

Keywords: autophagy, carbohydrate oxidation, colorectal cancer, energy expenditure, 5-FU
Abstract
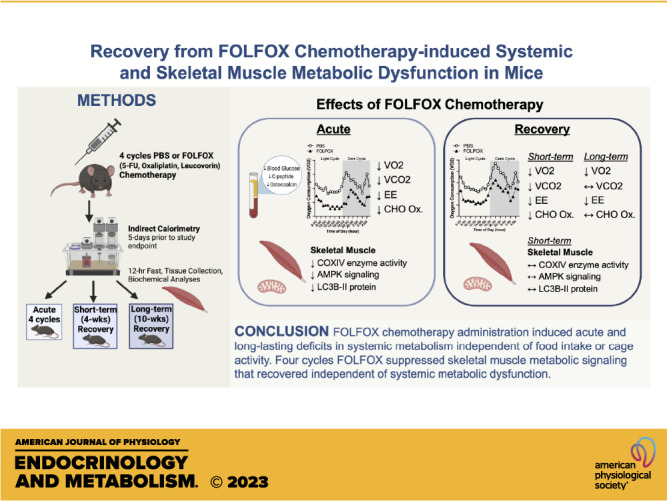
FOLFOX (5-fluorouracil, leucovorin, oxaliplatin) chemotherapy is used to treat colorectal cancer and can acutely induce metabolic dysfunction. However, the lasting effects on systemic and skeletal muscle metabolism after treatment cessation are poorly understood. Therefore, we investigated the acute and lasting effects of FOLFOX chemotherapy on systemic and skeletal muscle metabolism in mice. Direct effects of FOLFOX in cultured myotubes were also investigated. Male C57BL/6J mice completed four cycles (acute) of FOLFOX or PBS. Subsets were allowed to recover for 4 wk or 10 wk. Comprehensive Laboratory Animal Monitoring System (CLAMS) metabolic measurements were performed for 5 days before study endpoint. C2C12 myotubes were treated with FOLFOX for 24 hr. Acute FOLFOX attenuated body mass and body fat accretion independent of food intake or cage activity. Acute FOLFOX decreased blood glucose, oxygen consumption (V̇o2), carbon dioxide production (V̇co2), energy expenditure, and carbohydrate (CHO) oxidation. Deficits in V̇o2 and energy expenditure remained at 10 wk. CHO oxidation remained disrupted at 4 wk but returned to control levels after 10 wk. Acute FOLFOX reduced muscle COXIV enzyme activity, AMPK(T172), ULK1(S555), and LC3BII protein expression. Muscle LC3BII/I ratio was associated with altered CHO oxidation (r = 0.75, P = 0.03). In vitro, FOLFOX suppressed myotube AMPK(T172), ULK1(S555), and autophagy flux. Recovery for 4 wk normalized skeletal muscle AMPK and ULK1 phosphorylation. Our results provide evidence that FOLFOX disrupts systemic metabolism, which is not readily recoverable after treatment cessation. FOLFOX effects on skeletal muscle metabolic signaling did recover. Further investigations are warranted to prevent and treat FOLFOX-induced metabolic toxicities that negatively impact survival and life quality of patients with cancer.
NEW & NOTEWORTHY The present study demonstrates that FOLFOX chemotherapy induces long-lasting deficits in systemic metabolism. Interestingly, FOLFOX modestly suppressed skeletal muscle AMPK and autophagy signaling in vivo and in vitro. The FOLFOX-induced suppression of muscle metabolic signaling recovered after treatment cessation, independent of systemic metabolic dysfunction. Future research should investigate if activating AMPK during treatment can prevent long-term toxicities to improve health and quality of life of patients with cancer and survivors.
INTRODUCTION
Chemotherapy is a mainstay treatment option for patients with cancer despite the development of severe side effects that can last long after treatment completion. Aside from the known toxic effects on tumor cells, 5-fluorouracil (5-FU)-based chemotherapies can induce systemic toxicities in clinical and preclinical models (1–5). These toxicities can occur acutely or develop late after treatment cessation (6–8). The FOLFOX (5-FU, leucovorin, oxaliplatin) regimen is widely used for colorectal cancer treatment (9). Although many preclinical and clinical studies have evaluated tumor-related toxicities, safety, efficacy, and patient survival outcomes, few have investigated the mechanisms underlying systemic and tissue metabolic complications associated with FOLFOX chemotherapy (8). Moreover, cancer-induced metabolic alterations can complicate identifying chemotherapy-specific effects on metabolism. We recently reported that FOLFOX treatment without cancer is sufficient to induce long-lasting weakness and fatigue in mice (6). However, gaps remain in our understanding of FOLFOX chemotherapy’s effects on systemic and tissue metabolism and the subsequent recovery following treatment completion.
Maintaining systemic metabolic homeostasis requires coordinated regulation of processes involving the gut, kidney, brain, pancreas, adipose tissue, liver, and skeletal muscle (10). Disruptions to metabolic regulation can involve body composition changes, inflammation, and behaviors related to feeding, fasting, and physical activity (11, 12). In preclinical models, physical inactivity and inflammation can contribute to cancer cachexia and metabolic dysfunction involving carbohydrate and lipid oxidation (11, 13). In patients with colorectal cancer, altered body composition, inflammation, and physical inactivity remain crucial prognostic markers for poor survival and increased risk of developing chemotherapy treatment-related toxicities (14–18). Many chemotherapeutic agents can damage endocrine organs resulting in whole body metabolic dysfunction involving altered energy expenditure, glucose metabolism, and insulin resistance (8, 19, 20). Metabolic dysfunction can also include alterations in diurnal metabolic flexibility involving the oxidation of lipids and carbohydrates, which can be analyzed by indirect calorimetry assessing oxygen consumption (V̇o2) and carbon dioxide production (V̇co2) (12, 21–23). Reported chemotherapy-induced changes in energy expenditure are equivocal, being found to be increased and decreased in patients with cancer (16, 24). However, whether increased or decreased, abnormal energy expenditure in patients with cancer is associated with a higher risk of chemotherapy treatment-related toxicities (24). Although colorectal cancer has established effects on metabolic homeostasis in patients and preclinical models, the contribution of 5-FU-based chemotherapies on systemic metabolic dysfunction, including energy expenditure, metabolic substrate flexibility, and insulin resistance, warrants further investigation.
The importance of skeletal muscle mass maintenance for cancer patient survival and quality of life is well established (25). However, alterations in muscle biochemical properties that impact chemotherapy toxicities during treatment and recovery remain poorly understood. Skeletal muscle has critical metabolic functions encompassing glucose uptake, lipid metabolism, and amino acid storage, which are tightly regulated to maintain systemic metabolic homeostasis (21). There is potential for cancer and chemotherapy to disrupt skeletal muscle’s ability to respond to the systemic environment in patients with cancer, which would create a destructive cascade of events affecting multiple tissues and organ systems (8, 19). Low muscle mass in patients with colorectal cancer receiving chemotherapy is associated with poor prognosis and reduced quality of life (15, 26, 27). It has been shown that colorectal cancer and a 5-FU-based chemotherapy, FOLFIRI (5-FU, leucovorin, irinotecan), can disrupt systemic and skeletal muscle metabolism involving glycolysis and tricarboxylic acid (TCA) intermediates in mice (28, 29). Furthermore, FOLFIRI administration in mice is sufficient to induce muscle mass loss and mitochondria dysfunction (29, 30). Skeletal muscle cellular metabolic regulation involves the mammalian target of rapamycin complex 1 (mTORC1) and 5′AMP-activated protein kinase (AMPK) signaling pathways (31). mTORC1 is a critical integration site of nutrient, mechanical, and growth factor-related stimuli that promote muscle protein synthesis, ribosome biogenesis, fatty acid, and nucleotide synthesis (32). As a cellular energy sensor, AMPK signaling can promote autophagy, mitochondria biogenesis, and fatty acid oxidation and negatively regulate mTORC1 (33). Disrupted mTORC1 and AMPK regulation of protein synthesis and autophagy has been widely examined in preclinical cancer cachexia models (34–36). These signaling cascades also regulate macroautophagy (herein called autophagy) activation by cell stress or nutrient deprivation (37). Further investigation is needed to establish FOLFOX chemotherapy effects on skeletal muscle metabolic signaling involving mTORC1, AMPK, and the processes of protein synthesis and autophagy.
There has been considerable scientific interest in understanding acute chemotherapy toxicities on systemic metabolism, skeletal muscle mass, and metabolic function (38). This interest has led to numerous preclinical studies in mice investigating several types of chemotherapy drugs, with considerable variation in drug doses and dosing regimens (38). This heterogeneity of the research designs, treatment regimens, and drug doses has contributed to an inadequate understanding of the acute effects of several clinically relevant chemotherapy agents. Furthermore, due to the scope and design of these prior studies, there is a minimal understanding of the long-term chemotherapy toxicities in preclinical models after the cessation of treatment, which has clear clinical significance for cancer survivors. We have reported suppressed physical function involving fatigue and volitional strength 10 wk after the cessation of FOLFOX chemotherapy in mice (6). However, the effects of FOLFOX treatment on systemic and muscle metabolism and the subsequent recovery after cessation of treatment are poorly understood. Therefore, we investigated the acute effects and recovery from a physiologically relevant FOLFOX chemotherapy dosing regimen on systemic and skeletal muscle metabolism in male mice. Male C57BL/6J mice completed four cycles of either FOLFOX or PBS administration. Subsets of mice were monitored for either 4 or 10 wk after treatment cessation to examine recovery. Cultured myotubes were used to examine the direct effects of FOLFOX on metabolic signaling and autophagy regulation.
METHODS
Cell Culture
C2C12 myoblasts [American Type Culture Collection (ATCC) CRL-1772)] were seeded on type I collagen-coated six-well plates in growth media (GM): Dulbecco’s modified Eagle Media (DMEM; Gibco No. 11995-065) supplemented with 10% fetal bovine serum (FBS), 50 U/mL penicillin, and 50 µg/mL streptomycin. Cells were maintained at 37°C, 5% CO2, and used before passage 15. To induce differentiation, myoblasts were switched to differentiation media (DM) at ∼90% confluence (DM: DMEM supplemented with 2% horse serum, 50 U/mL penicillin, and 50 µg/mL streptomycin). To reduce the effects of proliferating myoblasts, day 3-differentiated myotubes were treated with 20 µM cytosine β-d-arabinofuranoside (AraC; Sigma No. C1768) for 24 h as described (39). DM was replenished every 48 h and experiments were conducted as described (40). Briefly, on day 5, differentiated myotubes were rinsed with phosphate-buffered saline (PBS) and maintained in GM diluted 1:2 with serum-free DMEM. Day 6-differentiated myotubes were treated with vehicle (0.1% DMSO) or FOLFOX (5-FU: 50 µg/mL, Oxaliplatin: 4 µg/mL, Leucovorin: 10 µg/mL) as described (30, 41) for 24 h. Autophagy flux was assessed by dosing 100 nM Bafilomycin A1 (Sigma No. 19–148), a specific inhibitor of the lysosomal proton pump V-ATPase, 4 h before cell harvest (42). Cells were harvested in ice-cold RIPA buffer (Thermo Scientific No. 89901) plus protease phosphatase inhibitor cocktail (Thermo Scientific No. 78442). Cell lysates were centrifuged at 14,000 rpm for 15 min at 4°C, and the supernatant was collected for protein. A minimum of three replicates from at least two independent experiments were used for analysis.
Animals
Ten-week-old male C57BL/6J mice (Strain No.: 000664) were purchased from The Jackson Laboratory. Mice were group housed, kept on a 12:12-h light/dark cycle beginning at 0600, and given standard rodent chow ad libitum (Harlan Teklad Rodent Diet No. 7912). Before euthanasia, all mice were fasted for 12 h overnight. Experiments were approved by the University of Tennessee Health Science Center Animal Care and Use Committee.
