

Summary
Merkel cell carcinoma (MCC), a rare but aggressive skin cancer, remains a challenge in the era of precision medicine. Immune checkpoint inhibitors (ICIs), the only approved therapy for advanced MCC, are impeded by high primary and acquired resistance. Hence, we dissect transcriptomic heterogeneity at single-cell resolution in a panel of patient tumors, revealing phenotypic plasticity in a subset of treatment-naive MCC. The tumor cells in a “mesenchymal-like” state are endowed with an inflamed phenotype that portends a better ICI response. This observation is also validated in the largest whole transcriptomic dataset available from MCC patient tumors. In contrast, ICI-resistant tumors predominantly express neuroepithelial markers in a well-differentiated state with “immune-cold” landscape. Importantly, a subtle shift to “mesenchymal-like” state reverts copanlisib resistance in primary MCC cells, highlighting potential strategies in patient stratification for therapeutics to harness tumor cell plasticity, augment treatment efficacy, and avert resistance.
Keywords: Merkel cell carcinoma, skin cancer, neuroendocrine carcinoma, tumor cell state, cellular plasticity, single cell RNA-seq, tumor heterogeneity, immunotherapy resistance, copanlisib, epigenetics
Graphical abstract
Highlights
-
•
Tumor cell states and distinct immune landscapes in MCC patient tumors
-
•
“Mesenchymal-like” state with an inflamed phenotype
-
•
“Well-differentiated neuroepithelial” state in “immune-cold” ICI-resistant MCC
-
•
Subtle shift to “mesenchymal-like” state reverts copanlisib insensitivity
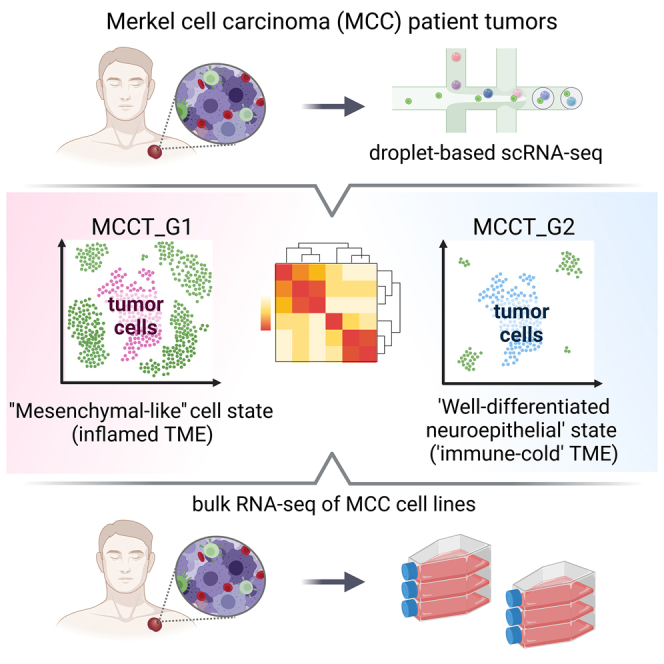
Das et al. utilize single-cell RNA sequencing to uncover transcriptomic heterogeneity with distinct immune landscapes in Merkel cell carcinoma patient tumors. The inherent cell states are present in patient-derived cell lines devoid of tumor microenvironment, underscoring intrinsic cellular plasticity amenable to manipulation to avert and revert therapeutic resistance, a potential clinically actionable finding.
Introduction
Merkel cell carcinoma (MCC) is an aggressive neuroendocrine cancer of the skin that has quadrupled in incidence during the past 20 years.1 Its disease-associated mortality rate exceeds that of melanoma, with a dismal 0%–18% 5-year survival rate in advanced disease.2 The clinical success of immune checkpoint inhibitors (ICIs) in MCC has been astounding yet still encumbered by a high rate of primary and acquired resistance.3 Critically, there is still no curative therapy for patients who are ineligible for ICIs due to comorbidities and for those failing out of ICIs. Given its aggressive and immunosuppressive nature, MCC remains poorly understood and difficult to treat. Vexing challenges include limited experimental models, scarcity of relapse, and treatment-resistant tissue samples due to its rarity, and lack of in-depth understanding of tumor heterogeneity and resistance mechanisms. In contrast to other human cancers for which biomarker selection has dramatically improved treatment response, advanced MCC is treated as a single entity. Classically, MCC tumor heterogeneity has been attributed to variant disease etiologies, mediated by either UV exposure or Merkel cell polyomavirus (MCPyV) since its discovery in 2008.4 Compared to MCPyV-positive MCCs, tumors without detectable MCPyV harbor a high tumor mutation burden with UV signatures.5 However, over the years it has become clear that these two groups are largely similar in clinical presentation, prognosis, and treatment response to ICIs.5,6 Clinically, MCC is considered as a neuroendocrine carcinoma of the skin expressing both epithelial and neuroendocrine markers; however, its cell of origin has been under constant debate, exerting another layer of complexity to tumor heterogeneity.2 Moreover, clinical trials for MCC patients who failed ICI therapy have largely focused on unselected patient populations and have yielded disappointing results,7 highlighting the need to better understand heterogeneous biology and uncover novel approaches that complement and extend current therapy.
Rapid advances in next-generation sequencing have provided robust tools to elucidate tumor heterogeneity and complexity in the tumor microenvironment (TME). Microarray and bulk RNA sequencing (RNA-seq) have gleaned substantial information from heterogeneous tissues but obscure transcription of individual cells and their interactions. In contrast, single-cell RNA-seq (scRNA-seq) directly measures transcriptional outputs of individual cells and parses plasticity within a single tumor and its TME with unprecedented granularity.8 Thereby, applications of this innovative technique have revealed unanticipated levels of tumor heterogeneity and exquisite cancer cell plasticity underlying not only tumor development and progression but also resistance to and relapse after therapy.8,9,10,11 Moreover, scRNA-seq has defined immune cell composition and functional status and has identified the T cell exhaustion program as a major mechanism of tumor immune evasion.12,13 Furthermore, studies have demonstrated that tumor cells in a “mesenchymal-like” state are associated with an “inflamed phenotype” and benefit from ICI therapy in small cell lung cancer (SCLC) and glioblastoma multiforme (GBM).9,14 On the contrary, recent studies in melanoma and prostate cancer have shown that a “mesenchymal-like” state contributes to therapeutic resistance.15,16,17,18 Thus, it is imperative to conduct comprehensive analysis of MCC patient tumors and their TME to precisely characterize tumor cell plasticity and reveal novel therapeutic vulnerabilities.
Here, we conducted a comprehensive analysis of single-cell transcriptomic profiles of 11 MCC tumors from nine patients and incisively dissected the heterogeneity of tumors and their TME. Our studies have identified phenotypic plasticity and have gained insights into MCC ICI resistance mechanisms as well as potential strategies in manipulating the tumor cell state to augment therapeutic response and avert resistance, underscoring patient stratification upon initial diagnosis to match therapies and achieve long-lasting clinical benefits.
Results
scRNA-seq charts transcriptomic heterogeneity and unveils tumor cell plasticity in MCC
To comprehensively interrogate heterogeneity, we generated single-cell transcriptomic profiles for 11 freshly procured human MCC tumors from nine patients. The data compendium spanned nine treatment-naive tumors (no prior systemic or radiation treatment) from seven MCC patients. Treatment-naive samples included one parotid gland tumor, two primary skin tumors, two lymph node metastases, and two primary skin tumors with matched lymph node metastases (Table S1). Additionally, two ICI-resistant (ICI-R) tumors that progressed on the ICI pembrolizumab were included in the study (Table S1). MCC tumors from four out of seven treatment-naive patients harbored detectable MCPyV (57%). A total of 46,027 cells from nine treatment-naive MCC tumors (Table S1 and Figure S1A) were integrated into one dataset by application of Seurat’s “anchor-based” strategy and corrected for batch effect.19,20 Unsupervised principal component analysis (PCA) followed by graph-based clustering and non-linear uniform manifold approximation and projection (UMAP) delineated 15 distinct cell clusters (Figure 1A).
Figure 1.

scRNA-seq identifies phenotypic plasticity in treatment-naive MCC
(A) UMAP visualization of 15 distinct clusters (n = 46,027 [all cells]).
(B and C) Distribution of cell types and heatmap of top 20 variable genes. MC, macrophage/monocytes; CAF, cancer-associated fibroblasts.
(D and E) (D) UMAP visualization of five tumor cell clusters (n = 22,978 cells) and (E) heatmap of top 20 variable genes in each cluster.
(F) Pearson correlation heatmap of pseudo-bulk data from tumor cells with unsupervised hierarchical clustering.
(G) Heatmap of differentially expressed genes between these two groups (log2 fold change >0.25) with unsupervised clustering.
(H) Scaled dot plot of epithelial and neuroendocrine marker genes in MCC tumor cells.
(I) Hallmark Pathway by GSEA of MCCT_G1 and MCCT_G2 tumor cells.
(J) Scaled dot plot of EMT transcription factors (TFs) and mesenchymal marker genes in MCC tumor cells.
The vertical dotted line demarcates MCCT_G1 from MCCT_G2 in (H) and (J).
To identify cell types, we annotated each cluster based on well-established markers and validated them using SingleR (Figures 1B, S1B, and S1C).21 MCC tumors express specific neuroendocrine markers such as neuron-specific enolase (ENO2), neural cell adhesion molecule-1 (NCAM1, CD56), chromogranin (CHGA), and synaptophysin (SYP).1,2,6 Utilizing these canonical markers, we identified seven clusters as tumor cells and the remaining eight clusters as immune and other cell types. Clusters 2, 4, and 9 harbored CD3 T cells and clusters 3, 11, and 13 represented CD19+/PAX5+ B cells, whereas cluster 12 was composed of CD14+/CD68+ monocytes/macrophages (MC) (Figures 1A and 1B). As cancer-associated fibroblasts (CAFs) have not been characterized in MCC, we curated a CAF signature gene list from a previous study (Table S2).22 Upon scoring our tumor subset, we identified cluster 14 with a high CAF score (CAF1) and activation-associated marker genes (Figures 1B, S1B, and S1D). A small number of natural killer (NK) cells and dendritic cells (DCs) were dispersed among CD3 T cell and CD19+/CD20+ B cell clusters (Figure S1B). MCC tumor cells displayed a distinct gene expression pattern compared to other cell types (Figure 1C). Notably, there was variability in the percentage of tumor and immune cell composition among the nine treatment-naive tumors; however, this could not be attributed to capture bias (Figures S1A, S1C, and S1E). We further subclustered tumor cells into five clusters (Figures 1D and 1E). Expression of top variable genes (n = 20) highlighted prominent intertumoral heterogeneity (Figure S1F). To understand sample-to-sample differences and assess transcriptomic heterogeneity, we generated a pseudo-bulk dataset for each tumor by aggregating the raw count of genes across cell types.23 PCA followed by correlation-based Euclidean distance measurements on the normalized tumor cell pseudo-bulk data hierarchically grouped T99, T101, and T102 under one clade, separated from the remaining six samples (Figure 1F). The separation was further accentuated by concordant expression of differentially expressed genes (DEGs) between these two groups in scRNA-seq data (Figure 1G and Table S3). Samples did not cluster together based on the sites of biopsy (skin vs. lymph node) or MCPyV status, suggesting that grouping was not constrained by tumor stages and was virus independent. The primary tumors and matched lymph node tumors (T98 A and B, T112 A and B) were the nearest neighbors, implying that tumors from distinct sites of the same patient shared similar gene expression to a great extent (Figures 1F and 1G).
