

Abstract
53BP1 is a chromatin‐binding protein that promotes DNA double‐strand break repair through the recruitment of downstream effectors including RIF1, shieldin, and CST. The structural basis of the protein–protein interactions within the 53BP1‐RIF1‐shieldin‐CST pathway that are essential for its DNA repair activity is largely unknown. Here, we used AlphaFold2‐Multimer (AF2) to predict all possible pairwise combinations of proteins within this pathway and provide structural models of seven previously characterized interactions. This analysis also predicted an entirely novel binding interface between the HEAT‐repeat domain of RIF1 and the eIF4E‐like domain of SHLD3. Extensive interrogation of this interface through both in vitro pulldown analysis and cellular assays supports the AF2‐predicted model and demonstrates that RIF1‐SHLD3 binding is essential for shieldin recruitment to sites of DNA damage, and for its role in antibody class switch recombination and PARP inhibitor sensitivity. Direct physical interaction between RIF1 and SHLD3 is therefore essential for 53BP1‐RIF1‐shieldin‐CST pathway activity.
Keywords: 53BP1, AlphaFold2, DNA double‐strand breaks, protein interaction prediction, shieldin
Subject Categories: DNA Replication, Recombination & Repair; Structural Biology
The 53BP1‐RIF1‐shieldin pathway regulates DNA double‐strand break repair. This study uses AlphaFold2 to map the protein–protein interactions in this pathway and verifies that the predicted SHLD3–RIF1 interaction is essential for shieldin activity.
Introduction
The nucleolytic processing and fill‐in synthesis of the ends of DNA double‐strand breaks (DSBs) is under the control of the chromatin‐binding protein 53BP1, which is recruited to RNF168‐ubiquitylated chromatin in a large domain that flanks the DSB (Panier & Boulton, 2014). The regulation of DNA end processing (or stability) impacts the type of DSB repair employed to mend the break, with 53BP1 promoting DNA end stability, which in turn favors nonhomologous end‐joining (NHEJ). 53BP1 carries out this function by acting as a platform for the recruitment of downstream factors that include RIF1 and PTIP (Callen et al, 2013; Chapman et al, 2013; Escribano‐Díaz et al, 2013; Zimmermann et al, 2013). RIF1 then recruits the shieldin complex to facilitate DNA repair (Dev et al, 2018; Gao et al, 2018; Ghezraoui et al, 2018; Gupta et al, 2018; Mirman et al, 2018; Noordermeer et al, 2018; Setiaputra & Durocher, 2019). The 53BP1‐RIF1‐shieldin pathway promotes NHEJ and DSB end‐stability by opposing the 5′‐3′ nuclease degradation of DSB ends associated with homologous recombination (HR) repair. 53BP1‐RIF1‐shieldin‐stimulated NHEJ is essential for antibody class switch recombination in B cells (CSR; Ward et al, 2004; Chapman et al, 2013; Ghezraoui et al, 2018; Noordermeer et al, 2018). Its role in suppressing HR mediates the toxicity of poly(ADP) ribose polymerase 1 inhibitor (PARPi) in BRCA1‐mutated tumors (Noordermeer et al, 2018). Furthermore, 53BP1 accumulates in large nuclear bodies in G1 following DNA replication stress during the preceding cell cycle and suppresses toxic HR‐mediated repair at these lesions (Spies et al, 2019). Although the precise mechanism underlying HR suppression by this pathway remains under investigation, shieldin recruits the CTC1‐STN1‐TEN1 (CST)‐Pol α‐Primase complex to DSBs to perform fill‐in DNA synthesis at resected breaks (Mirman et al, 2018, 2022b; Schimmel et al, 2021). Shieldin has also been shown to recruit the structure‐specific endonuclease ASTE1 to DSBs where it cleaves single‐stranded DNA (ssDNA) overhangs (Zhao et al, 2021). Understanding the molecular basis underlying the recruitment of 53BP1 and its downstream effectors to DSBs is essential to determine how this pathway regulates DNA repair.
53BP1 is recruited to DNA‐damage sites by directly binding methylated histone H4 and DNA‐damage‐induced histone H2A lysine 15 ubiquitylation through its tandem Tudor and ubiquitin‐dependent recruitment domains, respectively (Fradet‐Turcotte et al, 2013). RIF1 localizes to DNA breaks by binding three doubly‐phosphorylated 53BP1 epitopes that are characterized by an LxL dileucine motif (x signifies any residue) preceding the phosphoresidues (Setiaputra et al, 2022). RIF1 recognizes phosphorylated 53BP1 using its N‐terminal HEAT‐repeat domain (Setiaputra et al, 2022). Precisely how shieldin is recruited to DSBs is unknown, although RIF1 and shieldin can interact (Noordermeer et al, 2018; Setiaputra et al, 2022), suggesting that a hitherto uncharacterized RIF1‐shieldin binding interface mediates shieldin localization to and function at DNA break sites. Shieldin consists of four subunits: SHLD1, SHLD2, SHLD3, and REV7 (Setiaputra & Durocher, 2019). SHLD3 is required for DSB localization of all other shieldin subunits, making it the primary candidate for RIF1‐mediated recruitment (Noordermeer et al, 2018). SHLD3 binds REV7 through its N terminus, and REV7 in turn binds the SHLD2 N terminus (Ghezraoui et al, 2018; Dai et al, 2020; Liang et al, 2020). The SHLD2 C terminus consists of three tandem oligonucleotide‐binding folds (OB‐folds) that bind ssDNA and serve as the binding site of SHLD1 (Noordermeer et al, 2018). Yeast‐two‐hybrid experiments found that CST binds shieldin at multiple sites, with SHLD1 providing a key interaction interface (Mirman et al, 2018, 2022b). Aside from the nucleosome‐53BP1 and SHLD3‐REV7‐SHLD2 subcomplexes (Wilson et al, 2016; Liang et al, 2020), the structural basis for the protein–protein interactions outlined above are unknown.
In this study, we used the protein–protein interaction prediction algorithm AlphaFold2‐Multimer (AF2; preprint: Evans et al, 2021; Jumper et al, 2021; Mirdita et al, 2022) to probe the 53BP1‐RIF1‐shieldin‐CST pathway. This approach accurately predicts known binding interfaces and provides the structural basis for multiple previously characterized interactions within the 53BP1 pathway. Additionally, AF2 analysis predicted a hitherto undescribed interaction between the eIF4E‐like domain of SHLD3 and the HEAT‐repeat domain of RIF1. We confirmed this prediction both in vitro and in cellular assays measuring shieldin recruitment to sites of DNA damage by RIF1 and show that mutating this interface disrupts shieldin‐dependent CSR and PARPi sensitivity. Shieldin recruitment to DSB sites and its ability to modulate DSB repair are therefore dependent on a direct SHLD3–RIF1 interaction.
Results
Modeling the 53BP1‐RIF1‐shieldin‐CST interaction network using AlphaFold2
We sought to understand the structural basis for the interactions facilitating 53BP1‐RIF1‐shieldin‐CST recruitment to sites of DNA damage. Despite a good understanding of the domains involved in these interactions, the large size and propensity for disorder within the proteins of this pathway make them refractory for purification and structural determination. We instead used AlphaFold2‐Multimer (AF2) to predict the heterodimeric structure for every unique pairwise combination for all known components of this pathway (53BP1, RIF1, SHLD1, SHLD2, SHLD3, REV7, ASTE1, CTC1, STN1, TEN1; Fig 1A). Due to graphical memory limitations, we divided large proteins into multiple fragments. Five models were generated without using templates for each prediction and we scored each model based on pDockQ and mean interface‐predicted aligned error (PAE), two parameters that discriminate against incorrect AF2 predictions (Fig 1A, Table EV1; Bryant et al, 2022; Yin et al, 2022). Out of 1,775 models from 355 unique protein pairs, 138 models (7.8%) from 51 pairs satisfied the pDockQ and mean interface PAE cutoffs of > 0.23 and < 15 Å, respectively (Fig EV1A). To further remove false positives, we assigned higher prediction confidence to interaction pairs in which ≥ 4 out of five models were consistent with each other. Predictions meeting this cutoff are depicted in Fig 1B, with the addition of the 53BP1 oligomerization domain, which only has 3/5 consistent models but is supported by experimental evidence (Zgheib et al, 2009; Sundaravinayagam et al, 2019). Where possible, we merged multiple complementary pairwise predictions to depict larger macromolecular complexes (SHLD1‐SHLD2‐CTC1, CTC1‐STN1‐TEN1; Fig 1B panels v and viii). This analysis successfully recapitulated experimentally‐determined structures of SHLD3‐REV7 (PDB ID:6KTO; Liang et al, 2020) and the CST complex consisting of CTC1‐STN1‐TEN1 (PDB ID:8D0K; He et al, 2022), further supporting the utility of this approach (Fig 1B panels ix and viii). Interestingly, the AF2 analysis predicted multiple unusual REV7 structures. REV7 is known to form head‐to‐head dimers (Xie et al, 2021), but AF2 predicted an extensively domain‐swapped dimer instead (Fig EV1B). Furthermore, REV7 is predicted to interact with four unique RIF1 and 53BP1 fragments, and all four predict the same mode of HORMA domain seatbelt‐mediated interaction seen in REV7‐SHLD3 (Fig EV1C; Liang et al, 2020). Since we previously showed that REV7 requires SHLD3 for 53BP1 or RIF1 colocalization at DSBs (Noordermeer et al, 2018), and repeating the same AF2 prediction in the presence of the REV7‐interacting region of SHLD3 displaces these RIF1 and 53BP1 fragments (Appendix Fig S1), we excluded these models from further analysis. ASTE1 was not predicted to interact with any proteins tested (Fig EV1A).
Figure 1. Exhaustive AlphaFold2‐Multimer prediction of pairwise protein–protein interactions within the 53BP1‐RIF1‐shieldin‐CST pathway.
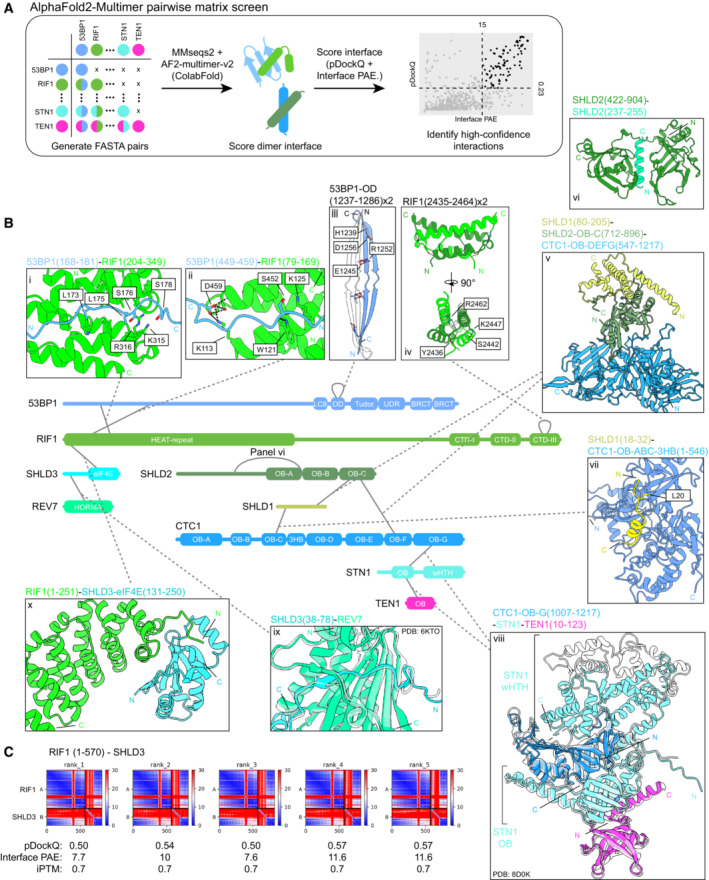
- Schematic of the AF2 pairwise matrix screen for protein–protein interactions within the 53BP1‐RIF1‐shieldin‐CST pathway. Scatter plot of the interface‐predicted aligned error (PAE) vs pDockQ scores for the models predicted in this analysis is shown, along with the cutoff values of 15 Å and 0.23 for PAE and pDockQ, respectively. Models meeting the cutoff are represented as black points and those that do not are represented as gray points.
- High‐confidence interactions predicted by AF2. Structures of interfaces are shown, with corresponding experimental structures overlaid (translucent) if available (CST complex and SHLD3‐REV7; PDB ID: 8D0K, 6KTO). Modeled hydrogen bonds and salt bridges are shown as black and red dashed lines, respectively. See also Fig EV1.
- PAE plots of RIF1 (1–570) and SHLD3, with calculated pDockQ, interface PAE, and interface‐predicted template modeling (iPTM) scores.
Source data are available online for this figure.
Figure EV1. Data supporting the AlphaFold2‐Multimer pairwise matrix screen for protein–protein interactions in the 53BP1‐RIF1‐shieldin‐CST pathway.