Chemotherapy Treatment and Recovery
At 12–13 wk of age, mice were randomized into treatment groups and administered intraperitoneal injections of either phosphate-buffered saline (PBS) or FOLFOX chemotherapy as described (6) [5-Fluorouracil (5-FU): 30 mg/kg body mass, Oxaliplatin: 6 mg/kg body mass, Leucovorin: 90 mg/kg body mass (Sigma #F8423, #O9512, #PHR1541)]. Body mass was measured before each injection to dose chemotherapy. One cycle of chemotherapy consisted of an injection followed by 2 wk of monitoring, which was completed four times (i.e., four cycles). Mice were euthanized for tissue collection after the fourth cycle (week 8; Fig. 1A) to examine acute toxicities (PBS n = 8, FOLFOX n = 8). Additional subsets were allowed to recover for either 4 wk (short-term recovery: PBS n = 11, FOLFOX n = 12) or 10 wk after the fourth injection (long-term recovery: PBS n = 8, FOLFOX n = 13). Body mass was monitored in all mice before each chemotherapy injection at pre (week 0; before cycle 1), mid (week 4; before cycle 3), and post four cycles (week 8). Body mass was also measured in subsets of mice at study endpoints for short-term recovery (week 10) and long-term recovery (week 16).
Figure 1.

Four cycles FOLFOX chemotherapy induces deficits in systemic oxygen consumption (V̇o2), energy expenditure, and carbohydrate (CHO) oxidation independent of cage activity. A: study design. Male C57BL/6J mice were administered four cycles of either PBS or FOLFOX (5-FU, oxaliplatin, leucovorin) over 8 wk. Mice were individually housed in CLAMS metabolic cages for 5 days before tissue collection. B: body mass at Pre (0 wk), Mid (4 wk), and Post (8 wk) (PBS: n = 27, FOLFOX: n = 33) and body fat percentage in PBS and FOLFOX mice (C). D: food intake of individually housed mice during CLAMS measurements. Cage activity (E) and relative (normalized to kg body weight) oxygen consumption (V̇o2; F), carbon dioxide production (V̇co2; G), and energy expenditure expressed as the average of the light cycle (H) (0600 to 1700) and dark cycle (1800 to 0500). CHO oxidation (I) and CHO oxidation metabolic flexibility (J). Lipid oxidation (K) and lipid oxidation metabolic flexibility (L). PBS: n = 8, FOLFOX: n = 8. Data are presented as means ± SE. Data are analyzed using two-way repeated-measures ANOVA with post hoc analysis when appropriate (B, E–H, I, K) or unpaired t test (C, D, J, L). Statistical significance was set to P < 0.05. Bold and italic text denotes significance. a,b,cSignificant difference between groups; *different from PBS; #main effect of time/cycle; $main effect of FOLFOX. CHO, carbohydrate; CLAMS, Comprehensive Laboratory Animal Monitoring System; FOLFOX, 5-fluorouracil, leucovorin, oxaliplatin; g, grams; hr, hour; kg, kilograms; kcal, kilocalorie; mL, milliliters; ME, main effect; PBS, phosphate-buffered saline.
Body Composition
Body composition was measured noninvasively by magnetic resonance imaging (EchoMRI 1100 Body Composition Analyzer). The machine was calibrated before experimental measurements using canola oil. Each mouse was individually processed through the Echo MRI, and lean and fat mass measurements were recorded.
Indirect Calorimetry
Indirect calorimetry was performed as described (11). Subsets of mice were individually housed and placed in Comprehensive Laboratory Animal Monitoring System (CLAMS; Columbus Instruments) cages for five consecutive days at ambient temperature (22°C) on a 12-h light/dark cycle and mice were given standard rodent chow ad libitum. For long-term recovery, metabolic outcomes were assessed in a subset of mice based on experimental constraints. PBS control CLAMS metabolic outcomes for short-term (n = 11) and long-term recovery (n = 3) were not significantly different and thus combined for analyses. Group N’s for CLAMS cage analyses are as follows: four cycles PBS n = 8, FOLFOX n = 8; 4-wk recovery: PBS n = 14, FOLFOX n = 12; 10-wk recovery: PBS n = 14, FOLFOX n = 4. The first 24 h were removed from analyses to allow for acclimatization. Oxygen consumption (V̇o2), carbon dioxide production (V̇co2), respiratory exchange ratio (RER; V̇co2/V̇o2), energy expenditure, XY ambulatory activity (minimum three different consecutive horizontal beam breaks in counts), and food intake were measured. Carbohydrate (CHO) and lipid oxidation were calculated using equations assuming negligible protein oxidation (43). V̇o2 and V̇co2 measurements were converted to absolute values (mL/min) before use in the equation. Equations are as follows: carbohydrate oxidation (g/min): (4.585 × V̇co2) − (3.226 × V̇o2) and lipid oxidation (g/min): (1.695 × V̇o2) − (1.701 × V̇co2).
Glucose Tolerance Test and Fasting Metabolic Hormones
Mice were fasted for 5 h at the start of the light cycle, and then glucose tolerance test (GTT) was performed. Mice received an intraperitoneal injection of dextrose solution (d-glucose; 2 mg/kg). Blood glucose was determined using a standard glucometer (Contour Next, Parsippany, NJ). Blood glucose was monitored at time 0, before injection, and after injection at 30, 60, 90, and 120 min. Blood (10–20 µL) was collected at each time point with heparinized capillary tubes, centrifuged at 10,000 g for 10 min at 4°C, and the supernatant was collected. Plasma insulin concentrations were determined using enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions (Mercodia No. 10–1247). Blood glucose and plasma insulin were collected and measured as above after a 12-h overnight fast, before euthanasia. The homeostatic model assessment for insulin resistance (HOMA-IR) was calculated by the following formula: fasting glucose (mmol/L) × fasting insulin (mU/L)/22.5. Fasting (12-h) plasma metabolic hormones (C-peptide, Ghrelin, GIP, PYY, and Resistin) were evaluated using Luminex Magpix multiplex analyzer (MILLIPLEX MAP Hormone Bead Panel, Millipore Sigma). Plasma uncarboxylated osteocalcin (Glu-OC) (Biomatik No. EKF57818) and adiponectin (R&D Systems No. MRP300) concentrations were determined using commercially available ELISA kits according to the manufacturer’s instructions.
Protein Synthesis Measurement
The surface sensing of translation (SUnSET) technique was used to determine muscle protein synthesis rates as previously described (44). Briefly, puromycin (Millipore Sigma, No. 540411) was dissolved in sterile saline and delivered via intraperitoneal injection (0.04 µmol/g BW) 30-min before euthanasia. Puromycin incorporation into newly synthesized peptides was assessed by Western blot. Gastrocnemius muscle homogenate samples from mice not labeled with puromycin were used as negative controls.
Tissue Collection
All mice were fasted 12 h overnight and euthanized for tissue collection at the start of the light cycle. At the time of euthanasia, mice were fully anesthetized (3–5% isoflurane) and then underwent cervical dislocation. Hindlimb muscles and organs were rapidly excised, cleared of excessive connective tissue, weighed, and snap frozen in liquid nitrogen and then stored at −80°C. Hindlimb muscles included the soleus, plantaris, gastrocnemius, extensor digitorum longus, and tibialis anterior. The gastrocnemius muscle was cut into the visually red lateral head, and the white intermediate portion of the muscle was located between the medial and lateral gastrocnemius heads.
Cytochrome c Oxidase Enzyme Activity
Cytochrome c oxidase (COXIV) enzyme activity was measured as described (45). Briefly, soleus and plantaris muscles were homogenized in extraction buffer (0.1 M KH2PO4/Na2HPO4, 2 mM EDTA, pH 7.2). COXIV enzyme activity was determined colorimetrically by measuring the oxidation rate of fully reduced cytochrome c at 550 nm (BioVision #K287).
Mitochondrial DNA PCR
DNA was isolated from rectus femoris and liver tissue sections using DNAzol reagent (Invitrogen #10503-027) as described (46). Briefly, tissues (∼20–30 mg) were homogenized in 1-mL DNAzol, pelleted with 100% ethanol, and resuspended in 8 mM NaOH. Quantitative real-time PCR analysis was performed in 20 μL reactions using 2XPowerTrack SYBR Green master mix, 2 μL of DNA, 1 μL of forward and reverse primers (500 nM), and nuclease-free water. Mitochondrially encoded genes included 16S rRNA and mitochondrial NADH dehydrogenase subunit 1 (MT-ND1); hexokinase 2 (HK2) was used for nuclear-encoded gene (nDNA) expression (47). The mitochondrial DNA (mtDNA)/nDNA ratio analysis was calculated using the 2−ΔΔCT method with HK2 as the housekeeping gene, as described (47).
RNA Isolation, cDNA Synthesis, and Real-Time PCR
RNA isolation, cDNA synthesis, and real-time PCR (RT-PCR) were performed as described (6). Total RNA was isolated from ∼20 mg of frozen tissue sections using TRIzol reagent (Invitrogen #15596018) per manufacturer’s guidelines. After phenol-chloroform extraction, RNA was purified using the PureLink RNA Mini Kit (Invitrogen #12183018) and eluted in nuclease-free water. Total RNA concentration (260 nm) and purity (260/280 ratio) were measured using spectrophotometry. cDNA was reverse transcribed from 1 μg of total RNA using Superscript IV Reverse Transcriptase and random hexamers according to manufacturer guidelines (Invitrogen #18090200). RT-PCR was carried out in 20 μL reactions using 2XPowerTrack SYBR Green master mix, 2 μL of cDNA, 1 μL of forward and reverse primers (500 nM), and nuclease-free water. Forward and reverse primer sequences (Supplemental Table S1) were synthesized by Integrated DNA Technologies (IDT) and validated by agarose gel electrophoresis. Melt-curve analysis ensured that the PCR reaction produced only one amplicon. RT-PCR was carried out on the Applied Biosystems QuantStudio3 system. Reactions were incubated for 2-min at 95°C, followed by 40 cycles of a 15-s denaturation step at 95°C, and 1-min annealing/extension at 60°C. The 2−ΔΔCt method was used to determine gene expression changes between treatment groups with the GAPDH Ct as the correction factor.
Western Blotting
Western blot analysis was performed as described (46). Briefly, frozen gastrocnemius muscle sections were homogenized in Mueller buffer, and protein concentration was determined using the Bradford method (48). Muscle homogenate was fractionated on 10–15% SDS-polyacrylamide gels and transferred to PVDF membranes. Ponceau S (Fisher #501037137) stain was used to ensure equal loading of each gel. Membranes were blocked for 1-h in 5% milk in Tris-buffered saline with 0.1% Tween-20 (TBST) at room temperature. Primary antibodies were purchased from Cell Signaling for: OPA1 (#80471), MFN2 (#9482), p-DRP1(S616) (#3455), DRP1 (#5391), p-AMPK(T172) (#2535), AMPK (#2603), p-ULK1(S555) (#5869), p-ULK1(S757) (#14202), ULK1 (#8054), p-rpS6(S240/244) (#5364), rpS6 (#2217), LC3B (#2775), SQSTM1/p62 (#23214), and from Millipore Sigma for: FIS1 (#HPA017430), and Puromycin (#MABE343). Primary antibodies (phosphorylated proteins 1:1,000 dilution; total proteins 1:2,000 dilution) were incubated overnight in 5% milk-TBST. Membranes were washed and incubated at a 1:4,000 dilution in 5% milk-TBST containing anti-rabbit (#7074) or anti-mouse (#7076) (Cell Signaling) IgG horseradish-peroxidase conjugated secondary antibodies for 1 h at room temperature. Enhanced chemiluminescence (Prometheus Pro-Signal Femto, Genesee Scientific #20–302) was used to visualize the antibody-antigen interactions. Immunoblot images were digitally imaged using the iBright 1500 system (Invitrogen, Carlsbad, CA), and blots were quantified by densitometry using imaging software (ImageJ, NIH). After imaging, membranes were washed and stained with Coomassie Blue to visualize equal protein loading.
Statistical Analysis
Results are reported as mean ± SEM. Body mass measurements over time and CLAMS metabolic outcomes were analyzed (GraphPad Prism 9) using two-way repeated-measures ANOVA with Šídák's multiple comparisons test when appropriate. Body composition, food intake, metabolic flexibility, plasma hormones, muscle COXIV enzyme activity, mRNA, and protein expression data were analyzed using an unpaired t test. For associations, the Shapiro–Wilk test was used to determine normal distribution; if normally distributed, the correlation was measured using the Pearson r coefficient. Spearman’s rank correlation coefficient was used if samples were not normally distributed. For all analyses, statistical significance was set to P < 0.05.
