

Summary
The rapidly emerging field of computational pathology has demonstrated promise in developing objective prognostic models from histology images. However, most prognostic models are either based on histology or genomics alone and do not address how these data sources can be integrated to develop joint image-omic prognostic models. Additionally, identifying explainable morphological and molecular descriptors from these models that govern such prognosis is of interest. We use multimodal deep learning to jointly examine pathology whole slide images and molecular profile data from 14 cancer types. Our weakly-supervised, multimodal deep learning algorithm is able to fuse these heterogeneous modalities to predict outcomes and discover prognostic features that correlate with poor and favorable outcomes. We present all analyses for morphological and molecular correlates of patient prognosis across the 14 cancer types at both a disease and patient-level in an interactive open-access database (http://pancancer.mahmoodlab.org) to allow for further exploration, biomarker discovery and feature assessment.
eTOC Blurb
Chen et al. present a pan-cancer analysis that uses deep learning to integrate whole slide pathology images and molecular features to predict cancer prognosis, with multimodal interpretability used to elucidate morphologic and molecular correlates of prognosis.
Graphical Abstract
Introduction
Cancer is defined by hallmark histopathological, genomic, and transcriptomic heterogeneity in the tumor and tissue microenvironment that contributes towards variability in treatment response rates and patient outcomes (Marusyk et al. 2012). The current clinical paradigm for many cancer types involves the manual assessment of histopathologic features such as tumor invasion, anaplasia, necrosis, and mitoses, which are then used as grading and staging criteria for stratifying patients into distinct risk groups for therapeutic decision-making. For instance, in the Tumor, Nodes, and Metastases (TNM) staging system primary tumors are categorized into stages based on tumor severity (e.g. - size, growth, atypia), which are then used in treatment planning, eligibility for surgical operations, radiation dosage, and other treatment decisions (Amin et al.,2017). However, the subjective interpretation of histopathologic features has been demonstrated to suffer from large inter- and intra-observer variability and patients in the same grade or stage still have significant variability in outcomes. While many histopathologic biomarkers have been established for diagnostic tasks, most are based on the morphology and location of tumor cells alone, and lack a fine-grained understanding of how the spatial organization of stromal, tumor, and immune cells in the broader tumor microenvironment contributes toward patient risk (Marusyk et al.,2012;Chang et al.,2013;Heindl et al.,2015;Kather et al.,2018,Tarantino et al.,2021). Recent advancements made in deep learning for computational pathology have enabled the use of whole slide images (WSIs) for automated cancer diagnosis and quantification of morphologic phenotypes in the tumor microenvironment. Using weakly-supervised learning, slide-level clinical annotations can be used to guide deep learning algorithms in recapitulating routine diagnostic tasks such as cancer detection, grading and subtyping (Campanella et al.,2019;Lu et al.,2021). Though such algorithms can reach performance on-par with human experts for narrowly-defined problems, the quantification of novel prognostic morphological features is limited as training with subjective human annotations may fail to extract heretofore unrecognized properties that could be used to improve patient prognostication (Echle et al.,2020). To capture more objective and prognostic morphological features not extracted in routine clinical workflows, recent deep learning-based approaches propose supervision using outcome-based labels such as disease-free and overall survival times as ground truth (Harder et al.,2019;Courtiol et al.,2019;Kather et al.,2019;Kulkarni et al.,2020;Wuclzyn et al.,2021). Indeed, recent work has shown there is enormous potential in using deep learning for automated biomarker discovery of novel and prognostic morphological determinants (Beck et al.,2011;Echle et al.,2020;Diao et al.,2021).
Though prognostic morphological biomarkers may potentially be elucidated using outcome-based labels as supervision in WSIs, in the broader context, cancer prognostication is a multimodal problem that is driven by markers in histology, clinical data, and genomics (Ludwig and Weinstein,2005;Gentzler et al.,2014;Fridman et al.,2017; Mobadersany et al., 2018). From the emergence of next generation sequencing and development of targeted molecular therapies, therapeutic decision-making processes for many cancer types have become increasingly complex due to the inclusion of molecular biomarkers in prognostication (Hyman et al., 2015). For instance, the presence of Epidermal Growth Factor Receptor (EGFR) exon 19 deletions and exon 21 p.Leu858Arg substitutions are indications for the use of targeted therapies such as erlotinib in EGFR mutant lung and pancreatic cancers (Mayekar and Bivona,2017;Zhou et al.,2021). In combination with histological assessment, joint image-omic biomarkers such as oligodendroglioma and astrocytoma histologies with IDH1 mutation and 1p/19q-codeletion status is able to perform fine-grained stratification of patients into low-, intermediate-, and high-risk groups (Louis et al.,2016;Bai et al.,2016;Cloughesy et al.,2019) and determing the presence or absence of these integrated biomarkers has become standard of care in assessment of brain tumors by pathologists. Using deep learning, multimodal fusion of molecular biomarkers and extracted morphological features from WSIs has potential clinical application in not only improving precision in patient risk stratification, but also assist in the discovery and validation of multimodal biomarkers where combinatory effects of histology and genomic biomarkers are not known (Bera et al.,2019). Recent multimodal studies performed on the TCGA have focused on learning genotype-phenotype associations via predicting molecular aberrations using histology, which can assist in deciding targeted molecular therapies for patients without next-generation sequencing (Coudray et al.,2018; Kather et al.,2019;Fu et al.,2020;Kather et al.,2020). Though multimodal, in this direction of work, feature extraction of WSIs is guided using molecular aberrations as a supervisory signal, rather than multimodal integration of histology and genomics guided using outcome-based labels.
Results
Deep learning-based Multimodal Integration
In order to address the challenges in developing joint image-omic biomarkers that can be used for cancer prognosis, we propose a deep learning-based multimodal fusion (MMF) algorithm that uses both H&E whole slide images and molecular profile features (mutation status, copy number variation, RNA-Seq expression) to measure and explain relative risk of cancer death (Figure 1A). Our multimodal network is capable of not only integrating these two modalities in weakly-supervised learning tasks such as survival outcome prediction, but also explaining how histopathology features, molecular features, and their interactions contribute locally towards low- and high-risk patients (Figure 1B, 1C, 1D, 1E). After risk assessment within a patient cohort, our network uses both attention- and attribution-based interpretability as an untargeted approach for estimating prognostic markers across all patients (Figure 1B, 1C, 1D, 1E, 1F). Our study uses 6,592 gigapixel WSIs from 5,720 patient samples across 14 cancer types from the TCGA (Table S1). For each cancer type, we trained our multimodal model in a five-fold cross-validation using our weakly-supervised paradigm and conducted ablation analyses comparing with the performance of unimodal prognostic models. Following training and model evaluation, we conducted extensive analyses on the interpretability of our networks, investigating local- and global-level image-omic explanations for each cancer type, quantifying the tissue microarchitecture corresponding relevant morphology and also investigating shifts in feature importance when comparing unimodal interpretability versus multimodal interpretability.
Figure 1: Pathology-Omic Research Platform for Integrative Survival Estimation (PORPOISE) Workflow.

A. Patient data in the form of digitized high-resolution FFPE H&E histology glass slides (known as WSIs) with corresponding molecular data are used as input in our algorithm. Our multimodal algorithm consists of three neural network modules together: 1) an attention-based multiple instance learning (AMIL) network for processing WSIs, 2) a self-normalizing network (SNN) for processing molecular data features, and 3) a multimodal fusion layer that computes the Kronecker Product to model pairwise feature interactions between histology and molecular features. B. For WSIs, per-patient local explanations are visualized as high-resolution attention heatmaps using attention-based interpretability, in which high attention regions (red) in the heatmap correspond to morphological features that contribute to the model’s predicted risk score. C. Global morphological patterns are extracted via cell quantification of high attention regions in low- and high-risk patient cohorts. D. For molecular features, per-patient local explanations are visualized using attribution-based interpretability in Integrated Gradients. E. Global interpretability for molecular features is performed via analyzing the directionality, feature value and magnitude of gene attributions across all patients. F. Kaplan-Meier analysis is performed to visualize patient stratification of low- and high-risk patients for individual cancer types.
See also Table S1.
We additionally developed a research tool that uses model explanations of both whole slide image and molecular profile data to drive the discovery of new prognostic biomarkers. Using our multimodal network, we developed the Pathology-Omics Research Platform for Integrative Survival Estimation (PORPOISE), an interactive platform that directly yields prognostic markers learned by our model for thousands of patients across multiple cancer types, available at http://pancancer.mahmoodlab.org (Interactive Demo). Specifically, PORPOISE allows the user to visualize: 1) raw H&E image pyramidal TIFFs overlayed with attention-based interpretability from both unimodal and multimodal training, 2) local explanations of molecular features using attribution-based interpretability for each patient, and 3) global patterns of morphological and molecular feature importance for each disease model. To validate that PORPOISE can be used to drive the discovery of human-identifiable prognostic biomarkers, we analyzed high attention morphological regions in WSIs and further quantified the tumor microenvironment to quantify morphologic correlates of high and low risk patients.
Multimodal Integration Improves Patient Risk Stratification
We first evaluated our proposed multimodal fusion deep learning model (MMF) using 5-fold cross validation on the paired WSI-molecular datasets from 14 cancer types. We also compared our model with unimodal deep learning models trained with one modality: an Attention-based Multiple Instance Learning (AMIL) model that uses only WSIs, and a Self-Normalizing Network (SNN) model that uses only molecular features. To compare the performances of these models, we used cross-validated concordance index (c-Index) to measure the predictive performance of each model, Kaplan-Meier curves to visualize the quality of patient stratification between predicted low- and high-risk patient populations, and the logrank test to assess patient stratification statistical significance in distinguishing low- and high-risk groups at the 50% percentile of predicted risk scores (Figure 2A, 2B, S1, and Table S2). In addition to c-Index, we also report Dynamic AUC (termed Survival AUC) which corrects for optimistic bias from censorship in computing model performance (Table S3).
Figure 2: Model performances of PORPOISE and understanding impact of multimodal training.

A. Kaplan-Meier analysis of patient stratification of low- and high-risk patients via MMF across all 14 cancer types. Low- and high-risks are defined by the median 50% percentile of hazard predictions via MMF. Logrank test was used to test for statistical significance in survival distributions between low- and high-risk patients (with * marked if P-Value < 0.05). B. c-Index performance of SNN, AMIL and MMF in each cancer type in a five-fold cross-validation (n=5,720). Horizontal line for each model shows average c-Index performance across all cancer types. Box plots correspond to c-indices of 1000 bootstrap replicates on the aggregated risk predictions. C. Distribution of WSI attribution across 14 cancer types. Each dot represents the proportion of feature attribution given to the WSI modality input compared to molecular feature input. Attributions were computed on the aggregated risk predictions in each disease model.
Across the 14 cancer types, MMF achieved an overall c-Index of 0.645, whereas AMIL and SNN had overall c-Indices of 0.585 and 0.607 respectively. On Survival AUC, similar improvement in multimodal integration was found with an overall performance of 0.662 in comparison to 0.616 and 0.598 in SNN and AMIL respectively (Table S2). In one-versus-all model performance comparisons on individual cancer types, MMF achieved the highest c-Index on 12 out of 14 (12/14) cancer types, with models for 10/14 cancer types demonstrating statistical significance in binary patient stratification (Figure 2A,2B, 2C). In comparison to SNN which uses only molecular features, MMF also demonstrated consistent performance in both c-Index and Survival AUC across all cancer types. Though SNN had comparable performance on some cancer types, we observed both substantial improvement in model performance and patient stratification for BRCA, COADREAD, LUAD, PAAD, UCEC, which did not show significance in SNN patient stratification (Figure 2B, Table S2). In comparison to AMIL, MMF showed improvement on all cancer types except LUSC and UCEC, with improvement in patient stratification significance in 7/14 cancer types. We note that SKCM has an admixture of easily-distinguished disease forms (e.g. containing both primary and metastatic cases), which may optimistically bias model performances. Overall, however, model performances were found to improve following multimodal integration for almost all cancer types (Figure 2B). In examining unimodal models that were close to MMF performance, SNN showed significance in stratifying KIRC and KIRP (though predicted risk groups are better delineated in MMF, and AMIL showed significance in stratifying LIHC, STAD, and UCEC.