To examine MCC markers, we found that classical neuroendocrine transcription factors and neuroendocrine markers, such as ATOH1, SOX2, ENO2, SYP, and CHGA, were expressed at lower levels in T99, T101, and T102 (Figure 1H). Interestingly, HES6 and INSM1 were highly expressed in all MCC samples. Therefore, we broadly subtyped T99, T101, and T102 tumors as group 1 MCC (MCCT_G1) and the remaining tumors as group 2 MCC (MCCT_G2) for further characterization. To gain further insights, we performed differential gene expression analysis to determine specific transcriptomic programs for these two groups. Pre-ranked gene set enrichment analysis (GSEA) of top variable genes revealed that MCCT_G1 tumors had significant enrichment of genes involved in tumor necrosis factor α (TNF-α) signaling via nuclear factor (NF)-κB pathways, epithelial-to-mesenchymal transition (EMT), and cell-cycle progression (Figure 1I). In contrast, MCCT_G2 tumors were marked by enrichment of genes participating in energy metabolism-related pathways, including oxidative phosphorylation (OXPHOS), tricarboxylic acid (TCA) or citric acid cycle, and fatty acid breakdown (Figure 1I and Table S4). Other notably enriched pathways in MCCT_G1 included the p53 pathway, inflammation, KRAS signaling, and transforming growth factor β (TGF-β) signaling.
Although EMT is the most commonly cited mechanism for cancer metastasis and therapeutic resistance, recent studies have identified partial mesenchymal phenotypes lying between the fully epithelial and fully mesenchymal poles,22 and this “mesenchymal-like” state in tumor cells is linked to inflamed phenotype, treatment response, and resistance in human cancers.9,14,15,16,17,18 Informed by the GSEA data that EMT was one of the top pathways involved, we carefully examined core EMT transcription factors (EMT_TFs) and markers, and found higher expression of ZEB1, JUNB, VIM, S100A11, EMP3, and especially RHOB in MCCT_G1 tumors (Figure 1J). Congruent with published reports,22 MES gene signals were strongest in CAFs, with tumor cells expressing EMT markers typically at lower levels, suggestive of a partially mesenchymal phenotype in MCCT_G1 tumors (Figure S2A). By immunohistochemistry staining, we found that MCCT_G1 tumor cells were enriched in VIM expression, implying a “mesenchymal-like” state in MCCT_G1 tumor cells (Figure S2B). Our analysis until now has hinted that a subset of MCC tumors might inherit phenotypic plasticity in the primary tumors and carry through metastasis; this was supported by the observation that “mesenchymal-like” MCCs were composed of both primary skin tumors and lymph node metastases (Table S1). Therefore, characterization of MCC tumor cell states at the initial diagnosis may have significant implication for treatment response and resistance.
“Mesenchymal-like” state is likely determined by non-genomic mechanisms
Next, we investigated whether this separation was indirectly reflected in genetic alterations of individual tumors. As whole-genome sequencing data of these samples was not available, we inferred copy-number variations (CNVs) using the scRNA-seq dataset. Detection of genetic mutations within individual cells from scRNA-seq data is limited by partial coverage of the transcriptome. However, large-scale CNVs can be robustly detected based on the average up- or downregulation of large sets of genes within each chromosome region.8,24,25 Recent studies have demonstrated that CNVs inferred from scRNA-seq are highly concordant with DNA-based CNV results.26 Inferred aberrations in our tumor samples aligned with MCPyV status; MCPyV-positive tumors carried fewer aberrations compared to MCPyV-negative tumors (Figure S2C).5 Moreover, MCCs in the “mesenchymal-like” state encompassed both MCPyV-negative and MCPyV-positive tumors, implying that tumor cell state was unlikely to be determined by genetic alterations. Furthermore, chromosome 6p amplification has been previously reported in MCC.27,28 However, our data indicated that genes residing in the specific chromosome 6p region, including human leukocyte antigen (HLA) loci, were prominently under-represented and agreed with the expression levels of HLA molecules in both MCCT_G1 (T99) and MCCT_G2 (T100, T112A, T112B) tumors (Figures S2C and S2D), suggesting a common deletion of the region in both groups.
Recent studies have suggested that cell state heterogeneity arises largely independent of genetic variation.29,30 To decipher the underlying MCC gene regulatory networks and mine the key transcription factors (TFs) in treatment-naive MCC tumors, we applied pySCENIC, a Python implementation of the SCENIC (Single-Cell Regulatory Network Inference and Clustering) pipeline that reconstructs regulons (TFs and their target genes) from scRNA-seq data (STAR Methods).31,32 Enrichment analysis was based on the Z score and calculated by the mean regulon specificity score (RSS) of all single cells/standard deviation of RSS of all cells to identify regulons in MCCT_G1 tumors. We have identified ZEB1, ALX1, and MEF2C as top regulons in MCCT_G1 tumors (Table S5). Similar to ZEB1, ALX1 and MEF2C have been involved in EMT in a TGF-β-dependent manner.33,34
scRNA-seq discloses potential therapeutic vulnerabilities to overcome ICI resistance
As acquired resistance is common in MCC, a better understanding of the molecular underpinning of ICI resistance is of high clinical relevance. scRNA-seq data from MCC remains scarce, particularly data from ICI-R tumors, mainly due to tissue unavailability, since surgery is usually not the next option for patients who have developed ICI resistance.
For further characterization, two ICI-R MCCs were merged with nine treatment-naive tumors in our scRNA-seq dataset (Table S1). A total of ∼35,800 tumor cells were reanalyzed at higher resolution forming six meta-clusters (Figure 2A and Table S1), and we observed a distinct differential gene expression pattern between treatment-naive and ICI-R MCCs (Figure 2B). Pre-ranked GSEA analysis of ICI-R and MCCT_G1 tumor cells using the Hallmark database revealed prominent downregulation of type I and type II interferon response, as well as TNF-α signaling by NF-κB, inflammation, EMT, and TGF-β signaling in ICI-R tumor cells (Figure 2C and Table S4). Recent studies in prostate cancer and melanoma suggest that the “mesenchymal-like” state in tumor cells is associated with metastasis and therapeutic resistance.15,18 To our surprise, tumor cells from ICI-R samples displayed a well-differentiated phenotype with higher expression of neuroepithelial markers and significantly lower levels of mesenchymal marker genes, similar to MCCT_G2 tumors (Figures 2D and 2E). To confirm transcriptomic resemblance, we performed pseudo-bulk analysis on tumor cells from MCCT_G1, MCCT_G2, and ICI-R groups. PCA followed by correlation-based Euclidean distance measurement validated our observation, placing both MCCT_G2 and ICI-R tumor cells in proximity under a single clade (Figure 2F). In comparison to MCCT_G2, ICI-R tumor cells display significant downregulation of major histocompatibility complex class I molecule expression: HLA-A, HLA-B, HLA-C, and HLA-E (Figure 2G).
Figure 2.

scRNA-seq reveals a well-differentiated neuroepithelial state in ICI-R MCC associated with distinct pathways
(A) UMAP clusters of tumor cells from nine treatment-naive and two ICI-R tumors (n = 35,796 cells), split based on treatment status.
(B) Heatmap of top 100 variable genes in treatment-naive and ICI-R tumor cells (n = 35,796 cells).
(C) Hallmark Pathway by GSEA of ICI-R and MCCT_G1 tumor cells.
(D and E) Scaled dot plots of (D) epithelial and neuroendocrine markers, and (E) EMT transcription factors (TFs) and mesenchymal markers in MCCT_G1, MCCT_G2, and ICI-R tumor cells, and the ICI-R dataset as reported by Paulson et al.35 (UW).
(F) Pearson correlation heatmap of pseudo-bulk data from MCCT_G1, MCCT_G2, and ICI-R tumor cells.
(G) Volcano plot of differentially expressed genes between ICI-R and MCCT_G2 tumor cells.
scRNA-seq reveals that the “mesenchymal-like” state is endowed with inflamed phenotype
To date, comprehensive single-cell analysis of immune profiling in the MCC TME has not been elucidated. Next, we enquired whether the “mesenchymal-like” state was also echoed by enriched tumor-associated immune cells, and we first focused on the treatment-naive MCCs. A total of ∼12,800 CD45+ cells from nine treatment-naive tumors were positively selected from batch effect-corrected composite object, and unsupervised clustering identified seven distinct immune cell populations (Figures 3A and 3B; Table S1). The number of immune cells was markedly higher in MCCT_G1 tumors in the “mesenchymal-like” state, which could not be ascribed to capture bias (Figures S1A, S1C, and S1F). CD3 T cells constituted ∼70% of total CD45+ cells, of which ∼21.7% and ∼17.6% were CD8 and CD4 T cells, respectively. While there was a high percentage of CD19+ or CD20+ B cells (25%), the remaining cells were NCAM1+/KLRC2+ NK cells (2.7%), CD14+/CD68+ MC (1.1%), and DCs (1.0%). Interestingly, immune cell composition displayed a high degree of heterogeneity in treatment-naive MCCs (Figure S3A).
Figure 3.

scRNA-seq charts heterogeneity of tumor-associated immune cells in MCC
(A) Left: UMAP visualization of CD45+ cells (n = 12,796 cells) with seven distinct cell populations, namely B cells, CD4 T cells, CD8 T cells, CD4 and CD8 double-negative T cells (DNTC), natural killer cells (NK), dendritic cells (DC), and macrophage/monocytes (MC), and their percentages of distribution (color-matched histogram). Right: split UMAPs depicting distribution of cell types in MCCT_G1 and MCCT_G2 tumors and color-matched histograms of relative abundance.
(B) Heatmap of top 20 variable genes across cell types identified in (A).
(C) Left: UMAP visualization of nine distinct T cell subtypes in CD4/CD8 T cell population (n = 3,169 cells) from the dataset in (A). Middle: split UMAPs depicting groupwise distribution and color-matched histograms of relative abundance. Right: scaled dot plot of marker genes for the T cell subtypes.
(D) Left (bar graph): relative abundance of DNTCs in MCCT_G1 and MCCT_G2 tumors. Right (violin plots): expression of canonical naive, effector, exhaustion, and memory markers in MCCT_G1 and MCCT_G2 tumors.
(E) Split UMAP visualization of seven CD45+ immune cell subtypes (n = 14,895 cells) and color-matched histograms depicting their relative abundance (bottom) in MCCT_G1, MCCT_G2, and ICI-R tumors.
(F) Violin plots of exhaustion-associated immune checkpoint molecules and cytokines in CD3 T cells.