RESULTS
Four Cycles FOLFOX Administration: Body Composition, Food Intake, and Cage Activity
Mice were administered four cycles of either PBS or FOLFOX chemotherapy (Fig. 1A), and body mass was measured in all mice before the FOLFOX administration (Pre), after the second FOLFOX cycle (Mid), and after the fourth FOLFOX cycle (Post). Both PBS and FOLFOX mice significantly increased body mass Mid and Post compared with Pre (Fig. 1B). There were no differences in body mass between PBS and FOLFOX mice at Pre or Mid time points. However, FOLFOX mice had lower body mass Post compared with PBS mice (Fig. 1B). Body mass was also examined at the end of the fourth cycle following a 12-h fast (means ± SE) (post-fast body mass: PBS: 27.9 ± 0.94 g, FOLFOX: 27.1 ± 2.5 g; P = 0.420). FOLFOX mice lost less body mass after the fast compared with PBS (PBS: −12 ± 1.0%, FOLFOX: −8.2 ± 0.63%; P = 0.012). Lean and fat mass were measured via Echo MRI at the Post timepoint and expressed as a percentage of body mass. FOLFOX-treated mice had increased percent lean mass (PBS: 82.9 ± 0.8, FOLFOX: 87.2 ± 0.5; P < 0.001) and significantly reduced percent body fat (Fig. 1C) compared with PBS. Food intake (Fig. 1D) and cage activity (Fig. 1E) were not different between PBS and FOLFOX mice. These data indicate that FOLFOX chemotherapy treatment in mice can induce deficits in fat mass accretion, which was independent of changes in food intake or cage activity after four FOLFOX cycles.
Four Cycles FOLFOX Administration: Systemic Oxygen Consumption, Energy Expenditure, and CHO Oxidation
Mice were administered four cycles of either PBS or FOLFOX chemotherapy and underwent metabolic measurements in CLAMS cages for 5 days after the completion of the fourth cycle (Fig. 1A). There were significant main effects for both PBS and FOLFOX mice to increase cage activity (Fig. 1E), relative (normalized to body weight) V̇o2 (Fig. 1F), V̇co2 (Fig. 1G), and energy expenditure (Fig. 1H) when switching from the light to the dark cycle. There were significant main effects for FOLFOX to have decreased relative V̇o2 (Fig. 1F), V̇co2 (Fig. 1G), and energy expenditure (Fig. 1H) in the light and dark cycle compared with PBS. In addition, four cycles of FOLFOX reduced daily average absolute and lean body mass normalized V̇o2, V̇co2, and energy expenditure when compared with PBS (Table 1). Daily average RER was not different between PBS and FOLFOX (PBS: 0.839 ± 0.007, FOLFOX: 0.829 ± 0.003; P = 0.282). There was a significant effect of time for increased CHO (Fig. 1I) and decreased lipid oxidation (Fig. 1K) in PBS and FOLFOX mice when switching from the light to dark cycle. However, FOLFOX mice exhibited an impaired ability to increase CHO oxidation in the dark cycle (Fig. 1J). Metabolic flexibility for lipid oxidation was not different between PBS and FOLFOX mice (Fig. 1L). Compared to PBS, FOLFOX treatment significantly reduced CHO oxidation in the dark cycle (hours: 1800, 1900, 2000, 0400) (Fig. 1I) and lipid oxidation in the light cycle (hour: 1100) (Fig. 1K). There was a significant positive association between hindlimb muscle mass and daily average CHO oxidation in FOLFOX-treated mice (Table 2), indicating that skeletal muscle may be important in regulating CHO oxidation after FOLFOX treatment. These data indicate that FOLFOX chemotherapy treatment in mice can induce deficits in systemic metabolism independent of changes in food intake or cage activity after four cycles.
Table 1.
Effect of FOLFOX treatment and recovery on daily average absolute and relative V̇o2, V̇co2, and energy expenditure
| Four Cycles |
Short-Term Recovery |
Long-Term Recovery |
||||
|---|---|---|---|---|---|---|
| PBS | FOLFOX | PBS | FOLFOX | PBS | FOLFOX | |
| V̇o2, mL/h | 123.9 (2.7) | 101.1 (3.6)* | 120.3 (4.4) | 102.5 (1.8)* | 120.3 (4.4) | 97.2 (1.5)* |
| V̇o2, mL/kg lean/h | 4,688 (78) | 3,965 (60)* | 4,543 (114) | 3,960 (60)* | 4,543 (114) | 3,842 (76)* |
| V̇co2, mL/h | 104.2 (2.0) | 84.2 (3.1)* | 102.0 (4.0) | 83.2 (1.8)* | 102.0 (4.0) | 85.3 (2.6)* |
| V̇co2, mL/kg lean/h | 3,945 (75) | 3,299 (46)* | 3,852 (107) | 3,220 (89)* | 3,852 (107) | 3,369 (63)* |
| Energy expenditure, kcal/h | 0.600 (0.01) | 0.489 (0.02)* | 0.580 (0.02) | 0.494 (0.01)* | 0.580 (0.02) | 0.477 (0.01)* |
| Energy expenditure, kcal/kg lean/h | 22.7 (0.39) | 19.1 (0.28)* | 21.9 (0.50) | 19.1 (0.35)* | 21.9 (0.50) | 18.9 (0.35)* |
Data are presented as means (SE). Daily average values for oxygen consumption (V̇o2), carbon dioxide production (V̇co2), and energy expenditure. Values are represented as absolute or relative to lean body mass (kg). Four cycles: PBS n = 8, FOLFOX n = 8; short-term (4 wk) Recovery: PBS n = 14, FOLFOX n = 12; long-term (10 wk) recovery: PBS n = 14, FOLFOX n = 4. Data are analyzed using unpaired t test between PBS and FOLFOX treatments at each timepoint. Statistical significance is set to P < 0.05. *Significant P value. FOLFOX, 5-fluorouracil, leucovorin, oxaliplatin; h, hour; kcal, kilocalorie; mL, milliliter.
Table 2.
Pearson correlation reveals skeletal muscle mass is positively associated with CHO oxidation in FOLFOX chemotherapy treated mice
| Relative V̇o2, mL/kg BW/h |
Energy Expenditure, kcal/kg BW/h |
CHO Oxidation, g/min |
Lipid Oxidation, g/min |
|||||
|---|---|---|---|---|---|---|---|---|
| PBS | FOLFOX | PBS | FOLFOX | PBS | FOLFOX | PBS | FOLFOX | |
| Hindlimb muscle mass, mg | 0.296 | −0.088 | 0.294 | 0.025 | −0.049 | 0.867* | 0.383 | 0.452 |
| Body fat % | −0.675 | −0.855* | −0.644 | −0.869* | −0.117 | −0.277 | −0.612 | −0.471 |
| Fasting glucose, mg/dL | 0.694 | 0.437 | 0.670 | 0.392 | 0.522 | −0.043 | 0.200 | 0.559 |
| Fasting insulin, ng/mL | −0.013 | 0.293 | −0.091 | 0.337 | −0.153 | 0.546 | 0.395 | −0.092 |
Pearson correlation coefficients for associations between daily average oxygen consumption (V̇o2), energy expenditure (EE), carbohydrate (CHO), and lipid oxidation in the four cycle PBS- or FOLFOX-treated mice (n = 8/group). Values are Pearson r coefficients. Statistical significance is set to P < 0.05. bold with * denotes significant P value. BW, body weight; dL, deciliter; FOLFOX, 5-fluorouracil, leucovorin, oxaliplatin; g, grams; kcal, kilocalories; kg, kilograms; mg, milligrams; min, minutes; ng, nanograms.
Four Cycles FOLFOX Administration: Systemic Glucose Metabolism and Plasma Hormones
FOLFOX effects on systemic glucose metabolism were examined by fasting plasma concentrations of various metabolic hormones and a glucose tolerance test after four cycles. Following a 12-h fast, blood glucose levels were significantly lower in FOLFOX than in PBS (Fig. 2A). However, this was independent of changes in fasting plasma insulin (Fig. 2B) or HOMA-IR (Fig. 2C). Fasting plasma concentrations of C-peptide, gastric inhibitory peptide (GIP), and uncarboxylated osteocalcin (Glu-OC) were significantly reduced in FOLFOX compared with PBS (Table 3). Fasting plasma ghrelin, peptide YY (PYY), resistin, and adiponectin levels were not different between PBS and FOLFOX (Table 3). A glucose tolerance test (GTT) was performed in mice after the completion of the fourth cycle, following a 5-h fast. There was a significant effect of FOLFOX to have reduced blood glucose during the GTT compared with PBS (Fig. 2D). Blood glucose area under the curve (AUC) was significantly lower in FOLFOX (Fig. 2D); however, plasma insulin AUC was not different compared with PBS (Fig. 2E). Red and white gastrocnemius muscle and epidydimal white adipose tissue (eWAT) glucose transporter type 4 (GLUT4) mRNA expression was not different between PBS and FOLFOX (Fig. 2F). Alterations in body composition and metabolic hormone concentrations has been linked to systemic metabolic dysfunction. Body fat percentage was negatively associated with relative V̇o2 and energy expenditure in FOLFOX mice (Table 2). Body fat, fasting glucose, and insulin were not associated with relative V̇o2, energy expenditure, CHO, or lipid oxidation in PBS or FOLFOX mice (Table 2). These data indicate that four cycles FOLFOX chemotherapy induces hypoglycemia but does not induce insulin resistance or alterations in tissue expression of GLUT4 mRNA.
Figure 2.

Four cycles of FOLFOX chemotherapy does not induce insulin resistance or glucose intolerance after fasting. Blood glucose (A) and plasma insulin (B) following a 12-h fast. Homeostatic model assessment for insulin resistance (HOMA-IR) (C). Glucose tolerance test (GTT) blood glucose (D) and plasma insulin curves (E) and area under the curve (AUC) values; GTT was performed after a 5-h fast. F: glucose transporter type 4 (GLUT4) mRNA expression in red and white gastrocnemius skeletal muscle and epidydimal white adipose tissue (eWAT). PBS: n = 8, FOLFOX: n = 8. Data are presented as means ± SE. Data are analyzed using unpaired t test or two-way repeated-measures ANOVA (GTT curves D–E). Statistical significance was set to P < 0.05. Bold and italic text denotes significance. *Different from PBS. dL, deciliter; FOLFOX, 5-fluorouracil, leucovorin, oxaliplatin; mg, milligrams; mL, milliliters; ng, nanograms.
Table 3.
Effect of four cycles FOLFOX on fasting plasma concentrations of metabolic hormones
| PBS | FOLFOX | |
|---|---|---|
| C-peptide, pg/mL | 220 (22) | 146 (18)* |
| Ghrelin, pg/mL | 26.3 (4.4) | 33.9 (9.1) |
| GIP, pg/mL | 148 (48) | 71.9 (9.2)* |
| PYY, pg/mL | 120 (11) | 171 (49) |
| Resistin, pg/mL | 7,092 (1,059) | 6,385 (1,182) |
| Glu-OC, ng/mL | 9.2 (0.81) | 6.6 (0.34)* |
| Adiponectin, µg/mL | 11.2 (0.76) | 9.5 (0.57) |
Data are presented as means (SE). n = 5–8/group. Data are analyzed using unpaired t test. Statistical significance is set to P < 0.05. *Significant P value. FOLFOX, 5-fluorouracil, leucovorin, oxaliplatin; GIP, gastric inhibitory peptide; Glu-OC, uncarboxylated osteocalcin; mL, milliliter; ng, nanogram; pg, picogram; PYY, peptide tyrosine tyrosine; µg, microgram.
Four Cycles FOLFOX Administration: Skeletal Muscle COXIV Enzyme Activity, mtDNA, and Mitochondria Quality Control Gene and Protein Expression
FOLFOX effects on skeletal muscle mitochondria-related gene and protein expression were examined after four FOLFOX cycles. Mitochondria cytochrome c oxidase (COXIV) enzyme activity was measured in the soleus and plantaris muscles. FOLFOX significantly reduced COXIV activity in both muscle types (soleus −29%, plantaris −26%) (Fig. 3A). Mitochondrial DNA (mtDNA) content was estimated using the ratio of mitochondrially encoded genes (MT-ND1 and 16S rRNA) to a nuclear-encoded gene (HK2). There were no significant differences between PBS and FOLFOX for muscle (Fig. 3B) or liver (Fig. 3C) mtDNA content. Gastrocnemius muscle mRNA expression of mitochondria uncoupling proteins 2 and 3 (UCP2, UCP3) was not different between PBS and FOLFOX (Fig. 3D). FOLFOX significantly reduced muscle PGC-1α mRNA expression (Fig. 3E); however, there were no differences for NRF1 or TFAM expression compared with PBS (Fig. 3E). There were no differences between PBS and FOLFOX for mitochondria quality control related genes (Fig. 3F). FOLFOX reduced MFN2 protein expression compared with PBS (Fig. 3G). There were no differences for red gastrocnemius protein expression of OPA1, FIS1, or phosphorylated to total DRP1(S616) (Fig. 3G). These data indicate that while FOLFOX induces alterations in systemic metabolism and muscle COXIV enzyme activity, the transcription of many mitochondria-related genes may not be affected by FOLFOX treatment in skeletal muscle.