Amongst all single cancer types included in our study, KIRP had the largest performance increase with multimodal training, reaching a c-Index performance of 0.816 (95% CI: 0.705–0.880, P-Value = 3.83 × 10−4, logrank test), compared to 0.539 (95% CI: 0.408–0.625, P-Value = 5.86 × 10−1, logrank test) using AMIL and 0.779 (95% CI: 0.678–0.857, P-Value = 2.27 × 10−3, logrank test) using SNN (Table S2). Following correction of potential optimistic bias with high censorship via Survival AUC evaluation, we observed similar model performances with MMF reaching an AUC of 0.791 (SD: 0.102) compared to 0.530 (SD: 0.082) in AMIL and 0.743 (SD: 0.095). PAAD demonstrated substantial improvement with multimodal training, with a c-Index of 0.653 (95% CI: 0.571–0.696, P-Value = 1.69 × 10−3, logrank test), compared to 0.580 (95% CI: 0.485–0.613, P-Value = 2.30 × 10−1, logrank test) using AMIL and 0.593 (95% CI: 0.507–0.656, P-Value = 5.59 × 10−2, logrank test) using SNN (Table S2). For PAAD, we observed that training unimodal models using either only histology or genomics did not show statistical significance as Kaplan-Meier survival curves demonstrate poor stratification of predicted low- and high-risk groups of patients with low survival in these two cancer types. However, these groups were disentangled following multimodal integration, which is in line with our observed improvement in MMF performance in PAAD. We demonstrate similar stratification results in BRCA, COADREAD, and LUAD in separating low survival groups using MMF (Figure S2, S3, S6, and S11).
In addition to conducting ablation studies in comparing unimodal and multimodal models, we also assessed Cox proportional hazard models using age, gender, and tumor grade covariates as baselines, which were still outperformed by MMF (Table S3). In head-to-head comparisons on cancer types with only available grade information, AMIL outperforms Cox models with an average c-Index of 0.601 compared to 0.592, which suggests that current subjective assessments for tumor grade in cancer prognosis may be refined using objective, deep learning-based phenotyping for evaluating prognosis.
We additionally quantify the prognostic importance of each modality, giving us the ability to determine which cancer types warrant development of multimodal prognostic models and for which tumor type histology or genomics alone may be enough to build sufficient prognostic models. In quantifying the contribution of using WSIs in cancer prognosis, WSIs on average accounted for 16.8% of input attributions in MMF for all cancer types, which suggests that molecular features drive most of the risk prediction in MMF (Figure 2C, Table S3). This substantiates the observation that molecular profiles are more prognostic for survival than WSIs in most cancer types (in comparing the performances of SNN and AMIL). However, we note that for MMF models evaluated on UCEC, WSIs contributed to 55.1% of all input attributions, which corroborates with high AMIL performance on this cancer type. We also observe relatively larger average WSI contributions in HNSC, STAD, and LIHC as well, which corroborates with the cancer types in which AMIL outperformed SNN. For LUSC, in which AMIL also outperformed SNN, we observe a relatively low average WSI attribution of 5.8%, which potentially corroborates MMF under-performance as the model was unable to attribute feature importance to prognostic information in WSIs. Interestingly, WSIs contributed to 32.4% of input attributions in PAAD, despite AMIL performing worse than SNN, which may suggest that MMF is able to extract prognostic morphological features not captured in molecular biomarkers via SNN or unimodal feature extraction via AMIL.
Multimodal Interpretability for Joint Image-Omic Biomarker Discovery
For interpretation and further validation of our models, we applied attention- and gradient-based interpretability to our trained SNN, AMIL, and MMF models in order to explain how WSI and molecular features are respectively used to predict prognosis. For WSIs, we used a custom visualization tool that overlays attention weights computed from AMIL (and the AMIL subnetwork from MMF) onto the H&E diagnostic slide, which is displayed as a high-resolution attention heatmap that shows relative prognostic relevance of image regions used to predict risk (Figure 3A, 4A, 5A, 6A). For molecular features, we used Shapley Additive Explanation (SHAP)-styled attribution decision plots to visualize the attribution weight and direction of each molecular feature calculated by Integrated Gradients in SNN (and the SNN subnetwork of MMF) (Figure 3B, 4B, 5B, 6B and Figures 3D, 4D, 5D, 6D). These interpretation and visualization techniques were then used to build our discovery platform, PORPOISE, which we then applied to each of our models and across all patients, yielding attention heatmaps and attribution decision plots for all 6,592 WSIs and 5,720 patients. Visualizations for analyses for individual patient model explanations in PORPOISE are termed local interpretability, with analyses performed on cancer-wide patient populations termed global interpretability. Figures 3–6 show local and global interpretability for KIRC, KIRP, LGG, and PAAD. Global interpretability results for the rest of the cancer types, as well as local interpretability results for all patients in the best validation splits are made available in Figures S2-S11.
Figure 3: Quantitative performance, local model explanation, and global interpretability analyses of PORPOISE on clear cell renal cell carcinoma (KIRC).
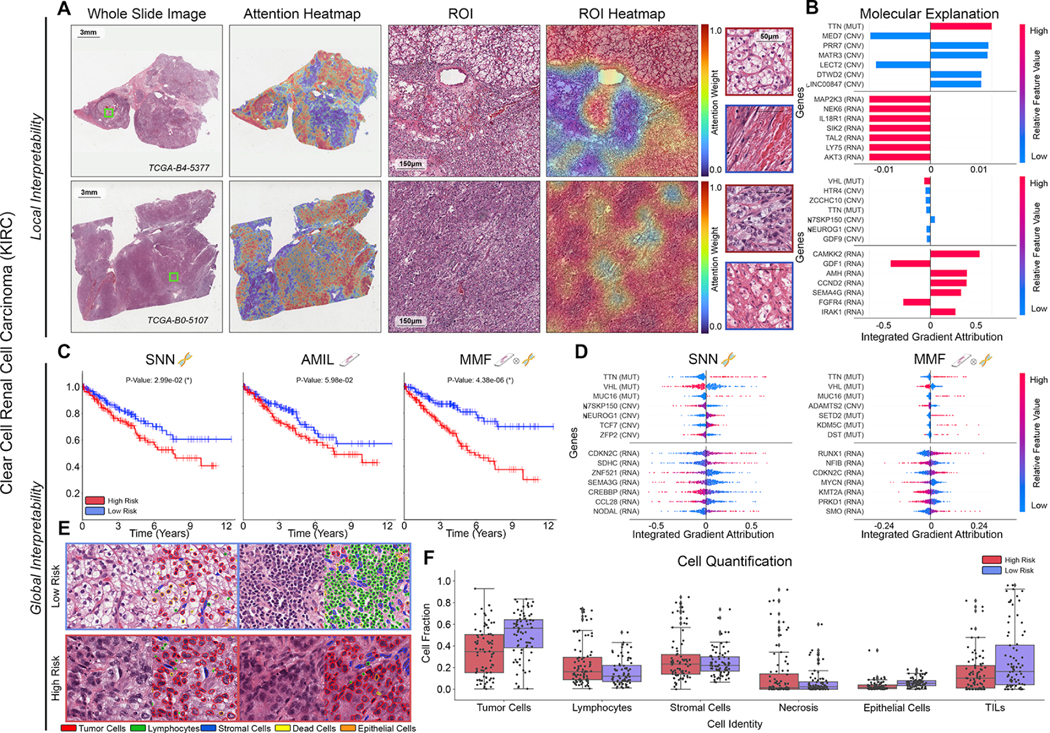
A. For KIRC (n=345), high attention for low-risk cases (top, n=80) tends to focus on classic clear cell morphology while in high-risk cases (bottom, n=80), high attention often corresponds to areas with decreased cytoplasm or increased nuclear to cytoplasmic ratio. B. Local gene attributions for the corresponding low-risk (top) and high-risk (bottom) cases. C. Kaplan–Meier curves for omics-only (left, “SNN”), histology-only (center, “AMIL”) and multimodal fusion (right, “MMF”), showing improved separation using MMF. D. Global gene attributions across patient cohorts according to unimodal interpretability (left, “SNN”), and multimodal interpretability (right, “MMF”). SNN and MMF were both able to identify immune-related and prognostic markers such as CDKN2C and VHL in KIRC. MMF additionally attributes to other immune-related / prognostic genes such as RUNX1 and NFIB in KIRC. E. Exemplar high attention patches from low-risk (top) and high-risk (bottom) cases with corresponding cell labels. F. Quantification of cell types in high attention patches for each disease overall, showing increased tumor and TIL presence.
Figure 4: Quantitative performance, local model explanation, and global interpretability analyses of PORPOISE in papillary renal cell carcinoma (KIRP).
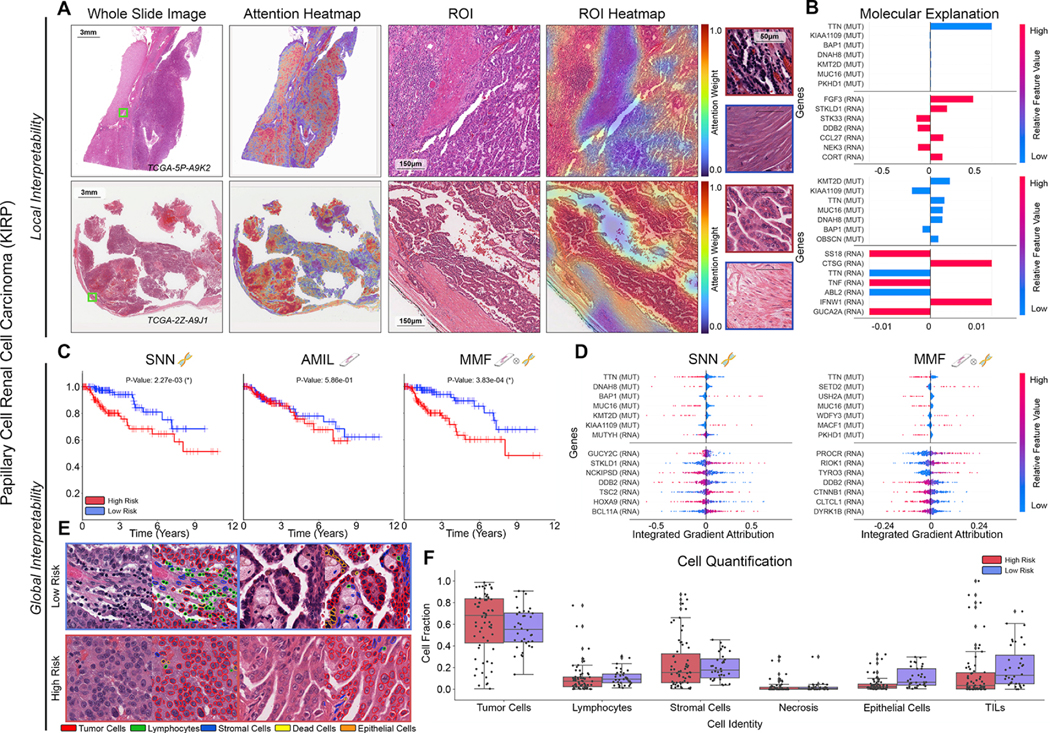
A. For KIRP (n=253), low-risk cases (top, n=36) often have high attention paid to complex and curving papillary architecture while for high-risk cases (bottom, n=63), high attention is paid to denser areas of tumor cells. B. Local gene attributions for the corresponding low-risk (top) and high-risk (bottom) cases. C. Kaplan–Meier curves for omics-only (left, “SNN”), histology-only (center, “AMIL”) and multimodal fusion (right, “MMF”), showing improved separation using MMF. D. Global gene attributions across patient cohorts according to unimodal interpretability (left, “SNN”), and multimodal interpretability (right, “MMF”). SNN and MMF were both able to identify prognostic markers such as BAP1 in KIRP. MMF additionally attributes to other immune-related / prognostic genes such as PROCR and RIOK1 in KIRP. E. Exemplar high attention patches from low-risk (top) and high-risk (bottom) cases with corresponding cell labels. F. Quantification of cell types in high attention patches for each disease overall, showing increased epithelial cell and TIL presence.
