

Keywords: inflammation, mitochondria, mitochondrial dynamics, mitochondrial dysfunction, mitophagy
Abstract
Mitochondria are well known as organelles responsible for the maintenance of cellular bioenergetics through the production of ATP. Although oxidative phosphorylation may be their most important function, mitochondria are also integral for the synthesis of metabolic precursors, calcium regulation, the production of reactive oxygen species, immune signaling, and apoptosis. Considering the breadth of their responsibilities, mitochondria are fundamental for cellular metabolism and homeostasis. Appreciating this significance, translational medicine has begun to investigate how mitochondrial dysfunction can represent a harbinger of disease. In this review, we provide a detailed overview of mitochondrial metabolism, cellular bioenergetics, mitochondrial dynamics, autophagy, mitochondrial damage-associated molecular patterns, mitochondria-mediated cell death pathways, and how mitochondrial dysfunction at any of these levels is associated with disease pathogenesis. Mitochondria-dependent pathways may thereby represent an attractive therapeutic target for ameliorating human disease.
CLINICAL HIGHLIGHTS.
Mitochondria are the primary site of cellular energy production in eukaryotes.
Mitochondria are also crucial for maintaining cellular and organismal homeostasis through the integral role they play in cellular bioenergetics, metabolic precursor synthesis, calcium regulation, ROS production, immune signaling, and apoptosis.
Recent data have shown that mitochondria also function as a reservoir of critical second messengers and effector molecules, such as DAMPs, inflammasomes, and sirtuins, that mediate important cellular and physiological processes.
Mitochondrial dysfunction, caused by environmental conditions or injurious stimuli, contributes to the initiation and/or pathogenesis of human diseases and aging.
Within this review, we discuss how derangement of key mitochondrial pathways contributes to the pathogenesis of cardiovascular disease, pulmonary arterial hypertension, chronic obstructive lung disease, idiopathic pulmonary fibrosis, sepsis, and neurodegenerative diseases.
There is an anticipation of continued discovery of mitochondrial pathways important to the pathogenesis of human disease. These discoveries may lead to molecular targets as diagnostics and therapeutics for patients with mitochondria pathway-based medical disorders.
1. MITOCHONDRIAL DYSFUNCTION: A HARBINGER OF HUMAN DISEASE PATHOGENESIS
1.1. Introduction
Mitochondria serve as the principal site of cellular energy production (bioenergetics) in eukaryotes. Because of their vital importance as a nexus for key cellular metabolic pathways, these complex double membrane-bound organelles also act as sentinels for cellular and organismal homeostasis. Mitochondria contribute to fatty acid (FA) and amino acid (AA) metabolism, synthesis of biomolecules (i.e., heme and iron-sulfur clusters), calcium (Ca2+) regulation, antiviral defenses, and cell signaling (1–4). Conversely, in their failure to perform these functions because of decline in integrity or numbers, often resulting from exposure to adverse environmental conditions or injurious stimuli, mitochondria may represent a harbinger of disease pathogenesis. An emergent hypothesis, based on experimental evidence, is that deficits in mitochondrial integrity, bioenergetics, or other metabolic or regulatory functions may contribute mechanistically to the initiation and/or pathogenesis of many human diseases as well as the aging process (5–10). Additionally, mitochondria also impact cellular processes such as programmed cell death (apoptosis) and inflammation, which can modify disease outcomes (4, 11, 12).
Mitochondria are unique organelles in that, unlike other cellular components, they harbor their own genome, consisting of mitochondrial DNA (mtDNA), a closed circular loop of double-stranded DNA of 16,569 kb, which encodes 37 genes on both strands. The products of these genes encode 13 proteins involved in mitochondrial electron transfer (e.g., subunits of NADH dehydrogenase 1, cytochrome b, cytochrome-c oxidase I, ATP synthase 6), 2 ribosomal RNAs, and 22 mitochondrial transfer RNAs involved in the synthesis of mitochondrial proteins (13, 14). The mitochondrial genome is highly susceptible to mutation. A D-loop region of ∼1 kb contains two hypervariable regions with higher mutation frequency. Increased mutation frequency of mtDNA (heteroplasmy) may also be associated with select human diseases. Mammalian mtDNA replication is operated by nuclear-encoded DNA polymerase-γ (Polg). A mouse model of accelerated mtDNA mutation (i.e., mice homozygous for a Polg NH2-terminal exonuclease domain missense mutation, Polg D257A) exhibited accelerated aging, progressive anemia, as well as defective mitophagy during erythrocyte maturation (15, 16).
Mutations in either mitochondrial or nuclear-encoded genes affecting bioenergetics (e.g., subunits of electron transport chain components) can elicit rare and distinct human genetic diseases, including various encephalopathies, neuropathies, and cardiomyopathies (17–20). Mutations in nuclear-encoded genes affecting mitochondrial quality can also cause disease; for example, LRRK2, PARK2, PARK7, and PINK1 are attributed to familial Parkinson’s disease (21). Mitochondria also serve as reservoirs of critical second messengers and key effector molecules in mediating important cellular and physiological processes, with overexpression or deficiencies of these key molecules contributing to the pathogenesis of human diseases (22–24). Although mitochondria are essential for normal physiological processes, in nongenetic diseases of complex or unclear etiology it is not always evident whether mitochondrial injury is causative of a particular disease or sustained as the consequence of disease pathogenesis (25). Hence, the relative importance of mitochondrial dysfunction to disease may vary in a disease- and model-specific fashion. As not all human diseases are associated with mitochondrial processes, this review examines the propathogenic role of mitochondrial dysfunction in select human diseases, for which evidence has accrued in experimental animal models or human samples.
Mitochondria populations are tightly regulated by processes that ensure the maintenance of required numbers and their quality. These processes include the proliferation (biogenesis) of mitochondria and the culling of dysfunctional mitochondria by autophagy (mitophagy) (26–28). Additionally, mitochondria represent dynamic structures that are highly mobile within the cytosol and subject to genetically programmed regulation of their morphology, also for overall maintenance of quality (29, 30). Among these, the processes of fission and fusion represent not only physiological mechanisms but also adaptive mechanisms in response to environmental stress.
The following sections provide an introduction to various mitochondria-associated processes that can impact human disease progression, including bioenergetics, mitochondrial metabolism, Ca2+ regulation, mitophagy, mitochondrial dynamics (i.e., fission, fusion), and mitochondrial biogenesis. This review aims to provide a background for researchers across multiple disciplines seeking to gain knowledge in mitochondrial processes. Subsequently, this review explores the roles of these processes in specific pathologies, which will serve as a reference for translational researchers focused on understanding the role of mitochondrial processes in the pathogenesis of specific diseases as well as their potential for development as therapeutic targets. The diseases that are considered are largely represented by cardiovascular, lung, infectious/inflammatory, and neurodegenerative diseases. The role of mitochondria in cancer progression has been reviewed elsewhere (12).
1.2. Morphology, Bioenergetics, and Metabolism
The recognition of mitochondria as distinct organelles dates to early light microscopic observations, including those of the Swiss physiologist Albert von Köliker (ca. 1857). After advances in histological staining, Richard Altmann (ca. 1886) coined the term “bioblasts” for filamentous structures he believed to be of prokaryotic origin (31, 32). His observations predated later hypotheses by evolutionary biologists that mitochondria may have arisen as an endosymbiotic fusion event between eubacteria and a primitive eukaryotic cell (33). The term “mitochondria,” which is derived from the Greek mitos, meaning thread, and chondros, meaning granule, was later introduced by biologist Carl Benda in 1898 during the study of spermatogenesis (34). The German biochemist Leonor Michaelis introduced the first mitochondria-specific vital stain, Janus Green-B, in 1900 (35). The first electron micrographs of mitochondria were published by Claude and Fullam in 1945 (36). Further ultrastructural analyses by George Palade (37) elucidated fine details of membrane structures, including the inner membrane cristae. The recognition of the functional role of mitochondria in energy production dates to 1946, coincident with the localization of cytochrome-c oxidase and succinate oxidase enzymes to mitochondrial particles (38).
In the modern view, mitochondria are distinct organelles that are encased by the outer mitochondrial membrane (OMM), which in turn defines an intermembrane space (IMS) and encloses a separate inner mitochondrial membrane (IMM). Invaginations of IMM, termed cristae, serve as host for the respiratory apparatus. An IMM protein complex, the mitochondrial contact and cristae organizing site (MICOS) is essential for mitochondrial ultrastructural integrity, as deletion of components of this complex ablates crista formation (39, 40).
The IMM encloses a central compartment termed the matrix. The cellular energy currency generated by mitochondria is manifest in the production of the high-energy phosphate carrier adenosine-5′-triphosphate (ATP), in a process called oxidative phosphorylation (OXPHOS). Tissues with high metabolic demand, such as skeletal muscle, have significantly more mitochondrial content to meet the energy requirements of the tissue.
During oxidative cellular metabolism (FIGURE 1), the oxidation of glucose during glycolysis culminates in the generation of pyruvate, which, upon import to the mitochondria via the pyruvate transporter, is converted to acetyl-CoA (AcCoA) that drives the Krebs cycle in the mitochondrial matrix. Nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) generated in the Krebs cycle serve as electron donors for mitochondrial OXPHOS. This process involves transfer of electrons across protein complexes that comprise the mitochondrial electron transport chain (ETC). The ETC consists of four complexes (Complexes 1–4) that facilitate electron transport to molecular oxygen (O2) (41): Complex 1 (CI: NADH dehydrogenase, NADH:ubiquinone oxidoreductase); Complex 2 (CII: succinate dehydrogenase), which also resides in the Krebs cycle and uses the Krebs cycle product succinate as substrate; Complex 3 [CIII: coenzyme Q (CoQ):cytochrome c oxidoreductase, cytochrome bc1); Complex 4 (CIV: cytochrome-c oxidase, cytochrome aa3]. These protein complexes include cofactors, heme and flavin, and interact with small molecular electron carriers: ubiquinone and cytochrome c (Cyt-c). Terminal fixation of oxygen by CIV activity converts O2 to water by a four-electron reduction process (1/2O2 + NADH + H+ → H2O + NAD+). Bulk electron flow through the ETC generates a proton efflux gradient across the IMM, which in turn drives ATP production via ATP synthase (FoF1-ATPase) (1, 19). A theoretical maximal yield of 36–38 ATP molecules is generated per mole of glucose oxidized, of which 34–36 molecules are attributed to OXPHOS and the balance to glycolysis and the Krebs cycle.
FIGURE 1.

Mitochondrial bioenergetics. A: normal mitochondria serve essential functions for the cell including ATP generation as a result of a functioning electron transport chain, calcium handling and metabolism regulated through the mitochondrial transition pore, energy generation through the oxidation of acetyl-CoA in the Krebs cycle, and maintenance of mitochondrial integrity through mitochondrial dynamics, including mitochondrial fission and fusion. B: loss of mitochondrial bioenergetics has been linked to human disease pathogenesis. Mitochondrial reactive oxygen species (mtROS) can induce cyclophilin D, leading to mitochondrial permeability transition pore-driven necrosis and loss of mitochondrial bioenergetics, a process previously linked to amyotrophic lateral sclerosis and Parkinson’s disease onset. Mitochondrial membrane depolarization or loss of membrane potential (−ΔΨm) can ultimately lead to mitochondrial membrane rupture with the release of mitochondrial DAMPs, with implications for sepsis and fibrotic lung disease. Failure of mitofission-related mitochondrial maintenance has been linked to Alzheimer’s disease and fibrotic lung disease, whereas failure to properly dispose of abnormal mitochondria through mitophagy has been linked to neurodegenerative diseases as well as chronic lung disease. Cell death processes, including intrinsic and extrinsic apoptosis and necroptosis, cross talk to mitochondrial energetics, with implications for human disease. Cell stress secondary to inflammatory signaling can increase mitochondrial bioenergetics, leading to mtROS-driven mitochondrial DNA release (mtDNA) that mediates inflammatory diseases including sepsis and fibrotic lung disease. See glossary for abbreviations. Image created with BioRender.com, with permission.
Besides pyruvate, mitochondria can also utilize other metabolic substrates. In the process of FA β-oxidation (FAO), long-chain FAs are activated in the cytosol to acyl-CoA derivatives, which are imported across both mitochondrial membranes into the mitochondrial matrix by carnitine acyltransferases. FAO is catalyzed at the IMM in a series of enzymatic steps initiated by acyl-CoA dehydrogenase. This process results in regeneration of AcCoA and generates FADH2 and NADH for electron transfer reactions (19). Glutamine, another alternate substrate, is imported to mitochondria and converted to glutamate by glutaminase and then to α-ketoglutarate by glutamate dehydrogenase and transaminases, which is then utilized in the Krebs cycle (1). During anaerobic glycolysis (Warburg effect), ATP is formed independently of the Krebs cycle, with the by-product of lactate generation. Current thinking suggests that lactate can be reconverted to metabolic substrates but, however, that mitochondria do not directly oxidize lactate (42).
In addition to their cardinal role in bioenergetics, mitochondria are active participants in other catabolic and anabolic pathways (43). During starvation, when availability of carbohydrates and insulin levels is low, increased mitochondrial β-oxidation in the liver generates ketone bodies as alternative fuel. In this pathway, AcCoA is converted to acetoacetate and β-hydroxybutyrate, which are then used to regenerate AcCoA to drive the Krebs cycle in extrahepatic tissues (44). Activation of the ketogenic pathway may promote mitochondrial homeostasis by activating the antioxidant response (44). Although FA synthesis via fatty acid synthase (FASN) is primarily a cytosolic process, mitochondria are also believed to harbor their own apparatus for FA synthesis (mtFAS), which generates lipoic acid and long acyl chains (45, 46). The products of this incompletely understood pathway may regulate ETC assembly and Krebs cycle enzyme activities (46, 47).
Mitochondrial oxidation of AAs takes place within the mitochondrial matrix. AAs are deaminated and then converted to various products including pyruvate, AcCoA, or Krebs cycle intermediates (19, 48). The branched-chain amino acids (BCAAs) (i.e., leucine, isoleucine, and valine) are essential for anabolic and catabolic processes. After import into the matrix, BCAAs are catabolized by branched-chain aminotransferases (BCATs), which include a mitochondrial isoform BCAT2, and then converted into acyl-CoA derivatives by branched-chain α-ketoacid dehydrogenase (BCKDH). BCAA can influence the regulation of bioenergetics, stimulate mitochondrial biogenesis (see sect. 1.7), activate the mammalian target of rapamycin (mTOR) pathway, potentially inhibiting autophagy, and alter insulin sensitivity (49). BCAA metabolism has been recently implicated in the pathogenesis of cardiovascular disease (50).
Mitochondria are also host to the one-carbon (1-C) pathway (folate metabolism), which maintains the homeostasis of select AAs (serine, glycine, methionine) and drives the synthesis of nucleotides (i.e., purines, pyrimidines) (43, 51). Furthermore, mitochondria maintain redox balance by regenerating the reduced forms of NADH/NADPH in the matrix from their oxidized forms by mitochondrial dehydrogenases (i.e., isocitrate, methylene tetrahydrofolate dehydrogenase, respectively) (43, 52). Finally, mitochondria are also involved in the biosynthesis of heme, which occurs via a sequence of enzymatic steps that begin in the mitochondria with the formation of δ-aminolevulinic acid (ALA) and end in mitochondria with the incorporation of iron into protoporphyrin-IX by ferrochelatase. Heme is a cofactor required for proteins involved in O2 transport and cellular metabolism, as well as mitochondrial respiratory chain cytochromes (53).
In conclusion, mitochondrial bioenergetics, and its deregulation, has been widely studied as an important contributor to human disease, such as neurodegenerative diseases (see sect. 6). The ETC is a major contributor of reactive oxygen species, which have been implicated in both physiological and pathophysiological processes (see sect. 1.4). Current challenges remain to define the cross talk between bioenergetics and regulation of other mitochondria-dependent processes, such as permeability transition, mitophagy, and regulation of dynamics (see sects. 1.3, 1.4, 1.6, and 1.7). As discussed in subsequent sections, aberrations in mitochondria-specific metabolic pathways such as AA catabolism, FA handling, and Ca2+ handling may influence specific disease pathogenesis. Global studies of mitochondria-dependent metabolic changes may ultimately lead to the identification of metabolic signatures of distinct diseases.
Finally, as discussed in sects. 2 and 3, mitochondria can influence cellular processes such as inflammation and cell death through the release of soluble mediators and damage-associated molecular patterns (DAMPs). For example, signaling pathways culminating in mitochondrial dysfunction, or disruption of endoplasmic reticulum (ER)-mitochondrial interactions, can trigger apoptosis, which is ultimately mediated by the release of the ETC component Cyt-c (11, 54).
1.3. Mitochondrial Ca2+ Regulation
Mitochondria exert a crucial cellular homeostatic function by buffering intracellular Ca2+ levels through the uptake, storage, and release of Ca2+. Within mitochondria, Ca2+ is utilized for energy production and metabolism and can activate enzymes of the Krebs cycle [i.e., pyruvate dehydrogenase (PDH), isocitrate dehydrogenase, α-ketoglutarate dehydrogenase] as well as activate ATP synthase (29, 55–57). Relationships between changes in matrix Ca2+ and increases in ATP production have been shown in isolated skeletal mitochondria (58).
The OMM is permeable to Ca2+ uptake from the cytosol, which is mediated by regulation of voltage-dependent anion channels (VDACs) (59, 60). Ca2+ ions traverse the IMM into the matrix via the mitochondrial Ca2+ uniporter complex (MCUC). This complex consists of pore-forming units, MCU, its dominant-negative form, MCUb, and additional regulatory subunits: mitochondrial calcium uptake-1, -2, and -3 (MICU1, MICU2, MICU3), the essential MCU regulator (EMRE), and the mitochondrial Ca2+ uniporter regulator 1 (MCUR1) (55, 61–63). Regulated efflux of Ca2+ from the matrix is facilitated by the Na+/Ca2+ exchanger (NCLX) and the H+/Ca2+ antiporter (64).
Under conditions of excessive Ca2+ uptake, mitochondrial Ca2+ triggers apoptosis by opening the mitochondrial permeability transition pore (mPTP), which is implicated in cell death regulation (65, 66). The mPTP is a transmembrane protein complex residing in the IMM that, when opened, allows bulk efflux of particles < 1.5 kDa, including protons and Ca2+, from the mitochondrial matrix (67–69). The structural components of the mPTP remain incompletely delineated but historically include VDAC, adenine nucleotide transporter (ANT), and cyclophilin D (CypD). However, recent studies suggest that the Ca2+-bound form of the F1Fo-ATP synthase may constitute a newly recognized major regulator of mPTP (70). mPTP opening can be stimulated by several factors, including reduction of adenine nucleotide pools, phosphate accumulation, accumulation of matrix Ca2+, and mtROS production (see sect. 1.4). CypD regulates pore opening via intrinsic peptidyl-prolyl cis-trans isomerase activity, whereas cyclosporin-A (CsA) acts as an inhibitor of CypD and mPTP opening (71). Transient openings of the mPTP may act as a physiological mechanism to release ions in a controlled manner, whereas sustained high-conductance opening of the mPTP is associated with mitochondrial dysfunction, collapse of membrane potential (see sect. 1.4), and triggering of regulated cell death (RCD) pathways, including necrosis and apoptosis (see sect. 3) (67).
Both mitochondria and endoplasmic reticulum (ER) represent major stores of intracellular Ca2+. Mitochondria interact with the ER through distinct contact points termed mitochondria-associated ER membranes (MAMs) and are essential for Ca2+ uptake and regulation. The MAM is a subdomain of the ER that connects with the OMM through tethering complexes, such as the complex between vesicle-associated membrane protein-associated protein B (VAPB) on the ER and protein tyrosine phosphatase interacting protein 51 (PTPIP51) on the mitochondria (i.e., VAPB-PTPIP51); the inositol triphosphate receptor (IP3R)/glucose-regulated protein 75 (Grp75)/voltage-dependent anion (VDAC1) and Parkinson protein DJ1 complex; mitofusin-2 (Mfn2) on the ER with Mfn1/Mfn2 on the OMM; Rho GTPases Miro1 and Miro2; and others (72–76). The MAMs are important in the regulation of mitochondrial dynamics (see sect. 1.7) and are also essential for mitochondrial Ca2+ uptake via the IP3R/Grp75/DJ1/VDAC1 complex. PDZ domain-containing protein 8 (PDZD8) has been identified as another tethering complex relevant for mitochondrial Ca2+ uptake in neurons (77). The disruption of MAMs may drive pathological processes. For example, excess Ca2+ transfer from ER to mitochondria has been linked to proinflammatory processes including mtROS generation and inflammasome activation (see sect. 2) and regulation of cell death (see sect. 3) (78).
In conclusion, Ca2+ dysregulation, including ER MAM disruption and the accumulation of matrix Ca2+, is associated with mtROS generation and mPTP opening. This leads to increased inflammation and cell death, which may play an important role in pathological conditions such as ischemia-reperfusion injury (IRI) and in the pathogenesis of various diseases, in particular cardiovascular diseases and neurodegenerative disorders (see sect. 6) (67). Current challenges include deducing the molecular structures as well as the regulation of mPTP, MCU, and MAMs. Progress in these areas will facilitate future design of therapeutics targeting mitochondrial Ca2+ regulation, for which there is a current deficiency (67).
1.4. Mitochondrial Dysfunction, Membrane Depolarization, and Reactive Oxygen Species Formation
Acute cellular injury may result in loss or decline of key mitochondrial functions. The term “mitochondrial dysfunction” is associated with impairment of bioenergetics and loss of ATP production (79). Mitochondrial dysfunction may also encompass dysregulation of Ca2+ homeostasis, mitochondrial membrane depolarization, or loss of membrane potential (−ΔΨm), and aberrant production of reactive oxygen species (ROS) (79).
Mitochondrial membrane potential (ΔΨm) is a primary indicator of mitochondrial function. ΔΨm and the proton gradient, ΔpH, are generated by proton efflux from Complexes I, III, and IV and used to generate ATP (80). The absolute reported value of the electrochemical membrane potential across the IMM has been estimated at −180 mV for isolated respiring mitochondria (81). ΔΨm measurements vary between cell types and display considerable intracellular heterogeneity (82, 83). For example, in HepG2 cells a mean value of −131.33 ± 10.37 mV was reported for ΔΨm, with an intracellular heterogeneity of 20.73 ± 3.33 mV (82), whereas in rat cortical neurons a resting value of −139 mV was reported, which ranged between −108 and −158 mV with stimulation (84). ΔΨm is stable in resting cells, whereas both increases (hyperpolarization) and decreases (depolarization) may be associated with pathological conditions (80). Collapse in ΔΨm can be elicited by mPTP opening and Ca2+ release (80). Mitochondria are depolarized by exposure to exogenous oxidants including tert-butyl-hydroperoxide (t-BuOOH) or OXPHOS uncoupling agents, such as carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (80, 85), trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP), 2,4-dinitrophenol (DNP), and others (86).
In isolated rat liver mitochondria, pro-oxidants such as t-BuOOH and diamide induced mitochondrial membrane permeability transition (MPT) resulting in Ca2+ efflux, collapse of ΔΨm, and mitochondrial swelling (87). The mPTP inhibitor cyclosporin-A (CsA) reduced mitochondrial swelling but not oxidant-induced collapse of ΔΨm (87). In this study, oxidant-induced −ΔΨm was linked to redox regulation of a CsA-insensitive low-conductance Ca2+ channel (87). Increases in ΔΨm in nonstressed neural cells were elicited by the MPT inhibitor CsA or by the Ca2+ chelator BAPTA and were also observed in PC12 cells overexpressing antiapoptotic protein Bcl-2 (88). In neural cells, however, CsA inhibited loss of ΔΨm induced by t-BuOOH (88).
mtROS represent a major component of total cellular ROS and are initially represented by the production of superoxide anion (O2•−), a free radical generated by univalent reduction of O2 (89). O2•− undergoes spontaneous or enzymatic dismutation to produce the oxidant hydrogen peroxide (H2O2), which diffuses across cellular membranes and may affect the redox state of proteins at distal sites (89).
The generation of mtROS is site specific and variable with stimuli. Substrate-derived electrons can reduce O2 to O2•− at eight mitochondrial sites, primarily represented by CI and CIII (90). In isolated mitochondria, O2•− produced at CI by forward electron transfer is enhanced by the CI inhibitor rotenone and can increase in mitochondria with reduced energy production, high ΔpH, higher ratio of reduced to oxidized CoQ, or high NADH-to-NAD+ ratio (90). O2•− production at CI leaks into the matrix. O2•− generated at CIII may originate at the ubiquinol oxidation site (center P, Qo site), particularly in response to hypoxia, and can enter the IMS (91, 92).
Significant mtROS generation at CI may also be achieved by mitochondrial reverse electron transport (RET) (93). In RET, electrons from ubiquinol are transported in reverse direction to CI, which results in reduction of NAD+ to generate NADH. RET is driven by succinate accumulation and hyperreduction of CII and may also respond to other substrates such as FAs (93, 94). Studies on IRI have suggested potential physiological relevance of succinate-driven RET in mtROS generation leading to cell death in vivo (95).
The relationship between mtROS production, generated by electron leakage from the respiratory chain, and ΔΨm, remains incompletely understood. mtROS production may be initiated during mitochondrial membrane hyperpolarization. Excessive ROS increases may coincide with mPTP openings and lead to collapse of ΔΨm (96). A catalytic cycle of amplification of ROS production triggered by mtROS has been termed “ROS-induced ROS release” (RIRR) and may lead to maladaptive outcomes (96).
Primary generation of mtROS may lead to secondary generation of other reactive oxygen or nitrogen species (ROS/RNS). Metal catalysis of H2O2 generates the highly reactive hydroxyl radical (•OH). This species can abstract electrons from organic substrates such as unsaturated FAs, leading to the generation of toxic metabolites such as malondialdehyde and 4-hydroxynonenal. O2•− can also recombine with nitric oxide (NO) to form peroxynitrite (ONOO−). Cellular ROS can also arise from enzymatic activities, namely NADPH:oxidases (NOX), which generate O2•− at the expense of reducing equivalents from NADPH. These are typically associated with phagocyte production of ROS for bactericidal purposes.
Among these, the NOX4 isoform has been characterized as localizing to mitochondria in cardiomyocytes and myofibroblasts and may contribute to cardiac injury (97–99). Cardiac-specific NOX4-knockout mice displayed reduced cardiomyocyte O2•− production, mitochondrial dysfunction, and morphological aberrations in response to cardiac pressure overload (99). Additionally, these mice displayed reduced cardiac hypertrophy, fibrosis, and apoptosis and improved cardiac function (99). NOX4 also localizes to other cellular membranes such as ER and plasma membrane in a cell type-specific manner (100).
Eukaryotic cells have evolved with enzymatic and chemical systems for mitigating the excess production of cellular and mtROS. These include superoxide dismutases (SODs), which catalyze the dismutation of O2•−to generate H2O2. SOD1 (Cu-Zn) localizes to the cytosol and IMS, SOD2 (Mn) localizes to the matrix, and SOD3 (Cu-Zn) localizes to the extracellular space (91). In mammalian cells, mtROS levels are buffered against the cellular reducing potential, determined by the ratios of reduced/oxidized forms of glutathione (GSH/GSSG), thioredoxin-1 and -2 (TXN1, TXN2), and nicotinamide adenine dinucleotide (phosphate) NAD(P)H/NAD(P)+. NADPH from the pentose phosphate pathway is used to regenerate cytosolic or mitochondrial GSH (mGSH) via glutathione reductase and to regenerate TXN1 or TXN2, in the cytosol and mitochondrial matrix, respectively, via respective thioredoxin reductases (101). H2O2 is detoxified by catalase in peroxisomes, whereas H2O2 in the cytosol and mitochondrial matrix is metabolized by GSH peroxidases (GPXs) and peroxiredoxins (Prxs), which are maintained by GSH and TXN, respectively (101).
mGSH serves several important functions in the mitochondria, in conjunction with mitochondrial thioredoxin. These functions include general antioxidant protection, redox buffering of free thiol groups as found in critical metabolic and respiratory chain enzymes, and maintenance and activation of iron-sulfur clusters (102). GSH is synthesized in the cytoplasm, whereas mGSH originates from the cytoplasmic GSH pool and is imported to the mitochondria from the cytosol. Recent studies identify SLC25A39 as an active transporter of GSH into the mitochondria (103, 104). Deficiency in mGSH may be associated with destabilization of iron-sulfur clusters, impaired ETC activity, increased mtROS burden, and inhibition of cell proliferation and may also promote regulated cell death (103).
Excessive ROS production that supersedes cellular reducing capacity and antioxidant defenses may cause cellular injury via random oxidation of lipids, DNA, and proteins, which collectively are believed to contribute to the aging process. Production of mtROS may lead to the regulation of downstream cellular functions, including inflammation (see sect. 2) and apoptosis (see sect. 3). For example, mtROS generated at CIII stabilize hypoxia-inducible factor-1α (HIF-1α), leading to increases in HIF-1-mediated signaling (51, 91, 105, 106). mtROS signaling is also implicated in the upstream regulation of the cellular autophagy program, a homeostatic adaptive response, whereas collapse of ΔΨm is considered an initiating signal for mitophagy (see sect. 1.6) (85, 91).
In conclusion, the mechanisms that regulate ΔΨm and mtROS production and their relationships in various pathological conditions remain incompletely understood. Future progress will depend on enhanced methods to deduce these relationships in vivo. Since mtROS generation can occur at multiple sites, whose regulation and functional significance may differ in a cell type- and inducer-specific fashion, the site-specific modulation of mtROS production by selective modulators may represent a future approach to therapeutics development.
1.5. Regulation of Mitochondrial Processes by Sirtuins
Energy-consuming cellular activities, including inflammation, cause considerable mitochondrial stress leading to energy consumption, oxidative stress, ROS generation, and cell death. Sirtuins are intracellular enzymes that exert their prosurvival effects through posttranslational mechanisms. Sirtuins are a group of histone deacetylases dependent on NAD+ to regulate cellular life span, inflammation, glucose homeostasis, and age-related disease such as cancer. The requirement of NAD+ as a cofactor and the mitochondrial localization of select sirtuins (Sirt3, Sirt5, Sirt7) imply an important role for sirtuins in regulating cellular energy consumption. First discovered in yeast, silent information regulator 2 (Sir2) is a nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylase that controls longevity in lower eukaryotes (107). In multicellular organisms, sirtuins act through chromatin dynamics (histone deacetylases) and transcriptional regulation to fine-tune expression of metabolic and cell stress proteins. For this review, we focus on the role of sirtuins in mitochondrial processes including inflammation (sect. 2) as well as the role of sirtuins in modulating mitochondria-driven cell death responses (sect. 3). The impact of sirtuins in disease pathogenesis is discussed in sect. 4. The role of sirtuins in glucose homeostasis and age-related diseases such as cancer has been reviewed elsewhere (108, 109).
The deacetylase activity of sirtuins is NAD+ dependent. Sirtuin uses NAD+ as a catalyst to transfer the acetyl group from proteins and peptides to the 2′-OH of nicotinamide ribose, yielding 2′-O-acetyl-ADP-ribose. The nicotinamide ribosyl bond is cleaved, and one net water molecule is added to nicotinamide ribose (110). The sirtuin family of histone deacetylases (HDACs) was named after their homology to Sir2, and they comprise group III of HDACs that are NAD+ dependent (107, 111, 112). There are seven human proteins homologous to Sir2 (SIRT1 through SIRT7). These proteins have a highly conserved NAD-dependent sirtuin core domain, have diverse cellular locations, target multiple substrates, and affect a broad range of cellular functions (113). SIRT1, SIRT6, and SIRT7 are localized to the nucleus. Only SIRT2 is predominantly cytoplasmic, and SIRT1 is reported to be cytoplasmic in certain types of cells. SIRT3–5 primarily reside in mitochondria (112). Deacetylation and ADP-ribosylation reactions by sirtuins are similar, and thus sirtuins posttranslationally regulate various biological processes (110, 113–115). SIRT4 is mostly involved in glucose metabolism, and SIRT6 and SIRT7 are less well characterized; therefore, these three sirtuins are not discussed here.
The NAD dependence of sirtuins links them to the metabolic activity of cells. Sirtuins interact with metabolic enzymes and transcription factors to influence bioenergetic pathways and meet the cellular energy demand following sensing alterations in cellular NAD+ levels (115). Sirtuin 1 (SIRT1) is by far the most-studied member of the Sirtuin family, and although it is not physically associated with mitochondria, it impacts mitochondrial oxidative stress. Changes in the NAD+-to-NADH ratio or increased ROS production can enhance SIRT1 activity. In response to high intracellular ROS, SIRT1 increases FOXO3a-mediated increase in the antioxidant enzyme catalase, whose increased activity metabolizes H2O2, leading to reduction in oxygen consumption and decreased ROS generation (116). Similarly, SIRT1 can alter the transcriptional activity of the mitochondrial biogenesis coactivator PGC-1α, reducing oxygen consumption by up to 25% in SIRT1-overexpressing cell lines (117).
Sirtuin 2 (SIRT2) can similarly alter metabolic mitochondrial proteins in response to changes in the cellular energetic state. SIRT2 is the primary cytoplasmic sirtuin, although it has also been found in the nucleus, and its function has only recently been related to mitochondria. SIRT2 expression is detected in a wide range of mouse tissues but particularly in metabolically relevant organs, such as muscle, liver, testes, pancreas, kidney, and adipose tissue; however, the highest expression is found in the brain (118, 119). SIRT2 was found to associate with the IMM in central nervous system (CNS) cells (120). The acetylation of several metabolic mitochondrial proteins was altered in Sirt2-deficient mice, and the loss of Sirt2 increased oxidative stress, decreased ATP levels, and altered mitochondrial morphology (120). SIRT2 expression is also regulated in response to changes in cellular energetic state (121) and may participate in the regulation of cellular antioxidant defenses (122).
SIRT2 protects against ROS by activating FOXO3a, leading to increased expression of the antioxidant protein SOD2, therefore decreasing ROS levels (122–122).
Sirtuins can also directly regulate mitochondrial processes. Sirtuin 3 (SIRT3) was the first sirtuin found to be localized to the mitochondrial matrix (123, 124). SIRT3 appears to regulate mitochondrial functions, as its overexpression increases respiration and decreases ROS production (125). Its deacetylase activity is reported to be required for the induction of uncoupling protein-1 (UCP1) (125). SIRT3 may regulate the activity of acetyl-CoA synthetase (AceCS). AceCS uses acetate, CoA, and ATP to form acetyl-CoA, which is an intermediate in the Krebs cycle and is required for cholesterol and FA synthesis. Acetylation of mitochondrial AceCS (AceCS2) inactivates the enzyme, whereas deacetylation by SIRT3 activates it. SIRT1 can deacetylate and activate the cytosolic form of AceCS (AceCS1). These data suggest that SIRT3 may play a role in regulating the entry of carbons from acetate into central metabolism. SIRT3 may be especially important under conditions of energy limitation (i.e., during fasting or caloric restriction) to ensure full incorporation of dietary or ketone-derived acetate into metabolism (126). SIRT3 can also boost the enzymatic activity of glutamate dehydrogenase (GDH) via deacetylation and thereby may contribute to enhanced glucose synthesis from AAs (126, 127).
Sirtuin 5 (SIRT5) has emerged as a central regulator of cellular energy metabolism through direct effects on the mitochondria (128). Unlike SIRT1 and SIRT3, SIRT5 displays strong lysine-desuccinylase/demalonylase/deglutarylase activity, with only very weak lysine-deacetylase activity (128–131). SIRT5 is located in both the mitochondria, where it plays a central role in mitochondrial processes, and additionally in the cytosol, where it elevates glycolysis (132–134). SIRT5 can directly interact with cardiolipin (CL) and may localize to the CL-rich domains on the IMM that are critical for mitochondrial functionality (135). SIRT5 can desuccinylate multiple subunits of all four respiratory chain complexes and ATP synthase (135). Previous studies have identified SIRT5 as a positive regulator of CII and a promoter of mitochondrial energy metabolism (135, 136). SIRT5 can promote glycolytic flux through demalonylation of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and other glycolytic enzymes and desuccinylation of pyruvate kinase M2 (PKM2) (134, 137). SIRT5 also suppresses pyruvate dehydrogenase complex (PDC), a regulator of acetyl-CoA production and Krebs cycle activity, in effect decreasing energy production of the cell (138–140). On the other hand, overexpression of SIRT5 enhanced ATP synthesis and O2 consumption in HepG2 cells, highlighting the possible cell type-specific function of SIRT5 (141). SIRT5 also promotes ROS detoxification. SIRT5 binds to, desuccinylates, and activates SOD1 (142). As a result, SOD1-mediated ROS detoxification is significantly increased when SOD1 is co-overexpressed with SIRT5 (142). SIRT5 desuccinylates and deglutarylates isocitrate dehydrogenase 2 (IDH2) and glucose-6-phosphate dehydrogenase (G6PD), respectively, to activate these proteins (143). Since both IDH2 and G6PD are major NADPH-producing enzymes, SIRT5 plays a key role in promoting NADPH production and attenuating cellular ROS levels.
In summary, sirtuins are key enzymatic regulators of mitochondrial processes, whether indirectly through their regulation of antioxidant molecules (e.g., SIRT1 and catalase) or through ROS-countering effects (SIRT1, SIRT2, SIRT3, and SIRT5) or directly through the promotion of more efficient energy generation (SIRT3 and SIRT5). Sirtuins have received extensive interest as potential therapeutics and remain an attractive target for investigators seeking to develop drugs to counter human diseases driven by mitochondrial dysfunction, inflammation, and aging.
1.6. Mitophagy and Mitochondrial Quality Control
Mitophagy constitutes a key cellular pathway in an aggregate of mitochondrial quality control (MQC) mechanisms (FIGURE 2), which collectively include biogenesis and dynamics as discussed below. The mitophagy pathway, a form of selective autophagy, functions to maintain cellular homeostasis by ensuring the health of the mitochondrial population, which is achieved through the targeting and turnover (degradation) of depolarized or dysfunctional mitochondria via the autophagy-lysosomal degradation pathway. In this pathway, mitochondria are selectively assimilated into double-membraned autophagosomes. Mitophagy is subsequently completed via fusion of autophagosomes to acidic lysosomes, where the mitochondria cargo is degraded (28). This process is believed to preclude the excessive release of mtROS and DAMPs from injured mitochondria (27, 28). In general, the autophagy program is regulated by the products of autophagy-related genes (ATGs), which include key homologs of ATG8 (i.e., microtubule-associated protein-1 light chain 3B, LC3B), ATG6 (Beclin 1), and ATG1 (ULK1) (144).
FIGURE 2.
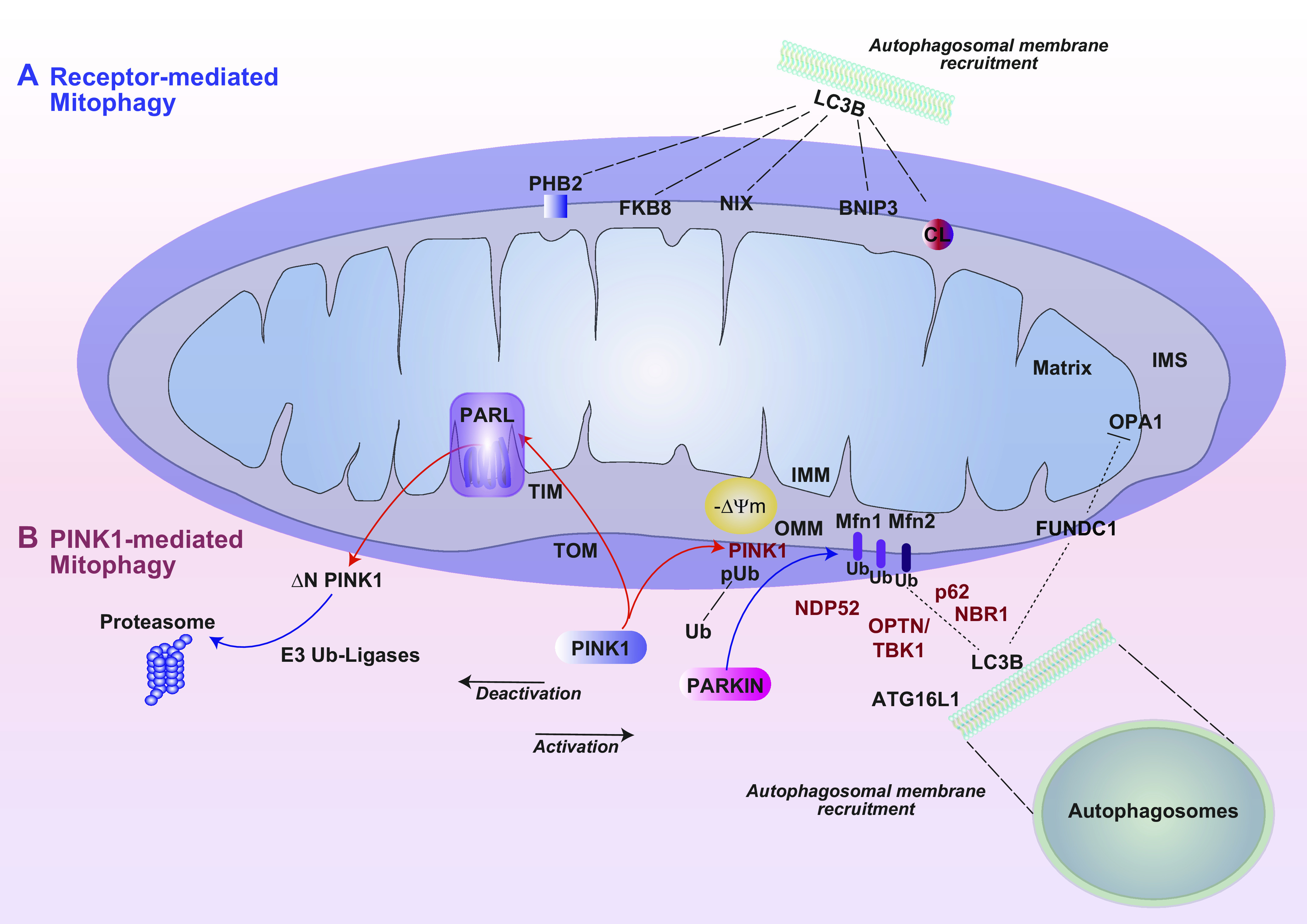
Mitophagy. Mitophagy is a selective process for the turnover of mitochondria by the lysosome-dependent autophagy pathway. Distinct modes of mitophagy include receptor-dependent mitophagy (A) and PINK1/Parkin-dependent mitophagy (B), the primary pathway by which dysfunctional or depolarizing mitochondria are cleared. Under resting conditions PINK1 is imported into the mitochondria via TIM and TOM and processed by the IMM rhomboid protease PARL. A truncated form of PINK1 (ΔPINK1) is exported from the mitochondria and processed for proteasomal degradation via E3 ubiquitin ligases. In response to mitochondrial depolarization, PINK1 is stabilized on the OMM, phosphorylates ubiquitin (UB), and recruits Parkin to the mitochondria. Parkin, an E3 ubiquitin ligase, ubiquitinates OMM proteins, including Mfn1/Mfn2 and others. Ubiquitinated OMM proteins interact with mitophagy cargo adaptors, which, through LIR domains, interact with LC3 on nascent autophagosome membranes. This process facilitates recruitment of additional autophagy initiating factors, including Atg16L, and others. Mitophagy cargo adaptors include NDP52, p62 (SQSTM1), NBR1, and OPTN activated by TBK1. Receptor-dependent mitophagy (A) is important for physiological regulation of mitochondrial populations and utilizes OMM receptor proteins that interact directly with autophagosomal membrane protein LC3. These include BNIP3, Nix1, the membrane lipid cardiolipin (CL), and others (i.e., PHB2, FKB8). FUNDC1 is a mitophagy receptor that is specifically activated by hypoxia and that can also inhibit fusion via interaction with Opa1. Targeting of mitochondria to autophagosome membranes ultimately results in their assimilation in mature autophagosomes and degradation in the lysosomal compartment. See glossary for abbreviations.
Like other mitochondrial processes, mitophagy has the potential to impact the pathogenesis of human diseases (145, 146). Mitophagy is triggered by stress stimuli, including hypoxia, radiation, exposure to oxidants, and other insults that may injure the mitochondria. Ser/Thr kinase phosphatase and tensin homolog (PTEN)-inducible kinase-1 (PINK1) forms a 700-kDa complex with the translocase of the outer membrane (TOM) (147). In fully polarized mitochondria TOM facilitates the mitochondrial import of PINK1 across the IMM and its transport to the translocase of the inner mitochondrial membrane (TIM). PINK1 is cleaved to an NH2-terminal-truncated 53-kDa protein (DN-PINK1) by the mitochondrial protease presenilin-associated rhomboid-like protein (PARL), which thereby serves as a critical regulator of mitophagy (148, 149). DN-PINK1 was not detected in Parl−/− MEFs (150). The PINK1 fragment is in turn retrotranslocated to the cytosol, processed by E3 ubiquitin ligases (i.e., UBR1, UBR2, and UBR4) via the arginine N-end rule degradative (Arg-N-degron) pathway, and ultimately degraded by the ubiquitin proteasome system (UPS) (151, 152). This degradative system ensures that PINK1 is not available to initiate mitophagy of healthy mitochondria. On depolarizing mitochondria, the translocase system is inhibited and PINK1 is retained on the OMM in a complex with TOM proteins (147).
Mitophagy is activated when PARL undergoes autocatalytic β-cleavage in response to mitochondrial ATP depletion, to generate PACT, which is less efficient at processing PINK1 (150). Recent studies suggest that this process may be under metabolic regulation by pyruvate dehydrogenase kinase 2 (PDK2), which phosphorylates pyruvate dehydrogenase in the matrix, thereby acting as an inhibitor of the Krebs cycle and AcCoA formation. PDK2 was shown to phosphorylate PARL and inhibit β-cleavage, thus acting as a negative regulator of the mitophagy pathway. Conversely, inhibition of PDK2 with its chemical inhibitor dichloroacetate was shown to promote β-cleavage of PARL and activate mitophagy. These associations were validated by gain- and loss-of-function experiments: overexpression of PDK2 inhibited PARL β-cleavage in HEK293 cells, whereas knockdown of PDK2 increased β-cleavage in PARL-overexpressing HEK293 cells. These events identify a mitophagy switch that is sensitive to energy production (150).
An alternate pathway for PINK1 degradation involves its accumulation at ER contact sites and its degradation by an ER-associated degradation pathway. PINK1 may be ubiquitinated by the E3 ligases gp78 and HRD1 at the ER-mitochondria interface. Ubiquitinated PINK1 forms a complex with valosin-containing protein and its adaptor UFD1, which facilitate its proteasomal degradation (153).
PINK1 catalyzes the phosphorylation of ubiquitin (Ub) to activate ubiquitin for the mitophagy process (154–156). On depolarizing mitochondria, stabilized PINK1 phosphorylates the ubiquitin E3 ligase Parkin (PRKN) at the ubiquitin-like domain (UBL), leading to its recruitment to the mitochondria (157, 158). Parkin in turn ubiquitinates OMM proteins including mitofusin-1 and -2 (Mfn-1, Mfn-2) and other targets with phosphorylated serine 65 Ub (pS65-Ub) (159, 160). Parkin-mediated ubiquitination and degradation of mitofusins serves as a mechanism to promote mitophagy in part through inhibition of mitochondrial fusion (161).
In Pink1/Parkin-dependent mitophagy, ubiquitinated mitochondria/OMM proteins may serve as binding targets for autophagic cargo adaptors that contain intrinsic LC3 interactive regions (LIRs). p62SQSTM1, a general cargo adaptor protein, was originally proposed as a cargo adaptor in Pink/Parkin-mediated mitophagy. Later studies implicated that the adaptor p62 is important in mitochondrial clustering but dispensable for mitophagy (162). Among the known cargo adaptors for mitophagy are nuclear dot protein 52 kDa (NDP52) and optineurin (OPTN). NDP52 and OPTN may be recruited to the mitochondria by PINK1 kinase activity and recognize ubiquitinated mitochondria via intrinsic ubiquitin binding regions. However, recruitment of these factors to depolarizing mitochondria was not entirely dependent on Parkin-dependent ubiquitination of mitochondria in some experimental models (163, 164). Furthermore, recruitment of mitophagy adaptors to ubiquitinated mitochondria occurred in parallel with activation of TBK1 kinase, which associates with OPTN, NDP52, and p62SQSTM1 (165). TBK1 phosphorylation of OPTN at S473 and S513 promotes OPTN mitochondrial retention and activation of mitophagy (165). NDP52 and OPTN may interact with LC3B by cargo adaptor complexes that in turn recruit autophagosome membranes, possibly originating in the ER, to the mitochondria, facilitating the assimilation of mitochondria into nascent autophagosomes. Mitochondria-localized NDP52 and OPTN recruit the autophagy-related factors ULK1, DFCP1, and WIPI1 to the proximity of the depolarizing mitochondria, leading to downstream recruitment of Atg16L1 and LC3 for autophagosome formation (146, 163, 166, 167).