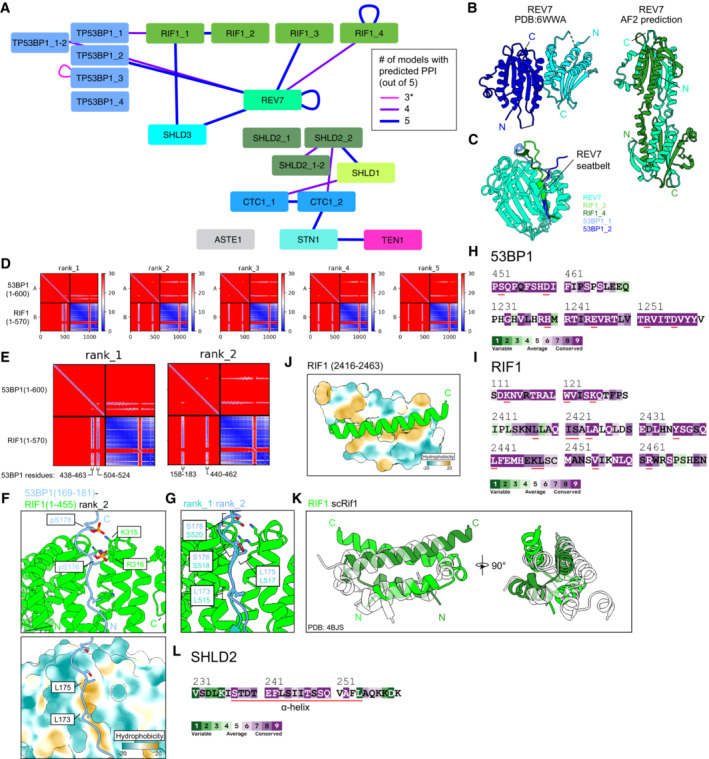
- Schematic of predictions that meet the cutoff scores of pDockQ > 0.23, interface PAE < 15 Å, and four or more repeated predictions. Nodes are proteins and fragments used for each prediction. Junction nodes are fragments spanning ± 200 residues from where large proteins are divided and are only shown if interactions meeting the score cutoffs are present (e.g., TP53BP1_1‐2). Edges link the two chains used for each prediction. Edge thickness and color correspond to the number of predictions (out of five for each pair) meeting the score cutoffs. *Self‐association of TP53BP1_3 is shown despite not meeting the ≥ 4 consistent model cutoff due to previous experimental results corroborating the predicted interaction.
- Comparison of REV7 dimer structures determined from X‐ray crystallography (Left, PDB ID: 6WWA) and from AlphaFold2 prediction (right).
- Superimposition of predicted heterodimeric structures between REV7 and RIF1 fragments 3 and 4 and 53BP1 fragments 1 and 2. See also Appendix Fig S1.
- PAE plots of the predicted 53BP1 (fragment 1; 1–600) and RIF1 (fragment 1; 1–570), ranked by predicted template model (pTM) scores.
- Close‐ups of two PAE plots from (D), with the 53BP1 regions of high PAE confidence labeled.
- Predicted structures of the 53BP1‐RIF1 interface. Top, phosphate groups modeled onto two serines of 53BP1 whose phosphorylation is known to be essential for interaction with RIF1. Bottom, surface representation of RIF1 colored by hydrophobicity.
- Superimposition of the 53BP1‐RIF1 interface from two different predicted models.
- ConSurf sequence conservation analysis of the indicated 53BP1 regions. Residues important for the predicted secondary RIF1‐53BP1 interface (53BP1 residues 440–462) or 53BP1 oligomerization (53BP1 residues 1,237–1,286) are underlined in red.
- ConSurf sequence conservation analysis of the indicated RIF1 regions. Residues important for the predicted RIF1 oligomerization (RIF1 residues 2,435–2,464) or the predicted secondary RIF1‐53BP1 interface (RIF1 residues 79–169) are underlined in red.
- Predicted structure of the RIF1 dimerization interface, one monomer is shown with surface representation colored by hydrophobicity.
- Superimposition of the RIF1 oligomerization domain and the experimentally‐determined structure (translucent) of the corresponding region in S. cerevisiae Rif1 (PDB ID: 4BJS).
- ConSurf sequence conservation analysis of the indicated SHLD2 region. Residues comprising the alpha helix predicted to bind SHLD2 OB‐A and OB‐B are underlined in red.
Structural basis for 53BP1–RIF1 interaction and oligomerization
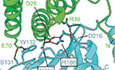
53BP1 is predicted to interact with RIF1 through its N‐terminal unstructured domain and with itself through its oligomerization domain (Fig 1B panels i–iii). We previously identified that RIF1 binds three 53BP1 phosphorylated epitopes that mediate its recruitment to DSB sites (Setiaputra et al, 2022). Strikingly, despite not being designed to identify post‐translational modifications, AF2 identified two of these motifs (Figs 1B panel i and EV1D–G). In both cases, the apposed serine and leucine residues are oriented towards a groove formed between two RIF1 α‐helical HEAT repeats (Fig EV1G). Remarkably, the two 53BP1 phosphoacceptor serine residues are oriented towards the RIF1 K315/R316 residues that we found to be essential for this interaction (Setiaputra et al, 2022). The predicted structures can accommodate phosphate groups modeled onto the putatively phosphorylated serines (Fig EV1F, top panel). The two leucine residues of the LxL motif, which are essential for RIF1‐53BP1 binding, are buried within a RIF1 hydrophobic cleft (Fig EV1F, bottom panel). Interestingly, all five AF2 models predict a second site of 53BP1–RIF1 interaction involving 53BP1 residues 440–462 (Figs 1B panel ii and EV1D and E). This region contains one predicted ATM phosphosite at S452 positioned adjacent to RIF1 K125, which are both highly conserved (Fig EV1H and I). 53BP1 serine 452 is not essential for RIF1 recruitment to DNA‐damage sites (Setiaputra et al, 2022), and potentially represents a secondary 53BP1–RIF1 interaction interface. This secondary interface is predicted to contain three hydrogen bonds between four highly conserved residues (Figs 1B panel ii and EV1H and I). These AF2 predictions provide a compelling structural explanation for the RIF1‐53BP1 phosphodependent interaction that we previously described.
53BP1 and RIF1 are both known to form higher‐ordered structures (Zgheib et al, 2009; Moriyama et al, 2018). 53BP1 multimerization through its oligomerization domain (OD) is important for its DNA‐damage localization and function (Zgheib et al, 2009; Sundaravinayagam et al, 2019). The AF2 prediction corresponding to this region predicted that the 53BP1‐OD dimerizes through an extended antiparallel beta sheet (Fig 1B panel iii) stabilized through backbone interactions and two salt bridges between highly conserved residues (D1256‐H1239 and E1245‐R1252; Fig EV1H). Notably, D1256 is essential for 53BP1 oligomerization and DNA‐damage recruitment, supporting this prediction (Zgheib et al, 2009). The 53BP1‐OD tetramerizes in vitro (Sundaravinayagam et al, 2019), but subsequent AF2 prediction using four copies of the domain did not recapitulate a tetrameric arrangement. Nevertheless, the predicted 53BP1‐OD dimer potentially represents a sub‐assembly within the tetrameric structure.
RIF1 is known to dimerize through both its N‐terminal HEAT repeats and its extreme C terminus (Moriyama et al, 2018). The AF2 analysis predicted C‐terminal dimerization through a head‐to‐head intercalated four‐helix bundle stabilized by a buried hydrophobic core and two hydrogen bonds (Figs 1B panel iv and EV1K). These interactions involve highly conserved hydrophobic and polar residues (L2417, I2421, L2424, L2441, L2448, V2455 and S2422, Y2436, R2462, K2447; Fig EV1I). This region is conserved in yeast Rif1, which oligomerizes in an analogous fashion (Fig EV1K; Shi et al, 2013). The biological importance of RIF1 dimerization is unclear and identifying specific residues necessary for this interaction will be essential for future efforts to address this question.
Predicted interactions between the shieldin and CST complexes
The molecular determinants of shieldin and CST complex assembly are relatively well understood. The AF2 analysis recapitulated the interaction between the SHLD3 N terminus and the REV7 HORMA domain (Liang et al, 2020; Fig 1B panel ix), though it did not detect the interaction between the SHLD2 N terminus and REV7. SHLD1 is known to interact with the SHLD2 C terminus, which contains three tandem OB‐folds, specifically through the third OB‐fold (OB‐C; Dev et al, 2018). Consistent with this experimental observation, AF2 predicts that SHLD1 binds the SHLD2 third OB‐fold (Fig 1B panel v). Furthermore, SHLD2 conserved residues 237–255 (Fig EV1L) form an alpha helix that is predicted to bind between the second and third OB‐folds of SHLD2 (Fig 1B panel vi). The CST complex structure has been solved by cryo‐electron microscopy (Lim et al, 2020; Cai et al, 2022; He et al, 2022), and AF2 correctly predicted the organization of the subcomplex consisting of the CTC1 C‐terminal OB‐fold, STN1 N‐terminal OB‐fold, and TEN1 (Fig 1B panel viii), though the position of STN1 C‐terminal winged helix‐turn‐helix (wHTH) domain bound to CTC1 was not consistent with any of the published structures of CST in either the “Head” or “Arm” conformations (Lim et al, 2020; He et al, 2022) (Appendix Fig S2A).
Shieldin is known to interact with the CST complex that facilitates the fill‐in of resected DNA by Pol α‐Primase (Mirman et al, 2022a). The AF2 analysis predicted two points of interaction between shieldin and CST: SHLD1‐CTC1 (Fig 1B panel vii, Appendix Fig S2B) and SHLD2‐CTC1 (Fig 1B panel v). The predicted SHLD1–CTC1 interaction occurs between the SHLD1 N terminus and the face of CTC1 consisting of OB‐ABC and the hinge three‐helix bundle. This binding site has been previously described (Mirman et al, 2022a) and disrupting this interface by mutating SHLD1 leucine 20 that is buried within the binding surface ablates this interaction (Mirman et al, 2022b). Our AF2 analysis also predicts an interaction between the third SHLD2 OB‐fold with CTC1 that is compatible with SHLD1‐SHLD2 binding (Fig 1B panel v), though this region is not sufficient for interaction in yeast‐two‐hybrid assays (Mirman et al, 2018). The predicted SHLD2‐CTC1 binding site overlaps with the ssDNA‐binding site in CTC1 but is not mutually exclusive with the POLA1 catalytic domain in the preinitiation complex (PIC) conformation of CST‐Pol α‐Primase (Appendix Fig S2C–E). However, the predicted SHLD2 binding site is sterically incompatible with the CST dimerization interface proximal to the ssDNA‐binding site (Lim et al, 2020). As the AF2 protein–protein interaction prediction recapitulated the most known interactions involving the shieldin complex, we turned our attention to the entirely novel prediction of the RIF1‐SHLD3 binding interface.
RIF1 HEAT repeats interact with the SHLD3 C‐terminal eIF4E‐like domain
The AF2 analysis predicted a high‐confidence interaction between the N‐terminal HEAT repeats of RIF1 and the C terminus of SHLD3. The C‐terminal half of SHLD3 is predicted to form a globular domain with structural homology to the translation initiation factor eIF4E (DALI search; Holm & Sander, 1995). All five models scored highly, with pDockQ scores ≥ 0.5, mean interface PAE ≤ 11.6 Å, and iPTM scores ≥ 0.7, a range of values that discriminates between accurate and inaccurate predictions by previous benchmarking studies (Fig 1C; Bryant et al, 2022; Yin et al, 2022). An additional AF2 prediction isolating the SHLD3 C terminus and the RIF1 residues 1–615 (Fig 2A) further improved scores to pDockQ ≥ 0.5, mean interface PAE ≤ 5.8 Å, and iPTM ≥ 0.91, with high pLDDT scores (parameter reflecting per‐residue AF2 prediction confidence; Jumper et al, 2021) throughout and a consistent interface across five separate models (Fig EV2A–C). To rule out a potential AF2 bias for predicting nonspecific RIF1 HEAT‐repeat binding to eIF4E‐like domains, we predicted models for RIF1 (1–615) with the three human eIF4E paralogues and two unrelated but structurally homologous proteins from the AF2 database (Fig EV2D and Table EV2). None of the predicted pairs satisfied the AF2 cutoff criterion used in this study.
Figure 2. The RIF1 N‐terminal HEAT repeats interact with the eIF4E‐like C terminus of SHLD3.
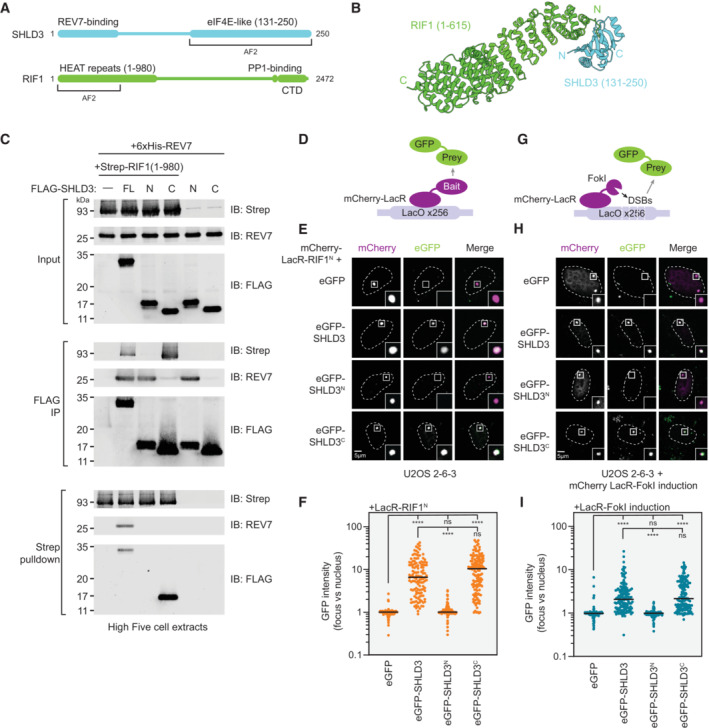
- Schematic of the domain organization of SHLD3 and RIF1, and the fragments used for additional AF2 analysis.
- Top‐ranked model predicted by AF2 between RIF1 HEAT‐repeat residues 1–615 and SHLD3 eIF4E‐like domain residues 131–250. See also Fig EV2A–D.
- Whole cell extracts of High Five insect cells individually expressing FLAG‐SHLD3, Strep‐RIF1, and 6xHis‐REV7 through baculovirus infection were combined and subjected to FLAG immunoprecipitation or streptactin pulldown and immunoblotted for REV7 or the Strep or FLAG epitopes. Results are representative of three biologically independent experiments. IB—immunoblot. FL—full‐length. For SHLD3, N and C correspond to residues 1–125 and 126–250, respectively.
- Schematic of the LacR/LacO assay using plasmid‐encoded mCherry‐LacR‐fused bait and eGFP‐fused prey proteins transfected in the U2OS 2‐6‐3 cell line containing ~256 lac operator (LacO) repeats.
- Representative micrographs of the LacR/LacO assay using mCherry‐LacR‐RIF1N as bait to evaluate chromatin recruitment of eGFP‐tagged SHLD3 variants. SHLD3: residues 2–250. SHLD3N: residues 2–125. SHLD3C: residues 126–250. RIF1N: residues 1–967. See also Fig EV2E–G.