Figure 5: Quantitative performance, local model explanation, and global interpretability analyses of PORPOISE on lower-grade gliomas (LGG).
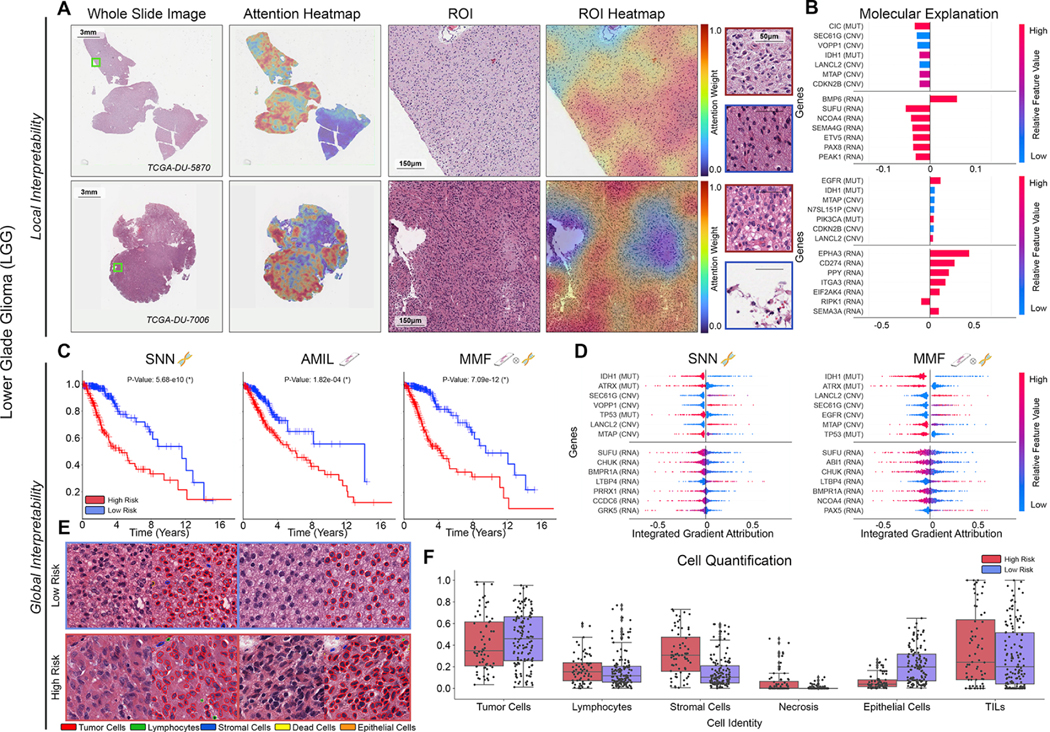
A. For LGG (n=404), high attention for low-risk cases (top, n=133) tends to focus on dense regions of tumor cells, while in high-risk cases (bottom, n=68), high attention focuses on both dense regions of tumor cells and areas of vascular proliferation. B. Local gene attributions for the corresponding low-risk (top) and high-risk (bottom) cases. C. Kaplan–Meier curves for omics-only (left, “SNN”), histology-only (center, “AMIL”) and multimodal fusion (right, “MMF”), demonstrating improvement in patient stratification in MMF. D. Global gene attributions across patient cohorts according to unimodal interpretability (left, “SNN”), and multimodal interpretability (right, “MMF”). SNN and MMF were both able to identify immune-related and prognostic markers such as IDH1, ATRX, EGFR, and CDKN2B in LGG. E. High attention patches from low-risk (top) and high-risk (bottom) cases with corresponding cell labels, showing oligodendroglioma and astrocytoma subtypes respectively. F. Quantification of cell types in high attention patches for each disease overall, with statistical significance for increased necrosis in high-risk patients.
Figure 6: Quantitative performance, local model explanation, and global interpretability analyses of PORPOISE on pancreatic adenocarcinoma (PAAD).
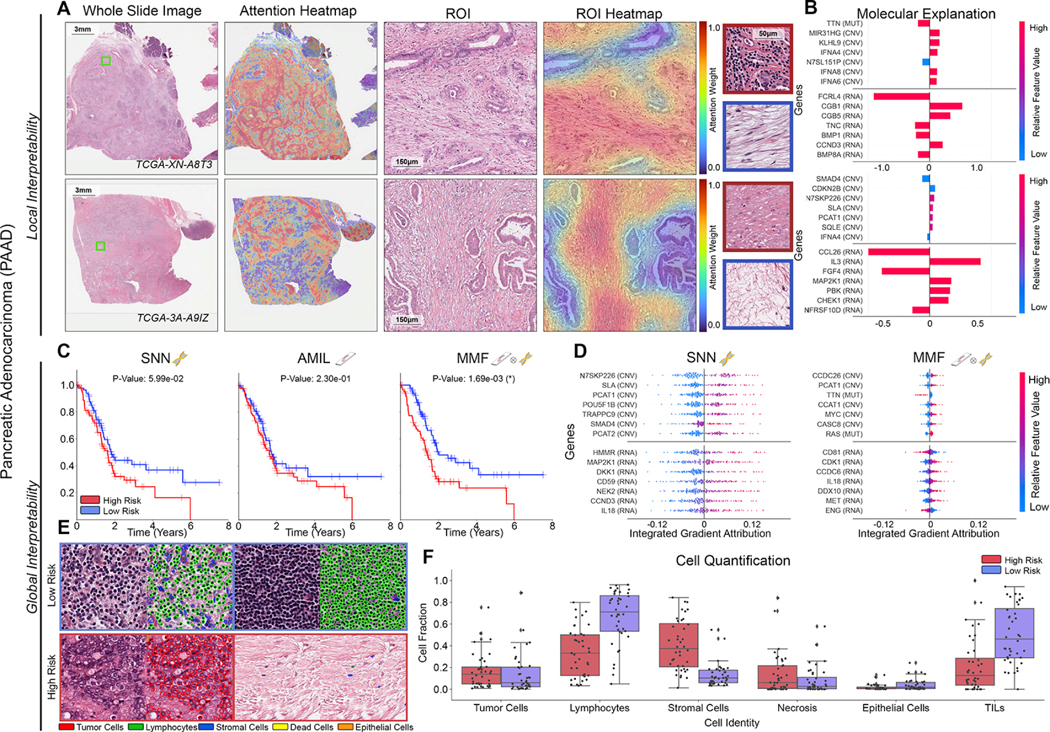
A. For PAAD (n=160), high attention for low-risk cases (top, n=40) tends to focus on stroma-contained dispersed glands and aggregates of lymphocytes, while in high-risk cases (bottom, n=40), high attention focuses on tumor-associated and myxoid stroma. B. Local gene attributions for the corresponding low-risk (top) and high-risk (bottom) cases from a and g. C. Kaplan–Meier curves for omics-only (left, “SNN”), histology-only (center, “AMIL”) and multimodal fusion (right, “MMF”), demonstrating SNN and AMIL showing poor separation of patients with low survival, with better stratification following multimodal integration. D. Global gene attributions across patient cohorts according to unimodal interpretability (left, “SNN”), and multimodal interpretability (right, “MMF”). SNN and MMF were both able to identify immune-related and prognostic markers such as IL8, EGFR, and MET in PAAD. MMF additionally shifts attribution to other immune-related / prognostic genes such as CD81, CDK1, and IL9. E. High attention patches from low-risk (top) and high-risk (bottom) cases with corresponding cell labels. F. Quantification of cell types in high attention patches for each disease overall, showing increased lymphocyte and TIL presence in low-risk patients, as well as increased necrosis presence in PAAD.
Patient stratification for unimodal and multimodal prognostic models is shown in Figures 3C, 4C, 5C, 6C, 7C and S2-S11. To obtain an understanding of how morphological features were used by the model, we assessed high attention regions of WSIs in the top 25% (high-risk group) and bottom 25% (low-risk group) of predicted patient risks for each cancer type, which reflect favorable and poor cancer prognosis respectively. In addition to visual inspection from two pathologists, we simultaneously segmented and classified cell type identities across high attention regions in our WSIs. Figure 3A, 4A, 5A, 6A show attention heatmaps with exemplar ROIs for low- and high-risk cases in KIRC, KIRP, LGG, and PAAD, and Figure 3E, 4E, 5E, and 6E shows semantic segmentation of cell types in high attention tissue regions in low- and high-risk cases. Figure 3F, 4F, 5F, and 6F compares quantitative cell type distributions in high attention patches of low- and high-risk cases in these cancer types. Across all cancer types, we generally observed that high attention regions in low-risk patients corresponded with greater immune cell presence and lower tumor grade than that of high-risk patients, with 8/14 cancer types demonstrating statistically significant differences in lymphocyte cell fractions in high attention regions (Figure 6F, S3-10). Furthermore, we also observed that high attention regions in high-risk patients corresponded with increased tumor presence, higher tumor grade and tumor invasion in certain cancer types, with 6/14 cancer types demonstrating statistically significance differences in tumor cell fractions (Figure S3, S5, S6, S7, S9, S11). Figure 6F, S3, and S7 show clear differences in cell fraction distributions in comparing tumor cell fraction (BRCA P-Value: 2.17 × 10−9, LUAD P-Value: 1.45 × 10−3) and lymphocyte cell fraction (BRCA P-Value: 6.79 × 10−14, LUAD P-Value: 1.06 × 10−9, PAAD P-Value: 2.04 × 10−8 t-test). Figure 3E, 4E, 5E and 6E show exemplar high attention regions in low- and high-risk respectively, with attention-based interpretability identifying dense immune cell infiltrates (green) in low-risk patients and nuclear pleomorphism and atypia in tumor cells (red) in high-risk patients. Interestingly, increased fractional tumor cell content in high attention regions were not discovered in high-risk patients for KIRC and KIRP (Figure 3F and 4F). However, visual inspection of high attention regions in these cancer types revealed that tumor cells in low-risk patients corresponded with lower tumor grade than that of high-risk patients. Figure 3A and 4A provide examples of attention heatmaps for low- and high-risk patients in KIRC and KIRP, in which high attention regions in high-risk KIRC patients corresponded with central necrosis, and high attention regions in high-risk KIRP correspond with tumor cells invading the renal capsule. To understand how attention shifts when conditioning on molecular features in multimodal interpretability, we also had two pathologists use PORPOISE to assess unimodal and multimodal attention heatmaps. For certain cancer types such as BRCA and KIRC, attention in MMF shifted way from tumor-only regions and towards both stroma and tumor regions, which demonstrates the prognostic relevance of stromal regions (Figure S12) (Beck et al.,2011;Bejnordi et al., 2017).