To gain further insights, a total of ∼3,100 CD4 and CD8 T cells were analyzed, resulting in identification of nine distinct subtypes (Figures 3C, S3B, and S3C). Two major cell populations were recognized: naive T cells (30%) and CD8 effector T cells (∼31%) (Figure 3C, vertical bar graph). Two additional CD4 T cell subtypes were also observed: exhausted (∼12%) and regulatory T cells (Tregs) (∼4%), both expressing CTLA4 and TIGIT (Figure 3C, dot plot). CD8 effector exhausted T cells (∼9%), effector memory cells (<2%), resident (∼10%), and CD83+ T cells (<1.0%) contributed to the remaining populations (Figure 3C, vertical bar graph). MCCT_G1 tumors in the “mesenchymal-like” state harbored more CD4/CD8 naive and resident T cells, whereas both CD4 Tregs and CD8 exhausted T cells were relatively higher in MCCT_G2 (Figure 3C), with comparable CD8 effector T cells in both groups. double-negative T cells (DNTCs) are a rare population in the peripheral blood and lymph organs36; however, to our surprise, ∼40% of CD3 T cells were DNTCs with >80% residing in MCCT_G1 tumors (Figure 3D, left column). Although T regulatory and T helper-like functions of DNTCs have been reported to play a crucial role in graft-versus-host and autoimmune diseases,36 their precise function in tumors remains poorly understood. Marker gene expression suggested that DNTCs existed in a naive-like inert state as supported by the expression of naive lymphocyte markers, such as LEF1, SELL, and CCR7 (Figure 3D); however, both immune stimulatory chemokine CCL-5 and suppressive cytokine TGF-β were expressed (Figure S3D). Unsupervised clustering of ∼2,300 B cells uncovered three distinct subsets (Figure S3E). The IgDlow/CD27+/CD99+ memory B cells constituted the largest population (∼73%), followed by IgMhigh/IgDhigh/CD24+ naive B cells (∼25%). The remaining cells were CD27−/CD99+ atypical memory B cells (∼4%). Thus, the majority of B cells were in memory or naive states in treatment-naive TME.
Consistently, ICI-R tumors mirrored MCCT_G2 tumors with an “immune-cold” landscape. Unsupervised clustering of ∼15,000 CD45+ cells from both treatment-naive and ICI-R tumors displayed a marked reduction in immune cell distribution and abundance in ICI-R MCC (Figure 3E). Upon curating available public databases, we were able to identify one ICI-R MCC with scRNA-seq data published by Paulson et al. (labeled as UW in Figures 2D and 2E)35 and observed that this case resembled our ICI-R samples with its tumor cells displaying a well-differentiated neuroepithelial state (Figures 2D and 2E). Notably, this tumor sample had very few immune cells, which agreed with the “immune-cold” phenotype in ICI-R MCC. Interestingly, TIGIT and CTLA4 were detected at markedly higher levels in ICI-R tumors (Figure 3F), supporting the application of anti-TIGIT and anti-CTLA4 antibodies in MCC patients who develop resistance to anti-PD1 therapy. As inflammation is one of the top pathways on the GSEA analysis of tumor cells, we then examined cytokines, chemokines, receptors, and inflammatory genes. As shown in Figures S4A and S4B, CXCR4, CCR7, CCL5, and STAT1 were significantly upregulated in “mesenchymal-like” state MCCs. CXCR4 expression was further confirmed in patient tumors by immunohistochemistry, while CCL5 expression was confirmed in MCC cell lines by qPCR (Figures S4C and S4D).
To further evaluate tumor-immune crosstalk in MCC TME, we analyzed the inferred “cell-cell” communication networks among tumor cells, different types of immune cells, and fibroblasts from our scRNA-seq dataset. Utilizing the recently developed R package CellChat, which can quantitatively analyze ligand-receptor pairs from its database containing >2,000 known molecular interactions to depict key incoming and outgoing autocrine and paracrine signals among different cell types,37 we have established active “cell-cell” communication patterns among seven cell types in the MCC TME, including tumor cells, T cells, NK cells, macrophages, DCs, B cells, and fibroblasts (Figure 4). As displayed in circle plots, the number and strength of interactions between different cell types were greater in MCCT_G1 tumors, reflected by the weight of chords connecting two cell types (Figures 4A, S5A, and S5B). The chord diagrams illustrated that the outgoing signals from tumor cells toward immune cells were largely mediated by macrophage migration inhibitory factor (MIF)-(CD74-CXCR4) and MIF-(CD74-CD44) signaling axes in both MCCT_G1 and MCCT_G2 tumors (Figure 4B). Interactions between tumor cells and immune cells (except T cells) were stronger in MCCT_G1 tumors, as highlighted by a longer arc in chord diagrams and shown in comparative bubble plot analysis of communication probability (Figures 4B and 4C). The outgoing interaction of tumors with immune cells through the midkine (MDK)-nucleolin (NCL) pathway was more prominent in MCCT_G1 tumors (Figures 4B and 4C). In contrast, tumor cell-fibroblast communication was only observed in MCCT_G2 tumors and was predominantly mediated by the MDK/NCL pathway in MCCT_G2 tumors. Given the role of MIF and MDK in the regulation of inflammation and T cells,38,39,40 these results were in line with our prior observation of the highly inflamed TME in “mesenchymal-like” state MCCT_G1 tumors. Additionally, the amyloid-β precursor protein (APP)/CD74 pathway is likely involved in tumor cell-B cell and tumor cell-NK cell interactions in MCCT_G1 tumors (Figures 4B and 4C).
Figure 4.

CellChat analysis reveals stronger interactions of tumor cells with other cell types in MCCT_G1 tumor microenvironment
(A) Circle plots representing the number (left) and weight/strength (right) of interactions between the seven cell types in MCCT_G1 (top) and MCCT_G2 (bottom) tumors. Outgoing signals from a cell type are represented by color-matched chords, with chord thickness proportional to the number/strength of the corresponding signal.
(B) Chord diagrams of outgoing signals from MCCT_G1 (left) and MCCT_G2 (right) tumor cells to other cell types in the tumor microenvironment. Outgoing signals are color matched to the origin (tumor cells), with each arc representing one pathway and arc length depicting the strength.
(C) Comparative bubble plot of communication probability for the top ligand-receptor pairs of outgoing signals from tumor cells to other cell types in the tumor microenvironment.
The top three enriched pathways are highlighted in (B) and in the red boxes in (C).
Taken together, these results highlighted that MCCT_G1 tumors in the “mesenchymal-like” state possessed an inflamed phenotype, which might be primed for ICI response and potentially derive greater benefit from ICIs. It also augurs for potential combinatorial strategies to induce a “mesenchymal-like” state in MCCT_G2 tumors to amend favorable ICI response; both observations warrant further confirmatory studies with a larger cohort of ICI-R tumors, ideally compared to ICI-responsive MCCs.
“Mesenchymal-like” state correlates with a better ICI response in MCC
To obtain an independent assessment, we utilized an 18-gene interferon-γ (IFN-γ) signature (IFNG18S) that has been reported to reliably predict clinical response to ICIs in multiple solid tumors, independent of tumor mutation burden.9,41 Consistently, IFNG18S scored higher in the “mesenchymal-like” MCCT_G1 tumors, positing a better response to ICI therapy (Figure 5A). This observation was further confirmed in our ICI-R tumors with the lowest score (Figure 5A). We next asked whether the “mesenchymal-like” cell state correlated with patient survival. While there are limited whole transcriptomic studies utilizing MCC patient tumors at the single-cell and bulk RNA-seq levels, we identified a recent study with the largest published cohort of MCC patient tumors to date.42 In the study, bulk RNA-seq was performed on 102 archival MCC tumors, and gene expression was correlated with clinical data, resulting in the identification of key marker genes associated with patient survival and death. These signatures, referred to as survival-associated genes (SAGs) and death-associated genes (DAGs), pointed toward an enrichment of oncogenic and immune-response pathways in conferring survival to MCC patients. Therefore we probed our scRNA-seq data using these signatures and queried whether either group displayed a preferential disposition toward survival. Since bulk RNA-seq in the aforementioned study entailed tumor cells as well as other cell types in the TME, scRNA-seq data encompassing all cell types was accordingly used to score for these two signatures. Interestingly, tumors in the “mesenchymal-like” state displayed higher expression of SAGs and corresponding low expression of DAGs (Figure 5A). Conversely, ICI-R tumors displayed high expression of DAGs and corresponding low expression of SAGs (Figure 5A).
Figure 5.

MCCT_G1 tumors with an inflamed phenotype correlate with better ICI response and survival-associated genes
(A) Violin plots of the 18-gene IFN-γ gene signature (IFNG18S), survival-associated gene signature (SAG), and death-associated gene signature (DAG) in MCCT_G1, MCCT_G2, and ICI-R tumors.
(B) Signature score of IFNG18S, MESI-19 (generated from MCC tumor cells), and SIG-14 (generated from entire cell populations) in the glioblastoma multiforme dataset with known ICI response (R, responder; NR, non-responder). Significance of enrichment between responder (R) and non-responder (NR) was calculated using a non-parametric Mann-Whitney test. Solid black and dotted lines within each violin represent the median and quartiles, respectively.
(C) Classification of 102 MCC patient tumor RNA-seq samples based on enrichment of survival-associated gene signature (SAG) and cross-validated against death-associated gene signature (DAG).
(D) Enrichment score for IFNG18S, MESI-19, and SIG-14 in SAGhigh and SAGlow patients identified in (C).
ns, not significant; ∗∗p < 0.005, ∗∗∗p < 0.0005, ∗∗∗∗p < 0.00005 as analyzed by one-way ANOVA for (A) and unpaired non-parametric Mann-Whitney test for (D).
Next, we generated signature gene lists using our scRNA-seq data from treatment-naive MCC patient tumors based on gene expression only in MCC tumor cells. We carefully curated an EMT signature gene (ESG) list including 456 genes from studies across human cancers (Table S6).22,23,43 According to the gene expression levels in our dataset, we then extracted 293 mesenchymal candidate genes from this ESG list (Table S6). Genes with higher expression in MCC tumor cells and CAFs (STAR Methods), but not in any other cell types, were used to generate a 19-gene mesenchymal-specific signature index (MESI-19, Table S2). Next, we attempted to create a signature based on gene expression from the entire cell population. GSEA was performed on DEGs between MCCT_G1 and MCCT_G2 tumors, followed by probing of Hallmark pathways. Significantly upregulated pathways were retrieved, and genes with normalized enrichment scores ≥1.5 were retained from each pathway to generate a 14-gene signature list (SIG-14) (Table S2). For further validation, we probed for studies with publicly available data where no further action was needed for data acquisition, focusing on two neuroendocrine tumors that closely resemble MCC: SCLC and GBM. We employed the search strategies previously published44 and identified an appropriate GBM dataset with ICI treatment response information.45 All three gene lists IFNG18S, MESI-19, and SIG-14 scored higher in the ICI responders in the glioblastoma dataset (Figure 5B). Although this trend was notable, significance was not reached in the GBM dataset, probably due to the small sample size. To investigate whether there was a correlation between these gene lists and patient survival, we stratified the dataset of 102 MCC patients according to relative expression of survival-associated gene signature (SAG); samples were grouped as SAGhigh, SAGmid, and SAGlow (STAR Methods), and this grouping was further confirmed against the death-associated gene signature (DAG)—e.g., the SAGhigh group had the lowest DAG score (Figure 5C). Interestingly, all three gene lists IFNG18S, MESI-19, and SIG-14 scored significantly higher in the SAGhigh group (Figure 5D). Taken together, our results have inferred potential coherence between the “mesenchymal-like” tumor cell state with better ICI response and high expression of SAGs, and such observations will benefit from further clinical validation.