- Quantification of LacR/LacO assay measuring chromatin recruitment of eGFP‐tagged SHLD3 variants to LacO arrays by mCherry‐LacR‐RIF1N. GFP intensities are presented as a ratio between the average fluorescence intensity within the mCherry‐labeled LacR focus and the average nuclear intensity. Bars represent mean values (n = 138, 126, 129, 137 for eGFP, eGFP‐SHLD3, eGFP‐SHLD3N, eGFP‐SHLD3C from three biologically independent experiments). Analysis was performed by the Kruskal–Wallis test followed by Dunn's multiple comparisons against empty vector control or full‐length eGFP‐SHLD3. ****P < 0.0001. ns P > 0.05.
- Schematic of the LacR‐FokI assay that directs FokI nuclease‐mediated DNA double‐strand breaks to the U2OS 2‐6‐3 LacO array to analyze DNA‐damage localization of eGFP‐fused prey proteins encoded by transfected plasmids.
- Representative micrographs of the LacR‐FokI assay to evaluate DNA‐damage recruitment of eGFP‐tagged SHLD3 variants after induction of LacR‐FokI expression. See also Fig EV2H–J.
- Quantification of LacR‐FokI assay evaluating recruitment of eGFP‐tagged SHLD3 variants to FokI‐induced DSBs at LacO arrays. GFP intensities are presented as a ratio between the average fluorescence intensity within the mCherry‐labeled LacR‐FokI focus and the average nuclear intensity. Bars represent mean (n = 143, 147, 147, 141 for eGFP, eGFP‐SHLD3, eGFP‐SHLD3N, eGFP‐SHLD3C from three biologically independent experiments). Analysis was performed by the Kruskal–Wallis test followed by Dunn's multiple comparisons against empty vector control or full‐length eGFP‐SHLD3. ****P < 0.0001. ns P > 0.05.
Source data are available online for this figure.
Figure EV2. Data supporting the SHLD3 C terminus as necessary and sufficient for RIF1 binding and recruitment to sites of DNA damage.
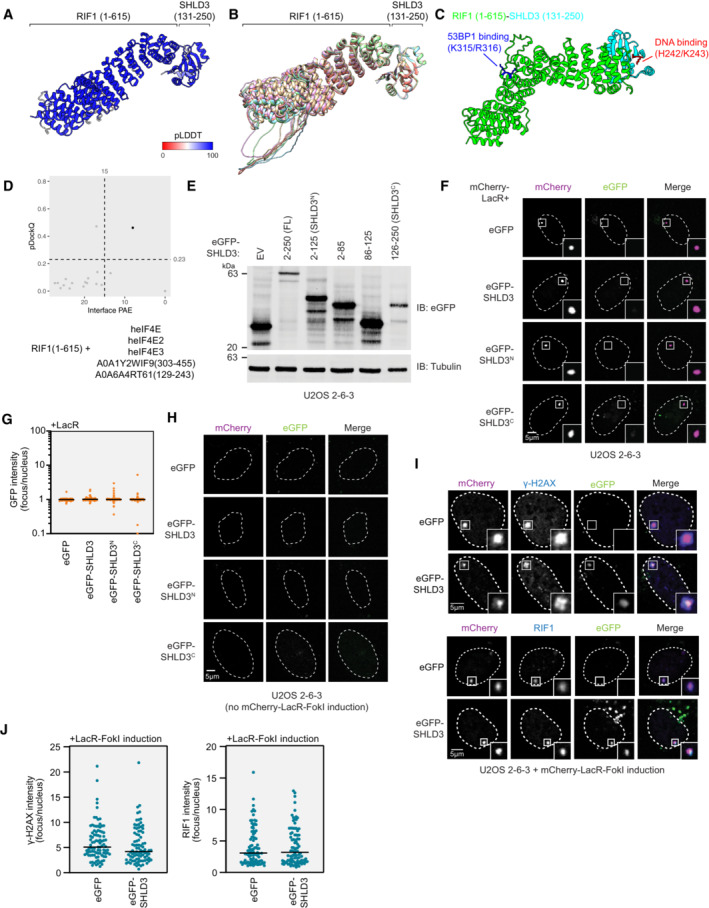
- Top‐ranking model of RIF1 (1–615) and SHLD3 (131–250) colored by pLDDT score.
- Five AF2‐predicted models of RIF1 (1–615) and SHLD3 (131–250) superimposed, aligned by the SHLD3 chain.
- Top‐ranking AF2 model of RIF1 (1–615) and SHLD3 (131–250), highlighting residues associated with SHLD3 DNA binding and RIF1 53BP1 phosphopeptide binding.
- Scatter plot of interface PAE vs pDockQ scores of AF2‐predicted models between RIF1 (1–615) with the indicated paralogues of the SHLD3 eIF4E‐like domain. Cutoff scores of 15 Å and 0.23 PAE and pDockQ scores are shown as dotted lines. Models meeting the cutoff are represented as black points and those that do not are represented as gray points.
- Immunoblot of whole cell extracts of U2OS 2‐6‐3 cells transfected with plasmids encoding the indicated eGFP‐tagged SHLD3. Lysates were probed for eGFP and tubulin (loading control). IB—immunoblot. EV—empty vector.
- Representative micrographs of the LacR/LacO assay using mCherry‐LacR as bait to evaluate chromatin recruitment of eGFP‐tagged SHLD3 variants as a control for the experiments using mCherry‐LacR‐RIF1N in Fig 2E and F.
- Quantification of F. GFP intensities are presented as a ratio between the average fluorescence intensity within the mCherry‐labeled LacR focus and the average nuclear intensity. Bars represent mean (n = 140, 138, 131, 133 for eGFP, eGFP‐SHLD3, eGFP‐SHLD3N, eGFP‐SHLD3C from three biologically independent experiments).
- Representative micrographs of control experiments for the LacR‐FokI assay in Fig 2H and I to evaluate DNA‐damage recruitment of eGFP‐tagged SHLD3 variants. LacR‐FokI expression was not induced, and no mCherry‐LacR‐FokI or eGFP‐SHLD3 foci were detected.
- Representative micrographs of the LacR‐FokI assay to evaluate DNA‐damage induction after mCherry‐LacR‐FokI expression. U2OS 2‐6‐3 cells were transfected with plasmids encoding eGFP‐SHLD3 and treated with 4‐hydroxytamoxifen and Shield‐1 peptide to induce mCherry‐LacR‐FokI expression. The cells were then analyzed for γ‐H2AX focus formation colocalizing with mCherry‐LacR‐FokI as a proxy for DNA double‐strand break formation through immunofluorescence (top). Colocalization of mCherry‐LacR‐FokI focus with endogenous RIF1 was also assessed (bottom).
- Quantification of I. Immunofluorescence intensities are presented as a ratio between the average fluorescence intensity within the mCherry‐labeled LacR focus and the average nuclear intensity. Bars represent mean (n = 95, 88, 90, 94 for γ‐H2AX eGFP, γ‐H2AX eGFP‐SHLD3, RIF1 eGFP, RIF1 eGFP‐SHLD3 from two biologically independent experiments).
Source data are available online for this figure.
The AF2‐predicted interface lies between the first two RIF1 HEAT‐repeat helices and the face of the SHLD3 eIF4E‐like domain β‐sheet with a total buried surface area of 1,808 Å2 (Fig 2B). The AF2 model of the RIF1 N terminus and the SHLD3 C terminus is sterically incompatible with a recently described SHLD3 DNA‐binding activity (preprint: Susvirkar & Faesen, 2022), suggesting that any DNA binding by SHLD3 is mutually exclusive with its RIF1 association (Fig EV2C). The RIF1 K315/R316 residues, which are essential for 53BP1 binding (Setiaputra et al, 2022), are distal from the predicted SHLD3 binding interface (Fig EV2C).
To validate the predicted RIF1–SHLD3 interaction in vitro, we first used baculovirus‐mediated transduction of insect cells to individually express Strep‐tagged human RIF1N (residues 1–980), FLAG‐tagged SHLD3, and His‐tagged REV7 and then performed reciprocal FLAG and Strep pulldowns from combined insect cell extracts to study their interactions. These experiments confirmed previous observations that the N terminus of SHLD3 (residues 2–125; SHLD3N) is necessary and sufficient for REV7 binding, while its eIF4E‐like domain‐containing C terminus (residues 126–250; SHLD3C) does not participate in this interaction (Fig 2C; Ghezraoui et al, 2018). Consistent with the AF2 prediction, the C terminus of SHLD3 is sufficient for interacting with the RIF1 N‐terminal HEAT‐repeat domain encompassed by RIF1N. Additionally, RIF1N is unable to bind REV7 in the absence of SHLD3, supporting the previous observation that SHLD3 bridges RIF1 and REV7 (Fig 2C; Noordermeer et al, 2018). Together these results show that recombinant SHLD3 interacts with REV7 and RIF1 through its N‐ and C‐terminal domains, respectively.
To determine whether this interaction is recapitulated in cells, we employed the U2OS 2‐6‐3 cell line that contains an array of ~256 Lac operator (LacO) repeats (Janicki et al, 2004). We expressed the RIF1 HEAT repeats (residues 1–967) fused to mCherry‐Lac repressor (LacR) and determined whether it can recruit GFP‐labeled SHLD3 to the LacO array (Figs 2D–F and EV2E–G). Consistent with the pulldown results, the C terminus of SHLD3 is necessary and sufficient for its robust recruitment to chromatin by RIF1 (Fig 2E and F). Notably, unlike 53BP1‐nucleosome or RIF1–53BP1 interactions, this recruitment occurs without exogenous DNA damage, suggesting that it is independent of DNA‐damage‐induced post‐translational modifications.
SHLD3 is responsible for recruiting shieldin to DSB sites (Noordermeer et al, 2018). Since RIF1 is essential for shieldin DNA‐damage localization, we hypothesized that the SHLD3 C terminus is necessary and sufficient for SHLD3 recruitment to DSBs. To test this possibility, we induced DSBs at the LacO array in U2OS 2‐6‐3 cells by expressing FokI endonuclease fused to mCherry‐LacR (Figs 2G–I and EV2H–J; Shanbhag et al, 2010) and determined which region of SHLD3 was essential for its DSB localization. We observed that the SHLD3 C terminus is essential for its recruitment to sites of FokI‐induced DNA breaks (Fig 2H and I). These observations show that the SHLD3 eIF4E‐like domain binds RIF1 HEAT repeats to recruit shieldin to sites of DNA damage.
SHLD3 interacts with RIF1 through a highly charged interface
The predicted SHLD3‐RIF1 interface contains several conserved basic and acidic residues that are poised to form multiple electrostatic contacts (Fig 3A). We identified five putative electrostatic interactions between conserved pairs of residues (Fig 3A) and expressed SHLD3C variants with alanine substitutions to test the contributions of these predicted electrostatic contacts to the SHLD3–RIF1 interaction. Recombinant SHLD3 variants with W132A, N201A, and D216A mutations were deficient in RIF1N binding in reciprocal pulldown experiments from insect cell extracts, while R166A mutation completely abolished interaction with RIF1N (Fig 3B). We next asked whether these point mutations also disrupt RIF1N–SHLD3C interaction in cells. Consistent with the pulldown experiments, the W132A and R166A variants failed to colocalize with LacR‐RIF1N at the LacO array, while the N201A and D216A variants were partially defective in colocalization, as shown by weaker intensity foci (Figs 3C and EV3A and B). Furthermore, the W132A and R166A variants were unable to accumulate at FokI‐induced DSBs, whereas the N201A and D216A variants retained weak localization, suggesting partially impaired interaction with RIF1 (Figs 3D and EV3C). By contrast, the S131A substitution did not impact interaction with RIF1N in pulldown assays and this SHLD3 variant was fully proficient in colocalizing with LacR‐RIF1N at the LacO array as well as being recruited to FokI‐induced DSBs. Transient expression of these SHLD3 variants did not affect the localization of endogenous RIF1 to FokI‐induced DSBs (Fig EV3D and E), consistent with shieldin being recruited downstream of RIF1. In summary, we evaluated the AF2 prediction that five SHLD3 residues facilitate RIF1–SHLD3 interaction through both in vitro pulldown and cellular recruitment assays and determined that R166 is essential for RIF1 binding, W132, N201, and D216 are important, while S131 is dispensable for this activity. We also tested whether the putative DNA‐binding residues of SHLD3C (H242 and K243; preprint: Susvirkar & Faesen, 2022) participate in RIF1N‐mediated recruitment. We determined through the LacR‐LacO assay that the SHLD3C H242A/K243A variant is efficiently recruited to chromatin by LacR‐RIF1N (Fig EV3F–H).
Figure 3. SHLD3 binds RIF1 through polar interactions that are essential for recruitment to DNA breaks.
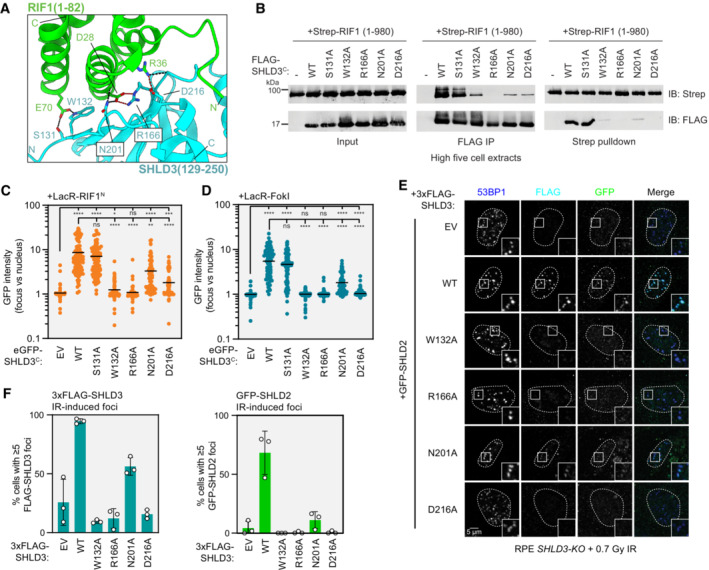
- Close‐up view of the hydrogen bond and salt bridge network between conserved RIF1 and SHLD3 residues in the AF2‐predicted model. Hydrogen bonds and salt bridges are depicted as black and red dashed lines, respectively.