Figure 3.

Effects of four cycles FOLFOX on skeletal muscle cytochrome c oxidase IV (COXIV) enzyme activity, mitochondrial DNA (mtDNA), mitochondria quality control gene and protein expression. A: cytochrome c oxidase IV (COXIV) enzyme activity in soleus and plantaris muscles. B: mitochondrial DNA content in rectus femoris skeletal muscle and liver tissue (C). Red gastrocnemius skeletal muscle mRNA expression of mitochondria uncoupling proteins UCP2 and UCP3 (D); mitochondria biogenesis and transcription: PGC-1α, NRF1, and TFAM (E); mitochondria quality control: OPA1, MFN2, FIS1, DRP1 (F), and protein expression of OPA1, MFN2, FIS1, and phosphorylated (p) to total ratio of DRP1 (S616) with representative Western blot images and membrane Ponceau S stain for total protein (G). PBS: n = 8, FOLFOX: n = 8. Data are presented as means ± SE. Data are analyzed using unpaired t test. Statistical significance was set to P < 0.05. *Different from PBS. DRP, dynamin-related protein 1; FIS1, mitochondrial fission protein 1; FOLFOX, 5-fluorouracil, leucovorin, oxaliplatin; MFN2, mitofusin 2; mtDNA, mitochondrial DNA; MT-ND1, mitochondrial NADH dehydrogenase subunit I; nDNA, nuclear DNA; NRF1, nuclear respiratory factor 1; OPA1, optic atrophy 1; PGC-1α, peroxisome proliferator-activated receptor-gamma coactivator 1 alpha; TFAM, mitochondrial transcription factor A; UCP, uncoupling protein; 16S rRNA, 16S ribosomal RNA.
Four Cycles FOLFOX Administration: Skeletal Muscle Metabolic and Autophagy Signaling
Skeletal muscle AMPK and mTORC1 signaling are critical for maintaining metabolic homeostasis. The Unc-51-like kinase 1 (ULK1) is a protein involved in autophagy regulation; it can be activated by AMPK phosphorylation at Ser555 or inhibited by mTORC1 phosphorylation at Ser757. Ribosomal protein S6 (rpS6) is a well-known downstream target of mTORC1 signaling in muscle and a component of the 40S ribosomal subunit (49). Red gastrocnemius muscle AMPK and ULK1 total protein expression was not altered by FOLFOX treatment. However, FOLFOX significantly reduced total rpS6 protein expression compared with PBS (Fig. 4, A and B). Gastrocnemius muscle AMPK (T172) and ULK1 (S555) phosphorylation were reduced in FOLFOX compared with PBS (Fig. 4C). It has been reported that adiponectin can activate skeletal muscle AMPK signaling (50). We observed a positive association between plasma adiponectin levels and muscle AMPK phosphorylation in PBS mice that was not present in the FOLFOX mice (Supplemental Table S2). ULK1 (S757) phosphorylation was not different between PBS and FOLFOX (Fig. 4C). Despite a reduction in total rpS6 protein expression, the ratio of phosphorylated rpS6 (S240/244) to total rpS6 was unchanged by FOLFOX (Fig. 4, A–C). Puromycin incorporation did not differ between PBS and FOLFOX (Fig. 4D). However, puromycin incorporation was positively associated with muscle rpS6 phosphorylation in FOLFOX mice (Fig. 4E). FOLFOX decreased muscle LC3BII protein expression (Fig. 4F) and the LC3BII/I ratio was negatively associated with puromycin incorporation in FOLFOX mice (Fig. 4G). Interestingly, LC3BII/I was positively associated with daily average CHO oxidation in FOLFOX mice (Fig. 4H). Autophagy-related gene expression was not altered by FOLFOX treatment (Supplemental Fig. S1, A–C). These data indicate that FOLFOX treatment suppresses skeletal muscle AMPK phosphorylation and its downstream target, ULK1 (S555). The suppression of this signaling pathway and reduced muscle LC3BII protein expression suggests that FOLFOX chemotherapy may negatively regulate skeletal muscle autophagy, which could be related to systemic metabolic dysfunction.
Figure 4.

Four cycles of FOLFOX chemotherapy disrupts skeletal muscle metabolic and autophagy signaling after a 12-h fast. A: representative Western blot images with Coomassie blue protein stain, dashed line represents different areas of the same blot. Red gastrocnemius skeletal muscle total protein expression of AMPK, ULK1, and rpS6 (B). C: phosphorylated to total ratio of AMPK (T172), ULK1 (S555), ULK1 (S757), and rpS6 (S240/244). D: puromycin incorporation with representative Western blot and Coomassie blue protein stain, dashed line represents different areas of the same blot. E: Pearson r correlation between phosphorylated (p) rpS6 (S240/244) red gastrocnemius protein expression to protein synthesis in FOLFOX mice. F: protein expression of LC3BI, LC3BII, and the LC3BII/I ratio. G: Pearson r correlation between LC3BII/I and protein synthesis and CHO oxidation in FOLFOX mice (H). PBS: n = 8, FOLFOX: n = 8. Data are presented as means ± SE. Data are analyzed using unpaired t test and correlation with Pearson r correlation coefficient. Statistical significance was set to P < 0.05. Bold and italic values denote significance. *Different from PBS. AMPK, AMP-activated protein kinase; FOLFOX, 5-fluorouracil, leucovorin, oxaliplatin; LC3B, microtubule-associated protein 1 light chain 3B; NC, negative control; rpS6, ribosomal protein S6; ULK1, unc-51-like kinase 1.
FOLFOX Administration to C2C12 Myotubes: AMPK Phosphorylation, Protein Synthesis, and Autophagy Flux
C2C12 myotube cultures were used to examine the direct effects of FOLFOX chemotherapy on muscle metabolic signaling involving the AMPK regulation of autophagy. Differentiated C2C12 myotubes were pretreated with AraC at day 3 of differentiation to reduce potential effects of proliferating myoblasts, and at day 6 of differentiation, FOLFOX or vehicle were added for 24 h. FOLFOX administration did not alter the total protein expression of AMPK; however, myotube ULK1 and rpS6 total protein expression was significantly reduced compared with vehicle (Fig. 5, A and B). FOLFOX treatment decreased the phosphorylated to total protein expression of AMPK (T172) and ULK1 (S555 and S757) compared with vehicle (Fig. 5C). Interestingly, FOLFOX had direct effects of reducing myotube protein synthesis (Fig. 5D). Autophagy flux was assessed using the specific vacuolar H+ ATPase (V-ATPase) inhibitor, bafilomycin A1. There was a significant interaction for FOLFOX to suppress LC3BII/I ratio compared with vehicle-treated myotubes dosed with bafilomycin (Fig. 5, E and F). There was a main effect for FOLFOX to increase p62 protein expression compared with vehicle controls (Fig. 5G), which accumulates when autophagy is inhibited. These results suggest that FOLFOX directly affects muscle cells to suppress AMPK signaling, protein synthesis, and autophagy flux.
Figure 5.

FOLFOX administration alters C2C12 myotube AMPK phosphorylation, protein synthesis, and autophagy flux. A: representative Western blot images with Coomassie blue protein stain. B: C2C12 myotube total protein expression of AMPK, ULK1, and rpS6 after 24-h treatment with vehicle (0.1% DMSO) or FOLFOX. C: phosphorylated to total ratio of AMPK (T172), ULK1 (S555), ULK1 (S757), and rpS6 (S240/244). D: puromycin incorporation with representative Western blot and Coomassie blue protein stain, dashed line represents different areas of the same blot. E: representative Western blot images with Coomassie blue protein stain. LC3BII/I ratio (F) and p62 protein expression (G) in vehicle and FOLFOX-treated myotubes treated with or without 100 nM Bafilomycin A1. Vehicle: n = 6, FOLFOX: n = 6, samples are collected from two independent experiments. Data are presented as means ± SE. Data are analyzed using unpaired t test (B–D) and two-way ANOVA with post hoc (F and G). Statistical significance set to P < 0.05. Bold and italic values denote significance. *Different from vehicle; †main effect of bafilomycin; $main effect of FOLFOX; a,b,csignificant difference between groups. AMPK, AMP-activated protein kinase; FOLFOX, 5-fluorouracil, leucovorin, oxaliplatin; LC3B, microtubule-associated protein 1 light chain 3B; ULK1, unc-51-like kinase 1.
Short-Term Recovery from FOLFOX Administration: Body Composition and Systemic Metabolism
Chemotherapy can induce functional deficits that are not readily reversible following treatment completion; however, the lasting effects on systemic metabolism are not well established. Mice that received four cycles of either PBS or FOLFOX were allowed to recover for 4 wk after the last injection and then placed in CLAMS cages to determine if the FOLFOX-induced suppression of systemic metabolism remains as a late effect of treatment (Fig. 6A). Body mass was reduced 7% in FOLFOX compared with PBS at 4 wk recovery (Fig. 6B). FOLFOX mice had increased percent lean mass (PBS: 82.0 ± 1.2, FOLFOX: 86.4 ± 0.59; P < 0.001), whereas percent body fat was reduced 31% in FOLFOX compared with PBS at 4 wk recovery (Fig. 6C). Food intake and cage activity were not different between PBS and FOLFOX (Fig. 6, D and E). Fasting blood glucose was similar between PBS and FOLFOX (PBS: 106.6 ± 11.2, FOLFOX: 90.1 ± 5.2; P = 0.186). There were significant main effects for PBS and FOLFOX mice to increase cage activity (Fig. 6E), relative (normalized to body weight) V̇o2 (Fig. 6F), V̇co2 (Fig. 6G), and energy expenditure (Fig. 6H) in the dark compared with the light cycle. There was a significant main effect for FOLFOX to have decreased relative V̇o2 (Fig. 6F), V̇co2 (Fig. 6G), and energy expenditure (Fig. 6H) in the light and dark cycle compared with PBS. At short-term recovery compared with PBS, FOLFOX reduced daily average absolute and lean body mass normalized V̇o2, V̇co2, and energy expenditure (Table 1). Daily average RER was lower in FOLFOX than in PBS at 4 wk recovery (PBS: 0.844 ± 0.008, FOLFOX: 0.809 ± 0.011; P = 0.017). There was a main effect of time for increased CHO oxidation (Fig. 6I) and decreased lipid oxidation (Fig. 6K) in PBS and FOLFOX mice when switching from the light to dark cycle. However, compared with PBS, FOLFOX treatment significantly reduced CHO oxidation (hours: 1500, 1700, 2000, 2100) (Fig. 6I). Lipid oxidation was not different between PBS and FOLFOX treatments at 4 wk recovery (Fig. 6K). Metabolic flexibility for CHO (Fig. 6J) and lipid oxidation (Fig. 6L) was not different between PBS and FOLFOX. These data indicate that FOLFOX chemotherapy treatment in mice can induce lasting deficits in fat accretion and systemic metabolism. Specifically, V̇o2, V̇co2, energy expenditure, and CHO oxidation were still reduced 4 wk after FOLFOX cessation.
Figure 6.