Analyses of the phenotypes of Pink1- or Parkin-deficient mice (Pink-1−/−, Prkn−/−) have provided insight into the physiological roles of these proteins, in particular in the nervous system. Although overall neural histology was reported unchanged, Pink-1−/− mice displayed defects in dopamine release in the striatum and impairments of corticostriatal long-term potentiation and long-term depression, which could be remediated by dopaminergic agonists (168). Pink-1−/− mice also displayed early-onset motor defects, including impaired limb motor skills and turning skills (169). In Pink-1−/− cortical neurons, a phenotype of loss, fragmentation, and altered mitochondrial trafficking was observed (170). Pink-1−/− mice were also found to display an age-dependent loss of mitochondrial function, including impaired bioenergetics and susceptibility to stress stimuli in neurons (171). Both Pink-1−/− mice and Prkn−/− mice displayed increased inflammation and expression of proinflammatory cytokines in response to excessive exercise (172). Prkn−/− (mutator) mice displayed a proinflammatory phenotype accompanied by increased incidence of mtDNA mutation (172). These proinflammatory phenotypes of respective knockout mice were found to depend on the stimulator of interferon response cGAMP interactor 1 (STING) pathway (172). However, Pink-1−/− and Prkn−/− mice do not fully recapitulate neurodegenerative symptoms of Parkinson’s disease (168, 171, 173).
Although PINK1/Parkin-mediated mitophagy provides a mechanism for the degradation of injured mitochondria, alternative mitophagy pathways can regulate mitochondrial number to match metabolic demand. A ubiquitin-independent receptor-dependent form of mitophagy operates the turnover of mitochondria in erythrocytes and reticulocytes (174, 175). In maturing erythrocytes, mitophagy may be initiated by the BH3-only protein Nix (i.e., Bnip3L). Nix localizes to the OMM and directly interacts with mammalian Atg8 homologs (i.e., LC3A, LC3B, GABARAP) through its LIR (176), although LC3-independent pathways have also been proposed. Removal of mitochondria by Nix-dependent mitophagy serves a required physiological function in reticulocyte differentiation (175).
Fun14 domain-containing protein 1 (FUNDC1) is another mitophagy receptor that has been identified in mammalian cells (177). The role of FUNDC1 in mitophagy regulation is specific to the hypoxia response (178). FUNDC1 is located on the OMM and recruits LC3 for autophagosome formation. FUNDC1 also accumulates at the ER MAM in association with calnexin. During mitophagy, attenuation of the FUNDC1-calnexin interaction association promotes dynamin-related protein-1 (Drp1) recruitment to the MAM and initiates mitochondrial fission (177).
Phosphoglycerate mutase family member 5 (PGAM5), a mitochondrially localized Ser/Thr protein phosphatase, regulates FUNDC1-dependent mitophagy. Under conditions of mitochondrial membrane depolarization, where PINK1 is stabilized on mitochondria, PGAM5 is differentially cleaved by the mitochondrial protease PARL during mitochondrial depolarization (179). The PGAM5 truncation product dephosphorylates FUNDC1 at Ser 13 to activate mitophagy in response to mitochondrial depolarization (180, 181). The dephospho-form of FUNDC1 then recruits LC3 to initiate autophagosome formation (180). BCL2L1/Bcl-XL inhibits FUNDC1-dependent mitophagy via its intrinsic BH3 domain. BCL2L1 interacts with and inhibits PGAM5, thus inhibiting the dephosphorylation of FUNDC1 at Ser 13 (182). The PGAM5-FUNDC1 axis is further regulated by the mitochondrial protein syntaxin 17 (Stx17) (183).
An IMM mitophagy receptor, prohibitin 2 (PHB2), was also identified that promotes Pink1/Parkin-dependent mitophagy. PHB2 depletion activates PARL-dependent mitophagy inhibition. PGAM5 can also promote PHB2-dependent mitophagy (184). Other putative regulators of mitophagy include SMURF1, an alternate ubiquitin ligase that may substitute for Parkin (185). High-mobility group box 1 (HMGB1) acting on heat shock protein-β1 was proposed as an independent trigger of mitophagy (186). Taken together, these observations suggest that mitophagy is a complex and tightly regulated process that is orchestrated via partially understood mechanisms, whose regulation may vary in a stimulus- or context-dependent fashion. In subsequent sections, the role of mitophagy is explored in the pathogenesis of specific diseases including pulmonary conditions (see sect. 4) and neurodegenerative disorders (see sect. 6). A thorough understanding of the mechanisms that regulate mitophagy in various pathophysiological contexts will determine whether this pathway can be effectively targeted for therapeutic gain without side effects.
1.7. Mitochondrial Dynamics
Mitochondria are dynamic organelles that are subject to genetically regulated programs for modification of their morphology, size, and distribution. Mitochondria are subject to spatial rearrangement and thus are actively trafficked through the cytosol on dynein and kinesin networks. The remodeling of mitochondrial size and shape into larger extended or smaller structures is referred to as “mitochondrial dynamics,” which includes the processes of fission and fusion (FIGURE 3). The balance between fission and fusion serves to maintain a healthy population of mitochondria (29). Both processes serve to dilute dysfunctional mitochondria. For example, fission subdivides mitochondria to facilitate turnover by mitophagy (29, 30). In contrast, fusion creates larger, healthier mitochondria by recombining dysfunctional mitochondria to diminish the impact of damaged regions. Mitochondrial dynamics are regulated by proteins of the dynamin family of GTPases (29, 187). Regulators of mitochondrial dynamics may also communicate with other mitochondrial processes, such as mitochondrial biogenesis and mitophagy. Other aspects of mitochondrial spatial regulation include mitochondrial motility and interactions with other organelles, specifically the ER at MAM sites, the lysosomes, and the actin cytoskeleton. Mitochondrial dynamics, including the balance between fission and fusion events, have emerging roles in influencing or altering the course of disease, which are discussed below for their relevance to specific diseases (187–192).
FIGURE 3.
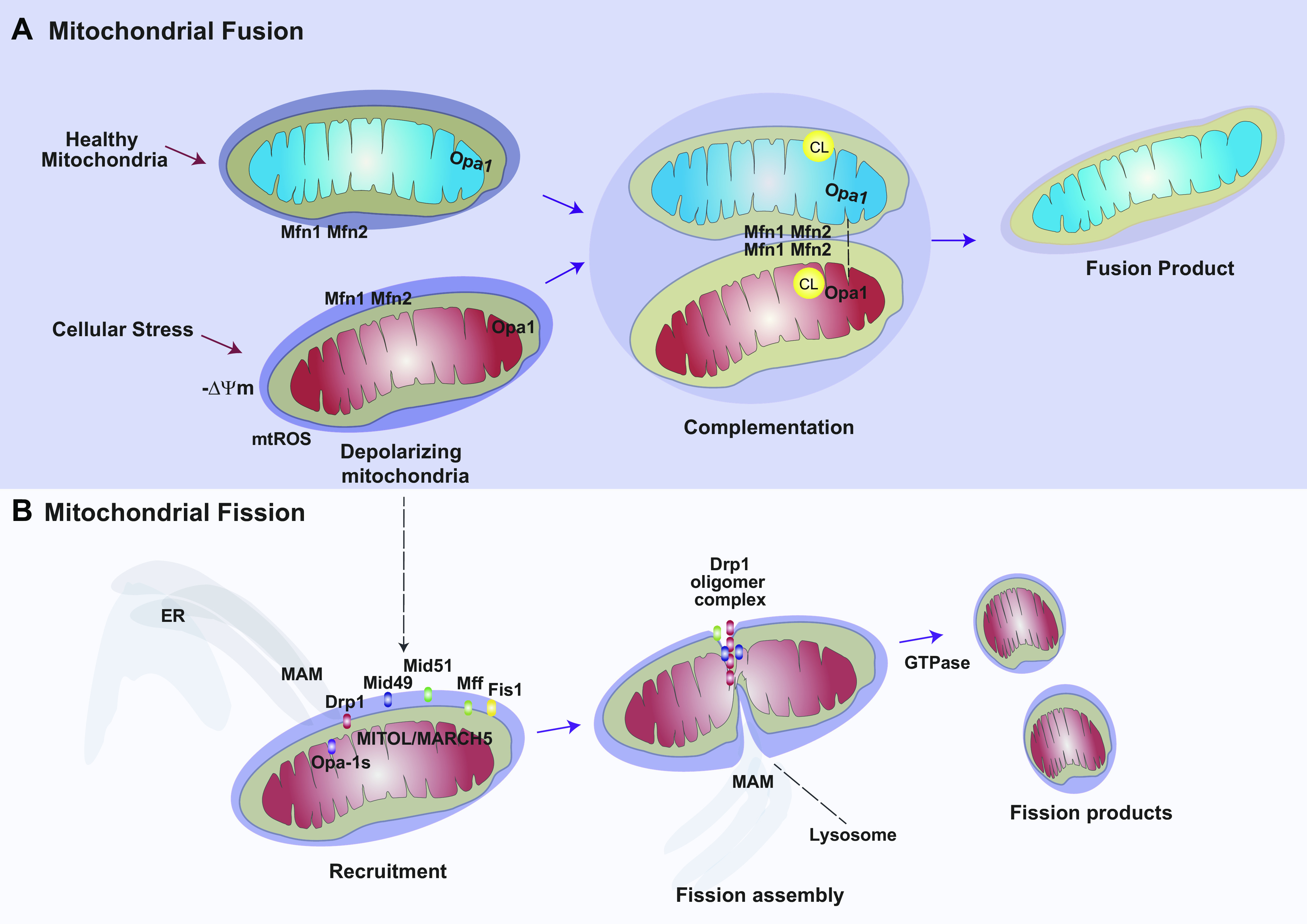
Mitochondrial dynamics. The mitochondrial population is subject to genetic regulation of its morphology and distribution, in processes referred to as mitochondrial dynamics. Mitochondrial biogenesis ensures the proliferation and maintenance of a healthy mitochondrial population via its principal regulatory components, PGC-1α, NRF1, and TFAM. Stress conditions lead to mitochondrial dysfunction including enhanced mtROS production and mitochondrial depolarization. A: the process of fusion creates larger mitochondria from 2 or more mitochondria. This process may serve to repair dysfunctional mitochondria via complementation with healthy mitochondria. B: the process of fission generates smaller mitochondria by dividing injured mitochondria. Drp1 is the principal regulator of fission. Drp1 is activated by phosphorylation at Ser637. Initiation of fission involves recruitment and oligomerization of Drp1 by accessory molecules including Mff, Fis1, and Mid59/Mid51. Opa-1s (short form) promotes fission. Fission is enabled by interactions with the ER MAM and requires GTPase activity for execution. See glossary for abbreviations.
1.7.1. Mitochondrial fission.
Mitochondrial fission is a process that leads to mitochondrial fragmentation or the generation of smaller mitochondria from larger precursors. Mitochondrial fission serves a physiological role in subdividing the mitochondrial population for cell replication. Furthermore, mitochondrial fission may serve to precondition depolarizing or dysfunctional mitochondria for active turnover of the fission products by mitophagy (29, 30). Emerging studies implicate that mitochondrial fission is regulated by multiorganelle contacts, which involve interaction with the actin cytoskeleton, and tripartite interactions between the ER, mitochondria, and lysosome, which have been collectively referred to as the “divisome” (193, 194).
During mitochondrial fission, the ER preconstricts the mitochondrial membrane via contact with the MAM. Drp1, a primary regulator of fission, oligomerizes into ringlike structures around mitochondrial fission sites, to further constrict the organelle in a GTP-dependent manner (192, 195). After Drp1 oligomerization, subsequent mitochondrial constriction causes the recruitment of dynamin 2 (Dmn2), which terminates mitochondrial membrane scission via hydrolysis of GTP (196, 197).
In human cells, the ratio of S616 to S637 phosphorylation regulates Drp1 activation (human, isoform 1). Ser 616 phosphorylation (activating) is mediated by cyclin B1/CDK1 in response to activating stimuli such as hypoxia and hypoxia-mimetic compounds (198) and by calmodulin-dependent protein kinase II (CaMKII) in response to ionizing radiation (199). Drp1 Ser 637 phosphorylation (inactivating) is mediated by cyclic AMP (cAMP)-dependent protein kinase A (PKA) and reversed by calcineurin (29, 200). In mice, phosphoregulation of Drp1 is dependent on S600 and S579 (201). Drp1 is recruited to the OMM, where it forms oligomers. The localization of Drp1 to a fission assembly at the mitochondria is regulated by other OMM-localizing proteins that serve as mitochondrial receptors for Drp1. These include mitochondrial fission protein 1 (Fis1), mitochondrial fission factor (MFF), and mitochondrial elongation factors (MIEF1 and MIEF2; also known as MiD51, MiD49) (202–206). MIEF1 and MIEF2 facilitate the association of Drp1 and MFF. The balance of MIEFs versus MFF is believed to regulate fission, such that low to moderate levels of MIEFs are ideal to promote mitochondrial fission. In contrast, high levels of MIEFs sequester Drp1 on the mitochondrial surface, which results in mitochondrial elongation (fusion) (207). The nucleotidyltransferase domain of MiD51 binds ADP to promote fission (208, 209).
Fis1, which promotes mitochondrial fission, also inhibits the activities of profusion GTPases (202, 210). Human Fis1 (hFis1)-mediated mitochondrial fragmentation can occur in the absence of Drp1 and Dyn2 (210). Fis1 and Mff were found to be important for the regulation of the formation of Drp1 puncta on mitochondria but were not totally indispensable for fission (211). Mitochondrial fission process-1,18 kDa (MTP18) has been identified as a newly characterized IMM-localizing fission factor identified in central nervous system (CNS) nerve development (212). The OMM-associated protein mitochondrial ubiquitin ligase (MITOL/MARCH5), of the membrane-associated RING-CH E3 ubiquitin ligase (MARCH) family, was identified as a factor that also regulates mitochondrial fission via the ubiquitination of Drp1 (213–215). PGAM5 may participate in Drp1 activation and initiation of fission in response to mitochondrial depolarization (181).
Emerging studies suggest that lysosome-mitochondria contact also mediates fission. Lysosome-mitochondria contact formation was enhanced by active GTP-bound lysosomal RAB7, and release of this interaction was mediated by Fis1-dependent recruitment of RAB7 GTPase-activating protein TBC1D15 to mitochondria to activate RAB7 GTP hydrolysis (216). Recent studies also suggest that ER-generated phosphatidylinositol-4-phosphate (PI4P) may also act as a signal for mitochondrial fission. ER vesicle-associated membrane protein (VAMP)-associated proteins recruit lysosomes to the site of mitochondrial division to promote mitochondrial constriction. In this model, the lysosomal lipid transfer protein ORP1L mediates transfer of PI4P from the ER to the mitochondria, which is required for fission (217). Taken together, these observations suggest that mitochondrial fission is orchestrated by a complex series of events that involve multiorganelle interactions, signaling intermediates, and protein effectors.
Further research will reveal additional mechanisms that regulate fission as well as cross talk between fission and other mitochondria-dependent processes. Aberrations in mitochondrial fission were associated with mitophagy in lung disorders (see sect. 4) and may play a contributory role in neurodegenerative diseases (see sect. 6).
1.7.2. Mitochondrial fusion.
Mitochondrial fusion is a process that generates larger, elongated mitochondria from smaller mitochondria. This process, which allows for distribution of mitochondrial components within the network, is believed to be largely beneficial in contributing to mitochondrial homeostasis and MQC. Fusion is associated with increased mitochondrial Krebs cycle activity and ATP production. Fusion may alleviate mitochondrial injury by recombining injured mitochondria with healthy components and normalize mitochondrial membrane potential in damaged mitochondria (30, 218). The fusion process is a composite event that begins with the OMM and progresses to the IMM. Fusion is mediated by GTPases mitofusin-1 (Mfn-1), mitofusin-2 (Mfn-2), and optic atrophy 1 (Opa-1) protein (219). Mfn1 or Mfn2 genetic deficiencies in mice, which are lethal, cause an early embryonic phenotype of impaired fusion and mitochondrial fragmentation (220). Mfn1/Mfn2 isoforms localize to the OMM and regulate OMM fusion. Mfn1/Mfn2 can tether adjacent mitochondria, and MFN1 can function to tether mitochondria to the ER membrane (221). IMM fusion is regulated by Opa-1, which requires the assistance of Mfn1 but not Mfn2 (222). Recent studies suggest that membrane-associated Opa1 (l-Opa1) executes IMM fusion via transmembrane interactions with CL (223, 224). A short form of Opa1 (s-Opa1) can promote or inhibit membrane fusion in a concentration-dependent manner (223, 225). Additionally, Opa1 also has a function in maintenance of crista structure (223, 224). Human Fis1 (hFis1), a regulator of fission, was found to bind to Mfn1, Mfn2, and OPA1 to inhibit intrinsic GTPase activity, thereby negatively regulating fusion (210). Recent evidence suggests that MAM contact points between the ER and mitochondria are responsible for regulated balance between fission and fusion events (218). Pink1/Parkin, key regulators of mitophagy, may regulate fusion through enhancing the ubiquitination of Mfn1 and Mfn2 (29, 159). Parkin or PINK1 knockdown reversed Mfn1/Mfn2 ubiquitination in response to promitophagy stimuli (159). Ubiquitination of fusion proteins by Parkin is believed to prevent damaged mitochondria from engaging in fusion, thereby selecting them for mitophagy (28).
In humans, mutations in mitochondrial fusion genes (MFN2 and OPA1) cause neurodegenerative disease [Charcot–Marie–Tooth type 2A and Kjer disease/autosomal dominant optic atrophy (188, 226, 227)]. Opa1 deficiency in retinal ganglion cells was reported to cause significant fragmentation of mitochondrial morphology, activation of mitochondrial motility, and impaired respiratory function (228).
Mitochondrial fusion is believed to represent an adaptive or repair process. Recent studies suggest that deficiencies in mitochondrial fusion may impact the progression of disease, including pulmonary diseases (see sect. 4) and neurodegenerative diseases (see sect. 6). Further understanding of the regulation of fusion, and the interplay of fusion with other mitochondrial processes, will determine whether the fusion apparatus may provide an effective therapeutic target in disease.
1.7.3. Mitochondrial motility.
The homeostasis of the mitochondrial network is maintained by dynamic processes, such as fission and fusion, as well as by factors that govern the spatiotemporal organization of mitochondria within individual cells, a process referred to as mitochondrial motility. Mitochondria are transported bidirectionally within cells along cytoskeletal filaments by the action of the cytoplasmic motor proteins dynein and kinesin (ATP hydrolases). Dyneins (i.e., dynein1-dynactin complex) move cargoes toward the cell center (retrograde transport), whereas kinesins (i.e., KIF5) move cargoes toward the cell exterior (anterograde transport) (229).
Mitochondrial mobility via motor proteins is regulated by transport adaptors of the mitochondrial Rho GTPase family, Miro1 and Miro2 (Rhot1/Rhot2), which are OMM-associated proteins with two Ca2+ binding and two GTPase domains (230). Miro1, the dominant isoform, forms complexes with motor proteins (i.e., kinesin) via additional adaptor proteins, the trafficking kinesin-binding proteins Trak-1/2 (mammalian homologs of Milton). Miro1 can also interact with fusion proteins, Mfn1/2, and ER MAM sites (230). Thus, Miro1 may have contributory functions in mitochondrial Ca2+ regulation and fusion (231, 232). Binding of Ca2+ to Miro1 at its intrinsic EF hand domains negatively regulates mitochondrial motility by triggering release of motor complexes from the cytoskeleton (233). The Ca2+ binding sites of Miro1 are also responsible for glutamate regulation of mitochondrial arrest in neurons (234). Additional anchor adaptors such as syntaphilin, responsible for Ca2+-dependent mitochondrial arrest in axons, can impair mitochondrial motility by directly interacting with motor proteins (229). Pink1/Parkin can arrest the motility of dysfunctional mitochondria before mitophagy by respectively phosphorylating and ubiquitinating Miro (235). Miro1 can also reciprocally act as a mitophagy regulator (230). For example, Miro1 deletion results in impaired Parkin recruitment to dysfunctional mitochondria in neurons (236).
Mitochondrial motility is also subject to metabolic regulation. Mobility is impaired by glucose levels via posttranslation modification of Trak (Milton) via O-GlcNAcylation by the action of O-GlcNAc transferase (OGT), which acts as the glucose sensor (237). Four and a half LIM domains protein 2 (FHL2) functions as an anchor to secure mitochondria to F-actin via recognition of O-GlcNAcylated Trak in motor-adaptor complexes (238).
The regulation of mitochondrial motility by motor-adaptor complexes and the balance between anterograde or retrograde trafficking and cytoskeletal anchoring remain incompletely understood. Further work will identify the identity of additional adaptor proteins and their regulation by discrete signaling networks. The function of mitochondrial motility in disease also remains incompletely delineated. The process may serve to distribute mitochondria to sites of increased energy demand or to deliver mitochondria to appropriate intracellular sites for their processing via fission, fusion, or mitophagy as required under specific conditions (229, 238).
1.7.4. Intercellular transport of mitochondria.
Mitochondria may undergo intercellular transport, which can serve homeostatic functions by exchanging mitochondria between cells. Intercellular transport of mitochondria may be accomplished via physical connective pathways termed tunneling nanotubes (TNTs). The TNTs have an F-actin cytoskeleton and can contain microtubules, and thus also support bidirectional motor protein complex-dependent trafficking of mitochondria. Miro proteins participate in regulating mitochondrial intercellular transport via TNTs (239).
Mitochondria can also be transported between cells in extracellular vesicles (EVs) of various sizes (240). Within EVs, mitochondria may be packaged into subcompartments termed mitochondria-derived vesicles (MDVs) (239). Opa1- and Snx9-dependent MDVs prepare functional mitochondria for export, whereas Parkin selects against dysfunctional mitochondria for export (239). Alternately, transfer of mitochondria through gap junctions or by cell fusion events has been described (230). The function of intercellular transport of mitochondria in disease is incompletely understood but may serve as a survival mechanism, to compensate stressed cells with functional mitochondria derived from healthy cells (241). In disease, such transfer may be propathogenic in the case of promoting cancer cell survival and tumorigenesis. Further studies will determine the potential of both intra- and intercellular mitochondrial trafficking as targets for therapeutic development (230, 242).
1.8. Mitochondrial Biogenesis
Mitochondrial biogenesis is a genetically regulated program for the proliferation of mitochondria during cell division and cellular stress (FIGURE 4) (26). A number of physiological and environmental cues can trigger mitochondrial biogenesis, which include starvation, exercise, hypothermia, inflammation, and oxidative stress (243). This process contributes to MQC processes, in part by replenishing functional mitochondria after their turnover by mitophagy. Mitochondrial biogenesis is tightly regulated by several coactivators and transcription factors, including transcriptional coactivator peroxisome proliferator-activated receptor gamma (PPARγ) coactivator 1α (PGC-1α), PGC-1β, and PGC-1-related coactivator (PRC) (244). PGC-1α is considered the master inducible regulator of mitochondrial biogenesis (244).
FIGURE 4.
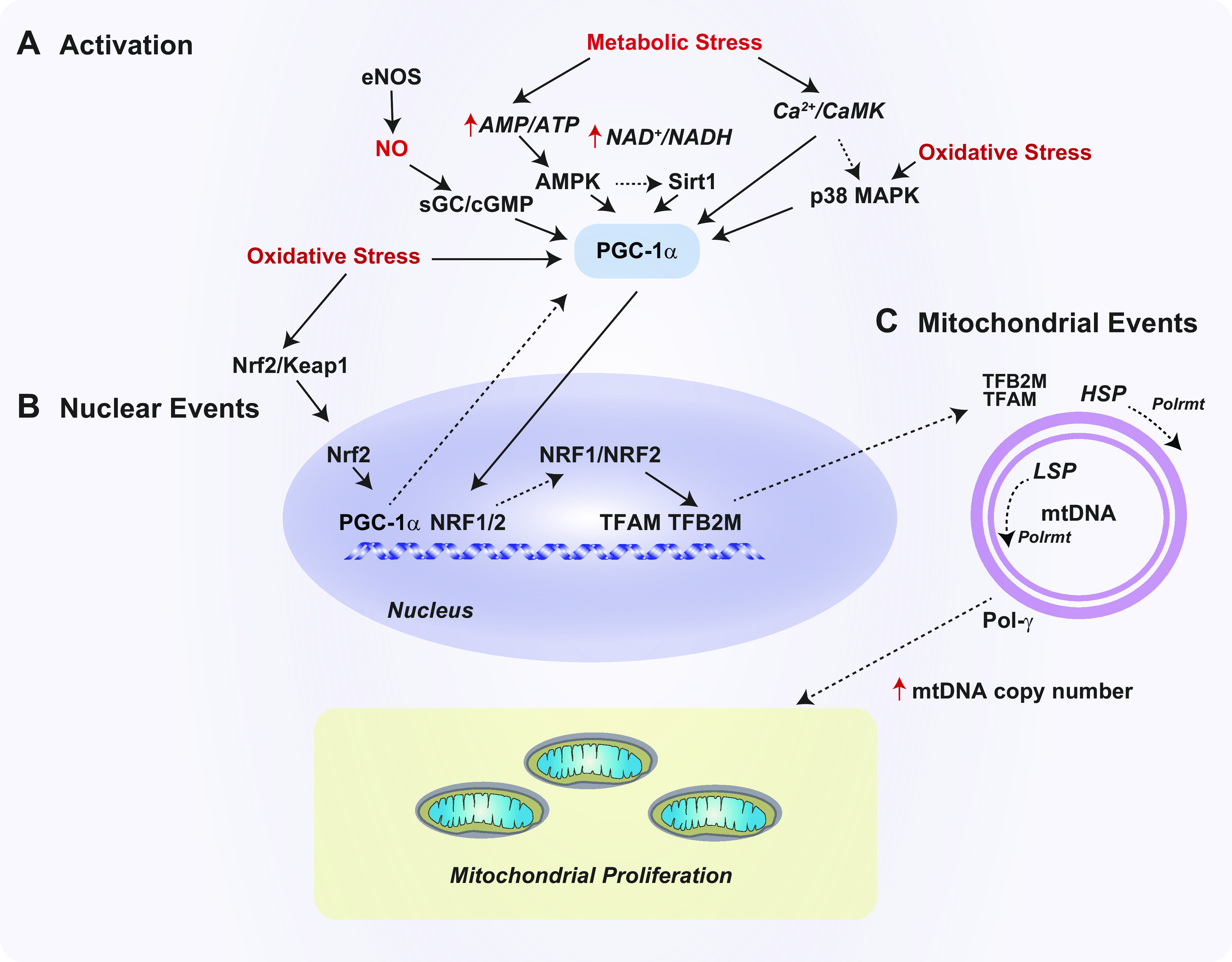
Regulation of mitochondrial biogenesis. The mitochondrial biogenesis pathway governs mitochondrial DNA (mtDNA) replication and mitochondrial proliferation. This process is regulated by peroxisome proliferator-activated receptor gamma (PPARγ) coactivator 1-α (PGC-1α) and related factors. A: several signaling pathways can trigger PGC-1α activation and downstream processes. These include the sirtuin-1 (Sirt1)-5′ adenosine monophosphate-activated protein kinase (AMPK) axis and endothelial nitric oxide synthase (eNOS)-derived nitric oxide (NO), production which activates the soluble guanylate cyclase (sGC)/guanosine 3′,5′ cyclic monophosphate (cGMP) pathway. Metabolic signals can also activate PGC-1α via the Ca2+/calmodulin-dependent kinase (CaMK) pathway and/or increased oxidative stress and mtROS production, which culminate in p38 mitogen-activated protein kinase (p38 MAPK) activation. Redox imbalance can activate PGC-1α transcription via the nuclear factor erythroid 2-related factor-2 (Nrf2)/Kelch-like ECH-associated protein 1 (Keap1) pathway. B: in the nucleus, PGC-1α drives the transcription of nuclear respiratory factors (NRF1 and NRF2). NRF1 regulates the expression of mitochondrial transcription factor-A (mitochondrial) (TFAM) and transcription factor-B2 (mitochondrial) (TFB2M). C: these nuclear-encoded factors regulate mtDNA transcription in the mitochondria via activation of mitochondrial RNA polymerase (Polrmt) at 2 promoters, the light strain promoter (LSP) and the heavy strand promoter (HSP). TFAM can also promote mtDNA replication via activation of mtDNA polymerase-gamma (Pol-γ). Increased mtDNA replication supports mitochondrial proliferation.
PGC-1α is responsive to metabolic regulation by multiple transcriptional and posttranscriptional mechanisms (245). For example, PGC-1α is regulated by phosphorylation via p38 mitogen-activated protein kinase (p38 MAPK), 5′ adenosine monophosphate-activated protein kinase (AMPK), and glycogen synthase kinase 3β (GSK3β). PGC-1α is also regulated by deacetylation via the AMPK-Sirt1 axis (246–248). Activation of PGC-1α by AMPK represents a response to cellular energy depletion (i.e., increased AMP-to-ATP ratio), whereas direct regulation by Sirt1 responds to NAD+-to-NADH ratio. Recent evidence suggests that PGC-1α is regulated by BCAAs (i.e., leucine) (249). For example, leucine supplementation in vivo can upregulate markers of mitochondrial biogenesis in muscle tissues (249). Transgenic overexpression of PGC-1α in skeletal muscle regulates the expression of enzymes involved in BCAA metabolism in skeletal muscle mitochondria (250).
PGC-1α regulates several downstream transcription factors. Of these, PGC-1α-regulated targets related to mitochondrial biogenesis include nuclear respiratory factor-1 and -2 (NRF1, NRF2) and estrogen‐related receptor‐α (ERR‐α). Activation of these factors leads to upregulation of mitochondrial transcription factor A (TFAM), a nuclear-encoded gene, which is regarded as the master regulator of mtDNA replication (26). TFAM factor binds to mtDNA and facilitates recruitment of the mtDNA polymerase (POLRMT) as well as recruitment of the accessory transcription factor B2 (TFT2BM) (251). TFAM primes mtDNA replication executed by DNA polymerase-γ. These factors also drive mtDNA transcription at two major promoters, the light strand promoter (LSP) and the heavy strand promoter 1 (HSP1) (252).
Under oxidative stress conditions, mitochondrial biogenesis is also regulated by transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2, NFE2L2), a master regulator of the antioxidant response. Chemical activators of Nrf2 such as sulforaphane have been shown to increase mitochondrial biogenesis (253). Nrf2 induced by various stimuli may regulate the downstream transcription of PGC-1α and NRF1. Nrf2 regulates the expression of heme oxygenase-1, which has also been shown to influence mitochondrial biogenesis programs (254).
Emerging studies suggest regulatory relationships between PGC-1α-dependent mitochondrial biogenesis, mitochondrial dynamics, and mitophagy. For example, in a model of rotenone-induced dopamine neurotoxicity, genetic downregulation of fission or promotion of fusion proteins (i.e., Mfn2) enhanced neural protection and stimulated PGC-1α-dependent mitochondrial biogenesis (255). Conversely, gain- and loss-of-function experiments established a role for PGC-1α in downregulating fission and promoting fusion (255). Similar associations were observed with resveratrol, which counteracted rotenone toxicity by upregulating mitochondrial biogenesis and reciprocally regulating fission and fusion (256). PGC-1α upregulation was also associated with downregulation of Pink1-dependent mitophagy in this model, whereas PINK1 expression downregulated TFAM and PGC-1α expression (257).
In conclusion, mitochondrial biogenesis responds to cellular energy needs and is generally considered a prosurvival mechanism that is co- or reciprocally regulated with other mitochondrial processes, such as fission, fusion, and mitophagy. Deregulation of mitochondrial biogenesis and/or its underlying regulatory mechanisms has been described in numerous human diseases (see sects. 5 and 6). Pharmacological targeting of this pathway may show promise in neurodegenerative and metabolic diseases.
2. MITOCHONDRIAL DYSFUNCTION AND METABOLISM IN THE REGULATION OF INFLAMMATION
In addition to their vital functions in regulating bioenergetics and apoptosis, mitochondria have emerged as crucial signaling platforms for the regulation of inflammation and innate immune responses (4, 12, 258, 259). Pathogen-associated molecular patterns (PAMPs) are characteristic features of many pathogens, including bacteria, fungi, protozoa, and viruses (260). In the innate immune system, molecular sensing of PAMPs and DAMPs occurs through interactions with pattern recognition receptors (PRRs). The PRRs include members of the Toll-like receptor (TLR), retinoic acid-inducible gene (RIG)-I-like receptor (RLR), and nucleotide binding domain leucine-rich repeat-containing receptor or “NOD-like receptor” (NLR) families. Upon pattern recognition, activated PRRs trigger downstream signal transduction pathways that regulate inflammatory and innate immune responses (261). Mitochondria have long been known to play a functional role in TLR signal generation via metabolic production of mtROS, which are known to signal to NF-κB-dependent regulation of proinflammatory cytokines. Emerging studies have also established that mitochondria-associated factors can signal distinct and specialized innate immune mechanisms that include inflammasome-dependent inflammatory cytokine responses and interferon (IFN)-dependent antiviral responses, as discussed below, which contribute to modulation of host defenses. Thus, mitochondrial signaling to the regulation of inflammation and innate immunity represents an important underlying mechanism in the pathogenesis of diseases where inflammation or pathogen infection plays a key role.
2.1. Mitochondrial DAMPs
2.1.1. Mitochondrial DNA.
To fully appreciate how mitochondria modulate the innate immune system, specifically through the release of DAMPs like mitochondrial DNA (mtDNA) (FIGURE 5), the discussion must begin with the endosymbiotic theory of mitochondria. Eons ago, α-proteobacterium fused with either an archaean or eukaryotic host to form a novel cellular organism (262). The reason for this event is unclear, with some theorizing that the impetus was a symbiotic exchange of hydrogen ions between a methanotrophic α-proteobacterium and a methanogenic host while others postulate that the incorporation of an aerobic α-proteobacterium into an anaerobic host conferred a survival advantage (263–265). Irrespective of why this occurred, genome reduction ensued, with a significant portion of the proteobacterium’s DNA being lost or incorporated into the nuclear genome as it evolved into modern-day mitochondria (262).
FIGURE 5.
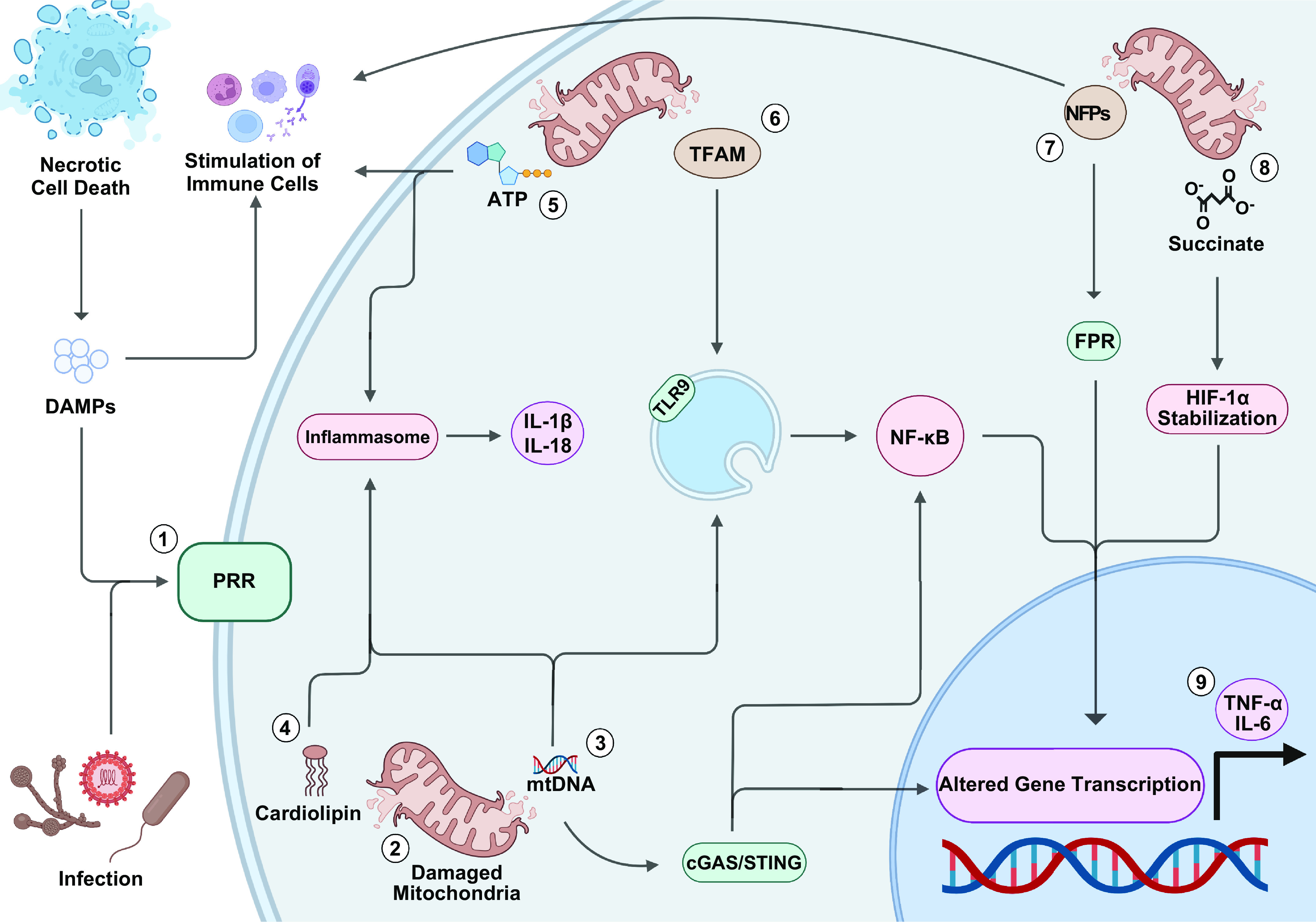
Mitochondrial damage-associated molecular patterns (DAMPs). 1: DAMPs released by necrotic cells or pathogen-associated molecular patterns (PAMPs) released during infection bind pattern recognition receptors (PRRs). Once activated, PRRs initiate a signal transduction cascade causing increased production of mitochondrial reactive oxygen species (mtROS). 2: The increased oxidative stress can fragment the mitochondrial genome and damage mitochondria, allowing mitochondrial DAMPs to enter the cytosol. 3: Cytosolic mitochondrial DNA (mtDNA) can bind toll-like receptor 9 (TLR-9), leading to activation of NF-κB, inflammasomes leading to caspase-mediated production of IL-1β and IL-18 from their zymogens, or cGAS/STING-mediated activation of NF-κB and interferon-stimulated genes (ISGs). 4: Cardiolipin can facilitate organization of the inflammasome. 5: ATP can activate inflammasome or facilitate recruitment and activation of immune cells if released to the extracellular space. 6: Mitochondrial transcription factor A (TFAM) facilitates internalization of mtDNA by endosomes where it can activate TLR-9. 7: N-formyl peptides (NFPs) can stimulate immune cells or alter gene expression through binding formyl peptide receptors (FPRs). 8: Succinate can alter gene expression by stabilizing Hypoxia-inducible factor-1α (HIF-1α). 9: Mitochondrial DAMPs can alter gene transcription, resulting in the production of tumor necrosis factor-α (TNF-α), IL-6, and other proinflammatory cytokines. Image created with BioRender.com, with permission.
Today, human mitochondria contain a circular genome composed of 16,569 base pairs encoding 37 genes (266, 267). Because of their ancestral origin, the mitochondrial genome contains unmethylated CpG motifs (268). These motifs, a common feature of bacterial DNA, are minimally, if at all, found within the human genome. This dichotomy enabled the evolution of pattern recognition receptors (PRRs) by the innate immune system capable of recognizing unmethylated CpG motifs as a pathogen-associated molecular pattern (PAMP) (268). However, innate immunity lacks the sophistication to distinguish unmethylated CpG motifs of bacterial origin from those of mitochondrial origin. Consequently, extramitochondrial DNA stimulates PRRs no differently than bacterial DNA; albeit as a DAMP rather than a PAMP. Under normal physiological conditions this does not pose a problem, as mtDNA is confined to mitochondria. However, in the setting of mitochondrial stress, cellular damage, or necrosis mtDNA can enter the cytoplasm or extracellular space, imposing an immunostimulatory effect (269, 270).
To expound on this process, toll-like receptors (TLRs) are a family of PRRs responsible for the rapid detection of extracellular and intracellular pathogens by the innate immune system (271). In humans, investigators have identified 10 different TLRs, which are differentially expressed according to cell type (271, 272). Among this family, human TLR-9 is primarily expressed by B cells and plasmacytoid dendritic cells (DCs), where it is responsible for recognition of oligonucleotides containing unmethylated CpG motifs (271). Internalization of DNA containing these motifs will stimulate endolysosomal TLR-9 to initiate a complex proinflammatory response mediated by adaptor molecule myeloid protein 88 (MyD88), interleukin-1 receptor-associated kinase-1 (IRAK1), interferon regulatory factor-7 (IRF7), and tumor necrosis factor-α receptor-activated factor-6 (TRAF6) (271, 273–276). The strength of the ensuing response is contingent on the structure of the oligonucleotide as determined by the number of CpG motifs, the spacing between motifs, the oligonucleotide backbone, and the presence of certain flanking sequences (277).
The effect of TLR-9 stimulation is the induction of NF-κB and other intermediaries to promote the secretion of IL-6, TNF-α, type I interferons, and IL-10 (271). TLR-9-activated B cells display increased antigen sensitivity and differentiate into antibody-producing plasma cells, providing a link between innate and adaptive immune responses (271, 278). Furthermore, TLR-9 activation stimulates migration, maturation, and expression of costimulatory molecules or antigens by plasmacytoid dendritic cells, facilitating T-cell activation (271, 279).
Besides TLR-9, mtDNA can trigger the innate immune system through interaction with multimeric cytosolic protein complexes known as inflammasomes (269). Numerous studies implicate mtDNA as a potent agonist of the NLRP3, NLRC4, and AIM2 inflammasomes (270, 280, 281). Inflammasomes are discussed in greater detail in sect. 2.3.
Additionally, cytosolic mtDNA binds the indiscriminate cyclic GMP-AMP synthase (cGAS) to produce cyclic guanosine monophosphate-adenosine monophosphate (cGAMP). This cyclic dinucleotide is an important intracellular signaling molecule that activates the ER protein stimulator of interferon genes (STING) to induce a type I interferon (IFN1) response (282). This occurs via STING-mediated activation of TANK-binding kinase 1 (TBK1) and I-κB kinase (IKK). These protein kinases phosphorylate interferon regulatory factor 3 (IRF3) and the inhibitory I-κBα, allowing IRF3 and NF-κB to migrate to the nucleus, where they induce expression of IFNβ, interferon-stimulated genes (ISGs), and other cytokines. Stimulation of the cGAS-STING pathway by mtDNA has been connected to the development of obesity-induced metabolic dysfunction, acute kidney injury, and amyotrophic lateral sclerosis (ALS) (283–285).
2.1.2. N-formyl peptide.
Another category of mitochondrial DAMPs that owe their function to the bacterial origin of mitochondria are N-formylated peptides (NFPs). Like bacteria, mitochondria require N-formylated methionine (fMet), a constituent of NFPs, for translation initiation (286). Normally, endogenous NFPs are confined to mitochondria (286). However, during periods of cellular injury or mitochondrial stress NFPs, like mtDNA, can be released into the cytosol or extracellular space (286, 287). When this occurs, NFPs interact with either formyl peptide receptor (FPR), FPR-like 1 (FPRL-1), or FPRL-2 on phagocytic leukocytes to direct an immune response (288).
Initially, FPRs were identified as PRRs predominantly expressed by neutrophils and monocytes that drove leukocyte chemotaxis, degranulation, cytokine production (IL-1α, IL-1β, IL-6, and IL-8), and ROS production in response to bacterial NFPs (289, 290). Further research involving mitochondrial NFPs proved they could elicit a similar response (291, 292). Since then, FRPs have been found in a multitude of cells and tissue types (288). Furthermore, FRPs appear to be promiscuous receptors, responding not only to NFPs with varying affinities but also to HIV-1 envelope peptides, annexins, and lipids (288, 293). Even though the signaling pathway for these Gi protein-coupled receptors has been described, our understanding of the functional role of FPRs remains limited because of challenges imposed by the diversity of receptor agonists and the absence of clear human-murine orthologs (288, 294).
2.1.3. Adenosine triphosphate.
In 1929, Karl Lohmann (295) as well as Cyrus Hartwell Fiske and Yellagaprada SubbaRow (296) independently discovered adenosine triphosphate (ATP). Their findings fostered decades of research into the unique biochemical and enzymatic mechanisms encompassing the role of ATP as the conveyor of cellular energy. In tandem with that body of research, a detailed understanding of purinergic signaling developed, revealing an unexpected role for ATP as an extracellular ligand essential for neurotransmission, endothelium-mediated vasodilation, platelet aggregation, and inflammation (297, 298). Relevant to the discussion of mitochondrial DAMPs is how ATP regulates the activity of neutrophils, macrophages, and other immune cells.
Under normal physiological conditions, intracellular ATP exists at millimolar concentrations (299). In contrast, ATP circulates in the extracellular environment at submicromolar concentrations (299). This compartmental difference is vital to ATP’s ability to function as a DAMP. When ATP is released into the extracellular environment during cell injury, mechanical stress, bacterial infection, or necrotic cell death, the spike in plasma ATP concentration is promptly recognized by purinergic receptors (P2Rs) (299). There are two distinct families of P2Rs. The P2YR family is composed of eight G protein-coupled receptors that recognize ATP and other adenine nucleotides (300). In contrast, the P2XR family is composed of seven ATP-gated inotropic receptors (300).
For neutrophils, extracellular ATP promotes upregulation of endothelial E-selectin via P2X7 activation to support neutrophil tethering (301, 302). As neutrophils roll along the endothelium toward the site of injury they are primed by ATP and other agents, causing rapid upregulation of the adhesion receptor macrophage-1 antigen (Mac-1) (299, 303). These steps are accompanied by ATP-mediated changes to the vascular endothelium aiding neutrophil transmigration (304). After extravasation, neutrophils travel along a chemotactic gradient toward the site of injury (299). Once they arrive at the inflammatory site, their bactericidal functions are further enhanced by the presence of ATP within the inflammatory milieu. Through P2Y2 receptors, ATP stimulates neutrophil degranulation and augments production of ROS, boosting oxidative burst (305, 306). Moreover, extracellular ATP can influence the duration of the proinflammatory response by working alongside granulocyte macrophage colony-stimulating factor (GM-CSF) to delay neutrophil apoptosis (307). Interestingly, adenosine forms a negative feedback loop with ATP, offsetting its proinflammatory characteristics. Both TNF-α and oxidative stress inhibit ectonucleoside triphosphate diphosphohydrolase-1 (NTDase1 or CD39) and 5′ nucleotidase (5′-NT or CD73), slowing the breakdown of ATP to adenosine during inflammation.
Like neutrophils, macrophage adhesion benefits from ATP-mediated pathways upregulating endothelial E-selectin and monocyte Mac-1 (301, 302). One notable difference is that monocyte adhesion is further facilitated by ATP enhancing endothelial expression of VCAM-1 through P2Y2 activity (308). After monocytes have firmly adhered to the endothelium, extracellular ATP activates shedding of monocyte L-selectin (CD62L) through P2X7, thus stimulating transmigration (309). This process is furthered by ATP inducing endothelial shrinkage or apoptosis through P2Y receptors, permitting monocyte passage through the endothelial barrier (299). After transmigration, ATP stimulates monocyte chemotaxis. To orchestrate the inflammatory response, activated macrophages release a variety of cytokines and chemokines. Whether it be through P2X7 activation of the NF-κB pathway or other avenues, ATP stimulates the production of IL-1, IL-1β, IL-6, IL-18, and TNF-α (310). Furthermore, ATP has been shown to enhance LPS-induced NO production via inducible nitric oxide synthase (iNOS or NOS2), which is essential for the creation of reactive nitrogen species (RNS) (311). ATP also stimulates ROS generation. Finally, prolonged exposure to ATP triggers cell death pathways via P2X7 activation (312); it has been hypothesized these pathways exist to protect tissues from an aberrant, uncontrolled inflammatory response.
For dendritic cells (DCs), the effect of ATP on their migration depends on the molecule’s concentration. At low micromolar concentrations, ATP stimulates DC migration to the inflammatory site. However, upon arrival the higher concentrations of ATP inhibit DC movement via P2Y11, prolonging their exposure to the nidus of inflammation where it can phagocytose potential antigens (313). Additionally, stimulation of P2Y11 allows ATP to act synergistically with TNF-α, LPS, CD40L, and other inflammatory mediators to promote DC maturation (314). The production of cytokines is also regulated by ATP concentrations. Low concentrations appear to inhibit the production of TNF-α, IL-1α, IL-1β, IL-6, and IL-12 (299). Meanwhile, high concentrations of ATP are associated with increased production of IL-1β, TNF-α, and IL-12 (299). Mirroring their cytokine production, DCs exposed to low levels of ATP appear to initiate a Th2 response whereas DCs exposed to high ATP levels initiate a Th1 response (299). Prolonged ATP exposure is also associated with P2X7-mediated cytotoxicity, which leads to DC apoptosis.
Relatively less is known about ATP-mediated regulation of lymphocytes (299, 315). It has been accepted that ATP stimulates or inhibits lymphocyte activity based on nucleotide concentration and cell type. As an example, intermediate levels of ATP enhance CD4+ T cell activity and boosts their proliferation through upregulation of IL-2 (316). However, high concentrations of ATP not only induce CD4+ T cell apoptosis but also enhance Treg activity and proliferation (316). Additionally, ATP inhibits IL-2-mediated proliferation of natural killer cells. Furthermore, activated T cells release ATP, suggesting that the nucleotide is involved with autocrine and/or paracrine signaling.