- Whole cell extracts of High Five insect cells individually expressing the wild‐type (WT) or indicated alanine substitution FLAG‐SHLD3C (residues 126–250) variants and Strep‐RIF1N (residues 1–980) through baculovirus infection were combined and subjected to FLAG immunoprecipitation or streptactin pulldown and immunoblotted for the Strep or FLAG epitopes. The results are representative of two biologically independent experiments. IB—immunoblot. IP—immunoprecipitation.
- Quantification of LacR/LacO assay measuring recruitment of eGFP‐SHLD3C wild‐type (WT) or alanine substitution variants to LacO arrays in U2OS 2‐6‐3 cells by mCherry‐LacR‐RIF1N. GFP intensities are presented as a ratio between the average fluorescence intensity within the mCherry‐labeled LacR‐RIF1N focus and the average nuclear intensity. Bars represent mean values (n = 134, 125, 120, 130, 104, 129, 117 for EV, WT, S131A, W132A, R166A, N201A, D216A from three biologically independent experiments). EV: empty vector. Analysis was performed by the Kruskal–Wallis test followed by Dunn's multiple comparisons against empty vector and WT controls. ****P < 0.0001. ***P = 0.0004. **P = 0.002. *P = 0.02. ns P > 0.05. See also Fig EV3A and B.
- Quantification of LacR‐FokI assay measuring recruitment of eGFP‐SHLD3C wild‐type (WT) or alanine substitution variants to sites of DNA double‐strand breaks induced by mCherry‐LacR‐FokI in U2OS 2‐6‐3 cells. GFP intensities are presented as a ratio between the average fluorescence intensity within the mCherry‐labeled LacR‐FokI focus and the average nuclear intensity. Bars represent mean values (n = 103, 136, 159, 151, 139, 145, 140 for EV, WT, S131A, W132A, R166A, N201A, D216A from three biologically independent experiments). Analysis was performed by the Kruskal–Wallis test followed by Dunn's multiple comparisons against empty vector and WT controls. ****P < 0.0001. ns P > 0.05. See also Fig EV3C and E.
- Representative micrographs of immunofluorescence experiments (from three biologically independent experiments) analyzing localization of SHLD3 and SHLD2 to ionizing radiation‐induced foci. RPE SHLD3‐KO cells were complemented by transduction of lentivirus encoding the indicated 3xFLAG‐SHLD3 variants, transfected with eGFP‐SHLD2‐encoding plasmids, treated with 0.7 Gy X‐ray irradiation, and processed for immunofluorescence microscopy after 1 h using antibodies against 53BP1, FLAG, and GFP.
- Quantification of immunofluorescence experiment analyzing localization of SHLD3 and SHLD2 to ionizing radiation‐induced foci (0.7 Gy) in RPE SHLD3‐KO cells. The percentage of cells containing ≥ 5 53BP1‐colocalizing 3xFLAG‐SHLD3 (top) or eGFP‐SHLD2 (bottom) foci were manually counted. Bars represent mean values ± s.d. (n = 3 biologically independent experiments with ≥ 61 cells imaged each). See also Fig EV3I and J.
Source data are available online for this figure.
Figure EV3. Data supporting the polar interactions that are essential for SHLD3‐RIF1 binding.
- Immunoblot of whole cell extracts of U2OS 2‐6‐3 cells transfected with plasmids encoding the indicated eGFP‐tagged SHLD3C (residues 126–250) variants. Lysates were probed for eGFP and tubulin (loading control). IB—immunoblot. EV—empty vector. WT—wild‐type.
- Representative micrographs of the LacR/LacO assay using mCherry‐LacR‐RIF1N as bait to evaluate chromatin recruitment of eGFP‐tagged SHLD3C alanine substitution variants shown in Fig 3C. SHLD3C: residues 126–250. RIF1N: residues 1–967.
- Representative micrographs of the LacR‐FokI assay to evaluate DNA‐damage recruitment of eGFP‐tagged SHLD3C alanine substitution variants after induction of LacR‐FokI expression shown in Fig 3D. SHLD3C: residues 126–250.
- Representative micrographs of the LacR‐FokI assay to evaluate DNA‐damage recruitment of endogenous RIF1 after induction of LacR‐FokI expression in the presence of exogenously expressed eGFP‐tagged SHLD3C alanine substitution variants. SHLD3C: residues 126–250.
- Quantification of D. RIF1 immunofluorescence intensities are presented as a ratio between the average fluorescence intensity within the mCherry‐labeled LacR focus and the average nuclear intensity. Bars represent mean values (n = 99, 92, 88, 97, 90, 92, 100 for EV, WT, S131A, W132A, R166A, N201A, D216A from two biologically independent experiments).
- Immunoblot of whole cell extracts of U2OS 2‐6‐3 cells transfected with plasmids encoding the indicated eGFP‐tagged SHLD3C (residues 126–250) variants. Lysates were probed for eGFP and tubulin (loading control).
- Representative micrographs of the LacR/LacO assay using mCherry‐LacR‐RIF1N as bait to evaluate chromatin recruitment of eGFP‐tagged SHLD3 H242A/K243A.
- Quantification of G. GFP intensities are presented as a ratio between the average fluorescence intensity within the mCherry‐labeled LacR‐RIF1N focus and the average nuclear intensity. Bars represent mean values (n = 127, 129, 136 for EV, WT, H242A/K243A from three biologically independent experiments). Analysis was performed using the Kruskal–Wallis test followed by Dunn's multiple comparisons against empty vector control. ****P < 0.0001.
- Immunoblot of whole cell extracts of RPE SHLD3‐KO cells stably transduced with lentivirus encoding the indicated 3xFLAG‐tagged SHLD3 alanine substitution variants. Lysates were probed for FLAG and tubulin (loading control). IB—immunoblot.
- Immunoblot of whole cell extracts of complemented RPE SHLD3‐KO cells shown in (I) that were transfected with plasmid encoding eGFP‐SHLD2. Lysates were probed for eGFP and tubulin (loading control).
Source data are available online for this figure.
53BP1 and its downstream effectors are recruited to DSBs induced by ionizing radiation (IR; Escribano‐Díaz et al, 2013). We tested whether the recruitment of shieldin to IR‐induced foci is dependent on the predicted RIF1‐SHLD3 interface by stably expressing SHLD3 variants in RPE1 hTERT p53‐KO SHLD3‐KO FLAG‐Cas9 (RPE SHLD3‐KO) cells via lentiviral transduction (Fig EV3I) and X‐irradiating them with 0.7 Gy dose prior to analysis by immunofluorescence microscopy (Fig 3E and F). We also monitored IR‐induced focus formation of SHLD2, a shieldin subunit downstream of SHLD3, through transient transfection of eGFP‐SHLD2‐encoding plasmids, to facilitate SHLD2 focus visualization (Fig EV3J). Consistent with the FokI‐induced DSB localization experiments, mutating SHLD3 residues essential for RIF1–SHLD3 interaction results in defective IR‐induced focus formation of both SHLD3 and SHLD2, with mutations of W132, R166, and D216 causing near‐complete loss of IR‐induced focus formation.
SHLD3 is recruited to 53BP1 bodies through its interaction with RIF1
In addition to focal accumulation around DSBs, 53BP1 marks DNA lesions in G1 associated with replication stress in the preceding cell cycle (Lukas et al, 2011). These 53BP1 bodies are large and their formation requires many of the same DNA‐damage signaling factors involved in 53BP1 localization to DSBs such as the kinase ATM, histone H2AX phosphorylation, and MDC1 (Harrigan et al, 2011). 53BP1 bodies arise from the mitotic passage of under‐replicated DNA at difficult‐to‐replicate loci such as common fragile sites (Harrigan et al, 2011). A recent study implicated RIF1 and shieldin in the prevention of aberrant HR at 53BP1 bodies, leading to the delayed resolution of these lesions (Spies et al, 2019). However, the presence of shieldin at such bodies has not been described. We induced mild DNA replication stress with a low dose (0.2 μM) of the B‐family DNA polymerase inhibitor aphidicolin (Wright & Brown, 1990) in RPE SHLD3‐KO cells stably expressing SHLD3 variants (Fig EV3I) and analyzed SHLD3 localization into 53BP1 bodies by immunofluorescence using Pearson Correlation Coefficient measurements between pixels within each nucleus (Fig 4A and B). FLAG‐SHLD3 shows robust colocalization with 53BP1 bodies. Measurements of FLAG‐SHLD3 intensity within 53BP1 bodies found that SHLD3 variants defective in RIF1 binding were deficient in 53BP1 colocalization (Fig 4C and D). Since RIF1 promotes the formation of 53BP1 bodies in response to aphidicolin‐induced replication stress (Watts et al, 2020), we investigated whether loss of SHLD3 affects 53BP1 body formation. In both the absence and presence of aphidicolin, RPE SHLD3‐KO cells do not display significant differences in the number of 53BP1 bodies compared with wild‐type RPE (Fig 4E), consistent with SHLD3 recruitment being downstream of 53BP1. These observations suggest that similar mechanisms underlie shieldin recruitment to both 53BP1 bodies and DSB sites, and that the importance of RIF1 for 53BP1 body formation is independent of shieldin accumulation at these sites.
Figure 4. RIF1 binding is essential for SHLD3 localization to 53BP1 bodies.
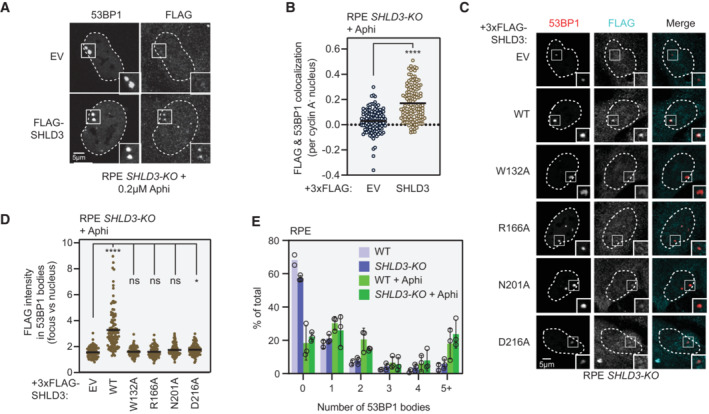
- Representative micrographs of immunofluorescence experiments (from three biologically independent experiments) analyzing localization of SHLD3 to 53BP1 bodies in RPE SHLD3‐KO cells. 3xFLAG‐SHLD3‐complemented RPE SHLD3‐KO cells (see Fig EV3I) were treated with 200 nM aphidicolin (Aphi) for 24 h and processed for immunofluorescence microscopy using antibodies against 53BP1, FLAG, and cyclin A. 53BP1 bodies are defined as distinct foci visible in cyclin A‐negative cells.
- Quantification of FLAG and 53BP1 colocalization from (A) performed in CellProfiler. Pearson's correlation coefficients (PCC) were calculated for pixels within each cyclin A‐negative, 53BP1 body‐positive nucleus between the 53BP1 and FLAG channels. Each point represents the PCC value of an individual nucleus. Bars represent mean values (n = 120, 130 for EV, SHLD3 from three biologically independent experiments). Analysis was performed using the two‐tailed Mann–Whitney test. ****P < 0.0001.
- Representative micrographs of immunofluorescence experiments analyzing localization of FLAG‐SHLD3 alanine substitution variants stably expressed in RPE SHLD3‐KO cells by lentivirus transduction. Cells were treated with 200 nM Aphi for 24 h and processed for immunofluorescence microscopy using antibodies raised against 53BP1, FLAG, and cyclin A. 53BP1 bodies are defined as distinct foci visible in cyclin A‐negative cells.
- Quantification of immunofluorescence experiments analyzing the recruitment of FLAG‐SHLD3 variants to 53BP1 bodies in RPE SHLD3‐KO cells. Individual points represent the ratio between average FLAG intensity within 53BP1 bodies and the average nuclear intensity. Bars represent mean values (n = 89, 102, 91, 95, 102, 95 for EV, WT, W132A, R166A, N201A, D216A from three biologically independent experiments). Analysis was performed using the Kruskal–Wallis test followed by Dunn's multiple comparisons against empty vector control. ****P < 0.001, *P = 0.03, ns P > 0.05. See also Fig EV3I.
- Quantification of the percentage of cells containing the indicated number of 53BP1 bodies in RPE WT and SHLD3‐KO cells with or without 24 h 200 nM Aphi treatment. Bars represent mean ± s.d. (n = 3 biologically independent experiments with ≥ 30 nuclei imaged each).
Source data are available online for this figure.
Dissecting the RIF1 surface that facilitates SHLD3 binding
Next, we sought to validate the region of RIF1 that interacts with SHLD3. AF2 analysis predicts that the two N‐terminal α‐helices of the HEAT‐repeat domain participate in SHLD3 interactions (Fig 5A, left panel). Consistent with this prediction, LacR fused to a truncated form of the RIF1 HEAT‐repeat domain containing only the N‐terminal 567 residues was fully proficient in recruiting full‐length SHLD3 to LacO arrays (Figs 5A right panel and EV4A and B). However, the deletion of the first 173 residues from this protein (yielding RIF1 174–567), which includes the predicted SHLD3‐binding region, abolished its ability to interact with SHLD3 as determined by their lack of cellular colocalization at the LacO array (Figs 5A right panel and EV4A and B). To further interrogate the predicted RIF1‐SHLD3 binding interface, we expressed LacR‐RIF1N proteins containing alanine substitutions at residues E70, D28, and R36 that are predicted to form hydrogen bonds or salt bridges with SHLD3 residues W132, both R166 and N201, and D216, respectively (Fig 3A). The LacR‐RIF1N D28A and R36A variants were highly defective in recruiting SHLD3 to LacO arrays (Figs 5B and EV4C and D). Unexpectedly, LacR‐RIF1 E70A was fully proficient in interacting with SHLD3 despite the importance of SHLD3 W132 in both LacO and DSB recruitment (Fig 3C and D). This observation indicates that the SHLD3 W132 residue facilitates RIF1‐SHLD3 binding through interactions other than the predicted E70‐W132 hydrogen bond. We then individually expressed SHLD3C with the D28A and R36A RIF1N variants in insect cells and tested their ability to interact through reciprocal pulldown experiments in combined cell extracts. Consistent with the LacR experiments, both D28A and R36A RIF1N variants were deficient in copurifying with SHLD3C in vitro, with D28A having the most severe defect (Fig 5C). These results point to the predicted RIF1D28–SHLD3R166 electrostatic interaction as an essential component of the binding interface and indicate that the predicted RIF1R36–SHLD3D216 interaction also contributes to the SHLD3–RIF1 interaction.