Deficits in V̇o2, V̇co2, energy expenditure, and CHO oxidation remain following 4-weeks recovery from FOLFOX. A: study design. Male C57BL/6J mice were administered four cycles of either PBS or FOLFOX and allowed to recover for 4 wk after the last injection. Mice were individually housed in CLAMS metabolic cages for 5 days before tissue collection. Body mass (B) and body fat percentage (C) at Post (week 10). D: food intake of individually housed mice during CLAMS measurements. Cage activity (E) and relative (normalized to kg body weight) V̇o2 (F), V̇co2 (G), and energy expenditure (H) expressed as the average of the light cycle (0600–1700) and dark cycle (1800–0500). CHO oxidation (I) and metabolic flexibility (J). Lipid oxidation (K) and metabolic flexibility (L). PBS: n = 14, FOLFOX: n = 12. Data are presented as means ± SE. Data are analyzed using unpaired t test (B–D, J, L) or two-way repeated-measures ANOVA with post hoc analysis (E–I, K). Statistical significance set to P < 0.05. Bold and italic values denote significance. *Different from PBS; #main effect of time/cycle; $main effect of FOLFOX. CHO, carbohydrate; CLAMS, Comprehensive Laboratory Animal Monitoring System; FOLFOX, 5-fluorouracil, leucovorin, oxaliplatin; V̇co2, carbon dioxide production; V̇o2, oxygen consumption.
Short-Term Recovery from FOLFOX Administration: Skeletal Muscle COXIV Enzyme Activity and Metabolic Signaling
Short-term recovery of skeletal muscle metabolic signaling was assessed in mice that received four cycles of PBS or FOLFOX and then allowed to recover for 4 wk. Soleus and plantaris muscle COXIV enzyme activity were not different between PBS and FOLFOX (Fig. 7A). At 4 wk recovery, FOLFOX gastrocnemius muscle AMPK and rpS6 total protein expression were reduced compared with PBS. Total ULK1 protein expression was not different between FOLFOX and PBS (Fig. 7, B–C). The phosphorylated to total protein ratio of AMPK (T172), ULK1 (S555, S757), and rpS6 (S240/244) was not different between PBS and FOLFOX at 4 wk recovery (Fig. 7D). Puromycin incorporation was not different between PBS and FOLFOX (Fig. 7E). LC3B protein expression recovered and was not different between PBS and FOLFOX 4 wk posttreatment cessation (Fig. 7F). There was a trend (P = 0.08) for the LC3BII/I ratio to be correlated to CHO oxidation in FOLFOX mice at 4 wk recovery (Fig. 7G). These data indicate that despite reductions in systemic metabolism, skeletal muscle metabolic signaling mostly recovers 4 wk post-FOLFOX cessation. Interestingly, skeletal muscle rpS6 total protein expression remained reduced in FOLFOX-treated mice at 4 wk recovery.
Figure 7.

Skeletal muscle COXIV enzyme activity and metabolic signaling following 4 wk recovery from FOLFOX. A: COXIV enzyme activity in soleus and plantaris skeletal muscle. B: representative Western blot images with Coomassie blue protein stain, dashed line represents different areas of the same blot. Red gastrocnemius skeletal muscle total protein expression of AMPK, ULK1, and rpS6 (C). D: phosphorylated to total ratio of AMPK (T172), ULK1 (S555), ULK1 (S757), and rpS6 (S240/244). E: puromycin incorporation with representative Western blot and Coomassie blue protein stain, dashed line represents different areas of the same blot. F: protein expression of LC3BI, LC3BII, and the LC3BII/I ratio. G: Spearman r correlation between LC3BII/I and CHO oxidation in FOLFOX mice at 4 wk recovery. PBS: n = 11, FOLFOX: n = 11 or 12. Data are presented as means ± SE. Data are analyzed using unpaired t test and correlation with Spearman r correlation coefficient. Statistical significance set to P < 0.05. Bold and italic values denote significance. *Different from PBS. AMPK, AMP-activated protein kinase; COXIV, cytochrome c oxidase; FOLFOX, 5-fluorouracil, leucovorin, oxaliplatin; LC3B, microtubule-associated protein 1 light chain 3B; rpS6, ribosomal protein S6; ULK1, unc-51-like kinase 1.
Long-Term Recovery from FOLFOX Administration: Body Composition and Systemic Metabolism
Long-term recovery was assessed in mice that received four cycles of PBS or FOLFOX and allowed to recover for 10 wk; subsets of mice were placed in CLAMS metabolic cages (Fig. 8A). Body mass (Fig. 8B), percent lean mass (PBS: 82.8 ± 2.1, FOLFOX: 86.0 ± 0.79; P = 0.113), and percent body fat (Fig. 8C) were not different between PBS and FOLFOX at 10 wk recovery. Food intake and cage activity remained unchanged between PBS and FOLFOX (Fig. 8, D and E). There was a significant main effect for PBS and FOLFOX to increase cage activity (Fig. 8E) in the dark compared with the light cycle. There were significant interactions for relative (normalized to body weight) V̇o2 (Fig. 8F), V̇co2 (Fig. 8G), and energy expenditure (Fig. 8H). Specifically, relative V̇o2, V̇co2, and energy expenditure were higher in the dark compared with the light cycle in both PBS and FOLFOX mice (Fig. 8, F–H). FOLFOX significantly reduced relative V̇o2 (Fig. 8F) and energy expenditure (Fig. 8H) in the light cycle compared with PBS. Relative V̇co2 was not different between PBS and FOLFOX in the light and dark cycles (Fig. 8G). During the long-term recovery, daily average absolute and lean body mass normalized V̇o2, V̇co2, and energy expenditure remained reduced by FOLFOX treatment (Table 1). Daily average RER was not different between PBS and FOLFOX at 10 wk recovery (PBS: 0.844 ± 0.008, FOLFOX: 0.871 ± 0.013; P = 0.112). There was a significant effect of time for increased CHO (Fig. 8I) and decreased lipid oxidation (Fig. 8K) in PBS and FOLFOX mice when switching from the light to dark cycle. Compared with PBS, FOLFOX CHO oxidation was reduced (hour: 1300) in the light cycle (Fig. 8I). There was a main effect for FOLFOX to have reduced lipid oxidation compared with PBS at 10 wk recovery (Fig. 8K). Metabolic flexibility for CHO (Fig. 8J) and lipid oxidation (Fig. 8L) was not different between PBS and FOLFOX. In summary, at 10 wk recovery, body and percent fat mass recovered in FOLFOX-treated mice. However, FOLFOX-induced deficits in systemic metabolism, specifically V̇o2 and energy expenditure, did not readily recover following treatment cessation. Data supplements can be accessed here: https://doi.org/10.6084/m9.figshare.22357213.v1.
Figure 8.

FOLFOX chemotherapy-induced deficits in systemic metabolism are not fully recovered after 10 wk. A: study design. Male C57BL/6J mice were administered four cycles of either PBS or FOLFOX and allowed to recover for 10 wk after the last injection. Mice were individually housed in CLAMS metabolic cages for 5 days before tissue collection. Body mass (B) and body fat percentage (C) at post (16 wk). D: food intake of individually housed mice during CLAMS measurements. Cage activity (E) and relative (normalized to kg body weight) V̇o2 (F), V̇co2 (G), and energy expenditure (H) expressed as the average of the light cycle (0600–1700) and dark cycle (1800–0500). CHO oxidation (I) and metabolic flexibility (J). Lipid oxidation (K) and metabolic flexibility (L). PBS: n = 14, FOLFOX: n = 4. Data are presented as means ± SE. Data are analyzed using unpaired t test (B–D, J, L) or two-way repeated-measures ANOVA with post hoc analysis (E–I, K). Statistical significance set to P < 0.05. Bold and italic values denote significance. *Different from PBS; ^different from light cycle; #main effect of time/cycle; CHO, carbohydrate; CLAMS, Comprehensive Laboratory Animal Monitoring System; FOLFOX, 5-fluorouracil, leucovorin, oxaliplatin; V̇co2, carbon dioxide production; V̇o2, oxygen consumption.
DISCUSSION
Over the past decade, colorectal cancer survivorship has dramatically improved, highlighting the importance of patient health and quality of life following chemotherapy treatment. Despite the rise in survivorship, many patients suffer debilitating side effects of chemotherapy long after treatment completion (51). We previously demonstrated that repeated cycles of FOLFOX chemotherapy can induce prolonged fatigue and weakness in mice (6). We have extended these findings in the current study to elucidate the effects of FOLFOX chemotherapy on systemic metabolism and skeletal muscle metabolic signaling during treatment and recovery. We report that repeated cycles of FOLFOX are sufficient to disrupt systemic metabolism in mice, which was not readily reversible after treatment cessation. Specifically, FOLFOX reduced V̇o2, V̇co2, energy expenditure, and CHO oxidation acutely and at 4 wk of recovery. Notably, V̇o2 and energy expenditure were still reduced in FOLFOX-treated mice after 10 wk of recovery. Together, we present evidence that FOLFOX chemotherapy is sufficient to induce critical disruptions to systemic metabolism that are not readily reversible following treatment completion.
As skeletal muscle is critical to systemic metabolic homeostasis and health, we examined muscle metabolic signaling during FOLFOX chemotherapy and recovery. We investigated FOLFOX chemotherapy effects on skeletal muscle metabolic signaling involving oxidative metabolism, AMPK, mTORC1, protein synthesis, and autophagy processes. Four cycles of FOLFOX treatment suppressed muscle COXIV activity, AMPK phosphorylation, and downstream signaling involving ULK1, which were restored after short-term recovery. Related to autophagy, the LC3BII/I ratio in muscle was not altered by acute FOLFOX treatment. However, the muscle LC3BII/I ratio was positively associated with CHO oxidation in FOLFOX-treated mice. Muscle protein synthesis rate, measured by puromycin incorporation, was not changed by FOLFOX but was inversely associated with the LC3BII/I protein expression. Interestingly, we investigated the direct effects of FOLFOX in C2C12 myotube cultures, and FOLFOX suppressed AMPK and autophagy flux. The relationship between FOLFOX chemotherapy’s acute suppression of AMPK signaling and downstream processes for rescuing long-lasting FOLFOX-induced disruptions to systemic metabolism warrants further investigation.
Resting energy expenditure (REE), or basal metabolic rate, describes the energy required to maintain vital physiological functions at rest (52). Whether elevated or reduced, abnormal energy metabolism is linked to a higher risk of developing chemotherapy treatment-related toxicities in patients (16, 24). Over half of the patients with cancer receiving chemotherapy demonstrate elevated REE, whereas ∼20% present decreased REE (24). Tumor type, stage, treatment regimen, and patient health status can contribute differentially to systemic metabolic aberrations (24). Reduced REE in patients with cancer is often linked to the loss of lean mass, reduced food intake, and physical inactivity; however, the underlying cellular mechanisms remain poorly understood (16, 24). We report that FOLFOX treatment alone was sufficient to reduce relative and absolute oxygen consumption and energy expenditure in mice, independent of changes in food intake and cage activity. Importantly, these metabolic changes were not readily reversible after treatment cessation, and the driver of these metabolic changes remains to be elucidated. We previously reported that absolute lean mass and hindlimb skeletal muscle mass were not different between PBS and FOLFOX treatments at four cycles and recovery, which others have reported using FOLFOX and 5-FU chemotherapy (30, 53). Interestingly, the long-lasting systemic metabolic disruptions caused by FOLFOX cannot fully be explained by a loss of lean mass, changes in food intake, or regular cage activity.
FOLFOX treatment was sufficient to disrupt body composition; male mice that received FOLFOX exhibited decreased body fat percentage compared with PBS controls. Altered body composition was related to suppressed fat mass accretion, which is interesting considering the FOLFOX-induced reduction in energy expenditure. We previously reported that absolute fat mass increased over time in PBS control male mice (6), therefore our data suggest that FOLFOX chemotherapy treatment suppressed the normal gain of fat mass in male mice. Low adipose tissue mass is frequently reported in preclinical and clinical models of colorectal cancer with and without chemotherapy (6, 14, 29, 30). Some studies suggest that the loss of adipose mass may occur before measurable muscle mass loss during the progression of cancer cachexia (54, 55). Although reduced adipose tissue mass is typically linked to increased lipolysis, suppressed lipogenesis may also contribute to this phenotype. Indeed, 5-FU + irinotecan chemotherapy treatment in rats significantly reduced adipocyte size (56). The authors report that adipose tissue expression of factors related to mitochondrial function, lipogenesis, and fatty acid oxidation was reduced following 5-FU + irinotecan treatment, which may contribute to reducing adipocyte size (56). Furthermore, adipose tissue can regulate systemic and skeletal muscle metabolism through adipokine secretion, such as adiponectin. Adipose tissue is also a known source of systemic inflammation (57, 58). FOLFOX-induced changes to adipose tissue function merit further investigation for having a regulatory role in disrupted systemic metabolism with cancer treatment.