Figure 7: Tumor Infiltrating Lymphocyte Quantification in Patient Risk Groups.

TIL quantification in high attention regions of predicted low- (BLCA n=90, BRCA n=220, COADREAD n=74, HNSC n=96, KIRC n=80, KIRP n=36, LGG n=133, LIHC n=85, LUAD n=105, LUSC n=97, PAAD n=40, SKCM n=29, STAD n=53, UCEC=104) and high-risk patient cases (BLCA n=93, BRCA n=223, COADREAD n=80, HNSC n=103, KIRC n=80, KIRP n=63, LGG n=68, LIHC n=84, LUAD n=89, LUSC n=103, PAAD n=40, SKCM n=55, STAD n=78, UCEC=125) across 14 cancer types. For each patient, the top 1% of scored high attention regions (512 × 512 40× image patches) were segmented and analyzed for tumor and immune cell presence. Image patches with high tumor-immune co-localization were indicated as positive for TIL presence (and negative otherwise). Across all patients, the fraction of high attention patches containing TIL presence was computed and visualized in the box plots. A two-sample t-test was computed for each cancer type to test the if the means of the TIL fraction distributions of low- and high-risk patients had a statistically significant difference (with * marked if P-Value < 0.05).
In parallel with assessing WSI interpretability, we also interrogated important model explanations in our molecular feature inputs. Figure 3B, 4B, 5B, 6B shows local interpretability and 3D, 4D, 5D, and 6D shows global importance of molecular features for KIRC, KIRP, LGG, and PAAD. Across all cancer types, gradient-based interpretability was able to identify many well-known oncogenes and immune-related genes established in existing biomedical literature and used in targeted molecular therapies (Uhlen et al.,2017). In the LGG cohort which has distinct molecular signatures, gradient-based interpretability identifies IDH1 mutation (P-Value: 2.31 × 10−89, t-test) status as the most attributed gene feature, which has important functions in cellular metabolism, epigenetic regulation and DNA repair and defines the IDH1-wildtype astrocytoma, IDH1-mutant astrocytoma and IDH1-mutant oligodendroglioma molecular subtypes (Louis et al., 2016). In addition, IDH1 mutation is associated with lower grade gliomas and thus favorable prognosis in comparison with IDH1-wildtype gliomas, which successfully corroborates with the attribution direction of IDH1 mutation in the attribution decision plot, in which the distribution of IDH1 mutation attributions has only negative attribution values (low-risk) (Figure 5D). The model also uses several other key oncogenes in other cancer types such as PIK3CA mutation (P-Value, 4.04 × 10−118, t-test) in BRCA, SOX9 mutation (P-Value, 7.56 × 10−64, t-test) and SOX11 mutation (P-Value, 1.65 × 10−58, t-test) in COADREAD, KRAS mutation in LUAD (P-Value, 1.98 × 10−63, t-test) and PAAD (P Value, 9.00 × 10−12, t-test), VHL (P-Value, 1.76 × 10−62, t-test) and BAP1 (P-Value, 5.57 × 10−18, t-test) mutations in KIRC (Figure 3D, 5D, S3, S6). In PAAD, we additionally observe attributions of immune-related genes such as CD81, CDK1, IL8, and IL9 RNA-Seq expression are found to be the most highly attributed, which corroborates with their roles in innate immunity and inflammatory cell signaling (Network et al.,2013;Uhlen et al.,2015;Chevrier et al.,2017;Uhlen et al.,2017,2019) (Figure 6D). Moreover, we note following conditioning on WSIs, MMF models for PAAD show a relative decrease in attribution for many genes, which corroborates with our previous observation of much higher WSI attribution in PAAD for MMF patient stratification. Across most cancer types, gene mutations that encode for extremely large proteins such as TTN, OBSCN, RYR3, and DNA5 were frequently found to be highly attributed. Though many of these genes are not explicitly cancer-associated and prognostic due to heterogeneity in the mutational processes of each cancer type, genomic instabilities in these large protein-coding domains may be an indirect measure of tumor mutational load (Lawrence et al.,2013;Rizvi et al.,2015;Shi et al.,2020;Oh et al.,2020). Attributions for all gene features for SNN and MMF can be found in Table S4.
Immune response as a prognostic marker
Lastly, we used the interpretability of PORPOISE as a mechanism to test the hypothesis that TIL presence corroborates with favorable cancer prognosis. Figure 7 shows the fractional distribution of TILs in the high attention regions for all 14 cancer types across the previously defined risk groups. In comparing TIL presence between low-risk and high-risk patients, we found that 9 out of 14 cancer types had a statistically significant increase in TIL presence amongst patients with predicted low-risk, indicating that model attention was localized to more immune-hot regions. For cancer types in our dataset that have been FDA approved immune checkpoint inhibitor therapies, TIL presence was used as model explanations for favorable prognosis in BRCA (P-Value, 5.17×10−11, t-test), HNSC (P-Value, 1.97×10−18, t-test), KIRC (P-Value, 1.86×10−3, t-test), LIHC (P-Value, 2.54 × 10−17, t-test), LUAD (P-Value, 1.54 × 10−21, t-test), LUSC (P-Value, 2.92 × 10−12, t-test), PAAD (P-Value, 3.77×10−6, t-test), STAD (P-Value, 1.09×10−9, t-test), and SKCM (P-Value, 6.29×10−10, t-test). This suggests that our trained models use morphological features for immune response as markers for predicting cancer prognosis, and supports a growing body of evidence that TILs have a prognostic role in many cancer types (Thorsson et al.,2018;Saltz et al.,2018;Shaban et al.,2019;AbdulJabbar et al.,2020). In breast cancer, Maley et al. performed hotspot analysis on the co-localization of immune cancer cells in WSIs and showed that immune-cancer co-localization was a significant predictive factor of long-term survival (Maley et al.,2015). In Oral Squamous Cell Carcinoma, Shaban et al. proposed a co-localization score for quantifying TIL density that showed similar findings (Shaban et al.,2019). In lung cancer, AbdulJabbar et al. proposed a deep learning framework for spatially profiling immune infiltration in H&E and IHC WSIs, and similarly found that tumors with more than one immune cold region had a higher risk of relapse (AbdulJabbar et al.,2020). Saltz et al. performed a pan-cancer analysis on the spatial organization of TILs in the TCGA, and showed how different phenotypes of TIL infiltrates correlates with survival (Saltz et al.,2018). The distinction of these analyses in comparison to PORPOISE, is that our method is not specifically trained in identifying TILs and correlating TILs with outcome. Rather, dissection of model interpretability reveals that TIL presence is used as prognostic morphological features in stratifying low- and high-risk patients.
Discussion
There is much promise that incorporating computational-derived, histomorphological biomarkers into clinical staging systems will allow for better risk stratification of patients (Echle et al.,2020;Bera et al.,2019). Current cancer staging systems, such as the TNM classification system struggle with precision and consistency, leading to subsequent variation in patient management and patient outcomes (Nicholson et al.,2001;Novara et al.,2007;Rabe et al.,2019). In this study, we present a method for interpretable, weakly-supervised, multimodal deep learning that integrates WSIs and molecular profile data for cancer prognosis, which we trained and validated on 6,592 WSIs from 5,720 patients with paired molecular profile data across 14 cancer types, and compared our method with unimodal deep learning models as well as Cox models with clinical covariates, achieving the highest c-Index performance on 12 out of 14 cancer types in a one-versus-all comparison. Our method also explores multimodal interpretability in explaining how features from WSIs and molecular features contribute towards risk. We developed PORPOISE, an interactive, freely available platform that directly yields both WSI and molecular feature explanations made by our model in our 14 cancer types. Our goal with PORPOISE is to begin making current black-box state-of-the-art methods in computational pathology, especially emerging multimodal methods, more transparent, explainable and usable for the broader biomedical research community. In making heatmaps and decision plots available for each cancer type, we hope that our tool would allow clinicians and researchers to devise their own hypotheses and investigate the discoveries explained using deep learning.
Though we find that multimodal integration benefits patient risk stratification for most cancer types, our results also suggest that for some cancer types, training unimodal algorithms using either WSIs or molecular features alone may achieve comparable stratification performance, as variance of cancer outcomes can be equally captured in either modality. Many practical settings may lack paired diagnostic slide or high-throughput sequencing data for the same tissue specimen, and here, unimodal deep learning-based cancer prognosis algorithms may have reduced barriers to clinical deployment. Though multimodal learning has been successful in technical domains such as the integration of audio, visual and language modalities, for clinical translational tasks, we note that the basis of improvement from multimodal integration needs to be grounded in the biology of each cancer type, as phenotypic manifestations in the tumor microenvironment that are entirely explained by contributions from genotype have high mutual information (Kather et al.,2020). In establishing unimodal and multimodal baselines for 14 diverse cancer types, our results advocate that the application of multimodal integration should be determined on a per-cancer basis, which may aid in introspecting clinical problems for unimodal or multimodal biomarkers on single disease models.
A limitation of our approach is that though our approach can point to “what” and “where”, it cannot always explain “why” for discovered features which must be further quantified and introspected based on human knowledge. For example, though TILs were found in most cancer types to distinguish low- and high-risk patients, post-hoc analyses still had to be done to quantify TIL presence and assess statistically significance between the two risk groups. In analyses on feature shift in WSIs, we observe that high attention often shifted way from tumor regions to stroma, normal tissue and other morphological regions in some cancer types. We speculate this observation is a result of the intrinsic differences between WSIs and molecular profile data, in which training dynamics may be biased towards using more information from the simpler modality (Wang et al.,2020). In molecular features, the genotypic information from gene mutation, copy number variation, and RNA-Seq abundances have no spatial resolution are averaged across cells in the tumor biopsies, whereas phenotypic information such as normal tissue, tumor cells, and other morphological determinants are spatially represented in WSIs. As a result, when our multimodal algorithm is already conditioned with tumor-related features (e.g. - TP53 mutation status, PTEN loss) in the molecular profile, it can attend to morphological regions with non-tumor information such as stroma to explain subtle differences in survival outcomes (Beck et al.,2011). In other words, feature importance does not need to be attributed to oncogenes in molecular features and tumor-containing image regions in WSIs which have high collinearity, which allows the multimodal network to learn other histology-specific prognostic information beyond tumor grades.
In addition to characterizing known human-identifiable phenotypes such as TILs in cancer, PORPOISE can potentially be used by the research community in further characterization of phenotypes that are currently not well understood via robust quantification of cell-type populations and tissue architecture (Diao et al.,2021). Moreover, multimodal networks for general disease prognostication and outcome prediction adapted to larger and well-curated clinical trial data can be used to aid in both discovery and validation of human-interpretable image-omic biomarkers in guiding treatment decisions. Tangential research directions in similar pursuit of this goal are the prediction of molecular biomarkers from WSIs and other genotype-phenotype correlations, which would decrease complexity of routine clinical workflows that require molecular assays for therapeutic decision-making. Though multimodal and elucidating morphological biomarkers that would predict molecular aberrations, there may exist orthogonal morphological biomarkers that are prognostic but do not have correlation with molecular features. As observed in PAAD, though AMIL is not prognostic for survival outcomes and performs worse than SNN, we demonstrate not only performance increase in multimodal integration, but also relatively high attribution given to WSIs, suggesting the existence of prognostic information not currently captured using molecular features or unimodal feature extraction in WSIs. On the other hand, in cancer types such as BRCA, COADREAD, and LUAD which benefit from multimodal integration, MMF models have lower attribution given to WSIs, which may result from prognostic information also partially explained via genomics as aberrations such as ERBB2 or KRAS mutation and the presence of microsatellite instability can be determined from histology (Coudray et al., 2018;Kather et al.,2019,2020;Gamble et al.,2021). Towards the development of computational support systems for therapeutic decision-making, further work in genotype-phenotype correlation-based analyses would develop more formal intuition in understanding shared and modality-specific prognostic information for multimodal integration-based approaches, which may lead to clinical validation of either single-modality or joint image-omic computational biomarkers (per cancer type) for downstream prognostication.
