

ABSTRACT
Huntington disease (HD) is an inherited neurodegenerative disease with adult-onset clinical symptoms. However, the mechanism by which aging triggers the onset of neurodegeneration in HD patients remains unclear. Modeling the age-dependent progression of HD with striatal medium spiny neurons (MSNs) generated by direct reprogramming of fibroblasts from HD patients at different disease stages identifies age-dependent decline in critical cellular functions such as autophagy/macroautophagy and onset of neurodegeneration. Mechanistically, MSNs derived from symptomatic HD patients (HD-MSNs) are characterized by increased chromatin accessibility proximal to the MIR29B-3p host gene and its upregulation compared to MSNs from younger pre-symptomatic patients. MIR29B-3p in turn targets and represses STAT3 (signal transducer and activator of transcription 3) that controls the biogenesis of autophagosomes, leading to HD-MSN degeneration. Our recent study demonstrates age-associated microRNA (miRNA) and autophagy dysregulation linked to MSN degeneration, and potential approaches for protecting MSNs by enhancing autophagy in HD.
Abbreviations: HD: Huntington disease; mHTT: mutant HTT; MIR9/9*-124: MIR9/9* and MIR124; miRNA: microRNA; MSN: medium spiny neuron; STAT3: signal transducer and activator of transcription 3
KEYWORDS: Aging, autophagy/macroautophagy, Huntington disease, microRNA-mediated neuronal reprogramming, neurodegeneration
HD is a dominantly inherited neurodegenerative disorder characterized by the CAG expansion mutation with the HTT gene and the loss of MSNs during adulthood. How aging in the human lifespan drives the onset of neurodegeneration in HD patients remains unclear. To model the age-dependent progression of HD, direct neuronal reprogramming has been used to recapitulate adult-onset pathology of HD by converting patients’ fibroblasts directly into MSNs, in which the maintenance of age signatures in directly reprogrammed neurons has been postulated to be critical for modeling late-onset neuropathology.
In our study [1], MSNs were generated through direct neuronal reprogramming using miRNAs, MIR9/9* and MIR124 (MIR9/9*-124) that induce neurogenic chromatin remodeling, while additional transcription factors BCL11B/CTIP2, DLX1, DLX2, and MYT1L (CDM) guide the conversion towardMSNs. We performed MIR9/9*-124-CDM-based MSN reprogramming in a total of 24 fibroblast samples, including six fibroblast lines from independent HD patients before the onset of clinical symptoms, six fibroblasts from symptomatic patients, six control fibroblasts from healthy young adults, and six older control individuals (Figure 1A). Notably, neurodegeneration is selectively manifested in HD-MSNs reprogrammed from symptomatic patients’ fibroblasts, while healthy young and old MSNs (Ctrl-MSNs) and HD-MSNs from pre-symptomatic patients’ fibroblasts (pre-HD-MSNs) display a much lower degree of neuronal death. Pre-HD-MSNs display much lower levels of caspase activation and ANXA5/annexin V signal, further supporting the notion that reprogrammed MSNs from patients from different stages reliably recapitulate age-related neuropathology of HD.
Figure 1.
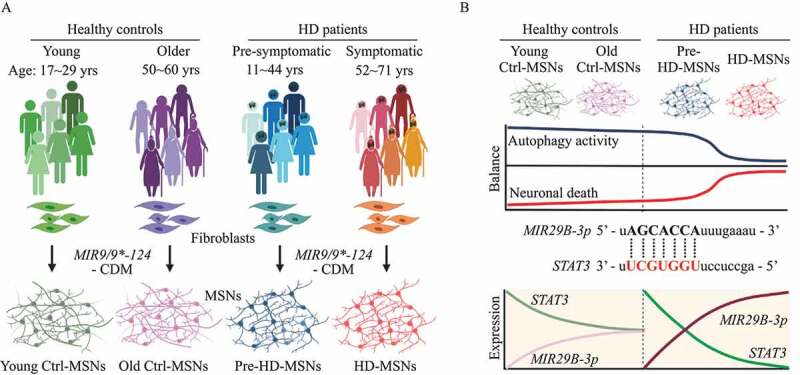
Modeling age-related Huntington disease progression through microRNA-mediated neuronal reprogramming. (A) Experimental scheme of MSNs conversion from fibroblasts of pre-symptomatic (pre-HD-MSNs), symptomatic HD patients (HD-MSNs), and respective healthy control individuals of similar ages (young or old-Ctrl-MSNs). MSNs were reprogrammed by overexpressing MIR9/9* and MIR124 (MIR9/9*-124), and MSN subtype-defining transcription factors, BCL11B/CTIP2, DLX1, DLX2, and MYT1L (CDM). (B) Top: Age-dependent changes in the balance between autophagy activity and neuronal death in patient-derived MSNs. Middle: the sequence of MIR29B-3p seed and seed-match in human STAT3 3′ UTR. Bottom: Aging-induced interaction between MIR29B-3p and STAT3 leading to autophagy impairment and neurodegeneration of HD-MSNs.