2.1.4. Mitochondrial transcription factor A.
As a high-mobility group (HMG) protein, mitochondrial transcription factor A (TFAM) is required for proper maintenance of the mitochondrial genome (252). TFAM performs this task by controlling transcription of mitochondrial promoter sequences to influence mtDNA copy number and through nonspecific binding to mtDNA to compact the genome into nucleoids (252). However, the function of TFAM is not limited to these regulatory responsibilities. Like its functional and morphological homolog HMGB1, TFAM acts synergistically with other DAMPs to influence the immune system (317, 318). After cell necrosis, TFAM remains bound to mtDNA (319). This facilitates internalization of mtDNA into endosomes of immune cells, where it augments mtDNA-mediated TLR-9 activation in a receptors for advanced glycation end products (RAGE)-dependent manner (319). In this fashion, TFAM has been shown to promote secretion of type I interferon by plasmacytoid dendritic cells and TNF-α by murine splenocytes (319, 320). Besides mtDNA, TFAM acts synergistically with NFPs to promote activation of human monocytes and release of IL-8 (290). Additionally, there are in vivo models implicating TFAM as a mitochondrial DAMP. Intra-cisterna magna injection of TFAM has been shown to upregulate expression of MCP-1, IL-1β, IL-6, TNF-α, and NF-κB inhibitor alpha (NFκBIA) in the hippocampus and frontal cortex of rats (321). Follow-up studies utilizing THP-1 monocytes, as models of human microglia, further implicate RAGE and Mac-1 as important mediators of a TFAM-induced inflammatory response (321). Finally, exposure of murine macrophages to recombinant TFAM has been shown to promote secretion of IL-6 and TNF-α in a dose-dependent manner (322). This response was replicated in healthy rats, where recombinant TFAM not only promoted the production of proinflammatory cytokines but was associated with neutrophil infiltration of the lungs and end-organ damage (322).
2.1.5. Cardiolipin.
Cardiolipin (CL) is a tetra-acylated phosphatidylglycerol lipid contributing to the cell membranes of most prokaryotes (286, 323). This phospholipid can also be found in eukaryotes, where it is a significant component of the IMM—another remnant of the mitochondrion’s bacterial ancestry (41, 286). CL is essential for protein transport across mitochondrial membranes, the organelle’s morphology including their iconic cristae, cellular respiration, biogenesis, apoptosis, and immune signaling (41, 286). The immunological role of CL is complex, displaying both anti-inflammatory and proinflammatory properties.
When mitochondria are damaged, CL is externalized from the inner to the outer mitochondrial membrane via phospholipid scramblase-3 (PLS3) (324, 325). This redistribution of CL allows it to bind LC3 so it can be incorporated into autophagosomes (324, 325) for mitophagy. Extracellular mitochondria are also phagocytosed through Cluster of Differentiation 36 (CD36) binding to CL (326). There is additional evidence suggesting that modification of CL can alter the affinity of CL for its receptors (323–325). Together, these findings suggest that the expression of CL can be manipulated as needed to control mitochondria-mediated inflammatory or cell death pathways.
Importantly, the anti-inflammatory properties of CL are not limited to mitophagy. Experiments involving unsaturated CL indicate that the lipid can also inhibit LPS binding to TLR4/Myeloid Differentiation 2 (TLR4/MD2) (327). By antagonizing TLR4/MD2, CL prevents LPS from stimulating production of TNF-α, IL-1β, IFN-γ, and other proinflammatory cytokines (327, 328). Supporting in vivo evidence is found in the bronchoalveolar lavage of mice with Escherichia coli pneumonia (323, 329). Mice with lower levels of unsaturated CL were observed to have higher levels of TNF-α, whereas higher levels of unsaturated CL correlated with lower levels of TNF-α, IL-1β, and IFN-γ. Furthermore, healthy mice exposed to exogenous intratracheal CL had lower levels of the proinflammatory IL-2 and IFN-γ in lavage fluid but higher levels of anti-inflammatory IL-10.
Finally, several proinflammatory roles of CL have been identified. LPS induces translocation of CL to the outer mitochondrial membrane in a ROS-dependent manner. This process, which may involve oxidization of CL, facilitates colocalization of caspase-1 and the NLRP3 inflammasome to the mitochondrial membrane (41, 330). By aiding organization of the NLRP3 inflammasome, CL promotes inflammasome activation by mtDNA and other ligands to produce IL-1β and IL-18. Furthermore, oxidized CL mobilizes intracellular Ca2+ stores in phagocytes, stimulating production of leukotriene B4 and 5-lipoxygenase (5-LOX) gene expression (331). In vascular endothelium, CL upregulates ICAM-1 and VCAM-1, facilitating intercellular adhesion by leukocytes (331). Finally, saturation of CL’s acyl chains significantly alters the relationship between CL and TLR4/MD2. Whereas unsaturated CL is anti-inflammatory, saturated CL appears proinflammatory, as it stimulates TLR4/MD2, like LPS, to trigger production of TNF-α, type 1 IFN, and IL-1β (323).
2.1.6. Succinate.
As part of the Krebs cycle, succinyl-CoA synthetase catalyzes the reaction of succinyl-coenzyme A (succinyl-CoA) with an inorganic phosphate and adenosine diphosphate (ADP) to generate succinate and ATP (332). From here, CII catalyzes the oxidation of succinate to fumarate alongside the reduction of ubiquinone. As these reactions illustrate, succinate is vital to cellular bioenergetics since it is directly involved in the production of ATP and functions as a link between the Krebs cycle and OXPHOS. Interestingly, succinate also functions as a mitochondrial DAMP. In macrophages, LPS upregulates glycolysis via a TLR4-mediated pathway (333). The subsequent rise in succinate is responsible for stabilization of HIF-1α. The downstream effects of HIF-1α include upregulation of IL-1β. Akin to other DAMPs, succinate is released into the extracellular space after tissue injury or necrotic cell death (334). Plus, in vitro inhibition of CIII by antimycin results in the secretion of succinate by muscle cells, suggesting that impairment of the ETC may also drive its secretion (335). Once in the extracellular space, succinate may bind GPR91, a G protein-coupled receptor, to stimulate chemotaxis, production of proinflammatory cytokines, and T cell activation by DCs (334).
2.1.7. Conclusion.
Although there are numerous mitochondrial DAMPs (MTDs), they all share a common feature. When these molecules are released into the intra- or extracellular environment, they can elicit an immune response, often because they resemble bacterial antigens. What remains to be seen is whether these molecules are merely passive bystanders, representing the sequela of disease, or they significantly contribute to acute or chronic inflammatory processes by propagating the initial inflammatory insult. Understanding this distinction in the context of disease has the potential to transform clinical practice. Using mtDNA as an example, investigators have observed an association between deceased donor plasma mtDNA levels and early allograft dysfunction in liver transplant recipients (336). If mtDNA actively contributes to the development of early allograft dysfunction, rather than simply being a marker of donor inflammation, then using deceased donor mtDNA to guide posttransplant immunotherapy or developing agents to temper pretransplant donor mtDNA levels may improve outcomes by decreasing rates of early allograft dysfunction. Addressing these concepts represents the future of MTD therapeutics.
2.2. Sirtuin Regulation of Inflammation
Sirtuins play an important role in the epigenetic regulation of inflammatory responses. By seeking to restore immunometabolic homeostasis during periods of cell stress and high metabolic demand, sirtuins promote cellular adaptation to inflammatory stimuli by influencing mitochondrial bioenergetics (337). The deacectylase activity of SIRT1 and SIRT3 is regulated in acute inflammation in response to the high-NAD+/low-cellular energy environment induced by sepsis (338, 339). Early induction of SIRT1 can be beneficial, with murine studies showing that early blockade of SIRT1 with the specific inhibitor EX-527 worsens survival in a murine model of polymicrobial sepsis (340). In vitro studies have clarified the critical cross talk between sirtuins and mitochondrial biogenesis that mediates this benefit. In monocytes, NAD+-dependent SIRT1, RELB, and SIRT6 nuclear proteins regulate a switch from the glycolysis-dependent acute inflammatory response to FAO-dependent sepsis adaptation. This switch is mediated by SIRT3 regulation of Krebs cycle, leading to increased mitochondrial O2 consumption. The subsequent induction of SIRT1 increases mitochondrial biogenesis and serves to help the cell adapt to this high mitochondrial energetic state (337). But SIRT1-mediated sepsis adaptation can become maladaptive, including the regulation of the late-phase hypoinflammatory sepsis phenotype. Late blockade of SIRT1 improves survival in a murine model of polymicrobial sepsis by restoring expression of leukocyte-endothelial interactions and improving peritoneal bacterial clearance, demonstrating the pleotropic effects of sirtuins in inflammatory conditions (340).
SIRT1 can also attenuate inflammatory responses by regulating NF-κB. Under high-cellular NAD+ conditions, SIRT1 interacts with transducing-like enhancer of split-1 (TLE1) to repress NF-κB activity (341). Similarly, SIRT1 has been shown to deacetylate the RelA/p65 subunit of NF-κB, leading to lower expression of inflammatory genes (339). Reflecting this regulation, the SIRT1 agonist resveratrol is capable of downregulating NF-κB-mediated proinflammatory cytokines (342). SIRT1 regulation of NF-κB has important implications for inflammatory diseases, including age-related cardiovascular disease, chronic lung disease, and neurodegenerative diseases. SIRT1 is protective against chronic inflammation that drives age related cardiovascular disease (343, 344). In chronic obstructive pulmonary disease (COPD), an inflammatory lung condition induced by chronic cigarette smoke exposure, SIRT1 is reduced in both the lungs and macrophages of humans with COPD (345, 346) and knockdown of SIRT1 in macrophages exposed to cigarette smoke extract is associated with increased IL-8 release. Among neurodegenerative diseases, SIRT1 has been shown to be neuroprotective (347, 348).
2.3. Inflammasome-Dependent Pathways
2.3.1. Inflammasomes.
Inflammasomes are specialized multiprotein inflammatory signaling platforms that regulate innate immune responses (349). In response to microbial infection, activation of the inflammasomes contributes to host protection by inducing immune responses that provide a defense mechanism against invading microorganisms. Dysregulated activation of inflammasomes, however, can also lead to tissue injury associated with hyperinflammation and also trigger gasdermin-D-dependent cell death (pyroptosis) (349–351). The cytosolic NLRs are characterized by a homologous region that includes an intrinsic nucleotide-binding oligomerization domain (NACHT) and a COOH-terminal leucine-rich repeat domain (LRR) (261, 349). NLRs also contain a variable NH2-terminal domain that may include a pyrin domain (PYD), a caspase-recruitment domain (CARD), or a baculovirus inhibitor of apoptosis repeat (BIR) domain (261). Among the NLRs, NLRP1, NLRP3, NLRC4, and NLRB1 can form multiprotein inflammasome complexes upon PRR stimulation (352). Furthermore, the pyrin and HIN domain-containing (PYHIN) family member absent in melanoma 2 (AIM2) can form inflammasome complexes in response to the presence of pathogen-derived dsDNA, and thereby acts as the primary sensor for this DAMP (353). Inflammasome complexes contain additional key protein constituents, including apoptosis-associated speck-like protein containing CARD (ASC), which functions as an adaptor, and the pro-form of caspase-1. Inflammasome complex formation promotes the proteolytic cleavage and activation of the cysteine protease caspase-1 (354). Activated caspase-1 catalyzes the proteolytic cleavage of proinflammatory cytokine precursors from their pro-forms into their active forms (e.g., IL-1β and IL-18), which are responsible for propagating inflammatory and innate immune responses (354, 355).
Among the known inflammasomes, the NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inflammasome has been the most widely studied in models of disease pathogenesis (261). Activation of the NLRP3 inflammasome proceeds via a canonical two-step mechanism. The first and priming step of NLRP3 inflammasome activation involves ligand-dependent activation of TLRs (e.g., TLR4), that stimulates NF-κB-dependent transcription of NLRP3. NLRP3 protein levels are rate-limiting for inflammasome activation (355). Subsequently, NLRP3 activation requires a second activating signal such as ATP-dependent P2X7 activation, leading to K+ efflux, and may involve other ion fluxes such as cytosolic Ca2+ accumulation and Cl− efflux (351). Many diverse agents such as bacterial components and toxins and crystalline or particulate substrates such as silica or asbestos can trigger inflammasome activation (350, 351). Endogenously derived NLRP3 inflammasome activators can also include diverse substances such as monosodium urate, cholesterol, cyclic GMP-AMP, oxidized phospholipids, oxidized mtDNA, as well as extracellular ATP (eATP), released during tissue injury (351).
2.3.2. Regulation of inflammasome activation by mitochondrial dysfunction.
Dysfunctional mitochondria may initiate signals underlying NLRP3 inflammasome activation by cellular stress (FIGURE 6). Several studies have revealed that mitochondrial dysfunction can increase NLRP3 inflammasome activation (270, 356). Nakahira et al. (270) demonstrated a relationship between mitochondrial release of DAMPS and NLRP3 activation. Using bone marrow-derived macrophages (BMDMs) stimulated with LPS and ATP as a model of NLRP3-inflammasome activation, these investigators showed that NLRP3 activation was associated with compromised mitochondrial integrity, membrane depolarization (−ΔΨm), and elevated mtROS generation. Importantly, loss of mitochondrial integrity was associated with mtDNA release into the cytosol and was required for NLRP3 inflammasome activation and, subsequently, increased maturation and secretion of IL-1β and IL-18 (270). Application of mitochondrial-targeted antioxidants such as mitoTEMPO to scavenge mtROS or reduction of cytosolic DNA by DNase transfection inhibited NLRP3 inflammasome activation and caspase-1-dependent IL-1β secretion (270). The translocation of mtDNA from the mitochondria to the cytosol was NLRP3 dependent as it was reduced in macrophages isolated from Nlrp3−/− mice (270). Application of the ETC inhibitor rotenone increased macrophage NLRP3 inflammasome activation, thereby implicating mtROS in the activation mechanism (356). In subsequent studies mtDNA was reported to act as a direct binding signal for NLRP3 inflammasome activation, preferentially in its oxidatively modified form (280). Oxidized mtDNA was found in association with NLRP3 in activated macrophages, and 8-hydroxy-guanosine (8-OHdG) competitively inhibited proinflammatory cytokine secretion in these cells (280). The mitochondrial membrane lipid CL implicated in apoptosis regulation was also proposed to directly activate NLRP3 via complex formation (330).
FIGURE 6.
Regulation of the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome by mitochondrial DAMPs. The NLRP3 inflammasome is activated by a 2-signal activation mechanism. A: first, a Toll-like receptor (TLR) ligand such as lipopolysaccharide (LPS) binds to TLR4 and induces inflammasome priming via nuclear factor-κB (NF-κB) activation. B: NF-κB drives NLRP3 transcription and the expression of pro-forms of caspase-1, IL-1β, and IL-18. C: a second activating signal, typically ATP, activates P2X purinoceptor 7 (P2Rx7) and induces K+ efflux. D: NLRP3 forms a complex with the adaptor molecule apoptosis-associated speck-like protein containing a COOH-terminal caspase recruitment domain (ASC) and pro-caspase-1. E: mitochondrial dysfunction by injurious stimuli will trigger the release of mitochondrial reactive oxygen species (mtROS) and mitochondrial DNA (mtDNA); the oxidized form of mtDNA (ox-mtDNA) has been implicated in NLRP3 inflammasome regulation. Pink1/Parkin-dependent mitophagy removes depolarizing mitochondria via the lysosomal degradation pathway and thereby acts as an inhibitor of NLRP3 inflammasome activation. F: inflammasome activation triggers the cleavage of caspase-1, which in turn promotes the maturation and secretion of inflammasome cytokines IL-1β and IL-18. G: activated caspase-1 also triggers membrane disruption and pyroptotic cell death via activation of gasdermin-D (GSDMD) via its active NH2 terminal (GSDMD-N). −ΔΨm, mitochondrial membrane potential (loss).
Intriguingly, NLRP3 activation by proinflammatory stimuli is negatively regulated by cellular macro-autophagy and by its mitochondria-specific selective form, mitophagy. Autophagy generally directs the lysosome-dependent degradation of cellular components, including mitochondria, via autophagosomal delivery (357). Autophagy gene Atg16-deficient mice and BMDMs isolated from LC3B-deficient mice showed a phenotype of increased caspase-1-dependent production of IL-1β and IL-18 in response to LPS and other inducing signals such as nigericin (270, 358). Application of the autophagy/mitophagy inhibitor 3-MA increased mtROS production and NLRP3 inflammasome activation (356). Thus, an emergent paradigm is that, in the absence of autophagy/mitophagy, depolarizing mitochondria can accumulate and generate excessive mtROS and release DAMPs such as mtDNA into the cytoplasm, events that trigger activation of the NLRP3 inflammasome (270, 356).
Aside from mitochondrial DAMPs, other mitochondria-dependent signaling molecules can cross talk to virus-dependent inflammasome activation (359). NLRP3 inflammasome signaling can be enhanced by genetic deficiency in PINK1 (360). Furthermore, MAVS was found to be required for NLRP3 inflammasome activation in response to activation by certain RNA viruses but not by other stimuli (361).
In addition to mtROS/mtDNA, inflammasome activation may also be linked to mitochondria-dependent metabolic pathways. For example, mitochondrial uncoupling protein-2 (UCP2) is an IMM protein that can regulate the ETC as well as mitochondrial export of metabolic precursors (362). Deficiency of UCP2 in astrocytes has been shown to increase ER stress and activate the NLRP3 inflammasome, and thereby promote neuroinflammation (363).
Taken together, these studies have uncovered profound metabolic components in macrophage inflammasome activation profiles, which may be significant in diseases involving heightened inflammatory responses.
2.4. Mitochondrial Regulation of Antiviral Responses
Mitochondria contribute significantly to the regulation of antiviral defenses (FIGURE 7). The mitochondrial antiviral signaling (MAVS) protein has emerged as an integral OMM protein that has a regulatory function in innate immune responses. MAVS can signal NF-κB as well as regulate IFN1 responses, which defend against a number of known viral infections including but not limited to coronaviruses, West Nile virus, Japanese encephalitis virus, influenza A virus, and Sendai virus (195, 364, 365). MAVS is alternately known as CARDIF, VISA, and IPF-1 (366, 367, 368, 369). MAVS localization is not exclusive to the mitochondria, as this protein has also been found associated with the ER-MAM and peroxisomes (259, 370, 371). The MAVS protein contains a COOH-terminal transmembrane domain, which anchors it to the OMM, and an NH2-terminal caspase recruitment domain (CARD), which facilitates interaction with other CARD domain-containing proteins. These include retinoic acid-inducible gene-I (RIG-I)-like receptor RNA helicases (RLRs) that bind to double-stranded (ds) viral RNA, such as RIG-I and melanoma differentiation-associated gene 5 (MDA5) (366, 372, 373). MAVS also contains a middle proline-rich region that facilitates interactions with TNFR-related factor (TRAF) family proteins responsible for downstream signaling events (366, 372).
FIGURE 7.
Mitochondria-dependent anti-viral responses. Mitochondrial antiviral-signaling protein (MAVS) is an OMM protein that serves as a platform for the regulation of antiviral responses. During viral infections, RIG-I (retinoic acid-inducible gene I) and MDA5 serve as cytosolic pattern recognition receptors (PRR) responsible for initiating the type-1 interferon (IFN1) response. Viral double-stranded RNA (v-dsRNA) binds to RIG1 (or MDA5) to activate the protein. RIG-I is covalently modified with K63-Ub chains by the E3 ligase tripartite motif 25 (TRIM25). The NH2-terminal caspase activation and recruitment domains (CARDs) of activated RIG-I target and bind the CARDs of MAVS. Activated MAVS forms filamentous structures that are prerequisite for downstream signaling. One arm of the MAVs pathway activates a canonical nuclear factor kappa-b (NF-kB)-dependent pathway that regulates pro-inflammatory cytokines (e.g., IL-1b) production. This pathway depends on complexes with TNF receptor-associated factors (TRAF)-2/-5/-6, TNFR1-associated death domain protein (TRADD), tripartite motif 14 (TRIM14) and other proteins such as FADD and RIPK1. A second arm of the MAVS pathway involves complexes of TRAFs-2/-3/-5/-6 to activate the IFN1 response. This pathway also requires NEMO/IKKe and TBK1, which phosphorylates and activates IRF3 and IRF7. Homodimerization of IRF3 and IRF7 triggers the transcription of IFN genes. MAVS can be stimulated via release of DAMPs from mitochondria, such as mtDNA, which activates MAVS via a cGAS/cGAMP/STING axis. MAVS activation can be inhibited by fusion protein MFN2 and interactions with the ER MAM, and by other factors including NLRX1 and gC1qR. MAVS and RIG-I are marked for proteasomal degradation via K48-Ub modification by PCB1/PCBP2 or RNFs, respectively. cGAS, cyclic GMP-AMP synthase; cGAMP, cyclic GMP-AMP; gC1qR, globular C1q receptor; DAMP, damage associated molecular pattern; ER, endoplasmic reticulum; FADD, Fas-associated protein with death domain; IFN1, interferon type 1; IKK, I-kB kinase (a, b, e); IRF-3/-7, interferon regulatory factor-3/-7; I-kBa, inhibitor of NF-kB (alpha); MAM, mitochondria-associated membrane; MDA5: melanoma differentiation-associated protein-5; MFN2: mitofusin-2; mtDNA, mitochondrial DNA; NEMO, NF-kB essential modulator; NLRX1, NLR family member X1; PCBP-1/-2, poly(RC)-binding protein (PCBP)-1,-2; RIPK1, receptor-interacting serine/threonine-protein kinase 1; RNF, ring finger proteins; STING, stimulator of interferon genes; TBK1, TANK-binding kinase-1; TRADD, TNFR1-associated death domain protein.
In response to viral infection, RIG-I functions as a PRR and binds to viral dsRNA and can be regulated by ubiquitination via several E3 ubiquitin ligases (374, 375). RNF125 inhibits RIG-I signaling by promoting its proteasomal degradation by ubiquitination at Lys 48 (376). Tripartite motif-containing protein-25 (TRIM25) and Riplet can activate RIG-I. TRIM25 catalyzes Lys 63 polyubiquitination at Lys 172, which promotes interaction with the MAVS CARD domain (374, 377, 378). RLR activation by viral dsRNA also triggers MAVS ubiquitination on lysine 7 and 10 by TRIM25, also priming MAVS for proteasomal degradation concomitant with downstream signaling (379). In the presence of K63 ubiquitin chains, RIG-I catalyzes the filamentous aggregation of MAVS on OMM to form prionlike aggregates (380, 381). The formation of MAVS aggregates is believed to be the cardinal initiating event in the activation of downstream signals (381).
MAVS operates two key signaling pathways in innate immunity culminating in either IFN1- or NF-κB-dependent responses. MAVS can form complexes with TRAF-2, -3,- 5, and -6. These in turn activate the TBK1 complex (TBK1, IKKι/ε, and NEMO). The TBK1 complex phosphorylates and promotes dimerization of IRF3 or IRF7, which in turn translocate to the nucleus. These factors bind IFN response elements and induce IFN1 gene expression (382, 383). Alternately, MAVS complexes with TRAF-2, -5, and -6 activate an IKK complex (IKKα/β and NEMO). This complex is responsible for NF-κB activation, which regulates proinflammatory gene expression (372, 384). Recent studies suggest that the mitochondria-localized deubiquitinase USP18 assists in the formation of K63-linked polyubiquitination of MAVS, via interactions with TRIM31 (385).
A role for PINK1-dependent mitophagy has recently emerged in the cross-regulation of MAVS, underscoring the interrelationship of mitochondrial processes. During mitochondrial dysfunction, PINK1 was found to form a complex MAVS that effectively inhibited multimeric MAVS aggregation. Both MAVS-mediated antiviral innate immune and NLRP3 inflammasome signaling were found to be enhanced by genetic deficiency in PINK1 (360). These studies evoke intriguing relationships between mitochondrial quality control mechanisms and activation of inflammatory pathways (360, 386).
Emerging studies also suggest cross-regulation of MAVS signaling with processes that regulate mitochondrial dynamics. MAVS can be activated by Mff, which senses mitochondrial energy charge. Mff mediates the formation of active MAVS clusters on mitochondria, independently of Drp1 and the process of mitochondrial fission. During mitochondrial dysfunction, the energy-sensing kinase AMPK can phosphorylate Mff, leading to the disorganization of MAVS clusters and repression of the acute antiviral response. Mff exerts a crucial function in MAVS-dependent innate immune responses by sensing mitochondrial energy metabolism via AMPK (387). Conversely, recent studies suggest that upon activation of MAVS signaling TBK1 can directly phosphorylate DRP1/DNM1L, which impairs DRP1 oligomerization and its functioning in mitochondrial fission. The TBK1-DRP1 axis was essential for assembly of large MAVS aggregates. Interference with TBK1-DRP1 signaling compromised antiviral responses (388).
Some studies have also demonstrated a relationship between mitochondrial fusion and antiviral signaling pathways. For example, mouse embryo fibroblasts (MEFs) deficient in mitochondrial fusion as the result of targeted deletion of both Mfn1 and Mfn2 exhibited impaired induction of IFNs and proinflammatory cytokines in response to viral infection, resulting in increased viral replication (389). MEFs with null mutations in either Mfn1 or Mfn2, however, retained antiviral responses. Reduced mitochondrial membrane potential (−ΔΨm) correlated with reduced antiviral response (389). In contrast, overexpression of Mfn2 was shown to inhibit RIG-I-, MDA-5-, and MAVS-dependent activation of IRF-3 and NF-κB. Genetic deficiency of Mfn2 augmented infection-associated IFN-β and thereby improved antiviral responses. Thus, Mfn2 may modulate antiviral signaling in a manner independent of its role in mitochondrial fusion (390).
Mitochondrial activation of MAVS signaling may significantly contribute to host defense in infectious diseases, whereas deficiencies in this pathway may increase susceptibility to disease. Increased understanding of mitochondria signaling to inflammatory and host defense processes may inform therapeutic development in infectious diseases.
3. MITOCHONDRIAL DYSFUNCTION AND METABOLISM IN THE REGULATION OF CELL DEATH
Mitochondrial dysfunction can influence the regulation of distinct cell death pathways, which in turn may contribute to the pathogenesis of specific diseases. Cell death is generally divided into two overarching categories: regulated cell death (RCD), involving coordinated, genetically regulated programs, and nonregulated forms defined as cataclysmic or accidental cell death (ACD) (391–394) Apoptosis represents the most widely studied, classical form of RCD. This mode of cell death is nonlytic, requiring the activation of a cascade of protease activities within an intact plasma membrane, and is morphologically characterized by cytosolic shrinkage, plasma membrane blebbing, chromatin condensation, and DNA fragmentation (395). In contrast, necrosis, a classical form of ACD, is defined by loss of energy charge (i.e., increased AMP-to-ATP ratio), cell swelling, plasma membrane damage, cell lysis, and leakage of cytosolic constituents, which may subsequently trigger inflammatory responses in surrounding tissue.
Newly recognized forms of RCD, which bear features of necrosis but are regulated by cellular programs, have been defined by their distinct morphological and biochemical characteristics. Of RCD, the major types include MPT-dependent necrosis, necroptosis, pyroptosis, and others (392). These subtypes differ in their biochemical characterization but represent inflammatory, lytic forms of RCD, each with potential significance in human disease pathogenesis. Although apoptosis represents a cardinal example of mitochondria involvement in programmed cell death regulation, altered mitochondrial functions, including bioenergetics, turnover, and dynamics, can also affect the regulation or outcome of other nonapoptotic RCD pathways. This section explores how mitochondrial dysfunction can impact signaling networks and the evidence for their regulation of apoptosis and other specific forms of RCD.
3.1. Apoptosis
Mitochondria are integral players in the regulation of apoptosis (396, 397). Cells can trigger apoptosis via several alternate signaling pathways, which ultimately converge to mitochondria-dependent events. The “intrinsic” apoptotic pathway represents a major mechanism by which exposure to adverse environmental agents triggers apoptosis via signals that converge on mitochondrial integrity. This pathway is dependent on activation of a proteolytic cascade for both regulation and execution via cysteinyl-aspartate specific proteases (caspases). The intrinsic apoptotic pathway is regulated by multiple protein factors of the Bcl-2 family. These include proapoptotic proteins involved in mitochondrial outer membrane permeabilization (MOMP), a key trigger of apoptosis (i.e., Bax, Bak, Bok), the proapoptotic BH3-only proteins [i.e., Bid, Bim, Bik, Bmf, Bad, Puma, Noxa, and Mcl1-s (short variant)], and antiapoptotic proteins [Bcl-2, Bcl-XL, Bcl-W, and Mcl1-l (long variant)] (398–401). These proteins can form homodimers and heterodimers via their Bcl-2 homology (BH) domains. The balance of Bcl-2 family protein abundance and interactions is critical in cell fate determination (402). In response to diverse stimuli, proapoptotic Bcl-2 family proteins such as Bax initiate the intrinsic apoptotic pathway by forming channels in the OMM, thereby facilitating the release of Cyt-c and other proapoptotic mediators from the IMS (403–405). CL exerts an important function in apoptosis signaling (41). CL is also implicated as a platform that facilitates the recruitment of Bax/Bak oligomers to the OMM for initiation of apoptosis. Cyt-c-specific peroxidase activity can trigger CL oxidation, leading to the release of Cyt-c (406). The release of Cyt-c during apoptosis activation is accompanied by substantial mitochondrial crista remodeling (398–401).
Once released to the cytosol, Cyt-c forms an oligomeric complex termed the “apoptosome” with Apaf-1, dATP, and pro-caspase-9. This complex activates caspase-9 and, in turn, its downstream caspase-3, resulting in the activation of downstream caspases (i.e., caspase-3, -7) and the apoptotic program. Additional mitochondria-derived factors that are released into the cytosol include second mitochondria-derived activator of caspases/direct inhibitor of apoptosis (IAP)-binding protein with low pI (Smac/Diablo), Omi/HtrA2, and apoptosis-inducing factor (AIF) (407–410). In apoptotic cells, Smac/Diablo released into the cytosol promotes caspase activity by antagonizing inhibitors of apoptosis (IAPs), which include X-linked IAP (XIAP), c-IAP1, c-IAP2, and survivin (407).
In contrast, the extrinsic apoptotic pathway initiates when a death-inducing ligand, such as FasL, interacts with its cell surface receptor (i.e., Fas), forming a death-inducing signal complex (DISC) (411). Activation of Fas triggers its oligomerization and the rapid recruitment of FADD (Fas-associated death domain protein) and caspase-8 to the cytoplasmic death domain of Fas, which activates caspase-8. Active caspase-8 subsequently cleaves Bid into its truncated form (tBid), which translocates to the mitochondrial membrane, where it triggers Cyt-c release and subsequent caspase-9 activation (411).
Accumulated evidence suggests that cross-regulation of mitochondrial dynamics with that of apoptosis can occur. The relationships between mitochondrial fission, crista remodeling, and apoptosis remain incompletely understood. PARL, which is implicated in the regulation of mitophagy and mitochondrial dynamics, also differentially regulates apoptosis. PARL-knockout MEFs displayed enhanced cristate remodeling and faster rate of Cyt-c release. The IMM-resident lipid CL enhances PARL activity in vitro. In addition to regulating Pink1 turnover, PARL was found to cleave PGAM5 and Smac/Diablo (412, 413). PARL-dependent cleavage of Smac in the IMM exposes an NH2-terminal IAP-binding motif, which permits its proapoptotic inhibition of XIAP (414, 415). Loss of PARL was shown to prevent the proapoptotic function of Smac by preventing Smac maturation (414).
The Drp1 GTPase that regulates fission also cross talks to apoptosis. Drp1-dependent mitochondrial fission requiring MiD49/MiD51 was shown to regulate crista remodeling during intrinsic apoptosis (416). Although both Drp1- and MiD49/MiD51-knockout cells were refractory to crista remodeling, the phenotype in MiD49/MiD51-knockout cells was sensitive to Opa1 disruption (416). Drp1 was found to participate in Cyt-c release during stimulation with activators of intrinsic apoptosis. Drp1−/− MEFs were protected from oxidant- and Bid-induced apoptosis. Cyt-c release was found to be dependent on Drp1 but not specifically on mitochondrial fragmentation during intrinsic apoptosis. Drp1−/− MEFs also displayed reduced crista remodeling in the context of apoptosis resistance. This study concluded that Drp-1 promotes apoptosis in part by regulating crista remodeling independently of mitochondrial fission (417). In contrast, deficiency of MIEF1/MiD51, which acts as a recruitment factor for Drp1 during fission, sensitized cancer cells to mitochondrial dysfunction, PINK1/Parkin-dependent mitophagy, and triggered Bax-mediated apoptosis (418). Recent interactome studies revealed putative interactions of the Bcl-2-family protein Bok with Drp-1, further suggesting cross-regulation of fission and apoptosis (419). Drp1 was also implicated as a downstream effector of BH2-only mimetic-induced apoptosis, independently of its role in regulating fission (420). Decline in Mcl1-l-to-Mcl1-s ratio is known as a proapoptotic signal, though recent studies indicate cross talk to regulation of mitochondrial dynamics (398). Artificial oligonucleotide-driven reduction of Mcl1-l-to-Mcl1-s ratio, a proapoptotic signal, was associated with mitochondrial hyperfusion, which was also found to be dependent on Drp1 regulation (398).
Opa1, a GTPase implicated in the regulation of fusion, also contributes to the regulation of apoptosis (421). OPA1 functions to sequester Cyt-c in mitochondrial cristae, independently of its regulation of the fusion process (422). Cleavage of L-Opa1 by the protease Oma1 inhibits mitochondrial fusion and primes cells for apoptosis (421). Oma1 turnover is regulated both by CL binding and by the prohibitin-1/2 (PHB-1/2) complex of the IMM. PHB exerts antiapoptotic effects in neurons by decreasing tB499-dependent Cyt-c release and inhibiting caspase-9 activation (421). The mitochondrial protease PARL also regulates Opa1. PARL−/− MEFs were refractory to antiapoptotic effects of Opa1 and had reduced Opa1 levels in the IMS (423).
Genetic deficiency of hFis1 (an inhibitor of Opa1 GTPase) inhibited cell death more potently than deficiency of Drp1. Opa1-deficient cells were sensitized to exogenous apoptosis induction, in a hFis1-dependent manner. Opa1 was proposed as an antiapoptotic inhibitor of Fis1. The authors concluded that mitochondrial fission itself is not responsible for apoptosis but regulators of fission and fusion can positively and negatively cross-regulate apoptosis (424). A chemical inhibitor of DRP1 was shown to prevent mitochondria division and Bax-mediated OMM permeabilization during apoptosis (425).
The relationship between mitophagy and apoptosis remains controversial. Activation of mitophagy as an initial cellular protective stratagem may prevent propagation of mitochondrial dysfunction, by turning over depolarizing or damaged mitochondria, thus precluding intrinsic apoptosis (426). PINK1 and associated mitophagy is implicated in neuroprotection for its antiapoptotic effects (427). PINK1 was found to cross-regulate apoptotic programs via the phosphorylation of the antiapoptotic protein Bcl-XL, which prevents its cleavage (428). PINK1 may also exert an antiapoptotic function by phosphorylating Bad at Ser 112/136 (429). PINK1 also phosphorylates the mitochondria-released death effector HtrA2 (430). Furthermore, Parkin may exert an antiapoptotic effect on the OMM via the ubiquitination of Bax at Lys 128 (431). A proapoptotic role for Parkin was implied by ubiquitination of Mcl1-l (432). Contrasting evidence from select model systems also suggests that PINK1 or Parkin may promote cell death. Prolonged overexpression of PINK1 or Parkin triggered a caspase-independent cell death, independent of mitochondrial turnover (433). PINK1 deletion in epithelial cells protected epithelial cells from cell death with features of nonapoptotic RCD (necroptosis) in response to cigarette smoke (CS) exposures, whereas knockdown of the autophagy protein LC3 protected against apoptosis in this model (434). The protective or detrimental role of Pink1-dependent mitophagy, and autophagy in general, with regard to RCD pathways may thus vary in a context-specific fashion and may be related to the degree of mitochondrial injury.
3.2. Regulated Necrotic Cell Death
Necrotic cell death is now understood to represent independent RCD programs, including cyclophilin D (CypD)-dependent MPT and receptor interacting protein kinase-3 (RIPK3)-dependent necroptosis (391). These divergent cell death processes converge on a necrotic morphology characterized by organelle swelling, including mitochondria, increased cellular volume, and plasma membrane rupture with the release of immune activating ligands termed DAMPs, with implications for human disease (435). Both MPT-dependent necrosis and RIPK3-dependent necroptosis are associated with accumulation of ROS, impaired OXPHOS resulting in decreased production of ATP, and loss of cellular bioenergetics and resultant cell death (436–438). Yet mitochondria are required for MPT whereas mitochondrial depletion does not interfere with RIPK3-dependent necroptosis, highlighting the independence of these seemingly related forms of regulated necrotic cell death.
3.2.1. Mitochondrial permeability transition-driven necrosis.
Under normal physiological conditions, the mPTP complex assembled between the IMM and OMM establishes a transient throughfare for small solutes such as Ca2+ and mtROS to maintain cellular health. However, under specific intramitochondrial perturbation, such as excessive mitochondrial matrix ROS or Ca2+, prolonged increases in the permeability of the IMM can lead to MPT, with loss of mitochondrial matrix proteins into the cytosol and disrupted cellular bioenergetics, directly leading to necrotic cell death. This process is dependent on CypD, which controls the sensitivity of the mPTP opening in response to various stimuli, including Ca2+ (439, 440). Beyond CypD, the other essential regulators and components of the mPTP complex are still under investigation. CypD has been directly linked to relevant IMM proteins including the ADP/ATP transporter adenine nucleotide translocase (ANT) and the mitochondrial phosphate carrier (PiC), but genetic studies of ANT and PiC have failed to demonstrate that loss of these proteins significantly alters mPTP opening (441–444). Furthermore, the F1Fo-ATP synthase, which couples protein translocation to ATP synthesis, has also been implicated, as CypD modulates the F1Fo-ATP synthase and ATP synthase dimers and monomers recapitulate mPTP function, but this requires further study (445–447). Additionally, VDAC was hypothesized to contribute to mPTP, as it shares similar properties to mPTP, but genetic studies did not support VDAC as essential to mPTP function (448).
Diverse regulators of mitochondrial health can stimulate the mPTP, including apoptosis effectors, fission proteins, and Ca2+ transporter complex molecules. Under ischemia-reperfusion conditions, the central apoptosis regulators Bax and Bak facilitate pore opening irrespective of their role in apoptosis. Loss of Bax and Bak resulted in resistance to mitochondrial membrane permeability, illustrating the interplay between apoptotic and necrotic cell death processes (449). Similarly, Drp1 facilitates mPTP opening, irrespective of its role in mitochondrial fission, when phosphorylated under conditions of β-adrenergic stimulation (450). Additionally, disruption of the MCU can lead to mPTP opening by altering mitochondrial Ca2+ metabolism. The IMM protein MICU1 is a calcium gatekeeper capable of sensing extracellular Ca2+. In hepatocytes deficient in MICU1, Ca2+ overload-induced mPTP was accelerated, highlighting how regulators of cellular health can also induce MTP-driven necrosis (451).
3.2.2. RIPK3-dependent necrosis.
RIPK3-dependent necrosis is a form of RCD that results from the sequential activation of signal transduction molecules and is mediated by necrotic cell death proteins RIPK3 and mixed-lineage kinase domain-like pseudokinase (MLKL) (391). Often referred to as nonapoptotic cell death, necroptosis was known to share the upstream mediator receptor interacting protein kinase-1 (RIPK1) with apoptotic cell death and NF-κB activation (452–454). Upon ligation of the Fas or TNF-R1 death receptors, RIPK1 and tumor necrosis factor receptor type 1-associated death domain (TRADD) are recruited to these death receptors (455, 456). RIPK1 and TRADD share a death domain, which allows for interaction to form complex 1 (457, 458, 459). Under conditions of caspase-8 depletion or cIAP deficiency, the kinase RIPK3 is recruited, setting in motion the early stages of necrotic cell death (460). Necroptosis is dependent on RIPK3 and the formation of the regulatory necrosome complex with RIPK1 and MLKL. The RIPK3-mediated phosphorylation of MLKL within the necrosome is the terminal step in necroptosis execution leading to cell membrane lysis and the release of DAMPs.
Early necroptosis investigations linked it to mitochondrial dysfunction through the observed increase in ROS in cells undergoing TNF-α-induced necrotic cell death. TNF-α induced, RIPK1-dependent necroptosis was found to result in mtROS accumulation, and both ROS and necroptosis are attenuated by the genetic depletion of RIPK1, highlighting the role of RIPK1 as an upstream initiator of both necroptosis and mitochondrial bioenergetics (453, 461). Although RIPK1 lies at the intersection of NF-κB activation and TNF-α-induced apoptosis and necroptosis, it is only RIPK1-dependent necroptosis that results in ROS accumulation. Under experimental conditions of caspase inhibition (zVAD), a model of exaggerated necroptosis, RIPK1 colocalizes to the mitochondria and is required for TNF-α-induced inhibition of ADP/ATP exchange, leading to increased ROS and decreased cellular ATP levels, and ultimately necrotic cell death (462). TNF-α-induced ROS can also indirectly induce necrotic cell death, and the administration of ROS scavengers attenuates this necroptosis response (453). RIPK1 is also essential for TNF to induce ROS, as RIPK1-deficient cells do not accumulate ROS in response to TNF (453).
The identification of the essential necroptosis protein RIPK3 led to more specific understanding of the cross talk between RIPK3-mediated necroptosis and mitochondrial bioenergetics (460, 463, 464). RIPK3 regulates TNF-induced ROS through interaction with mitochondrial-derived metabolic enzymes, including PYGI, GLUL, and GLUD1, and this is attenuated by genetic depletion of RIPK3, although this may be cell type specific (460, 464). Although the inner mitochondrial protein PGAM5 was previously linked to RIPK3 necroptosis through mobilization of Drp1, subsequent studies have shown this is not essential for necroptosis execution (465, 466). Despite these observed associations between RIPK3-mediated necrosis and mitochondrial proteins, the requirement of mitochondria for canonical necroptosis has been called into question. Using parkin-induced mitochondrial depleted cells, Tait et al. (467) demonstrated that RIPK3-mediated necrosis can proceed, highlighting that mitochondrial dysfunction is associated with but not essential for RIPK3-mediated necrosis.
Diverse stimuli can induce necroptosis through mitochondrial mechanisms. Nitration of NDUFB8 through NO administration induces RIPK1/RIPK3-mediated cell death and is neutralized by the mitochondrial antioxidant SOD2 (468). Similarly, cystine starvation induces mitochondrial fragmentation, dysfunction, and ROS production leading to necroptosis induction, a process reversed by the ROS scavenger Necrox-5 (469). Shikonin, a necroptosis inducer in cancer cells, triggers necroptosis by inducing overproduction of intracellular ROS, leading to RIPK1 and RIPK3 induction in a dose-dependent manner (470). Similarly, Mycobacterium tuberculosis toxin tuberculosis necrotizing toxin (TNT) is a NAD+ glycohydrolase capable of depleting NAD+ in macrophages, leading to activation of RIPK3 and MLKL, depolarized mitochondria, impaired ATP synthesis, and necrotic cell death (471).
Necrotic stimuli can lead to release of mitochondrial DAMPs (MTDs) into the circulation with functionally important immune consequences. MTDs include NFPs and mtDNA, which activate human polymorphonuclear neutrophils (PMNs) through FPR-1 and TLR-9, respectively (472). Circulating MTDs can elicit neutrophil-mediated organ injury. The mechanism of mtDNA release has been further elucidated. PUMA, a proapoptotic BH3-only Bcl-2 family member, is induced and plays a role in necroptotic death. PUMA induction enhances necroptotic signaling by promoting the release of mtDNA and activation of cytosolic DNA sensors (473). On induction, PUMA promotes the cytosolic release of mtDNA and activation of the DNA sensors DAI/Zbp1 and STING, leading to enhanced RIPK3 and MLKL phosphorylation in a positive feedback loop. Deletion of PUMA partially rescues necroptosis-mediated developmental defects in FADD-deficient embryos (473).
3.3. Alternate Cell Death Pathways: Pyroptosis
Alternate lytic, inflammatory cell death pathways, including caspase-mediated pyroptosis, have been mechanistically linked to mitochondrial processes. Pyroptosis is a form of regulated cell death that requires the inflammasome signaling platform to produce caspases capable of activating gasdermin proteins (specifically GSDMD and GSDME), pore-forming executioner proteins capable of cytolysis and release of inflammatory mediators (474–476). Until recently, the regulation of GSDMD oligomerization and pore formation was poorly understood. After caspase-mediated processing of GSDMD into the pore-forming NH2-terminal GSDMD (NT-GSDMD), NT-GSDMD translocates to the cell surface, where oligomerization occurs to form the lytic pore. This oligomerization has now been shown to depend on mtROS. mTOR Complex 1 (mTORC1), a metabolic signaling complex that interacts with the Ragulator-Rag complex on the surface of the lysosome to regulate protein synthesis in response to amino acid levels, also regulates ROS production in mitochondria. Either deficiency or inhibition of Rag proteins or mTORC1 elements as well as ROS inhibition reduced GSDMD pore formation and lytic cell death (477). Pyroptosis also cross talks to other cell death processes, including apoptosis, through mitochondrial permeabilization (478). NT-GSDMD/E capable of cell membrane pore formation can also target the mitochondria to promote Cyt-c release to augment mitochondrial apoptotic pathways.
3.4. Sirtuin Regulation of Programmed Cell Death
As cell survival enzymes working in part through mitochondrial processes, sirtuins regulate numerous genes that counter cell death programs. SIRT1 upregulates the deacetylation of FOXO, which in turn activates pro-survival pathways, including promoting cell cycle arrest, limiting oxidative stress by increasing expression of antioxidant molecules, and regulating DNA damage genes, such as RAD51, increasing DNA damage repair (479, 480). Excessive induction of FOXO can paradoxically increase cell death through the induction of proapoptotic genes and interference with cell cycle progression. To limit this excessive impact of FOXO on cell survival, FOXO is degraded by ubiquitination, restoring balance to this senescence pathway by limiting FOXO-induced cell death (481, 482).
SIRT1 regulation of NF-κB is also pro-survival, leading to increased expression of anti-apoptosis-related genes, including inhibitor of apoptosis proteins (IAPs), the Bcl-2 family, TNFR-associated factor (TRAF-1, TRAF-2), and JNK (483), thereby controlling apoptosis (479, 484). SIRT1 also regulates the expression of the master mitochondrial regulator, PGC-1α. SIRT1-dependent acetylation of PGC-1α and the subsequent interaction with several downstream transcription factors can profoundly influence apoptosis through mitochondrial mechanisms, including modulation of the anti-apoptotic Bcl-2 family proteins and controlling redox homeostasis through transcriptional coactivation of nuclear respiratory factors (NRF-1 and NRF-2) and mitochondrial transcription factor A (TFAM), promoting the transcription of genes involved in mitochondrial biogenesis. This SIRT1 regulation of PGC-1α endows SIRT1 with the ability to control mitochondrial function and in turn indirectly influences cellular apoptosis (479).
The mitochondrial sirtuin SIRT3 also regulates programmed cell death, including apoptosis and mPTP opening, through deacetylation of mitochondrial proteins. This regulation is cell type and context dependent (485). In metabolically active tissues such as the heart, SIRT3 promotes cell survival by deacetylating CypD (486). In cardiac myocytes, NAD+-dependent SIRT3 deacetylation of CypD lysine 166 attenuates mPTP opening and mitochondrial swelling, a phenotype reversed by the CypD inhibitor cyclosporin-A (486). SIRT3 can similarly regulate apoptosis under stress conditions in the heart (487). During stress, SIRT3 levels are increased in murine cardiac myocytes, where deacetylate of Ku70 impedes translocation of the proapoptotic protein Bax to mitochondria. This contrasts with the role of SIRT3 in other disease states, such as malignancy, where SIRT3’s regulation of cell survival supports its function as a tumor promoter or suppressor, depending on tumor and cell type (485).
4. MITOCHONDRIAL DYSFUNCTION IN CARDIOVASCULAR AND PULMONARY DISEASE
4.1. Age-Related Cardiovascular Disease
Cardiovascular disease (CVD), an umbrella term encompassing coronary heart disease, aortic atherosclerosis, peripheral artery disease, and cerebrovascular disease, is the leading cause of death globally (488). In 2019, 18.6 million deaths worldwide were attributed to CVD, representing a 17.1% increase from 2010 (488). The global age-adjusted mortality decreased by 11.1% during this period, suggesting that significant progress has been made in the prevention and treatment of CVD (488). This accomplishment has been attributed to addressing CVD risk factors such as hypertension, diabetes, dyslipidemia, obesity, inactivity, and smoking (489).
However, age represents a major unmodifiable risk factor for CVD. This age-related disease progression is driven by a series of structural and functional changes that compromise arterial elasticity and endothelial homeostasis (490). As part of the aging process, endothelial cells lose their ability to maintain a balanced redox state and exhibit a proinflammatory phenotype (490). As the center of oxidative metabolism, mitochondria play a decisive role in age-related endothelial dysfunction and the subsequent development of CVD (FIGURE 8).
FIGURE 8.