Altered food intake is often attributed to body composition and body mass changes. Both cancer and some chemotherapeutic agents can have an anorexic effect. Importantly, preclinical studies examining chemotherapy toxicities often do not account for the potential suppression of food intake caused by the treatment, which could have profound effects on the measured outcomes. Body mass changes can also be attributed to other factors including edema and hypovolemia, which should not be discounted. In the current study, we allowed mice to receive several chemotherapy cycles and found that food intake did not differ between PBS and FOLFOX at the study endpoints. However, food intake may have been reduced shortly after each chemotherapy injection. In fact, Ebadi et al. (56) reported reduced food intake 2–3 days after each chemotherapy cycle (one cycle = one wk), but food consumption returned to baseline and was not different compared with controls at the end of the study. Reduced food intake can lead to a negative energy balance, triggering fatty acid oxidation. A deeper understanding of how FOLFOX chemotherapy suppresses fat mass accretion in the context of specific factors that affect body mass should provide valuable insight into the overall systemic metabolic dysfunction.
Plasma levels of several metabolic-regulating growth factors, hormones, and signaling proteins have been linked to the development of metabolic dysfunction (59). We report that FOLFOX chemotherapy reduces fasting blood glucose, GIP, and C-peptide. However, fasting plasma insulin, ghrelin, adiponectin, and resistin remained unchanged in FOLFOX chemotherapy-treated mice compared with PBS controls. The skeletal system and bone-derived hormones also play a critical role in regulating systemic metabolism (60). Osteocalcin is a bone-derived hormone that contains three γ-carboxyglutamic acid residues; however, only the uncarboxylated form is biologically active (61). We previously reported myelosuppression in FOLFOX-treated mice, evidenced by reduced red and white blood cells, a common side effect of chemotherapy (6). In the current study, we observed reduced plasma uncarboxylated osteocalcin (Glu-OC) following four cycles of FOLFOX chemotherapy. Studies have demonstrated that Glu-OC acts as a hormone to regulate insulin sensitivity, glucose metabolism, and energy expenditure, and Glu-OC increases following aerobic exercise (60, 62–64). Interestingly, Glu-OC can regulate skeletal muscle metabolic homeostasis through GPRC6A receptor activation (63, 65). The results from the current study suggest that FOLFOX chemotherapy induces toxicities to both bone and skeletal muscle tissues in mice. Future research should determine if Glu-OC activation of skeletal muscle GPRC6A signaling could prevent FOLFOX chemotherapy-induced metabolic dysfunction.
At a molecular level, variations in energy expenditure can be impacted by cell size, mitochondria coupling efficiency and proton leak, the chemical composition of cell membranes, metabolic enzyme activity, and macromolecule biosynthesis (e.g., protein synthesis) (66). Skeletal muscle is one of the largest tissues in the body and displays remarkable plasticity in response to changes in the external environment. mTORC1 and AMPK signaling are critical regulators of muscle’s response to the systemic environment and are regulated by nutrient and energy availability. We report that FOLFOX did not alter rpS6 phosphorylation downstream of mTORC1 in vivo and in vitro. However, total rpS6 protein expression was reduced in mouse skeletal muscle and myotubes treated with FOLFOX, which could have implications for translation efficiency as it is a component of the 40S ribosomal subunit. Classically, chemotherapeutic agents exert cytotoxic effects by altering DNA metabolism and through the chemical induction of DNA damage (67). Interestingly, recent evidence strongly suggests that the antimetabolite 5-FU alters various aspects of RNA metabolism (68). Specifically, Therizols et al. (69) showed that 5-FU disrupts RNA processing and protein synthesis and produces fluorinated ribosomes that exhibit altered translational activity in tumor cells. Puromycin incorporation, which reflects muscle protein synthesis rate, was not changed in mice at the end of the study. However, FOLFOX had direct effects on reducing protein synthesis in C2C12 myotubes. Further research is required to determine if 5-FU or the combined FOLFOX chemotherapy regimen induces skeletal muscle ribosomal damage, which could contribute to muscle and systemic metabolic dysfunction.
Skeletal muscle mitochondria dysfunction is often reported in preclinical models using 5-FU-based chemotherapies (30, 70, 71). We previously reported that FOLFOX treatment in mice suppressed oxidative and glycolytic skeletal muscle mitochondria OXPHOS complex protein expression. However, it recovered 10 wk after treatment cessation (6). We demonstrate similar results in the current study. COXIV is the last complex in the electron transport chain that couples electron transfer from cytochrome c to molecular oxygen. Reduced skeletal muscle COXIV activity is implicated in various metabolic disorders (72–74). In the current study, FOLFOX chemotherapy reduced soleus and plantaris muscle COXIV enzyme activity. However, this recovered in both muscles 4 wk posttreatment cessation. Increased expression of mitochondrial uncoupling proteins (UCPs) could be linked to dysregulated mitochondrial function, as they uncouple oxygen consumption from ATP synthesis (75). Following four cycles of FOLFOX, there were no differences in muscle gene expression of UCP2 and UCP3. In addition, FOLFOX did not alter the transcription of mitochondria quality control-related genes, which regulate mitochondria biogenesis and size through fusion and fission events (76). 5-FU-based chemotherapies can damage metabolic tissues, including skeletal muscle and the liver (77), which may be linked to reduced mitochondria content. However, we did not find differences in mitochondrial DNA content in skeletal muscle or liver tissues. Therefore, following FOLFOX treatment, muscle mitochondria number may be maintained. Although mitochondria number and mitochondria-related gene expression were largely unchanged, further research is required to determine if mitochondrial function (e.g., oxygen consumption) is altered by FOLFOX chemotherapy treatment and recovery.
Autophagy is a cellular recycling system strongly activated in response to nutrient deprivation (37). The autophagosome initiates the capture of targeted cellular components, which are subsequently degraded in the lysosome. Products of this catabolic process can be made available for de novo protein synthesis (78). Skeletal muscle autophagy is required to maintain muscle function. Mice with autophagy defects exhibit impaired mitochondria function, an accumulation of protein aggregates, and altered muscle morphology (79, 80). During prolonged fasting, skeletal muscle AMPK is required to maintain systemic metabolism and skeletal muscle autophagy flux (81). Following a 12-h fast, we report that FOLFOX reduced AMPK phosphorylation at T172, a primary determinant of AMPK activity (82). We also demonstrate the direct effects of FOLFOX to decrease AMPK T172 phosphorylation and autophagy in C2C12 myotubes. Bujak et al. (81) reported that the loss of skeletal muscle AMPK (AMPK-MKO) lowered fasting blood glucose in mice, which was not due to changes in muscle or liver glycogen content. Fasting-induced hypoglycemia in AMPK-MKO mice resulted from an inability to supply the liver with gluconeogenic substrates (e.g., alanine). Skeletal muscle is a primary amino acid source for systemic metabolism. During prolonged fasting, gluconeogenic demands can be maintained by activating skeletal muscle catabolism (81). Myofiber size remained unchanged in response to 48-h fasting in AMPK-MKO mice compared with the wild type, where fiber cross-sectional area (CSA) was reduced by 18% (81). We report that FOLFOX treatment induces hypoglycemia and suppresses skeletal muscle AMPK signaling in mice. Moreover, FOLFOX mice lost less body weight than controls following a 12-h fast. The FOLFOX-induced suppression of blood glucose and skeletal muscle AMPK signaling recovered 4 wk postchemotherapy cessation. Therefore, suppressing skeletal muscle AMPK signaling cannot fully explain long-lasting deficits in systemic metabolism. However, early disruptions to skeletal muscle metabolism should still be considered a therapeutic target to prevent long-term chemotherapy-induced metabolic dysfunction.
The ubiquitin-proteasome system (UPS) and autophagy lysosomal systems are considered primary pathways through which muscle proteins are broken down (83). UPS primarily breaks down small, short-lived proteins, whereas autophagy facilitates bulk protein breakdown through lysosomal degradation (31). We have shown that FOLFOX does not alter muscle-specific E3 ubiquitin ligase Atrogin-1 and MuRF1 gene expression, which are involved in the UPS (6). mTORC1 and AMPK converge on the protein ULK1 to regulate autophagy. mTORC1 phosphorylation of ULK1 at S757 inhibits ULK1 complex formation with FIP200 and Atg13 to inactivate autophagy (84). Conversely, increased AMPK activation promotes phosphorylation of ULK1 at S555, which allows for the ULK1 complex formation, a critical step in autophagy initiation (84–86). We report that in addition to reduced skeletal muscle AMPK phosphorylation, the downstream phosphorylation of ULK1 S555 is reduced by FOLFOX in vivo and in vitro. FOLFOX-treated mice have decreased muscle LC3BII protein expression. LC3I lipidation generates LC3II, which is incorporated into the autophagosome membrane and correlates to autophagosome number (79, 87). Reduced AMPK (T172), ULK1 (S555), and LC3BII protein expression suggest that FOLFOX suppresses skeletal muscle autophagy. Autophagy inhibition in C2C12 myotubes using bafilomycin A1 revealed that 24 h FOLFOX treatment suppressed autophagy flux, confirmed by the reduction of LC3B II/I and increased p62 protein expression, which accumulates when autophagy is inhibited (88). In FOLFOX-treated mice, the LC3BII/I ratio was positively correlated to CHO oxidation, linking muscle autophagy to systemic metabolic dysfunction. However, LC3B protein expression was not different between control and chemotherapy-treated groups at 4 wk of recovery while CHO oxidation remained reduced; therefore, other factors may be at play.
In conclusion, our study provides novel evidence that repeated cycles of FOLFOX chemotherapy can induce long-lasting deficits in systemic metabolism and disrupt skeletal muscle metabolic signaling in mice. The reduction in systemic metabolism was not readily reversible 10 wk after treatment completion. The findings suggest that the FOLFOX-induced disruptions to systemic metabolism occur independent of changes in lean mass, food intake, or cage activity. However, the FOLFOX-induced suppression of fat mass accretion and acute changes in food intake or cage activity immediately following chemotherapy injections cannot be disregarded and may play a role in the suppression of systemic metabolism. FOLFOX suppressed skeletal muscle metabolic signaling involving AMPK and autophagy in vivo and in vitro. Although these aberrations may contribute to long-term chemotherapy toxicities, the suppression of this signaling pathway in muscle recovered 4 wk after treatment completion. Overall, the results provide valuable insights into the mechanisms responsible for chemotherapy-related toxicities that can and cannot readily recover following treatment cessation. We previously demonstrated that short-duration treadmill exercise could offset long-lasting fatigue in FOLFOX-treated mice. Further research is required to determine if exercise or clinically relevant AMPK activators could be used during chemotherapy treatment to prevent long-lasting metabolic disturbances.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Fig. S1 and Supplemental Tables S1 and S2: https://figshare.com/s/e2d07f508781072286b8.
GRANTS
This work was supported by the National Institutes of Health (National Cancer Institute) grant R21 CA-231131 to J.A.C. and the NIH (National Heart, Lung, and Blood Institute) grant R01 HL146443 to A.J.S.
DISCLAIMERS
The study results are presented clearly, honestly, and without fabrication, falsification, or inappropriate data manipulation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.A.C. conceived and designed research; J.L.H., B.R.C., H.G.P., and D.R.B. performed experiments; J.L.H., J.A.C., B.R.C., H.G.P., D.R.B., and Q.Z. analyzed data; J.L.H., S.E.A., B.R.C., H.G.P., D.R.B., Q.Z., J.S.M., E.S.G., M.J.P., A.J.S., and J.A.C. interpreted results of experiments; J.L.H., B.R.C., H.G.P., D.R.B., and J.A.C. prepared figures; J.L.H., B.R.C., D.R.B., J.S.M., E.S.G., M.J.P., A.J.S., S.E.A., and J.A.C. drafted manuscript; J.L.H., B.R.C., H.G.P., D.R.B., Q.Z., J.S.M., E.S.G., M.J.P., A.J.S., S.E.A., and J.A.C. edited and revised manuscript; J.L.H., B.R.C., H.G.P., D.R.B., Q.Z., J.S.M., E.S.G., M.J.P., A.J.S., S.E.A., and J.A.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The graphical abstract was created with BioRender.com.