Weighted gene co-expression network analysis (WGCNA) comparing the transcriptome from pre-HD-MSNs, HD-MSNs, and young or old-Ctrl-MSNs by RNA-seq identified gene modules with altered gene expression that correlate with donor ages and disease stages. The gene module most significantly associated with downregulated gene networks with increasing ages and disease stages is enriched for genes associated with cell death, protein folding, senescence, and autophagy. Therefore, the differential cell death phenotype between pre-HD-MSNs and HD-MSNs is also reflected at the transcriptome level.
One of the genes downregulated in HD-MSNs compared to pre-HD-MSNs is STAT3, which regulates the balance between autophagy and cell death. Furthermore, upstream regulator analysis of downregulated gene module in HD-MSNs predicts autophagy inhibitors as upstream effectors, identifying changes in gene networks associated with age-related autophagy dysfunction in HD-MSNs.
Reflecting the gene expression differences, HD-MSNs show severely impaired autophagy function compared to pre-HD-MSNs and Ctrl-MSNs as inferred by the reduced number of autophagosomes and autolysosomes and the increased level of SQSTM1/p62. Interestingly, the link between autophagy and the onset of neurodegeneration becomes evident as treating pre-HD-MSNs with LY294002, a PtdIns3K and autophagy inhibitor, induces neuronal death while having no effect in Ctrl-MSNs. These results underscore the detrimental effect of autophagy inhibition specifically in HD patient-derived neurons. In addition, autophagic reduction in pre-HD-MSNs also increases mutant HTT (mHTT) aggregation whereas the glibenclamide analog G2, an autophagy activator, promotes HD-MSNs’ survival that correlates with increased autophagy activity and reduced mHTT aggregation. Thus, chemical inhibition or activation of autophagy in pre- or HD-MSNs reveals the link between the age-associated decline of autophagy in HD and MSN degeneration (Figure 1B).
To identify mechanisms responsible for the age-associated reduction in autophagy, we compared the chromatin accessibility landscape in reprogrammed MSNs between pre- and symptomatic stages. Interestingly, among the identified differentially accessible regions (DARs) were chromatin regions proximally located around miRNA precursors. Of these, the chromatin locus proximal to MIR29B1, a host gene of MIR29B-3p, becomes more accessible in HD-MSNs compared to pre-HD-MSNs. Interestingly, we found that albeit mildly, MIR29B-3p is upregulated in general with aging in the human postmortem striatum from elderly, cognitively normal individuals (83–91 years of age) over the young healthy individuals (8–19 years of age). In the context of HD, however, the MIR29B-3p upregulation is much more exaggerated HD-MSNs compared to pre-HD-MSNs, and the MIR29B-3p upregulation is also evident in the basal ganglia sections of HD patients even when compared to age-matched healthy individuals. Additionally, overexpressing MIR29B-3p in pre-HD-MSNs induces neuronal death and mHTT aggregation, whereas inhibiting MIR29B-3p promotes HD-MSNs’ survival and reduces mHTT aggregation. These results together demonstrated the detrimental effect of MIR29B-3p on HD-MSNs.
Among the predicted targets of MIR29B-3p, STAT3 was identified as a critical gene because many DARs closed in HD-MSNs occur near genes important for autophagy such as ATG5 and ATG7, which contain binding sites for STAT3. The 3ʹUTR of STAT3 harbors a functional seed-match sequence for MIR29B-3p, which interestingly, is absent in mice. In addition, knocking down STAT3 in pre-HD-MSNs reduces the autophagy activity and increases mHTT aggregation and neuronal death, whereas overexpressing STAT3 cDNA in HD-MSNs rescues cells from neurodegeneration. Moreover, treating HD-MSNs with a MIR29B-3p inhibitor reduces neuronal death while this effect is reversed by knocking down STAT3 in the presence of a MIR29B-3p inhibitor. Our results, thus, revealed the aging-induced interaction between MIR29B-3p and STAT3 contributing to autophagy dysfunction and HD-MSN degeneration.
In summary, we found that the genetic pathways and differential neurodegenerative states correlate with age-related stages of HD in patient-derived MSNs. Reversing the age-associated decline of autophagy chemically or via miRNA inhibition is sufficient to rescue HD-MSNs from neurodegeneration. Based on these findings, further investigations into using autophagy enhancer compounds, such as the G2 analog or antisense oligos against MIR29B-3p, may offer a therapeutic approach that will protect MSNs against neurodegeneration in HD.
Funding Statement
This study was supported by the following grants: R01NS107488 (National Institute of Neurological Disorders and Stroke), RF1AG056296 (National Institute on Aging), CHDI Fund (CHDI Foundation), HDF Grant (Hereditary Disease Foundation), CAF Grant (Cure Alzheimer’s Fund), and Mallinckrodt Scholar Award (Edward Mallinckrodt, Jr. Foundation).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Reference
- [1].Oh YM, Lee SW, Kim WK, et al. Age-related Huntington’s disease progression modeled in directly reprogrammed patient-derived striatal neurons highlights impaired autophagy. Nat Neurosci. 2022;25:1420–1433. PMID: 36303071. [DOI] [PMC free article] [PubMed] [Google Scholar]