Mitochondrial dysfunction in age-related cardiovascular disease (CVD). 1: The proximity of the mitochondrial genome to the ETC is associated with the age-related accrual of mutations, which ultimately impair mitochondrial bioenergetics and increase mtROS. 2: To manage the rise in mtROS, superoxide dismutase 2 (SOD2) converts O2•− to H2O2, an uncharged lipophilic molecule that transverses mitochondrial membranes to enter the cytosol. Once in the cytosol, H2O2 can impair cell function by causing oxidative damage to cellular components (DNA, proteins, and lipids) 3: Alternatively, H2O2 can stimulate NF-κB nuclear translocation to promote the expression of proinflammatory genes such as TNF-α, IL-6, VCAM-1, ICAM-1, and MMP-9 that are important for atherosclerotic plaque formation. By upregulating proinflammatory cytokines, the NF-κB signal transduction cascade can become self-sustaining, resulting in a chronic inflammatory state. Of note, several other pathways, such as the renin-angiotensin system and TNF-α, can contribute to the development of age-related CVD by converging on NF-κB (not shown). 4: During aging, NAD+ levels decline, impairing sirtuin function. Decreased sirtuin 1 (SIRT1) activity is associated with downregulation of endothelial nitric oxide synthase (eNOS) and increased production of lipids, and inflammatory cytokines. 5: In contrast, decreased sirtuin 3 (SIRT3) activity is associated with downregulation of SOD2 and PGC-1α, which further compromises mitochondrial function, bioenergetics, and biogenesis. See glossary for abbreviations. Image created with BioRender.com, with permission.
The mitochondrial genome is particularly susceptible to mutation and oxidative damage given the proximity of the ETC and inefficient mtDNA repair mechanisms (282). Over time, mtDNA damage impairs ETC function, thereby compromising mitochondrial bioenergetics and increasing mtROS (491, 492). To mitigate the ensuing rise in O2•−, mitochondria rely on SOD2 for the conversion of O2•− to H2O2. As an uncharged lipophilic molecule, H2O2 freely traverses mitochondrial membranes to enter the cytosol, causing oxidative damage to cellular components (DNA, proteins, and lipids) (493). Other noteworthy sources of age-related O2•− production include upregulation of NOX and endothelial nitric oxide synthase (eNOS) uncoupling (494, 495).
As a further complication, intracellular SOD production does not increase with age (494, 496). The absence of compensatory mechanisms to increase SOD2 expression effectively places mitochondrial antioxidant defense at a loss. Consequently, excess O2•− is free to damage mitochondrial components via H2O2 generation or oxidize NO to ONOO− (490). The significance of this reaction is twofold. First, NO depletion promotes the development of hypertension by divesting endothelial cells of a potent vasodilator. Second, ONOO− causes nitrosative damage to cellular components. Markers of oxidative or nitrosative stress (i.e., glutathionylation, nitrotyrosine, 4-hydroxynonenal, and malondialdehyde) have been found in abundance in aged arteries, highlighting the significance of this phenomenon (497–499). This time-dependent accumulation of oxidative and nitrosative damage is often referred to as the oxidative stress theory of aging and is a major contributor to age-related CVD (493).
Cells often rely on autophagy, mitophagy, and mitochondrial fission or fusion to mitigate the harmful effects of oxidative damage. However, dysregulation of these processes during aging can accelerate CVD. As an example, autophagy is reduced over time, allowing oxidative damage to accumulate, which propagates further oxidative stress and inflammation (500). In contrast, overexpression of Atg5 in mice can increase their life span by inducing autophagy (501). Furthermore, impairment of endothelial cell autophagy by Atg3 knockdown was associated with increased oxidative stress and cytokine production in response to shear stress (502). Additionally, these endothelial cells displayed decreased activation of eNOS and diminished NO production. Mitophagy is of particular importance to the pathogenesis of myocardial ischemia-reperfusion injury, where the inciting event causes loss of cristae and mitochondrial swelling (503, 504). Ordinarily, mitophagy would be protective, but excessive Drp1 activation initiates a sequence of events that terminates with cardiomyocyte apoptosis and ventricular dysfunction (503, 504).
Besides causing cellular damage, mtROS provide a link between age-related oxidative stress and chronic inflammation. Oxidants, such as H2O2, activate IKK, causing I-κB degradation (493). This enables NF-κB nuclear translocation, which promotes a proinflammatory gene expression profile characterized by cytokine production (i.e., TNF-α and IL-6), upregulation of leukocyte adhesion molecules (i.e., VCAM-1 and ICAM-1), macrophage conversion to foam cells, and upregulation of matrix metalloprotease-9 (MMP-9) expression (493, 505–507). These changes are vital for atherosclerotic plaque formation and vascular remodeling. Several other pathways, such as the renin-angiotensin system and TNF-α, augment age-related CVD by converging on NF-κB (493). Moreover, by recruiting immune cells and upregulating proinflammatory cytokines, the NF-κB signal transduction cascade can become self-sustaining through continuous ROS exposure leading to a chronic proinflammatory state.
Another regulator of age-related CVD is mitochondrial adaptor protein p66Shc. Posttranslational phosphorylation by protein kinase C-β (PKC-β) promotes translocation of p66Shc to the IMS, where it complexes with TIM/TOM (492, 508, 509). Activated p66Shc transfers electrons from reduced Cyt-c to O2, generating H2O2 and promoting apoptosis via the MPTP (509). In this manner, p66Shc regulates oxidative stress and mitochondria-mediated apoptosis. Animal models have shown that p66Shc−/− mice are resistant to oxidative stress, are protected against age-related endothelial dysfunction, and exhibit an extended life span (499, 510, 511). This stems from decreased ROS/RNS production and increased NO bioavailability. Interestingly, H2O2 activates PKC-β, suggesting a possible feedforward mechanism.
In endothelial cells, oxidized low-density lipoprotein (ox-LDL) binds oxidized low-density lipoprotein receptor 1 [OLR1 or lectin-type oxidized LDL receptor 1 (LOX-1)], precipitating NOX-mediated ROS activation of PKC-β and p66Shc (512). Accordingly, p66Shc−/− mice exhibit less hyperlipidemia-induced oxidative stress and apoptosis than their wild-type counterparts despite equivocal lipid levels (512, 513). Additionally, p66Shc deletion protects apolipoprotein E-knockout mice from atherosclerotic plaque formation (512, 514). Furthermore, hypertension studies involving p66Shc have found that endothelial stretch and angiotensin II cannot promote hypertension or endothelial dysfunction in p66ShcRNAi mice (512, 515, 516).
During aging, NAD+ levels decline, thus depriving sirtuins of their requisite cofactor (517). In the case of SIRT1, decreased activity causes endothelial dysfunction through upregulation of p66Shc, downregulation of eNOS, possible upregulation of angiotensin II, as well as increased production of lipids and cytokines (512, 518). Furthermore, decreased SIRT1 activity is associated with a decline in Von Hippel–Lindau (VHL) E3 ubiquitin ligase, which ultimately permits normoxic HIF-1α stabilization (517). Besides SIRT1, the age-related decline in SIRT3 activity is associated with downregulation of SOD2 and PGC-1α (519). Collectively, these metabolic changes contribute to aging by compromising mitochondrial function, bioenergetics, and biogenesis while simultaneously promoting senescence. Importantly, endothelial senescence may propagate oxidative stress and vascular inflammation among healthy, nonsenescent neighboring cells via a senescence-associated secretory phenotype (SASP) (490).
As demonstrated by the above, age-related CVD is characterized by structural and functional changes emanating from time-dependent changes in mitochondrial function. Understanding how to ameliorate mitochondrial oxidative stress and the organelle’s functional decline over time is inherent to the treatment of age-related CVD.
4.2. Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH), one of five groups of pulmonary hypertension (PH), is a rare disease affecting the pulmonary vasculature. Although accurate estimates of PAH prevalence are limited by disease heterogeneity and the lack of a global registry, best estimates approximate PAH prevalence at 5–52 cases per million adults (520, 521). Globally, schistosomiasis is the most common cause of PAH; however, in regions where schistosomiasis is not endemic, idiopathic and inheritable causes (i.e., BMPR2, ALK1, ENG) predominate (522). Additional etiologies of PAH include congenital heart disease, persistent PH of the newborn syndrome, drugs or toxins (i.e., appetite suppressants, dasatinib, amphetamines), underlying medical conditions (i.e., connective tissue diseases, HIV, portal hypertension), and pulmonary veno-occlusive disease or pulmonary capillary hemangiomatosis (522).
Despite being etiologically diverse, PAH is characterized by pulmonary artery remodeling through hypertrophic changes, neovascularization, aberrant cell proliferation, neointimal fibrosis, and in situ thrombosis (523). As this proliferative vasculopathy develops, pulmonary vascular resistance (PVR) rises, placing undue stress on the right ventricle. Initially, the right ventricle maintains forward flow through adaptive hypertrophy (524). However, chronic exposure to elevated PVR will ultimately precipitate right heart failure (524). Clinically, this manifests as fatigue, dyspnea, hypoxia, and fluid retention in the form of hepatic congestion, ascites, and lower extremity edema. If untreated, PAH has a median survival of 2.8 yr (522).
Fundamental to the development of PAH is dysregulation of mitochondrial bioenergetics. Specifically, there is a shift from oxidative phosphorylation to glycolysis and lactic acid fermentation to maintain ATP production. This phenomenon, known as the Warburg effect, was first described in tumor cells and has paved the way for comparisons between diseases (525). As seen in cancer, glycolytic shift in PAH causes hyperpolarization of the mitochondrial membrane, which promotes apoptosis resistance by inhibiting the release of proapoptotic factors. The net result is the development of hyperproliferative, antiapoptotic cells that characterize the vasculopathy of PAH.
Numerous mitochondrial pathways contribute to glycolytic shift (FIGURE 9). Endothelial cells of PAH patients have higher levels of NOX-1, which increase cellular and mtROS production (526). Not only does this encourage vascular remodeling via ROS-mediated stimulation of sonic hedgehog and gremlin-1, but the increased oxidative stress damages mitochondria (526). As mitophagy ensues, endothelial cells become reliant on glycolysis to fulfill their energetic needs.
FIGURE 9.

Mitochondrial dysfunction in pulmonary hypertension. PAH is associated with a series of mitochondria-mediated metabolic derangements that contribute to vascular remodeling, apoptosis resistance, and bioenergetic compromise. 1: Increased NOX-1 encourages vascular remodeling via ROS-mediated stimulation of sonic hedgehog (SHH) and gremlin-1 (Grem1). Furthermore, the increased mtROS stimulates mitophagy. As mitochondria are removed, endothelial cells rely on glycolysis to meet their energetic demands. This glycolytic shift, also known as the Warburg effect, is associated with altered mitochondrial membrane polarization, which promotes apoptosis resistance by inhibiting the release of proapoptotic factors. 2: Decreased superoxide dismutase 2 (SOD2) is not only associated with increased mtROS but also with HIF-1α stabilization. 3: Additionally, decreased PGC-1α activity inhibits SIRT3 expression through impaired coactivation of ERR-α. This promotes HIF-1α stabilization and STAT3 activation, which promotes hyperproliferation and apoptosis resistance. 4: Mitochondrial calcium dysregulation, which can occur through multiple mechanisms in PAH, promotes pulmonary vasoconstriction and HIF-1α stabilization. See glossary for other abbreviations. Image created with BioRender.com, with permission.
Additionally, STAT3 is a transcription factor that upregulates provirus integration site for Moloney murine leukemia virus (Pim1), a proto-oncogene Ser/Thr-protein kinase (527). Pim1 deters apoptosis in pulmonary artery smooth muscle cells (PASMCs) by causing mitochondrial hyperpolarization and upregulating Bcl-2 through nuclear factor of activated T cells (NFAT) (527). Besides Pim1, STAT3 also directly upregulates NFAT and Krüppel-life factor 5 (KLF5) (528). The latter triggers PASMC proliferation through upregulation of cyclin B1 and prevents apoptosis through survivin-mediated caspase inhibition (528). Furthermore, STAT3 promotes vascular remodeling in PH by upregulating VEGF and other angiogenic factors (523, 527, 529, 530).
Another key player in the development of PAH is HIF-1α. Under normoxic conditions, H2O2 signaling opens voltage-gated K+ channels, suppressing stabilization of HIF-1α (525). However, if SOD2 levels are compromised, through impaired mitochondrial transport or increased degradation, the result will be lower H2O2 levels, leading to stabilization of HIF-1α (531, 532). Ordinarily, HIF-1α combats hypoxic conditions by upregulating the transcription of genes pertinent to erythropoiesis, vascular tone, and angiogenesis (523). However, numerous PH studies have observed low levels of SOD2 and elevated HIF-1α in the absence of hypoxia, indicating the presence of a pseudohypoxic state (532, 533).
Dysregulation of intracellular Ca2+ signaling is another pathological feature of PH. The MCUC is a transmembrane protein that controls the transport of cytosolic Ca2+ into mitochondria (534). In PASMCs, MCU downregulation impairs MCUC formation, causing mitochondria to lose their ability to buffer cytosolic Ca2+ levels and consequently their ability to limit vasoconstriction (534). Additionally, elevated cytosolic Ca2+ levels activate CaMK phosphorylation of Drp1, which encourages fragmentation of the mitochondrial network by increasing mitochondrial fission (29, 535). MCUC dysregulation also causes mitochondrial Ca2+ levels to decrease, preventing activation of Ca2+-sensitive PDH causing PASMCs to undergo glycolytic shift (534, 536).
Another important Ca2+ uniporter, UCP2, transports Ca2+ from the ER into mitochondria (536). In UCP2-knockout mice, the dysfunctional Ca2+ transport causes glycolytic shift, inhibition of Ca2+-sensitive PDH, decreased mtROS production, and activation of HIF-1α (536). Conversely, overexpression of UCP2 by cardiomyocytes is associated with apoptosis resistance through suppression of cell death markers (537). Additionally, transient receptor potential channels (TRPCs) are a family of nonselective cation channels involved in intracellular Ca2+ transport (538, 539). Within this family, TRPC3 localized to the IMM in PASMCs is upregulated in PAH, which facilitates mitochondrial Ca2+ influx and vasoconstriction (538, 540, 541). Furthermore, TRPC3 and TRPC6 upregulation is associated with PASMC proliferation (540, 541).
Importantly, Ca2+ dysregulation in PAH is not limited to aberrant Ca2+ channel activity. Neurite outgrowth inhibitor-B (Nogo-B), a member of the reticulon family, regulates the structure of the ER and facilitates efficient transport between the ER and mitochondria (542). Overexpression of Nogo-B is associated with mitochondrial hyperpolarization, stabilization of HIF-1α, and an antiapoptotic phenotype (542).
Sirtuins are also involved in the pathogenesis of PAH. Analysis of PASMCs in idiopathic PAH found normal SIRT1 gene expression but impaired SIRT1 signaling leading to higher acetylation of SIRT1 targets, such as PGC-1α and FOXO1. Inhibition of PGC-1α and its targets reduced mitochondrial mass and promoted glycolytic shift (543). Animal models designed to replicate SIRT1 inactivity or depletion displayed the vascular remodeling, hyperproliferation, glycolytic shift, and mitochondrial fragmentation characteristic of PAH (543). Furthermore, exposure to STAC-3, a SIRT1 activator, decreased PASMC proliferation and restored mitochondrial biogenesis.
Unlike SIRT1, SIRT3 gene expression is downregulated in PASMCs of idiopathic PAH (544). Presumably, decreased PGC-1α expression or activity inhibits SIRT3 expression through impaired coactivation of ERR-α. Failure of SIRT3 to deacetylate its downstream targets promotes PAH through HIF-1α stabilization, STAT3-Pim1-NFAT-survivin activation, PDH inhibition, and mitochondrial hyperpolarization (544). Importantly, restoration of SIRT3 activity reversed the hemodynamic consequences of PAH in mice by decreasing pulmonary artery proliferation and inducing apoptosis.
To summarize, mitochondrial dysfunction in PAH is a complex process involving impaired cellular bioenergetics, increased mtROS, inappropriate stabilization of HIF-1α, calcium dysregulation, and decreased PGC-1α expression. The unique interplay of these maladaptive processes encourages a hyperproliferative, antiapoptotic phenotype and vascular remodeling. To date, therapeutics for PAH are limited in both their scope and efficacy. Clinicians predominantly rely on monotherapy or combination therapy involving nitric oxide or prostacyclin agonists and endothelin antagonists to dilate the pulmonary vasculature. Although these interventions provide symptom relief, they are not curative. Interestingly, similarities between PAH and cancer have facilitated a reconceptualization of PAH therapy, namely, whether investigators can apply what they have learned from precision therapy in cancer to the development of small molecules or immunotherapy for PAH (545–547). Time will determine whether these investigative pursuits yield novel therapies that can be added to the PAH armamentarium.
4.3. Chronic Lung Diseases
Chronic lung diseases continue to pose a significant challenge to healthcare systems globally. Among these, chronic obstructive pulmonary disease (COPD) ranks third with respect to global burden of disease. COPD consists of mixed phenotypes involving emphysema (enlarged airspace and loss of alveoli) and upper airways disease (bronchitis, airway remodeling, and fibrosis). This disease arises from inhalation exposures to smoke and airborne particulates, with cigarette smoking (CS) representing the principal risk factor (548, 549). The primary pathogenic mechanisms include oxidative stress and hyperactivation of inflammatory pathways resulting from chronic CS exposure (548). In contrast, another chronic progressive lung disease, idiopathic pulmonary fibrosis (IPF), has unclear etiology, is age dependent, and affects ∼3 million people worldwide. IPF is characterized by progressive lung scarring, a high rate of mortality, and poor 5-yr survival rate (550). The etiology of IPF remains incompletely understood but may involve a combination of genetic risk factors and environmental factors (550). The primary pathogenic mechanisms may include epithelial cell activation and differentiation, suppression of autophagy, and activation of inflammation and senescence pathways (551).
Over the last decade, experimental evidence has accumulated that mitochondrial dysfunction plays a central role in the pathogenesis of chronic lung diseases, including COPD and IPF (10, 551–555) (FIGURE 10). These studies include genomic profiling implicating altered mitochondrial gene signatures, histopathological data indicating abnormal mitochondrial function and release of DAMPs, and biochemical evidence for altered mitochondrial metabolism in both animal models and cell culture studies, including major cell subtypes of the lung (e.g., epithelial, mesenchymal, and immune cells). Cellular changes associated with mitochondrial dysfunction include altered activation of autophagy and mitophagy and dysregulation of mitochondrial dynamics (i.e., fission, fusion). Furthermore, these changes have also been associated with dysregulation of RCD programs, including apoptosis, and necroptosis, as well as activation of cellular senescence programs. In this section we review the evidence in human and mouse model studies for mitochondrial dysfunction as a key pathological determinant in two major chronic lung diseases, COPD and IPF.
FIGURE 10.
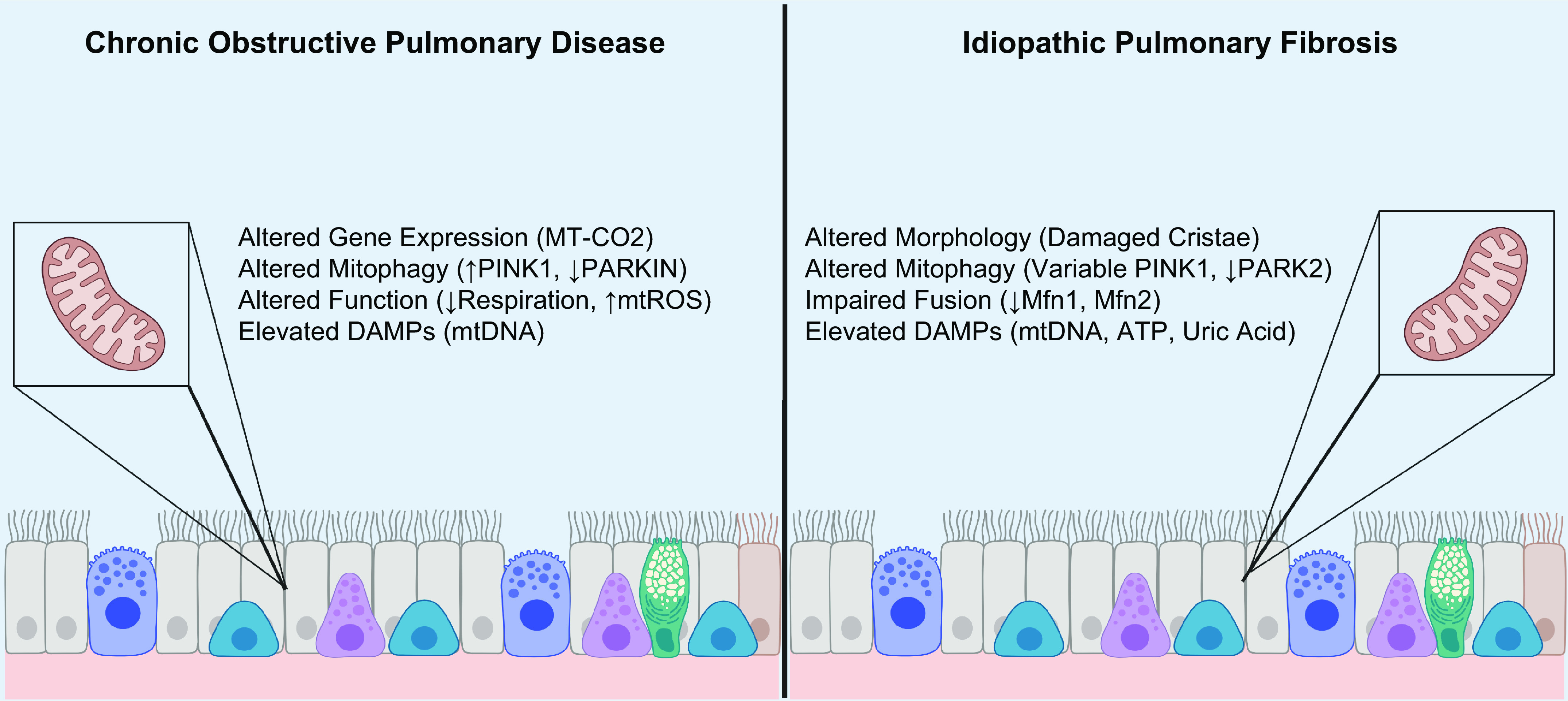
Mitochondrial dysfunction in chronic lung disease. Mitochondrial dysfunction plays a central role in the pathogenesis of chronic lung diseases, such as COPD and IPF. Genomic studies in COPD have identified differential gene expression among patients, with MT-CO2 being upregulated in emphysema. Additionally, mitophagy and cell death pathways appear to be dysregulated in COPD, with some studies noting increased PINK1 associated with upregulation of RIPK3-mediated necroptosis and others observing decreased PARKIN expression leading to mitophagy impairment and cellular senescence. COPD is also associated with impaired cellular respiration and increased generation of mtROS. In contrast, IPF is associated with morphological changes to mitochondria, such as the swelling of cristae. Mitophagy also appears dysregulated with variable expression of PINK1 and reduced expression of PARK2. Mitochondrial fusion is also involved in IPF, with Mfn1 and Mfn2 deficiency associated with lung fibrosis. Disease severity in both COPD and IPF has also been associated with elevated levels of circulating mitochondrial DAMPs. See glossary for abbreviations. Image created with BioRender.com, with permission.
4.3.1. Chronic obstructive pulmonary disease.
Several studies have uncovered mitochondria-related phenotypes from human COPD samples. Recent genomic studies have uncovered sex-specific transcriptional signatures in ex-smoking COPD patients. An RNAseq transcriptome analysis of bronchial brush cells was conducted on stringently defined ex-smoking COPD patients, which revealed genes differentially expressed between males and females. Among the many reported differences, a mitochondria-specific gene signature was identified in the emphysema dominant disease group in males. Among upregulated genes, the MT-CO2 (mitochondrially encoded cytochrome-c oxidase II) gene was validated as transcriptionally upregulated in bronchial epithelial cells (556). Future transcriptional profiling studies will further resolve transcriptional signatures of mitochondrial dysfunction that discriminate between COPD subtypes.
Furthermore, biochemical characterizations of human COPD lung samples indicate the involvement of mitochondrial processes. The possible contributions of autophagy and mitophagy to COPD pathogenesis have been deduced from analysis of human COPD lung tissue. Increased autophagosome numbers and increased expression of LC3B-II (active, lipidated form) were observed in human lung tissues from patients with severe COPD (557, 558). The expression of the mitophagy regulator protein PINK1 was increased in epithelial cells in emphysematous regions of human lung. Enhanced PINK1 expression in COPD lung tissue coincided with increased expression of the necroptosis regulator protein RIPK3 and was related to activation of regulated necrosis pathways (434). In contrast, elevated p62 expression and reduced Parkin expression were observed in the lung tissue of advanced COPD patients and attributed to autophagy/mitophagy impairment in association with the development of a senescence phenotype of bronchial epithelial cells (559, 560). Impaired autophagy was reported in the alveolar macrophages isolated from human smokers and in CS-exposed macrophages, which suggests compromised pathogen clearance by macrophages (561). Further studies determining autophagic flux and its relationships to cell fate in various cellular compartments will resolve apparent discrepancies in this field. Taken together, these observations suggest that mitophagy is significantly regulated in COPD and contributes to its pathogenesis via modulation of epithelial cell fate. In skeletal muscle of patients with COPD, relative to control patients an abnormal mitochondrial phenotype was described, which included mitochondrial functional alterations, reduced mitochondrial density, impaired respiratory function, and increased mtROS production (562). Further studies will be needed to define how mitochondrial dysfunction, mitophagy, and other mitochondrial quality control mechanisms may influence the pathogenesis of various subtypes of human COPD, in lung, and in other tissue such as skeletal muscle and kidney.
Experimental models of COPD include cultured target cells (i.e., epithelial cells) exposed to CS or its aqueous extract (CSE) as well as mice subjected to acute or chronic exposures to environmental CS. In studies that used cultured human pulmonary epithelial cells (e.g., A549, Beas-2B, and primary human lung epithelial cells), exposure to CSE resulted in significant dose-dependent changes in mitochondrial morphology and increased indexes of mitochondrial dysfunction (434, 563). Epithelial cells exposed to CSE displayed dose-dependent loss of ATP production, mitochondrial depolarization, increased mtROS production, and decline in mitochondrial respiration (434, 563). Similar studies reported loss of mitochondrial ETC function in response to CSE involving inhibition of CI and CII activities (564). CSE caused changes in mitochondrial morphology consistent with injury in Beas-2B and primary human epithelial cells and, furthermore, caused mitochondrial iron uptake in association with cellular injury (434, 563). Mice genetically deficient in iron regulatory protein 2 (IRP2), a COPD susceptibility locus, displayed reduced mitochondrial dysfunction and mitochondrial abnormalities in vivo following chronic CS exposure (563). Chronic CS exposure in mice also caused mitochondrial depolarization and mitochondrial abnormalities in lung tissue (434, 563).
In CS exposure models, activation of mitophagy was found to aggravate CS-induced epithelial cellular injury. Exposure to CSE induced functional mitophagy in pulmonary epithelial cells by inducing mitochondrial depolarization and stabilization of PINK1 (434). CSE induced dose-dependent increases in PINK1 expression and increased activation of the fission regulator DRP-1, by phosphorylation, which were dependent on the upregulation of mtROS, as these events were reversed by the mitochondria-targeted antioxidant mitoTEMPO (434). Mice genetically deficient in PINK1 displayed a resistance phenotype against mitochondrial dysfunction, airspace enlargement, and airway dysfunction during CS exposure in vivo (434). These studies suggest that CS-induced autophagy and selective autophagy (i.e., PINK1-dependent mitophagy) promote CS-induced airway dysfunction and airspace enlargement in mice (434). Further studies revealed that induction of the mitophagy pathway in epithelial cells was associated with secondary activation of the cellular necroptosis pathway and upregulation of RIPK3. The regulation of RIPK3 was dependent on PINK1, as determined by genetic deletion studies in vitro and in vivo (434). Furthermore, the chemical inhibitor of mitochondrial division/mitophagy Mdivi-1 protected against CS-induced mitochondrial dysfunction and necroptotic cell death (434). Consistently, the necroptosis inhibitor necrostatin-1 inhibited neutrophilic airway inflammation in CS-exposed mice (565). In contrasting studies, genetic interference of PINK1 or Parkin, which inhibited mitophagy and increased mtROS production, activated cellular senescence pathways in CS-exposed primary human bronchial epithelial cells, suggesting a protective role for these proteins (560). The reasons for discrepancies between studies remain unclear, although taken together they demonstrate active roles for PINK1 in determining cell fate in CS exposure models. Recent studies have also uncovered potential roles for fusion proteins in COPD. The long OPA1 isoform (Opa1-l) along with SLP2 and PH1/PH2 were identified as mediators of CS-induced lung damage, causing mitophagy/mitochondrial dysfunction in COPD (566).
Further studies will resolve the controversies related to the function of autophagy and mitophagy in disease progression, and their relationships to other mitochondrial processes such as fission and fusion, in the hope of developing autophagy/mitophagy-targeted therapies for this disease.
4.3.2. Idiopathic pulmonary fibrosis.
Evidence for a role for mitochondrial dysfunction in pulmonary fibrosis (PF) has been described in clinical samples derived from IPF patients, as well as in models of experimental PF. For example, electron microscopy studies have revealed an increased number of mitochondria with swollen morphology or damaged cristae in IPF lung samples compared with control lung (567, 568). Furthermore, increased mitochondrial proliferation was observed in alveolar type II (AT2) epithelial cells in highly fibrotic areas of human IPF lungs (567). Increased mitochondrial content was also associated with increased ER stress markers in AT2 cells of IPF patients (567). Lung tissues from IPF patients were deficient in autophagy as determined by LC3B-II expression and absence of autophagosomes (569). The activation of AMPK, as determined by the levels of p-AMPK, were reduced in myofibroblast foci, consistent with autophagy impairment (570).
Gene expression profiling studies have described significant downregulation of PINK1 gene expression in lung samples from IPF patients (567). These authors also reported accumulation of ATF3, a transcriptional inhibitor of PINK1, in aged or IPF lung tissue (571). In contrast, increased PINK1 expression was also reported in IPF lung tissue by immunofluorescence associated with increased numbers of damaged mitochondria (568). The reasons for these discrepancies in reported PINK1 expression in human IPF lung tissue remain unclear. Human IPF lung displayed reduced PARK2 (Parkin) expression in association with upregulated platelet-derived growth factor receptor (PDGFR) phosphorylation (572). The latter study related these changes to mitophagy impairment and increased myofibroblast differentiation in IPF (572).
In mouse models, Pink1−/− mice were found to be susceptible to bleomycin-induced lung fibrosis, supportive of an antifibrotic role for PINK1 in experimental models (567, 568). In contrast, genetic deficiency of the mitophagy regulator PGAM5, thereby inhibiting PGAM5-dependent mitophagy, resulted in protection from experimental PF (573). Taken together, these studies suggest that mitophagy may serve a protective function in PF but also a potential detrimental role if deregulated. Overexpression of the mitochondrial deacetylase SIRT3 in the airways was found to restore apoptosis sensitivity and promote resolution of age-dependent fibrosis in mice (574).
The mitochondrial fusion proteins Mfn1 and Mfn2 can also impact the profibrotic phenotype. Genetic deficiency of Mfn1 and Mfn2 in mice led to increased lung pathology and mortality. Mfn1-Mfn2 double-knockout mice displayed a phenotype of spontaneous lung fibrosis. Additionally, Mfn1 and Mfn2 were found to regulate the synthesis of phospholipids and cholesterol in AT2 cells, suggesting a novel link between the regulation of mitochondrial fusion and lipid metabolism (575). Genetic deficiency in FASN also aggravated experimental PF, underscoring the relationship between lipid metabolism, mitochondria-associated phenotypes, and fibrotic lung disease (575).
Cellular studies in IPF have focused on AT2 cells and fibroblasts, although other cell types, such as immune cells, may play a contributory role to IPF pathogenesis. Abnormal differentiation of AT2 cells has been explored as a central theme in the cellular pathogenesis of IPF. Metabolic reprogramming and activation of cellular senescence pathways in epithelial cells and other cell types may also contribute to the profibrotic phenotype. Accumulating evidence suggests a central role for mitochondrial dysfunction in contributing to AT2 cellular dysfunction in the context of IPF pathogenesis (551).
In cell culture studies, TGF-β1 stimulation of lung epithelial cells increased mitophagy, as demonstrated by increased LC3B and PINK1 colocalization, mitochondrial depolarization, mtROS production, and elevated expression of PINK1 and phospho-Drp1 (568). These phenomena were reversed by application of mitochondria-targeted antioxidants (i.e., mitoTEMPO). Genetic interference of PINK1 expression in pulmonary epithelial cells promoted TGF-β1-induced cell death (568). These experiments suggested that TGF-β1 can activate mitophagy as a protective response in epithelial cells. Epithelial cells isolated from Pink1−/− mice displayed increased mtROS production and cell death in response to TGF-β1, confirming a protective role for PINK1 in TGF-β1-dependent responses. Genetic interference of PINK1 expression in lung epithelial cells also resulted in mitochondrial depolarization and expression of profibrotic factors (567). PINK1 deficiency was also found to aggravate mtDNA release and activation of TLR9-dependent responses in experimental fibrosis (576). AT2 cells subjected to bleomycin challenge responded with activation of the NLRP3 inflammasome and subsequent production of IL-1β, which is known to respond to activation by mitochondrial DAMPs (577).
In cultured fibroblasts, knockdown of Parkin enhanced myofibroblast differentiation and proliferation, through a mechanism involving ROS-dependent activation of the PDGFR/phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway (572). Fibroblasts isolated from human IPF patients displayed reduced autophagy, associated with elevated activation of mTOR, and decreased activity of the energy-sensing kinase AMPK (570). AMPK-activating compounds inhibited profibrotic responses in TGF-β1-stimulated fibroblasts as well as stimulating mitochondrial biogenesis and restoring fibroblast sensitivity to apoptosis (570). Taken together, these studies revealed cell type-specific differences in the regulation and significance of autophagy/mitophagy responses in experimental fibrosis. As with COPD, further studies will resolve the controversies related to the function of autophagy/mitophagy in fibrotic disease progression, which may facilitate the development of autophagy/mitophagy-targeted therapies for IPF and related disorders.
4.3.3. Mitochondrial DAMPs in lung disease.
In an analysis of COPD patients and smokers (SPIROMICS cohort), urinary mtDNA levels were associated with increased respiratory symptom burden among non-COPD smokers. Significant sex differences in urinary mtDNA levels were observed, with females displaying higher urinary mtDNA levels than males across all study groups. Urinary mtDNA was associated with worse spirometry and emphysema in males only and with worse respiratory symptoms in females only. These studies suggested that mtDNA levels in fluids may identify distinct and sex-specific clinical phenotypes in pathophysiological responses in males versus females with COPD (578). Further research will ascertain the significance of plasma mtDNA levels in chronic lung disease. Circulating ATP and uric acid represent additional DAMPs associated with NLRP3 inflammasome activation that may be relevant in fibrotic lung disease. Fibrotic patients displayed elevated extracellular ATP content in bronchioalveolar lavage fluid (BALF) relative to control patients (579). Elevated extracellular ATP, which signals to P2RX7 to activate the NLRP3 inflammasome, was also observed in bleomycin-induced experimental fibrosis, consistent with a phenotype of increased lung IL-1β production (579). Similarly, elevated uric acid levels were found to aggravate the profibrotic phenotype in experimental fibrosis (580).
As the result of exposure to CS, cellular DAMPs released from necrotic epithelial cells and neutrophils were observed to stimulate proinflammatory cytokine and chemokine (e.g., IL-6, CXCL8) production in epithelial cells and neutrophil-dependent inflammation in mice (565, 581). Mitochondrial DAMPs include mtDNA, which may be released from depolarizing mitochondria in response to proinflammatory stress (270). mtDNA can act as a cellular DAMP to activate macrophage inflammatory pathways, including NLRP3 inflammasome activation, leading to enhanced secretion of proinflammatory cytokines (270). Circulating mtDNA levels in bronchioalveolar lavage fluid (BALF) and plasma were found to be increased and associated with disease progression and reduced survival in IPF patients (582). Enhanced mtDNA oxidation and damage were also reported in aging and IPF lungs and found elevated in other fibrotic lung disorders (576). In mechanistic studies, mtDNA was identified as a proinflammatory and profibrotic factor toward normal human lung fibroblasts, whereby exposure to exogenous mtDNA induced α-smooth muscle actin expression (582). The authors proposed that mtDNA could be used as a predictor of clinical outcomes in IPF.
Further research will determine whether DAMPs, including mtDNA, have potential to predict severity and mortality in chronic lung diseases.
5. MITOCHONDRIAL DYSFUNCTION IN INFECTIOUS AND INFLAMMATORY DISEASE
5.1. Sepsis
Despite decades of research, sepsis persists as an extremely morbid syndrome with an unacceptably high mortality (583). In the United States alone, 1.7 million adults are hospitalized with sepsis per annum, resulting in 270,000 deaths (584). Yet the significant in-hospital mortality of sepsis fails to approximate the true burden of disease. One in every five sepsis survivors experiences late mortality, dying within 2 yr of hospitalization (585). Furthermore, sepsis survivors must also contend with significant morbidity in the form of cognitive impairment, deterioration in their physical function, and decreased quality of life (586–588).
Even though sepsis research has yet to significantly attenuate the disease burden, remarkable strides have been made in our understanding of disease pathogenesis (589). Among these has been the redefining of sepsis as a dysregulated host response to infection that leads to life-threatening organ dysfunction (590). As this new definition implies, numerous host- and pathogen-specific factors are responsible for sepsis-induced organ dysfunction. Of the identified host factors, emerging evidence suggests that mitochondrial activity is a key determinant of sepsis.
A major goal of sepsis management is minimizing end-organ damage by optimizing tissue perfusion and oxygenation (591). However, animal models and clinical studies have found evidence of decreased O2 consumption, increased O2 tension, and decreased ATP production during sepsis despite adequate O2 delivery (592–595). The absence of significant cell death in these settings suggests that O2 utilization is compromised. This may account for why two randomized controlled trials designed to optimize hemodynamic parameters and tissue O2 delivery for critically ill patients failed to improve outcomes (592, 596, 597).
One of the proposed mechanisms for impaired O2 utilization in sepsis is NO-mediated inhibition of cellular respiration. During sepsis, TNF-α, IL-1, and INF-γ enhance NO production through iNOS (598). Even though NO is important for host defense against pathogens, its overproduction can be detrimental. Besides mediating vasodilatory shock, NO may impair cellular respiration as a competitive, reversible inhibitor of CIV. Accordingly, an acute rise in NO can impede ATP production while simultaneously increasing oxidative stress through production of mtROS.
Furthermore, NO operates as an indirect inhibitor of cellular respiration by reacting with O2•− to form ONOO−, a potent RNS (599, 600). ONOO− inhibits the ETC through nitrosylation of its protein complexes (599–601). Corroborating in vivo sepsis models have observed decreased O2 consumption in rat cardiomyocytes associated with decreased CI activity, decreased ATP synthesis, and increased NO and O2•− production without alteration of Ψm (602). Additionally, treatment of rats with an iNOS inhibitor and a ONOO− decomposition catalyst has been shown to attenuate the development of sepsis-induced AKI by decreasing nitrosative stress (603).
In a transcriptomic analysis of septic patients, OXPHOS gene expression was significantly downregulated in sepsis nonsurvivors relative to healthy control subjects (604). Although gene expression was also compromised in sepsis survivors, less than half of the studied transcripts were significantly affected. Moreover, sepsis survivors exhibited increased transcription of PGC-1α, NRF-1, and SOD2 relative to nonsurvivors. In fact, there was no appreciable difference in PGC-1α and NRF-1 levels between sepsis nonsurvivors and healthy control subjects. Collectively, these findings suggest that sepsis interferes with mitochondrial bioenergetics, potentially through oxidative stress, and that mitochondrial biogenesis is integral for survival.
Regardless of how cellular respiration is compromised, when it occurs cells are forced to prioritize activities to avoid critical ATP depletion, as this could trigger necrotic cell death. Slowing of cellular metabolism is protective, allowing cells to hibernate, which ultimately facilitates a more rapid recovery (605). Although energy-conserving mechanisms extend cell survival, they may do so at the expense of organ function (599). Suppression of energy-dependent activities in sepsis is believed to contribute to decreased cardiomyocyte contractility, a major precipitant of shock (606). In alveolar epithelial cells, the basolateral Na+-K+-ATPase is an energy-demanding enzyme; its downregulation during sepsis impairs alveolar fluid clearance, contributing to the development of pulmonary edema (599). Similarly, hepatocytes experience a decrease in synthetic function and clearance. Histology studies of septic patients have also found a discrepancy between the extent of kidney injury and renal dysfunction, suggesting that cell energetics, rather than cell death, is the driving factor (605).
Besides cell respiration, mitochondria-mediated apoptosis is also altered during sepsis. To prevent unwanted inflammation, the population of circulating neutrophils is tightly controlled by constitutive neutrophil apoptosis (607). Neutrophil exposure to the septic milieu increases the ΔΨm, causing sequestration of Cyt-c (608). The net result is an antiapoptotic phenotype intended to augment neutrophil half-life. Although this is an effective way to expand the neutrophil population against infection, the accumulation of neutrophils in tissues can be detrimental because the cytotoxic compounds they release are as injurious to surrounding tissues as to their intended target. The collateral tissue damage caused by delaying mitochondria-mediated apoptotic pathways promotes organ dysfunction, such as acute respiratory distress syndrome (ARDS) (609).
Additionally, pathogen virulence factors can alter mitochondria-mediated apoptotic pathways to diminish the production of proinflammatory cytokines, avoid intraphagocytic killing, and encourage infectious spread (610–612). This is exemplified by Streptococcus pyogenes and Listeria monocytogenes. These bacteria produce pore-forming cytolysins to escape phagosomes and damage mitochondrial membranes; the latter is associated with a decrease in Ψm and cellular respiration (613). These alterations slow cell metabolism and may precipitate apoptosis in both immune and nonimmune cells. illustrating how pathogens manipulate mitochondria to subvert the immune response and cause end-organ damage (612–616). In contrast, Chlamydia spp. block the release of Cyt-c and inhibit caspase-3 activity early on in an infection. By shutting down apoptotic pathways, this obligate intracellular bacterium is able to commandeer host machinery for an extended period of time, thereby facilitating its survival (612).
Over the past decade, there has been extensive speculation about whether mtDNA can be utilized as a biomarker for sepsis severity. Considering the biology, investigators have hypothesized that mtDNA is not only released in response to infection but can propagate the inflammatory response through interaction with TLR-9, inflammasomes, and other PRRs (617). If true, mtDNA could serve as a surrogate marker of inflammation providing clinicians with invaluable information about a patient’s severity of illness, response to treatment, and overall prognosis.
One of the landmark studies of mtDNA in sepsis was published by Nakahira et al. in 2013 (618). Admittedly, this two-cohort, observational study enrolled a heterogeneous population of critically ill patients from medical, surgical, and neuro intensive care units (ICUs), but most patients had sepsis as an underlying diagnosis. Not only did investigators find mtDNA to be significantly associated with 28-day mortality in both cohorts, but the odds of dying within 28 days of ICU admission increased with each log10-unit increase in mtDNA. This body of work provides exceptionally strong evidence that rising mtDNA levels reflect a worsening prognosis in a sepsis-predominant critically ill cohort. Moreover, mtDNA had a stronger association with 28-day mortality than lactate, a biomarker currently used to assess sepsis severity.
Even though numerous studies have found mtDNA to be significantly elevated in sepsis, they have not consistently found mtDNA to be associated with severity of illness or outcomes (619). Thus, one of the greatest challenges faced by the field is understanding the nature of these discrepancies. One possibility is that discrepant results stem from methodological differences: protocols for the isolation and measurement of mtDNA have yet to be fully standardized. Additionally, several studies have employed small cohorts, thereby limiting their power. Most studies are also designed with the intent of measuring mtDNA at a single time point. There may be greater utility in designing longitudinal studies to follow how mtDNA changes over time, akin to troponin. Furthermore, we do not have a clear sense of the variables that may confound mtDNA measurements.
In conclusion, sepsis has classically been conceived of as a systemic inflammatory syndrome; however, recent advancements recognize that sepsis is more aptly described as a dysregulated host response to infection. A significant contributing factor to this maladaptive response is mitochondrial dysfunction, specifically bioenergetic failure. By understanding how mitochondria fail to meet the energetic demands of their host, opportunities arise to redesign how sepsis management is approached. Perhaps the future of supportive care in sepsis begins at the cellular level, with therapeutics aimed at restoring mitochondrial function.
6. MITOCHONDRIAL DYSFUNCTION IN AGE-RELATED NEURODEGENERATIVE DISEASE
Emerging experimental evidence suggests that mitochondrial dysfunction may significantly contribute to the pathogenesis of human neurodegenerative disorders, the most common of which include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) or Lou Gehrig’s disease. These and related disorders are characterized by neuronal cell dysfunction and death that progressively lead to cognitive impairment and/or loss of motor control. Biochemically, these diseases share common pathological features including enhanced oxidative stress and disruption in neuronal proteostasis, attributed to accumulation of aggregated mutant proteins. The net result is cellular and mitochondrial dysfunction, the latter of which can exacerbate disease pathogenesis by activating cell death and inflammatory pathways (620, 621). Histological evidence for mitochondrial dysfunction in neurodegenerative disorders includes mitochondrial abnormalities in human postmortem brain tissue. Additionally, in vivo and in vitro studies have associated pathological end points with altered mitochondrial function involving mitochondrial morphology, bioenergetics, mitochondrial metabolism, Ca2+ handling, mitochondrial dynamics, mtROS production, biogenesis, mitophagy, and other biophysical processes such as axonal transport and ER MAM function (190, 191, 622–624) (FIGURE 11). These associations vary in their relative importance and characterization with each disease type and model system as discussed below.
FIGURE 11.
Mitochondrial dysfunction in neurodegenerative disease. Mitochondrial dysfunction is a hallmark of neurodegenerative diseases. Mutant proteins and their aggregates have been implicated in the pathogenesis of various neurodegenerative diseases, including AD, HD, PD, and ALS, by disrupting proteostasis and mitochondrial function. Mutant Aβ and hyperphosphorylated Tau (p-T), mutant Huntingtin (mHTT), and α-synuclein (α-Syn) aggregates can block electron transport chain (ETC) activities at various complexes. α-Syn and mHTT may also trigger mitochondrial dysfunction and mitochondrial reactive oxygen species (mtROS) production. mHTT, p-T, and mutant Aβ can interfere with the Sirt1-PGC-1α axis and its initiation of mitochondrial biogenesis. mHTT, p-T, and mutant Aβ can promote mitochondrial fragmentation via interactions with dynamin-related protein-1 (Drp1). Mutant forms of PTEN-induced kinase 1 (PINK1) can also interfere with PINK1-dependent Drp1 S616 phosphorylation. Autophagy and mitophagy processes may be disrupted by mutant proteins: mHTT can interfere with autophagosome formation, whereas p-T and mutant Aβ block autophagosome-lysosome fusion. Mutant PINK1 (mPINK1) and Parkin (mParkin) may cause mitophagy dysfunction. Superoxide dismutase 1 (SOD1) catalyzes the dismutation of superoxide generated by mitochondria, whereas in ALS the mutant form (mSOD1) is defective at removing superoxide, which favors peroxynitrite formation. mSOD1 can also interfere with mitochondrial axonal transport via inhibiting the transport regulator protein mitochondrial Rho GTPase (MIRO) and by promoting the autophagosome-dependent degradation of MIRO. −ΔΨm, mitochondrial membrane potential (loss); ER, endoplasmic reticulum; MAM, mitochondria-associated membrane; p62, SQSTM1 autophagy cargo adaptor protein. See glossary for other abbreviations.
6.1. Alzheimer’s Disease
Alzheimer’s disease (AD) is a common neurodegenerative disease primarily associated with progressive memory loss, synaptic dysfunction, and cognitive impairments. AD represents the sixth leading cause of death in the United States, with an estimated prevalence of 6.5 million people >65 yr of age (625). Although most cases are sporadic, familial AD (FAD) is associated with mutations in the amyloid precursor protein (APP) gene or in Presenilin genes, PSEN1 and PSEN2, that encode proteins that process APP and additional risk factors such as the APOEε4 allele (626, 627). AD is characterized by accumulation of plaques containing amyloid-β (Aβ), derived from APP, as well as neurofibrillary tangles consisting of the hyperphosphorylated form of microtubule-associated protein Tau (p-Tau), in the brains of AD patients (626).
Mitochondrial dysfunction is believed to play a major contributory role in AD pathogenesis based on biochemical and histological evidence (628–630). In human disease, gross mitochondrial abnormalities have been identified in histological analyses of AD brain (629). AD neurons were characterized by loss of mitochondrial numbers and leakage of mtDNA and COX into neural cytoplasm (631). Human studies have revealed altered or impaired activities of enzymes involved in glucose metabolism and bioenergetics, including CI, CIV, ATP synthase, and other metabolic enzymes (628). Of these, COX activity was consistently found impaired or reduced in AD brain, in part attributed to reduced expression of enzyme protein and altered enzyme kinetics (632–636). Recent studies also uncovered altered expression of proteins involved in Ca2+ exchange in postmortem AD brain (637).
Experimental modeling in AD has relied on the generation of transgenic mouse models including APP, APP/PS1, 5XFAD, Tg2576, and 3xTg-AD mice (638). Among these, the 3XTg-AD mice [Tg(APPSwe,tauP301L)1Lfa)] exhibit both Aβ- and Tau-dependent pathology (638). Data from these models have supported a central role for mitochondrial dysfunction and associated alterations in metabolism in AD pathogenesis.