Patient-derived primary MCC cell lines retain “mesenchymal-like” features with therapeutic vulnerability
Cell-based models are critical in examining and understanding the underlying biology of cancer.46 To expand our observations, we surveyed eight patient-derived primary MCC cell lines with bulk RNA-seq (STAR Methods). Seven patient-derived primary MCC cell lines were established and characterized in our laboratory as described previously, and MCPyV status was assessed (STAR Methods)47,48; the originating tumors for these cell lines were different from the tumors in the scRNA-seq dataset. Similar to the classic MKL-1 cell line, our MCPyV-negative MCC-2, -3, -5, -8, and -9 cell lines and MCPyV-positive cell lines MCC-16 and MCC-21 display classical MCC features, including the characteristic suspension cell clusters.48 PCA followed by correlation-based Euclidean distance measurement and hierarchical clustering on the eight MCC cell lines placed MCC-3, -5, -9, and -21 under one clade, whereas MCC-2, -8, -16, and MKL-1 formed the second node independent of MCPyV status (Figure 6A). To further classify whether these two nodes bore distinct transcriptomes, top up- and downregulated genes (log2 fold change >2) were subjected to pre-ranked GSEA analysis as described earlier. Similar to findings in “mesenchymal-like” state MCCT_G1 tumors, MCC-3, -5, -9, and -21 cell lines were enriched with genes in the IFN response, TNF-α signaling via NF-κB, inflammation, and EMT (Figure 6B and Table S4). Consistently, MCC-3, -5, -9, and -21 cell lines clustered under one node in the “mesenchymal-like” state (Figures 6C and 6D). In contrast to a previous study suggesting that EMT was observed only in atypical adherent MCC cell lines that have been questioned in several studies as representative MCC models,49 we found that “mesenchymal-like” cell state prevailed in the treatment-naive patient tumors as well as in our primary MCC cell lines with characteristic suspension cell clusters (Figure S6). Despite relatively lower expression of ATOH1, SYP, and CHGA, the “mesenchymal-like” state retained strong ENO2 expression (Figure 6C). Thus, we observed that recurrent transcriptomic programs dictate the “mesenchymal-like” state both in vivo and in vitro, lending further support to the notion that MCC cell lines mirrored transcriptomic heterogeneity inherited in patient tumors and are thus fit for purpose to address outstanding questions in the field.
Figure 6.

Patient-derived MCC cell lines retain “mesenchymal-like” state and transcriptomic plasticity
(A) Pearson correlation heatmap with unsupervised clustering based on sample distance among eight MCC cell lines.
(B) Hallmark Pathway enrichment by GSEA of MCC_G1 and MCC_G2 cell lines.
(C and D) Heatmaps with unsupervised hierarchical clustering depicting normalized expression across eight MCC cell lines of (C) selected epithelial and neuroendocrine genes and (D) selected EMT transcription factors (TFs) and mesenchymal marker genes.
It is well known that EMT-TFs can be regulated epigenetically50; therefore, we examined whether epigenetic regulation would affect cellular states in MCC. To test this hypothesis, we utilized domatinostat, a class I histone deacetylase (HDAC) inhibitor, which also demonstrated anti-MCC activity in a prior study.51 Similarly, we have shown that MKL-1 cells treated with domatinostat displayed dose-dependent HDAC inhibition and simultaneous acetylation of lysine 27 (K27) in histone 3 protein (Figure 7A). Moreover, GSEA on DEGs identified by bulk RNA-seq revealed that EMT is one of the top upregulated pathways in MKL-1 cells upon domatinostat treatment (Figure 7B and Table S4), which was further confirmed by qPCR (Figure 7C). Currently there is a lack of syngeneic MCC animal models with intact immune systems, preventing examination of whether a subtle shift of cell state could revert primary ICI resistance. Nonetheless, we examined whether a cell state shift would revert resistance to phosphatidylinositide-3-kinase (PI3K) inhibition in primary MCC cells described previously.47 Aberrant activation of the PI3K pathway is detected in many cancers and in up to 80% of MCCs, making it a potential therapeutic target.52,53 Importantly, we previously achieved clinical success of treating a stage IV MCC patient with idelalisib, the first Food and Drug Administration (FDA)-approved PI3K-δ inhibitor.54 More recently, we reported that copanlisib, the second FDA-approved PI3K-α/δ inhibitor, exhibited potent anti-MCC effects in pre-clinical studies.47 In contrast to “mesenchymal-like” MCC cell lines (MCC-3, -5, -9, and -21) which demonstrated sensitivity to copanlisib (MCC-5 shown in Figure S9), we observed primary resistance in MKL-1 cells with half-maximal growth inhibitory concentration (GI50) >10 μM.47 Here, we tested whether the cell state shift upon domatinostat treatment would revert resistance to copanlisib in MKL-1 cells. To this end, we sensitized MKL-1 cells with domatinostat (GI50 dose, ∼500 nM) and simultaneously measured copanlisib antitumor activities by colorimetric cell proliferation assay (CCK-8, Millipore Sigma). Remarkably, we found that domatinostat significantly reverted MKL-1 resistance to copanlisib and observed nearly 200-fold reduction of GI50 (47 nM), affirming our hypothesis that pre-emptively manipulating the intrinsic cell state can impart greater therapeutic susceptibility in MCC. To further confirm our findings in MKL-1 cells, we used the MCC-16 cell line with growth characteristics as well as gene expression patterns mimicking those in the G2 MCC cell lines (Figure S6). Similar findings were also observed in MCC-16 cells treated with domatinostat and copanlisib (Figures 7C and 7D). We also observed dose-dependent acetylation of lysine 27 (K27) in histone 3 protein in domatinostat-treated MCC-16 cells, indicating HDAC inhibition upon domatinostat treatment (Figure 7A). Additionally, we demonstrated that EMT-TFs can be regulated by TGF-β in both MKL-1 and MCC-16 cells (Figure S10).
Figure 7.

HDAC inhibition in MKL-1 and MCC-16 cells shifts intrinsic cell state and reverts PI3K therapeutic resistance
(A) Immunoblots showing dose-dependent HDAC inhibition in MKL-1 (left) and MCC-16 (right) cells upon domatinostat treatment. Ratios of H3K27-Ac to total H3 are presented as mean ± SD for each dose (n = 3).
(B) Hallmark Pathway by GSEA in MKL-1 cells treated with domatinostat by bulk RNA-seq.
(C) Relative mRNA expression of EMT transcription factors in MKL-1 and MCC-16 cells receiving either DMSO (vehicle control) or 10 μM domatinostat for 24 h. Data are presented as mean ± SD, and all samples were run in triplicate. ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 as analyzed by unpaired Student’s t test.
(D) Comparison of dose-dependent drug response in MKL-1 and MCC-16 cells treated with domatinostat or copanlisib alone (dose range 30 nM to 30 μM), or a combination of copanlisib (dose range 30 nM to 30 μM) with fixed dose of 500 nM domatinostat. Data are presented as mean ± SD for each dose, n = 6 per dose, with half-maximal growth inhibitory concentration as analyzed by non-linear regression model using GraphPad Prism.
Discussion
Intratumor heterogeneity (ITH) and the composition/functional status of tumor-associated immune cells in the TME play a pivotal role in cancer progression, metastasis, treatment response/resistance, and relapse. Owing to its capacity to create transcriptomic profiles of single cells in multicellular TME, scRNA-seq together with advanced computational pipelines has been intensively used to dissect the dynamics of ITH and immune landscapes in a variety of human cancers.8,9,10,26,55,56 Moreover, scRNA-seq has discovered unanticipated cellular plasticity associated with distinct functions without concomitant underlying changes in the genome.9,29,30 As a highly aggressive neuroendocrine cancer, MCC has proven a challenge in the era of personalized medicine, partly due to limited understanding of tumor heterogeneity. Overcoming the limitation of tumor sample availability, we apply this innovative technique and comprehensively dissect the transcriptomic heterogeneity of tumors and the TME. We have observed transcriptomic plasticity converging on an inflamed “mesenchymal-like” tumor cell state that portends favorable ICI response. In particular, we have demonstrated that shifting to a “mesenchymal-like” state by HDAC inhibition reverts therapeutic resistance to PI3K inhibition in MCC cells. Our data reveals a non-genomic resistance mechanism and opens therapeutic avenues to overcome ICI resistance, and proposes therapeutic strategies in manipulating cell state to augment efficacy and avert resistance.
Upon stringently excluding any confounding effect, single-cell transcriptomic profiling of tumor cells in treatment-naive patient tumors discloses phenotypic plasticity in a subset of MCC tumors, independent of MCPyV and likely mediated by non-genomic mechanisms. Consistent with previously reported association between EMT plasticity and immune-related gene expression in human cancers, MCC in the “mesenchymal-like” state is endowed with inflamed phenotype and captures several features that are predictive of ICI response.9,14 In the TME, interactions between tumor cells and immune cells are stronger in MCCT_G1 tumors, which is largely mediated by MIF-(CD74-CD44) and MIF-(CD74-CXCR4) signaling axes. The observation is further substantiated by findings in ICI-R tumors demonstrating a well-differentiated state with higher expression of neuroepithelial markers and “immune-cold” TME. While the “mesenchymal-like” state is linked to its inflammatory features in patient tumors, it is also retained in several primary MCC cell lines devoid of TME, suggesting that tumor intrinsic factors dictate transcriptomic plasticity. While ICIs are now the standard of care for advanced MCC, predictive biomarkers for this therapeutic class have remained elusive, without definitive evidence supporting MCPyV status, tumor mutation burden, and PD-L1 expression. Although similar correlation of the “mesenchymal-like” state with favorable ICI response has been described in two neuroendocrine tumors, SCLC and GBM, our observation will benefit from clinical validation in MCC patients.
Analyses of CD45+ cells in treatment-naive MCCs have shown that over 40% of CD3 T cells are DNTCs, in sharp contrast to other human cancers in which DNTCs account for less than 5% of CD3 T cells.36 Although most DNTCs are in naive and resident memory states, their precise role needs to be defined in MCC. When analyzed as an independent subset, ∼50% of CD4 and CD8 T cells are naive and resident T cells, with more abundant association with “mesenchymal-like” MCC. A recent study combining nanostring and immunohistochemistry in patient tumors suggests that increased CD27+ central memory CD8 T cells is predictive of favorable ICI response in MCC57; however, we found more abundant CD27+ CD8 T cells in ICI-R tumors (Figure S3F). In addition to an “immune-cold” landscape, TIGIT is a shared targetable immune checkpoint molecule in both MCCT_G2 and ICI-R MCC, providing the rationale for future clinical trial(s) of anti-TIGIT antibody alone or in combination with other ICIs as the first-line therapy or as the second line for patients who develop resistance to anti-PD1/PD-L1 therapies. Recently, tumor-associated B cells have been shown to induce melanoma cells with cancer stem cell-like activities.58 Moreover, a recent paper has demonstrated enrichment of cytotoxic T cells in TCGA (The Cancer Genome Atlas) mesenchymal tumors and activation of cytotoxic T cells by “mesenchymal-like” tumor cells in GBM and SCLC.9,14 Furthermore, tumor-associated macrophages induce the “mesenchymal-like” state in tumor cells, but it is not absolutely required, suggesting that cell-intrinsic factors are the main determinants of tumor cell states.14 The “mesenchymal-like” phenotype is also observed in patient-derived MCC cell lines, and we have shown that a subtle shift to “mesenchymal-like” state by epigenetic inhibition reverts resistance to PI3K inhibition in MKL-1 cells, warranting future studies to uncover potential therapeutic opportunities in MCC. The current study would also benefit from patient survival and treatment data to further confirm our results, which is hampered by the limited single-cell information in publicly available datasets. Additionally, a paucity of ICI-R MCC tumor samples bars definitive conclusion.