Overall, this study is a proof-of-concept showing the development multimodal prognostic models from orthogonal data streams using weakly supervised deep learning and subsequently identifying correlative features that drive such prognosis. Future work will focus on developing more focused prognostic models by curating larger multimodal datasets for individual disease models, adapting models to large independent multimodal test cohorts, and using multimodal deep learning for predicting response and resistance to treatment. As research advances in sequencing technologies such as single-cell RNA-Seq, mass cytometry, and spatial transcriptomics, these technologies continue to mature and gain clinical penetrance, in combination with whole slide imaging, and our approach to understanding molecular biology will become increasingly spatially-resolved and multimodal (Abdelmoula et al., 2016; Berglund et al., 2018; Giesen et al., 2014; Jackson et al., 2020; Schapiro et al., 2017, He et al.,2020). In using bulk molecular profile data, our multimodal learning algorithm is considered a “late fusion” architecture, in which unimodal WSI and molecular features are fused near the output end of the network (Baltrušaitis et al., 2018). However, spatially-resolved genomics and transcriptomics data in combination with whole slide imaging has the potential to enable “early fusion” deep learning techniques that integrate local histopathology image regions and molecular features with exact spatial correspondences, which will lead to more robust characterizations and spatial organization mappings of intratumoral heterogeneity, immune cell presence, and other morphological determinants.
Star Methods
Resource Availability
Lead Contact
Further information and requests regarding this manuscript should be sent to and will be fulfilled by the lead investigator, Faisal Mahmood (faisalmahmood@bwh.harvard.edu).
Materials Availability
This study did not generate any unique reagents.
Data and Code Availability
All diagnostic whole slide images (WSIs) and their corresponding molecular and clinical data were obtained from The Cancer Genome Atlas and are publicly accessible through the NIH Genomic Data Commons Data Portal https://portal.gdc.cancer.gov/. All code was implemented in Python, using PyTorch as the primary deep learning package. All code and scripts to reproduce the experiments of this paper are available at: https://github.com/mahmoodlab/PORPOISE.
Method Details
Dataset Description
6,592 H&E diagnostic WSIs with corresponding molecular and clinical data were collected from 5,720 patients across 14 cancer types from TCGA via the NIH Genomic Data Commons Data Portal. Sample inclusion criteria in dataset collection were defined by: 1) dataset size and balanced distribution of uncensored-to-censored patients in each TCGA project, and 2) availability of matching CNV, mutation, and RNA-Seq abundances for each WSI (WSI-CNV-MUT-RNA). To mitigate overfitting in modeling the survival distribution during survival analysis, TCGA projects with less than 150 patients (after WSI and molecular data alignment) and have poor uncensorship (less than 5% uncensored patients) were excluded from the study. For the gastrointestinal tract, cancer types from these organs were grouped together respectively, forming the combined TCGA project - COADREAD (colon (COAD) and rectal (READ) adenocarcinoma). For LGG, other cases in the TCGA glioma cohort (such as glioblastomas) were included during model training, with evaluation and interpretability only performed on LGG cases. For inclusion of Skin Cutaneous Melanoma (TCGA-SKCM) and Uterine Corpus Endometrial Carcinoma (TCGA-UCEC) that have large data missingness, criteria for data alignment were relaxed to include samples with only matching WSI-MUT-RNA and WSI-CNV-MUT respectively. We note that in TCGA-SKCM, we also included metastatic cases as very few primary tumors had matching molecular profile information. To decrease feature sparsity in molecular profile data, genes with greater than 10% CNV or 5% mutation frequency in each cancer study were used. In TCGA-SKCM and TCGA-UCEC, we used mutation frequency cutoffs of 10%, as using the cutoff for other cancer types resulted in few-to-zero gene features.. To limit the number of the features from RNA-Seq, we used gene sets from the gene family categories from the Molecular Signatures Database (Subramanian et al., 2005). Molecular and clinical data were obtained from quality-controlled files from the cBioPortal. Summary tables of cohort characteristics, censorship statistics, and feature alignment can be found in Table S1 and Table S3.
WSI Processing
For each WSI, automated segmentation of tissue was performed using the public CLAM (Lu et al., 2021) repository for WSI analysis. Following segmentation, image patches of size 256 × 256 were extracted without overlap at the 20× equivalent pyramid level from all tissue regions identified. Subsequently, a ResNet50 model pretrained on ImageNet is used as an encoder to convert each 256 × 256 patch into 1024-dimensional feature vector, via spatial average pooling after the 3rd residual block. To speed up this process, multiple GPUs were used to perform computation in parallel using a batch-size of 128 per GPU.
Deep Learning-based Survival Analysis for Integrating Whole Slide Images and Genomic Features
PORPOISE (Pathology-Omics Research Platform for Integrated Survival Estimation) uses a high-throughput, interpretable, weakly-supervised, multimodal deep learning algorithm (MMF) designed for integrating whole slide images and molecular profile data in weakly-supervised learning tasks such as patient-level cancer prognosis via survival analysis. Given 1) diagnostic WSIs as pyramidal files and 2) processed genomic and transcriptomic features for a single patient, MMF learns to jointly represent these two heterogenous data modalities. Though tasked for survival analysis, our algorithm is adaptable to any combination of modalities, and flexible for solving any learning tasks in computational pathology that have patient-level labels. Our algorithm consists of three components: 1) attention-based Multiple Instance Learning (AMIL) for processing WSIs, 2) Self-Normalizing Networks (SNN) for processing molecular profile data, and 3) a multimodal fusion layer (extended from Pathomic Fusion) for integrating WSIs and molecular profile data (Chen et al., 2020; Ilse et al., 2018; Klambauer et al., 2017; Lu et al., 2021).
AMIL.
To perform survival prediction from WSIs, we extend the attention-based multiple instance learning algorithm, which was originally proposed for weakly-supervised classification. Under the multiple instance learning framework, each gigapixel WSI is divided into smaller regions and viewed as a collection (bag) of patches (instances) with a corresponding slide-level label used for training. Accordingly, following WSI processing, each WSI bag is represented by a matrix tensor, where is the number of patches (bag size), which varies between slides, and C is the feature dimension and equals 1024 for the ResNet50 encoder we used. Since survival outcome information is available at the patient-level instead of for individual slides, we collectively treat all WSIs corresponding to a patient case as a single WSI bag during training and evaluation. Namely, for a patient case with WSIs with bag sizes respectively, the WSI bag corresponding the patient is formed by concatenating all bags, and has dimensions , where .
The model can be described by three components, the projection layer , the attention module , and the prediction layer . Incoming patch-level embeddings of each WSI bag, are first mapped into a more compact, dataset-specific 512-dimensional feature space by the fully-connected layer with weights and bias . For succinctness, from now on, we refer to layers using their weights only (the bias terms are implied). Subsequently, the attention module learns to score each region for its perceived relevance to patient-level prognostic prediction. Regions with high attention scores contribute more to the patient-level feature representation relative to regions assigned low attention scores, when information across all regions in the patient’s WSIs are aggregated, in an operation known as attention-pooling (Ilse et al., 2018). Specifically, consists of 3 fully-connected layers with weights , and . Given a patch embedding (the row entry of ), its attention score is computed by:
The attention-pooling operation then aggregates the patch-level feature representations into the patient representation using computed attention scores as weight coefficients, where is the vector of attention scores:
The final patient-level prediction scores 𝐬 are computed from the bag representation using the prediction layer with weights and sigmoid activation: . This architectural choice and the negative-log-likelihood function for discrete-time survival modeling, are described in detail in a proceeding section. The last fully-connected layer is used to learn a representation , which is then used as input to our multimodal fusion layer.
SNN.
For survival prediction using molecular features, we used the Self-Normalizing Network (SNN) which has previously been demonstrated to work well in high-dimensional low-sample size (HDLSS) scenarios (Klambauer et al., 2017). For learning scenarios such as genomics that have hundreds to thousands of features with relatively few training samples, traditional Feedforward networks are prone to overfitting, as well as training instabilities from current deep learning regularization techniques such as stochastic gradient descent and Dropout. To employ more robust regularization techniques on high-dimensional low sample size genomics data, we adopted the normalizing activation and dropout layers from the SNN architecture: 1) scaled exponential linear units (SeLU) and 2) Alpha Dropout. In comparison with rectified linear unit (ReLU) activations common in current Feedforward networks, SeLU activations would drive the outputs of every layer towards zero mean and unit variance during layer propagation. The SeLU activation is defined as:
where , . To main normalization after Dropout, instead of setting the activation value to be 0 with with probability for for a neuron in a given layer, the activation value is set to be , which ensures the self-normalization property with updated mean and variance:
The SNN architecture used for molecular feature input consists of 2 hidden layers of 256 neurons each, with SeLU activation and Alpha Dropout applied to every layer. The last fully-connected layer is used to learn a representation , which is then used as input to our multimodal fusion layer. We ablated performance of MMF using fully-connected layers without self-normalization and also without regularization, and found that self-normalization and regularization are important for multimodal training (Table S3 and S3)
Multimodal Fusion Layer.
Following the construction of unimodal feature representations from the AMIL and SNN subnetworks, we learn a multimodal feature representation using Kronecker Product Fusion, which would capture important interactions between these two modalities (Chen et al., 2020; Zadeh et al., 2017). Our joint multimodal tensor is computed by the Kronecker product of and , in which every neuron in is multiplied by every other neuron in to capture all bimodal interactions. To also preserve the unimodal features, we also append “1” to each unimodal feature representation before fusion, which is shown the equation below:
where is the Kronecker Product, and is a differentiable multimodal tensor that models all unimodal and biomodal interaction with computation. To decrease impact of noise unimodal features and to reduce feature collinearity between the WSI and molecular feature modalities, we used a gating-based attention mechanism that additionally controls the expressiveness of each modality:
For a modality with learned unimodal features , we learn a weight matrix that would score the relative importance of each feature in modality . After performing Softmax, can be interpreted as an attention score of how and attends to each feature in . We obtain the gated representation in taking the element-wise product of the original unimodal features and attention scores . In our implementation of gating-based attention, we applied gating to both modalities prior to fusion, with additional skip connections made to the penultimate hidden layer of our multimodal network. Following Kronecker Product Fusion, we propagate our multimodal tensor through two hidden layers of size 256, which is then subsequently supervised using a cross entropy-based loss function for survival analysis. Table S3 shows an ablation study in using multimodal gating for pathology gating only, genomic gating only, and both pathology and genomic gating prior to Kronecker Product Fusion. To assess multimodal performance with other fusion mechanisms, we compared vector concatenation and a low-rank implementation of Kronecker Product Fusion, which similarly outperform unimodal approaches (Table S3) (Liu et al., 2018).
Survival Loss Function.