Figure 5. RIF1 binds SHLD3 through polar residues within its extreme N terminus.
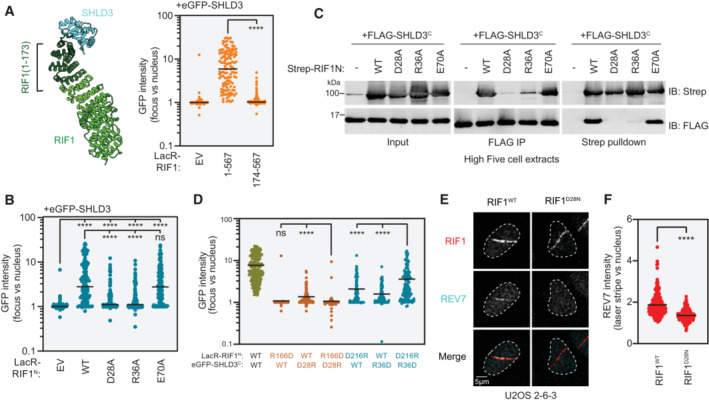
- Left: Representation of the region of the RIF1 N‐terminal HEAT repeats that are predicted to bind the SHLD3 eIF4E‐like domain. Residues 1–173 are highlighted. Right: Quantification of LacR/LacO assay measuring eGFP‐SHLD3 recruitment to LacO arrays in U2OS 2‐6‐3 cells by the indicated truncated mCherry‐LacR‐RIF1N variants. GFP intensities are presented as a ratio between the average fluorescence intensity within the mCherry‐labeled LacR‐RIF1N focus and the average nuclear intensity. Bars represent mean values (n = 148, 123, 122 for EV, 1–567, 174–567 from three biologically independent experiments). Analysis was performed using the Mann–Whitney two‐tailed test. ****P < 0.001. See also Fig EV4A and B. EV—empty vector.
- Quantification of LacR/LacO assay measuring eGFP‐SHLD3 recruitment to LacO arrays in U2OS 2‐6‐3 cells by mCherry‐LacR‐RIF1N (residues 1–967) or the indicated alanine substitution variants. GFP intensities are presented as a ratio between the average fluorescence intensity within the mCherry‐labeled LacR focus and the average nuclear intensity. Bars represent mean values (n = 142, 121, 132, 135, 127 for EV, WT, D28A, R36A, E70A from three biologically independent experiments). Analysis was performed using the Kruskal–Wallis test followed by Dunn's multiple comparisons against empty vector (EV) and wild‐type RIF1N (WT) controls. ****P < 0.001, ns P > 0.05. See also Fig EV4C and D.
- Whole cell extracts of High Five insect cells individually expressing the FLAG‐SHLD3C (residues 126–250) and the indicated alanine substitution Strep‐RIF1 (residues 1–980) variants through baculovirus infection were combined and subjected to FLAG immunoprecipitation or streptactin pulldown and immunoblotted for the Strep or FLAG epitopes. The results are representative of two biologically independent experiments. IB—immunoblot. IP—immunoprecipitation.
- Quantification of LacR/LacO assay measuring eGFP‐SHLD3C recruitment to LacO arrays in U2OS 2‐6‐3 cells by mCherry‐LacR‐RIF1N with both transfected plasmids bearing charge‐reversal mutations. Bars represent mean values (n = 222, 128, 121, 135, 131, 127, 128 for WT/WT, R166D/WT, WT/D28R, R166D/D28R, D216R/WT, WT/R36D, D216R/R36D from five biologically independent experiments). Analysis was performed using the Kruskal–Wallis test followed by Dunn's multiple comparisons. ****P < 0.0001, ns P > 0.05. See also Fig EV4E and F.
- Representative micrographs of UV laser microirradiation experiments measuring DNA‐damage recruitment of REV7 in U2OS 2‐6‐3 cells with endogenously mutated RIF1. DNA damage was induced in U2OS 2‐6‐3 cells through irradiation in the form of linear stripes and analyzed by immunofluorescence microscopy with RIF1 and REV7 antibodies. See also Fig EV4G and H.
- Quantification of UV laser microirradiation immunofluorescence experiment measuring DNA‐damage recruitment of REV7 in U2OS 2‐6‐3 cells with endogenously mutated RIF1. REV7 immunofluorescence intensities are presented as a ratio between the average fluorescence intensity within the RIF1‐labeled irradiation stripe and the average nuclear intensity. Only nuclei containing RIF1 stripes are quantified. Bars represent mean values (n = 206, 215 for RIF1WT, RIF1D28N from two biologically independent experiments). Analysis was performed using the Welch's t‐test. ****P < 0.0001.
Source data are available online for this figure.
Figure EV4. Data supporting the extreme N terminus of RIF1 binding SHLD3 through polar residues.
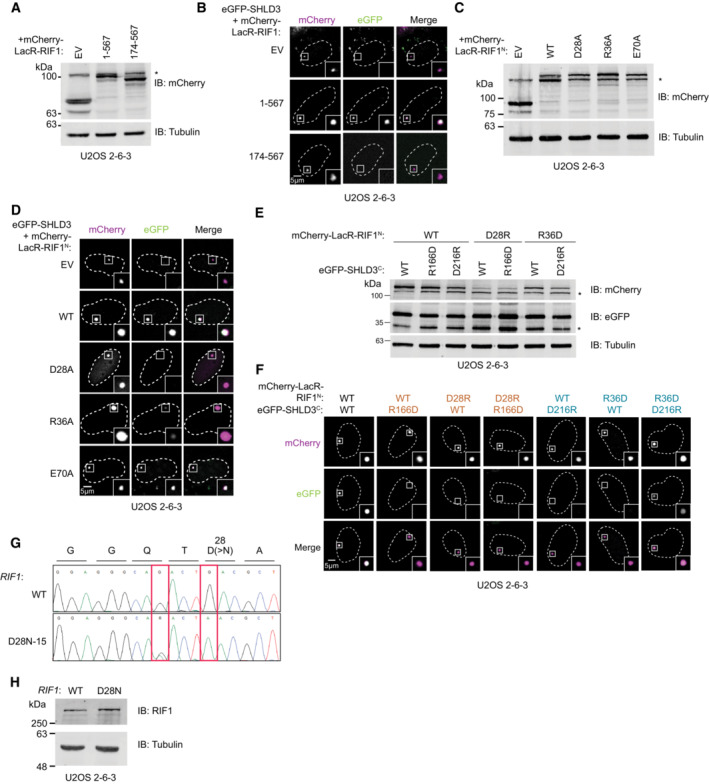
- Immunoblot of whole cell extracts of U2OS 2‐6‐3 cells transfected with plasmids encoding the indicated mCherry‐LacR‐fused RIF1 truncations. Lysates were probed for mCherry and tubulin (loading control). IB: immunoblot, EV: empty vector. *: nonspecific band.
- Representative micrographs (of three biologically independent experiments) of the LacR/LacO assay using the indicated mCherry‐LacR‐fused RIF1 variants to evaluate their ability to recruit eGFP‐SHLD3 to chromatin shown in Fig 5A.
- Immunoblot of whole cell extracts of U2OS 2‐6‐3 cells transfected with plasmids encoding the indicated mCherry‐LacR‐fused RIF1N (residues 1–967) alanine variants. Lysates were probed for mCherry and tubulin (loading control). *: nonspecific band.
- Representative micrographs (of three biologically independent experiments) of the LacR/LacO assay using the indicated mCherry‐LacR‐fused RIF1N variants to evaluate their ability to recruit eGFP‐SHLD3 to chromatin shown in Fig 5B. WT: wild‐type.
- Immunoblot of whole cell extracts of U2OS 2‐6‐3 cells transfected with plasmids encoding the indicated mCherry‐LacR‐RIF1N and eGFP‐SHLD3C variants. Lysates were probed for mCherry, eGFP, and tubulin (loading control). *: nonspecific band.
- Representative micrographs of the LacR/LacO assay using the indicated mCherry‐LacR‐RIF1N and eGFP‐SHLD3C variants to evaluate their colocalization at LacO arrays shown in Fig 5D.
- Sanger sequencing chromatograms of PCR products amplified from U2OS 2‐6‐3 cells with and without subjecting it to base editing to introduce endogenous D28N mutations. A single clone was isolated that contained the desired mutation (D28N‐15). Red boxes highlight induced mutations. The first heterozygous G > A mutation is silent. The second homozygous G > A mutation results in the desired D28N substitution.
- Immunoblot of whole cell extracts of U2OS 2‐6‐3 cells with or without base editing to introduce endogenous D28N mutation. Lysates were probed for RIF1 and tubulin (loading control).
Source data are available online for this figure.
We next probed the RIF1D28–SHLD3R166 and RIF1R36–SHLD3D216 electrostatic interaction pairs through charge‐reversal mutation using the LacO/LacR chromatin localization assay (Figs 5D and EV4E and F). As predicted, individual charge‐reversal mutations within the two electrostatic interaction pairs result in defective mCherry‐LacR‐colocalizing eGFP‐SHLD3C focus formation. Combining the RIF1N R166D and SHLD3C D28R mutations did not rescue SHLD3C focus formation, suggesting either that the combined mutations did not restore the electrostatic interaction, or that other essential interactions are associated with these residues. Indeed, RIF1D28 is also predicted to form a hydrogen bond with SHLD3N201 (Fig 3A). Strikingly, combining the RIF1N R36D and SHLD3C D216R variants partially rescues SHLD3C focus formation, providing strong evidence that this salt bridge was accurately predicted by AF2 and that it plays an important role in the RIF1–SHLD3 interaction.
Thus far the experiments we performed utilized cellular models overexpressing RIF1 and SHLD3 variants. To ensure that our observations apply to endogenously produced proteins, we used a dCas9‐guided base editor (Koblan et al, 2018) to generate U2OS 2‐6‐3 cells carrying homozygous D28N mutations at the endogenous RIF1 locus (Fig EV4G). The D28N mutation did not alter RIF1 expression (Fig EV4H). Since the isolated homozygous RIF1‐D28N clones lost the LacO array, we monitored REV7 recruitment to UV laser microirradiation sites instead of at FokI‐induced DSBs (Fig 5E) and found that RIF1D28N failed to recruit REV7 to sites of laser microirradiation (Fig 5E and F). We conclude that shieldin recruitment to DSB sites depends on the physical and direct interaction between SHLD3 and RIF1 modeled by AF2.
SHLD3–RIF1 interaction promotes class switch recombination
We next investigated whether mutations abolishing SHLD3‐RIF1 binding abrogate shieldin function. We evaluated antibody class switch recombination (CSR) in CH12F3 mouse B cell lymphoma cells. Upon stimulation with a cocktail of anti‐CD40 antibody, TGF‐β, and IL‐4, CH12F3 cells rapidly undergo CSR to convert its expressed immunoglobulin isotype from IgM to IgA (Nakamura et al, 2006). CSR involves the induction of DSBs at switch regions within the immunoglobulin heavy chain locus and ligation of distal breaks leading to exon recombination (Methot & Di Noia, 2017). This process is reliant on the 53BP1‐RIF1‐shieldin pathway, whose disruption leads to excessive resection of CSR‐induced DSBs into the coding regions of the immunoglobulin locus (Ghezraoui et al, 2018; Ling et al, 2020). Accordingly, CRISPR‐Cas9‐generated CH12F3 Shld3 −/− cells are severely deficient in CSR (Fig EV5A and B; Noordermeer et al, 2018).
Figure EV5. Data supporting the importance of SHLD3‐RIF1 binding for shieldin activity.
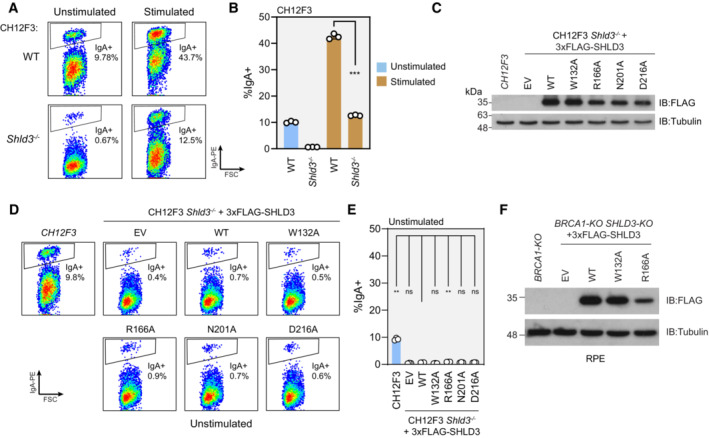
- Representative flow cytometry density plots measuring IgA expression in unstimulated and stimulated CH12F3 wild‐type or Shld3 −/− cells. Values shown are %IgA+ cells. FSC: forward scatter. WT—wild‐type.
- Quantification of class switch recombination data shown in (A). Bars represent mean ± s.d., n = 3 biologically independent experiments. Analysis was performed using the Welch's two‐tailed t‐test. ***P = 0.0002.
- Immunoblot of whole cell extracts of CH12F3 Shld3 −/− cells stably transduced with lentivirus encoding the indicated FLAG‐tagged SHLD3 alanine substitution variants. Lysates were probed for FLAG and tubulin (loading control). IB—immunoblot. EV—empty vector.