In summary, comprehensive scRNA-seq analysis of patient MCC tumors has revealed phenotypic plasticity at the transcriptomic level with distinct immune landscapes and therapeutic vulnerabilities. Experimentally, phenotypic plasticity is asserted in patient-derived MCC cell lines and regulated epigenetically. Clinically, phenotypic plasticity is indicative of favorable ICI response and prognosis. More broadly, recent studies in rare cancers (which in aggregate represent one-fourth of all cancer deaths)59 have yielded unique insights that can be generalized across human cancers. In a similar vein, MCC exemplifies cancers with great treatment challenges in the era of precision medicine, e.g., no targeted therapy and no treatment option after developing ICI resistance. Our data suggest clinical patient stratification to match baseline tumors to therapy and that manipulating tumor cell states can augment treatment efficacy and avert resistance in MCC. Finally, we present a non-genomic mechanism of ICI resistance to open therapeutic avenues for further exploitation.
Limitations of the study
We acknowledge that there are several limitations in the current study. First, a definitive conclusion cannot be drawn, as our observations that the “mesenchymal-like” state correlates with potentially favorable ICI treatment response are based on data from other cancer types. Our validation studies rely on scRNA-seq data from an appropriate GBM dataset that is available. Thus, our findings will benefit from clinical validation in MCC patients. Moreover, we have encountered one of the common challenges in rare cancer research: paucity of fresh tumor samples suitable for scRNA-seq. Our findings will be strengthened by ample patient tumor samples, especially well-defined primary or acquired ICI-resistant MCC. Recent technological breakthroughs with validated scRNA-seq assays in fixed tissues will greatly expand patient sample availability. Furthermore, definitive identification of TFs dictating cell states requires comprehensive functional characterization, which will be carried out in the future. Our observations may serve as a platform to invite future studies to characterize cellular plasticity in MCC, specific to therapeutic response and resistance.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti human CXCR4 | R&D Systems | Cat# MAB172; Clone: 44716; RRID: AB_2089399 |
| Mouse monoclonal anti neuron-specific enolase (NSE/ENO2) | Ventana Medical Systems | Cat#760-4786; Clone: MRQ-55; RRID: n/a |
| Mouse monoclonal anti vimentin | Ventana Medical Systems | Cat#790-2917; Clone: V9; RRID: n/a |
| Rabbit monoclonal anti histone H3 | Cell Signaling Technology | Cat#4499; Clone: D1H2; RRID: AB_10544537 |
| Rabbit monoclonal anti acetyl-histone H3 | Cell Signaling Technology | Cat#8173; Clone: D5E4; RRID: AB_10949503 |
| Mouse monoclonal anti α-tubulin | Millipore Sigma | Cat#T9026; Clone: DM1A; RRID: AB_477593 |
| Biological samples | ||
| Merkel cell carcinoma patient tumors | This paper (collected under relevant IRB protocols); Cooperative Human Tissue Network | https://www.chtn.org/ |
| Chemicals, peptides, and recombinant proteins | ||
| SYTOX Green | Invitrogen | Cat#S7020 |
| Domatinostat | Selleck Chemicals | Cat#S7555 |
| Copanlisib | Selleck Chemicals | Cat#S2802 |
| Critical commercial assays | ||
| DNeasy Blood & Tissue Kit | Qiagen | Cat#69506 |
| RNeasy Plus Kit | Qiagen | Cat#74134 |
| Chromium Single Cell Gene Expression Kits (v.3.1) | 10x Genomics | Cat#1000121 |
| Chromium Chip G kit | 10x Genomics | Cat#1000120 |
| RNA Nano Kit | Agilent | Cat#5067-1511 |
| High Sensitivity DNA Kit | Agilent | Cat#5067-4626 |
| Tumor Dissociation Kit, human | Miltenyi Biotec | Cat#130-095-929 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368814 |
| Cell Counting Kit - 8 | Millipore Sigma | Cat#96992 |
| Deposited data | ||
| Single-cell and bulk RNA-seq data | This paper | GEO: GSE226438, GSE223275, and GSE233454 |
| Human reference genome NCBI build 38, GRCh38 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/human |
| Glioblastoma RNA-seq dataset | Zhao et al.45 | BioProject PRJNA482620 |
| Merkel cell carcinoma RNA-seq dataset | Sundqvist et al.42 | BioProject PRJNA775071 |
| ICI-resistant MCC, scRNA dataset | Paulson et al.35 | GEO: GSE117988 |
| cisTarget (v10nr_clust) | Herrmann et al.60 | https://resources.aertslab.org/cistarget/ |
| gene order file, gencode_v21_gen_pos.complete.txt | Broad Institute repository | https://data.broadinstitute.org/Trinity/CTAT/cnv/ |
| Western blot images | This paper; Mendeley Data |
Figure 7; https://doi.org/10.17632/hb3gvgs4rr.1 |
| Experimental models: Cell lines | ||
| Patient derived MCC cell lines (MCC-2, MCC-3, MCC-5, MCC-8, MCC-9, MCC-16, MCC-21) | Generated in our lab | N/A |
| MKL-1 cell line | Gifted by Dr. Jurgen Becker (University Hospital Essen, Germany) | RRID: CVCL_2600 |
| Oligonucleotides | ||
| Primer for LT3 (MCPyV detection) Forward 5'-TTGTCTCGCCAGCATTGTAG-3' Reverse 5'-ATATAGGGGCCTCGTCAACC-3' |
Feng et al.4 | N/A |
| Primer for LT1 (MCPyV detection) Forward 5'-TACAAGCACTCCACCAAAGC-3' Reverse 5'-TCCAATTACAGCTGGCCTCT-3' |
Feng et al.4 | N/A |
| qPCR primer and probe, LT3 (MCPyV detection) Forward 5'-TCGCCAGCATTGTAGTCTAAAAAC-3' Reverse 5'-CCAAACCAAAGAATAAAGCACTGA-3' Probe FAM-AGCAAAAACACTCTCCCCACGT CAGACAG-BHQ |
Rodig et al.61 | N/A |
| qPCR primer and probe, Set9 (MCPyV detection) Forward 5'-TTAGCTGTAAGTTGTCTCGCC-3' Reverse 5'-CACCAGTCAAAACTTTCCCAAG-3' Probe FAM-AAACACTCTCCCCACGTCAGA CAG-BHQ |
Rodig et al.61 | N/A |
| qPCR primer and probe, SmallT (MCPyV detection) Forward 5'-GCAAAAAAACTGTCTGACGTGG-3' Reverse 5'-CCACCAGTCAAAACTTTCCCA-3' Probe FAM-TATCAGTGCTTTATTCTTTGGTT TGGATTTCCTCCT-BHQ |
Bhatia et al.62 | N/A |
| See Table S7 for full list of Taqman qPCR primers | Life Technologies | #4331182 |
| Software and algorithms | ||
| Graph Pad Prism v9 | Graphpad | www.graphpad.com |
| CellRanger software v6.1.2 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| FACSDiva v8.0.1 | BD Biosciences | https://www.bdbiosciences.com/en-us/products/software/instrument-software/bd-facsdiva-software |
| R v4.1.1 | R-project | https://www.r-project.org/ |
| Seurat v.4.2 – v4.3 | Satija et al.20 | https://satijalab.org/seurat/articles/install.html |
| Rsubread v2.8.2 | Liao et al.63 | https://bioconductor.org/packages/release/bioc/html/Rsubread.html |
| DESseq2 v1.34.0 | Love et al.64 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| SingleR v1.8.1 | Aran et al.21 | https://bioconductor.org/packages/release/bioc/html/SingleR.html |
| CellChat v1.5.0 | Jin et al.37 | https://github.com/sqjin/CellChat |
| SCENIC | Aibar et al.32 | https://scenic.aertslab.org/ |
| Gene set variation analysis (GSVA) 1.42.0 | Hänzelmann et al.65 | https://bioconductor.org/packages/devel/bioc/vignettes/GSVA/inst/doc/GSVA.html |
Resource availability
Lead contact
Further information and resource requests should be directed to and will be fulfilled by the lead contact, Dr Ling Gao (ling.gao@va.gov).
Materials availability
This study did not generate unique materials.
Experimental model and subject details
Patient tumor derived MCC cell lines
Patient tumor derived MCC cell lines (coded as MCC-2, MCC-3, MCC-5, MCC-8, MCC-9, MCC-16 and MCC-21) were established in our lab under a study protocol (#131586) first approved by the Institutional Review Board (IRB) at the University of Arkansas for Medical Sciences (UAMS) and currently maintained under an IRB-approved protocol (#1619070) at the Veterans Affairs Long Beach Healthcare System (VALBHS). MCC-2, -3, -8, and -9 originated from male patients and MCC-5, -16, and -21 originated from female patients; tumor tissues were collected after appropriate consent was obtained by the study teams at UAMS and the Cooperative Human Tissue Network (CHTN, a prospective collection service supported by the National Cancer Institute, which provides de-identified human biospecimens collected from routine procedures under protocols that are approved by local IRBs). MKL-1, a MCPyV-positive cell line from a male patient, was a kind gift from Dr. Jurgen Becker (University Hospital Essen, Germany). MCC cell lines were authenticated by STR-profiling performed by Genetica (Laboratory Corporation of America; Burlington, NC). Each MCC cell line was compared against its respective primary MCC tumor; MKL-1 cell line was compared against initial passage and confirmed in the associated STR profile in Cellosaurus (accession number CVCL_2600). Suspension cultures of primary human MCC cell lines used in this study were maintained in RPMI-1640 medium (#30-2001, American Type Culture Collection) supplemented with 10% fetal bovine serum, penicillin-streptomycin (100 U/mL) and L-glutamine (4mM) at 37°C in a humidified atmosphere with 5% CO2. Cells were fed with fresh complete media every 48h and split to maintain logarithmic growth.
Human tumor tissues
The eleven MCC patient tumors were obtained through CHTN and under study protocols approved by the VALBHS IRB (protocol #1619070 and #1654375), in accordance with local and federal regulations for human subject research. Tissues were collected after informed consent was obtained by CHTN and our study teams, respectively; basic information is included in Table S1.