To perform survival prediction from right-censored, patient-level survival data, we first 591 partition the continuous time scale of overall patient survival time in months, into 4 non-overlapping bins: , , , , where , and define the quartiles of event times for uncensored patients. Subsequently, for each patient entry in the dataset, indexed by with corresponding follow-up time , we define the discretized event time as the index of the bin interval that contains :
To avoid confusion, we refer to the discretized ground truth label of the patient as . For a given patient with bag-level representation , the prediction layer with weights models the hazard function defined as:
which relates to the survival function through:
To optimize the model parameters, we use the log likelihood function for a discrete survival model (Zadeh and Schmid, 2020), which given the binary censorship status , can be expressed as
In this formulation, we use for patients who have lived past the end of the follow-up period and in the event that the patient passed away precisely at time . During training, the contribution of uncensored patient cases can be emphasized by minimizing a weighted sum of and
The second term of the loss function corresponding uncensored patients, is defined by:
Training Details.
For each disease model studied patient cases were randomly split into non-overlapping training (80%) and test (20%) sets that were used to train models and evaluate the performance. These training and test sets were constructed at a patient case level i.e., all slides corresponding to a given patient case were only present in either the test or train set and slides from the same case were never simultaneously part of training and testing. We repeated the experiments for each disease model in a five-fold cross-validation reassigning patient cases into non-overlapping training and testing cohorts five times. The same procedure was adopted for training and evaluating MMF and unimodal models. Across all cancer types, MMF is trained end-to-end with AMIL subnetwork, SNN subnetwork and multimodal fusion layer, using Adam optimization with a learning rate of 2 × 10−4, coefficient of 0.9, coefficient of 0.999, weight decay of 1 × 10−5, and weight decay of 1 × 10−5 for 20 epochs. Because WSIs across patient samples have varying image dimension sizes, we randomly sampled paired WSIs and molecular features with a mini-batch size of 1. In performing comparative analyses with unimodal networks, AMIL and SNN were also trained independently using the same survival loss function and hyperparameters as MMF.
Multimodal Interpretability and Visualization
Local WSI Interpretability.
For a given WSI, to perform visual interpretation of the relative importance of different tissue regions towards the patient-level prognostic prediction, we first compute attention scores for 256 × 256 patches (without overlap) from all tissue regions in the slide. We refer to the attention score distribution across all patches from all WSIs from the patient case as the reference distribution. For fine-grained attention heatmaps, attention scores for each WSI are recomputed by increasing the tiling overlap to up to 90%. For visualization, the attention scores are converted to percentile scores between 0.0 (low attention) to 1.0 (high attention) using the initial reference distribution, and spatially registered onto the corresponding WSIs (scores from overlapping patches are averaged). The resulting heatmap, referred to as local WSI interpretability, is transformed to RGB values using a colormap and overlayed onto the original H&E image with a transparency value of 0.5. To interpret these heatmaps, note that in contrast with classification tasks in which attention heatmaps would localize areas of diagnostic relevance for predicting a discrete class, survival outcome prediction is an ordinal regression task in which high attention weights correspond to regions with high prognostic relevance in determining relative predicted risk. For example, WSIs with predicted high-risk scores would have high attention on high tumor grade or tumor invasion regions used in explaining poor survival, whereas WSIs with predicted low risk scores would have high attention on low tumor grade or lymphocyte-containing regions used in explaining favorable survival.
Global WSI Interpretability.
For sets of WSIs belonging to different patient cohorts, we performed global WSI interpretability by quantifying and characterizing the morphological patterns in the highest-attended image patches from each WSI. Since WSIs have differing image dimensions, we extracted a proportional amount of high attention image patches (1%) to the total image dimension. On average, each WSI contained 13,487 512 × 512 40 × images, with approximately 135 image patches used as high attention regions. These attention patches are analyzed using a HoverNet model pretrained for simultaneous cell instance segmentation and classification (Graham et al., 2019). Cells are classified as either tumor cells (red), lymphocytes (green), connective tissue (blue), dead cells (yellow), or non-neoplastic epithelial cells (orange). For each of these cell types, we analyzed the cell type frequency across all counted cells in the highest-attended image patches in a given patient, then analyzed the cell fraction distribution across all patients in low-risk and high-risk patients, defined as patients below and above the 25% and 75% predicted risk percentiles respectively.
Tumor-Infiltrating Lymphocyte Detection.
To detect Tumor-Infiltrating Lymphocyte (TIL) presence in image patches, similar to other work, we defined TIL presence as the co-localization of tumor and immune cells which reflects intratumoral TIL response (Maley et al., 2015; Shaban et al., 2019). Following cell instance segmentation and classification of tumor and immune cells in the highest-attended 512 × 512 20 × image patches, we defined a heuristic which classified an image patch as positive for TIL presence with high tumor-immune cell co-localization (patch containing more than 20 counted cells, and more than 10 detected lymphocytes and 5 detected tumor cells). Similar to computing cell fraction distributions, for the highest-attended image patches in a given patient, we computed the fraction of TIL positive image patches, and plotted its distribution in low and high risk patients.
Local and Global SNN Interpretability.
For a given set of molecular features belonging to a patient sample, to characterize feature importance magnitude and direction of impact, we used Integrated Gradients (IG), a gradient-based feature attribution method that attributes the prediction of deep networks to their inputs (Sundararajan et al., 2017). IG satisfies two axioms for interpretability: 1) Sensitivity, in which for every desired input and baseline that differ in one feature but have different predictions, the differing feature should be given a non-zero attribution, and 2) Implementation Invariance, which states that two networks are functionally equivalent if their outputs are equal for all inputs. For a given input , IG calculates the gradients of across different scales against a zero-scaled baseline , which then uses the Gauss-Legendre quadrature to approximate the integral of gradients.
Using IG, for each molecular feature in input belonging to a patient sample, we compute feature attribution values, which corresponds to the magnitude of how much varying that feature in will change the output. Features that have no impact on the output would have zero attribution, whereas features that affect the output would have larger magnitude (interpreted as feature importance). In the context of regression tasks such as survival analysis, features that are positive attribution contribute towards increasing the output value (high risk), whereas negative attribution corresponds with decreasing the output value (low risk). For individual samples, we can use IG to understand how molecular features contribute towards the model risk prediction, which we can visualize as bar plots (termed local interpretability), where the x-axis corresponds with attribution value, the y-axis ranks features in order of absolute attribution magnitude (in descending order), and color corresponds with feature value. For binary data such as mutation status, bar colors are either colored blue (feature value ‘0’, wild-type) or red (feature value ‘1’, or mutation). For categorical and continuous data such as copy number variation and RNA-Seq abundance, bar colors are visualized using heatmap colors, where blue is low feature value (copy number loss / low RNA-Seq abundance) and red is high feature value (copy number gain / high RNA-Seq abundance). For large cohorts of patients from a cancer type, we can visualize the distribution of feature attributions across all patients (termed global interpretability), where each dot represents the attribution and feature value of an individual feature of an individual patient sample. Plots and terminology for local and global interpretability were derived from decision plots in Shapley Additive Explanation-based methods (Lundberg et al., 2020).
Measuring WSI Contribution in Model Prediction.
To measure the contribution of WSIs in model predictions, for each patient sample, we compute the attributions for each modality at the penultimate hidden layer before multimodal fusion (last layer of the SNN and AMIL subnetworks). Then, we normalize the sum of absolute attribution values for each modality, to estimate percentage that each modality contributes towards the model prediction (Kokhlikyan et al., 2020).
Computational Hardware and Software
PORPOISE was built with the OpenSeaDragon API and is hosted on Google Cloud. Python (version 3.7.7) packages used by PORPOISE include PyTorch (version 1.3.0), Lifelines (version 0.24.6), NumPy (version 1.18.1), Pandas (version 1.1.3), PIL (version 7.0.0), and OpenSlide (version 1.1.1). All WSIs were processed on Intel Xeon multi-core CPUs (Central Processing Units) and a total of 16 2080 Ti GPUs (Graphics Processing Units) using our custom, publicly available CLAM (Lu et al., 2021) whole slide processing pipeline. The multimodal fusion layer for integrating WSIs and molecular profiles was implemented using our custom, publicly available Pathomic Fusion (Chen et al., 2020) software implemented in Python. Deep learning models were trained with Nvidia softwares CUDA 11.0 and cuDNN 7.5. Integrated Gradients was implemented using Captum (version 0.2.0) (Kokhlikyan et al., 2020). Cell instance segmentation and classification was implemented using the HoVerNet software (Graham et al., 2019). Statistical analyses such as two-sampled t-tests and logrank tests used implementations from SciPy (1.4.1) and Lifelines (version 0.24.6) respectively. Plotting and visualization packages were generated using Seaborn (0.9.0), Matplotlib (version 3.1.1), and Shap (0.35.0
Quantification and Statistical Analysis
To plot the Kaplan-Meier curves, we aggregated out-of-sample risk predictions from the validation folds and plotted them against their survival time (Mobadersany et al., 2018). For significance testing of patient stratification in Kaplan-Meier analysis, we use the logrank test to measure if the difference of two survival distributions is statistically significant (P-Value < 0.05) (Bland and Altman, 2004). Cross-validated c-Index performance is reported as the average c-Index over the 5-folds. To estimate 95% confidence intervals in cross-validation, we performed non-parametric bootstrapping using 1000 replicates on the out-of-sample predictions in the validation folds (LeDell et al., 2015; Tsamardinos et al., 2018). In addition to c-Index, we also report Cumulative / Dynamic AUC (termed Survival AUC), a time-dependent measure of model performance that evaluates how well the model stratifies patient risk across various time points, and additionally corrects for optimistic bias from censorship via computing an inverse probability of censor weighting. For assessing global morphological feature significance of individual cell type presence, two-sample t-tests were performed in evaluating the statistical significance of mean cell fraction distributions in the top 1% of high attention regions of low and high-risk patients (P-Value < 0.05). For assessing global molecular feature significance of individual gene features, two-sample t-tests were performed in evaluating the statistical significance of attribution distributions of low and high gene feature values (below and above median gene feature value respectively). For all box plots, boxes indicate the 1st, median, and 3rd quartile values of the data distribution, and whiskers extend to data points within 1.5× the interquartile range.
Supplementary Material
Table S3. Additional Data Tables and Ablation Studies. Related to Figure 2.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| Biological samples | ||
| Whole slide images (TCGA) | https://portal.gdc.cancer.gov/ | RRID:SCR_003193 |
| Chemicals, peptides, and recombinant proteins | ||
| Critical commercial assays | ||
| Deposited data | ||
| The Cancer Genome Atlas | https://portal.gdc.cancer.gov/ | RRID:SCR_003193 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and algorithms | ||
| PORPOISE | This paper; https://github.com/mahmoodlab/PORPOISE | |
| CLAM | https://github.com/mahmoodlab/CLAM | |
| Pathomic Fusion | https://github.com/mahmoodlab/PathomicFusion | |
| HoVeR-Net | https://github.com/vqdang/hover_net | |
| Python (3.7.7) | https://www.python.org/ | RRID:SCR_008394 |
| NVIDIA CUDA (11.0) | https://developer.nvidia.com/cuda-toolkit | |
| NVIDIA cuDNN (7.5) | https://developer.nvidia.com/cudnn | |
| PyTorch (1.6.0) | https://pytorch.org | RRID:SCR_018536 |
| Captum (0.2.0) | https://captum.ai | |
| NumPy (1.18.1) | http://www.numpy.org | RRID:SCR_008633 |
| Pandas (1.1.3) | https://pandas.pydata.org | RRID:SCR_018214 |
| PIL (7.0.0) | https://pillow.readthedocs.io/en/stable/ | |
| Openslide (1.1.1) | https://openslide.org/ | |
| Scipy (1.4.1) | http://www.scipy.org | RRID:SCR_008058 |
| Lifelines (0.24.6) | https://lifelines.readthedocs.io/ | |
| Seaborn (0.9.0) | https://seaborn.pydata.org/ | |
| Matplotlib (3.1.1) | https://matplotlib.org/ | RRID:SCR_008624 |
| Shap (0.35.0) | https://shap.readthedocs.io/en/latest/index.html | |
| Other | ||
Highlights.