- Representative flow cytometry density plots measuring IgA expression in unstimulated CH12F3 wild‐type or Shld3 −/− cells stably transduced with lentivirus encoding FLAG‐tagged SHLD3 alanine substitution variants. Control experiment for Fig 6B. Values shown are %IgA+ cells.
- Quantification of class switch recombination data shown in (D). Bars represent mean ± s.d., n = 3 biologically independent experiments. Analysis was performed using the Dunnett's multiple comparisons test compared against Shld3 −/− cells complemented with wild‐type SHLD3. **P < 0.01, ns P > 0.05.
- Immunoblot of whole cell extracts of RPE BRCA1‐KO SHLD3‐KO cells stably transduced with lentivirus encoding the indicated FLAG‐tagged SHLD3 alanine substitution variants. Lysates were probed for FLAG and tubulin (loading control).
Source data are available online for this figure.
We evaluated the ability of SHLD3 variants deficient in RIF1 interaction to undergo CSR (Figs 6A and B and EV5C–E). CH12F3 Shld3 −/− cells transduced with lentivirus expressing wild‐type human SHLD3 regained CSR activity. Consistent with DSB localization and in vitro pulldown results, the R166A variant that is completely unable to interact with RIF1 (Fig 3B and C) did not support CSR. Interestingly, the W132A, N201A, and D216A variants that retained partial interaction with RIF1 (Fig 3B and C) showed differing CSR activity. While the W132A variant did not rescue IgA class switching, both N201A and D216A variants showed comparable CSR to the wild‐type complementation (Fig 6A and B). Our results suggest that the RIF1–SHLD3 interaction is essential for shieldin‐dependent CSR and that there may be subtle differences in the manner by which mouse and human RIF1 interact with SHLD3.
Figure 6. RIF1–SHLD3 interaction is essential for shieldin function.
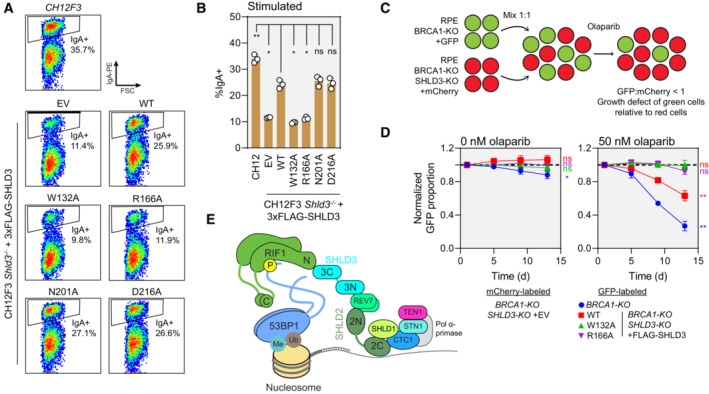
- Representative flow cytometry density plots (from three biologically independent experiments) measuring IgA expression in stimulated CH12F3 Shld3 −/− cells stably expressing FLAG‐tagged SHLD3 alanine substitution variants through lentiviral transduction. Values shown are %IgA+ cells. WT—wild‐type. EV—empty vector. FSC—forward scatter. See also Fig EV5C–E.
- Quantification of class switch recombination in stimulated CH12F3 Shld3 −/− cells stably expressing the indicated FLAG‐SHLD3 variants through lentiviral transduction. Bars represent mean ± s.d., n = 3 biologically independent experiments. Analysis was performed using one‐way ANOVA followed by the Dunnett's multiple comparisons test against Shld3 −/− cells complemented with wild‐type SHLD3. **P < 0.01, *P < 0.05, ns P > 0.05.
- Schematic of the two‐color competitive growth assay. The indicated RPE cell lines transduced with lentivirus encoding either eGFP or mCherry. eGFP‐ and mCherry‐labeled cells were mixed 1:1 and treated with olaparib, and the relative number of labeled cells was monitored over time by automated fluorescence microscopy. eGFP:mCherry ratios are normalized to the value 1‐day postplating. An eGFP:mCherry ratio < 1 indicates slower growth of the eGFP‐expressing cell line relative to the mCherry‐expressing cells.
- Competitive growth assay of RPE BRCA1‐KO SHLD3‐KO cells complemented with 3xFLAG‐SHLD3 variants. Growth of the indicated eGFP‐expressing cells was compared with mCherry‐expressing RPE BRCA1‐KO SHLD3‐KO cells transduced with empty vector after mixing 1:1 in 24‐well plates. Plates were imaged 1, 5, 9, and 13 days after plating. Dotted black lines represent an eGFP:mCherry ratio of 1. Points represent mean ± s.d. (n = 3 biologically independent experiments). Analysis was performed using a one‐sample t‐test against a hypothetical eGFP:mCherry ratio value of 1 for the day 13 time point. **P < 0.01, ns P > 0.05. See also Fig EV5F.
- Schematic of the molecular basis for 53BP1‐RIF1‐shieldin‐CST recruitment to sites of DNA damage.
Source data are available online for this figure.
SHLD3–RIF1 interaction promotes PARPi sensitivity
Loss of shieldin results in PARPi resistance in BRCA1‐mutated cells (Noordermeer et al, 2018). We evaluated whether SHLD3 is important for mediating PARPi sensitivity through a competitive growth assay between RPE1 hTERT p53‐KO BRCA1‐KO FLAG‐Cas9 (RPE BRCA1‐KO) cells and an isogenic SHLD3‐KO counterpart (RPE BRCA1‐KO SHLD3‐KO; Fig 6C and D). As expected, the RPE BRCA1‐KO SHLD3‐KO cell line outgrows RPE BRCA1‐KO cells in the presence of the PARPi olaparib (Fig 6D). We expressed FLAG‐SHLD3 variants in the RPE BRCA1‐KO SHLD3‐KO cell line through lentiviral transduction and tested their growth against an empty vector control (Figs 6D and EV5F). Complementation with wild‐type SHLD3 caused loss of cell fitness in the presence of olaparib, while SHLD3 variants W132A and R166A that are unable to interact with RIF1 did not and acted similar to the SHLD3‐KO (Fig 6D). These results suggest that the RIF1–SHLD3 interaction is essential for the function of shieldin in mediating PARPi toxicity in BRCA1‐mutated cells.
Discussion
The development of deep‐learning based protein structure prediction systems such as AlphaFold2‐Multimer and RoseTTAFold (Baek et al, 2021; preprint: Evans et al, 2021) made rapid, scalable, and highly accurate protein–protein structure prediction possible. A combination of these two methods was applied to comprehensively predict the protein structures of yeast protein complexes (Humphreys et al, 2021). However, this large‐scale approach has two primary obstacles: the exponential increase in the time necessary to compute the interactome of the larger human proteome, and the identification of false‐positive predictions. In this study, we bypassed these obstacles by applying exhaustive AF2‐pairwise protein interaction prediction on the 53BP1‐RIF1‐shieldin‐CST pathway. A focused set of proteins within the pairwise matrix enabled exhaustive analysis with minimal computational cost. Furthermore, there is a wealth of available literature on the protein–protein interactions within this pathway, thus providing a body of experimental validation for novel predicted structures.
Our analysis provides the structural basis for multiple known interactions, including 53BP1 phosphorylation‐dependent recognition by RIF1 (Setiaputra et al, 2022), 53BP1 dimerization through its oligomerization domain (Zgheib et al, 2009), RIF1 dimerization through its C terminus (Moriyama et al, 2018), and SHLD1 binding to CTC1 (Mirman et al, 2022b). Our analysis also revealed the novel prediction of the RIF1‐SHLD3 binding interface which we confirmed through extensive in vitro and cellular experiments. These findings enhance our understanding of the molecular basis for 53BP1‐RIF1‐shieldin‐CST assembly (Fig 6E) with putative structural information that shows remarkable agreement with experimental data and represent yet another validation of this approach to discover novel biologically relevant protein–protein interactions. The steady march of innovation in reducing the computational cost of protein structure prediction (Humphreys et al, 2021; Mirdita et al, 2022; preprint: Bryant & Noe, 2023) and in benchmarking scoring functions to detect accurate models (Bryant et al, 2022; Yin et al, 2022; preprint: Zhu et al, 2022) will increase the utility and accessibility of computational mining of the protein interactome.
While experimentally validating the predicted RIF1‐SHLD3 interface, we discovered several insights into shieldin function. Consistent with our previous experiments using recombinant RIF1N‐SHLD3‐REV7 (Setiaputra et al, 2022), our in vitro pulldown experiments confirm that this interaction occurs in the absence of DNA‐damage signaling. Tethering the RIF1 HEAT‐repeat domain to LacO arrays is sufficient for SHLD3 chromatin recruitment (Fig 2F). However, the existence of 53BP1 variants that localize RIF1 but not shieldin to DSBs (Setiaputra et al, 2022) suggests that this interface alone is insufficient for shieldin recruitment or that it is unavailable for SHLD3 binding in the majority of RIF1 molecules at break sites. The molecular details of shieldin recruitment by RIF1 are further complicated by the observation that the SHLD3 N201A and D216A variants that are impaired but not completely deficient in RIF1 binding can complement CSR to the same degree as wild‐type SHLD3 (Fig 6B). This, paired with the observation that shieldin is expressed at extremely low levels (Gupta et al, 2018) suggests that only a small number of the complex is required to mediate its function at DSBs and suggests the presence of additional hitherto uncharacterized mechanisms regulating shieldin or CST‐Polα‐primase activity at DSB sites (Noordermeer et al, 2018; Mirman et al, 2022b). The protein–protein interfaces within 53BP1‐RIF1‐shieldin‐CST explored in this study will greatly contribute to the ongoing efforts to characterize this enigmatic pathway.
Materials and Methods
Cell lines
U2OS 2‐6‐3 cells (Shanbhag et al, 2010) were cultured in McCoy's 5A (Modified) Medium (Gibco) supplemented with 10% fetal bovine serum (FBS; Wisent) and 50 IU/ml penicillin, 50 μg/ml streptomycin (Wisent). CH12F3‐2 cells (referred to as CH12F3) were cultured in RPMI 1640 (Gibco) supplemented with 10% FBS, 50 IU/ml penicillin, 50 μg/ml streptomycin, 5% NCTC‐109 (Gibco), 60 μM β‐mercaptoethanol (Sigma‐Aldrich). CH12F3‐2 Shld3 −/− Clone 2 was previously generated (Noordermeer et al, 2018). RPE1 hTERT p53‐KO FLAG‐Cas9 and 293T cells were cultured in Dulbecco's Modified Eagle Medium (DMEM; Gibco) supplemented with 10% FBS, 50 IU/ml penicillin, 50 μg/ml streptomycin, 1× GlutaMax (Gibco), 1xMEM nonessential amino acids (MEM‐NEAA; Gibco). Sf9 and High Five insect cells were maintained in suspension in I‐Max (Wisent). All mammalian cell lines were grown at 37°C, 5% CO2, and atmospheric O2 except for BRCA1‐mutated RPE cell lines, which were maintained at 3% O2. Insect cells were grown at 27°C shaking at 110 rpm.
RPE1 hTERT p53‐KO SHLD3‐KO FLAG‐Cas9 cells were generated by transfecting RPE1 hTERT p53‐KO FLAG‐Cas9 cells with in vitro‐transcribed sgRNA using Lipofectamine RNAiMAX (Thermo‐Fisher) according to the manufacturer's instructions using 0.6 μg each of two sgRNAs (sgSHLD3 #1: GGTGATCTTTTAGGTCTGAG, sgSHLD3 #2: TGAATTGTAGCATTACAAGA). sgRNAs were generated by in vitro transcription using TranscriptAid T7 High Yield Transcription Kit (Thermo Scientific) and cleaned using the Agencourt RNAClean XP Kit (Beckman‐Coulter) according to the manufacturer's instructions. After transfection, individual clones were collected and knockouts were confirmed by PCR amplification and the ICE CRISPR analysis tool (Synthego).
RPE1 hTERT p53‐KO BRCA1‐KO SHLD3‐KO FLAG‐Cas9 cells were generated by transfecting RPE1 hTERT p53‐KO BRCA1‐KO FLAG‐Cas9 cells (Zimmermann et al, 2018) with sgSHLD3 #1 as described above. Knockouts were confirmed by PCR amplification and the ICE CRISPR analysis tool.
U2OS 2‐6‐3 D28N were generated by electroporation of pCMV_BE4max (Addgene #112093; Koblan et al, 2018) and phU6‐gRNA expression cassette encoding the guide sequence AAGCGTCAGTCTGCCCTCCA (Addgene #53188; Kabadi et al, 2014). After 3 days of recovery, cells were plated at low density to isolate colonies. Editing was analyzed by PCR amplification of the edited region and Sanger sequencing.
Cell lines used in this study are routinely tested for mycoplasma contamination. Cell lines were not recently authenticated by STR profiling. All cell lines generated in this study are available upon request.
Plasmids
eGFP‐SHLD3 truncation‐expressing plasmids were generated using Gibson cloning to delete fragments from pcDNA5‐FRT/TO‐eGFP‐SHLD3 (Noordermeer et al, 2018). Amino acid substitutions in the C‐terminal (126–250) truncation were created via QuikChange site‐directed mutagenesis (Agilent). pDEST‐mCherry‐LacR‐RIF1 (1–567) truncations were generated by deletion PCR of the pDEST‐mCherry‐LacR‐RIF1 (1–967) plasmid. Amino acid substitutions in pDEST‐mCherry‐LacR‐RIF1 (1–967) were created using QuikChange site‐directed mutagenesis.
pAC8‐FLAG‐SHLD3 truncation plasmids were generated using Gibson cloning (New England Biolabs) and a pAC8‐FLAG‐SHLD3 template (Setiaputra et al, 2022). Amino acid substitutions of pAC8‐FLAG‐SHLD3 were made via site‐directed mutagenesis. Amino acid substitutions of Strep‐RIF1 (1–980) were generated using site‐directed mutagenesis on a pFastBac‐Strep‐RIF1 (1–980) template (Setiaputra et al, 2022).
The pHIV‐3xFLAG‐NAT plasmid was generated by inserting a 3xFLAG N‐terminal tag into the pHIV‐NAT‐hCD52 plasmid (Willis et al, 2018) by ligation of annealed oligos into the NotI and XmaI sites. The SHLD3 (2–250) coding sequence was ligated into the NheI and XmaI sites of pHIV‐3xFLAG‐NAT.