Method details
DNA extraction
Genomic DNA was extracted using the DNeasy Blood & Tissue Kit (#69506, Qiagen) per manufacturer’s instructions. The MCPyV status of each cell line was examined by PCR and qRT-PCR as described in other studies,4,61,62 using primers listed in the key resources table. PCR was carried out using iCycler Thermal Cycler (Bio-Rad) under the following conditions for LT3 and LT1: 94°C for 5min, followed by 35 cycles of 94°C for 30s, 53.5°C for 30s, 68°C for 30s, then final 68°C for 10min, followed by electrophoresis on 1.5% agarose gel and visualized under ultraviolet light. Quantitative PCR was carried out using CFX96 Touch Real-Time PCR Detection System (Bio-Rad) with an initial denaturation at 95°C for 10min, followed by 40 cycles of 95°C for 15s and 60°C for 1min.
scRNA library preparation and sequencing
Resected patient tumors were dissociated using a Human Tumor Dissociation kit (#130-095-929, Miltenyi Biotec) with the modified protocol reported by Sade-Feldman et al.,66 followed by red blood cell lysis, then washed and passed through 70μm sterile cell strainer to obtain single-cell suspensions. Dead cells were excluded by SYTOX staining (#S7020, Invitrogen) and live cells were collected by the FACSAria Fusion flow cytometer (BD Biosciences) and BD FACSDiva v8.0.1 software at the UCI Institute for Immunology flow cytometry core facility. Sorted cells were washed with PBS containing 0.04% BSA and resuspended at a final density of ∼1,000 cells/μL. Libraries were created targeting ∼10,000 cells/sample for capture using Chromium Single Cell 3’ Reagent Kits and Chromium Chip G kits (#1000121 and #1000120, 10X Genomics) following the CG00052 Rev B. user guide. Each library was subsequently sequenced on the Illumina NovaSeq 6000 platform, targeting an average of ∼50,000 reads/cell.
scRNA-seq data processing
FASTQ files were aligned to GRCh38 human reference genome using 10x Genomics Cell Ranger Count v6.1.2. Cell Ranger Aggr function was used to normalize the number of average mapped reads per cell across the libraries. Cell Ranger output files were read into R version 4.1.1 and processed using Seurat.20 Data was filtered for cells containing a minimum of 1,000 and no more than 10,000 unique genes and containing less than 20% mitochondrial genome. Additionally, genes detected in fewer than 4 cells were also removed from further analysis. For the ‘treatment-naive’ dataset, the 9 sample libraries were anchored and integrated using the top 2,000 variable features identified using the vst method. The integrated object was scaled, and principal component analysis (PCA) was performed. Thereafter, the first 15 PCs were used for Unifold Manifold Approximation and Projection (UMAP) non-linear dimensionality reduction followed by calculating the k-nearest neighbor graph and Louvain clustering at varying resolution (0.1-1.0). The marker genes per cluster were identified using the FindAllMarkers() function based on the Wilcoxon Rank Sum test. The DoHeatmap() function was used to generate heatmaps for the top variable genes based on scaled expression values. Marker gene expression and SingleR analysis21 were used to annotate cell-type specific clusters, and clusters with concurrent annotations were identified and merged to generate the cell-type annotated object. The ‘treatment-naive and ICI-R’ dataset was prepared in a similar manner integrating 11 samples (9 treatment-naive and 2 ICI-R tumors), followed by dimensionality reduction and cluster annotation using the first 19 PCs. Malignant cells were extracted to create a separate tumor object and clustered further on its ‘integrated’ assay with 15 PCs and resolution = 0.1. FeaturePlot() and Vlnplot() functions were used for visualizing feature expression in low-dimensional space. CD45+ immune cells, CD3+ T cells, CD4+/CD8+ T cells, CD19+/CD20+ B cells were identified based on marker gene expression and SingleR annotation in their corresponding parent objects and analyzed separately in a similar manner. Pseudo-bulk transcriptome for each sample was generated by summing the UMI (unique molecular identifier) counts across cells as recommended for DESeq2 (https://hbctraining.github.io/scRNA-seq_online/lessons/pseudobulk_DESeq2_scrnaseq.html). Finally, scRNAseq data from an MCC patient with MCPyV associated Merkel cell carcinoma, reported by Paulson et al.,35 was downloaded from GEO (GSE117988). After initial data processing and cell type annotation, the resulting Seurat object was merged with our MCC patient tumor dataset and integrated to remove batch effect. The combined dataset was scaled followed by PCA and clustering, and expression of marker genes were then retrieved and visualized.
mRNA-seq of MCC cell lines and validation datasets
Total RNA from eight MCC cell lines, viz. MCC-2, MCC-3, MCC-5, MCC-8, MCC-9, MCC-16, MCC-21 and MKL-1 was extracted using Qiagen RNeasy kit (#74106). RNA quality was evaluated (Agilent Bioanalyzer Nano RNA chip) and libraries were constructed based on Illumina TruSeq stranded mRNA protocol. Libraries were normalized, multiplexed, and sequenced using paired-end 100-cycle chemistry for the Illumina HiSeq 4000. Library construction was performed according to the Illumina TruSeq mRNA stranded protocol. mRNA was enriched using oligo dT magnetic beads with the recommended amount of total RNA, and later fragmented chemically. First strand complementary DNA (cDNA) synthesis was performed using random primers and reverse transcriptase. Post second strand synthesis, the double-stranded cDNA was cleaned using AMPure XP beads, subjected to end repair, and finally 3’ adenylated. Illumina barcoded adapters were ligated on to the ends, and the adapter ligated fragments were enriched by nine cycles of PCR. The resulting libraries were validated by qPCR on the QuantStudio7 Flex system (Applied Biosystems) and sized by Agilent Bioanalyzer DNA high sensitivity chip on the Agilent 2100 Bioanalyzer system. The concentrations for the libraries were normalized and then multiplexed together and sequenced using paired-end 100 cycles chemistry for the HiSeq 4000. The resulting reads from each cell line were then aligned and annotated to the human genome (GRCh38) using the align function in Rsubread package.63 The count-matrices from the corresponding bam files were retrieved using the featureCounts function.63 Paired-end sequencing for untreated MCC-2 and untreated MCC-8 cells, as well as MKL-1 cells treated with either DMSO or 10μM domatinostat (24h), was performed using Zymo Research sequencing services (Irvine, CA), and count-matrices were generated from the provided bam files as described earlier. DESeq2 package,64 which uses a negative binomial distribution model to test differential expression, was then used to carry out further analyses. Differential gene expression (DGE) analysis was performed on raw counts, using the DESeq function, setting appropriate reference groups. Log2FoldChange (LFC) shrinkage was carried out using the apeglm method in the lfcShrink() function.67 Distance measurements were computed on log normalized (log2(n+1)) counts using the dist() function and later plotted as a heatmap using pheatmap(). The calculated sample distances were used to perform hierarchical clustering across rows and columns.
For data validation, bulk RNAseq datasets of 102 MCC tumors42 and 17 glioblastoma multiforme45 were downloaded from Sequence Read Archive (SRA), NIH. Raw FASTQ files were processed to generate count matrices as described above. ICI response information for each GBM sample was retrieved from the SRA metadata file and added as coldata prior to analysis. In absence of individual survival and death information for the 102 MCC patient tumor dataset, patients were stratified based on the enrichment score of survival-associated gene signature (SAG) reported by Sundqvist et al.,42 as SAGhigh (third quartile), SAGlow (first quartile) and SAGmid (interquartile). The classification information was then added as coldata prior to analysis.
Differential gene expression (DGE) and GSEA
DGE analysis was calculated on the integrated object using Seurat’s FindMarkers() function on the ‘RNA’ assay with Wilcoxon Rank Sum test. The pheatmap() or EnhancedVolcano() functions were used for visualization. DGE list with log2 transformed scores were retrieved from Seurat using the FindMarker() function or the DESeq() from the mRNA-seq datasets and filtered with p<0.05, and ranked based on log2Fc score. GSEA was performed on a ‘pre-ranked’ list using web-based gene set analysis toolkit, WebGestalt (http://webgestalt.org/), selecting the appropriate database.
Generating gene signatures
We first curated an EMT signature gene list including 456 genes based on studies across human cancers (Table S6),22,23 which was then narrowed down to a candidate gene list of 293 mesenchymal genes (Table S6). Genes were trimmed with avg.exp > 0 and count > 0, distributed across Q3 and Q4 quartile of average expression, and were expressed across all cell types without compromising the retention of established lineage markers and pct.exp ≥ 50%. The resulting 45 genes were further trimmed down to 19, based on high expression in MCC tumor cells only (Table S2).
To generate an MCC specific gene signature that could distinguish inflamed and ‘immune-cold’ MCC tumors, and is predictive of ICI response, we performed pre-ranked GSEA analysis on differentially expressed genes between MCCT_G1 and MCCT_G2 tumor samples. To ensure signature applicability to both single and bulk RNA-seq datasets, the entire tumor microenvironment (containing tumor cells as well as other cell types) was used to generate the differential gene list. Genes involved in the top positively enriched pathways were tabulated and then trimmed based on the assigned normalized enrichment score (NES ≥ 1.5). A non-redundant gene list of 14 genes (SIG-14, Table S2) was thereafter obtained upon merging.
‘Cell-cell’ communication analyses
‘Cell-cell’ communication networks were inferred based on transcript abundance of ligand-receptor pairs with CellChat (version 1.5.0).37 To model these networks, an expression matrix containing SingleR annotated to normalized MCC tumor samples with the selected cell types were inputted into CellChat using the human database for ligand-receptor pairs. Over-expressed genes and ligand-receptor pairs were identified by running identifyOverExpressedGenes() and identifyOverExpressedInteractions() with default settings. The communication probabilities at pair and pathway levels were calculated by running computeCommunProb() and computeCommunProbPathway() functions under default parameters (threshold = 0.05). ‘Cell-cell’ communications with a minimum of 10 cells/group were filtered out and later aggregated using the aggregateNet() function with default parameters. netVisual_bubble(), netVisual_circle() and netVisual_chord() functions were thereafter used for visualization, setting the sources.use or targets.use parameters to the desired cell type.
Gene signature scoring
For scRNA-seq datasets, each signature score was calculated by generating the total gene expression for each gene and separating them into 25 bins of similar expression. For every gene from a signature, 100 ‘control’ genes were selected from its corresponding bin and added as a ‘control’ signature. The expression of the genes in the ‘target’ and ‘control’ was averaged across each cell to generate a ‘target’ and ‘control’ score. A cell’s score for the ‘target’ signature was then calculated as the difference between the ‘target’ and ‘control’ score. An unpaired Wilcox test at 95% confidence interval was used to determine statistical significance. Gene signature enrichment in bulk RNA-seq samples was calculated using GSVA package,65 which employs a non-parametric method for single-sample gene set enrichment. Normalized count matrices from the validation datasets were used alongside the desired gene signature to calculate the GSVA enrichment score.
Master regulator analysis
To generate an MCC specific gene regulatory network, the SCENIC pipeline was implemented in Python.32 A gene expression matrix containing the raw counts of MCC tumor cells was used in conjunction with a complete list of human TFs and motif annotation information (downloaded from https://resources.aertslab.org/cistarget/) to infer the gene regulatory network and co-expression modules. Cellular enrichment analysis was performed using AUCell to identify cells with active gene-networks in our scRNA-seq dataset. Cell metadata, defining each MCC subtype, was thereafter used to probe for subtype-specific regulon enrichment.
Inferred copy number variation (CNV) analysis
The count matrix for inferred CNV analysis was generated by extracting the ‘count’ slot of Seurat object using the GetAssayData() function. Cells were annotated as tumor or CD3+ T cells as reference cells. Gene order file ‘gencode_v21_gen_pos.complete.txt’ was downloaded from Broad Institute repository (https://data.broadinstitute.org/Trinity/CTAT/cnv/). CNV was estimated using inferCNV by aligning genes to their chromosomal location and applying a moving average to the relative expression, with a sliding window of 100 genes as previously described and analysis mode kept to default.24 Heatmaps for the inferred CNVs were generated using the plot_cnv() with hclust set to agglomerative hierarchical clustering method ‘ward.D’.
Immunohistochemistry
Consecutive five-micrometer-thick tissue slides were prepared from formalin-fixed paraffin-embedded (FFPE) MCC patient samples for immunohistochemistry (IHC). IHC staining for CXCR4 started with deparaffinization followed by antigen retrieval in heated 1X citrate buffer (#H3300, Vector Laboratories) for 20min, blocking in normal goat serum (#S1012, Vector Laboratories), and overnight incubation with mouse monoclonal anti-human CXCR4 primary antibody (1:100, Clone 44716, #MAB172, R&D Systems). Slides were incubated with goat anti-mouse secondary antibody (#ab6789, Abcam) for 1 hour at room temperature and detected using a horseradish peroxidase detection system with DAB as chromogen (#SK4105, Vector Laboratories). IHC staining for ENO2 and vimentin was performed with the BenchMark ULTRA IHC/ISH system (Roche Diagnostics) using standard automated protocols. The primary antibodies ENO2 (pre-diluted, Clone MRQ-55, #760-4786, Ventana Medical Systems) and vimentin (pre-diluted, V9, 790-2917, Ventana Medical Systems) were incubated for 24minand 16min, respectively, and detected using a horseradish peroxidase detection (HRP) system with DAB as chromogen. Slides were then visualized, and images were captured using a stereomicroscope with a digital camera (Discovery V12 and AxioCam; Carl Zeiss, Inc).