REFERENCES
- 1. Sougiannis AT, VanderVeen BN, Enos RT, Velazquez KT, Bader JE, Carson M, Chatzistamou I, Walla M, Pena MM, Kubinak JL, Nagarkatti M, Carson JA, Murphy EA. Impact of 5 fluorouracil chemotherapy on gut inflammation, functional parameters, and gut microbiota. Brain Behav Immun 80: 44–55, 2019. doi: 10.1016/j.bbi.2019.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. VanderVeen BN, Sougiannis AT, Velazquez KT, Carson JA, Fan D, Murphy EA. The acute effects of 5 fluorouracil on skeletal muscle resident and infiltrating immune cells in mice. Front Physiol 11: 593468, 2020. doi: 10.3389/fphys.2020.593468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. VanderVeen BN, Cardaci TD, McDonald SJ, Madero SS, Unger CA, Bullard BM, Enos RT, Velázquez KT, Kubinak JL, Fan D, Murphy EA. Obesity reduced survival with 5-fluorouracil and did not protect against chemotherapy-induced cachexia or immune cell cytotoxicity in mice. Cancer Biol Ther 23: 1–15, 2022. doi: 10.1080/15384047.2022.2108306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feng J-P, Yuan X-L, Li M, Fang J, Xie T, Zhou Y, Zhu Y-M, Luo M, Lin M, Ye D-W. Secondary diabetes associated with 5‐fluorouracil‐based chemotherapy regimens in non‐diabetic patients with colorectal cancer: results from a single‐centre cohort study. Colorectal Dis 15: 27–33, 2013. doi: 10.1111/j.1463-1318.2012.03097.x. [DOI] [PubMed] [Google Scholar]
- 5. Prado CMM, Baracos VE, McCargar LJ, Mourtzakis M, Mulder KE, Reiman T, Butts CA, Scarfe AG, Sawyer MB. Body composition as an independent determinant of 5-fluorouracil–based chemotherapy toxicity. Clin Cancer Res 13: 3264–3268, 2007. doi: 10.1158/1078-0432.CCR-06-3067. [DOI] [PubMed] [Google Scholar]
- 6. Halle JL, Counts BR, Zhang Q, Carson JA. Short duration treadmill exercise improves physical function and skeletal muscle mitochondria protein expression after recovery from FOLFOX chemotherapy in male mice. FASEB J 36: e22437, 2022. doi: 10.1096/fj.202200460R. [DOI] [PubMed] [Google Scholar]
- 7. Denlinger CS, Barsevick AM. The challenges of colorectal cancer survivorship. J Natl Compr Canc Netw 7: 883–893, 2009. doi: 10.6004/jnccn.2009.0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gebauer J, Higham C, Langer T, Denzer C, Brabant G. Long-term endocrine and metabolic consequences of cancer treatment: a systematic review. Endocr Rev 40: 711–767, 2019. doi: 10.1210/er.2018-00092. [DOI] [PubMed] [Google Scholar]
- 9. Neugut AI, Lin A, Raab GT, Hillyer GC, Keller D, O'Neil DS, Accordino MK, Kiran RP, Wright J, Hershman DL. FOLFOX and FOLFIRI use in stage IV colon cancer: analysis of SEER-medicare data. Clin Colorectal Cancer 18: 133–140, 2019. doi: 10.1016/j.clcc.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang Z, Ying Z, Bosy-Westphal A, Zhang J, Schautz B, Later W, Heymsfield SB, Müller MJ. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr 92: 1369–1377, 2010. doi: 10.3945/ajcn.2010.29885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Counts BR, Halle JL, Carson JA. Early onset physical inactivity and metabolic dysfunction in tumor-bearing mice is associated with accelerated cachexia. Med Sci Sports Exer 54: 77–88, 2022. doi: 10.1249/MSS.0000000000002772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Counts BR, Hardee JP, Fix DK, Vanderveen BN, Montalvo RN, Carson JA. Cachexia disrupts diurnal regulation of activity, feeding, and muscle mechanistic target of rapamycin complex 1 in mice. Med Sci Sports Exerc 52: 577–587, 2020. doi: 10.1249/MSS.0000000000002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Murphy KT, Chee A, Trieu J, Naim T, Lynch GS. Importance of functional and metabolic impairments in the characterization of the C-26 murine model of cancer cachexia. Dis Model Mech 5: 533–545, 2012. doi: 10.1242/dmm.008839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chung E, Lee HS, Cho E-S, Park EJ, Baik SH, Lee KY, Kang J. Changes in body composition during adjuvant FOLFOX chemotherapy and overall survival in non-metastatic colon cancer. Cancers (Basel) 12: 60, 2019. doi: 10.3390/cancers12010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cespedes Feliciano EM, Lee VS, Prado CM, Meyerhardt JA, Alexeeff S, Kroenke CH, Xiao J, Castillo AL, Caan BJ. Muscle mass at the time of diagnosis of nonmetastatic colon cancer and early discontinuation of chemotherapy, delays, and dose reductions on adjuvant FOLFOX: the C‐SCANS study. Cancer 123: 4868–4877, 2017. doi: 10.1002/cncr.30950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Demark-Wahnefried W, Hars V, Conaway MR, Havlin K, Rimer B, McElveen G, Winer E. Reduced rates of metabolism and decreased physical activity in breast cancer patients receiving adjuvant chemotherapy. Am J Clin Nutr 65: 1495–1501, 1997. doi: 10.1093/ajcn/65.5.1495. [DOI] [PubMed] [Google Scholar]
- 17. Lee CS, Ryan EJ, Doherty GA. Gastro-intestinal toxicity of chemotherapeutics in colorectal cancer: the role of inflammation. World J Gastroenterol 20: 3751–3761, 2014. doi: 10.3748/wjg.v20.i14.3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rossi S, Basso M, Strippoli A, Schinzari G, D'Argento E, Larocca M, Cassano A, Barone C. Are markers of systemic inflammation good prognostic indicators in colorectal cancer? Clin Colorectal Cancer 16: 264–274, 2017. doi: 10.1016/j.clcc.2017.03.015. [DOI] [PubMed] [Google Scholar]
- 19. Van Soom T, El Bakkali S, Gebruers N, Verbelen H, Tjalma W, van Breda E. The effects of chemotherapy on energy metabolic aspects in cancer patients: a systematic review. Clin Nutr 39: 1863–1877, 2020. doi: 10.1016/j.clnu.2019.07.028. [DOI] [PubMed] [Google Scholar]
- 20. Dieli-Conwright CM, Wong L, Waliany S, Mortimer JE. Metabolic syndrome and breast cancer survivors: a follow-up analysis after completion of chemotherapy. Diabetol Metab Syndr 14: 36, 2022. doi: 10.1186/s13098-022-00807-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goodpaster BH, Sparks LM. Metabolic flexibility in health and disease. Cell Metab 25: 1027–1036, 2017. doi: 10.1016/j.cmet.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lowell BB, Spiegelman BM. Towards a molecular understanding of adaptive thermogenesis. Nature 404: 652–660, 2000. doi: 10.1038/35007527. [DOI] [PubMed] [Google Scholar]
- 23. Ferrannini E. The theoretical bases of indirect calorimetry: a review. Metabolism 37: 287–301, 1988. doi: 10.1016/0026-0495(88)90110-2. [DOI] [PubMed] [Google Scholar]
- 24. Jouinot A, Vazeille C, Durand JP, Huillard O, Boudou-Rouquette P, Coriat R, Chapron J, Neveux N, De Bandt JP, Alexandre J, Cynober L, Goldwasser F. Resting energy expenditure in the risk assessment of anticancer treatments. Clin Nutr 37: 558–565, 2018. doi: 10.1016/j.clnu.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 25. Pin F, Couch ME, Bonetto A. Preservation of muscle mass as a strategy to reduce the toxic effects of cancer chemotherapy on body composition. Curr Opin Support Palliat Care 12: 420–426, 2018. doi: 10.1097/SPC.0000000000000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kurk S, Peeters P, Stellato R, Dorresteijn B, de Jong P, Jourdan M, Creemers G-J, Erdkamp F, de Jongh F, Kint P, Simkens L, Tanis B, Tjin-A-Ton M, Van Der Velden A, Punt C, Koopman M, May A. Skeletal muscle mass loss and dose‐limiting toxicities in metastatic colorectal cancer patients. J Cachexia Sarcopenia Muscle 10: 803–813, 2019. doi: 10.1002/jcsm.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jung H-W, Kim JW, Kim J-Y, Kim S-W, Yang HK, Lee JW, Lee K-W, Kim D-W, Kang S-B, Kim K-I, Kim C-H, Kim JH. Effect of muscle mass on toxicity and survival in patients with colon cancer undergoing adjuvant chemotherapy. Support Care Cancer 23: 687–694, 2015. doi: 10.1007/s00520-014-2418-6. [DOI] [PubMed] [Google Scholar]
- 28. Pin F, Barreto R, Couch ME, Bonetto A, O'Connell TM. Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism. J Cachexia Sarcopenia Muscle 10: 140–154, 2019. doi: 10.1002/jcsm.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barreto R, Mandili G, Witzmann FA, Novelli F, Zimmers TA, Bonetto A. Cancer and chemotherapy contribute to muscle loss by activating common signaling pathways. Front Physiol 7: 472, 2016. doi: 10.3389/fphys.2016.00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barreto R, Waning DL, Gao H, Liu Y, Zimmers TA, Bonetto A. Chemotherapy-related cachexia is associated with mitochondrial depletion and the activation of ERK1/2 and p38 MAPKs. Oncotarget 7: 43442–43460, 2016. doi: 10.18632/oncotarget.9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ottens F, Franz A, Hoppe T. Build-UPS and break-downs: metabolism impacts on proteostasis and aging. Cell Death Differ 28: 505–521, 2021. [Erratum in Cell Death Differ 29: 465, 2021]. doi: 10.1038/s41418-020-00682-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Battaglioni S, Benjamin D, Wälchli M, Maier T, Hall MN. mTOR substrate phosphorylation in growth control. Cell 185: 1814–1836, 2022. doi: 10.1016/j.cell.2022.04.013. [DOI] [PubMed] [Google Scholar]
- 33. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19: 121–135, 2018. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Geremia A, Sartori R, Baraldo M, Nogara L, Balmaceda V, Dumitras GA, Ciciliot S, Scalabrin M, Nolte H, Blaauw B. Activation of Akt–mTORC1 signalling reverts cancer‐dependent muscle wasting. J Cachexia Sarcopenia Muscle 13: 648–661, 2022. doi: 10.1002/jcsm.12854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Penna F, Ballaro R, Martinez-Cristobal P, Sala D, Sebastian D, Busquets S, Muscaritoli M, Argiles JM, Costelli P, Zorzano A. Autophagy exacerbates muscle wasting in cancer cachexia and impairs mitochondrial function. J Mol Biol 431: 2674–2686, 2019. doi: 10.1016/j.jmb.2019.05.032. [DOI] [PubMed] [Google Scholar]
- 36. White JP, Puppa MJ, Gao S, Sato S, Welle SL, Carson JA. Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. Am J Physiol Endocrinol Metab 304: E1042–E1052, 2013. doi: 10.1152/ajpendo.00410.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. He C. Balancing nutrient and energy demand and supply via autophagy. Curr Biol 32: R684–R696, 2022. doi: 10.1016/j.cub.2022.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Campelj DG, Goodman CA, Rybalka E. Chemotherapy-induced myopathy: the dark side of the cachexia sphere. Cancers (Basel) 13: 3615, 2021. doi: 10.3390/cancers13143615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. von Walden F, Liu C, Aurigemma N, Nader GA. mTOR signaling regulates myotube hypertrophy by modulating protein synthesis, rDNA transcription, and chromatin remodeling. Am J Physiol Cell Physiol 311: C663–C672, 2016. doi: 10.1152/ajpcell.00144.2016. [DOI] [PubMed] [Google Scholar]
- 40. Halle JL, Counts-Franch BR, Prince RM, Carson JA. The effect of mechanical stretch on myotube growth suppression by colon-26 tumor-derived factors. Front Cell Dev Biol 9: 690452, 2021. doi: 10.3389/fcell.2021.690452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rybalka E, Timpani CA, Cheregi BD, Sorensen JC, Nurgali K, Hayes A. Chemotherapeutic agents induce mitochondrial superoxide production and toxicity but do not alter respiration in skeletal muscle in vitro. Mitochondrion 42: 33–49, 2018. doi: 10.1016/j.mito.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 42. Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 11: 1437–1438, 2015. doi: 10.1080/15548627.2015.1066957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lusk G. Analysis of the oxidation of mixtures of carbohydrate and fat: a correction. J Biol Chem 59, 1924. [Google Scholar]