For example, 3xTg-AD mice displayed a phenotype of mitochondrial dysfunction in the brain involving increased oxidant stress, reduced respiration and decline of PDH protein and activity, and increased Aβ accumulation in mitochondria (639). Additionally, 3xTg-AD mice display a phenotype of depletion in Krebs cycle metabolites (640).
In APP transgenic mice (Tg2576), genetic profiling studies revealed upregulation of mitochondrial proteins involved in energy metabolism and apoptosis. Elevated ATPase-6 expression was associated with enhanced oxidative stress in neurons of Tg2576 mice (641). Both mutant APP and Aβ protein target mitochondrial function in Tg2576 mice, resulting in increased ROS production and reduced COX activity (642). Different mutations of presenilin-1 (PS1), the catalytic subunit of γ-secretase, that are associated with familial AD had variable impact on mitochondrial functions, including modulation of ER MAM contact and respiratory activity (643). In 5XFAD mice, full and COOH-terminal truncated forms of APP accumulate in mitochondria and promote mitochondrial dysfunction, including impaired respiration and mtROS production. Partial deletion of the β-site APP-cleaving enzyme-1 (heterozygote) reduced APP accumulation in mitochondria and improved cognitive function in 5XFAD mice (644).
Mechanistic studies support mitochondrial dysfunction as a driver of AD pathogenesis. For example, embryonic hippocampal neurons derived from 3xTg-AD mice had reduced mitochondrial respiration and increased glycolysis (639). Cybrids generated with mtDNA extracted from AD platelets displayed a phenotype of impaired bioenergetics, mtROS production, and increased apoptosis (645). Aβ can cause mitochondrial dysfunction, associated with enhanced mtROS formation, cyclophilin D-dependent mPTP opening, and neuronal cell death (646–648). APP mice cross-bred for cyclophilin deficiency displayed improved cognitive function (646). Mitochondrial dysfunction may also represent a primary cause of microglia activation, which drives neuroinflammation (649).
Mouse models of AD have also uncovered a major role for mitochondrial Ca2+ accumulation in disease pathogenesis, via promoting mtROS production, metabolic dysfunction, and neuronal cell death (637). Recent studies using 3xTg-AD mice demonstrated that Ca2+ accumulation in the mitochondrial matrix may precede mitochondrial dysfunction and neural pathology. Neuron-specific deletion of the mitochondrial Na+/Ca2+ exchanger (NCLX) in 3xTg-AD mice promoted cognitive decline and increased amyloidosis and tau pathology. Knockin of neuronal NCLX in 3xTg-AD mice rescued AD pathology and cognitive decline (637). In 3XTg-AD mice, inhibition of type 1 inositol trisphosphate receptor (InsP3R1) expression and associated Ca2+ signaling reduced Aβ accumulation and tau hyperphosphorylation and restored hippocampal long-term potentiation and cognitive defects (650). These observations have placed Ca2+ signaling alongside mitochondrial dysfunction as a primary driver of AD pathogenesis and suggest mitochondrial Ca2+ exchange as a potential therapeutic target in AD.
Accumulating research suggests that autophagy and mitophagy are altered in AD, based on observations of abnormal mitochondria. Autophagy/mitophagy defects are related to accumulations of Aβ and p-Tau during AD pathogenesis (651). Mitophagy was impaired in the brain tissue of AD patients, in induced pluripotent stem cell-derived human AD neurons, and in animal models of AD (652). In an APP/PS1 mouse model, mitophagy functioned to remove insoluble Aβ aggregates and prevent cognitive impairment by promoting phagocytosis of Aβ plaques and reduction of neuroinflammation. Increased levels of Aβ and APP in a transgenic APP model resulted in reduced levels of several autophagy and mitophagy proteins, including PINK1 and LC3 (653). In transgenic mAPP mice, PINK1 deficiency accelerated age-dependent cognitive impairments and increased brain Aβ accumulation and mitochondrial dysfunction (654). Conversely, PINK1 gene therapy in mAPP mice promoted mitophagy clearance of mitochondria and reduced neuropathology (654).
Age-dependent increased levels of Aβ and p-Tau were associated with a phenotype of impaired Parkin assimilation to mitochondria (655, 656). Compromised mitophagy may also involve inhibited autophagosome-lysosome fusion and impaired lysosomal acidification (657). Pharmacological enhancement of neuronal mitophagy using novel compounds (urolithin A, actinonin) reduced p-Tau accumulation in human neuronal cells and reversed memory impairment in transgenic mouse models of AD (652, 658). In contrast, recent studies also reported that Aβ overload increases autophagy and mitophagy in young APP/PS1 mice (659). Although interpretation of autophagic activity may vary between studies, mitophagy enhancers may represent a novel class of experimental therapeutics in AD (658, 660).
Furthermore, increasing evidence points to altered mitochondrial dynamics, including fission and motility, as contributing to AD pathogenesis (661, 662). Significant mitochondrial fragmentation and impaired axonal transport of mitochondria were reported in AD neurons, associated with increased GTPase activity in postmortem frontal cortex tissue (663). AD brain tissue also exhibited increased expression in fission regulatory proteins (i.e., Drp1, Fis1) and reduced expression of fusion-associated proteins (i.e., Mfn1/2, Opa1) (664). Immunohistochemical analyses of AD brain tissue revealed that both Aβ and p-Tau can directly interact with Drp1 and that these interactions were associated with increased mitochondrial fragmentation in AD (663, 664). Mitochondrial fission-linked GTPase activity was significantly elevated in cortical tissues from APP, APP/PS1, and 3xTg-AD mice (663). In the APP/PS1 mouse model, expression of mitochondrial fission proteins Drp1 and Fis1 was increased whereas fusion proteins (Mfn1, Mfn2, and Opa1) and biogenesis-related factors (PGC-1α, NRF1, NRF2, and TFAM) were decreased (653). Partial genetic reduction of Drp1 (heterozygote) in APP mice reduced Aβ production and mitochondrial dysfunction and increased mitochondrial biogenesis and synaptic activity (665). In contrast, genetic deletion of DRP1 in CA1 neurons promoted cognitive deficits in mice expressing mutant hAPP in neurons, associated with mitochondrial Ca2+ overload. These results suggested a protective role for Drp1 in this model (666). In addition to mitochondrial dysfunction, primary neurons from Tg2576 mice displayed increased mitochondrial fission and decreased fusion, mitochondrial fragmentation and damaged cristae, and impaired mobility. A mitochondria-targeted antioxidant restored mitochondrial transport and synaptic viability and decreased the number of damaged mitochondria in this model (667). In CRND8 APP transgenic mice, increased neuronal mitochondrial fission and impaired motility were observed. Treatment with the mitochondrial fission/mitophagy inhibitor Mdvi-1 ameliorated these effects and restored cognitive defects (668).
Finally, aberrations in the regulation of mitochondrial biogenesis have also been observed in models of AD. Decline in neuronal expression of PGC-1α has been found in general association with AD pathology (669). Expression of FAD mutant PS1 was associated with reduced PGC-1α expression in murine cells (670). Sirt1-dependent activation of PGC-1α was implicated in neuronal protection in mouse models of AD (671). Vector-driven overexpression of PGC-1α in cortex of APP/PS1 mice improved mitochondrial dysfunction and restored cognitive defects (672).
In conclusion, disruption of mitochondrial Ca2+ balance has emerged as a primary driver of AD pathogenesis, which is associated with mitochondrial and metabolic dysfunction and may precede Aβ accumulation, Tau pathology, and neuronal cell death. Alterations in mitophagy, mitochondrial dynamics, motility, and other MQC processes have been proposed as contributing to the pathogenic mechanism of AD (663, 664, 673). Further resolution of the hierarchies of these events may inform therapeutic development.
6.2. Parkinson’s Disease
Parkinson’s disease (PD) is the second most common progressive neurodegenerative disease. PD affects 1–2% of the population above 65 yr, and its prevalence increases to ∼4% in individuals above 85 yr (21). PD is characterized by progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the formation of Lewy bodies (674). The primary symptoms include primary motor deficits (i.e., bradykinesia, tremor, rigidity, and postural instability) and nonmotor symptoms (i.e., dyssomnia, depression, etc.) (674). PD occurs in familial (5–10%) and sporadic forms, the latter of which has unclear etiology.
Familial PD is attributed to mutations in distinct loci (PARK1–PARK23), which include five that are casually related to PD pathogenesis. These are represented by PARK1 (Snca) encoding α-synuclein, the main constituent of Lewy bodies, PARK2 (Parkin), PARK6 (Pink1), PARK7 (DJ1), and PARK8 (Lrrk2) (21, 675). Several of these genes encode proteins, (i.e., Pink1 and Parkin) that exert intimate functions in MQC processes, namely mitophagy (676). whereas DJ1 has a purported mitochondrial redox sensing and ROS scavenging function (677). Defects in mitochondrial bioenergetics and/or COX expression have also been observed in PD brain tissue (678, 679). Accumulations of α-synuclein can cause mitochondrial dysfunction, which may reciprocally exacerbate α-synuclein accumulation in the disease pathogenesis (680).
Overall, PD is described as a disease of abnormal or impaired mitophagy, and discoveries made in elucidating PD pathogenesis led to modern understanding of PINK1-Parkin-dependent mitophagy (28, 674). Mutations in DJ1 also contribute to impaired mitophagy and mitochondrial dynamics in PD (681).
Among the mouse models used to study PD, Pink1- or Parkin-null mice do not recapitulate the PD phenotype. Similarly, Parkin−/−/PolgAD257A/D257A mice did not produce a Parkinson phenotype, and the loss of Parkin did not exacerbate mitochondrial dysfunction over the mtDNA mutator genotype (682). A genetic model of PD (mitoPARK) based on neuronal specific deletion of TFAM displays respiratory chain deficiency in dopaminergic neurons (683). A more recently developed genetic model of PD (p.Thr61Ile knockin) exhibits neurological features of PD and aberrant mitochondria phenotypes in the brain (684). Mouse models of PD have also relied on the use of toxins, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which is metabolized to the neurotoxin 1-methyl-4-phenylpyridinium (MPP+) (685). In MPTP-treated mice, transcriptomic and proteomic analyses revealed a phenotype of enhanced mitochondrial dysfunction and oxidative stress (686). Pharmacological activation of mitophagy with natural flavonoids alleviated Parkinson-like symptoms in this model (687).
Pink1 deficiency, associated with familial PD, causes impairments in mitochondrial Ca2+ handling (688). Neuronal PINK1 deficiency causes mitochondrial Ca2+ accumulation, associated with mtROS production and mPTP opening leading to neuronal cell death (688). LRRK2 deficiency also led to impairment of NCLX-dependent Ca2+ efflux, leading to mitochondrial Ca2+ accumulation (689).
Mitochondrial dynamics, quality control, and mitophagy may also contribute to PD (690). The mitophagy regulator PINK1 directly phosphorylates Drp1 on S616. Drp1S616 phosphorylation is reduced in PINK1-deficient cells and mouse tissues but is independent of Parkin. Impaired PINK1-mediated Drp1S616 phosphorylation was associated with the pathogenesis of both familial and sporadic PD (691). Parkin interacts with and ubiquitinates Drp1 to promote its degradation by the proteasome. Mutation or knockdown of Parkin inhibits Drp1 ubiquitination and degradation, leading to increased levels of Drp1-dependent fission. These studies identified Drp1 as a novel substrate of Parkin and associated abnormal Parkin expression to mitochondrial dysfunction in the pathogenesis of PD (692). The dominant p.D620N variant of vacuolar protein sorting 35 ortholog (VPS35) gene is also associated with familial PD. Mutations of this gene were associated with increased mitochondrial depolarization and impaired initiation of Pink1/Parkin mitophagy (693).
PD pathogenesis also includes phenotypes of impaired mitochondrial biogenesis (680). SIRT1 expression is reduced in PD; SIRT1 maintains PGC-1α in a deacetylated state and thereby preserves PGC-1α levels. Thus, the SIRT1-PGC-1α axis may have therapeutic potential in PD and other neurodegenerative diseases (694).
In conclusion, the pathogenesis of PD also involves mitochondrial dysfunction, deficient mitophagy, and altered mitochondrial dynamics, which provide opportunities for therapeutic targeting in this disease.
6.3. Huntington’s Disease
Huntington’s disease (HD) arises from CAG repeats in exon 1 of huntingtin protein (HTT) that generate polyglutamine (polyQ) extensions of the protein (695). HD is characterized by progressive motor impairment, cognitive defects, and psychological abnormalities. Accumulations of mutant Huntingtin (mHTT) in this disease can impact mitochondrial functions including Ca2+ buffering and impaired respiratory chain activity at CII, CIII, and CIV (696, 697). Inhibition of COX and cytochrome b gene expression were reported in the striatum and frontal cortex of HD patients accompanied by increased 8-OHdG levels, a marker of oxidative DNA damage (698). Neural tissue from HD patients also exhibited increased expression of mitochondrial fission genes (Drp1, Fis1) and reduced expression of fusion genes (Mfn1, Mfn2, Opa1) (698). Toxic accumulation of mHtt directly in neuronal mitochondria was also documented (698). The role of mitochondrial Ca2+ handling in HD is incompletely characterized. mHTT can trigger mitochondrial Ca2+ overload and reduce the Ca2+ threshold for mPTP opening (699).
As in other neurodegenerative disorders, inefficient processing of damaged mitochondria via impaired mitophagy may play a pathogenic role in HD (700). Cellular models have confirmed that the polyQ tract of mHTT can interfere with the initiation of autophagy by obstructing the assembly of Ulk-1 and Beclin-1 complexes and furthermore by interfering with the functioning of selective autophagy adaptor proteins (701).
Mitochondrial fission processes driven by hyperactivation of Drp1 are also believed to play a major pathogenic role in HD (702). Mutant Huntingtin (mHTT) interacts with Drp1 to promote its GTPase activity, causing excessive fragmentation of mitochondria, promoting abnormal mitochondrial dynamics and neuronal damage in HD-affected neurons (703, 704). Aberrant Drp1 activity in HD neurons was also associated with defective anterograde mitochondrial movement and synaptic deficiencies (704). Enhanced Fis1-Drp1 interactions are implicit in Drp1 hyperactivation in HD. Inhibition of Drp1 hyperactivation by P110, a peptide inhibitor of Drp1-Fis1 interaction, was protective in the HD R6/2 mouse model, which expresses a fragment of mHTT (705). Application of P110 also improved the neurocognitive phenotype of HD-knockin (zQ175 KI) mice, which express full-length mHtt and exhibit progressive HD symptoms (706). Activation of SIRT3 conferred neuroprotection in HD models via normalization of aberrant Drp1 activation and fission (707). In parallel with fission abnormalities, deficiency in PGC-1α-dependent mitochondrial biogenesis may also contribute to HD pathogenesis (702). Pharmacological enhancement of PGC-1α expression improved the neuropathological phenotype in the R6/2 transgenic mouse and BCHD mouse models of HT (708).
6.4. Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a rare adult-onset progressive neurodegenerative disorder that causes muscle wasting and deformity and is associated with frontotemporal dementia. ALS originates both sporadically (90% of cases) and as an inherited disorder. Inherited cases partially associated with dominant mutations in the SOD1 gene and other loci including C9orf72 (chromosome 9 open reading frame 72), FUS (fused in sarcoma/translocated in liposarcoma or heterogeneous nuclear ribonucleoprotein P2), and TARDBP (transactive response DNA binding protein 43) (709). SOD1G93A mutant mice display a neurological phenotype that recapitulates human ALS and have been used extensively to model ALS in vivo.
Mitochondrial dysfunction is considered a key pathological determinant of experimental and human motor neuron degeneration in ALS (710). As in other neurodegenerative diseases, disruptions of mitochondrial structure, dynamics, bioenergetics, and Ca2+ buffering have been reported in ALS (710). This is corroborated by recent human studies using magnetic resonance spectroscopy, which revealed loss of bioenergetic capacity in brain and muscle of human ALS patients. Loss of brain muscle bioenergetic capacity correlated to pathophysiological indexes (711).
Double transgenic mice expressing expression SOD1G93A and the mitoflash indicator probe mt-cpYFP were used to detect oxidative stress status in ALS. In these mice, ROS-associated fluorescence increased in skeletal muscle at the time of onset of ALS symptoms. Onset of ALS symptoms was also associated with increased CypD, a regulator of mPTP opening. Transient overexpression of SOD1G93A in skeletal muscle of wild-type mice promoted mtROS production and CypD expression, whereas the CypD inhibitor cyclosporin-A reduced the mtROS production (712). SOD1G93A mice also exhibit bone loss, whereas osteoclasts derived from these mice displayed dysregulated mitochondrial dynamics, a phenotype that was partially reversed by Mdivi-1 (713).
Altered mitochondrial Ca2+ handling may also contribute to ALS pathogenesis (699). SOD1G93A-expressing motor neurons displayed increased mitochondrial Ca2+ accumulation, −ΔΨ, as well as increases in cytosolic and ER Ca2+ (714).
Mouse models also reveal disruptions in mitochondrial mobility and organelle interactions. SOD1G93A transgenic mice displayed a phenotype of impaired axonal transport of mitochondria in motor neurons. The phenotype was attributed to increased PINK1/Parkin-dependent degradation of MIRO, independent of cytosolic Ca2+ flux. The OMM protein mitochondrial Rho GTPase 1 (MIRO1) is a regulator of mitochondrial axonal transport in response to cytosolic Ca2+ levels and mitochondrial injury. MIRO1 is regulated by Pink1/Parkin-dependent mitophagy. The study concluded that SOD1G93A inhibits axonal transport of mitochondria by inducing PINK1/Parkin-dependent Miro1 degradation (715). Recent studies have deduced that disruption of ER-MAM integrity is a common pathological feature of familial ALS. SOD1G93A mutation was shown to disrupt ER-MAM integrity, as determined by novel MAM probes (i.e., MAM tracker dyes) (716).
In addition to SOD, mutations in Fus are also associated with familial ALS (717). Fus colocalized with the mitochondrial tethering protein syntaphilin (SNPH) in rat primary neurons and thus may exert a homeostatic function in mitochondria, whereas expression of mutant Fus R514G in rat primary neurons disrupted this interaction and caused mitochondrial abnormalities and loss of mitochondrial motility (718).
Activation of mitochondrial biogenesis was found to improve the phenotype of ALS. Overexpression of PGC-1α in SOD1 transgenic mouse (Tg-SOD1G93A/PGC-1α) improved motor function and survival relative to SOD1-G93A mice and improved mitochondrial ETC activity in the spinal cord of transgenic mice (719). Conversely, genetic silencing of PGC-1α aggravates the phenotypes of SOD1G93A mice (720). Chronic application of R13 [a precursor of 7,8-dihydroxyflavone and tyrosine kinase receptor B (TrkB)] agonist partially ameliorated the phenotype of SOD1G93A mice, including improvement of motor performance and reduction of motor neuron pathology. The beneficial effects of R13 were associated with promotion of OXPHOS-related gene expression in neural tissues including spinal cord, stimulation of AMPK and of PGC-1α/NRF1/TFAM-dependent mitochondrial biogenesis, and amelioration of mitochondrial dysfunction (721). Neuroprotection in SOD1G93A mice via PGC-1α-dependent responses was also achieved by application of a novel metal chelator compound (722). Taken together these observations indicate that ALS is associated with a strong phenotype of mitochondrial dysfunction in motor neurons.
6.5. Conclusions
Bioenergetic failure, Ca2+ dysregulation, mtROS, mitochondrial depolarization, and mPTP opening may play an important role in the early stages of various neurodegenerative disorders by contributing to neuronal cell death. Furthermore, aberrations in mitophagy and mitochondrial dynamics occur in parallel with mitochondrial dysfunction and may accelerate pathophysiology. Enhancement of mitophagy and mitochondrial biogenesis may present possible avenues for therapeutic targeting (see sect. 7) (660). Common elements of mitochondrial dysfunction such as loss of energy charge, mitophagy impairment, altered fission and biogenesis, or mobility defects may appear across different types of neurodegenerative disease, though their relative contribution to the pathogenesis of specific diseases may be different. The importance of the contributing mechanisms underlying mitochondrial dysfunction may also potentially vary in discrete neuronal cell populations, which may display differential susceptibility to initiating stimuli as well as differential roles in disease pathogenesis. Further experimental work aimed at improving our understanding of the neuronal cell type-specific regulation and cross talk of processes involved in mitochondrial homeostasis and dysregulation, using multiomics or systems biology approaches, may enhance understanding of neurodegenerative disease pathogenesis and also provide strategies for selective therapeutic targeting of these pathways.
7. THERAPEUTIC CONSIDERATIONS
The mitochondria, and the cellular and molecular processes that support their function and homeostatic regulation as summarized in this review, have revealed numerous potential targets for therapeutic intervention. Since no therapies specifically targeting mitochondria have been approved for clinical use to date, the therapeutic avenues remain largely in preclinical and clinical experimental stages (723).
Among the most widely cited therapeutic classes for mitochondrial medicine are mitochondria-targeted compounds, specifically antioxidants, that aim to reduce pathological accumulation of mtROS as well as preserve mitochondrial function (724, 725). These include natural compounds such as coenzyme Q (Q10) and its synthetic derivatives, idebenone and triphenylphosphonium (TPP)-conjugated MitoQ, and other TTP-conjugated antioxidants such as Mito-vitamin-E and mitoTEMPO (726–728). Idebenone has been tested in multiple clinical trials and showed promise as a treatment for Duchenne muscular dystrophy (727). MitoQ has been studied in several human clinical trials, and although the compound showed positive effects in hepatitis C infection, it failed to slow neurodegeneration in PD (729, 730). Recent preclinical studies suggest that mitoTEMPO can confer protection in acute liver injury (728).
A mitochondria-targeting oligopeptide, Szeto-Schiller-31 peptide (SS-31, elamipretide), confers mitochondrial protective effects by stabilizing the IMM, inhibiting mtROS formation and membrane depolarization, and by preventing mitochondrial swelling and mPTP opening (731, 732). Furthermore, SS-31 inhibits CL-dependent activation of Cyt-c peroxidase activity, preserves mitochondrial cristae, and enhances OXPHOS (733). SS-31 also promotes Sirt3-dependent mitochondrial fusion (734). In mouse models, SS-31 protected against LPS-induced cognitive decline (735). In addition to direct antioxidants, mitochondrial iron chelation also shows therapeutic potential in experimental models (736).
Mitophagy represents a major mitochondrial homeostatic mechanism that can be modulated for therapeutic gain. However, the significance of the process as a pro- or antipathogenic mediator may vary in a context-specific fashion; therefore care must be taken when targeting mitophagy in select diseases. Mitophagy-stimulating compounds may represent a therapeutic strategy in neurodegenerative disorders (658). Similarly, the machinery regulating mitochondrial dynamics may be targeted for therapeutic gain (190, 737). The Drp1 mitochondrial fission inhibitor Mdivi-1 has potential for protection in experimental models, including neurodegenerative diseases, though the specificity of this drug has been challenged (668, 737–741). Synergistic effects of SS-31 and Mdivi-1 combination treatment were reported in cellular models of AD (738). A Drp1 antagonist peptide showed therapeutic effects in an in vitro model of PD (742).
Mitochondrial biogenesis may be targeted for homeostatic control, by promoting mitochondrial replication in deficiency states. Therapeutic strategies targeting the upregulation of the master regulator PGC-1α show promise (743). These are exemplified by 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR), an AMPK activator, and natural compounds such as resveratrol, which activates sirtuin-1 (744–747). NAD+-replenishing compounds such as nicotinamide mononucleotide (NMN) also represent therapeutic candidates that can activate PGC-1α (747, 748). The PPAR-γ agonist pioglitazone showed positive effects on mitochondrial biogenesis in human diabetes and conferred neuroprotection in the APP/PS1 AD mouse (749–751).
Recent studies have also proposed therapeutic targeting of mPTP opening as a selective therapeutic strategy, exemplified by CsA, and advanced by newly identified small-molecule cyclophilin inhibitors (752, 753).
These examples point to a foundation for mitochondria-dependent molecular medicine, although further preclinical and clinical validation studies, as well as improved understanding of off-target effects and toxicity, will be needed to arrive at specific therapies (723, 727).
8. CONCLUSIONS AND FUTURE DIRECTIONS
The tenet that the mitochondria are functioning more than as an organelle serving as a source of ATP production and cellular respiration is intriguing and exciting. Accumulating data in recent years have provided the evidence that the mitochondria serve as a reservoir of critical second messengers and key effector molecules in mediating important cellular and physiological processes in the pathogenesis of human diseases. In the era of precision medicine and health, we anticipate a continued trajectory of new discoveries of mitochondrial pathways important in improved understanding of the role of mitochondrial biology in the pathogenesis of human disease. Importantly, these discoveries will hopefully lead to molecular targets as diagnostics and therapeutics in the approaches to confer impact on our patients with mitochondria pathway-based medical disorders.
GLOSSARY
- ΔΨm
Mitochondrial membrane potential (change)
- Aβ
Amyloid-beta protein
- AcCoA
Acetyl-CoA
- ACD
Accidental cell death
- AD
Alzheimer’s disease
- ALS
Amyotrophic lateral sclerosis
- AMP
Adenosine monophosphate
- AMPK
Adenosine monophosphate-activated protein kinase
- APP
Amyloid precursor protein
- ATG
Autophagy-related gene
- ATP
Adenosine-5′-triphosphate
- Beclin 1
Mammalian homolog of yeast Atg6/Vsp30
- Ca2+
Calcium
- CARD
Caspase-recruitment domain
- CL
Cardiolipin
- COPD
Chronic obstructive pulmonary disease
- COX
Cytochrome-c oxidase
- CS
Cigarette smoke
- CSE
Cigarette smoke extract (aqueous)
- CVD
Cardiovascular disease
- Cyt-c
Cytochrome c
- DAMP
Damage-associated molecular pattern
- Drp1
Dynamin-related protein-1
- ETC
Electron transport chain
- ER
Endoplasmic reticulum
- FA
Fatty acid
- FAO
Fatty acid β-oxidation
- FASN
Fatty acid synthase
- Fis1
Fission protein-1 (mitochondrial)
- FOXO
Forkhead family of transcription factors
- FUNDC1
Fun14 domain-containing protein 1
- GSDMD/E
Gasdermin-D/E
- GTP
Guanosine 5′-triphosphate
- GTPase
Guanosine 5′-triphosphate hydrolase
- HD
Huntington’s disease
- HIF-1α
Hypoxia-inducible factor-1 alpha
- HMGB1
High-mobility group box 1
- HTT
Huntingtin protein
- IFN
Interferon
- IMM
Inner mitochondrial membrane (mitochondrial)
- IMS
Inner membrane space (mitochondrial)
- IPF
Idiopathic pulmonary fibrosis
- LC3
Microtubule-associated protein-1 light chain 3
- LIR
LC3 interacting region
- LPS
Lipopolysaccharide
- MAM
Mitochondria-associated ER membrane
- MAVS
Mitochondrial antiviral signaling
- MCUC
Mitochondrial Ca2+ uniporter complex
- MEF
Mouse embryo fibroblast
- MFF
Mitochondrial fission factor
- MFN-1/-2
Mitofusin-1, mitofusin-2
- mHTT
Mutant Huntingtin protein
- MIEF-1/-2
Mitochondrial elongation factors-1, -2
- MITOL
Mitochondrial ubiquitin ligase
- MLKL
Mixed-lineage kinase domain-like pseudokinase
- MOMP
Mitochondrial outer membrane permeabilization
- MPAP
Mean pulmonary artery pressure
- MPT
Mitochondria permeability transition
- mPTP
Mitochondrial permeability transition pore
- MQC
Mitochondrial quality control
- mtDNA
Mitochondrial DNA
- mtROS
Mitochondrial reactive oxygen species
- NDP52
Nuclear dot protein, 52 kDa
- NFP
N-formylated peptide
- NLRP3
Nucleotide-binding oligomerization domain (NOD)-, leucine-rich repeat (LRR)-, and pyrin domain-containing protein 3
- NRF-1/-2
Nuclear respiratory factor-1 and -2
- Nrf-2
Nuclear factor erythroid 2-related factor 2
- OMM
Outer mitochondrial membrane (mitochondrial)
- OPA-1
Optic atrophy-1 (s, short form; l, long form)
- OPTN
Optineurin
- OXPHOS
Oxidative phosphorylation
- P2R
Purinergic receptor
- p62SQSTM1
Autophagy cargo adaptor protein
- PAH
Pulmonary arterial hypertension
- PAMP
Pathogen-associated molecular pattern
- Parkin
Parkinson’s protein 2; E3 ubiquitin ligase
- PARL
Presenilin-associated rhomboid-like protein
- PCWP
Pulmonary capillary wedge pressure
- PD
Parkinson’s disease
- PGAM5
Phosphoglycerate mutase family member 5
- PGC-1α
Peroxisome proliferator-activated receptor gamma (PPARγ) coactivator 1α
- PHB2
Prohibitin 2
- PI4P
Phosphatidylinositol-4-phosphate
- PINK1
Phosphatase and tensin homolog (PTEN)-inducible kinase-1
- PRR
Pattern recognition receptor
- RCD
Regulated cell death
- RIG-I
Retinoic acid-inducible gene-I
- RIPK3
Receptor-interacting protein kinase-3
- RLR
RIG-I-like receptor RNA helicases
- RNS
Reactive nitrogen species
- Sirt
Sirtuin; NAD+-dependent deacetylase
- STING
Stimulator of interferon genes
- TBK1
Tank binding kinase-1
- TFAM
Mitochondrial transcription factor A
- TGF-β1
Transforming growth factor-beta 1
- TIM
Translocase of the inner mitochondrial membrane
- TLR
Toll-like receptor
- TOM
Translocase of the outer mitochondrial membrane
- TRIM25
Tripartite motif-containing protein-25
- UCP2
Uncoupling protein-2 (mitochondrial)
GRANTS
This work was supported by National Institutes of Health Grants 5 T32 HL 134629-04 (to J.S.H.) and P01 HL114501 (to A.M.K.C.). This work was also supported by the Weill Cornell Department of Medicine Pre-Career Award (to M.P.) and by the Parker B. Francis Foundation and the Stony Wold-Herbert Fund (to D.R.P.)
DISCLOSURES
S.W.R. is a current employee and stockholder of Proterris Inc. and a former Weill Cornell employee. A.M.K.C. is a cofounder and equity stockholder of Proterris, which develops therapeutic uses for carbon monoxide. A.M.K.C. has a use patent on CO. Additionally, A.M.K.C. has a patent in COPD. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
J.S.H., S.W.R., and D.R.P. prepared figures; J.S.H., S.W.R., M.P., D.R.P., and A.M.K.C. drafted manuscript; J.S.H., S.W.R., D.R.P., and A.M.K.C. edited and revised manuscript; J.S.H., S.W.R., M.P., D.R.P., and A.M.K.C. approved final version of manuscript.
REFERENCES
- 1. Vakifahmetoglu-Norberg H, Ouchida AT, Norberg E. The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun 482: 426–431, 2017. doi: 10.1016/j.bbrc.2016.11.088. [DOI] [PubMed] [Google Scholar]
- 2. Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13: 566–578, 2012. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 3. Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 4: 552–565, 2003. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 4. Jin HS, Suh HW, Kim SJ, Jo EK. Mitochondrial control of innate immunity and inflammation. Immune Netw 17: 77–88, 2017. doi: 10.4110/in.2017.17.2.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell 120: 483–495, 2005. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 6. Zhunina OA, Yabbarov NG, Grechko AV, Starodubova AV, Ivanova E, Nikiforov NG, Orekhov AN. The role of mitochondrial dysfunction in vascular disease, tumorigenesis, and diabetes. Front Mol Biosci 8: 671908, 2021. doi: 10.3389/fmolb.2021.671908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Duchen MR, Szabadkai G. Roles of mitochondria in human disease. Essays Biochem 47: 115–137, 2010. doi: 10.1042/bse0470115. [DOI] [PubMed] [Google Scholar]
- 8. Schumacker PT, Gillespie MN, Nakahira K, Choi AM, Crouser ED, Piantadosi CA, Bhattacharya J. Mitochondria in lung biology and pathology: more than just a powerhouse. Am J Physiol Lung Cell Mol Physiol 306: L962–L974, 2014. doi: 10.1152/ajplung.00073.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou WC, Qu J, Xie SY, Sun Y, Yao HW. Mitochondrial dysfunction in chronic respiratory diseases: implications for the pathogenesis and potential therapeutics. Oxid Med Cell Longev 2021: 5188306, 2021. doi: 10.1155/2021/5188306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ryter SW, Rosas IO, Owen CA, Martinez FJ, Choi ME, Lee CG, Elias JA, Choi AM. Mitochondrial dysfunction as a pathogenic mediator of chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Ann Am Thorac Soc 15: S266–S272, 2018. doi: 10.1513/AnnalsATS.201808-585MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86: 147–157, 1996. doi: 10.1016/S0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 12. Missiroli S, Genovese I, Perrone M, Vezzani B, Vitto VA, Giorgi C. The role of mitochondria in inflammation: from cancer to neurodegenerative disorders. J Clin Med 9: 740, 2020. doi: 10.3390/jcm9030740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heuer B. Mitochondrial DNA: Unraveling the “other” genome. J Am Assoc Nurse Pract 33: 673–675, 2021. doi: 10.1097/JXX.0000000000000646. [DOI] [PubMed] [Google Scholar]
- 14. Clayton DA. Vertebrate mitochondrial DNA—a circle of surprises. Exp Cell Res 255: 4–9, 2000. doi: 10.1006/excr.1999.4763. [DOI] [PubMed] [Google Scholar]
- 15. Li-Harms X, Milasta S, Lynch J, Wright C, Joshi A, Iyengar R, Neale G, Wang X, Wang YD, Prolla TA, Thompson JE, Opferman JT, Green DR, Schuetz J, Kundu M. Mito-protective autophagy is impaired in erythroid cells of aged mtDNA-mutator mice. Blood 125: 162–174, 2015. doi: 10.1182/blood-2014-07-586396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen ML, Logan TD, Hochberg ML, Shelat SG, Yu X, Wilding GE, Tan W, Kujoth GC, Prolla TA, Selak MA, Kundu M, Carroll M, Thompson JE. Erythroid dysplasia, megaloblastic anemia, and impaired lymphopoiesis arising from mitochondrial dysfunction. Blood 114: 4045–4053, 2009. doi: 10.1182/blood-2008-08-169474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kanungo S, Morton J, Neelakantan M, Ching K, Saeedian J, Goldstein A. Mitochondrial disorders. Ann Transl Med 6: 475, 2018. doi: 10.21037/atm.2018.12.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koopman WJ, Willems PH, Smeitink JA. Monogenic mitochondrial disorders. N Engl J Med 366: 1132–1141, 2012. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- 19. Koopman WJ, Distelmaier F, Smeitink JA, Willems PH. OXPHOS mutations and neurodegeneration. EMBO J 32: 9–29, 2013. doi: 10.1038/emboj.2012.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Niyazov DM, Kahler SG, Frye RE. Primary mitochondrial disease and secondary mitochondrial dysfunction: importance of distinction for diagnosis and treatment. Mol Syndromol 7: 122–137, 2016. doi: 10.1159/000446586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 31: 763–780, 2010. doi: 10.1002/humu.21277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255, 2012. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA 105: 14447–14452, 2008. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y, Zhang D, Scadden DT, Chen D. SIRT3 reverses aging-associated degeneration. Cell Rep 3: 319–327, 2013. doi: 10.1016/j.celrep.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frezza C, Gottlieb E. Mitochondria in cancer: not just innocent bystanders. Semin Cancer Biol 19: 4–11, 2009. doi: 10.1016/j.semcancer.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 26. Popov LD. Mitochondrial biogenesis: an update. J Cell Mol Med 24: 4892–4899, 2020. doi: 10.1111/jcmm.15194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 28: R170–R185, 2018. doi: 10.1016/j.cub.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12: 9–14, 2011. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Archer SL. Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N Engl J Med 369: 2236–2251, 2013. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 30. Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 337: 1062–1065, 2012. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O’Rourke B. From bioblasts to mitochondria: ever expanding roles of mitochondria in cell physiology. Front Physiol 1: 7, 2010. doi: 10.3389/fphys.2010.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Altmann R. Die Elementarorganismen und Ihre Beziehungen zu den Zellen. Leipzig: Veit, 1890. [Google Scholar]
- 33. Youle RJ. Mitochondria—striking a balance between host and endosymbiont. Science 365, 2019. doi: 10.1126/science.aaw9855. [DOI] [PubMed] [Google Scholar]
- 34. Ernster L, Schatz G. Mitochondria: a historical review. J Cell Biol 91: 227s–255s, 1981. doi: 10.1083/jcb.91.3.227s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Michaelis L. Die vitale Farbung, eine Darstellungsmethode der Zellgranula. Arch Mikrosk Anat 55: 558–575, 1900. [Google Scholar]
- 36. Claude A, Fullam EF. An electron microscope study of isolated mitochondria: method and preliminary results. J Exp Med 81: 51–62, 1945. doi: 10.1084/jem.81.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Palade GE. The fine structure of mitochondria. Anat Rec 114: 427–451, 1952. doi: 10.1002/ar.1091140304. [DOI] [PubMed] [Google Scholar]
- 38. Hogeboom GH, Claude A, Hotch-Kiss RD. The distribution of cytochrome oxidase and succinoxidase in the cytoplasm of the mammalian liver cell. J Biol Chem 165: 615–629, 1946. doi: 10.1016/S0021-9258(17)41175-6. [DOI] [PubMed] [Google Scholar]
- 39. Hoppins S, Collins SR, Cassidy-Stone A, Hummel E, Devay RM, Lackner LL, Westermann B, Schuldiner M, Weissman JS, Nunnari J. A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J Cell Biol 195: 323–340, 2011. doi: 10.1083/jcb.201107053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Anand R, Reichert AS, Kondadi AK. Emerging roles of the MICOS complex in cristae dynamics and biogenesis. Biology (Basel) 10: 600, 2021. doi: 10.3390/biology10070600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dudek J. Role of cardiolipin in mitochondrial signaling pathways. Front Cell Dev Biol 5: 90, 2017. doi: 10.3389/fcell.2017.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Glancy B, Kane DA, Kavazis AN, Goodwin ML, Willis WT, Gladden LB. Mitochondrial lactate metabolism: history and implications for exercise and disease. J Physiol 599: 863–888, 2021. doi: 10.1113/JP278930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol 20: 745–754, 2018. doi: 10.1038/s41556-018-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kolb H, Kempf K, Röhling M, Lenzen-Schulte M, Schloot NC, Martin S. Ketone bodies: from enemy to friend and guardian angel. BMC Med 19: 313, 2021. doi: 10.1186/s12916-021-02185-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nowinski SM, Solmonson A, Rusin SF, Maschek JA, Bensard CL, Fogarty S, Jeong MY, Lettlova S, Berg JA, Morgan JT, Ouyang Y, Naylor BC, Paulo JA, Funai K, Cox JE, Gygi SP, Winge DR, DeBerardinis RJ, Rutter J. Mitochondrial fatty acid synthesis coordinates oxidative metabolism in mammalian mitochondria. Elife 9: e58041, 2020. doi: 10.7554/eLife.58041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kastaniotis AJ, Autio KJ, Kerätär JM, Monteuuis G, Mäkelä AM, Nair RR, Pietikäinen LP, Shvetsova A, Chen Z, Hiltunen JK. Mitochondrial fatty acid synthesis, fatty acids and mitochondrial physiology. Biochim Biophys Acta Mol Cell Biol Lipids 1862: 39–48, 2017. doi: 10.1016/j.bbalip.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 47. Van Vranken JG, Nowinski SM, Clowers KJ, Jeong MY, Ouyang Y, Berg JA, Gygi JP, Gygi SP, Winge DR, Rutter JA. Acylation is an acetyl-CoA-dependent modification required for electron transport chain assembly. Mol Cell 71: 567–580.e4, 2018. doi: 10.1016/j.molcel.2018.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27: 441–464, 2011. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 49. Ye Z, Wang S, Zhang C, Zhao Y. Coordinated modulation of energy metabolism and inflammation by branched-chain amino acids and fatty acids. Front Endocrinol (Lausanne) 11: 617, 2020. doi: 10.3389/fendo.2020.00617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xiong Y, Jiang L, Li T. Aberrant branched-chain amino acid catabolism in cardiovascular diseases. Front Cardiovasc Med 9: 965899, 2022. doi: 10.3389/fcvm.2022.965899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ducker GS, Rabinowitz JD. One-carbon metabolism in health and disease. Cell Metab 25: 27–42, 2017. doi: 10.1016/j.cmet.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xie N, Zhang L, Gao W, Huang C, Huber PE, Zhou X, Li C, Shen G, Zou B. NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct Target Ther 5: 227, 2020. doi: 10.1038/s41392-020-00311-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ryter SW. Significance of heme and heme degradation in the pathogenesis of acute lung and inflammatory disorders. Int J Mol Sci 22: 5509, 2021. doi: 10.3390/ijms22115509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li S, Zhang J, Liu C, Wang Q, Yan J, Hui L, Jia Q, Shan H, Tao L, Zhang M. The role of mitophagy in regulating cell death. Oxid Med Cell Longev 2021: 6617256, 2021. doi: 10.1155/2021/6617256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Finkel T, Menazza S, Holmström KM, Parks RJ, Liu J, Sun J, Liu J, Pan X, Murphy E. The ins and outs of mitochondrial calcium. Circ Res 116: 1810–1819, 2015. doi: 10.1161/CIRCRESAHA.116.305484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Denton RM, McCormack JG. On the role of the calcium transport cycle in heart and other mammalian mitochondria. FEBS Lett 119: 1–8, 1980. doi: 10.1016/0014-5793(80)80986-0. [DOI] [PubMed] [Google Scholar]
- 57. Territo PR, Mootha VK, French SA, Balaban RS. Ca2+ activation of heart mitochondrial oxidative phosphorylation: role of the F0/F1-ATPase. Am J Physiol Cell Physiol 278: C423–C435, 2000. doi: 10.1152/ajpcell.2000.278.2.C423. [DOI] [PubMed] [Google Scholar]
- 58. Glancy B, Willis WT, Chess DJ, Balaban RS. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 52: 2793–2809, 2013. doi: 10.1021/bi3015983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Báthori G, Csordás G, Garcia-Perez C, Davies E, Hajnóczky G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J Biol Chem 281: 17347–17358, 2006. doi: 10.1074/jbc.M600906200. [DOI] [PubMed] [Google Scholar]
- 60. Sander P, Gudermann T, Schredelseker J. A calcium guard in the outer membrane: is VDAC a regulated gatekeeper of mitochondrial calcium uptake? Int J Mol Sci 22: 946, 2021. doi: 10.3390/ijms22020946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Omu AE, Unuigbe JA. Acceptance of contraceptive practice by grandmultiparae in Benin City, Nigeria. Int J Gynaecol Obstet 24: 145–150, 1986. doi: 10.1016/0020-7292(86)90009-3. [DOI] [PubMed] [Google Scholar]
- 62. Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342: 1379–1382, 2013. doi: 10.1126/science.1242993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Garbincius JF, Elrod JW. Mitochondrial calcium exchange in physiology and disease. Physiol Rev 102: 893–992, 2022. doi: 10.1152/physrev.00041.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. De Stefani D, Rizzuto R, Pozzan T. Enjoy the trip: calcium in mitochondria back and forth. Annu Rev Biochem 85: 161–192, 2016. doi: 10.1146/annurev-biochem-060614-034216. [DOI] [PubMed] [Google Scholar]
- 65. Marchi S, Bittremieux M, Missiroli S, Morganti C, Patergnani S, Sbano L, Rimessi A, Kerkhofs M, Parys JB, Bultynck G, Giorgi C, Pinton P. Endoplasmic reticulum-mitochondria communication through Ca2+ signaling: the importance of mitochondria-associated membranes (MAMs). Adv Exp Med Biol 997: 49–67, 2017. doi: 10.1007/978-981-10-4567-7_4. [DOI] [PubMed] [Google Scholar]
- 66. Godoy JA, Rios JA, Picón-Pagès P, Herrera-Fernández V, Swaby B, Crepin G, Vicente R, Fernández-Fernández JM, Muñoz FJ. Mitostasis, calcium and free radicals in health, aging and neurodegeneration. Biomolecules 11: 1012, 2021. doi: 10.3390/biom11071012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bonora M, Giorgi C, Pinton P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat Rev Mol Cell Biol 23: 266–285, 2022. doi: 10.1038/s41580-021-00433-y. [DOI] [PubMed] [Google Scholar]
- 68. Marcu R, Kotha S, Zhi Z, Qin W, Neeley CK, Wang RK, Zheng Y, Hawkins BJ. The mitochondrial permeability transition pore regulates endothelial bioenergetics and angiogenesis. Circ Res 116: 1336–1345, 2015. doi: 10.1161/CIRCRESAHA.116.304881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 46: 821–831, 2009. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 70. Nesci S. What happens when the mitochondrial H+-translocating F1FO-ATP(hydrol)ase becomes a molecular target of calcium? The pore opens. Biochimie 198: 92–95, 2022. doi: 10.1016/j.biochi.2022.03.012. [DOI] [PubMed] [Google Scholar]
- 71. Parks RJ, Murphy E, Liu JC. Mitochondrial permeability transition pore and calcium handling. Methods Mol Biol 1782: 187–196, 2018. doi: 10.1007/978-1-4939-7831-1_11. [DOI] [PubMed] [Google Scholar]
- 72. Gao P, Yang W, Sun L. Mitochondria-associated endoplasmic reticulum membranes (MAMs) and their prospective roles in kidney disease. Oxid Med Cell Longev 2020: 3120539, 2020. doi: 10.1155/2020/3120539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 175: 901–911, 2006. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liu Y, Zhu X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl Neurodegener 6: 21, 2017. doi: 10.1186/s40035-017-0092-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Liu Y, Ma X, Fujioka H, Liu J, Chen S, Zhu X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc Natl Acad Sci USA 116: 25322–25328, 2019. doi: 10.1073/pnas.1906565116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Modi S, López-Doménech G, Halff EF, Covill-Cooke C, Ivankovic D, Melandri D, Arancibia-Cárcamo IL, Burden JJ, Lowe AR, Kittler JT. Miro clusters regulate ER-mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat Commun 10: 4399, 2019. doi: 10.1038/s41467-019-12382-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hirabayashi Y, Kwon SK, Paek H, Pernice WM, Paul MA, Lee J, Erfani P, Raczkowski A, Petrey DS, Pon LA, Polleux F. ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 358: 623–630, 2017. doi: 10.1126/science.aan6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Degechisa ST, Dabi YT, Gizaw ST. The mitochondrial associated endoplasmic reticulum membranes: a platform for the pathogenesis of inflammation-mediated metabolic diseases. Immun Inflamm Dis 10: e647, 2022. doi: 10.1002/iid3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J 435: 297–312, 2011. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD, Balakireva AV, Juhaszova M, Sollott SJ, Zorov DB. Mitochondrial membrane potential. Anal Biochem 552: 50–59, 2018. doi: 10.1016/j.ab.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kamo N, Muratsugu M, Hongoh R, Kobatake Y. Membrane potential of mitochondria measured with an electrode sensitive to tetraphenyl phosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J Membr Biol 49: 105–121, 1979. doi: 10.1007/BF01868720. [DOI] [PubMed] [Google Scholar]
- 82. Rovini A, Heslop K, Hunt EG, Morris ME, Fang D, Gooz M, Gerencser AA, Maldonado EN. Quantitative analysis of mitochondrial membrane potential heterogeneity in unsynchronized and synchronized cancer cells. FASEB J 35: e21148, 2021. doi: 10.1096/fj.202001693R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Collins TJ, Berridge MJ, Lipp P, Bootman MD. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J 21: 1616–1627, 2002. doi: 10.1093/emboj/21.7.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gerencser AA, Chinopoulos C, Birket MJ, Jastroch M, Vitelli C, Nicholls DG, Brand MD. Quantitative measurement of mitochondrial membrane potential in cultured cells: calcium-induced de- and hyperpolarization of neuronal mitochondria. J Physiol 590: 2845–2871, 2012. doi: 10.1113/jphysiol.2012.228387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Georgakopoulos ND, Wells G, Campanella M. The pharmacological regulation of cellular mitophagy. Nat Chem Biol 13: 136–146, 2017. doi: 10.1038/nchembio.2287. [DOI] [PubMed] [Google Scholar]
- 86. Demine S, Renard P, Arnould T. Mitochondrial uncoupling: a key controller of biological processes in physiology and diseases. Cells 8: 795, 2019. doi: 10.3390/cells8080795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kushnareva YE, Sokolove PM. Prooxidants open both the mitochondrial permeability transition pore and a low-conductance channel in the inner mitochondrial membrane. Arch Biochem Biophys 376: 377–388, 2000. doi: 10.1006/abbi.2000.1730. [DOI] [PubMed] [Google Scholar]
- 88. Kowaltowski AJ, Smaili SS, Russell JT, Fiskum G. Elevation of resting mitochondrial membrane potential of neural cells by cyclosporin A, BAPTA-AM, and bcl-2. Am J Physiol Cell Physiol 279: C852–C859, 2000. doi: 10.1152/ajpcell.2000.279.3.C852. [DOI] [PubMed] [Google Scholar]
- 89. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol 21: 363–383, 2020. doi: 10.1038/s41580-020-0230-3. [DOI] [PubMed] [Google Scholar]