Quantitative real-time PCR
Total RNAs were purified using RNeasy Plus kit (#74134, Qiagen) according to the manufacturer’s protocol. The cDNAs were synthesized from 0.5 to 1μg of total RNA using the Applied Biosystems High Capacity cDNA Reverse Transcription Kit (#4368814, Applied Biosystems) according to manufacturer’s instructions. Gene expression was examined using TaqMan Gene Expression Assay primers (referred in key resources and listed in Table S7) and Taqman Gene Expression MasterMix (#4369510, Applied Biosystems) on the CFX96 Touch Real-Time PCR Detection System (Bio-Rad), with an initial denaturation at 95°C for 10min, followed by 40 cycles of 95°C for 15s and 60°C for 1min. Samples were run in triplicate and relative mRNA expression was calculated by normalizing samples against the mitochondrial gene MRPS2 using the ΔCt method.
Western blot analysis
One million MCC-16 or MKL-1 cells were plated in 3ml of medium/well in 6-well culture plates, respectively. After incubation at 37°C and 5% CO2 for 2 hours, cells were treated with DMSO (#2650, Millipore Sigma) or domatinostat (#S7555, Selleck Chemicals) in dose range 0.3-30μM for 24 hours. Cells were washed twice with ice-cold PBS and lysed in 1× radioimmunoprecipitation assay (RIPA) buffer (#R-0278, Millipore Sigma) containing cOmplete Mini EDTA-free protease inhibitor cocktail (#04693159001, Roche, obtained from Millipore Sigma). After incubation on ice for 30min, cell lysates were clarified by centrifugation at 14,000 rpm for 15min at 4°C. 25 μg of total proteins were resolved by 12% SDS-PAGE gels and transferred electrophoretically onto polyvinylidene difluoride membrane (#IPVH00010, Millipore Sigma) using a semi-dry blotting system (Bio-Rad). Membranes were blocked in 5% fat-free milk/Tris-buffered saline containing 0.1% Tween 20 (TBS-T) for 1 hour and incubated with primary antibodies (histone H3: Clone D1H2, #4499, Cell Signaling Technology; acetyl-histone H3: Clone D5E4, #8173, Cell Signaling Technology; α-tubulin: Clone DM1A, #T9026, Millipore Sigma) at 4°C overnight. Membranes were then washed three times with TBS-T and incubated with HRP-conjugated secondary antibodies (Cell Signaling Technology) for 1 hour at room temperature. After rinsing three times with TBS-T, membranes were incubated with enhanced chemiluminescent detection reagents (#WBKLS0100, Millipore Sigma) for 5min. The proteins were visualized by exposing membrane to X-Ray film (#BX57, Midwest Scientific).
Cell viability assay
Cell viabilities were measured by Cell Counting Kit-8 (CCK-8) (#96992, Millipore Sigma) following manufacturer’s protocol. In brief, cells were plated at 1×105 cells/well in 96-well plates and incubated at 37°C and 5% CO2 for 4 hours before treatment with serial concentrations of copanlisib (#S2802, Selleck Chemicals), domatinostat (#S7555, Selleck Chemicals), or in combination for 72 hours. CCK-8 reagent (10% of well volume) was added to each well and incubated for 2-4hours at 37°C before measuring optical density (OD) at 450nm using a spectrophotometer. Half-maximal growth inhibitory dose (GI 50) was calculated by plotting dose-response curve, normalized against vehicle-treated cells, using GraphPad Prism.
Quantification and statistical analysis
Data are presented as the mean ± standard error of the means (SEM) or standard deviation. In addition to the analytical packages described above, GraphPad Prism 9.0 was used for statistical analysis. The significance threshold was set to FDR = 0.05 unless otherwise stated.
Acknowledgments
This work was made possible only through the resources and support provided by the generous donation of Mitchell and Joan Markow of the Two Sisters Foundation, Southern California Institute for Research and Education, Cooperative Human Tissue Network (CHTN), UC-Irvine Chao Family Comprehensive Cancer Center (CFCCC), VA Long Beach Pathology Laboratory, and UC-Irvine Department of Dermatology. We also wish to acknowledge support from the CFCCC’s shared research resources, Institute for Immunology Flow Core and the Genomics Research and Technology Hub, supported by the National Cancer Institute of the National Institutes of Health under award number P30CA062203, and NIH shared instrumentation grants 1S10RR025496-01, 1S10OD010794-01, and 1S10OD021718-01. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was supported by grants from the U.S. Department of Veterans Affairs (Biomedical Laboratory Research & Development Merit Review Award I01BX004841 to L.G.), the National Institutes of Health (National Cancer Institute R01CA266514-01 to L.G.), the Chao Family Comprehensive Cancer Center (Anti-Cancer Challenge Pilot Project award to L.G.), and the American Cancer Society (Research Scholar grant RSG-17-069-01 to L.G.).
Author contributions
Conceptualization, B.K.D., H.Z., and L.G.; methodology, B.K.D., A.K., G.J.V., M.D.K., N.L., Q.N., H.Z., J.W., and L.G.; investigation, B.K.D., A.K., G.J.V., M.D.K., N.L., Q.N., H.Z., J.W., and L.G.; visualization, B.K.D., A.K., M.D.K., N.L., Q.N., H.Z., J.W., and L.G.; funding acquisition, L.G.; project administration, H.Z. and L.G.; supervision, L.G.; writing – original draft, B.K.D., A.K., H.Z., and L.G.; writing – review & editing, B.K.D., A.K., M.D.K., Q.N., H.Z., J.W., and L.G.
Declaration of interests
The authors declare no competing interests.
Published: July 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101101.
Supplemental information
Data and code availability
-
•
Single-cell RNA-seq and bulk RNA-seq data have been deposited at GEO and are publicly available under accession numbers GEO: GSE226438, GEO: GSE223275, and GEO: GSE233454. Original western blot images have been deposited at Mendeley Data (https://doi.org/10.17632/hb3gvgs4rr.1) and are publicly available as of the date of publication.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in the paper is available from the lead contact upon request.
References
- 1.Becker J.C., Stang A., DeCaprio J.A., Cerroni L., Lebbé C., Veness M., Nghiem P. Merkel cell carcinoma. Nat. Rev. Dis. Primers. 2017;3 doi: 10.1038/nrdp.2017.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harms P.W., Harms K.L., Moore P.S., DeCaprio J.A., Nghiem P., Wong M.K.K., Brownell I., International Workshop on Merkel Cell Carcinoma Research IWMCC Working Group The biology and treatment of Merkel cell carcinoma: current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018;15:763–776. doi: 10.1038/s41571-018-0103-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.D'Angelo S.P., Lebbé C., Mortier L., Brohl A.S., Fazio N., Grob J.J., Prinzi N., Hanna G.J., Hassel J.C., Kiecker F., et al. First-line avelumab in a cohort of 116 patients with metastatic Merkel cell carcinoma (JAVELIN Merkel 200): primary and biomarker analyses of a phase II study. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2021-002646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng H., Shuda M., Chang Y., Moore P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knepper T.C., Montesion M., Russell J.S., Sokol E.S., Frampton G.M., Miller V.A., Albacker L.A., McLeod H.L., Eroglu Z., Khushalani N.I., et al. The Genomic Landscape of Merkel Cell Carcinoma and Clinicogenomic Biomarkers of Response to Immune Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2019;25:5961–5971. doi: 10.1158/1078-0432.Ccr-18-4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeCaprio J.A. Molecular Pathogenesis of Merkel Cell Carcinoma. Annu. Rev. Pathol. 2021;16:69–91. doi: 10.1146/annurev-pathmechdis-012419-032817. [DOI] [PubMed] [Google Scholar]
- 7.Knepper T.C., Panchaud R.A., Muradova E., Cohen L., DeCaprio J.A., Khushalani N.I., Tsai K.Y., Brohl A.S. An analysis of the use of targeted therapies in patients with advanced Merkel cell carcinoma and an evaluation of genomic correlates of response. Cancer Med. 2021;10:5889–5896. doi: 10.1002/cam4.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gay C.M., Stewart C.A., Park E.M., Diao L., Groves S.M., Heeke S., Nabet B.Y., Fujimoto J., Solis L.M., Lu W., et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell. 2021;39:346–360.e7. doi: 10.1016/j.ccell.2020.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stewart C.A., Gay C.M., Xi Y., Sivajothi S., Sivakamasundari V., Fujimoto J., Bolisetty M., Hartsfield P.M., Balasubramaniyan V., Chalishazar M.D., et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat. Cancer. 2020;1:423–436. doi: 10.1038/s43018-019-0020-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen S., Zhu G., Yang Y., Wang F., Xiao Y.T., Zhang N., Bian X., Zhu Y., Yu Y., Liu F., et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat. Cell Biol. 2021;23:87–98. doi: 10.1038/s41556-020-00613-6. [DOI] [PubMed] [Google Scholar]
- 12.Zheng L., Qin S., Si W., Wang A., Xing B., Gao R., Ren X., Wang L., Wu X., Zhang J., et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science. 2021;374 doi: 10.1126/science.abe6474. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y., Vu T., Palmer D.C., Kishton R.J., Gong L., Huang J., Nguyen T., Chen Z., Smith C., Livák F., et al. A T cell resilience model associated with response to immunotherapy in multiple tumor types. Nat. Med. 2022;28:1421–1431. doi: 10.1038/s41591-022-01799-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hara T., Chanoch-Myers R., Mathewson N.D., Myskiw C., Atta L., Bussema L., Eichhorn S.W., Greenwald A.C., Kinker G.S., Rodman C., et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell. 2021;39:779–792.e11. doi: 10.1016/j.ccell.2021.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan J.M., Zaidi S., Love J.R., Zhao J.L., Setty M., Wadosky K.M., Gopalan A., Choo Z.N., Persad S., Choi J., et al. Lineage plasticity in prostate cancer depends on JAK/STAT inflammatory signaling. Science. 2022;377:1180–1191. doi: 10.1126/science.abn0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng S., Wang C., Wang Y., Xu Y., Li X., Johnson N.A., Mukherji A., Lo U.G., Xu L., Gonzalez J., et al. Ectopic JAK-STAT activation enables the transition to a stem-like and multilineage state conferring AR-targeted therapy resistance. Nat. Cancer. 2022;3:1071–1087. doi: 10.1038/s43018-022-00431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh M.K., Altameemi S., Lares M., Newton M.A., Setaluri V. Role of dual specificity phosphatases (DUSPs) in melanoma cellular plasticity and drug resistance. Sci. Rep. 2022;12 doi: 10.1038/s41598-022-18578-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G., et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R., et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tyler M., Tirosh I. Decoupling epithelial-mesenchymal transitions from stromal profiles by integrative expression analysis. Nat. Commun. 2021;12:2592. doi: 10.1038/s41467-021-22800-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joanito I., Wirapati P., Zhao N., Nawaz Z., Yeo G., Lee F., Eng C.L.P., Macalinao D.C., Kahraman M., Srinivasan H., et al. Single-cell and bulk transcriptome sequencing identifies two epithelial tumor cell states and refines the consensus molecular classification of colorectal cancer. Nat. Genet. 2022;54:963–975. doi: 10.1038/s41588-022-01100-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L., et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tirosh I., Venteicher A.S., Hebert C., Escalante L.E., Patel A.P., Yizhak K., Fisher J.M., Rodman C., Mount C., Filbin M.G., et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539:309–313. doi: 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Puram S.V., Tirosh I., Parikh A.S., Patel A.P., Yizhak K., Gillespie S., Rodman C., Luo C.L., Mroz E.A., Emerick K.S., et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017;171:1611–1624.e24. doi: 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paulson K.G., Lemos B.D., Feng B., Jaimes N., Peñas P.F., Bi X., Maher E., Cohen L., Leonard J.H., Granter S.R., et al. Array-CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L-Myc. J. Invest. Dermatol. 2009;129:1547–1555. doi: 10.1038/jid.2008.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Gele M., Speleman F., Vandesompele J., Van Roy N., Leonard J.H. Characteristic pattern of chromosomal gains and losses in Merkel cell carcinoma detected by comparative genomic hybridization. Cancer Res. 1998;58:1503–1508. [PubMed] [Google Scholar]
