch B., Garrido Lopez P., Isla D., Aguado C., Callejo Perez A., Marse Fabregat R., Garcia Campelo M.R., Blasco Cordellat A., Sanchez Torres J.M., et al. 1010P Assessment of early resistance mechanisms to first-line osimertinib in EGFR-mutant NSCLC using spatial transcriptomics. Ann. Oncol. 2022;33:S1015–S1016. doi: 10.1016/j.annonc.2022.07.1136. [DOI] [Google Scholar]
- 34.Vogel C., Marcotte E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coleman N., Hong L., Zhang J., Heymach J., Hong D., Le X. Beyond epidermal growth factor receptor: MET amplification as a general resistance driver to targeted therapy in oncogene-driven non-small-cell lung cancer. ESMO Open. 2021;6:100319. doi: 10.1016/j.esmoop.2021.100319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartmaier R.J., Markovets A.A., Ahn M.J., Sequist L.V., Han J.Y., Cho B.C., Yu H.A., Kim S.W., Yang J.C.H., Lee J.S., et al. Osimertinib + savolitinib to overcome acquired MET-mediated resistance in epidermal growth factor receptor-mutated, MET-amplified non-small cell lung cancer: TATTON. Cancer Discov. 2023;13:98–113. doi: 10.1158/2159-8290.CD-22-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Husain H., Pavlick D.C., Fendler B.J., Madison R.W., Decker B., Gjoerup O., Parachoniak C.A., McLaughlin-Drubin M., Erlich R.L., Schrock A.B., et al. Tumor fraction correlates with detection of actionable variants across > 23,000 circulating tumor DNA samples. JCO Precis. Oncol. 2022;6:e2200261. doi: 10.1200/PO.22.00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sadiq A.A., Salgia R. MET as a possible target for non-small-cell lung cancer. J. Clin. Oncol. 2013;31:1089–1096. doi: 10.1200/JCO.2012.43.9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsubara D., Ishikawa S., Oguni S., Aburatani H., Fukayama M., Niki T. Molecular predictors of sensitivity to the MET inhibitor PHA665752 in lung carcinoma cells. J. Thorac. Oncol. 2010;5:1317–1324. doi: 10.1097/JTO.0b013e3181e2a409. [DOI] [PubMed] [Google Scholar]
- 40.Aguado C., Teixido C., Román R., Reyes R., Giménez-Capitán A., Marin E., Cabrera C., Viñolas N., Castillo S., Muñoz S., et al. Multiplex RNA-based detection of clinically relevant MET alterations in advanced non-small cell lung cancer. Mol. Oncol. 2021;15:350–363. doi: 10.1002/1878-0261.12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lv H., Shan B., Tian Z., Li Y., Zhang Y., Wen S. Soluble c-Met is a reliable and sensitive marker to detect c-Met expression level in lung cancer. Biomed Res. Int. 2015;2015:626578. doi: 10.1155/2015/626578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koivunen J.P., Mermel C., Zejnullahu K., Murphy C., Lifshits E., Holmes A.J., Choi H.G., Kim J., Chiang D., Thomas R., et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin. Cancer Res. 2008;14:4275–4283. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heuckmann J.M., Balke-Want H., Malchers F., Peifer M., Sos M.L., Koker M., Meder L., Lovly C.M., Heukamp L.C., Pao W., et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin. Cancer Res. 2012;18:4682–4690. doi: 10.1158/1078-0432.CCR-11-3260. [DOI] [PubMed] [Google Scholar]
- 44.Gower A., Hsu W.H., Hsu S.T., Wang Y., Giaccone G. EMT is associated with, but does not drive resistance to ALK inhibitors among EML4-ALK non-small cell lung cancer. Mol. Oncol. 2016;10:601–609. doi: 10.1016/j.molonc.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smit E., Dooms C., Raskin J., Nadal E., Tho L.M., Le X., Mazieres J., Hin H., Morise M., Zhu V., et al. Insight 2: a phase II study of tepotinib plus osimertinib in MET-amplified NSCLC and first-line osimertinib resistance. Future Oncol. 2022;18:1039–1054. doi: 10.2217/fon-2021-1406. [DOI] [PubMed] [Google Scholar]
- 46.Sequist L.V., Han J.Y., Ahn M.J., Cho B.C., Yu H., Kim S.W., Yang J.C.H., Lee J.S., Su W.C., Kowalski D., et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020;21:373–386. doi: 10.1016/S1470-2045(19)30785-5. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki S., Yonesaka K., Teramura T., Takehara T., Kato R., Sakai H., Haratani K., Tanizaki J., Kawakami H., Hayashi H., et al. KRAS inhibitor resistance in MET-amplified KRAS (G12C) non-small cell lung cancer induced by RAS- and non-RAS-mediated cell signaling mechanisms. Clin. Cancer Res. 2021;27:5697–5707. doi: 10.1158/1078-0432.CCR-21-0856. [DOI] [PubMed] [Google Scholar]
- 48.Elamin Y.Y., Robichaux J.P., Carter B.W., Altan M., Tran H., Gibbons D.L., Heeke S., Fossella F.V., Lam V.K., Le X., et al. Poziotinib for EGFR exon 20-mutant NSCLC: clinical efficacy, resistance mechanisms, and impact of insertion location on drug sensitivity. Cancer Cell. 2022;40:754–767.e6. doi: 10.1016/j.ccell.2022.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vives-Usano M., García Pelaez B., Román Lladó R., Garzón Ibañez M., Aldeguer E., Rodriguez S., Aguilar A., Pons F., Viteri S., Cabrera C., et al. Analysis of copy number variations in solid tumors using a next generation sequencing custom panel. J. Mol. Pathol. 2021;2:123–134. [Google Scholar]
- 50.Aguado C., Giménez-Capitán A., Román R., Rodríguez S., Jordana-Ariza N., Aguilar A., Cabrera-Gálvez C., Rivas-Corredor C., Lianes P., Viteri S., et al. RNA-based multiplexing assay for routine testing of fusion and splicing variants in cytological samples of NSCLC patients. Diagnostics. 2020;11:15. doi: 10.3390/diagnostics11010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reguart N., Teixidó C., Giménez-Capitán A., Paré L., Galván P., Viteri S., Rodríguez S., Peg V., Aldeguer E., Viñolas N., et al. Identification of ALK, ROS1, and RET fusions by a multiplexed mRNA-based assay in formalin-fixed, paraffin-embedded samples from advanced non-small-cell lung cancer patients. Clin. Chem. 2017;63:751–760. doi: 10.1373/clinchem.2016.265314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Raw NGS and nCounter data related to Figure 1 and Tables S1 and S2 are deposited in the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI), under the following project codes and accession numbers (the single accession numbers are listed in the key resources table):
SRA: PRJNA975361 (https://www.ncbi.nlm.nih.gov/bioproject/975361): SAMN35325436 - SAMN35325439.
SRA: PRJNA975367 (https://www.ncbi.nlm.nih.gov/bioproject/975367): SAMN35325464 - SAMN35325467.
SRA: PRJNA975431 (https://www.ncbi.nlm.nih.gov/bioproject/975431): SAMN35326201 - SAMN35326210.
SRA: PRJNA975444 (https://www.ncbi.nlm.nih.gov/bioproject/975444): SAMN35326335 - SAMN35326344.
SRA: PRJNA975456 (https://www.ncbi.nlm.nih.gov/bioproject/975456): SAMN35326492 - SAMN35326503.
Microscopy images and original western blot images reported in this paper will be shared by the lead contact upon request.
-
•
This study did not analyze codes.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.