- 90. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 417: 1–13, 2009. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48: 158–167, 2012. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bleier L, Dröse S. Superoxide generation by complex III: from mechanistic rationales to functional consequences. Biochim Biophys Acta 1827: 1320–1331, 2013. doi: 10.1016/j.bbabio.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 93. Scialò F, Fernández-Ayala DJ, Sanz A. Role of mitochondrial reverse electron transport in ROS signaling: potential roles in health and disease. Front Physiol 8: 428, 2017. doi: 10.3389/fphys.2017.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Onukwufor JO, Berry BJ, Wojtovich AP. Physiologic implications of reactive oxygen species production by mitochondrial Complex I reverse electron transport. Antioxidants (Basel) 8: 285, 2019. doi: 10.3390/antiox8080285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515: 431–435, 2014. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94: 909–950, 2014. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res 106: 1253–1264, 2010. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci USA 106: 14385–14390, 2009. doi: 10.1073/pnas.0906805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA 107: 15565–15570, 2010. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ago T, Matsushima S, Kuroda J, Zablocki D, Kitazono T, Sadoshima J. The NADPH oxidase Nox4 and aging in the heart. Aging (Albany NY) 2: 1012–1016, 2010. doi: 10.18632/aging.100261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bradshaw PC. Cytoplasmic and mitochondrial NADPH-coupled redox systems in the regulation of aging. Nutrients 11: 504, 2019. doi: 10.3390/nu11030504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Calabrese G, Morgan B, Riemer J. Mitochondrial glutathione: regulation and functions. Antioxid Redox Signal 27: 1162–1177, 2017. doi: 10.1089/ars.2017.7121. [DOI] [PubMed] [Google Scholar]
- 103. Wang Y, Yen FS, Zhu XG, Timson RC, Weber R, Xing C, Liu Y, Allwein B, Luo H, Yeh HW, Heissel S, Unlu G, Gamazon ER, Kharas MG, Hite R, Birsoy K. SLC25A39 is necessary for mitochondrial glutathione import in mammalian cells. Nature 599: 136–140, 2021. doi: 10.1038/s41586-021-04025-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mårtensson J, Lai JC, Meister A. High-affinity transport of glutathione is part of a multicomponent system essential for mitochondrial function. Proc Natl Acad Sci USA 87: 7185–7189, 1990. doi: 10.1073/pnas.87.18.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab 1: 401–408, 2005. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 106. Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 275: 25130–25138, 2000. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 107. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403: 795–800, 2000. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 108. Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13: 225–238, 2012. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Rajendran R, Garva R, Krstic-Demonacos M, Demonacos C. Sirtuins: molecular traffic lights in the crossroad of oxidative stress, chromatin remodeling, and transcription. J Biomed Biotechnol 2011: 368276, 2011. doi: 10.1155/2011/368276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem 75: 435–465, 2006. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 111. Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J. Sirtuins: the ‘magnificent seven’, function, metabolism and longevity. Ann Med 39: 335–345, 2007. doi: 10.1080/07853890701408194. [DOI] [PubMed] [Google Scholar]
- 112. Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell 16: 4623–4635, 2005. doi: 10.1091/mbc.e05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun 273: 793–798, 2000. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 114. Kupis W, Pałyga J, Tomal E, Niewiadomska E. The role of sirtuins in cellular homeostasis. J Physiol Biochem 72: 371–380, 2016. doi: 10.1007/s13105-016-0492-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Shahgaldi S, Kahmini FR. A comprehensive review of Sirtuins: with a major focus on redox homeostasis and metabolism. Life Sci 282: 119803, 2021. doi: 10.1016/j.lfs.2021.119803. [DOI] [PubMed] [Google Scholar]
- 116. Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, Washida N, Tokuyama H, Hayashi K, Itoh H. Sirt1 protects against oxidative stress-induced renal tubular cell apoptosis by the bidirectional regulation of catalase expression. Biochem Biophys Res Commun 372: 51–56, 2008. doi: 10.1016/j.bbrc.2008.04.176. [DOI] [PubMed] [Google Scholar]
- 117. Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1alpha. J Biol Chem 280: 16456–16460, 2005. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 118. Jing E, Gesta S, Kahn CR. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab 6: 105–114, 2007. doi: 10.1016/j.cmet.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Maxwell MM, Tomkinson EM, Nobles J, Wizeman JW, Amore AM, Quinti L, Chopra V, Hersch SM, Kazantsev AG. The Sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging CNS. Hum Mol Genet 20: 3986–3996, 2011. doi: 10.1093/hmg/ddr326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Liu G, Park SH, Imbesi M, Nathan WJ, Zou X, Zhu Y, Jiang H, Parisiadou L, Gius D. Loss of NAD-dependent protein deacetylase Sirtuin-2 alters mitochondrial protein acetylation and dysregulates mitophagy. Antioxid Redox Signal 26: 849–863, 2017. doi: 10.1089/ars.2016.6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6: 505–514, 2007. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- 122. Elkhwanky MS, Hakkola J. Extranuclear sirtuins and metabolic stress. Antioxid Redox Signal 28: 662–676, 2018. doi: 10.1089/ars.2017.7270. [DOI] [PubMed] [Google Scholar]
- 123. Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci USA 99: 13653–13658, 2002. doi: 10.1073/pnas.222538099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol 158: 647–657, 2002. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem 280: 13560–13567, 2005. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- 126. Haigis MC, Guarente LP. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev 20: 2913–2921, 2006. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 127. Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol 382: 790–801, 2008. doi: 10.1016/j.jmb.2008.07.048. [DOI] [PubMed] [Google Scholar]
- 128. Haschler TN, Horsley H, Balys M, Anderson G, Taanman JW, Unwin RJ, Norman JT. Sirtuin 5 depletion impairs mitochondrial function in human proximal tubular epithelial cells. Sci Rep 11: 15510, 2021. doi: 10.1038/s41598-021-94185-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Peng C, Lu Z, Xie Z, Cheng Z, Chen Y, Tan M, Luo H, Zhang Y, He W, Yang K, Zwaans BM, Tishkoff D, Ho L, Lombard D, He TC, Dai J, Verdin E, Ye Y, Zhao Y. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics 10: M111.012658, 2011. doi: 10.1074/mcp.M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334: 806–809, 2011. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, Park J, Chen Y, Huang H, Zhang Y, Ro J, Wagner GR, Green MF, Madsen AS, Schmiesing J, Peterson BS, Xu G, Ilkayeva OR, Muehlbauer MJ, Braulke T, Mühlhausen C, Backos DS, Olsen CA, McGuire PJ, Pletcher SD, Lombard DB, Hirschey MD, Zhao Y. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab 19: 605–617, 2014. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Boylston JA, Sun J, Chen Y, Gucek M, Sack MN, Murphy E. Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J Mol Cell Cardiol 88: 73–81, 2015. doi: 10.1016/j.yjmcc.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, Guo A, Gut P, Sahu AK, Li B, Uppala R, Fitch M, Riiff T, Zhu L, Zhou J, Mulhern D, Stevens RD, Ilkayeva OR, Newgard CB, Jacobson MP, Hellerstein M, Goetzman ES, Gibson BW, Verdin E. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab 18: 920–933, 2013. doi: 10.1016/j.cmet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Nishida Y, Rardin MJ, Carrico C, He W, Sahu AK, Gut P, Najjar R, Fitch M, Hellerstein M, Gibson BW, Verdin E. SIRT5 Regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol Cell 59: 321–332, 2015. doi: 10.1016/j.molcel.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Zhang Y, Bharathi SS, Rardin MJ, Lu J, Maringer KV, Sims-Lucas S, Prochownik EV, Gibson BW, Goetzman ES. Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J Biol Chem 292: 10239–10249, 2017. doi: 10.1074/jbc.M117.785022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Li F, He X, Ye D, Lin Y, Yu H, Yao C, Huang L, Zhang J, Wang F, Xu S, Wu X, Liu L, Yang C, Shi J, He X, Liu J, Qu Y, Guo F, Zhao J, Xu W, Zhao S. NADP+-IDH mutations promote hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Mol Cell 60: 661–675, 2015. doi: 10.1016/j.molcel.2015.10.017. [DOI] [PubMed] [Google Scholar]
- 137. Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, Xu Y, Zhou L, Chu Y, Zhang C, Qin X, Yang P, Yu H. SIRT5 desuccinylates and activates pyruvate kinase M2 to block macrophage IL-1beta production and to prevent DSS-induced colitis in mice. Cell Rep 19: 2331–2344, 2017. doi: 10.1016/j.celrep.2017.05.065. [DOI] [PubMed] [Google Scholar]
- 138. Kumar S, Lombard DB. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathobiology. Crit Rev Biochem Mol Biol 53: 311–334, 2018. doi: 10.1080/10409238.2018.1458071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BM, Skinner ME, Lombard DB, Zhao Y. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell 50: 919–930, 2013. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Xu YS, Liang JJ, Wang Y, Zhao XJ, Xu L, Xu YY, Zou QC, Zhang JM, Tu CE, Cui YG, Sun WH, Huang C, Yang JH, Chin YE. STAT3 undergoes acetylation-dependent mitochondrial translocation to regulate pyruvate metabolism. Sci Rep 6: 39517, 2016. doi: 10.1038/srep39517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Buler M, Aatsinki SM, Izzi V, Uusimaa J, Hakkola J. SIRT5 is under the control of PGC-1alpha and AMPK and is involved in regulation of mitochondrial energy metabolism. FASEB J 28: 3225–3237, 2014. doi: 10.1096/fj.13-245241. [DOI] [PubMed] [Google Scholar]
- 142. Lin ZF, Xu HB, Wang JY, Lin Q, Ruan Z, Liu FB, Jin W, Huang HH, Chen X. SIRT5 desuccinylates and activates SOD1 to eliminate ROS. Biochem Biophys Res Commun 441: 191–195, 2013. doi: 10.1016/j.bbrc.2013.10.033. [DOI] [PubMed] [Google Scholar]
- 143. Zhou L, Wang F, Sun R, Chen X, Zhang M, Xu Q, Wang Y, Wang S, Xiong Y, Guan KL, Yang P, Yu H, Ye D. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep 17: 811–822, 2016. doi: 10.15252/embr.201541643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Yoshii SR, Mizushima N. Autophagy machinery in the context of mammalian mitophagy. Biochim Biophys Acta 1853: 2797–2801, 2015. doi: 10.1016/j.bbamcr.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 145. Doblado L, Lueck C, Rey C, Samhan-Arias AK, Prieto I, Stacchiotti A, Monsalve M. Mitophagy in human diseases. Int J Mol Sci 22: 3903, 2021. doi: 10.3390/ijms22083903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Bakula D, Scheibye-Knudsen M. MitophAging: mitophagy in aging and disease. Front Cell Dev Biol 8: 239, 2020. doi: 10.3389/fcell.2020.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 22: 320–333, 2012. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, Abramov AY, Wood NW. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 20: 867–879, 2011. doi: 10.1093/hmg/ddq526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Meissner C, Lorenz H, Hehn B, Lemberg MK. Intramembrane protease PARL defines a negative regulator of PINK1- and PARK2/Parkin-dependent mitophagy. Autophagy 11: 1484–1498, 2015. doi: 10.1080/15548627.2015.1063763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Shi G, McQuibban GA. The mitochondrial rhomboid protease PARL is regulated by PDK2 to integrate mitochondrial quality control and metabolism. Cell Rep 18: 1458–1472, 2017. doi: 10.1016/j.celrep.2017.01.029. [DOI] [PubMed] [Google Scholar]
- 151. Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy 9: 1758–1769, 2013. doi: 10.4161/auto.24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Eldeeb MA, Ragheb MA. N-degron-mediated degradation and regulation of mitochondrial PINK1 kinase. Curr Genet 66: 693–701, 2020. doi: 10.1007/s00294-020-01062-2. [DOI] [PubMed] [Google Scholar]
- 153. Guardia-Laguarta C, Liu Y, Lauritzen KH, Erdjument-Bromage H, Martin B, Swayne TC, Jiang X, Przedborski S. PINK1 content in mitochondria is regulated by ER-associated degradation. J Neurosci 39: 7074–7085, 2019. doi: 10.1523/JNEUROSCI.1691-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166, 2014. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 155. Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J 460: 127–139, 2014. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205: 143–153, 2014. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803, 2008. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298, 2010. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 19: 4861–4870, 2010. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Truban D, Hou X, Caulfield TR, Fiesel FC, Springer W. PINK1, Parkin, and mitochondrial quality control: what can we learn about Parkinson’s disease pathobiology? J Parkinsons Dis 7: 13–29, 2017. doi: 10.3233/JPD-160989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Karbowski M, Youle RJ. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol 23: 476–482, 2011. doi: 10.1016/j.ceb.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6: 1090–1106, 2010. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314, 2015. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Padman BS, Nguyen TN, Uoselis L, Skulsuppaisarn M, Nguyen LK, Lazarou M. LC3/GABARAPs drive ubiquitin-independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat Commun 10: 408, 2019. doi: 10.1038/s41467-019-08335-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60: 7–20, 2015. doi: 10.1016/j.molcel.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci 125: 1488–1499, 2012. doi: 10.1242/jcs.094110. [DOI] [PubMed] [Google Scholar]
- 167. Yamano K, Kikuchi R, Kojima W, Hayashida R, Koyano F, Kawawaki J, Shoda T, Demizu Y, Naito M, Tanaka K, Matsuda N. Critical role of mitochondrial ubiquitination and the OPTN-ATG9A axis in mitophagy. J Cell Biol 219: e201912144, 2020. doi: 10.1083/jcb.201912144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci USA 104: 11441–11446, 2007. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Kelm-Nelson CA, Brauer AF, Barth KJ, Lake JM, Sinnen ML, Stehula FJ, Muslu C, Marongiu R, Kaplitt MG, Ciucci MR. Characterization of early-onset motor deficits in the Pink1-/- mouse model of Parkinson disease. Brain Res 1680: 1–12, 2018. doi: 10.1016/j.brainres.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Das Banerjee T, Dagda RY, Dagda M, Chu CT, Rice M, Vazquez-Mayorga E, Dagda RK. PINK1 regulates mitochondrial trafficking in dendrites of cortical neurons through mitochondrial PKA. J Neurochem 142: 545–559, 2017. doi: 10.1111/jnc.14083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Gispert S, Ricciardi F, Kurz A, Azizov M, Hoepken HH, Becker D, Voos W, Leuner K, Müller WE, Kudin AP, Kunz WS, Zimmermann A, Roeper J, Wenzel D, Jendrach M, García-Arencíbia M, Fernández-Ruiz J, Huber L, Rohrer H, Barrera M, Reichert AS, Rub U, Chen A, Nussbaum RL, Auburger G. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS One 4: e5777, 2009. doi: 10.1371/journal.pone.0005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, Cai H, Borsche M, Klein C, Youle RJ. Parkin and PINK1 mitigate STING-induced inflammation. Nature 561: 258–262, 2018. doi: 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 173. Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278: 43628–43635, 2003. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- 174. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dötsch V, Ney PA, Dikic I. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11: 45–51, 2010. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Novak I. Mitophagy: a complex mechanism of mitochondrial removal. Antioxid Redox Signal 17: 794–802, 2012. doi: 10.1089/ars.2011.4407. [DOI] [PubMed] [Google Scholar]
- 176. Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 285: 27879–27890, 2010. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Wu W, Lin C, Wu K, Jiang L, Wang X, Li W, Zhuang H, Zhang X, Chen H, Li S, Yang Y, Lu Y, Wang J, Zhu R, Zhang L, Sui S, Tan N, Zhao B, Zhang J, Li L, Feng D. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J 35: 1368–1384, 2016. doi: 10.15252/embj.201593102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Zhang W. The mitophagy receptor FUN14 domain-containing 1 (FUNDC1): a promising biomarker and potential therapeutic target of human diseases. Genes Dis 8: 640–654, 2021. doi: 10.1016/j.gendis.2020.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Sekine S, Kanamaru Y, Koike M, Nishihara A, Okada M, Kinoshita H, Kamiyama M, Maruyama J, Uchiyama Y, Ishihara N, Takeda K, Ichijo H. Rhomboid protease PARL mediates the mitochondrial membrane potential loss-induced cleavage of PGAM5. J Biol Chem 287: 34635–34645, 2012. doi: 10.1074/jbc.M112.357509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C, Duan L, Wang X, Liu L, Liu X, Shen Y, Zhu Y, Chen Q. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell 54: 362–377, 2014. doi: 10.1016/j.molcel.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 181. Ma K, Zhang Z, Chang R, Cheng H, Mu C, Zhao T, Chen L, Zhang C, Luo Q, Lin J, Zhu Y, Chen Q. Dynamic PGAM5 multimers dephosphorylate BCL-xL or FUNDC1 to regulate mitochondrial and cellular fate. Cell Death Differ 27: 1036–1051, 2020. doi: 10.1038/s41418-019-0396-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Wu H, Xue D, Chen G, Han Z, Huang L, Zhu C, Wang X, Jin H, Wang J, Zhu Y, Liu L, Chen Q. The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy 10: 1712–1725, 2014. doi: 10.4161/auto.29568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Sugo M, Kimura H, Arasaki K, Amemiya T, Hirota N, Dohmae N, Imai Y, Inoshita T, Shiba-Fukushima K, Hattori N, Cheng J, Fujimoto T, Wakana Y, Inoue H, Tagaya M. Syntaxin 17 regulates the localization and function of PGAM5 in mitochondrial division and mitophagy. EMBO J 37, 2018. doi: 10.15252/embj.201798899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Yan C, Gong L, Chen L, Xu M, Abou-Hamdan H, Tang M, Désaubry L, Song Z. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 16: 419–434, 2020. doi: 10.1080/15548627.2019.1628520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Orvedahl A, Sumpter R Jr, Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, Forst CV, Wrana JL, Zhang YE, Luby-Phelps K, Xavier RJ, Xie Y, Levine B. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480: 113–117, 2011. doi: 10.1038/nature10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Tang D, Kang R, Livesey KM, Kroemer G, Billiar TR, Van Houten B, Zeh HJ 3rd, Lotze MT. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab 13: 701–711, 2011. doi: 10.1016/j.cmet.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Singh S, Sharma S. Dynamin-related protein-1 as potential therapeutic target in various diseases. Inflammopharmacology 25: 383–392, 2017. doi: 10.1007/s10787-017-0347-y. [DOI] [PubMed] [Google Scholar]
- 188. Liesa M, Palacín M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89: 799–845, 2009. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 189. Eisner V, Picard M, Hajnóczky G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol 20: 755–765, 2018. doi: 10.1038/s41556-018-0133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol 15: 235–259, 2020. doi: 10.1146/annurev-pathmechdis-012419-032711. [DOI] [PubMed] [Google Scholar]
- 191. Yapa NMB, Lisnyak V, Reljic B, Ryan MT. Mitochondrial dynamics in health and disease. FEBS Lett 595: 1184–1204, 2021. doi: 10.1002/1873-3468.14077. [DOI] [PubMed] [Google Scholar]
- 192. Breitzig MT, Alleyn MD, Lockey RF, Kolliputi N. A mitochondrial delicacy: dynamin-related protein 1 and mitochondrial dynamics. Am J Physiol Cell Physiol 315: C80–C90, 2018. doi: 10.1152/ajpcell.00042.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Kraus F, Roy K, Pucadyil TJ, Ryan MT. Function and regulation of the divisome for mitochondrial fission. Nature 590: 57–66, 2021. doi: 10.1038/s41586-021-03214-x. [DOI] [PubMed] [Google Scholar]
- 194. Hatch AL, Gurel PS, Higgs HN. Novel roles for actin in mitochondrial fission. J Cell Sci 127: 4549–4560, 2014. doi: 10.1242/jcs.153791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol 16: R551–R560, 2006. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 196. Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem 62: 341–360, 2018. doi: 10.1042/EBC20170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Lee JE, Westrate LM, Wu H, Page C, Voeltz GK. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540: 139–143, 2016. doi: 10.1038/nature20555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, Chen Y, Morrow E, Weir EK, Rehman J, Archer SL. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 110: 1484–1497, 2012. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Bo T, Yamamori T, Suzuki M, Sakai Y, Yamamoto K, Inanami O. Calmodulin-dependent protein kinase II (CaMKII) mediates radiation-induced mitochondrial fission by regulating the phosphorylation of dynamin-related protein 1 (Drp1) at serine 616. Biochem Biophys Res Commun 495: 1601–1607, 2018. doi: 10.1016/j.bbrc.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 200. Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8: 939–944, 2007. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Valera-Alberni M, Joffraud M, Miro-Blanch J, Capellades J, Junza A, Dayon L, Núñez Galindo A, Sanchez-Garcia JL, Valsesia A, Cercillieux A, Söllner F, Ladurner AG, Yanes O, Cantó C. Crosstalk between Drp1 phosphorylation sites during mitochondrial remodeling and their impact on metabolic adaptation. Cell Rep 36: 109565, 2021. doi: 10.1016/j.celrep.2021.109565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Yu R, Lendahl U, Nistér M, Zhao J. Regulation of mammalian mitochondrial dynamics: opportunities and challenges. Front Endocrinol (Lausanne) 11: 374, 2020. doi: 10.3389/fendo.2020.00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep 12: 565–573, 2011. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J Biol Chem 288: 27584–27593, 2013. doi: 10.1074/jbc.M113.479873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R, Ryan MT. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J Cell Sci 129: 2170–2181, 2016. doi: 10.1242/jcs.185165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191: 1141–1158, 2010. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Yu R, Liu T, Jin SB, Ning C, Lendahl U, Nistér M, Zhao J. MIEF1/2 function as adaptors to recruit Drp1 to mitochondria and regulate the association of Drp1 with Mff. Sci Rep 7: 880, 2017. doi: 10.1038/s41598-017-00853-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Atkins K, Dasgupta A, Chen KH, Mewburn J, Archer SL. The role of Drp1 adaptor proteins MiD49 and MiD51 in mitochondrial fission: implications for human disease. Clin Sci (Lond) 130: 1861–1874, 2016. doi: 10.1042/CS20160030. [DOI] [PubMed] [Google Scholar]
- 209. Richter V, Palmer CS, Osellame LD, Singh AP, Elgass K, Stroud DA, Sesaki H, Kvansakul M, Ryan MT. Structural and functional analysis of MiD51, a dynamin receptor required for mitochondrial fission. J Cell Biol 204: 477–486, 2014. doi: 10.1083/jcb.201311014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Yu R, Jin SB, Lendahl U, Nistér M, Zhao J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J 38: e99748, 2019. doi: 10.15252/embj.201899748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Losón OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24: 659–667, 2013. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Kreymerman A, Buickians DN, Nahmou MM, Tran T, Galvao J, Wang Y, Sun N, Bazik L, Huynh SK, Cho IJ, Boczek T, Chang KC, Kunzevitzky NJ, Goldberg JL. MTP18 is a novel regulator of mitochondrial fission in CNS neuron development, axonal growth, and injury responses. Sci Rep 9: 10669, 2019. doi: 10.1038/s41598-019-46956-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Xu S, Cherok E, Das S, Li S, Roelofs BA, Ge SX, Polster BM, Boyman L, Lederer WJ, Wang C, Karbowski M. Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol Biol Cell 27: 349–359, 2016. doi: 10.1091/mbc.E15-09-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Nagashima S, Tokuyama T, Yonashiro R, Inatome R, Yanagi S. Roles of mitochondrial ubiquitin ligase MITOL/MARCH5 in mitochondrial dynamics and diseases. J Biochem 155: 273–279, 2014. doi: 10.1093/jb/mvu016. [DOI] [PubMed] [Google Scholar]
- 215. Shiiba I, Takeda K, Nagashima S, Yanagi S. Overview of mitochondrial E3 ubiquitin ligase MITOL/MARCH5 from molecular mechanisms to diseases. Int J Mol Sci 21: 3781, 2020. doi: 10.3390/ijms21113781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Wong YC, Ysselstein D, Krainc D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554: 382–386, 2018. doi: 10.1038/nature25486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Boutry M, Kim PK. ORP1L mediated PI4P signaling at ER-lysosome-mitochondrion three-way contact contributes to mitochondrial division. Nat Commun 12: 5354, 2021. doi: 10.1038/s41467-021-25621-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Abrisch RG, Gumbin SC, Wisniewski BT, Lackner LL, Voeltz GK. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J Cell Biol 219, 2020. doi: 10.1083/jcb.201911122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell 20: 3525–3532, 2009. doi: 10.1091/mbc.e09-03-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Murata D, Arai K, Iijima M, Sesaki H. Mitochondrial division, fusion and degradation. J Biochem 167: 233–241, 2020. doi: 10.1093/jb/mvz106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605–610, 2008. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 222. Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA 101: 15927–15932, 2004. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Ban T, Ishihara T, Kohno H, Saita S, Ichimura A, Maenaka K, Oka T, Mihara K, Ishihara N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol 19: 856–863, 2017. doi: 10.1038/ncb3560. [DOI] [PubMed] [Google Scholar]
- 224. Ban T, Kohno H, Ishihara T, Ishihara N. Relationship between OPA1 and cardiolipin in mitochondrial inner-membrane fusion. Biochim Biophys Acta Bioenerg 1859: 951–957, 2018. doi: 10.1016/j.bbabio.2018.05.016. [DOI] [PubMed] [Google Scholar]
- 225. Ge Y, Shi X, Boopathy S, McDonald J, Smith AW, Chao LH. Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. Elife 9: e50973, 2020. doi: 10.7554/eLife.50973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schröder JM, Vance JM. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36: 449–451, 2004. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 227. Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26: 211–215, 2000. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 228. Sun S, Erchova I, Sengpiel F, Votruba M. Opa1 deficiency leads to diminished mitochondrial bioenergetics with compensatory increased mitochondrial motility. Invest Ophthalmol Vis Sci 61: 42, 2020. doi: 10.1167/iovs.61.6.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Barnhart EL. Mechanics of mitochondrial motility in neurons. Curr Opin Cell Biol 38: 90–99, 2016. doi: 10.1016/j.ceb.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 230. Nahacka Z, Novak J, Zobalova R, Neuzil J. Miro proteins and their role in mitochondrial transfer in cancer and beyond. Front Cell Dev Biol 10: 937753, 2022. doi: 10.3389/fcell.2022.937753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Kay L, Pienaar IS, Cooray R, Black G, Soundararajan M. Understanding Miro GTPases: implications in the treatment of neurodegenerative disorders. Mol Neurobiol 55: 7352–7365, 2018. doi: 10.1007/s12035-018-0927-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnóczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci USA 105: 20728–20733, 2008. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Klosowiak JL, Focia PJ, Chakravarthy S, Landahl EC, Freymann DM, Rice SE. Structural coupling of the EF hand and C-terminal GTPase domains in the mitochondrial protein Miro. EMBO Rep 14: 968–974, 2013. doi: 10.1038/embor.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61: 541–555, 2009. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Shlevkov E, Kramer T, Schapansky J, LaVoie MJ, Schwarz TL. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc Natl Acad Sci USA 113: E6097–E6106, 2016. doi: 10.1073/pnas.1612283113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. López-Doménech G, Howden JH, Covill-Cooke C, Morfill C, Patel JV, Bürli R, Crowther D, Birsa N, Brandon NJ, Kittler JT. Loss of neuronal Miro1 disrupts mitophagy and induces hyperactivation of the integrated stress response. EMBO J 40: e100715, 2021. doi: 10.15252/embj.2018100715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Pekkurnaz G, Trinidad JC, Wang X, Kong D, Schwarz TL. Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell 158: 54–68, 2014. doi: 10.1016/j.cell.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Basu H, Pekkurnaz G, Falk J, Wei W, Chin M, Steen J, Schwarz TL. FHL2 anchors mitochondria to actin and adapts mitochondrial dynamics to glucose supply. J Cell Biol 220: e201912077, 2021. doi: 10.1083/jcb.201912077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Todkar K, Chikhi L, Desjardins V, El-Mortada F, Pépin G, Germain M. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat Commun 12: 1971, 2021. doi: 10.1038/s41467-021-21984-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Amari L, Germain M. Mitochondrial extracellular vesicles—origins and roles. Front Mol Neurosci 14: 767219, 2021. doi: 10.3389/fnmol.2021.767219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Michailov ML, Schad H, Dahlheim H, Jacob IC, Brechtelsbauer H. Renin-angiotensin system responses of acute graded hemorrhage in dogs. Circ Shock 21: 217–224, 1987. [PubMed] [Google Scholar]
- 242. Ottonelli I, Caraffi R, Tosi G, Vandelli MA, Duskey JT, Ruozi B. Tunneling nanotubes: a new target for nanomedicine? Int J Mol Sci 23: 2237, 2022. doi: 10.3390/ijms23042237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Cherry AD, Piantadosi CA. Regulation of mitochondrial biogenesis and its intersection with inflammatory responses. Antioxid Redox Signal 22: 965–976, 2015. doi: 10.1089/ars.2014.6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Gureev AP, Shaforostova EA, Popov VN. Regulation of mitochondrial biogenesis as a way for active longevity: interaction between the Nrf2 and PGC-1alpha signaling pathways. Front Genet 10: 435, 2019. doi: 10.3389/fgene.2019.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 1813: 1269–1278, 2011. doi: 10.1016/j.bbamcr.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem 47: 69–84, 2010. doi: 10.1042/bse0470069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434: 113–118, 2005. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 248. Villena JA. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J 282: 647–672, 2015. doi: 10.1111/febs.13175. [DOI] [PubMed] [Google Scholar]
- 249. Hinkle JS, Rivera CN, Vaughan RA. Branched-chain amino acids and mitochondrial biogenesis: an overview and mechanistic summary. Mol Nutr Food Res 66: e2200109, 2022. doi: 10.1002/mnfr.202200109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Hatazawa Y, Tadaishi M, Nagaike Y, Morita A, Ogawa Y, Ezaki O, Takai-Igarashi T, Kitaura Y, Shimomura Y, Kamei Y, Miura S. PGC-1alpha-mediated branched-chain amino acid metabolism in the skeletal muscle. PLoS One 9: e91006, 2014. doi: 10.1371/journal.pone.0091006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Chew K, Zhao L. Interactions of mitochondrial transcription factor A with DNA damage: mechanistic insights and functional implications. Genes (Basel) 12: 1246, 2021. doi: 10.3390/genes12081246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Ngo HB, Lovely GA, Phillips R, Chan DC. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat Commun 5: 3077, 2014. doi: 10.1038/ncomms4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med 88: 179–188, 2015. doi: 10.1016/j.freeradbiomed.2015.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res 103: 1232–1240, 2008. doi: 10.1161/01.RES.0000338597.71702.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Peng K, Yang L, Wang J, Ye F, Dan G, Zhao Y, Cai Y, Cui Z, Ao L, Liu J, Zou Z, Sai Y, Cao J. The interaction of mitochondrial biogenesis and fission/fusion mediated by PGC-1alpha regulates rotenone-induced dopaminergic neurotoxicity. Mol Neurobiol 54: 3783–3797, 2017. doi: 10.1007/s12035-016-9944-9. [DOI] [PubMed] [Google Scholar]
- 256. Peng K, Tao Y, Zhang J, Wang J, Ye F, Dan G, Zhao Y, Cai Y, Zhao J, Wu Q, Zou Z, Cao J, Sai Y. Resveratrol regulates mitochondrial biogenesis and fission/fusion to attenuate rotenone-induced neurotoxicity. Oxid Med Cell Longev 2016: 6705621, 2016. doi: 10.1155/2016/6705621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Peng K, Xiao J, Yang L, Ye F, Cao J, Sai Y. Mutual antagonism of PINK1/Parkin and PGC-1alpha contributes to maintenance of mitochondrial homeostasis in rotenone-induced neurotoxicity. Neurotox Res 35: 331–343, 2019. doi: 10.1007/s12640-018-9957-4. [DOI] [PubMed] [Google Scholar]
- 258. Bahat A, MacVicar T, Langer T. Metabolism and innate immunity meet at the mitochondria. Front Cell Dev Biol 9: 720490, 2021. doi: 10.3389/fcell.2021.720490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Vazquez C, Horner SM. MAVS coordination of antiviral innate immunity. J Virol 89: 6974–6977, 2015. doi: 10.1128/JVI.01918-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol 7: 131–137, 2006. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 261. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21: 677–687, 2015. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Dyall SD, Brown MT, Johnson PJ. Ancient invasions: from endosymbionts to organelles. Science 304: 253–257, 2004. doi: 10.1126/science.1094884. [DOI] [PubMed] [Google Scholar]
- 263. Martin W, Müller M. The hydrogen hypothesis for the first eukaryote. Nature 392: 37–41, 1998. doi: 10.1038/32096. [DOI] [PubMed] [Google Scholar]
- 264. Moreira D, Lopez-Garcia P. Symbiosis between methanogenic archaea and delta-proteobacteria as the origin of eukaryotes: the syntrophic hypothesis. J Mol Evol 47: 517–530, 1998. doi: 10.1007/pl00006408. [DOI] [PubMed] [Google Scholar]
- 265. Andersson SG, Karlberg O, Canbäck B, Kurland CG. On the origin of mitochondria: a genomics perspective. Philos Trans R Soc Lond B Biol Sci 358: 165–177, 2003. doi: 10.1098/rstb.2002.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Shadel GS, Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem 66: 409–435, 1997. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- 267. Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature 290: 457–465, 1981. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 268. West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol 11: 389–402, 2011. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Nakahira K, Hisata S, Choi AM. The roles of mitochondrial damage-associated molecular patterns in diseases. Antioxid Redox Signal 23: 1329–1350, 2015. doi: 10.1089/ars.2015.6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2011. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov 5: 471–484, 2006. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 272. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol 5: 987–995, 2004. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 273. Schnare M, Holt AC, Takeda K, Akira S, Medzhitov R. Recognition of CpG DNA is mediated by signaling pathways dependent on the adaptor protein MyD88. Curr Biol 10: 1139–1142, 2000. doi: 10.1016/s0960-9822(00)00700-4. [DOI] [PubMed] [Google Scholar]
- 274. Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-alpha induction. J Exp Med 201: 915–923, 2005. doi: 10.1084/jem.20042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S, Takeuchi O, Akira S. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 5: 1061–1068, 2004. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 276. Häcker H, Vabulas RM, Takeuchi O, Hoshino K, Akira S, Wagner H. Immune cell activation by bacterial CpG-DNA through myeloid differentiation marker 88 and tumor necrosis factor receptor-associated factor (TRAF)6. J Exp Med 192: 595–600, 2000. doi: 10.1084/jem.192.4.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Hartmann G, Weeratna RD, Ballas ZK, Payette P, Blackwell S, Suparto I, Rasmussen WL, Waldschmidt M, Sajuthi D, Purcell RH, Davis HL, Krieg AM. Delineation of a CpG phosphorothioate oligodeoxynucleotide for activating primate immune responses in vitro and in vivo. J Immunol 164: 1617–1624, 2000. doi: 10.4049/jimmunol.164.3.1617. [DOI] [PubMed] [Google Scholar]
- 278. Bernasconi NL, Onai N, Lanzavecchia A. A role for Toll-like receptors in acquired immunity: up-regulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood 101: 4500–4504, 2003. doi: 10.1182/blood-2002-11-3569. [DOI] [PubMed] [Google Scholar]
- 279. Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol 5: 1219–1226, 2004. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 280. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: 401–414, 2012. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Jabir MS, Hopkins L, Ritchie ND, Ullah I, Bayes HK, Li D, Tourlomousis P, Lupton A, Puleston D, Simon AK, Bryant C, Evans TJ. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy 11: 166–182, 2015. doi: 10.4161/15548627.2014.981915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol 17: 363–375, 2017. doi: 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, Louis C, Low RR, Moecking J, De Nardo D, Balka KR, Calleja DJ, Moghaddas F, Ni E, McLean CA, Samson AL, Tyebji S, Tonkin CJ, Bye CR, Turner BJ, Pepin G, Gantier MP, Rogers KL, McArthur K, Crouch PJ, Masters SL. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell 183: 636–649.e18, 2020. doi: 10.1016/j.cell.2020.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H, Fujii R, Ishidate F, Tanaka T, Tanaka Y, Hirokawa N, Nangaku M, Inagi R. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep 29: 1261–1273.e6, 2019. doi: 10.1016/j.celrep.2019.09.050. [DOI] [PubMed] [Google Scholar]
- 285. Bai J, Cervantes C, Liu J, He S, Zhou H, Zhang B, Cai H, Yin D, Hu D, Li Z, Chen H, Gao X, Wang F, O'Connor JC, Xu Y, Liu M, Dong LQ, Liu F. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc Natl Acad Sci USA 114: 12196–12201, 2017. doi: 10.1073/pnas.1708744114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Banoth B, Cassel SL. Mitochondria in innate immune signaling. Transl Res 202: 52–68, 2018. doi: 10.1016/j.trsl.2018.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Lee HY, Lee M, Bae YS. Formyl peptide receptors in cellular differentiation and inflammatory diseases. J Cell Biochem 118: 1300–1307, 2017. doi: 10.1002/jcb.25877. [DOI] [PubMed] [Google Scholar]
- 288. Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev 17: 501–519, 2006. doi: 10.1016/j.cytogfr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 289. Panaro MA, Acquafredda A, Sisto M, Lisi S, Maffione AB, Mitolo V. Biological role of the N-formyl peptide receptors. Immunopharmacol Immunotoxicol 28: 103–127, 2006. doi: 10.1080/08923970600625975. [DOI] [PubMed] [Google Scholar]
- 290. Crouser ED, Shao G, Julian MW, Macre JE, Shadel GS, Tridandapani S, Huang Q, Wewers MD. Monocyte activation by necrotic cells is promoted by mitochondrial proteins and formyl peptide receptors. Crit Care Med 37: 2000–2009, 2009. doi: 10.1097/CCM.0b013e3181a001ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Rabiet MJ, Huet E, Boulay F. Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur J Immunol 35: 2486–2495, 2005. doi: 10.1002/eji.200526338. [DOI] [PubMed] [Google Scholar]
- 292. Carp H. Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J Exp Med 155: 264–275, 1982. doi: 10.1084/jem.155.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Weiß E, Kretschmer D. Formyl-peptide receptors in infection, inflammation, and cancer. Trends Immunol 39: 815–829, 2018. doi: 10.1016/j.it.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 294. Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol 23: 541–548, 2002. doi: 10.1016/s1471-4906(02)02316-5. [DOI] [PubMed] [Google Scholar]
- 295. Lohmann K. Über die Pyrophosphatfraktion im Muskel. Naturwissenschaften 17: 624–625, 1929. [Google Scholar]
- 296. Fiske CH, Subbarow Y. Phosphorus compounds of muscle and liver. Science 70: 381–382, 1929. doi: 10.1126/science.70.1816.381.b. [DOI] [PubMed] [Google Scholar]
- 297. Burnstock G, Fredholm BB, North RA, Verkhratsky A. The birth and postnatal development of purinergic signalling. Acta Physiol (Oxf) 199: 93–147, 2010. doi: 10.1111/j.1748-1716.2010.02114.x. [DOI] [PubMed] [Google Scholar]
- 298. Burnstock G, Knight GE. Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol 240: 31–304, 2004. doi: 10.1016/S0074-7696(04)40002-3. [DOI] [PubMed] [Google Scholar]
- 299. Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5'-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther 112: 358–404, 2006. doi: 10.1016/j.pharmthera.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 300. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity 47: 15–31, 2017. doi: 10.1016/j.immuni.2017.06.020. [DOI] [PubMed] [Google Scholar]
- 301. von Albertini M, Palmetshofer A, Kaczmarek E, Koziak K, Stroka D, Grey ST, Stuhlmeier KM, Robson SC. Extracellular ATP and ADP activate transcription factor NF-kappa B and induce endothelial cell apoptosis. Biochem Biophys Res Commun 248: 822–829, 1998. doi: 10.1006/bbrc.1998.9055. [DOI] [PubMed] [Google Scholar]
- 302. Goepfert C, Imai M, Brouard S, Csizmadia E, Kaczmarek E, Robson SC. CD39 modulates endothelial cell activation and apoptosis. Mol Med 6: 591–603, 2000. [PMC free article] [PubMed] [Google Scholar]
- 303. Akbar GK, Mills DC, Kunapuli SP. Characterization of extracellular nucleotide-induced Mac-1 (alphaM beta2 integrin) surface expression on peripheral blood leukocytes. Biochem Biophys Res Commun 233: 71–75, 1997. doi: 10.1006/bbrc.1997.6396. [DOI] [PubMed] [Google Scholar]
- 304. Tanaka N, Kawasaki K, Nejime N, Kubota Y, Nakamura K, Kunitomo M, Takahashi K, Hashimoto M, Shinozuka K. P2Y receptor-mediated Ca2+ signaling increases human vascular endothelial cell permeability. J Pharmacol Sci 95: 174–180, 2004. doi: 10.1254/jphs.fpj03036x. [DOI] [PubMed] [Google Scholar]
- 305. Zhang Y, Palmblad J, Fredholm BB. Biphasic effect of ATP on neutrophil functions mediated by P2U and adenosine A2A receptors. Biochem Pharmacol 51: 957–965, 1996. doi: 10.1016/0006-2952(95)02403-4. [DOI] [PubMed] [Google Scholar]
- 306. Meshki J, Tuluc F, Bredetean O, Ding Z, Kunapuli SP. Molecular mechanism of nucleotide-induced primary granule release in human neutrophils: role for the P2Y2 receptor. Am J Physiol Cell Physiol 286: C264–C271, 2004. doi: 10.1152/ajpcell.00287.2003. [DOI] [PubMed] [Google Scholar]
- 307. Gasmi L, McLennan AG, Edwards SW. The diadenosine polyphosphates Ap3A and Ap4A and adenosine triphosphate interact with granulocyte-macrophage colony-stimulating factor to delay neutrophil apoptosis: implications for neutrophil: platelet interactions during inflammation. Blood 87: 3442–3449, 1996. [PubMed] [Google Scholar]
- 308. Merz J, Albrecht P, von Garlen S, Ahmed I, Dimanski D, Wolf D, Hilgendorf I, Härdtner C, Grotius K, Willecke F, Heidt T, Bugger H, Hoppe N, Kintscher U, von Zur Mühlen C, Idzko M, Bode C, Zirlik A, Stachon P. Purinergic receptor Y2 (P2Y2)- dependent VCAM-1 expression promotes immune cell infiltration in metabolic syndrome. Basic Res Cardiol 113: 45, 2018. doi: 10.1007/s00395-018-0702-1. [DOI] [PubMed] [Google Scholar]
- 309. Gu B, Bendall LJ, Wiley JS. Adenosine triphosphate-induced shedding of CD23 and L-selectin (CD62L) from lymphocytes is mediated by the same receptor but different metalloproteases. Blood 92: 946–951, 1998. [PubMed] [Google Scholar]
- 310. Liu Y, Xiao Y, Li Z. P2X7 receptor positively regulates MyD88-dependent NF-kappaB activation. Cytokine 55: 229–236, 2011. doi: 10.1016/j.cyto.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 311. Guerra AN, Fisette PL, Pfeiffer ZA, Quinchia-Rios BH, Prabhu U, Aga M, Denlinger LC, Guadarrama AG, Abozeid S, Sommer JA, Proctor RA, Bertics PJ. Purinergic receptor regulation of LPS-induced signaling and pathophysiology. J Endotoxin Res 9: 256–263, 2003. doi: 10.1179/096805103225001468. [DOI] [PubMed] [Google Scholar]
- 312. Bidula S, Dhuna K, Helliwell R, Stokes L. Positive allosteric modulation of P2X7 promotes apoptotic cell death over lytic cell death responses in macrophages. Cell Death Dis 10: 882, 2019. doi: 10.1038/s41419-019-2110-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Schnurr M, Toy T, Stoitzner P, Cameron P, Shin A, Beecroft T, Davis ID, Cebon J, Maraskovsky E. ATP gradients inhibit the migratory capacity of specific human dendritic cell types: implications for P2Y11 receptor signaling. Blood 102: 613–620, 2003. doi: 10.1182/blood-2002-12-3745. [DOI] [PubMed] [Google Scholar]
- 314. Wilkin F, Duhant X, Bruyns C, Suarez-Huerta N, Boeynaems JM, Robaye B. The P2Y11 receptor mediates the ATP-induced maturation of human monocyte-derived dendritic cells. J Immunol 166: 7172–7177, 2001. doi: 10.4049/jimmunol.166.12.7172. [DOI] [PubMed] [Google Scholar]
- 315. Przybyła T, Sakowicz-Burkiewicz M, Pawełczyk T. Purinergic signaling in B cells. Acta Biochim Pol 65: 1–7, 2018. doi: 10.18388/abp.2017_1588. [DOI] [PubMed] [Google Scholar]
- 316. Trabanelli S, Ocadliková D, Gulinelli S, Curti A, Salvestrini V, Vieira RP, Idzko M, Di Virgilio F, Ferrari D, Lemoli RM. Extracellular ATP exerts opposite effects on activated and regulatory CD4+ T cells via purinergic P2 receptor activation. J Immunol 189: 1303–1310, 2012. doi: 10.4049/jimmunol.1103800. [DOI] [PubMed] [Google Scholar]
- 317. Parisi MA, Clayton DA. Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science 252: 965–969, 1991. doi: 10.1126/science.2035027. [DOI] [PubMed] [Google Scholar]
- 318. Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 8: 487–496, 2007. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 319. Julian MW, Shao G, Bao S, Knoell DL, Papenfuss TL, VanGundy ZC, Crouser ED. Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. J Immunol 189: 433–443, 2012. doi: 10.4049/jimmunol.1101375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Julian MW, Shao G, Vangundy ZC, Papenfuss TL, Crouser ED. Mitochondrial transcription factor A, an endogenous danger signal, promotes TNFalpha release via RAGE- and TLR9-responsive plasmacytoid dendritic cells. PLoS One 8: e72354, 2013. doi: 10.1371/journal.pone.0072354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Schindler SM, Frank MG, Annis JL, Maier SF, Klegeris A. Pattern recognition receptors mediate pro-inflammatory effects of extracellular mitochondrial transcription factor A (TFAM). Mol Cell Neurosci 89: 71–79, 2018. doi: 10.1016/j.mcn.2018.04.005. [DOI] [PubMed] [Google Scholar]
- 322. Chaung WW, Wu R, Ji Y, Dong W, Wang P. Mitochondrial transcription factor A is a proinflammatory mediator in hemorrhagic shock. Int J Mol Med 30: 199–203, 2012. doi: 10.3892/ijmm.2012.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Pizzuto M, Pelegrin P. Cardiolipin in immune signaling and cell death. Trends Cell Biol 30: 892–903, 2020. doi: 10.1016/j.tcb.2020.09.004. [DOI] [PubMed] [Google Scholar]
- 324. Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Wang KZ, Zhu J, Klein-Seetharaman J, Balasubramanian K, Amoscato AA, Borisenko G, Huang Z, Gusdon AM, Cheikhi A, Steer EK, Wang R, Baty C, Watkins S, Bahar I, Bayir H, Kagan VE. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15: 1197–1205, 2013. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Antón Z, Landajuela A, Hervás JH, Montes LR, Hernández-Tiedra S, Velasco G, Goñi FM, Alonso A. Human Atg8-cardiolipin interactions in mitophagy: Specific properties of LC3B, GABARAPL2 and GABARAP. Autophagy 12: 2386–2403, 2016. doi: 10.1080/15548627.2016.1240856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Balasubramanian K, Maeda A, Lee JS, Mohammadyani D, Dar HH, Jiang JF, St Croix CM, Watkins S, Tyurin VA, Tyurina YY, Klöditz K, Polimova A, Kapralova VI, Xiong Z, Ray P, Klein-Seetharaman J, Mallampalli RK, Bayir H, Fadeel B, Kagan VE. Dichotomous roles for externalized cardiolipin in extracellular signaling: promotion of phagocytosis and attenuation of innate immunity. Sci Signal 8: ra95, 2015. doi: 10.1126/scisignal.aaa6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Pizzuto M, Lonez C, Baroja-Mazo A, Martínez-Banaclocha H, Tourlomousis P, Gangloff M, Pelegrin P, Ruysschaert JM, Gay NJ, Bryant CE. Saturation of acyl chains converts cardiolipin from an antagonist to an activator of Toll-like receptor-4. Cell Mol Life Sci 76: 3667–3678, 2019. doi: 10.1007/s00018-019-03113-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol 14: 546–558, 2014. doi: 10.1038/nri3713. [DOI] [PubMed] [Google Scholar]
- 329. Ray NB, Durairaj L, Chen BB, McVerry BJ, Ryan AJ, Donahoe M, Waltenbaugh AK, O'Donnell CP, Henderson FC, Etscheidt CA, McCoy DM, Agassandian M, Hayes-Rowan EC, Coon TA, Butler PL, Gakhar L, Mathur SN, Sieren JC, Tyurina YY, Kagan VE, McLennan G, Mallampalli RK. Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat Med 16: 1120–1127, 2010. doi: 10.1038/nm.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL, Sutterwala FS. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39: 311–323, 2013. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Wan M, Hua X, Su J, Thiagarajan D, Frostegård AG, Haeggström JZ, Frostegård J. Oxidized but not native cardiolipin has pro-inflammatory effects, which are inhibited by Annexin A5. Atherosclerosis 235: 592–598, 2014. doi: 10.1016/j.atherosclerosis.2014.05.913. [DOI] [PubMed] [Google Scholar]
- 332. Moosavi B, Berry EA, Zhu XL, Yang WC, Yang GF. The assembly of succinate dehydrogenase: a key enzyme in bioenergetics. Cell Mol Life Sci 76: 4023–4042, 2019. doi: 10.1007/s00018-019-03200-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, , et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496: 238–242, 2013. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido-Perrig N, Schwärzler C, Junt T, Voshol H, Meingassner JG, Mao X, Werner G, Rot A, Carballido JM. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol 9: 1261–1269, 2008. doi: 10.1038/ni.1657. [DOI] [PubMed] [Google Scholar]