All plasmids generated in this study are available upon request.
AlphaFold2‐Multimer pairwise matrix screen for protein–protein interactions
Amino acid sequences for human 53BP1, RIF1, SHLD1, SHLD2, SHLD3, REV7, CTC1, STN1, TEN1, and ASTE1 were retrieved from UniProt. The default sequences were used except for SHLD2, where the longer isoform (Q86V20‐2) was used. Longer proteins were manually split—avoiding cutting at structured sites—to accommodate graphical memory limitations. 53BP1 was divided into four fragments (1–600, 601–1,200, 1,201–1,715, 1,716–1,927), RIF1 into four fragments (1–570, 571–1,200, 1,200–1,800, 1,800–2,472), SHLD2 into two fragments (1–420, 421–904), and CTC1 into two fragments (1–546, 547–1,217). Joint FASTA files for every unique pair (including self‐pairs) were generated using a Python script and used as input files for AlphaFold2‐Multimer (AF2) prediction. Analysis was performed in a template‐free mode.
The LocalColabFold v1.4 implementation of AF2 was used (Mirdita et al, 2022) on a cloud 8xTesla V100 16GB GPU instance from Lambda Labs, using the following command:
colabfold_batch –num‐recycle 3 –num‐models 5 –model‐type alphafold2_multimer_v2 <fasta input folder>/ <output folder>/.
A second round of prediction was performed using the LocalColabFold v1.5 implementation on a cloud A10 Tensor Core 24GB GPU instance from Lambda Labs, this time analyzing fragments of proteins representing ± 200 residues from the junctions of proteins split in the first analysis. Pairs that were analyzed in the first experiment were not repeated. The following command was used:
colabfold_batch –num‐recycle 3 –num‐models 5 –model‐type alphafold2_multimer_v2 <fasta input folder>/ <output folder>/.
The confidence of each predicted interface was analyzed by a Python script measuring pDockQ (Bryant et al, 2022) and mean interface‐predicted aligned error (PAE). Mean interface‐predicted aligned error was determined by identifying every pair of residues whose Cβ atoms (or Cα if glycine) were within 9 Å (identification of interface residues was adapted from pDockQ.py; https://gitlab.com/ElofssonLab/FoldDock/‐/tree/main/src from Bryant et al, 2022), then calculating the average PAE value across all residue pairs. Individual models that have pDockQ scores > 0.23 and mean interface PAE scores < 15 Å were classified as potential interactors. Unique protein pairs with at least 4/5 models meeting this cutoff or having previous supporting experimental evidence were classified as high‐confidence interactors. Protein structures were visualized with UCSF ChimeraX (Goddard et al, 2018). Modeling phosphate groups, H‐bond prediction (0.4 Å distance tolerance, 21° angle tolerance), surface hydrophobicity calculation, and multi‐model alignments were performed using built‐in ChimeraX functions. Additional individual AF2 predictions were performed using the ColabFold Google Colab AlphaFold2_mmseqs2 sheet (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb). For analysis that displays side chains, an additional AMBER relaxation step was performed on the models using the relax_amber.ipynb Google Colab sheet (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/beta/relax_amber.ipynb) which is part of the ColabFold suite.
Lentivirus generation and infection
Lentiviruses were generated in 293T cells by cotransfecting the pHIV‐3xFLAG‐NAT viral targeting vector with plasmids encoding RRE, REV, and VSV‐G using the TransIT‐LT1 Transfection Reagent (Mirus). Viral supernatants were collected, filtered through a 0.45 μm syringe filter, and snap‐frozen in liquid nitrogen. Viral infections were performed in the presence of 8 μg/ml polybrene (Sigma‐Aldrich) for 24 h, after which the media was refreshed and selection with 0.2 μg/ml nourseothricin (Gold Biotechnology) was performed until uninfected controls no longer survived, upon which selection was no longer maintained.
SHLD3‐RIF1 co‐immunoprecipitations
Baculoviruses were generated in Spodoptera frugiperda Sf9 cells (Thermo‐Fisher) using the Bac‐to‐Bac method for the pFastBac‐derived vectors or, for the pAC8‐derived vectors, cotransfecting them with linearized viral DNA (Abdulrahman et al, 2009). The proteins were expressed in Trichoplusia ni High Five cells (Expression Systems) by baculoviral expression. Individual High Five cell cultures were infected, so that they would express a single recombinant protein.
36‐h postinfection, the cells were harvested by centrifugation. The cells were resuspended in lysis buffer (50 mM Tris–HCl pH 8, 200 mM NaCl, 1 mM PMSF, 1 mM TCEP, 1× SIGMAFAST protease inhibitor tablet) and lysed by sonication prior to being centrifuged at high speed (21,000 g, 20 min, 4°C) and the supernatant collected. Lysate containing SHLD3 recombinant protein was mixed with lysate containing RIF1 recombinant protein, REV7 recombinant protein, or both. The lysate mixtures were applied to FLAG M2 beads (Sigma‐Aldrich) or Strep‐Tactin Superflow high‐capacity resin (IBA Lifesciences) and incubated with rotation at 4°C for 1 h. The resin was washed twice, each wash including a 20‐min incubation with rotation at 4°C. The pulldowns were eluted from the FLAG and Strep resins using 0.1 mg/ml 3xFLAG peptide (GlpBio) or 2.5 mM desthiobiotin (Sigma‐Aldrich), respectively. The results were analyzed by immunoblotting. Table EV3 lists the antibodies used in this study.
U2OS‐2‐6‐3 SHLD3‐RIF1 colocalization assay
2.5–3.0 × 105 U2OS‐2‐6‐3 cells containing a LacO array of 256 repeated LacO sequences (Shanbhag et al, 2010) were plated on glass coverslips in 6‐well plates. The next day, the cells were transfected as follows: 1 μg eGFP‐SHLD3 constructs, 1 μg mCherry‐LacR‐RIF1 constructs, and 6 μl Lipofectamine 2000 (Thermo Scientific) were incubated in Opti‐MEM (Gibco) for 20 min at room temperature before being added to the cells whose media had been changed to McCoy's with 10% FBS and no antibiotics. Two hours later, the cells were washed in PBS and the transfection media was replaced by fresh McCoy's with 10% FBS and no antibiotics. Two days after transfection, the cells were washed in PBS, then incubated for 10 min on ice in 1 ml nuclear pre‐extraction buffer (20 mM HEPES pH 7.4, 20 mM NaCl, 5 mM MgCl2, 0.5% NP‐40, 1 mM DTT, and 1× cOmplete EDTA‐free protease inhibitor cocktail [Roche]), and washed again in PBS. The coverslips were then incubated for 10 min at room temperature in 1 ml 4% formaldehyde (Thermo Scientific) in PBS and washed three times in PBS.
Images for this and other microscopy experiments were acquired on a Zeiss LSM780 laser scanning confocal microscope on either a 40× or 63× Plan‐Apochromat objective lens (specific to the experiment) using the Zen Black software (Zeiss). Images were quantified using ImageJ to determine the average fluorescence intensities of the nuclear region colocalizing with the mCherry‐LacR signal and the average fluorescence intensity within the nuclear boundary. One was added to both values before calculating the ratio to minimize the confounding effects of dividing with extremely low‐intensity values.
Any adjustments to image brightness or contrast were applied to the entire image and to the same extent between all images of the same experiment.
U2OS‐2‐6‐3 FokI focus recruitment assay
2.5 × 105 U2OS‐2‐6‐3 cells containing a LacO array of 256 repeated LacO sequences and an inducible mCherry‐LacR‐FokI (Shanbhag et al, 2010) were plated on glass coverslips in 6‐well plates. The next day, the cells were transfected as follows: 2 μg eGFP‐SHLD3 constructs and 6 μl Lipofectamine 2000 (Thermo Scientific) were incubated in Opti‐MEM for 20 min at room temperature before being added to the cells whose media had been changed to McCoy's with 10% FBS and no antibiotics. Two hours later, the cells were washed in PBS and the media was changed to fresh McCoy's with 10% FBS and no antibiotics. Two days after transfection, the expression of mCherry‐LacR‐FokI was induced by the addition of 10 μg/ml 4‐hydroxytamoxifen (Sigma‐Aldrich) and 1 μM of Shield‐1 peptide (Clontech, Mountain View CA) for 4 h. Cells were washed in PBS, then incubated for 10 min on ice in 1 ml nuclear pre‐extraction buffer (20 mM HEPES pH 7.4, 20 mM NaCl, 5 mM MgCl2, 0.5% NP‐40, 1 mM DTT, and 1× cOmplete EDTA‐free protease inhibitor cocktail [Roche]), and washed again in PBS. The coverslips were then incubated for 10 min at room temperature in 1 ml of 4% formaldehyde in PBS and washed three times in PBS. Images were acquired and analyzed as described above.
Immunofluorescence
Fixed cells cultured on glass coverslips were placed in a humidified chamber and incubated in a blocking solution (PBS + 0.2% cold water fish gelatin + 0.5% BSA) for 30 min. The blocking solution was replaced with a primary antibody diluted in blocking solution and incubated for 2 h at room temperature. The coverslips were washed three times for a total of 15 min in PBS then incubated for 1 h at room temperature with a secondary antibody diluted in blocking solution. The coverslips were washed three times for a total of 15 min in PBS then mounted onto glass slides using ProLong Gold Antifade mounting media with DAPI (Invitrogen). Coverslips were imaged as described above. Table EV3 lists the antibodies used in this study.
Ionizing radiation‐induced focus formation
1.5–1.75 × 105 RPE SHLD3‐KO cells expressing SHLD3 variants were plated per well of a 6‐well plate containing glass coverslips. 24‐h postplating, the cells were transfected by incubating 2 μg eGFP‐SHLD2 construct and 6 μl Lipofectamine 2000 (Thermo Scientific) in Opti‐MEM for 20 min at room temperature before adding the mixture to the cells whose media had been changed to fresh DMEM. Two hours later, the cells were washed in PBS and the media was changed to fresh DMEM. 48‐h post‐transfection cells were treated with 0.7 Gy X‐irradiation using an X‐ray cabinet (Faxitron), incubated for 1 h at 37°C, washed in PBS, then incubated for 10 min on ice in 1 ml nuclear pre‐extraction buffer (20 mM HEPES pH 7.4, 20 mM NaCl, 5 mM MgCl2, 0.5% NP‐40, 1 mM DTT, and 1× cOmplete EDTA‐free protease inhibitor cocktail [Roche]), and washed again in PBS. The coverslips were then incubated for 10 min at room temperature in 1 ml 4% formaldehyde (Thermo Scientific) in PBS and washed three times in PBS. The cells were then stained for immunofluorescence and imaged. Images were blinded and quantified by counting the percentage of cells with at least five colocalizing SHLD3 and 53BP1 foci and colocalizing SHLD2 and 53BP1 foci.
Analyzing localization to 53BP1 bodies
RPE cells were grown on glass coverslips and treated with 200 nM aphidicolin (Sigma‐Aldrich) for 24 h. Coverslips were then washed with PBS, fixed with 4% formaldehyde for 10 min at room temperature, washed again 3× with PBS, permeabilized with 0.3% Triton X‐100 in PBS for 30 min at room temperature, and washed 3× with PBS. Coverslips were stained with antibodies to FLAG, 53BP1, and cyclin A. Cyclin A‐positive cells were discarded from the analysis. For Pearson Correlation Coefficient calculation, the MeasureColocalization functionality of CellProfiler v4.2.5 (Stirling et al, 2021) was used within each masked nucleus to calculate the pixel correlation between the 53BP1 and FLAG channels. For FLAG intensity in 53BP1 bodies measurement, 53BP1 foci in cyclin A‐negative nuclei were manually masked in ImageJ, and the mean FLAG fluorescence intensity within the mask was divided by the mean nuclear FLAG fluorescence intensity. Number of 53BP1 bodies were counted automatically by CellProfiler.
Laser microirradiation
Laser microirradiation experiments were performed as previously described (Setiaputra et al, 2022) except that coverslips were harvested 1 h after irradiation.
Class switch recombination assay
Class switch recombination assays were performed essentially as previously described (Noordermeer et al, 2018). 1 × 105 CH12F3‐2 cells were plated in 24‐well plates in growth medium supplemented with 1 μg/ml anti‐CD40 antibody (eBioscience), 1 ng/ml TGF‐β (R&D Systems), and 10 ng/ml mIL‐4 (R&D Systems). After 48 h, cells were harvested, stained with anti‐IgA‐PE (Southern Biotech), and fixed with 4% formaldehyde. Fluorescence signal was acquired on an Attune NxT Flow Cytometer (Thermo‐Fisher). Data were analyzed using FlowJo software (BD Biosciences).
Two‐color competitive growth assay
Competitive growth assays were modified from the previously described (Noordermeer et al, 2018). The indicated cell lines were labeled with either mCherry or eGFP through lentiviral transduction with LentiGuide‐Puro‐NLS‐GFP or ‐mCherry (Noordermeer et al, 2018) without sgRNA. After puromycin selection, mCherry‐ and eGFP‐expressing cells were mixed 1:1 and plated in 24‐well plates (1,250 cells each for a total of 2,500 per well, three wells each sample) and treated with or without 50 nM olaparib (SelleckChem). Cells were subcultured throughout the experiment as they approach confluency. The cells were imaged for eGFP and mCherry signal at 1, 5, 9, and 13 days after plating using the InCell Analyzer, imaging the entire well with 16 images per well (GE Healthcare) with a 4× objective. Quantification of eGFP‐ and mCherry‐positive cells was performed through an Acapella script (PerkinElmer). eGFP:mCherry ratio was normalized to the day 1 value.