- 44. Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 6: 275–277, 2009. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- 45. Fix DK, Hardee JP, Gao S, VanderVeen BN, Velázquez KT, Carson JA. Role of gp130 in basal and exercise-trained skeletal muscle mitochondrial quality control. J Appl Physiol (1985) 124: 1456–1470, 2018. doi: 10.1152/japplphysiol.01063.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. White JP, Baltgalvis KA, Puppa MJ, Sato S, Baynes JW, Carson JA. Muscle oxidative capacity during IL-6-dependent cancer cachexia. Am J Physiol Regul Integr Comp Physiol 300: R201–R211, 2011. doi: 10.1152/ajpregu.00300.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Quiros PM, Goyal A, Jha P, Auwerx J. Analysis of mtDNA/nDNA ratio in mice. Curr Protoc Mouse Biol 7: 47–54, 2017. doi: 10.1002/cpmo.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254, 1976. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 49. Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, Dor Y, Zisman P, Meyuhas O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev 19: 2199–2211, 2005. doi: 10.1101/gad.351605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yoon MJ, Lee GY, Chung J-J, Ahn YH, Hong SH, Kim JB. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator–activated receptor α. Diabetes 55: 2562–2570, 2006. doi: 10.2337/db05-1322. [DOI] [PubMed] [Google Scholar]
- 51. Nurgali K, Jagoe RT, Abalo R. Adverse effects of cancer chemotherapy: anything new to improve tolerance and reduce sequelae? Front Pharmacol 9: 245, 2018. doi: 10.3389/fphar.2018.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heymsfield SB, Smith B, Dahle J, Kennedy S, Fearnbach N, Thomas DM, Bosy‐Westphal A, Müller MJ. Resting energy expenditure: from cellular to whole‐body level, a mechanistic historical perspective. Obesity (Silver Spring) 29: 500–511, 2021. doi: 10.1002/oby.23090. [DOI] [PubMed] [Google Scholar]
- 53. Campelj DG, Timpani CA, Cree T, Petersen AC, Hayes A, Goodman CA, Rybalka E. Metronomic 5-fluorouracil delivery primes skeletal muscle for myopathy but does not cause cachexia. Pharmaceuticals 14: 478, 2021. doi: 10.3390/ph14050478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Halle JL, Pena GS, Paez HG, Castro AJ, Rossiter HB, Visavadiya NP, Whitehurst MA, Khamoui AV. Tissue-specific dysregulation of mitochondrial respiratory capacity and coupling control in colon-26 tumor-induced cachexia. Am J Physiol Regul Integr Comp Physiol 317: R68–R82, 2019. doi: 10.1152/ajpregu.00028.2019. [DOI] [PubMed] [Google Scholar]
- 55. Lim S, Brown JL, Washington TA, Greene NP. Development and progression of cancer cachexia: perspectives from bench to bedside. Sports Med Health Sci 2: 177–185, 2020. doi: 10.1016/j.smhs.2020.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ebadi M, Field CJ, Lehner R, Mazurak VC. Chemotherapy diminishes lipid storage capacity of adipose tissue in a preclinical model of colon cancer. Lipids Health Dis 16: 1–12, 2017. doi: 10.1186/s12944-017-0638-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Słomian G, Świętochowska E, Malinowska-Borowska J, Kasperczyk S, Rogalska A, Nowak P. Association between chemotherapy and plasma adipokines in patients with colorectal cancer. Pharmacol Rep 66: 902–907, 2014. doi: 10.1016/j.pharep.2014.05.015. [DOI] [PubMed] [Google Scholar]
- 58. Kawai T, Autieri MV, Scalia R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol 320: C375–C391, 2021. doi: 10.1152/ajpcell.00379.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Barchetta I, Cimini F, Ciccarelli G, Baroni M, Cavallo M. Sick fat: the good and the bad of old and new circulating markers of adipose tissue inflammation. J Endocrinol Invest 42: 1257–1272, 2019. doi: 10.1007/s40618-019-01052-3. [DOI] [PubMed] [Google Scholar]
- 60. Riquelme-Gallego B, García-Molina L, Cano-Ibáñez N, Sánchez-Delgado G, Andújar-Vera F, García-Fontana C, González-Salvatierra S, García-Recio E, Martínez-Ruiz V, Bueno-Cavanillas A, Muñoz-Torres M, García-Fontana B. Circulating undercarboxylated osteocalcin as estimator of cardiovascular and type 2 diabetes risk in metabolic syndrome patients. Sci Rep 10: 1840, 2020. doi: 10.1038/s41598-020-58760-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Booth SL, Centi A, Smith SR, Gundberg C. The role of osteocalcin in human glucose metabolism: marker or mediator? Nat Rev Endocrinol 9: 43–55, 2013. doi: 10.1038/nrendo.2012.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hill H, Grams J, Walton R, Liu J, Moellering D, Garvey W. Carboxylated and uncarboxylated forms of osteocalcin directly modulate the glucose transport system and inflammation in adipocytes. Horm Metab Res 46: 341–347, 2014. doi: 10.1055/s-0034-1368709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mera P, Laue K, Ferron M, Confavreux C, Wei J, Galán-Díez M, Lacampagne A, Mitchell SJ, Mattison JA, Chen Y, Bacchetta J, Szulc P, Kitsis RN, de Cabo R, Friedman RA, Torsitano C, McGraw TE, Puchowicz M, Kurland I, Karsenty G. Osteocalcin signaling in myofibers is necessary and sufficient for optimum adaptation to exercise. Cell Metab 23: 1078–1092, 2016. [Erratum in Cell Metab 25: 218, 2017]. doi: 10.1016/j.cmet.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Razny U, Fedak D, Kiec‐Wilk B, Goralska J, Gruca A, Zdzienicka A, Kiec‐Klimczak M, Solnica B, Hubalewska‐Dydejczyk A, Malczewska‐Malec M. Carboxylated and undercarboxylated osteocalcin in metabolic complications of human obesity and prediabetes. Diabetes Metab Res Rev 33: e2862, 2017. doi: 10.1002/dmrr.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pi M, Nishimoto SK, Quarles LD. GPRC6A: Jack of all metabolism (or master of none). Mol Metab 6: 185–193, 2017. doi: 10.1016/j.molmet.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Konarzewski M, Książek A. Determinants of intra-specific variation in basal metabolic rate. J Comp Physiol B 183: 27–41, 2013. doi: 10.1007/s00360-012-0698-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. DeVita VT Jr, Chu E. A history of cancer chemotherapy. Cancer Res 68: 8643–8653, 2008. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- 68. Chalabi-Dchar M, Fenouil T, Machon C, Vincent A, Catez F, Marcel V, Mertani HC, Saurin J-C, Bouvet P, Guitton J, Venezia ND, Diaz J-J. A novel view on an old drug, 5-fluorouracil: an unexpected RNA modifier with intriguing impact on cancer cell fate. NAR Cancer 3: zcab032, 2021. doi: 10.1093/narcan/zcab032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Therizols G, Bash-Imam Z, Panthu B, Machon C, Vincent A, Ripoll J, Nait-Slimane S, Chalabi-Dchar M, Gaucherot A, Garcia M, Laforets F, Marcel V, Baubaker-Vitre J, Monet M-A, Bouclier C, Vanbelle C, Souahlia G, Berthel E, Albaret MA, Mertani HC, Prudhomme M, Bertrand M, David A, Saurin J-C, Bouvet P, Rivals E, Ohlmann T, Guitton J, Venezia ND, Pannequin J, Catez F, Diaz J-J. Alteration of ribosome function upon 5-fluorouracil treatment favors cancer cell drug-tolerance. Nat Commun 13: 173, 2022. doi: 10.1038/s41467-021-27847-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ballarò R, Beltrà M, De Lucia S, Pin F, Ranjbar K, Hulmi JJ, Costelli P, Penna F. Moderate exercise in mice improves cancer plus chemotherapy‐induced muscle wasting and mitochondrial alterations. FASEB J 33: 5482–5494, 2019. doi: 10.1096/fj.201801862R. [DOI] [PubMed] [Google Scholar]
- 71. Ballarò R, Lopalco P, Audrito V, Beltrà M, Pin F, Angelini R, Costelli P, Corcelli A, Bonetto A, Szeto HH, O’Connell TM, Penna F. Targeting mitochondria by SS-31 ameliorates the whole body energy status in cancer-and chemotherapy-induced cachexia. Cancers 13: 850, 2021. doi: 10.3390/cancers13040850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu X, Zhang J, Ming Y, Chen X, Zeng M, Mao Y. The aggravation of mitochondrial dysfunction in nonalcoholic fatty liver disease accompanied with type 2 diabetes mellitus. Scand J Gastroenterol 50: 1152–1159, 2015. doi: 10.3109/00365521.2015.1030687. [DOI] [PubMed] [Google Scholar]
- 73. Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura‐Clapier R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol 551: 491–501, 2003. doi: 10.1113/jphysiol.2003.045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schiffer TA, Peleli M, Sundqvist ML, Ekblom B, Lundberg JO, Weitzberg E, Larsen FJ. Control of human energy expenditure by cytochrome c oxidase subunit IV-2. Am J Physiol Cell Physiol 311: C452–C461, 2016. doi: 10.1152/ajpcell.00099.2016. [DOI] [PubMed] [Google Scholar]
- 75. Ricquier D, Bouillaud F. Mitochondrial uncoupling proteins: from mitochondria to the regulation of energy balance. J Physiol 529: 3–10, 2000. doi: 10.1111/j.1469-7793.2000.00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sligar J, DeBruin DA, Saner NJ, Philp AM, Philp A. The importance of mitochondrial quality control for maintaining skeletal muscle function across health span. Am J Physiol Cell Physiol 322: C461–C467, 2022. doi: 10.1152/ajpcell.00388.2021. [DOI] [PubMed] [Google Scholar]
- 77. Robinson S, Mann J, Vasilaki A, Mathers J, Burt A, Oakley F, White S, Mann D. Pathogenesis of FOLFOX induced sinusoidal obstruction syndrome in a murine chemotherapy model. J Hepatol 59: 318–326, 2013. doi: 10.1016/j.jhep.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 16: 461–472, 2015. doi: 10.1038/nrm4024. [DOI] [PubMed] [Google Scholar]
- 79. Neel BA, Lin Y, Pessin JE. Skeletal muscle autophagy: a new metabolic regulator. Trends Endocrinol Metab 24: 635–643, 2013. doi: 10.1016/j.tem.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Castets P, Lin S, Rion N, Di Fulvio S, Romanino K, Guridi M, Frank S, Tintignac LA, Sinnreich M, Rüegg MA. Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab 17: 731–744, 2013. doi: 10.1016/j.cmet.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 81. Bujak AL, Crane JD, Lally JS, Ford RJ, Kang SJ, Rebalka IA, Green AE, Kemp BE, Hawke TJ, Schertzer JD, Steinberg GR. AMPK activation of muscle autophagy prevents fasting-induced hypoglycemia and myopathy during aging. Cell Metab 21: 883–890, 2015. doi: 10.1016/j.cmet.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Willows R, Sanders MJ, Xiao B, Patel BR, Martin SR, Read J, Wilson JR, Hubbard J, Gamblin SJ, Carling D. Phosphorylation of AMPK by upstream kinases is required for activity in mammalian cells. Biochem J 474: 3059–3073, 2017. doi: 10.1042/BCJ20170458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sandri M. Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int J Biochem Cell Biol 45: 2121–2129, 2013. doi: 10.1016/j.biocel.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141, 2011. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear LJ, Kundu M, Yan Z. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun 8: 548, 2017. doi: 10.1038/s41467-017-00520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Fan X-Y, Tian C, Wang H, Xu Y, Ren K, Zhang B-Y, Gao C, Shi Q, Meng G, Zhang L-B, Zhao Y-J, Shao Q-X, Dong X-P. Activation of the AMPK-ULK1 pathway plays an important role in autophagy during prion infection. Sci Rep 5: 14728, 2015. doi: 10.1038/srep14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 3: 542–545, 2007. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 88. Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 452: 181–197, 2009. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1 and Supplemental Tables S1 and S2: https://figshare.com/s/e2d07f508781072286b8.
Data Availability Statement
Data will be made available upon reasonable request.