- 335. Shaham O, Slate NG, Goldberger O, Xu Q, Ramanathan A, Souza AL, Clish CB, Sims KB, Mootha VK. A plasma signature of human mitochondrial disease revealed through metabolic profiling of spent media from cultured muscle cells. Proc Natl Acad Sci USA 107: 1571–1575, 2010. doi: 10.1073/pnas.0906039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Pollara J, Edwards RW, Lin L, Bendersky VA, Brennan TV. Circulating mitochondria in deceased organ donors are associated with immune activation and early allograft dysfunction. JCI Insight 3: e121622, 2018. doi: 10.1172/jci.insight.121622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Liu TF, Vachharajani V, Millet P, Bharadwaj MS, Molina AJ, McCall CE. Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J Biol Chem 290: 396–408, 2015. doi: 10.1074/jbc.M114.566349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, McCall CE. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J Biol Chem 286: 9856–9864, 2011. doi: 10.1074/jbc.M110.196790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23: 2369–2380, 2004. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Vachharajani VT, Liu T, Brown CM, Wang X, Buechler NL, Wells JD, Yoza BK, McCall CE. SIRT1 inhibition during the hypoinflammatory phenotype of sepsis enhances immunity and improves outcome. J Leukoc Biol 96: 785–796, 2014. doi: 10.1189/jlb.3MA0114-034RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Ghosh HS, Spencer JV, Ng B, McBurney MW, Robbins PD. Sirt1 interacts with transducin-like enhancer of split-1 to inhibit nuclear factor kappaB-mediated transcription. Biochem J 408: 105–111, 2007. doi: 10.1042/BJ20070817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Meng Y, Ma QY, Kou XP, Xu J. Effect of resveratrol on activation of nuclear factor kappa-B and inflammatory factors in rat model of acute pancreatitis. World J Gastroenterol 11: 525–528, 2005. doi: 10.3748/wjg.v11.i4.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Zeng HT, Fu YC, Yu W, Lin JM, Zhou L, Liu L, Wang W. SIRT1 prevents atherosclerosis via liverXreceptor and NFkappaB signaling in a U937 cell model. Mol Med Rep 8: 23–28, 2013. doi: 10.3892/mmr.2013.1460. [DOI] [PubMed] [Google Scholar]
- 344. Toniolo A, Warden EA, Nassi A, Cignarella A, Bolego C. Regulation of SIRT1 in vascular smooth muscle cells from streptozotocin-diabetic rats. PLoS One 8: e65666, 2013. doi: 10.1371/journal.pone.0065666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 177: 861–870, 2008. doi: 10.1164/rccm.200708-1269OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Yang SR, Chida AS, Bauter MR, Shafiq N, Seweryniak K, Maggirwar SB, Kilty I, Rahman I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol 291: L46–L57, 2006. doi: 10.1152/ajplung.00241.2005. [DOI] [PubMed] [Google Scholar]
- 347. Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305: 1010–1013, 2004. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- 348. Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J 26: 3169–3179, 2007. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13: 325–332, 2012. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell 165: 792–800, 2016. doi: 10.1016/j.cell.2016.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19: 477–489, 2019. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann NY Acad Sci 1319: 82–95, 2014. doi: 10.1111/nyas.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458: 514–518, 2009. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Schroder K, Tschopp J. The inflammasomes. Cell 140: 821–832, 2010. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 355. Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol 28: 137–161, 2012. doi: 10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- 356. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225, 2011. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 357. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 368: 651–662, 2013. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 358. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456: 264–268, 2008. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 359. Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc Natl Acad Sci USA 110: 17963–17968, 2013. doi: 10.1073/pnas.1312571110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Kim SH, Shin HJ, Yoon CM, Lee SW, Sharma L, Dela Cruz CS, Kang MJ. PINK1 inhibits multimeric aggregation and signaling of MAVS and MAVS-dependent lung pathology. Am J Respir Cell Mol Biol 64: 592–603, 2021. doi: 10.1165/rcmb.2020-0490OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, Yu JW, Alnemri ES. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol 191: 4358–4366, 2013. doi: 10.4049/jimmunol.1301170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Giardina TM, Steer JH, Lo SZ, Joyce DA. Uncoupling protein-2 accumulates rapidly in the inner mitochondrial membrane during mitochondrial reactive oxygen stress in macrophages. Biochim Biophys Acta 1777: 118–129, 2008. doi: 10.1016/j.bbabio.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 363. Lu M, Sun XL, Qiao C, Liu Y, Ding JH, Hu G. Uncoupling protein 2 deficiency aggravates astrocytic endoplasmic reticulum stress and nod-like receptor protein 3 inflammasome activation. Neurobiol Aging 35: 421–430, 2014. doi: 10.1016/j.neurobiolaging.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 364. Chen Y, Shi Y, Wu J, Qi N. MAVS: A two-sided CARD mediating antiviral innate immune signaling and regulating immune homeostasis. Front Microbiol 12: 744348, 2021. doi: 10.3389/fmicb.2021.744348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Kell AM, Gale M Jr.. RIG-I in RNA virus recognition. Virology 479-480: 110–121, 2015. doi: 10.1016/j.virol.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122: 669–682, 2005. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 367. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437: 1167–1172, 2005. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 368. Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 19: 727–740, 2005. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 369. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6: 981–988, 2005. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 370. Horner SM, Liu HM, Park HS, Briley J, Gale M Jr.. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci USA 108: 14590–14595, 2011. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Bender S, Reuter A, Eberle F, Einhorn E, Binder M, Bartenschlager R. Activation of type I and III interferon response by mitochondrial and peroxisomal MAVS and inhibition by hepatitis C virus. PLoS Pathog 11: e1005264, 2015. doi: 10.1371/journal.ppat.1005264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Ren Z, Ding T, Zuo Z, Xu Z, Deng J, Wei Z. Regulation of MAVS expression and signaling function in the antiviral innate immune response. Front Immunol 11: 1030, 2020. doi: 10.3389/fimmu.2020.01030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Yoneyama M, Jogi M, Onomoto K. Regulation of antiviral innate immune signaling by stress-induced RNA granules. J Biochem 159: 279–286, 2016. doi: 10.1093/jb/mvv122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Oshiumi H, Miyashita M, Inoue N, Okabe M, Matsumoto M, Seya T. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 8: 496–509, 2010. doi: 10.1016/j.chom.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 375. Thoresen D, Wang W, Galls D, Guo R, Xu L, Pyle AM. The molecular mechanism of RIG-I activation and signaling. Immunol Rev 304: 154–168, 2021. doi: 10.1111/imr.13022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci USA 104: 7500–7505, 2007. doi: 10.1073/pnas.0611551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446: 916–920, 2007. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 378. Gack MU, Kirchhofer A, Shin YC, Inn KS, Liang C, Cui S, Myong S, Ha T, Hopfner KP, Jung JU. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc Natl Acad Sci USA 105: 16743–16748, 2008. doi: 10.1073/pnas.0804947105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Castanier C, Zemirli N, Portier A, Garcin D, Bidère N, Vazquez A, Arnoult D. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol 10: 44, 2012. doi: 10.1186/1741-7007-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Peisley A, Wu B, Xu H, Chen ZJ, Hur S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 509: 110–114, 2014. doi: 10.1038/nature13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146: 448–461, 2011. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Liu S, Chen J, Cai X, Wu J, Chen X, Wu YT, Sun L, Chen ZJ. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife 2: e00785, 2013. doi: 10.7554/eLife.00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Fang R, Jiang Q, Zhou X, Wang C, Guan Y, Tao J, Xi J, Feng JM, Jiang Z. MAVS activates TBK1 and IKKepsilon through TRAFs in NEMO dependent and independent manner. PLoS Pathog 13: e1006720, 2017. doi: 10.1371/journal.ppat.1006720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Loo YM, Gale M Jr.. Immune signaling by RIG-I-like receptors. Immunity 34: 680–692, 2011. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Hou J, Han L, Zhao Z, Liu H, Zhang L, Ma C, Yi F, Liu B, Zheng Y, Gao C. USP18 positively regulates innate antiviral immunity by promoting K63-linked polyubiquitination of MAVS. Nat Commun 12: 2970, 2021. doi: 10.1038/s41467-021-23219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Rai P, Fessler MB. The MAVS and MAV-nots: PINK1 clears prion-like MAVS aggregates to extinguish mitochondrial inflammatory signaling. Am J Respir Cell Mol Biol 64: 528–530, 2021. doi: 10.1165/rcmb.2021-0055ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Hanada Y, Ishihara N, Wang L, Otera H, Ishihara T, Koshiba T, Mihara K, Ogawa Y, Nomura M. MAVS is energized by Mff which senses mitochondrial metabolism via AMPK for acute antiviral immunity. Nat Commun 11: 5711, 2020. doi: 10.1038/s41467-020-19287-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Chen S, Liu S, Wang J, Wu Q, Wang A, Guan H, Zhang Q, Zhang D, Wang X, Song H, Qin J, Zou J, Jiang Z, Ouyang S, Feng XH, Liang T, Xu P. TBK1-mediated DRP1 targeting confers nucleic acid sensing to reprogram mitochondrial dynamics and physiology. Mol Cell 80: 810–827.e7, 2020. doi: 10.1016/j.molcel.2020.10.018. [DOI] [PubMed] [Google Scholar]
- 389. Koshiba T, Yasukawa K, Yanagi Y, Kawabata S. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci Signal 4: ra7, 2011. doi: 10.1126/scisignal.2001147. [DOI] [PubMed] [Google Scholar]
- 390. Yasukawa K, Oshiumi H, Takeda M, Ishihara N, Yanagi Y, Seya T, Kawabata S, Koshiba T. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci Signal 2: ra47, 2009. doi: 10.1126/scisignal.2000287. [DOI] [PubMed] [Google Scholar]
- 391. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, , et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25: 486–541, 2018. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G. Cell death modalities: classification and pathophysiological implications. Cell Death Differ 14: 1237–1243, 2007. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- 393. Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 146: 3–15, 1995. [PMC free article] [PubMed] [Google Scholar]
- 394. Tummers B, Green DR. The evolution of regulated cell death pathways in animals and their evasion by pathogens. Physiol Rev 102: 411–454, 2022. doi: 10.1152/physrev.00002.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol 60: 619–642, 1998. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- 396. Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science 305: 626–629, 2004. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 397. Parsons MJ, Green DR. Mitochondria in cell death. Essays Biochem 47: 99–114, 2010. doi: 10.1042/bse0470099. [DOI] [PubMed] [Google Scholar]
- 398. Morciano G, Giorgi C, Balestra D, Marchi S, Perrone D, Pinotti M, Pinton P. Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Mol Biol Cell 27: 20–34, 2016. doi: 10.1091/mbc.E15-01-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ 25: 65–80, 2018. doi: 10.1038/cdd.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9: 47–59, 2008. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 401. Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288: 1053–1058, 2000. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 402. Boise LH, Gottschalk AR, Quintáns J, Thompson CB. Bcl-2 and Bcl-2-related proteins in apoptosis regulation. Curr Top Microbiol Immunol 200: 107–121, 1995. doi: 10.1007/978-3-642-79437-7_8. [DOI] [PubMed] [Google Scholar]
- 403. Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol 2: 63–67, 2001. doi: 10.1038/35048069. [DOI] [PubMed] [Google Scholar]
- 404. Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J 345: 271–278, 2000. doi: 10.1042/bj3450271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Huang DC, Strasser A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell 103: 839–842, 2000. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- 406. Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1: 223–232, 2005. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 407. van Loo G, Saelens X, van Gurp M, MacFarlane M, Martin SJ, Vandenabeele P. The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ 9: 1031–1042, 2002. doi: 10.1038/sj.cdd.4401088. [DOI] [PubMed] [Google Scholar]
- 408. van Loo G, van Gurp M, Depuydt B, Srinivasula SM, Rodriguez I, Alnemri ES, Gevaert K, Vandekerckhove J, Declercq W, Vandenabeele P. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ 9: 20–26, 2002. doi: 10.1038/sj.cdd.4400970. [DOI] [PubMed] [Google Scholar]
- 409. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102: 33–42, 2000. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 410. Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, Vandenabeele P. Toxic proteins released from mitochondria in cell death. Oncogene 23: 2861–2874, 2004. doi: 10.1038/sj.onc.1207523. [DOI] [PubMed] [Google Scholar]
- 411. Walsh CM, Luhrs KA, Arechiga AF. The “fuzzy logic” of the death-inducing signaling complex in lymphocytes. J Clin Immunol 23: 333–353, 2003. doi: 10.1023/a:1025313415487. [DOI] [PubMed] [Google Scholar]
- 412. Lysyk L, Brassard R, Arutyunova E, Siebert V, Jiang Z, Takyi E, Morrison M, Young HS, Lemberg MK, O’Donoghue AJ, Lemieux MJ. Insights into the catalytic properties of the mitochondrial rhomboid protease PARL. J Biol Chem 296: 100383, 2021. doi: 10.1016/j.jbc.2021.100383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Lysyk L, Brassard R, Touret N, Lemieux MJ. PARL protease: a glimpse at intramembrane proteolysis in the inner mitochondrial membrane. J Mol Biol 432: 5052–5062, 2020. doi: 10.1016/j.jmb.2020.04.006. [DOI] [PubMed] [Google Scholar]
- 414. Saita S, Nolte H, Fiedler KU, Kashkar H, Venne AS, Zahedi RP, Krüger M, Langer T. PARL mediates Smac proteolytic maturation in mitochondria to promote apoptosis. Nat Cell Biol 19: 318–328, 2017. doi: 10.1038/ncb3488. [DOI] [PubMed] [Google Scholar]
- 415. Ishihara N, Mihara K. PARL paves the way to apoptosis. Nat Cell Biol 19: 263–265, 2017. doi: 10.1038/ncb3504. [DOI] [PubMed] [Google Scholar]
- 416. Otera H, Miyata N, Kuge O, Mihara K. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J Cell Biol 212: 531–544, 2016. doi: 10.1083/jcb.201508099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Oettinghaus B, D’Alonzo D, Barbieri E, Restelli LM, Savoia C, Licci M, Tolnay M, Frank S, Scorrano L. DRP1-dependent apoptotic mitochondrial fission occurs independently of BAX, BAK and APAF1 to amplify cell death by BID and oxidative stress. Biochim Biophys Acta 1857: 1267–1276, 2016. doi: 10.1016/j.bbabio.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 418. Xian H, Liou YC. Loss of MIEF1/MiD51 confers susceptibility to BAX-mediated cell death and PINK1-PRKN-dependent mitophagy. Autophagy 15: 2107–2125, 2019. doi: 10.1080/15548627.2019.1596494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Szczesniak LM, Bonzerato CG, Wojcikiewicz RJ. Identification of the Bok interactome using proximity labeling. Front Cell Dev Biol 9: 689951, 2021. doi: 10.3389/fcell.2021.689951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Milani M, Beckett AJ, Al-Zebeeby A, Luo X, Prior IA, Cohen GM, Varadarajan S. DRP-1 functions independently of mitochondrial structural perturbations to facilitate BH3 mimetic-mediated apoptosis. Cell Death Discov 5: 117, 2019. doi: 10.1038/s41420-019-0199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Anderson CJ, Kahl A, Fruitman H, Qian L, Zhou P, Manfredi G, Iadecola C. Prohibitin levels regulate OMA1 activity and turnover in neurons. Cell Death Differ 27: 1896–1906, 2020. doi: 10.1038/s41418-019-0469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126: 177–189, 2006. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 423. Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D’Adamio L, Derks C, Dejaegere T, Pellegrini L, D’Hooge R, Scorrano L, De Strooper B. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 126: 163–175, 2006. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 424. Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell 15: 5001–5011, 2004. doi: 10.1091/mbc.e04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 14: 193–204, 2008. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Wanderoy S, Hees JT, Klesse R, Edlich F, Harbauer AB. Kill one or kill the many: interplay between mitophagy and apoptosis. Biol Chem 402: 73–88, 2020. doi: 10.1515/hsz-2020-0231. [DOI] [PubMed] [Google Scholar]
- 427. Petit A, Kawarai T, Paitel E, Sanjo N, Maj M, Scheid M, Chen F, Gu Y, Hasegawa H, Salehi-Rad S, Wang L, Rogaeva E, Fraser P, Robinson B, St George-Hyslop P, Tandon A. Wild-type PINK1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease-related mutations. J Biol Chem 280: 34025–34032, 2005. doi: 10.1074/jbc.M505143200. [DOI] [PubMed] [Google Scholar]
- 428. Arena G, Gelmetti V, Torosantucci L, Vignone D, Lamorte G, De Rosa P, Cilia E, Jonas EA, Valente EM. PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ 20: 920–930, 2013. doi: 10.1038/cdd.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Wan H, Tang B, Liao X, Zeng Q, Zhang Z, Liao L. Analysis of neuronal phosphoproteome reveals PINK1 regulation of BAD function and cell death. Cell Death Differ 25: 904–917, 2018. doi: 10.1038/s41418-017-0027-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430. Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, Wood NW, Martins LM, Downward J. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat Cell Biol 9: 1243–1252, 2007. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- 431. Cakir Z, Funk K, Lauterwasser J, Todt F, Zerbes RM, Oelgeklaus A, Tanaka A, van der Laan M, Edlich F. Parkin promotes proteasomal degradation of misregulated BAX. J Cell Sci 130: 2903–2913, 2017. doi: 10.1242/jcs.200162. [DOI] [PubMed] [Google Scholar]
- 432. Carroll RG, Hollville E, Martin SJ. Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep 9: 1538–1553, 2014. doi: 10.1016/j.celrep.2014.10.046. [DOI] [PubMed] [Google Scholar]
- 433. Akabane S, Matsuzaki K, Yamashita S, Arai K, Okatsu K, Kanki T, Matsuda N, Oka T. Constitutive activation of PINK1 protein leads to proteasome-mediated and non-apoptotic cell death independently of mitochondrial autophagy. J Biol Chem 291: 16162–16174, 2016. doi: 10.1074/jbc.M116.714923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, Hashimoto S, Ryter SW, Choi AM. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 124: 3987–4003, 2014. doi: 10.1172/JCI74985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Choi ME, Price DR, Ryter SW, Choi AM. Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight 4: e128834, 2019. doi: 10.1172/jci.insight.128834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436. Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, Cai Q, Yang ZH, Huang D, Wu R, Han J. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun 8: 14329, 2017. doi: 10.1038/ncomms14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA, Herman B. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1366: 177–196, 1998. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- 438. Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev 27: 1640–1649, 2013. doi: 10.1101/gad.223321.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439. Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434: 658–662, 2005. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 440. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434: 652–658, 2005. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 441. Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427: 461–465, 2004. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442. Varanyuwatana P, Halestrap AP. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 12: 120–125, 2012. doi: 10.1016/j.mito.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443. Woodfield K, Rück A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J 336: 287–290, 1998. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Leung AW, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem 283: 26312–26323, 2008. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem 284: 33982–33988, 2009. doi: 10.1074/jbc.M109.020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446. Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA Jr, Jonas EA. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci USA 111: 10580–10585, 2014. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447. Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabó I, Lippe G, Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA 110: 5887–5892, 2013. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448. Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9: 550–555, 2007. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449. Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez-Caballero S, Osinska H, Cheng EH, Robbins J, Kinnally KW, Molkentin JD. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife 2: e00772, 2013. doi: 10.7554/eLife.00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450. Xu S, Wang P, Zhang H, Gong G, Gutierrez Cortes N, Zhu W, Yoon Y, Tian R, Wang W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nat Commun 7: 13189, 2016. doi: 10.1038/ncomms13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451. Antony AN, Paillard M, Moffat C, Juskeviciute E, Correnti J, Bolon B, Rubin E, Csordás G, Seifert EL, Hoek JB, Hajnóczky G. MICU1 regulation of mitochondrial Ca2+ uptake dictates survival and tissue regeneration. Nat Commun 7: 10955, 2016. doi: 10.1038/ncomms10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15: 135–147, 2014. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 453. Lin Y, Choksi S, Shen HM, Yang QF, Hur GM, Kim YS, Tran JH, Nedospasov SA, Liu ZG. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J Biol Chem 279: 10822–10828, 2004. doi: 10.1074/jbc.M313141200. [DOI] [PubMed] [Google Scholar]
- 454. Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P. RIP1, a kinase on the crossroads of a cell's decision to live or die. Cell Death Differ 14: 400–410, 2007. doi: 10.1038/sj.cdd.4402085. [DOI] [PubMed] [Google Scholar]
- 455. Stanger BZ, Leder P, Lee TH, Kim E, Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 81: 513–523, 1995. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- 456. Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4: 387–396, 1996. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 457. MacEwan DJ. TNF receptor subtype signalling: differences and cellular consequences. Cell Signal 14: 477–492, 2002. doi: 10.1016/s0898-6568(01)00262-5. [DOI] [PubMed] [Google Scholar]
- 458. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104: 487–501, 2001. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 459. Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol 11: 372–377, 2001. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 460. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325: 332–336, 2009. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 461. Vanlangenakker N, Vanden Berghe T, Bogaert P, Laukens B, Zobel K, Deshayes K, Vucic D, Fulda S, Vandenabeele P, Bertrand MJ. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ 18: 656–665, 2011. doi: 10.1038/cdd.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462. Temkin V, Huang Q, Liu H, Osada H, Pope RM. Inhibition of ADP/ATP exchange in receptor-interacting protein-mediated necrosis. Mol Cell Biol 26: 2215–2225, 2006. doi: 10.1128/MCB.26.6.2215-2225.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK-M. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137: 1112–1123, 2009. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464. He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137: 1100–1111, 2009. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 465. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RC, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39: 443–453, 2013. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 466. Remijsen Q, Goossens V, Grootjans S, Van den Haute C, Vanlangenakker N, Dondelinger Y, Roelandt R, Bruggeman I, Goncalves A, Bertrand MJ, Baekelandt V, Takahashi N, Berghe TV, Vandenabeele P. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis 5: e1004, 2014. doi: 10.1038/cddis.2013.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467. Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, Karvela M, Ichim G, Yatim N, Albert ML, Kidd G, Wakefield R, Frase S, Krautwald S, Linkermann A, Green DR. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep 5: 878–885, 2013. doi: 10.1016/j.celrep.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468. Davis CW, Hawkins BJ, Ramasamy S, Irrinki KM, Cameron BA, Islam K, Daswani VP, Doonan PJ, Manevich Y, Madesh M. Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free Radic Biol Med 48: 306–317, 2010. doi: 10.1016/j.freeradbiomed.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469. Chen MS, Wang SF, Hsu CY, Yin PH, Yeh TS, Lee HC, Tseng LM. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2alpha-ATF4 pathway. Oncotarget 8: 114588–114602, 2017. doi: 10.18632/oncotarget.23055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470. Lu B, Gong X, Wang ZQ, Ding Y, Wang C, Luo TF, Piao MH, Meng FK, Chi GF, Luo YN, Ge PF. Shikonin induces glioma cell necroptosis in vitro by ROS overproduction and promoting RIP1/RIP3 necrosome formation. Acta Pharmacol Sin 38: 1543–1553, 2017. doi: 10.1038/aps.2017.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471. Pajuelo D, Gonzalez-Juarbe N, Tak U, Sun J, Orihuela CJ, Niederweis M. NAD+ depletion triggers macrophage necroptosis, a cell death pathway exploited by Mycobacterium tuberculosis. Cell Rep 24: 429–440, 2018. doi: 10.1016/j.celrep.2018.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107, 2010. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473. Chen D, Tong J, Yang L, Wei L, Stolz DB, Yu J, Zhang J, Zhang L. PUMA amplifies necroptosis signaling by activating cytosolic DNA sensors. Proc Natl Acad Sci USA 115: 3930–3935, 2018. doi: 10.1073/pnas.1717190115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol 9: 113–114, 2001. doi: 10.1016/s0966-842x(00)01936-3. [DOI] [PubMed] [Google Scholar]
- 475. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA 113: 7858–7863, 2016. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476. Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, Wang L, Fu TM, Jacobson MP, Greka A, Lieberman J, Ruan J, Wu H. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 593: 607–611, 2021. doi: 10.1038/s41586-021-03478-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477. Evavold CL, Hafner-Bratkovič I, Devant P, D’Andrea JM, Ngwa EM, Boršić E, Doench JG, LaFleur MW, Sharpe AH, Thiagarajah JR, Kagan JC. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell 184: 4495–4511.e19, 2021. doi: 10.1016/j.cell.2021.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun 10: 1689, 2019. doi: 10.1038/s41467-019-09397-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479. Ren Z, He H, Zuo Z, Xu Z, Wei Z, Deng J. The role of different SIRT1-mediated signaling pathways in toxic injury. Cell Mol Biol Lett 24: 36, 2019. doi: 10.1186/s11658-019-0158-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303: 2011–2015, 2004. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 481. Wang YQ, Cao Q, Wang F, Huang LY, Sang TT, Liu F, Chen SY. SIRT1 protects against oxidative stress-induced endothelial progenitor cells apoptosis by inhibiting FOXO3a via FOXO3a ubiquitination and degradation. J Cell Physiol 230: 2098–2107, 2015. doi: 10.1002/jcp.24938. [DOI] [PubMed] [Google Scholar]
- 482. Giannakou ME, Partridge L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol 14: 408–412, 2004. doi: 10.1016/j.tcb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 483. Gu X, Han D, Chen W, Zhang L, Lin Q, Gao J, Fanning S, Han B. SIRT1-mediated FoxOs pathways protect against apoptosis by promoting autophagy in osteoblast-like MC3T3-E1 cells exposed to sodium fluoride. Oncotarget 7: 65218–65230, 2016. doi: 10.18632/oncotarget.11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484. Panday A, Inda ME, Bagam P, Sahoo MK, Osorio D, Batra S. Transcription factor NF-kappaB: an update on intervention strategies. Arch Immunol Ther Exp (Warsz) 64: 463–483, 2016. doi: 10.1007/s00005-016-0405-y. [DOI] [PubMed] [Google Scholar]
- 485. Alhazzazi TY, Kamarajan P, Verdin E, Kapila YL. SIRT3 and cancer: tumor promoter or suppressor? Biochim Biophys Acta 1816: 80–88, 2011. doi: 10.1016/j.bbcan.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486. Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2: 914–923, 2010. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487. Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol 28: 6384–6401, 2008. doi: 10.1128/MCB.00426-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488. Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, , et al. Heart Disease and Stroke Statistics—2021 Update: a Report from the American Heart Association. Circulation 143: e254–e743, 2021. doi: 10.1161/CIR.0000000000000950. [DOI] [PubMed] [Google Scholar]
- 489. Mensah GA, Wei GS, Sorlie PD, Fine LJ, Rosenberg Y, Kaufmann PG, Mussolino ME, Hsu LL, Addou E, Engelgau MM, Gordon D. Decline in cardiovascular mortality: possible causes and implications. Circ Res 120: 366–380, 2017. doi: 10.1161/CIRCRESAHA.116.309115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490. Donato AJ, Machin DR, Lesniewski LA. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ Res 123: 825–848, 2018. doi: 10.1161/CIRCRESAHA.118.312563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491. Pollard AK, Craig EL, Chakrabarti L. Mitochondrial Complex 1 activity measured by spectrophotometry is reduced across all brain regions in ageing and more specifically in neurodegeneration. PLoS One 11: e0157405, 2016. doi: 10.1371/journal.pone.0157405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492. Forte M, Stanzione R, Cotugno M, Bianchi F, Marchitti S, Rubattu S. Vascular ageing in hypertension: focus on mitochondria. Mech Ageing Dev 189: 111267, 2020. doi: 10.1016/j.mad.2020.111267. [DOI] [PubMed] [Google Scholar]
- 493. Csiszar A, Wang M, Lakatta EG, Ungvari Z. Inflammation and endothelial dysfunction during aging: role of NF-kappaB. J Appl Physiol (1985) 105: 1333–1341, 2008. doi: 10.1152/japplphysiol.90470.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494. Durrant JR, Seals DR, Connell ML, Russell MJ, Lawson BR, Folian BJ, Donato AJ, Lesniewski LA. Voluntary wheel running restores endothelial function in conduit arteries of old mice: direct evidence for reduced oxidative stress, increased superoxide dismutase activity and down-regulation of NADPH oxidase. J Physiol 587: 3271–3285, 2009. doi: 10.1113/jphysiol.2009.169771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495. Santhanam AV, d’Uscio LV, Smith LA, Katusic ZS. Uncoupling of eNOS causes superoxide anion production and impairs NO signaling in the cerebral microvessels of hph-1 mice. J Neurochem 122: 1211–1218, 2012. doi: 10.1111/j.1471-4159.2012.07872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496. Donato AJ, Walker AE, Magerko KA, Bramwell RC, Black AD, Henson GD, Lawson BR, Lesniewski LA, Seals DR. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell 12: 772–783, 2013. doi: 10.1111/acel.12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497. Oelze M, Kröller-Schön S, Steven S, Lubos E, Doppler C, Hausding M, Tobias S, Brochhausen C, Li H, Torzewski M, Wenzel P, Bachschmid M, Lackner KJ, Schulz E, Münzel T, Daiber A. Glutathione peroxidase-1 deficiency potentiates dysregulatory modifications of endothelial nitric oxide synthase and vascular dysfunction in aging. Hypertension 63: 390–396, 2014. doi: 10.1161/HYPERTENSIONAHA.113.01602. [DOI] [PubMed] [Google Scholar]
- 498. Ungvari Z, Bailey-Downs L, Gautam T, Sosnowska D, Wang M, Monticone RE, Telljohann R, Pinto JT, de Cabo R, Sonntag WE, Lakatta EG, Csiszar A. Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-kappaB activation in the nonhuman primate Macaca mulatta. J Gerontol A Biol Sci Med Sci 66: 866–875, 2011. doi: 10.1093/gerona/glr092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499. Francia P, Delli Gatti C, Bachschmid M, Martin-Padura I, Savoia C, Migliaccio E, Pelicci PG, Schiavoni M, Lüscher TF, Volpe M, Cosentino F. Deletion of p66shc gene protects against age-related endothelial dysfunction. Circulation 110: 2889–2895, 2004. doi: 10.1161/01.CIR.0000147731.24444.4D. [DOI] [PubMed] [Google Scholar]
- 500. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell 146: 682–695, 2011. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 501. Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, Jung S, Jung YK. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun 4: 2300, 2013. doi: 10.1038/ncomms3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502. Bharath LP, Mueller R, Li Y, Ruan T, Kunz D, Goodrich R, Mills T, Deeter L, Sargsyan A, Anandh Babu PV, Graham TE, Symons JD. Impairment of autophagy in endothelial cells prevents shear-stress-induced increases in nitric oxide bioavailability. Can J Physiol Pharmacol 92: 605–612, 2014. doi: 10.1139/cjpp-2014-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503. Jin JY, Wei XX, Zhi XL, Wang XH, Meng D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol Sin 42: 655–664, 2021. doi: 10.1038/s41401-020-00518-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504. Yang Y, Li T, Li Z, Liu N, Yan Y, Liu B. Role of mitophagy in cardiovascular disease. Aging Dis 11: 419–437, 2020. doi: 10.14336/AD.2019.0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505. Donato AJ, Black AD, Jablonski KL, Gano LB, Seals DR. Aging is associated with greater nuclear NF kappa B, reduced I kappa B alpha, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging Cell 7: 805–812, 2008. doi: 10.1111/j.1474-9726.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506. Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA 97: 9052–9057, 2000. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507. Bond M, Chase AJ, Baker AH, Newby AC. Inhibition of transcription factor NF-kappaB reduces matrix metalloproteinase-1, -3 and -9 production by vascular smooth muscle cells. Cardiovasc Res 50: 556–565, 2001. doi: 10.1016/s0008-6363(01)00220-6. [DOI] [PubMed] [Google Scholar]
- 508. Lone A, Harris RA, Singh O, Betts DH, Cumming RC. p66Shc activation promotes increased oxidative phosphorylation and renders CNS cells more vulnerable to amyloid beta toxicity. Sci Rep 8: 17081, 2018. doi: 10.1038/s41598-018-35114-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509. Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122: 221–233, 2005. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 510. Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 402: 309–313, 1999. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- 511. Shi Y, Savarese G, Perrone-Filardi P, Lüscher TF, Camici GG. Enhanced age-dependent cerebrovascular dysfunction is mediated by adaptor protein p66Shc. Int J Cardiol 175: 446–450, 2014. doi: 10.1016/j.ijcard.2014.06.025. [DOI] [PubMed] [Google Scholar]
- 512. Kumar S. P66Shc and vascular endothelial function. Biosci Rep 39: BSR20182134, 2019. doi: 10.1042/BSR20182134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513. Napoli C, Martin-Padura I, de Nigris F, Giorgio M, Mansueto G, Somma P, Condorelli M, Sica G, De Rosa G, Pelicci P. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc Natl Acad Sci USA 100: 2112–2116, 2003. doi: 10.1073/pnas.0336359100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514. Martin-Padura I, de Nigris F, Migliaccio E, Mansueto G, Minardi S, Rienzo M, Lerman LO, Stendardo M, Giorgio M, De Rosa G, Pelicci PG, Napoli C. p66Shc deletion confers vascular protection in advanced atherosclerosis in hypercholesterolemic apolipoprotein E knockout mice. Endothelium 15: 276–287, 2008. doi: 10.1080/10623320802487791. [DOI] [PubMed] [Google Scholar]
- 515. Kim CS, Jung SB, Naqvi A, Hoffman TA, DeRicco J, Yamamori T, Cole MP, Jeon BH, Irani K. p53 impairs endothelium-dependent vasomotor function through transcriptional upregulation of p66shc. Circ Res 103: 1441–1450, 2008. doi: 10.1161/CIRCRESAHA.108.181644. [DOI] [PubMed] [Google Scholar]
- 516. Spescha RD, Glanzmann M, Simic B, Witassek F, Keller S, Akhmedov A, Tanner FC, Lüscher TF, Camici GG. Adaptor protein p66Shc mediates hypertension-associated, cyclic stretch-dependent, endothelial damage. Hypertension 64: 347–353, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02129. [DOI] [PubMed] [Google Scholar]
- 517. Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155: 1624–1638, 2013. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518. Thompson AM, Wagner R, Rzucidlo EM. Age-related loss of SirT1 expression results in dysregulated human vascular smooth muscle cell function. Am J Physiol Heart Circ Physiol 307: H533–H541, 2014. doi: 10.1152/ajpheart.00871.2013. [DOI] [PubMed] [Google Scholar]
- 519. Tseng AH, Shieh SS, Wang DL. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic Biol Med 63: 222–234, 2013. doi: 10.1016/j.freeradbiomed.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 520. Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, Barst RJ, Benza RL, Liou TG, Turner M, Giles S, Feldkircher K, Miller DP, McGoon MD. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest 137: 376–387, 2010. doi: 10.1378/chest.09-1140. [DOI] [PubMed] [Google Scholar]
- 521. Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J 30: 104–109, 2007. doi: 10.1183/09031936.00092306. [DOI] [PubMed] [Google Scholar]
- 522. Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ 360: j5492, 2018. doi: 10.1136/bmj.j5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523. Marshall JD, Bazan I, Zhang Y, Fares WH, Lee PJ. Mitochondrial dysfunction and pulmonary hypertension: cause, effect, or both. Am J Physiol Lung Cell Mol Physiol 314: L782–L796, 2018. doi: 10.1152/ajplung.00331.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524. Konstam MA, Kiernan MS, Bernstein D, Bozkurt B, Jacob M, Kapur NK, Kociol RD, Lewis EF, Mehra MR, Pagani FD, Raval AN, Ward C; American Heart Association Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; Council on Cardiovascular Surgery and Anesthesia. Evaluation and management of right-sided heart failure: a Scientific Statement from the American Heart Association. Circulation 137: e578–e622, 2018. doi: 10.1161/CIR.0000000000000560. [DOI] [PubMed] [Google Scholar]
- 525. Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol 294: H570–H578, 2008. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 526. Ghouleh IA, Sahoo S, Meijles DN, Amaral JH, de Jesus DS, Sembrat J, Rojas M, Goncharov DA, Goncharova EA, Pagano PJ. Endothelial Nox1 oxidase assembly in human pulmonary arterial hypertension; driver of Gremlin1-mediated proliferation. Clin Sci (Lond) 131: 2019–2035, 2017. doi: 10.1042/CS20160812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527. Paulin R, Meloche J, Bonnet S. STAT3 signaling in pulmonary arterial hypertension. JAKSTAT 1: 223–233, 2012. doi: 10.4161/jkst.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528. Courboulin A, Tremblay VL, Barrier M, Meloche J, Jacob MH, Chapolard M, Bisserier M, Paulin R, Lambert C, Provencher S, Bonnet S. Krüppel-like factor 5 contributes to pulmonary artery smooth muscle proliferation and resistance to apoptosis in human pulmonary arterial hypertension. Respir Res 12: 128, 2011. doi: 10.1186/1465-9921-12-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529. Stenmark KR, Tuder RM, El Kasmi KC. Metabolic reprogramming and inflammation act in concert to control vascular remodeling in hypoxic pulmonary hypertension. J Appl Physiol (1985) 119: 1164–1172, 2015. doi: 10.1152/japplphysiol.00283.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530. Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A, Drazba J, Anand-Apte B, Erzurum SC. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 293: L548–L554, 2007. doi: 10.1152/ajplung.00428.2006. [DOI] [PubMed] [Google Scholar]
- 531. Afolayan AJ, Teng RJ, Eis A, Rana U, Broniowska KA, Corbett JA, Pritchard K, Konduri GG. Inducible HSP70 regulates superoxide dismutase-2 and mitochondrial oxidative stress in the endothelial cells from developing lungs. Am J Physiol Lung Cell Mol Physiol 306: L351–L360, 2014. doi: 10.1152/ajplung.00264.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532. Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, Krishnamachary B, Zhen L, Mao T, Richter A, Erzurum SC, Tuder RM. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol 176: 1130–1138, 2010. doi: 10.2353/ajpath.2010.090832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533. Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113: 2630–2641, 2006. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 534. Hong Z, Chen KH, DasGupta A, Potus F, Dunham-Snary K, Bonnet S, Tian L, Fu J, Breuils-Bonnet S, Provencher S, Wu D, Mewburn J, Ormiston ML, Archer SL. MicroRNA-138 and microRNA-25 Down-regulate mitochondrial calcium uniporter, causing the pulmonary arterial hypertension cancer phenotype. Am J Respir Crit Care Med 195: 515–529, 2017. doi: 10.1164/rccm.201604-0814OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535. Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol 182: 573–585, 2008. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536. Dromparis P, Paulin R, Sutendra G, Qi AC, Bonnet S, Michelakis ED. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ Res 113: 126–136, 2013. doi: 10.1161/CIRCRESAHA.112.300699. [DOI] [PubMed] [Google Scholar]
- 537. Teshima Y, Akao M, Jones SP, Marbán E. Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ Res 93: 192–200, 2003. doi: 10.1161/01.RES.0000085581.60197.4D. [DOI] [PubMed] [Google Scholar]
- 538. Wang B, Xiong S, Lin S, Xia W, Li Q, Zhao Z, Wei X, Lu Z, Wei X, Gao P, Liu D, Zhu Z. Enhanced mitochondrial transient receptor potential channel, canonical type 3-mediated calcium handling in the vasculature from hypertensive rats. J Am Heart Assoc 6: e005812, 2017. doi: 10.1161/JAHA.117.005812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539. Feng S, Li H, Tai Y, Huang J, Su Y, Abramowitz J, Zhu MX, Birnbaumer L, Wang Y. Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc Natl Acad Sci USA 110: 11011–11016, 2013. doi: 10.1073/pnas.1309531110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540. Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA 101: 13861–13866, 2004. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541. Zhang S, Patel HH, Murray F, Remillard CV, Schach C, Thistlethwaite PA, Insel PA, Yuan JX. Pulmonary artery smooth muscle cells from normal subjects and IPAH patients show divergent cAMP-mediated effects on TRPC expression and capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol 292: L1202–L1210, 2007. doi: 10.1152/ajplung.00214.2006. [DOI] [PubMed] [Google Scholar]
- 542. Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, McMurtry MS, Michalak M, Vance JE, Sessa WC, Michelakis ED. The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med 3: 88ra55, 2011. doi: 10.1126/scitranslmed.3002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543. Zurlo G, Piquereau J, Moulin M, Pires Da Silva J, Gressette M, Ranchoux B, Garnier A, Ventura-Clapier R, Fadel E, Humbert M, Lemaire C, Perros F, Veksler V. Sirtuin 1 regulates pulmonary artery smooth muscle cell proliferation: role in pulmonary arterial hypertension. J Hypertens 36: 1164–1177, 2018. doi: 10.1097/HJH.0000000000001676. [DOI] [PubMed] [Google Scholar]
- 544. Paulin R, Dromparis P, Sutendra G, Gurtu V, Zervopoulos S, Bowers L, Haromy A, Webster L, Provencher S, Bonnet S, Michelakis ED. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab 20: 827–839, 2014. doi: 10.1016/j.cmet.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 545. Voelkel NF, Cool C, Lee SD, Wright L, Geraci MW, Tuder RM. Primary pulmonary hypertension between inflammation and cancer. Chest 114: 225S–230S, 1998. doi: 10.1378/chest.114.3_supplement.225s. [DOI] [PubMed] [Google Scholar]
- 546. Gurtu V, Michelakis ED. A paradigm shift is needed in the field of pulmonary arterial hypertension for its entrance into the precision medicine era. Circ Res 119: 1276–1279, 2016. doi: 10.1161/CIRCRESAHA.116.309689. [DOI] [PubMed] [Google Scholar]
- 547. Pullamsetti SS, Savai R, Seeger W, Goncharova EA. Translational advances in the field of pulmonary hypertension. From cancer biology to new pulmonary arterial hypertension therapeutics. targeting cell growth and proliferation signaling hubs. Am J Respir Crit Care Med 195: 425–437, 2017. doi: 10.1164/rccm.201606-1226PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548. Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med 35: 71–86, 2014. doi: 10.1016/j.ccm.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 549. Vogelmeier CF, Criner GJ, Martinez FJ, Anzueto A, Barnes PJ, Bourbeau J, Celli BR, Chen R, Decramer M, Fabbri LM, Frith P, Halpin DM, López Varela MV, Nishimura M, Roche N, Rodriguez-Roisin R, Sin DD, Singh D, Stockley R, Vestbo J, Wedzicha JA, Agusti A. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report: GOLD Executive Summary. Eur Respir J 49: 1700214, 2017. doi: 10.1183/13993003.00214-2017. [DOI] [PubMed] [Google Scholar]
- 550. Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, Swigris JJ, Taniguchi H, Wells AU. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers 3: 17074, 2017. doi: 10.1038/nrdp.2017.74. [DOI] [PubMed] [Google Scholar]
- 551. Rangarajan S, Bernard K, Thannickal VJ. Mitochondrial dysfunction in pulmonary fibrosis. Ann Am Thorac Soc 14: S383–S388, 2017. doi: 10.1513/AnnalsATS.201705-370AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552. Roque W, Cuevas-Mora K, Romero F. Mitochondrial quality control in age-related pulmonary fibrosis. Int J Mol Sci 21, 2020. doi: 10.3390/ijms21020643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553. Mora AL, Bueno M, Rojas M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J Clin Invest 127: 405–414, 2017. doi: 10.1172/JCI87440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554. Bueno M, Calyeca J, Rojas M, Mora AL. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol 33: 101509, 2020. doi: 10.1016/j.redox.2020.101509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555. Zank DC, Bueno M, Mora AL, Rojas M. Idiopathic pulmonary fibrosis: aging, mitochondrial dysfunction, and cellular bioenergetics. Front Med (Lausanne) 5: 10, 2018. doi: 10.3389/fmed.2018.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556. Esteve-Codina A, Hofer TP, Burggraf D, Heiss-Neumann MS, Gesierich W, Boland A, , et al. Gender specific airway gene expression in COPD sub-phenotypes supports a role of mitochondria and of different types of leukocytes. Sci Rep 11: 12848, 2021. doi: 10.1038/s41598-021-91742-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557. Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, Nakahira K, Pilewski JM, Lee JS, Zhang Y, Ryter SW, Choi AM. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One 3: e3316, 2008. doi: 10.1371/journal.pone.0003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558. Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, Choi AM. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci USA 107: 18880–18885, 2010. doi: 10.1073/pnas.1005574107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559. Vij N, Chandramani-Shivalingappa P, Van Westphal C, Hole R, Bodas M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am J Physiol Cell Physiol 314: C73–C87, 2018. doi: 10.1152/ajpcell.00110.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560. Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M, Hara H, Minagawa S, Wakui H, Fujii S, Kojima J, Shimizu K, Numata T, Kawaishi M, Odaka M, Morikawa T, Harada T, Nishimura SL, Kaneko Y, Nakayama K, Kuwano K. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 11: 547–559, 2015. doi: 10.1080/15548627.2015.1017190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561. Monick MM, Powers LS, Walters K, Lovan N, Zhang M, Gerke A, Hansdottir S, Hunninghake GW. Identification of an autophagy defect in smokers' alveolar macrophages. J Immunol 185: 5425–5435, 2010. doi: 10.4049/jimmunol.1001603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562. Gifford JR, Trinity JD, Kwon OS, Layec G, Garten RS, Park SY, Nelson AD, Richardson RS. Altered skeletal muscle mitochondrial phenotype in COPD: disease vs. disuse. J Appl Physiol (1985) 124: 1045–1053, 2018. doi: 10.1152/japplphysiol.00788.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563. Cloonan SM, Glass K, Laucho-Contreras ME, Bhashyam AR, Cervo M, Pabón MA, Konrad C, Polverino F, Siempos II, Perez E, Mizumura K, Ghosh MC, Parameswaran H, Williams NC, Rooney KT, Chen ZH, Goldklang MP, Yuan GC, Moore SC, Demeo DL, Rouault TA, D’Armiento JM, Schon EA, Manfredi G, Quackenbush J, Mahmood A, Silverman EK, Owen CA, Choi AM. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med 22: 163–174, 2016. doi: 10.1038/nm.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564. van der Toorn M, Slebos DJ, de Bruin HG, Leuvenink HG, Bakker SJ, Gans RO, Koëter GH, van Oosterhout AJ, Kauffman HF. Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. Am J Physiol Lung Cell Mol Physiol 292: L1211–L1218, 2007. doi: 10.1152/ajplung.00291.2006. [DOI] [PubMed] [Google Scholar]