- 29.Marjanovic N.D., Hofree M., Chan J.E., Canner D., Wu K., Trakala M., Hartmann G.G., Smith O.C., Kim J.Y., Evans K.V., et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell. 2020;38:229–246.e13. doi: 10.1016/j.ccell.2020.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shendy N.A.M., Zimmerman M.W., Abraham B.J., Durbin A.D. Intrinsic transcriptional heterogeneity in neuroblastoma guides mechanistic and therapeutic insights. Cell Rep. Med. 2022;3 doi: 10.1016/j.xcrm.2022.100632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van de Sande B., Flerin C., Davie K., De Waegeneer M., Hulselmans G., Aibar S., Seurinck R., Saelens W., Cannoodt R., Rouchon Q., et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020;15:2247–2276. doi: 10.1038/s41596-020-0336-2. [DOI] [PubMed] [Google Scholar]
- 32.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu W., Huang C., Wang Q., Huang T., Ding Y., Ma C., Ma H., Chen W. MEF2 transcription factors promotes EMT and invasiveness of hepatocellular carcinoma through TGF-β1 autoregulation circuitry. Tumour Biol. 2014;35:10943–10951. doi: 10.1007/s13277-014-2403-1. [DOI] [PubMed] [Google Scholar]
- 34.Yao W., Liu Y., Zhang Z., Li G., Xu X., Zou K., Xu Y., Zou L. ALX1 promotes migration and invasion of lung cancer cells through increasing snail expression. Int. J. Clin. Exp. Pathol. 2015;8:12129–12139. [PMC free article] [PubMed] [Google Scholar]
- 35.Paulson K.G., Voillet V., McAfee M.S., Hunter D.S., Wagener F.D., Perdicchio M., Valente W.J., Koelle S.J., Church C.D., Vandeven N., et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat. Commun. 2018;9:3868. doi: 10.1038/s41467-018-06300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu Z., Zheng Y., Sheng J., Han Y., Yang Y., Pan H., Yao J. CD3(+)CD4(-)CD8(-) (Double-Negative) T Cells in Inflammation, Immune Disorders and Cancer. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.816005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin S., Guerrero-Juarez C.F., Zhang L., Chang I., Ramos R., Kuan C.H., Myung P., Plikus M.V., Nie Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021;12:1088. doi: 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo X., Pan Y., Xiong M., Sanapala S., Anastasaki C., Cobb O., Dahiya S., Gutmann D.H. Midkine activation of CD8(+) T cells establishes a neuron-immune-cancer axis responsible for low-grade glioma growth. Nat. Commun. 2020;11:2177. doi: 10.1038/s41467-020-15770-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leng L., Metz C.N., Fang Y., Xu J., Donnelly S., Baugh J., Delohery T., Chen Y., Mitchell R.A., Bucala R. MIF signal transduction initiated by binding to CD74. J. Exp. Med. 2003;197:1467–1476. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartz V., Lue H., Kraemer S., Korbiel J., Krohn R., Ohl K., Bucala R., Weber C., Bernhagen J. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 2009;583:2749–2757. doi: 10.1016/j.febslet.2009.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ayers M., Lunceford J., Nebozhyn M., Murphy E., Loboda A., Kaufman D.R., Albright A., Cheng J.D., Kang S.P., Shankaran V., et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017;127:2930–2940. doi: 10.1172/jci91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sundqvist B., Kilpinen S., Böhling T., Koljonen V., Sihto H. Activation of Oncogenic and Immune-Response Pathways Is Linked to Disease-Specific Survival in Merkel Cell Carcinoma. Cancers. 2022;14 doi: 10.3390/cancers14153591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan T.Z., Miow Q.H., Miki Y., Noda T., Mori S., Huang R.Y.J., Thiery J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014;6:1279–1293. doi: 10.15252/emmm.201404208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kovács S.A., Győrffy B. Transcriptomic datasets of cancer patients treated with immune-checkpoint inhibitors: a systematic review. J. Transl. Med. 2022;20:249. doi: 10.1186/s12967-022-03409-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao J., Chen A.X., Gartrell R.D., Silverman A.M., Aparicio L., Chu T., Bordbar D., Shan D., Samanamud J., Mahajan A., et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019;25:462–469. doi: 10.1038/s41591-019-0349-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kinker G.S., Greenwald A.C., Tal R., Orlova Z., Cuoco M.S., McFarland J.M., Warren A., Rodman C., Roth J.A., Bender S.A., et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat. Genet. 2020;52:1208–1218. doi: 10.1038/s41588-020-00726-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fang B., Kannan A., Zhao S., Nguyen Q.H., Ejadi S., Yamamoto M., Camilo Barreto J., Zhao H., Gao L. Inhibition of PI3K by copanlisib exerts potent antitumor effects on Merkel cell carcinoma cell lines and mouse xenografts. Sci. Rep. 2020;10:8867. doi: 10.1038/s41598-020-65637-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shao Q., Kannan A., Lin Z., Stack B.C., Jr., Suen J.Y., Gao L. BET protein inhibitor JQ1 attenuates Myc-amplified MCC tumor growth in vivo. Cancer Res. 2014;74:7090–7102. doi: 10.1158/0008-5472.Can-14-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gravemeyer J., Lange A., Ritter C., Spassova I., Song L., Picard D., Remke M., Horny K., Sriram A., Gambichler T., et al. Classical and Variant Merkel Cell Carcinoma Cell Lines Display Different Degrees of Neuroendocrine Differentiation and Epithelial-Mesenchymal Transition. J. Invest. Dermatol. 2021;141:1675–1686.e4. doi: 10.1016/j.jid.2021.01.012. [DOI] [PubMed] [Google Scholar]
- 50.Skrypek N., Goossens S., De Smedt E., Vandamme N., Berx G. Epithelial-to-Mesenchymal Transition: Epigenetic Reprogramming Driving Cellular Plasticity. Trends Genet. 2017;33:943–959. doi: 10.1016/j.tig.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 51.Song L., Bretz A.C., Gravemeyer J., Spassova I., Muminova S., Gambichler T., Sriram A., Ferrone S., Becker J.C. The HDAC Inhibitor Domatinostat Promotes Cell-Cycle Arrest, Induces Apoptosis, and Increases Immunogenicity of Merkel Cell Carcinoma Cells. J. Invest. Dermatol. 2021;141:903–912.e4. doi: 10.1016/j.jid.2020.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nardi V., Song Y., Santamaria-Barria J.A., Cosper A.K., Lam Q., Faber A.C., Boland G.M., Yeap B.Y., Bergethon K., Scialabba V.L., et al. Activation of PI3K signaling in Merkel cell carcinoma. Clin. Cancer Res. 2012;18:1227–1236. doi: 10.1158/1078-0432.Ccr-11-2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iwasaki T., Matsushita M., Nonaka D., Kuwamoto S., Kato M., Murakami I., Nagata K., Nakajima H., Sano S., Hayashi K. Comparison of Akt/mTOR/4E-BP1 pathway signal activation and mutations of PIK3CA in Merkel cell polyomavirus-positive and Merkel cell polyomavirus-negative carcinomas. Hum. Pathol. 2015;46:210–216. doi: 10.1016/j.humpath.2014.07.025. [DOI] [PubMed] [Google Scholar]
- 54.Shiver M.B., Mahmoud F., Gao L. Response to Idelalisib in a Patient with Stage IV Merkel-Cell Carcinoma. N. Engl. J. Med. 2015;373:1580–1582. doi: 10.1056/NEJMc1507446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neftel C., Laffy J., Filbin M.G., Hara T., Shore M.E., Rahme G.J., Richman A.R., Silverbush D., Shaw M.L., Hebert C.M., et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 2019;178:835–849.e21. doi: 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chan J.M., Quintanal-Villalonga Á., Gao V.R., Xie Y., Allaj V., Chaudhary O., Masilionis I., Egger J., Chow A., Walle T., et al. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell. 2021;39:1479–1496.e18. doi: 10.1016/j.ccell.2021.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spassova I., Ugurel S., Kubat L., Zimmer L., Terheyden P., Mohr A., Björn Andtback H., Villabona L., Leiter U., Eigentler T., et al. Clinical and molecular characteristics associated with response to therapeutic PD-1/PD-L1 inhibition in advanced Merkel cell carcinoma. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2021-003198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Somasundaram R., Zhang G., Fukunaga-Kalabis M., Perego M., Krepler C., Xu X., Wagner C., Hristova D., Zhang J., Tian T., et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat. Commun. 2017;8:607. doi: 10.1038/s41467-017-00452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schott A.F., Welch J.J., Verschraegen C.F., Kurzrock R. The National Clinical Trials Network: Conducting Successful Clinical Trials of New Therapies for Rare Cancers. Semin. Oncol. 2015;42:731–739. doi: 10.1053/j.seminoncol.2015.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Herrmann C., Van de Sande B., Potier D., Aerts S. i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Res. 2012;40 doi: 10.1093/nar/gks543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rodig S.J., Cheng J., Wardzala J., DoRosario A., Scanlon J.J., Laga A.C., Martinez-Fernandez A., Barletta J.A., Bellizzi A.M., Sadasivam S., et al. Improved detection suggests all Merkel cell carcinomas harbor Merkel polyomavirus. J. Clin. Invest. 2012;122:4645–4653. doi: 10.1172/jci64116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhatia K., Goedert J.J., Modali R., Preiss L., Ayers L.W. Merkel cell carcinoma subgroups by Merkel cell polyomavirus DNA relative abundance and oncogene expression. Int. J. Cancer. 2010;126:2240–2246. doi: 10.1002/ijc.24676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liao Y., Smyth G.K., Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47 doi: 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sade-Feldman M., Yizhak K., Bjorgaard S.L., Ray J.P., de Boer C.G., Jenkins R.W., Lieb D.J., Chen J.H., Frederick D.T., Barzily-Rokni M., et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell. 2018;175:998–1013.e20. doi: 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu A., Ibrahim J.G., Love M.I. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics. 2019;35:2084–2092. doi: 10.1093/bioinformatics/bty895. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Single-cell RNA-seq and bulk RNA-seq data have been deposited at GEO and are publicly available under accession numbers GEO: GSE226438, GEO: GSE223275, and GEO: GSE233454. Original western blot images have been deposited at Mendeley Data (https://doi.org/10.17632/hb3gvgs4rr.1) and are publicly available as of the date of publication.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in the paper is available from the lead contact upon request.