Multimodal data fusion improves prognostic models for a majority of cancer types.
Multimodal attribution elucidates the importance of individual modalities.
Model interpretability elucidates morphologic and molecular correlates of prognosis.
Acknowledgements
This work was supported in part by BWH President’s Fund, MGH Pathology, Google Cloud Research Grant, Nvidia GPU Grant Program, NIGMS R35GM138216 (to F.M.). R.J.C. was additionally supported by the National Science Foundation (NSF) Graduate Fellowship. M.S. was additionally supported by the National Institutes of Health (NIH) National Library of Medicine (NLM) Biomedical Informatics and Data Science Research Training Program, T15LM007092. M.W. was additionally supported by the NIH National Human Genome Research Institute (NHGRI) Ruth L. Kirschstein National Research Service Award Bioinformatics Training Grant, T32HG002295. T.Y.C. was additionally supported by the NIH National Cancer Institute (NCI) Ruth L. Kirschstein National Service Award, T32CA251062. The content is solely the responsibility of the authors and does not reflect the official views of the NIH, NIGMS, NHGRI, NLM or the NCI.
Footnotes
Declaration of Interests
R.C. and F.M. are inventors on a patient which has been filed corresponding multimodal data fusion using deep learning. The authors declare no other competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdelmoula WM, Balluff B, Englert S, Dijkstra J, Reinders MJ, Walch A, McDonnell LA and Lelieveldt BP, (2016). Data-driven identification of prognostic tumor subpopulations using spatially mapped t-SNE of mass spectrometry imaging data. Proceedings of the National Academy of Sciences, 113(43), pp.12244–12249. 10.1073/pnas.1510227113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- AbdulJabbar K, Raza SEA, Rosenthal R, Jamal-Hanjani M, Veeriah S, Akarca A, Lund T, Moore DA, Salgado R, Al Bakir M. and Zapata L, (2020). Geospatial immune variability illuminates differential evolution of lung adenocarcinoma. Nature Medicine, 26(7), pp.1054–1062. 10.1038/s41591-020-0900-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin MB, Greene FL, Edge SB, Compton CC, Gershenwald JE, Brookland RK, Meyer L, Gress DM, Byrd DR and Winchester DP, (2017). The eighth edition AJCC cancer staging manual: continuing to build a bridge from a population‐ based to a more “personalized” approach to cancer staging. CA: a cancer journal for clinicians, 67(2), pp.93–99. 10.3322/caac.21388 [DOI] [PubMed] [Google Scholar]
- Bai H, Harmancı AS, Erson-Omay EZ, Li J, Coşkun S, Simon M, Krischek B, Özduman K, Omay SB, Sorensen EA and Turcan Ş, (2016). Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nature genetics, 48(1), pp.59–66. 10.1038/ng.3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltrušaitis T, Ahuja C. and Morency LP, (2018). Multimodal machine learning: A survey and taxonomy. IEEE transactions on pattern analysis and machine intelligence, 41(2), pp.423–443. 10.1109/tpami.2018.2798607 [DOI] [PubMed] [Google Scholar]
- Beck AH, Sangoi AR, Leung S, Marinelli RJ, Nielsen TO, Van De Vijver MJ, West RB, Van De Rijn M. and Koller D, (2011). Systematic analysis of breast cancer morphology uncovers stromal features associated with survival. Science translational medicine, 3(108), pp.108ra113–108ra113. 10.1126/scitranslmed.3002564 [DOI] [PubMed] [Google Scholar]
- Bejnordi BE, Lin J, Glass B, Mullooly M, Gierach GL, Sherman ME, Karssemeijer N, Van Der Laak J. and Beck AH, (2017), April. Deep learning-based assessment of tumor-associated stroma for diagnosing breast cancer in histopathology images. In 2017 IEEE 14th international symposium on biomedical imaging (ISBI 2017) (pp. 929–932). IEEE. 10.1109/isbi.2017.7950668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bera K, Schalper KA, Rimm DL, Velcheti V. and Madabhushi A, (2019). Artificial intelligence in digital pathology—new tools for diagnosis and precision oncology. Nature reviews Clinical oncology, 16(11), pp.703–715. 10.1038/s41571-019-0252-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund E, Maaskola J, Schultz N, Friedrich S, Marklund M, Bergenstråhle J, Tarish F, Tanoglidi A, Vickovic S, Larsson L. and Salmen F, (2018). Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nature communications, 9(1), pp.1–13. 10.1038/s41467-018-04724-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland JM and Altman DG, 2004. The logrank test. Bmj, 328(7447), p.1073. 10.1136/bmj.328.7447.1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH and Beroukhim R, (2013). The somatic genomic landscape of glioblastoma. Cell, 155(2), pp.462–477. 10.1016/j.cell.2014.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella G, Hanna MG, Geneslaw L, Miraflor A, Werneck Krauss Silva V, Busam KJ, Brogi E, Reuter VE, Klimstra DS and Fuchs TJ, (2019). Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nature medicine, 25(8), pp.1301–1309. 10.1038/s41591-019-0508-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Borowsky A, Spellman P. and Parvin B, (2013). Classification of tumor histology via morphometric context. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (pp. 2203–2210). 10.1109/cvpr.2013.286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RJ, Lu MY, Wang J, Williamson DF, Rodig SJ, Lindeman NI and Mahmood F, (2020). Pathomic fusion: an integrated framework for fusing histopathology and genomic features for cancer diagnosis and prognosis. IEEE Transactions on Medical Imaging. 10.1109/tmi.2020.3021387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, Ries CH, Ailles L, Jewett MAS, Moch H. and van den Broek M, (2017). An immune atlas of clear cell renal cell carcinoma. Cell, 169(4), pp.736–749. 10.1016/j.cell.2017.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA, Sanders CM and Kawaguchi ES, (2019). Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nature medicine, 25(3), pp.477–486. 10.1038/s41591-018-0337-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudray N, Ocampo PS, Sakellaropoulos T, Narula N, Snuderl M, Fenyö D, Moreira AL, Razavian N. and Tsirigos A, (2018). Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning. Nature medicine, 24(10), pp.1559–1567. 10.1038/s41591-018-0177-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtiol P, Maussion C, Moarii M, Pronier E, Pilcer S, Sefta M, Manceron P, Toldo S, Zaslavskiy M, Le Stang N. and Girard N, (2019). Deep learning-based classification of mesothelioma improves prediction of patient outcome. Nature medicine, 25(10), pp.1519–1525. 10.1038/s41591-019-0583-3 [DOI] [PubMed] [Google Scholar]
- Diao JA, Wang JK, Chui WF, Mountain V, Gullapally SC, Srinivasan R, Mitchell RN, Glass B, Hoffman S, Rao SK and Maheshwari C, (2021). Human-interpretable image features derived from densely mapped cancer pathology slides predict diverse molecular phenotypes. Nature communications, 12(1), pp.1–15. 10.1038/s41467-021-21896-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echle A, Rindtorff NT, Brinker TJ, Luedde T, Pearson AT and Kather JN, (2021). Deep learning in cancer pathology: a new generation of clinical biomarkers. British journal of cancer, 124(4), pp.686–696. 10.1038/s41416-020-01122-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman WH, Zitvogel L, Sautès–Fridman C. and Kroemer G, (2017). The immune contexture in cancer prognosis and treatment. Nature reviews Clinical oncology, 14(12), pp.717–734. 10.1038/nrclinonc.2017.101 [DOI] [PubMed] [Google Scholar]
- Fu Y, Jung AW, Torne RV, Gonzalez S, Vöhringer H, Shmatko A, Yates LR, Jimenez-Linan M, Moore L. and Gerstung M, (2020). Pan-cancer computational histopathology reveals mutations, tumor composition and prognosis. Nature Cancer, 1(8), pp.800–810. 10.1038/s43018-020-0085-8 [DOI] [PubMed] [Google Scholar]
- Gamble P, Jaroensri R, Wang H, Tan F, Moran M, Brown T, Flament-Auvigne I, Rakha EA, Toss M, Dabbs DJ and Regitnig P, (2021). Determining breast cancer biomarker status and associated morphological features using deep learning. Communications Medicine, 1(1), pp.1–12. 10.1038/s43856-021-00013-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentzler RD, Yentz SE, Johnson ML, Rademaker AW and Patel JD, (2014). The changing landscape of phase II/III metastatic NSCLC clinical trials and the importance of biomarker selection criteria. Cancer, 120(24), pp.3853–3858. 10.1002/cncr.28956 [DOI] [PubMed] [Google Scholar]
- Giesen C, Wang HA, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, Schüffler PJ, Grolimund D, Buhmann JM, Brandt S. and Varga Z, (2014). Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nature methods, 11(4), pp.417–422. 10.1038/nmeth.2869 [DOI] [PubMed] [Google Scholar]
- Graham S, Vu QD, Raza SEA, Azam A, Tsang YW, Kwak JT and Rajpoot N, (2019). Hover-net: Simultaneous segmentation and classification of nuclei in multi-tissue histology images. Medical Image Analysis, 58, p.101563. 10.1016/j.media.2019.101563 [DOI] [PubMed] [Google Scholar]
- Harder N, Schönmeyer R, Nekolla K, Meier A, Brieu N, Vanegas C, Madonna G, Capone M, Botti G, Ascierto PA and Schmidt G, (2019). Automatic discovery of image-based signatures for ipilimumab response prediction in malignant melanoma. Scientific reports, 9(1), pp.1–19. 10.1038/s41598-019-43525-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Bergenstråhle L, Stenbeck L, Abid A, Andersson A, Borg Å, Maaskola J, Lundeberg J. and Zou J, (2020). Integrating spatial gene expression and breast tumour morphology via deep learning. Nature biomedical engineering, 4(8), pp.827–834. 10.1038/s41551-020-0578-x [DOI] [PubMed] [Google Scholar]
- Heindl A, Nawaz S. and Yuan Y, (2015). Mapping spatial heterogeneity in the tumor microenvironment: a new era for digital pathology. Laboratory investigation, 95(4), pp.377–384. 10.1038/labinvest.2014.155 [DOI] [PubMed] [Google Scholar]
- Hyman DM, Solit DB, Arcila ME, Cheng DT, Sabbatini P, Baselga J, Berger MF and Ladanyi M, (2015). Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug discovery today, 20(12), pp.1422–1428. 10.1016/j.drudis.2015.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilse M, Tomczak J. and Welling M, (2018), July. Attention-based deep multiple instance learning. In International conference on machine learning (pp. 2127–2136). PMLR 80:2127–2136. (PMLR does not have a DOI) [Google Scholar]
- Jackson HW, Fischer JR, Zanotelli VR, Ali HR, Mechera R, Soysal SD, Moch H, Muenst S, Varga Z, Weber WP and Bodenmiller B, (2020). The single-cell pathology landscape of breast cancer. Nature, 578(7796), pp.615–620. 10.1038/s41586-019-1876-x [DOI] [PubMed] [Google Scholar]