- 565. Pouwels SD, Zijlstra GJ, van der Toorn M, Hesse L, Gras R, Ten Hacken NH, Krysko DV, Vandenabeele P, de Vries M, van Oosterhout AJ, Heijink IH, Nawijn MC. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am J Physiol Lung Cell Mol Physiol 310: L377–L386, 2016. doi: 10.1152/ajplung.00174.2015. [DOI] [PubMed] [Google Scholar]
- 566. Maremanda KP, Sundar IK, Rahman I. Role of inner mitochondrial protein OPA1 in mitochondrial dysfunction by tobacco smoking and in the pathogenesis of COPD. Redox Biol 45: 102055, 2021. doi: 10.1016/j.redox.2021.102055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567. Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS, Duncan SR, Rojas M, Shiva S, Chu CT, Mora AL. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest 125: 521–538, 2015. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568. Patel AS, Song JW, Chu SG, Mizumura K, Osorio JC, Shi Y, El-Chemaly S, Lee CG, Rosas IO, Elias JA, Choi AM, Morse D. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS One 10: e0121246, 2015. doi: 10.1371/journal.pone.0121246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569. Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, Rosas IO, Morse D. Autophagy in idiopathic pulmonary fibrosis. PLoS One 7: e41394, 2012. doi: 10.1371/journal.pone.0041394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570. Rangarajan S, Bone NB, Zmijewska AA, Jiang S, Park DW, Bernard K, Locy ML, Ravi S, Deshane J, Mannon RB, Abraham E, Darley-Usmar V, Thannickal VJ, Zmijewski JW. Metformin reverses established lung fibrosis in a bleomycin model. Nat Med 24: 1121–1127, 2018. doi: 10.1038/s41591-018-0087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571. Bueno M, Brands J, Voltz L, Fiedler K, Mays B, St Croix C, Sembrat J, Mallampalli RK, Rojas M, Mora AL. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 17: e12720, 2018. doi: 10.1111/acel.12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572. Kobayashi K, Araya J, Minagawa S, Hara H, Saito N, Kadota T, Sato N, Yoshida M, Tsubouchi K, Kurita Y, Ito S, Fujita Y, Takasaka N, Utsumi H, Yanagisawa H, Hashimoto M, Wakui H, Kojima J, Shimizu K, Numata T, Kawaishi M, Kaneko Y, Asano H, Yamashita M, Odaka M, Morikawa T, Nakayama K, Kuwano K. Involvement of PARK2-mediated mitophagy in idiopathic pulmonary fibrosis pathogenesis. J Immunol 197: 504–516, 2016. doi: 10.4049/jimmunol.1600265. [DOI] [PubMed] [Google Scholar]
- 573. Ganzleben I, He GW, Günther C, Prigge ES, Richter K, Rieker RJ, Mougiakakos D, Neurath MF, Becker C. PGAM5 is a key driver of mitochondrial dysfunction in experimental lung fibrosis. Cell Mol Life Sci 76: 4783–4794, 2019. doi: 10.1007/s00018-019-03133-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574. Rehan M, Kurundkar D, Kurundkar AR, Logsdon NJ, Smith SR, Chanda D, Bernard K, Sanders YY, Deshane JS, Dsouza KG, Rangarajan S, Zmijewski JW, Thannickal VJ. Restoration of SIRT3 gene expression by airway delivery resolves age-associated persistent lung fibrosis in mice. Nat Aging 1: 205–217, 2021. doi: 10.1038/s43587-021-00027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575. Chung KP, Hsu CL, Fan LC, Huang Z, Bhatia D, Chen YJ, Hisata S, Cho SJ, Nakahira K, Imamura M, Choi ME, Yu CJ, Cloonan SM, Choi AM. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat Commun 10: 3390, 2019. doi: 10.1038/s41467-019-11327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576. Bueno M, Zank D, Buendia-Roldán I, Fiedler K, Mays BG, Alvarez D, Sembrat J, Kimball B, Bullock JK, Martin JL, Nouraie M, Kaufman BA, Rojas M, Pardo A, Selman M, Mora AL. PINK1 attenuates mtDNA release in alveolar epithelial cells and TLR9 mediated profibrotic responses. PLoS One 14: e0218003, 2019. doi: 10.1371/journal.pone.0218003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577. Tian R, Zhu Y, Yao J, Meng X, Wang J, Xie H, Wang R. NLRP3 participates in the regulation of EMT in bleomycin-induced pulmonary fibrosis. Exp Cell Res 357: 328–334, 2017. doi: 10.1016/j.yexcr.2017.05.028. [DOI] [PubMed] [Google Scholar]
- 578. Zhang WZ, Rice MC, Hoffman KL, Oromendia C, Barjaktarevic IZ, Wells JM, Hastie AT, Labaki WW, Cooper CB, Comellas AP, Criner GJ, Krishnan JA, Paine R, Hansel NN, Bowler RP, Barr RG, Peters SP, Woodruff PG, Curtis JL, Han MK, Ballman KV, Martinez FJ, Choi AM, Nakahira K, Cloonan SM, Choi ME; SPIROMICS Investigators. Association of urine mitochondrial DNA with clinical measures of COPD in the SPIROMICS cohort. JCI Insight 5, 2020. doi: 10.1172/jci.insight.133984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579. Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L, Kanellopoulos J, Quesniaux VF, Marchand-Adam S, Crestani B, Ryffel B, Couillin I. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med 182: 774–783, 2010. doi: 10.1164/rccm.201003-0359OC. [DOI] [PubMed] [Google Scholar]
- 580. Gasse P, Riteau N, Charron S, Girre S, Fick L, Pétrilli V, Tschopp J, Lagente V, Quesniaux VF, Ryffel B, Couillin I. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med 179: 903–913, 2009. doi: 10.1164/rccm.200808-1274OC. [DOI] [PubMed] [Google Scholar]
- 581. Heijink IH, Pouwels SD, Leijendekker C, de Bruin HG, Zijlstra GJ, van der Vaart H, ten Hacken NH, van Oosterhout AJ, Nawijn MC, van der Toorn M. Cigarette smoke-induced damage-associated molecular pattern release from necrotic neutrophils triggers proinflammatory mediator release. Am J Respir Cell Mol Biol 52: 554–562, 2015. doi: 10.1165/rcmb.2013-0505OC. [DOI] [PubMed] [Google Scholar]
- 582. Ryu C, Sun H, Gulati M, Herazo-Maya JD, Chen Y, Osafo-Addo A, Brandsdorfer C, Winkler J, Blaul C, Faunce J, Pan H, Woolard T, Tzouvelekis A, Antin-Ozerkis DE, Puchalski JT, Slade M, Gonzalez AL, Bogenhagen DF, Kirillov V, Feghali-Bostwick C, Gibson K, Lindell K, Herzog RI, Dela Cruz CS, Mehal W, Kaminski N, Herzog EL, Trujillo G. Extracellular mitochondrial DNA is generated by fibroblasts and predicts death in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 196: 1571–1581, 2017. doi: 10.1164/rccm.201612-2480OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer S, Fleischmann-Struzek C, Machado FR, Reinhart KK, Rowan K, Seymour CW, Watson RS, West TE, Marinho F, Hay SI, Lozano R, Lopez AD, Angus DC, Murray CJ, Naghavi M. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet 395: 200–211, 2020. doi: 10.1016/S0140-6736(19)32989-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584. Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, Kadri SS, Angus DC, Danner RL, Fiore AE, Jernigan JA, Martin GS, Septimus E, Warren DK, Karcz A, Chan C, Menchaca JT, Wang R, Gruber S, Klompas M; CDC Prevention Epicenter Program. Incidence and trends of sepsis in US hospitals using clinical vs claims data, 2009-2014. JAMA 318: 1241–1249, 2017. doi: 10.1001/jama.2017.13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585. Prescott HC, Osterholzer JJ, Langa KM, Angus DC, Iwashyna TJ. Late mortality after sepsis: propensity matched cohort study. BMJ 353: i2375, 2016. doi: 10.1136/bmj.i2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586. Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 304: 1787–1794, 2010. doi: 10.1001/jama.2010.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587. Perl TM, Dvorak L, Hwang T, Wenzel RP. Long-term survival and function after suspected gram-negative sepsis. JAMA 274: 338–345, 1995. [PubMed] [Google Scholar]
- 588. Prescott HC, Angus DC. Enhancing recovery from sepsis: a review. JAMA 319: 62–75, 2018. doi: 10.1001/jama.2017.17687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589. Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers 2: 16045, 2016. doi: 10.1038/nrdp.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315: 801–810, 2016. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ 353: i1585, 2016. doi: 10.1136/bmj.i1585. [DOI] [PubMed] [Google Scholar]
- 592. Brealey D, Karyampudi S, Jacques TS, Novelli M, Stidwill R, Taylor V, Smolenski RT, Singer M. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am J Physiol Regul Integr Comp Physiol 286: R491–R497, 2004. doi: 10.1152/ajpregu.00432.2003. [DOI] [PubMed] [Google Scholar]
- 593. Kreymann G, Grosser S, Buggisch P, Gottschall C, Matthaei S, Greten H. Oxygen consumption and resting metabolic rate in sepsis, sepsis syndrome, and septic shock. Crit Care Med 21: 1012–1019, 1993. doi: 10.1097/00003246-199307000-00015. [DOI] [PubMed] [Google Scholar]
- 594. Boekstegers P, Weidenhöfer S, Pilz G, Werdan K. Peripheral oxygen availability within skeletal muscle in sepsis and septic shock: comparison to limited infection and cardiogenic shock. Infection 19: 317–323, 1991. doi: 10.1007/BF01645355. [DOI] [PubMed] [Google Scholar]
- 595. Rosser DM, Stidwill RP, Jacobson D, Singer M. Oxygen tension in the bladder epithelium rises in both high and low cardiac output endotoxemic sepsis. J Appl Physiol (1985) 79: 1878–1882, 1995. doi: 10.1152/jappl.1995.79.6.1878. [DOI] [PubMed] [Google Scholar]
- 596. Hayes MA, Timmins AC, Yau EH, Palazzo M, Hinds CJ, Watson D. Elevation of systemic oxygen delivery in the treatment of critically ill patients. N Engl J Med 330: 1717–1722, 1994. doi: 10.1056/NEJM199406163302404. [DOI] [PubMed] [Google Scholar]
- 597. Gattinoni L, Brazzi L, Pelosi P, Latini R, Tognoni G, Pesenti A, Fumagalli R. A trial of goal-oriented hemodynamic therapy in critically ill patients. SvO2 Collaborative Group. N Engl J Med 333: 1025–1032, 1995. doi: 10.1056/NEJM199510193331601. [DOI] [PubMed] [Google Scholar]
- 598. De Cruz SJ, Kenyon NJ, Sandrock CE. Bench-to-bedside review: the role of nitric oxide in sepsis. Expert Rev Respir Med 3: 511–521, 2009. doi: 10.1586/ers.09.39. [DOI] [PubMed] [Google Scholar]
- 599. Arulkumaran N, Deutschman CS, Pinsky MR, Zuckerbraun B, Schumacker PT, Gomez H, Gomez A, Murray P, Kellum JA; ADQI XIV Workgroup. Mitochondrial function in sepsis. Shock 45: 271–281, 2016. doi: 10.1097/SHK.0000000000000463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600. Fink MP. Cytopathic hypoxia. Mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit Care Clin 17: 219–237, 2001. doi: 10.1016/s0749-0704(05)70161-5. [DOI] [PubMed] [Google Scholar]
- 601. Murray J, Taylor SW, Zhang B, Ghosh SS, Capaldi RA. Oxidative damage to mitochondrial complex I due to peroxynitrite: identification of reactive tyrosines by mass spectrometry. J Biol Chem 278: 37223–37230, 2003. doi: 10.1074/jbc.M305694200. [DOI] [PubMed] [Google Scholar]
- 602. Vanasco V, Magnani ND, Cimolai MC, Valdez LB, Evelson P, Boveris A, Alvarez S. Endotoxemia impairs heart mitochondrial function by decreasing electron transfer, ATP synthesis and ATP content without affecting membrane potential. J Bioenerg Biomembr 44: 243–252, 2012. doi: 10.1007/s10863-012-9426-3. [DOI] [PubMed] [Google Scholar]
- 603. Seija M, Baccino C, Nin N, Sánchez-Rodríguez C, Granados R, Ferruelo A, Martínez-Caro L, Ruíz-Cabello J, de Paula M, Noboa O, Esteban A, Lorente JA. Role of peroxynitrite in sepsis-induced acute kidney injury in an experimental model of sepsis in rats. Shock 38: 403–410, 2012. doi: 10.1097/SHK.0b013e31826660f2. [DOI] [PubMed] [Google Scholar]
- 604. Carré JE, Orban JC, Re L, Felsmann K, Iffert W, Bauer M, Suliman HB, Piantadosi CA, Mayhew TM, Breen P, Stotz M, Singer M. Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med 182: 745–751, 2010. doi: 10.1164/rccm.201003-0326OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605. Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 348: 138–150, 2003. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 606. Levy RJ, Piel DA, Acton PD, Zhou R, Ferrari VA, Karp JS, Deutschman CS. Evidence of myocardial hibernation in the septic heart. Crit Care Med 33: 2752–2756, 2005. doi: 10.1097/01.ccm.0000189943.60945.77. [DOI] [PubMed] [Google Scholar]
- 607. Luo HR, Loison F. Constitutive neutrophil apoptosis: mechanisms and regulation. Am J Hematol 83: 288–295, 2008. doi: 10.1002/ajh.21078. [DOI] [PubMed] [Google Scholar]
- 608. Taneja R, Parodo J, Jia SH, Kapus A, Rotstein OD, Marshall JC. Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit Care Med 32: 1460–1469, 2004. doi: 10.1097/01.ccm.0000129975.26905.77. [DOI] [PubMed] [Google Scholar]
- 609. Suzuki T, Moraes TJ, Vachon E, Ginzberg HH, Huang TT, Matthay MA, Hollenberg MD, Marshall J, McCulloch CA, Abreu MT, Chow CW, Downey GP. Proteinase-activated receptor-1 mediates elastase-induced apoptosis of human lung epithelial cells. Am J Respir Cell Mol Biol 33: 231–247, 2005. doi: 10.1165/rcmb.2005-0109OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610. Lelubre C, Vincent JL. Mechanisms and treatment of organ failure in sepsis. Nat Rev Nephrol 14: 417–427, 2018. doi: 10.1038/s41581-018-0005-7. [DOI] [PubMed] [Google Scholar]
- 611. Pinheiro da Silva F, Nizet V. Cell death during sepsis: integration of disintegration in the inflammatory response to overwhelming infection. Apoptosis 14: 509–521, 2009. doi: 10.1007/s10495-009-0320-3. [DOI] [PubMed] [Google Scholar]
- 612. Gao LY, Kwaik YA. The modulation of host cell apoptosis by intracellular bacterial pathogens. Trends Microbiol 8: 306–313, 2000. doi: 10.1016/s0966-842x(00)01784-4. [DOI] [PubMed] [Google Scholar]
- 613. Stavru F, Bouillaud F, Sartori A, Ricquier D, Cossart P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc Natl Acad Sci USA 108: 3612–3617, 2011. doi: 10.1073/pnas.1100126108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614. Carrero JA, Calderon B, Unanue ER. Listeriolysin O from Listeria monocytogenes is a lymphocyte apoptogenic molecule. J Immunol 172: 4866–4874, 2004. doi: 10.4049/jimmunol.172.8.4866. [DOI] [PubMed] [Google Scholar]
- 615. Guzmán CA, Domann E, Rohde M, Bruder D, Darji A, Weiss S, Wehland J, Chakraborty T, Timmis KN. Apoptosis of mouse dendritic cells is triggered by listeriolysin, the major virulence determinant of Listeria monocytogenes. Mol Microbiol 20: 119–126, 1996. doi: 10.1111/j.1365-2958.1996.tb02494.x. [DOI] [PubMed] [Google Scholar]
- 616. Rogers HW, Callery MP, Deck B, Unanue ER. Listeria monocytogenes induces apoptosis of infected hepatocytes. J Immunol 156: 679–684, 1996. [PubMed] [Google Scholar]
- 617. Harrington JS, Choi AM, Nakahira K. Mitochondrial DNA in sepsis. Curr Opin Crit Care 23: 284–290, 2017. doi: 10.1097/MCC.0000000000000427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618. Nakahira K, Kyung SY, Rogers AJ, Gazourian L, Youn S, Massaro AF, Quintana C, Osorio JC, Wang Z, Zhao Y, Lawler LA, Christie JD, Meyer NJ, McCausland FR, Waikar SS, Waxman AB, Chung RT, Bueno R, Rosas IO, Fredenburgh LE, Baron RM, Christiani DC, Hunninghake GM, Choi AM. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med 10: e1001577, 2013. doi: 10.1371/journal.pmed.1001577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619. Harrington JS, Huh JW, Schenck EJ, Nakahira K, Siempos II, Choi AM. Circulating mitochondrial DNA as predictor of mortality in critically ill patients: a systematic review of clinical studies. Chest 156: 1120–1136, 2019. doi: 10.1016/j.chest.2019.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620. Islam MT. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res 39: 73–82, 2017. doi: 10.1080/01616412.2016.1251711. [DOI] [PubMed] [Google Scholar]
- 621. Elfawy HA, Das B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: etiologies and therapeutic strategies. Life Sci 218: 165–184, 2019. doi: 10.1016/j.lfs.2018.12.029. [DOI] [PubMed] [Google Scholar]
- 622. Reddy PH, Reddy TP, Manczak M, Calkins MJ, Shirendeb U, Mao P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res Rev 67: 103–118, 2011. doi: 10.1016/j.brainresrev.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623. Bertholet AM, Delerue T, Millet AM, Moulis MF, David C, Daloyau M, Arnauné-Pelloquin L, Davezac N, Mils V, Miquel MC, Rojo M, Belenguer P. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol Dis 90: 3–19, 2016. doi: 10.1016/j.nbd.2015.10.011. [DOI] [PubMed] [Google Scholar]
- 624. Wu Y, Chen M, Jiang J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 49: 35–45, 2019. doi: 10.1016/j.mito.2019.07.003. [DOI] [PubMed] [Google Scholar]
- 625.2021 Alzheimer’s disease facts and figures. Alzheimers Dement 17: 327–406, 2021. doi: 10.1002/alz.12328. [DOI] [PubMed] [Google Scholar]
- 626. Chen XQ, Mobley WC. Alzheimer disease pathogenesis: insights from molecular and cellular biology studies of oligomeric Abeta and Tau species. Front Neurosci 13: 659, 2019. doi: 10.3389/fnins.2019.00659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627. Calvo-Rodriguez M, Kharitonova EK, Bacskai BJ. Therapeutic strategies to target calcium dysregulation in Alzheimer’s disease. Cells 9: 2513, 2020. doi: 10.3390/cells9112513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628. Weidling IW, Swerdlow RH. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp Neurol 330: 113321, 2020. doi: 10.1016/j.expneurol.2020.113321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629. Silva DF, Selfridge JE, Lu J, Lezi E, Cardoso SM, Swerdlow RH. Mitochondrial abnormalities in Alzheimer’s disease: possible targets for therapeutic intervention. Adv Pharmacol 64: 83–126, 2012. doi: 10.1016/B978-0-12-394816-8.00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630. Dhapola R, Sarma P, Medhi B, Prakash A, Reddy DH. Recent advances in molecular pathways and therapeutic implications targeting mitochondrial dysfunction for Alzheimer’s disease. Mol Neurobiol 59: 535–555, 2022. doi: 10.1007/s12035-021-02612-6. [DOI] [PubMed] [Google Scholar]
- 631. Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21: 3017–3023, 2001. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632. Kish SJ, Bergeron C, Rajput A, Dozic S, Mastrogiacomo F, Chang LJ, Wilson JM, DiStefano LM, Nobrega JN. Brain cytochrome oxidase in Alzheimer’s disease. J Neurochem 59: 776–779, 1992. doi: 10.1111/j.1471-4159.1992.tb09439.x. [DOI] [PubMed] [Google Scholar]
- 633. Parker WD Jr. Cytochrome oxidase deficiency in Alzheimer’s disease. Ann NY Acad Sci 640: 59–64, 1991. doi: 10.1111/j.1749-6632.1991.tb00191.x. [DOI] [PubMed] [Google Scholar]
- 634. Beck SJ, Guo L, Phensy A, Tian J, Wang L, Tandon N, Gauba E, Lu L, Pascual JM, Kroener S, Du H. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat Commun 7: 11483, 2016. doi: 10.1038/ncomms11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635. Manczak M, Park BS, Jung Y, Reddy PH. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med 5: 147–162, 2004. doi: 10.1385/NMM:5:2:147. [DOI] [PubMed] [Google Scholar]
- 636. Cottrell DA, Blakely EL, Johnson MA, Ince PG, Turnbull DM. Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology 57: 260–264, 2001. doi: 10.1212/wnl.57.2.260. [DOI] [PubMed] [Google Scholar]
- 637. Jadiya P, Kolmetzky DW, Tomar D, Di Meco A, Lombardi AA, Lambert JP, Luongo TS, Ludtmann MH, Praticò D, Elrod JW. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat Commun 10: 3885, 2019. doi: 10.1038/s41467-019-11813-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638. Yokoyama M, Kobayashi H, Tatsumi L, Tomita T. Mouse models of Alzheimer’s disease. Front Mol Neurosci 15: 912995, 2022. doi: 10.3389/fnmol.2022.912995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639. Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’ disease. Proc Natl Acad Sci USA 106: 14670–14675, 2009. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640. Sancheti H, Kanamori K, Patil I, Díaz Brinton R, Ross BD, Cadenas E. Reversal of metabolic deficits by lipoic acid in a triple transgenic mouse model of Alzheimer’s disease: a 13C NMR study. J Cereb Blood Flow Metab 34: 288–296, 2014. doi: 10.1038/jcbfm.2013.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641. Reddy PH, McWeeney S, Park BS, Manczak M, Gutala RV, Partovi D, Jung Y, Yau V, Searles R, Mori M, Quinn J. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum Mol Genet 13: 1225–1240, 2004. doi: 10.1093/hmg/ddh140. [DOI] [PubMed] [Google Scholar]
- 642. Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15: 1437–1449, 2006. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 643. Han J, Park H, Maharana C, Gwon AR, Park J, Baek SH, Bae HG, Cho Y, Kim HK, Sul JH, Lee J, Kim E, Kim J, Cho Y, Park S, Palomera LF, Arumugam TV, Mattson MP, Jo DG. Alzheimer’s disease-causing presenilin-1 mutations have deleterious effects on mitochondrial function. Theranostics 11: 8855–8873, 2021. doi: 10.7150/thno.59776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644. Devi L, Ohno M. Mitochondrial dysfunction and accumulation of the beta-secretase-cleaved C-terminal fragment of APP in Alzheimer’s disease transgenic mice. Neurobiol Dis 45: 417–424, 2012. doi: 10.1016/j.nbd.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645. Khan SM, Cassarino DS, Abramova NN, Keeney PM, Borland MK, Trimmer PA, Krebs CT, Bennett JC, Parks JK, Swerdlow RH, Parker WD Jr, Bennett JP Jr.. Alzheimer’s disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann Neurol 48: 148–155, 2000. [PubMed] [Google Scholar]
- 646. Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med 14: 1097–1105, 2008. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647. Aleardi AM, Benard G, Augereau O, Malgat M, Talbot JC, Mazat JP, Letellier T, Dachary-Prigent J, Solaini GC, Rossignol R. Gradual alteration of mitochondrial structure and function by beta-amyloids: importance of membrane viscosity changes, energy deprivation, reactive oxygen species production, and cytochrome c release. J Bioenerg Biomembr 37: 207–225, 2005. doi: 10.1007/s10863-005-6631-3. [DOI] [PubMed] [Google Scholar]
- 648. Tillement L, Lecanu L, Papadopoulos V. Alzheimer’s disease: effects of beta-amyloid on mitochondria. Mitochondrion 11: 13–21, 2011. doi: 10.1016/j.mito.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 649. Li Y, Xia X, Wang Y, Zheng JC. Mitochondrial dysfunction in microglia: a novel perspective for pathogenesis of Alzheimer’s disease. J Neuroinflammation 19: 248, 2022. doi: 10.1186/s12974-022-02613-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 650. Shilling D, Müller M, Takano H, Mak DO, Abel T, Coulter DA, Foskett JK. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer's disease pathogenesis. J Neurosci 34: 6910–6923, 2014. doi: 10.1523/JNEUROSCI.5441-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651. Reddy PH, Oliver DM. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 8: 488, 2019. doi: 10.3390/cells8050488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 652. Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, Rocktäschel P, Croteau DL, Akbari M, Greig NH, Fladby T, Nilsen H, Cader MZ, Mattson MP, Tavernarakis N, Bohr VA. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci 22: 401–412, 2019. doi: 10.1038/s41593-018-0332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 653. Manczak M, Kandimalla R, Yin X, Reddy PH. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum Mol Genet 27: 1332–1342, 2018. doi: 10.1093/hmg/ddy042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 654. Du F, Yu Q, Yan S, Hu G, Lue LF, Walker DG, Wu L, Yan SF, Tieu K, Yan SS. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 140: 3233–3251, 2017. doi: 10.1093/brain/awx258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655. Martín-Maestro P, Gargini R, Perry G, Avila J, Garcia-Escudero V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum Mol Genet 25: 792–806, 2016. doi: 10.1093/hmg/ddv616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656. Goudarzi S, Hosseini A, Abdollahi M, Haghi-Aminjan H. Insights into Parkin-mediated mitophagy in Alzheimer’s disease: a systematic review. Front Aging Neurosci 13: 674071, 2021. doi: 10.3389/fnagi.2021.674071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 657. Chakravorty A, Jetto CT, Manjithaya R. Dysfunctional mitochondria and mitophagy as drivers of Alzheimer’s disease pathogenesis. Front Aging Neurosci 11: 311, 2019. doi: 10.3389/fnagi.2019.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 658. Pradeepkiran JA, Hindle A, Kshirsagar S, Reddy PH. Are mitophagy enhancers therapeutic targets for Alzheimer’s disease? Biomed Pharmacother 149: 112918, 2022. doi: 10.1016/j.biopha.2022.112918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 659. de la Cueva M, Antequera D, Ordoñez-Gutierrez L, Wandosell F, Camins A, Carro E, Bartolome F. Amyloid-beta impairs mitochondrial dynamics and autophagy in Alzheimer’s disease experimental models. Sci Rep 12: 10092, 2022. doi: 10.1038/s41598-022-13683-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660. Kaur S, Sharma N, Kumar V, Sharma D, Devi B, Kapil L, Singh C, Singh A. The role of mitophagy in various neurological diseases as a therapeutic approach. Cell Mol Neurobiol. In Press. doi: 10.1007/s10571-022-01302-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661. Medala VK, Gollapelli B, Dewanjee S, Ogunmokun G, Kandimalla R, Vallamkondu J. Mitochondrial dysfunction, mitophagy, and role of dynamin-related protein 1 in Alzheimer’s disease. J Neurosci Res 99: 1120–1135, 2021. doi: 10.1002/jnr.24781. [DOI] [PubMed] [Google Scholar]
- 662. Cai Q, Tammineni P. Alterations in mitochondrial quality control in Alzheimer’s disease. Front Cell Neurosci 10: 24, 2016. doi: 10.3389/fncel.2016.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663. Manczak M, Reddy PH. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet 21: 2538–2547, 2012. doi: 10.1093/hmg/dds072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 664. Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet 20: 2495–2509, 2011. doi: 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 665. Manczak M, Kandimalla R, Fry D, Sesaki H, Reddy PH. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum Mol Genet 25: 5148–5166, 2016. doi: 10.1093/hmg/ddw330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 666. Shields LY, Li H, Nguyen K, Kim H, Doric Z, Garcia JH, Gill TM, Haddad D, Vossel K, Calvert M, Nakamura K. Mitochondrial fission is a critical modulator of mutant APP-induced neural toxicity. J Biol Chem 296: 100469, 2021. doi: 10.1016/j.jbc.2021.100469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667. Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet 20: 4515–4529, 2011. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 668. Wang W, Yin J, Ma X, Zhao F, Siedlak SL, Wang Z, Torres S, Fujioka H, Xu Y, Perry G, Zhu X. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum Mol Genet 26: 4118–4131, 2017. doi: 10.1093/hmg/ddx299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669. Panes JD, Wendt A, Ramirez-Molina O, Castro PA, Fuentealba J. Deciphering the role of PGC-1alpha in neurological disorders: from mitochondrial dysfunction to synaptic failure. Neural Regen Res 17: 237–245, 2022. doi: 10.4103/1673-5374.317957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 670. Robinson A, Grösgen S, Mett J, Zimmer VC, Haupenthal VJ, Hundsdörfer B, Stahlmann CP, Slobodskoy Y, Müller UC, Hartmann T, Stein R, Grimm MO. Upregulation of PGC-1alpha expression by Alzheimer’s disease-associated pathway: presenilin 1/amyloid precursor protein (APP)/intracellular domain of APP. Aging Cell 13: 263–272, 2014. doi: 10.1111/acel.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 671. Dong YT, Cao K, Xiang J, Shan L, Guan ZZ. Silent Mating-Type Information Regulation 2 Homolog 1 attenuates the neurotoxicity associated with Alzheimer Disease via a mechanism which may involve regulation of peroxisome proliferator-activated receptor gamma coactivator 1-alpha. Am J Pathol 190: 1545–1564, 2020. doi: 10.1016/j.ajpath.2020.03.015. [DOI] [PubMed] [Google Scholar]
- 672. Wang J, Liu WJ, Shi HZ, Zhai HR, Qian JJ, Zhang WN. A role for PGC-1a in the control of abnormal mitochondrial dynamics in Alzheimer’s disease. Cells 11: 2849, 2022. doi: 10.3390/cells11182849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 673. Su B, Wang X, Bonda D, Perry G, Smith M, Zhu X. Abnormal mitochondrial dynamics—a novel therapeutic target for Alzheimer’s disease? Mol Neurobiol 41: 87–96, 2010. doi: 10.1007/s12035-009-8095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 674. Liu J, Liu W, Li R, Yang H. Mitophagy in Parkinson’s disease: from pathogenesis to treatment. Cells 8: 712, 2019. doi: 10.3390/cells8070712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 675. Larsen SB, Hanss Z, Krüger R. The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res 373: 21–37, 2018. doi: 10.1007/s00441-017-2768-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676. Verstraeten A, Theuns J, Van Broeckhoven C. Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trends Genet 31: 140–149, 2015. doi: 10.1016/j.tig.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 677. Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B, Ko HS, Sasaki M, Ischiropoulos H, Przedborski S, Dawson TM, Dawson VL. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci USA 104: 14807–14812, 2007. doi: 10.1073/pnas.0703219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 678. Itoh K, Weis S, Mehraein P, Müller-Höcker J. Defects of cytochrome c oxidase in the substantia nigra of Parkinson’s disease: and immunohistochemical and morphometric study. Mov Disord 12: 9–16, 1997. doi: 10.1002/mds.870120104. [DOI] [PubMed] [Google Scholar]
- 679. Qadri R, Goyal V, Behari M, Subramanian A, Datta SK, Mukhopadhyay AK. Alteration of mitochondrial function in oxidative stress in parkinsonian neurodegeneration: a cross-sectional study. Ann Indian Acad Neurol 24: 506–512, 2021. doi: 10.4103/aian.AIAN_392_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 680. Grünewald A, Kumar KR, Sue CM. New insights into the complex role of mitochondria in Parkinson’s disease. Prog Neurobiol 177: 73–93, 2019. doi: 10.1016/j.pneurobio.2018.09.003. [DOI] [PubMed] [Google Scholar]
- 681. Krebiehl G, Ruckerbauer S, Burbulla LF, Kieper N, Maurer B, Waak J, Wolburg H, Gizatullina Z, Gellerich FN, Woitalla D, Riess O, Kahle PJ, Proikas-Cezanne T, Krüger R. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS One 5: e9367, 2010. doi: 10.1371/journal.pone.0009367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682. Scott L, Karuppagounder SS, Neifert S, Kang BG, Wang H, Dawson VL, Dawson TM. The absence of Parkin does not promote dopamine or mitochondrial dysfunction in PolgA(D257A/D257A) mitochondrial mutator mice. J Neurosci 42: 9263–9277, 2022. doi: 10.1523/JNEUROSCI.0545-22.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 683. Ekstrand MI, Galter D. The MitoPark Mouse— an animal model of Parkinson’s disease with impaired respiratory chain function in dopamine neurons. Parkinsonism Relat Disord 15, Suppl 3: S185–S188, 2009. doi: 10.1016/S1353-8020(09)70811-9. [DOI] [PubMed] [Google Scholar]
- 684. Fan L, Zhang S, Li X, Hu Z, Yang J, Zhang S, Zheng H, Su Y, Luo H, Liu X, Fan Y, Sun H, Zhang Z, Miao J, Song B, Xia Z, Shi C, Mao C, Xu Y. CHCHD2 p.Thr61Ile knock-in mice exhibit motor defects and neuropathological features of Parkinson’s disease. Brain Pathol 33: e13124, 2022. doi: 10.1111/bpa.13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 685. Maegawa H, Niwa H. Generation of mitochondrial toxin rodent models of Parkinson’s disease using 6-OHDA, MPTP, and rotenone. Methods Mol Biol 2322: 95–110, 2021. doi: 10.1007/978-1-0716-1495-2_10. [DOI] [PubMed] [Google Scholar]
- 686. Chin MH, Qian WJ, Wang H, Petyuk VA, Bloom JS, Sforza DM, Laćan G, Liu D, Khan AH, Cantor RM, Bigelow DJ, Melega WP, Camp DG 2nd, Smith RD, Smith DJ. Mitochondrial dysfunction, oxidative stress, and apoptosis revealed by proteomic and transcriptomic analyses of the striata in two mouse models of Parkinson’s disease. J Proteome Res 7: 666–677, 2008. doi: 10.1021/pr070546l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 687. Liu X, Liu W, Wang C, Chen Y, Liu P, Hayashi T, Mizuno K, Hattori S, Fujisaki H, Ikejima T. Silibinin attenuates motor dysfunction in a mouse model of Parkinson’s disease by suppression of oxidative stress and neuroinflammation along with promotion of mitophagy. Physiol Behav 239: 113510, 2021. doi: 10.1016/j.physbeh.2021.113510. [DOI] [PubMed] [Google Scholar]
- 688. Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, Duchen MR, Abramov AY. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell 33: 627–638, 2009. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 689. Ludtmann MH, Kostic M, Horne A, Gandhi S, Sekler I, Abramov AY. LRRK2 deficiency induced mitochondrial Ca2+ efflux inhibition can be rescued by Na+/Ca2+/Li+ exchanger upregulation. Cell Death Dis 10: 265, 2019. doi: 10.1038/s41419-019-1469-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 690. Eldeeb MA, Thomas RA, Ragheb MA, Fallahi A, Fon EA. Mitochondrial quality control in health and in Parkinson’s disease. Physiol Rev 102: 1721–1755, 2022. doi: 10.1152/physrev.00041.2021. [DOI] [PubMed] [Google Scholar]
- 691. Han H, Tan J, Wang R, Wan H, He Y, Yan X, Guo J, Gao Q, Li J, Shang S, Chen F, Tian R, Liu W, Liao L, Tang B, Zhang Z. PINK1 phosphorylates Drp1S616 to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep 21: e48686, 2020. doi: 10.15252/embr.201948686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692. Wang H, Song P, Du L, Tian W, Yue W, Liu M, Li D, Wang B, Zhu Y, Cao C, Zhou J, Chen Q. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem 286: 11649–11658, 2011. doi: 10.1074/jbc.M110.144238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 693. Ma KY, Fokkens MR, Reggiori F, Mari M, Verbeek DS. Parkinson’s disease-associated VPS35 mutant reduces mitochondrial membrane potential and impairs PINK1/Parkin-mediated mitophagy. Transl Neurodegener 10: 19, 2021. doi: 10.1186/s40035-021-00243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 694. Li X, Feng Y, Wang XX, Truong D, Wu YC. The Critical role of SIRT1 in Parkinson’s disease: mechanism and therapeutic considerations. Aging Dis 11: 1608–1622, 2020. doi: 10.14336/AD.2020.0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 695.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72: 971–983, 1993. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 696. Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol 39: 385–389, 1996. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 697. Arenas J, Campos Y, Ribacoba R, Martín MA, Rubio JC, Ablanedo P, Cabello A. Complex I defect in muscle from patients with Huntington’s disease. Ann Neurol 43: 397–400, 1998. doi: 10.1002/ana.410430321. [DOI] [PubMed] [Google Scholar]
- 698. Shirendeb U, Reddy AP, Manczak M, Calkins MJ, Mao P, Tagle DA, Reddy PH. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum Mol Genet 20: 1438–1455, 2011. doi: 10.1093/hmg/ddr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 699. Matuz-Mares D, González-Andrade M, Araiza-Villanueva MG, Vilchis-Landeros MM, Vázquez-Meza H. Mitochondrial calcium: effects of its imbalance in disease. Antioxidants (Basel) 11: 801, 2022. doi: 10.3390/antiox11050801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700. Šonský I, Vodička P, Vodičková Kepková K, Hansíková H. Mitophagy in Huntington’s disease. Neurochem Int 149: 105147, 2021. doi: 10.1016/j.neuint.2021.105147. [DOI] [PubMed] [Google Scholar]
- 701. Franco-Iborra S, Plaza-Zabala A, Montpeyo M, Sebastian D, Vila M, Martinez-Vicente M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 17: 672–689, 2021. doi: 10.1080/15548627.2020.1728096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 702. Reddy PH. Increased mitochondrial fission and neuronal dysfunction in Huntington’s disease: implications for molecular inhibitors of excessive mitochondrial fission. Drug Discov Today 19: 951–955, 2014. doi: 10.1016/j.drudis.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 703. Song W, Chen J, Petrilli A, Liot G, Klinglmayr E, Zhou Y, Poquiz P, Tjong J, Pouladi MA, Hayden MR, Masliah E, Ellisman M, Rouiller I, Schwarzenbacher R, Bossy B, Perkins G, Bossy-Wetzel E. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat Med 17: 377–382, 2011. doi: 10.1038/nm.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 704. Shirendeb UP, Calkins MJ, Manczak M, Anekonda V, Dufour B, McBride JL, Mao P, Reddy PH. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum Mol Genet 21: 406–420, 2012. doi: 10.1093/hmg/ddr475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 705. Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J Clin Invest 123: 5371–5388, 2013. doi: 10.1172/JCI70911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 706. Zhao Y, Sun X, Qi X. Inhibition of Drp1 hyperactivation reduces neuropathology and behavioral deficits in zQ175 knock-in mouse model of Huntington’s disease. Biochem Biophys Res Commun 507: 319–323, 2018. doi: 10.1016/j.bbrc.2018.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 707. Naia L, Carmo C, Campesan S, Fão L, Cotton VE, Valero J, Lopes C, Rosenstock TR, Giorgini F, Rego AC. Mitochondrial SIRT3 confers neuroprotection in Huntington’s disease by regulation of oxidative challenges and mitochondrial dynamics. Free Radic Biol Med 163: 163–179, 2021. doi: 10.1016/j.freeradbiomed.2020.11.031. [DOI] [PubMed] [Google Scholar]
- 708. Chandra A, Sharma A, Calingasan NY, White JM, Shurubor Y, Yang XW, Beal MF, Johri A. Enhanced mitochondrial biogenesis ameliorates disease phenotype in a full-length mouse model of Huntington's disease. Hum Mol Genet 25: 2269–2282, 2016. doi: 10.1093/hmg/ddw095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 709. Obrador E, Salvador-Palmer R, Lopez-Blanch R, Jihad-Jebbar A, Valles SL, Estrela JM. The link between oxidative stress, redox status, bioenergetics and mitochondria in the pathophysiology of ALS. Int J Mol Sci 22, 2021. doi: 10.3390/ijms22126352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 710. Smith EF, Shaw PJ, De Vos KJ. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett 710: 132933, 2019. doi: 10.1016/j.neulet.2017.06.052. [DOI] [PubMed] [Google Scholar]
- 711. Sassani M, Alix JJ, McDermott CJ, Baster K, Hoggard N, Wild JM, Mortiboys HJ, Shaw PJ, Wilkinson ID, Jenkins TM. Magnetic resonance spectroscopy reveals mitochondrial dysfunction in amyotrophic lateral sclerosis. Brain 143: 3603–3618, 2020. doi: 10.1093/brain/awaa340. [DOI] [PubMed] [Google Scholar]
- 712. Xiao Y, Karam C, Yi J, Zhang L, Li X, Yoon D, Wang H, Dhakal K, Ramlow P, Yu T, Mo Z, Ma J, Zhou J. ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharmacol Res 138: 25–36, 2018. doi: 10.1016/j.phrs.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 713. Wang H, Yi J, Li X, Xiao Y, Dhakal K, Zhou J. ALS-associated mutation SOD1G93A leads to abnormal mitochondrial dynamics in osteocytes. Bone 106: 126–138, 2018. doi: 10.1016/j.bone.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 714. Tradewell ML, Cooper LA, Minotti S, Durham HD. Calcium dysregulation, mitochondrial pathology and protein aggregation in a culture model of amyotrophic lateral sclerosis: mechanistic relationship and differential sensitivity to intervention. Neurobiol Dis 42: 265–275, 2011. doi: 10.1016/j.nbd.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 715. Moller A, Bauer CS, Cohen RN, Webster CP, De Vos KJ. Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum Mol Genet 26: 4668–4679, 2017. doi: 10.1093/hmg/ddx348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 716. Sakai S, Watanabe S, Komine O, Sobue A, Yamanaka K. Novel reporters of mitochondria-associated membranes (MAM), MAMtrackers, demonstrate MAM disruption as a common pathological feature in amyotrophic lateral sclerosis. FASEB J 35: e21688, 2021. doi: 10.1096/fj.202100137R. [DOI] [PubMed] [Google Scholar]
- 717. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH Jr.. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323: 1205–1208, 2009. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 718. Salam S, Tacconelli S, Smith BN, Mitchell JC, Glennon E, Nikolaou N, Houart C, Vance C. Identification of a novel interaction of FUS and syntaphilin may explain synaptic and mitochondrial abnormalities caused by ALS mutations. Sci Rep 11: 13613, 2021. doi: 10.1038/s41598-021-93189-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 719. Zhao W, Varghese M, Yemul S, Pan Y, Cheng A, Marano P, Hassan S, Vempati P, Chen F, Qian X, Pasinetti GM. Peroxisome proliferator activator receptor gamma coactivator-1alpha (PGC-1alpha) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol Neurodegener 6: 51, 2011. doi: 10.1186/1750-1326-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 720. Qi Y, Yin X, Wang S, Jiang H, Wang X, Ren M, Su XP, Lei S, Feng H. PGC-1alpha silencing compounds the perturbation of mitochondrial function caused by mutant SOD1 in skeletal muscle of ALS mouse model. Front Aging Neurosci 7: 204, 2015. doi: 10.3389/fnagi.2015.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 721. Li X, Chen C, Zhan X, Li B, Zhang Z, Li S, Xie Y, Song X, Shen Y, Liu J, Liu P, Liu GP, Yang X. R13 preserves motor performance in SOD1G93A mice by improving mitochondrial function. Theranostics 11: 7294–7307, 2021. doi: 10.7150/thno.56070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 722. Golko-Perez S, Amit T, Bar-Am O, Youdim MB, Weinreb O. A novel iron chelator-radical scavenger ameliorates motor dysfunction and improves life span and mitochondrial biogenesis in SOD1G93A ALS mice. Neurotox Res 31: 230–244, 2017. doi: 10.1007/s12640-016-9677-6. [DOI] [PubMed] [Google Scholar]
- 723. Murphy MP, Hartley RC. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov 17: 865–886, 2018. doi: 10.1038/nrd.2018.174. [DOI] [PubMed] [Google Scholar]
- 724. Zielonka J, Joseph J, Sikora A, Hardy M, Ouari O, Vasquez-Vivar J, Cheng G, Lopez M, Kalyanaraman B. Mitochondria-targeted triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem Rev 117: 10043–10120, 2017. doi: 10.1021/acs.chemrev.7b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 725. Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J 19: 1088–1095, 2005. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 726. Tippairote T, Bjørklund G, Gasmi A, Semenova Y, Peana M, Chirumbolo S, Hangan T. Combined supplementation of Coenzyme Q10 and other nutrients in specific medical conditions. Nutrients 14: 4383, 2022. doi: 10.3390/nu14204383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 727. Singh A, Faccenda D, Campanella M. Pharmacological advances in mitochondrial therapy. EBioMedicine 65: 103244, 2021. doi: 10.1016/j.ebiom.2021.103244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 728. Wang PF, Xie K, Cao YX, Zhang A. Hepatoprotective effect of mitochondria-targeted antioxidant Mito-TEMPO against lipopolysaccharide-induced liver injury in mouse. Mediators Inflamm 2022: 6394199, 2022. doi: 10.1155/2022/6394199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 729. Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RA, Murphy MP. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int 30: 1019–1026, 2010. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- 730. Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM; Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord 25: 1670–1674, 2010. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 731. Ding XW, Robinson M, Li R, Aldhowayan H, Geetha T, Babu JR. Mitochondrial dysfunction and beneficial effects of mitochondria-targeted small peptide SS-31 in diabetes mellitus and Alzheimer’s disease. Pharmacol Res 171: 105783, 2021. doi: 10.1016/j.phrs.2021.105783. [DOI] [PubMed] [Google Scholar]
- 732. Zhu Y, Luo M, Bai X, Li J, Nie P, Li B, Luo P. SS-31, a mitochondria-targeting peptide, ameliorates kidney disease. Oxid Med Cell Longev 2022: 1295509, 2022. doi: 10.1155/2022/1295509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 733. Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 171: 2029–2050, 2014. doi: 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 734. Suo M, Qi Y, Liu L, Zhang C, Li J, Yan X, Zhang C, Ti Y, Chen T, Bu P. SS31 Alleviates Pressure Overload-Induced Heart Failure caused by Sirt3-mediated mitochondrial fusion. Front Cardiovasc Med 9: 858594, 2022. doi: 10.3389/fcvm.2022.858594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 735. Zhao W, Xu Z, Cao J, Fu Q, Wu Y, Zhang X, Long Y, Zhang X, Yang Y, Li Y, Mi W. Elamipretide (SS-31) improves mitochondrial dysfunction, synaptic and memory impairment induced by lipopolysaccharide in mice. J Neuroinflammation 16: 230, 2019. doi: 10.1186/s12974-019-1627-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 736. Fouché B, Turner S, Gorham R, Stephenson EJ, Gutbier S, Elson JL, García-Beltrán O, Van Der Westhuizen FH, Pienaar IS. A novel mitochondria-targeting iron chelator neuroprotects multimodally via HIF-1 modulation against a mitochondrial toxin in a dopaminergic cell model of Parkinson’s disease. Mol Neurobiol 60: 749–767, 2023. doi: 10.1007/s12035-022-03107-8. [DOI] [PubMed] [Google Scholar]
- 737. Rosdah AA, K Holien J, Delbridge LMD, Dusting GJ, Lim SY. Mitochondrial fission—a drug target for cytoprotection or cytodestruction? Pharmacol Res Perspect 4: e00235, 2016. doi: 10.1002/prp2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 738. Reddy PH, Manczak M, Yin X, Reddy AP. Synergistic protective effects of Mitochondrial Division Inhibitor 1 and mitochondria-targeted small peptide SS31 in Alzheimer’s disease. J Alzheimers Dis 62: 1549–1565, 2018. doi: 10.3233/JAD-170988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 739. Fan LF, He PY, Peng YC, Du QH, Ma YJ, Jin JX, Xu HZ, Li JR, Wang ZJ, Cao SL, Li T, Yan F, Gu C, Wang L, Chen G. Mdivi-1 ameliorates early brain injury after subarachnoid hemorrhage via the suppression of inflammation-related blood-brain barrier disruption and endoplasmic reticulum stress-based apoptosis. Free Radic Biol Med 112: 336–349, 2017. doi: 10.1016/j.freeradbiomed.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 740. Smith G, Gallo G. To mdivi-1 or not to mdivi-1: is that the question? Dev Neurobiol 77: 1260–1268, 2017. doi: 10.1002/dneu.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 741. Liu X, Song L, Yu J, Huang F, Li Y, Ma C. Mdivi-1: a promising drug and its underlying mechanisms in the treatment of neurodegenerative diseases. Histol Histopathol 37: 505–512, 2022. doi: 10.14670/HH-18-443. [DOI] [PubMed] [Google Scholar]
- 742. Qi X, Qvit N, Su YC, Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci 126: 789–802, 2013. doi: 10.1242/jcs.114439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 743. Aggarwal R, Potel KN, McFalls EO, Butterick TA, Kelly RF. Novel therapeutic approaches enhance PGC1-alpha to reduce oxidant stress-inflammatory signaling and improve functional recovery in hibernating myocardium. Antioxidants (Basel) 11: 2155, 2022. doi: 10.3390/antiox11112155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 744. Valero T. Mitochondrial biogenesis: pharmacological approaches. Curr Pharm Des 20: 5507–5509, 2014. doi: 10.2174/138161282035140911142118. [DOI] [PubMed] [Google Scholar]
- 745. Zheng M, Bai Y, Sun X, Fu R, Liu L, Liu M, Li Z, Huang X. Resveratrol reestablishes mitochondrial quality control in myocardial ischemia/reperfusion injury through Sirt1/Sirt3-Mfn2-Parkin-PGC-1alpha pathway. Molecules 27: 5545, 2022. doi: 10.3390/molecules27175545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 746. Zhou J, Yang Z, Shen R, Zhong W, Zheng H, Chen Z, Tang J, Zhu J. Resveratrol improves mitochondrial biogenesis function and activates PGC-1alpha Pathway in a preclinical model of early brain injury following subarachnoid hemorrhage. Front Mol Biosci 8: 620683, 2021. doi: 10.3389/fmolb.2021.620683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 747. Yang K, Yang M, Shen Y, Kang L, Zhu X, Dong W, Lei X. Resveratrol attenuates hyperoxia lung injury in neonatal rats by activating SIRT1/PGC-1alpha signaling pathway. Am J Perinatol. In Press. doi: 10.1055/a-1787-3396. [DOI] [PubMed] [Google Scholar]
- 748. Reiten OK, Wilvang MA, Mitchell SJ, Hu Z, Fang EF. Preclinical and clinical evidence of NAD+ precursors in health, disease, and ageing. Mech Ageing Dev 199: 111567, 2021. doi: 10.1016/j.mad.2021.111567. [DOI] [PubMed] [Google Scholar]
- 749. Chang KL, Wong LR, Pee HN, Yang S, Ho PC. Reverting metabolic dysfunction in cortex and cerebellum of APP/PS1 mice, a model for Alzheimer’s disease by pioglitazone, a peroxisome proliferator-activated receptor gamma (PPARgamma) agonist. Mol Neurobiol 56: 7267–7283, 2019. doi: 10.1007/s12035-019-1586-2. [DOI] [PubMed] [Google Scholar]
- 750. Bogacka I, Xie H, Bray GA, Smith SR. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes 54: 1392–1399, 2005. doi: 10.2337/diabetes.54.5.1392. [DOI] [PubMed] [Google Scholar]
- 751. Coletta DK, Sriwijitkamol A, Wajcberg E, Tantiwong P, Li M, Prentki M, Madiraju M, Jenkinson CP, Cersosimo E, Musi N, Defronzo RA. Pioglitazone stimulates AMP-activated protein kinase signalling and increases the expression of genes involved in adiponectin signalling, mitochondrial function and fat oxidation in human skeletal muscle in vivo: a randomised trial. Diabetologia 52: 723–732, 2009. doi: 10.1007/s00125-008-1256-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752. Peterson AA, Rangwala AM, Thakur MK, Ward PS, Hung C, Outhwaite IR, Chan AI, Usanov DL, Mootha VK, Seeliger MA, Liu DR. Discovery and molecular basis of subtype-selective cyclophilin inhibitors. Nat Chem Biol 18: 1184–1195, 2022. doi: 10.1038/s41589-022-01116-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 753. Panel M, Ruiz I, Brillet R, Lafdil F, Teixeira-Clerc F, Nguyen CT, Calderaro J, Gelin M, Allemand F, Guichou JF, Ghaleh B, Ahmed-Belkacem A, Morin D, Pawlotsky JM. Small-molecule inhibitors of cyclophilins block opening of the mitochondrial permeability transition pore and protect mice from hepatic ischemia/reperfusion injury. Gastroenterology 157: 1368–1382, 2019. doi: 10.1053/j.gastro.2019.07.026. [DOI] [PubMed] [Google Scholar]