- Kather JN, Heij LR, Grabsch HI, Loeffler C, Echle A, Muti HS, Krause J, Niehues JM, Sommer KA, Bankhead P. and Kooreman LF, (2020). Pan-cancer image-based detection of clinically actionable genetic alterations. Nature Cancer, 1(8), pp.789–799. 10.1038/s43018-020-0087-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kather JN, Suarez-Carmona M, Charoentong P, Weis CA, Hirsch D, Bankhead P, Horning M, Ferber D, Kel I, Herpel E. and Schott S, (2018). Topography of cancer-associated immune cells in human solid tumors. Elife, 7, p.e36967. 10.7554/elife.36967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kather JN, Krisam J, Charoentong P, Luedde T, Herpel E, Weis CA, Gaiser T, Marx A, Valous NA, Ferber D. and Jansen L, (2019). Predicting survival from colorectal cancer histology slides using deep learning: A retrospective multicenter study. PLoS medicine, 16(1), p.e1002730. 10.1371/journal.pmed.1002730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kather JN, Pearson AT, Halama N, Jäger D, Krause J, Loosen SH, Marx A, Boor P, Tacke F, Neumann UP and Grabsch HI, (2019). Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nature medicine, 25(7), pp.1054–1056. 10.1038/s41591-019-0462-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kather JN and Calderaro J, (2020). Development of AI-based pathology biomarkers in gastrointestinal and liver cancer. Nature Reviews Gastroenterology & Hepatology, 17(10), pp.591–592. / 10.1038/s41575-020-0343-3 [DOI] [PubMed] [Google Scholar]
- Klambauer G, Unterthiner T, Mayr A. and Hochreiter S, (2017). Self-normalizing neural networks. Advances in neural information processing systems, 30. (NIPS’17 did not have a DOI at that year.) [Google Scholar]
- Kokhlikyan N, Miglani V, Martin M, Wang E, Alsallakh B, Reynolds J, Melnikov A, Kliushkina N, Araya C, Yan S. and Reblitz-Richardson O, (2020). Captum: A unified and generic model interpretability library for pytorch. Preprint at arXiv, 10.48550/arXiv.2009.07896. [DOI] [Google Scholar]
- Kulkarni PM, Robinson EJ, Pradhan JS, Gartrell-Corrado RD, Rohr BR, Trager MH, Geskin LJ, Kluger HM, Wong PF, Acs B. and Rizk EM, (2020). Deep learning based on standard H&E images of primary melanoma tumors identifies patients at risk for visceral recurrence and death. Clinical Cancer Research, 26(5), pp.1126–1134. 10.1158/1078-0432.ccr-19-1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA and Kiezun A, (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature, 499(7457), pp.214–218. 10.1038/nature12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDell E, Petersen M. and van der Laan M, (2015). Computationally efficient confidence intervals for cross-validated area under the ROC curve estimates. Electronic journal of statistics, 9(1), p.1583. 10.1214/15-ejs1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Shen Y, Lakshminarasimhan VB, Liang PP, Bagher Zadeh A, and Morency L-P (2018). Efficient Low-rank Multimodal Fusion With Modality-Specific Factors (Association for Computational Linguistics; ). 10.18653/v1/P18-1209 [DOI] [Google Scholar]
- Louis DN, Perry A, Reifenberger G, Von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P. and Ellison DW, (2016). The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta neuropathologica, 131(6), pp.803–820. 10.1007/s00401-016-1545-1 [DOI] [PubMed] [Google Scholar]
- Lu MY, Williamson DF, Chen TY, Chen RJ, Barbieri M. and Mahmood F, (2021). Data-efficient and weakly supervised computational pathology on whole-slide images. Nature biomedical engineering, 5(6), pp.555–570. 10.1038/s41551-020-00682-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig JA and Weinstein JN, (2005). Biomarkers in cancer staging, prognosis and treatment selection. Nature Reviews Cancer, 5(11), pp.845–856. 10.1038/nrc1739 [DOI] [PubMed] [Google Scholar]
- Lundberg SM, Erion G, Chen H, DeGrave A, Prutkin JM, Nair B, Katz R, Himmelfarb J, Bansal N. and Lee SI, (2020). From local explanations to global understanding with explainable AI for trees. Nature machine intelligence, 2(1), pp.56–67. 10.1038/s42256-019-0138-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maley CC, Koelble K, Natrajan R, Aktipis A. and Yuan Y, (2015). An ecological measure of immune-cancer colocalization as a prognostic factor for breast cancer. Breast Cancer Research, 17(1), pp.1–13. 10.1186/s13058-015-0638-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusyk A, Almendro V. and Polyak K, (2012). Intra-tumour heterogeneity: a looking glass for cancer?. Nature Reviews Cancer, 12(5), pp.323–334. 10.1038/nrc3261 [DOI] [PubMed] [Google Scholar]
- Mayekar MK and Bivona TG, (2017). Current landscape of targeted therapy in lung cancer. Clinical Pharmacology & Therapeutics, 102(5), pp.757–764. 10.1002/cpt.810 [DOI] [PubMed] [Google Scholar]
- Mobadersany P, Yousefi S, Amgad M, Gutman DA, Barnholtz-Sloan JS, Vega JEV, Brat DJ and Cooper LA, (2018). Predicting cancer outcomes from histology and genomics using convolutional networks. Proceedings of the National Academy of Sciences, 115(13), pp.E2970–E2979. 10.1101/198010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network, (2013). Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature, 499(7456), p.43. 10.1038/nature12222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson AG, Perry LJ, Cury PM, Jackson P, McCormick CM, Corrin B. and Wells AU, (2001). Reproducibility of the WHO/IASLC grading system for pre‐ invasive squamous lesions of the bronchus: a study of inter‐ observer and intra‐ observer variation. Histopathology, 38(3), pp.202–208. 10.1046/j.13652559.2001.01078.x [DOI] [PubMed] [Google Scholar]
- Novara G, Martignoni G, Artibani W. and Ficarra V, (2007). Grading systems in renal cell carcinoma. The Journal of urology, 177(2), pp.430–436. 10.1016/j.juro.2006.09.034 [DOI] [PubMed] [Google Scholar]
- Oh JH, Jang SJ, Kim J, Sohn I, Lee JY, Cho EJ, Chun SM and Sung CO, (2020). Spontaneous mutations in the single TTN gene represent high tumor mutation burden. NPJ genomic medicine, 5(1), pp.1–11. 10.1038/s41525-019-0107-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrelli F, Ghidini M, Cabiddu M, Pezzica E, Corti D, Turati L, Costanzo A, Varricchio A, Ghidini A, Barni S. and Tomasello G, (2019). Microsatellite instability and survival in stage II colorectal cancer: a systematic review and meta-analysis. Anticancer Research, 39(12), pp.6431–6441. 10.21873/anticanres.13857 [DOI] [PubMed] [Google Scholar]
- Rabe K, Snir OL, Bossuyt V, Harigopal M, Celli R. and Reisenbichler ES, (2019). Interobserver variability in breast carcinoma grading results in prognostic stage differences. Human pathology, 94, pp.51–57. 10.1016/j.humpath.2019.09.006 [DOI] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS and Miller ML, (2015). Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science, 348(6230), pp.124–128. 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltz J, Gupta R, Hou L, Kurc T, Singh P, Nguyen V, Samaras D, Shroyer KR, Zhao T, Batiste R. and Van Arnam J, (2018). Spatial organization and molecular correlation of tumor-infiltrating lymphocytes using deep learning on pathology images. Cell reports, 23(1), pp.181–193. 10.1016/j.celrep.2018.03.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapiro D, Jackson HW, Raghuraman S, Fischer JR, Zanotelli VR, Schulz D, Giesen C, Catena R, Varga Z. and Bodenmiller B, (2017). histoCAT: analysis of cell phenotypes and interactions in multiplex image cytometry data. Nature methods, 14(9), pp.873–876. 10.1038/nmeth.4391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaban M, Khurram SA, Fraz MM, Alsubaie N, Masood I, Mushtaq S, Hassan M, Loya A. and Rajpoot NM, (2019). A novel digital score for abundance of tumour infiltrating lymphocytes predicts disease free survival in oral squamous cell carcinoma. Scientific reports, 9(1), pp.1–13. 10.1038/s41598-019-49710-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi JY, Wang X, Ding GY, Dong Z, Han J, Guan Z, Ma LJ, Zheng Y, Zhang L, Yu GZ and Wang XY, (2021). Exploring prognostic indicators in the pathological images of hepatocellular carcinoma based on deep learning. Gut, 70(5), pp.951–961. 10.1136/gutjnl-2020-320930 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES and Mesirov JP, (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences, 102(43), pp.15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararajan M, Taly A. and Yan Q, (2017), July. Axiomatic attribution for deep networks. In International conference on machine learning (pp. 3319–3328). PMLR 70:3319–3328.( PMLR does not have a DOI.) [Google Scholar]
- Tarantino P, Mazzarella L, Marra A, Trapani D. and Curigliano G, (2021). The evolving paradigm of biomarker actionability: histology-agnosticism as a spectrum, rather than a binary quality. Cancer Treatment Reviews, 94, p.102169. 10.1016/j.ctrv.2021.102169 [DOI] [PubMed] [Google Scholar]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Yang THO, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA and Ziv E, (2018). The immune landscape of cancer. Immunity, 48(4), pp.812–830. 10.1016/j.immuni.2021.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsamardinos I, Greasidou E. and Borboudakis G, (2018). Bootstrapping the out-of-sample predictions for efficient and accurate cross-validation. Machine Learning, 107(12), pp.1895–1922. 10.1007/s10994-018-5714-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Karlsson MJ, Zhong W, Tebani A, Pou C, Mikes J, Lakshmikanth T, Forsström B, Edfors F, Odeberg J. and Mardinoglu A, 2019. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science, 366(6472), p.eaax9198. 10.1126/science.aax9198 [DOI] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F. and Sanli K, (2017). A pathology atlas of the human cancer transcriptome. Science, 357(6352), p.eaan2507. 10.1126/science.aan2507 [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A. and Olsson I, (2015). Tissue-based map of the human proteome. Science, 347(6220), p.1260419. 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- Wang W, Tran D. and Feiszli M, (2020). What makes training multi-modal classification networks hard?. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition (pp. 12695–12705). 10.1109/cvpr42600.2020.01271 [DOI] [Google Scholar]
- Zadeh A, Chen M, Poria S, Cambria E, and Morency L-P (2017). Tensor Fusion Network for Multimodal Sentiment Analysis (Association for Computational Linguistics; ). 10.18653/v1/D17-1115 [DOI] [Google Scholar]
- Zadeh SG and Schmid M, (2020). Bias in cross-entropy-based training of deep survival networks. IEEE Transactions on Pattern Analysis and Machine Intelligence, 43(9), pp.3126–3137. 10.1109/tpami.2020.2979450 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Xu CR, Cheng Y, Liu YP, Chen GY, Cui JW, Yang N, Song Y, Li XL, Lu S. and Zhou JY, (2021). Bevacizumab plus erlotinib in Chinese patients with untreated, EGFR-mutated, advanced NSCLC (ARTEMIS-CTONG1509): a multicenter phase 3 study. Cancer Cell, 39(9), pp.1279–1291. 10.1016/j.ccell.2021.07.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3. Additional Data Tables and Ablation Studies. Related to Figure 2.
Data Availability Statement
All diagnostic whole slide images (WSIs) and their corresponding molecular and clinical data were obtained from The Cancer Genome Atlas and are publicly accessible through the NIH Genomic Data Commons Data Portal https://portal.gdc.cancer.gov/. All code was implemented in Python, using PyTorch as the primary deep learning package. All code and scripts to reproduce the experiments of this paper are available at: https://github.com/mahmoodlab/PORPOISE.