Statistical analysis
No statistical methods were used to determine sample sizes; instead, we used sample sizes similar to previous similar publications. Blinding was performed on the level of image filenames when manually quantifying the presence or absence of ionizing radiation‐induced foci that can be subjective, otherwise blinding was not performed. Tests for normality were performed using the Kolmogorov–Smirnov test, except for sample sizes ≤ 5, which were assumed to be normally distributed. Nonparametric multiple comparison tests against a single control sample were performed by the Kruskal–Wallis one‐way analysis of variance (ANOVA) with the Dunn's multiple comparisons test. Parametric multiple comparison tests were performed by ordinary one‐way ANOVA with the Dunnett's multiple comparisons test. For the nonparametric comparison of two groups, the Mann–Whitney test was performed. All statistical analysis was performed in GraphPad Prism 8 version 8.4.3.
Reporting
No formal guidelines or checklists were followed in the preparation of this manuscript.
Author contributions
Chérine Sifri: Conceptualization; investigation; writing – review and editing. Lisa Hoeg: Software. Daniel Durocher: Conceptualization; supervision; funding acquisition; writing – original draft; writing – review and editing. Dheva Setiaputra: Conceptualization; formal analysis; supervision; investigation; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
DD is a shareholder and advisor of Repare Therapeutics. The remaining authors declare no competing interests.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
PDF+
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank R. Szilard for critical reading of this manuscript and other members of the Durocher lab for valuable discussion. We thank R. Greenberg for the U2OS 2‐6‐3 cell line and R. Scully for the pHIV‐NAT‐hCD52 plasmid. DS was funded by grant 26002 from the Cancer Research Society for most of this work. DD is a Canada Research Chair (Tier 1), and work in the DD lab was funded by grants from the Canadian Institutes of Health Research (CIHR, PJT‐180438) and the Krembil Foundation to DD.
EMBO reports (2023) 24: e56834
Contributor Information
Daniel Durocher, Email: durocher@lunenfeld.ca.
Dheva Setiaputra, Email: setiaputra@lunenfeld.ca.
Data availability
The datasets and computer code produced in this study are available in the following databases:
Python scripts: Github (https://github.com/Dsetiaputra/scripts/tree/main/2023_sifrietal).
AlphaFold2 dataset: Mendeley Data (https://doi.org/10.17632/dj2kv8zzxy).
References
- Abdulrahman W, Uhring M, Kolb‐Cheynel I, Garnier J‐M, Moras D, Rochel N, Busso D, Poterszman A (2009) A set of baculovirus transfer vectors for screening of affinity tags and parallel expression strategies. Anal Biochem 385: 383–385 [DOI] [PubMed] [Google Scholar]
- Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD et al (2021) Accurate prediction of protein structures and interactions using a three‐track neural network. Science 373: 871–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant P, Noe F (2023) Rapid protein‐protein interaction network creation from multiple sequence alignments with Deep Learning. bioRxiv 10.1101/2023.04.15.536993 [PREPRINT] [DOI]
- Bryant P, Pozzati G, Elofsson A (2022) Improved prediction of protein‐protein interactions using AlphaFold2. Nat Commun 13: 1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai SW, Zinder JC, Svetlov V, Bush MW, Nudler E, Walz T, de Lange T (2022) Cryo‐EM structure of the human CST–Polα/primase complex in a recruitment state. Nat Struct Mol Biol 29: 813–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen E, Di Virgilio M, Kruhlak MJ, Nieto‐Soler M, Wong N, Chen H‐T, Faryabi RB, Polato F, Santos M, Starnes LM et al (2013) 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 153: 1266–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier J‐B, Borel V, Steger M, Tomas‐Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ (2013) RIF1 is essential for 53BP1‐dependent nonhomologous end joining and suppression of DNA double‐strand break resection. Mol Cell 49: 858–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Zhang F, Wang L, Shan S, Gong Z, Zhou Z (2020) Structural basis for shieldin complex subunit 3–mediated recruitment of the checkpoint protein REV7 during DNA double‐strand break repair. J Biol Chem 295: 250–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dev H, Chiang T‐WW, Lescale C, de Krijger I, Martin AG, Pilger D, Coates J, Sczaniecka‐Clift M, Wei W, Ostermaier M et al (2018) Shieldin complex promotes DNA end‐joining and counters homologous recombination in BRCA1‐null cells. Nat Cell Biol 20: 954–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano‐Díaz C, Orthwein A, Fradet‐Turcotte A, Xing M, Young JTF, Tkáč J, Cook MA, Rosebrock AP, Munro M, Canny MD et al (2013) A cell cycle‐dependent regulatory circuit composed of 53BP1‐RIF1 and BRCA1‐CtIP controls DNA repair pathway choice. Mol Cell 49: 872–883 [DOI] [PubMed] [Google Scholar]
- Evans R, O'Neill M, Pritzel A, Antropova N, Senior A, Green T, Žídek A, Bates R, Blackwell S, Yim J et al (2021) Protein complex prediction with AlphaFold‐multimer bioinformatics. bioRixv 10.1101/2021.10.04.463034 [PREPRINT] [DOI]
- Fradet‐Turcotte A, Canny MD, Escribano‐Díaz C, Orthwein A, Leung CCY, Huang H, Landry M‐C, Kitevski‐LeBlanc J, Noordermeer SM, Sicheri F et al (2013) 53BP1 is a reader of the DNA‐damage‐induced H2A Lys 15 ubiquitin mark. Nature 499: 50–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Feng S, Ning S, Liu J, Zhao H, Xu Y, Shang J, Li K, Li Q, Guo R et al (2018) An OB‐fold complex controls the repair pathways for DNA double‐strand breaks. Nat Commun 9: 3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, Fischer R, Deobagkar‐Lele M, Sanchiz‐Calvo M, Fueyo‐Marcos E et al (2018) 53BP1 cooperation with the REV7–shieldin complex underpins DNA structure‐specific NHEJ. Nature 560: 122–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE (2018) UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci 27: 14–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, Typas D, Lammers M, Mailand N, Nussenzweig A et al (2018) DNA repair network analysis reveals Shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell 173: 972–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP (2011) Replication stress induces 53BP1‐containing OPT domains in G1 cells. J Cell Biol 193: 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Lin X, Chavez BL, Agrawal S, Lusk BL, Lim CJ (2022) Structures of the human CST‐Polα–primase complex bound to telomere templates. Nature 608: 826–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C (1995) Dali: a network tool for protein structure comparison. Trends Biochem Sci 20: 478–480 [DOI] [PubMed] [Google Scholar]
- Humphreys IR, Pei J, Baek M, Krishnakumar A, Anishchenko I, Ovchinnikov S, Zhang J, Ness TJ, Banjade S, Bagde SR et al (2021) Computed structures of core eukaryotic protein complexes. Science 374: eabm4805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav‐Tal Y, Bertrand E, Singer RH et al (2004) From silencing to gene expression: real‐time analysis in single cells. Cell 116: 683–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi AM, Ousterout DG, Hilton IB, Gersbach CA (2014) Multiplex CRISPR/Cas9‐based genome engineering from a single lentiviral vector. Nucleic Acids Res 42: e147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koblan LW, Doman JL, Wilson C, Levy JM, Tay T, Newby GA, Maianti JP, Raguram A, Liu DR (2018) Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat Biotechnol 36: 843–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Feng J, Zuo P, Yang J, Lu Y, Yin Y (2020) Molecular basis for assembly of the shieldin complex and its implications for NHEJ. Nat Commun 11: 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CJ, Barbour AT, Zaug AJ, Goodrich KJ, McKay AE, Wuttke DS, Cech TR (2020) The structure of human CST reveals a decameric assembly bound to telomeric DNA. Science 368: 1081–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling AK, Munro M, Chaudhary N, Li C, Berru M, Wu B, Durocher D, Martin A (2020) SHLD2 promotes class switch recombination by preventing inactivating deletions within the Igh locus. EMBO Rep 21: e49823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas C, Savic V, Bekker‐Jensen S, Doil C, Neumann B, Sølvhøj Pedersen R, Grøfte M, Chan KL, Hickson ID, Bartek J et al (2011) 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 13: 243–253 [DOI] [PubMed] [Google Scholar]
- Methot SP, Di Noia JM (2017) Chapter two – Molecular mechanisms of somatic hypermutation and class switch recombination. In Advances in Immunology, Alt FW (ed), pp 37–87. Cambridge, MA: Academic Press; [DOI] [PubMed] [Google Scholar]
- Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M (2022) ColabFold: making protein folding accessible to all. Nat Methods 19: 679–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D, de Lange T (2018) 53BP1–RIF1–shieldin counteracts DSB resection through CST‐ and Polα‐dependent fill‐in. Nature 560: 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirman Z, Cai S, de Lange T (2022a) CST/Polα/primase‐mediated fill‐in synthesis at DSBs. Cell Cycle 22: 379–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirman Z, Sasi NK, King A, Chapman JR, de Lange T (2022b) 53BP1–shieldin‐dependent DSB processing in BRCA1‐deficient cells requires CST–Polα–primase fill‐in synthesis. Nat Cell Biol 24: 51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama K, Yoshizawa‐Sugata N, Masai H (2018) Oligomer formation and G‐quadruplex‐binding by purified murine Rif1 protein, a key organizer of higher‐order chromatin architecture. J Biol Chem 293: 3607–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Sakai W, Kawamoto T, Bree RT, Lowndes NF, Takeda S, Taniguchi Y (2006) Genetic dissection of vertebrate 53BP1: a major role in non‐homologous end joining of DNA double strand breaks. DNA Repair 5: 741–749 [DOI] [PubMed] [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Álvarez‐Quilón A, Moatti N, Zimmermann M et al (2018) The shieldin complex mediates 53BP1‐dependent DNA repair. Nature 560: 117–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panier S, Boulton SJ (2014) Double‐strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 15: 7–18 [DOI] [PubMed] [Google Scholar]
- Schimmel J, Muñoz‐Subirana N, Kool H, van Schendel R, Tijsterman M (2021) Small tandem DNA duplications result from CST‐guided pol α‐primase action at DNA break termini. Nat Commun 12: 4843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setiaputra D, Durocher D (2019) Shieldin – the protector of dna ends. EMBO Rep 20: e47560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setiaputra D, Escribano‐Díaz C, Reinert JK, Sadana P, Zong D, Callen E, Sifri C, Seebacher J, Nussenzweig A, Thomä NH et al (2022) RIF1 acts in DNA repair through phosphopeptide recognition of 53BP1. Mol Cell 82: 1359–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbhag NM, Rafalska‐Metcalf IU, Balane‐Bolivar C, Janicki SM, Greenberg RA (2010) ATM‐dependent chromatin changes silence transcription In cis to DNA double‐Strand breaks. Cell 141: 970–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Bunker RD, Mattarocci S, Ribeyre C, Faty M, Gut H, Scrima A, Rass U, Rubin SM, Shore D et al (2013) Rif1 and Rif2 shape telomere function and architecture through multivalent Rap1 interactions. Cell 153: 1340–1353 [DOI] [PubMed] [Google Scholar]
- Spies J, Lukas C, Somyajit K, Rask M‐B, Lukas J, Neelsen KJ (2019) 53BP1 nuclear bodies enforce replication timing at under‐replicated DNA to limit heritable DNA damage. Nat Cell Biol 21: 487–497 [DOI] [PubMed] [Google Scholar]
- Stirling DR, Swain‐Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, Goodman A (2021) CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics 22: 433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaravinayagam D, Rahjouei A, Andreani M, Tupiņa D, Balasubramanian S, Saha T, Delgado‐Benito V, Coralluzzo V, Daumke O, Di Virgilio M (2019) 53BP1 supports immunoglobulin class switch recombination independently of its DNA double‐Strand break end protection function. Cell Rep 28: 1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susvirkar V, Faesen AC (2022) Molecular insights into human Shieldin complex assembly and recruitment to DSBs. bioRxiv 10.1101/2022.06.09.495453 [PREPRINT] [DOI]
- Ward IM, Reina‐San‐Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F et al (2004) 53BP1 is required for class switch recombination. J Cell Biol 165: 459–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts LP, Natsume T, Saito Y, Garzon J, Dong Q, Boteva L, Gilbert N, Kanemaki MT, Hiraga S‐I, Donaldson AD (2020) The RIF1‐long splice variant promotes G1 phase 53BP1 nuclear bodies to protect against replication stress. Elife 9: e58020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Panday A, Duffey EE, Scully R (2018) Rad51 recruitment and exclusion of non‐homologous end joining during homologous recombination at a Tus/Ter mammalian replication fork barrier. PLoS Genet 14: e1007486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MD, Benlekbir S, Fradet‐Turcotte A, Sherker A, Julien J‐P, McEwan A, Noordermeer SM, Sicheri F, Rubinstein JL, Durocher D (2016) The structural basis of modified nucleosome recognition by 53BP1. Nature 536: 100–103 [DOI] [PubMed] [Google Scholar]
- Wright GE, Brown NC (1990) Deoxyribonucleotide analogs as inhibitors and substrates of DNA polymerases. Pharmacol Ther 47: 447–497 [DOI] [PubMed] [Google Scholar]
- Xie W, Wang S, Wang J, de la Cruz MJ, Xu G, Scaltriti M, Patel DJ (2021) Molecular mechanisms of assembly and TRIP13‐mediated remodeling of the human Shieldin complex. Proc Natl Acad Sci U S A 118: e2024512118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin R, Feng BY, Varshney A, Pierce BG (2022) Benchmarking AlphaFold for protein complex modeling reveals accuracy determinants. Protein Sci 31: e4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgheib O, Pataky K, Brugger J, Halazonetis TD (2009) An oligomerized 53BP1 tudor domain suffices for recognition of DNA double‐strand breaks. Mol Cell Biol 29: 1050–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, Kim W, Gao H, Liu C, Zhang Y, Chen Y, Deng M, Zhou Q, Huang J, Hu Q et al (2021) ASTE1 promotes shieldin‐complex‐mediated DNA repair by attenuating end resection. Nat Cell Biol 23: 894–904 [DOI] [PubMed] [Google Scholar]
- Zhu W, Shenoy A, Kundrotas P, Elofsson A (2022) Evaluation of AlphaFold‐Multimer prediction on multi‐chain protein complexes. bioRxiv 10.1101/2022.12.08.519586 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T (2013) 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339: 700–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaitė Ž, Muir M, Fluteau A, Aregger M, McEwan A et al (2018) CRISPR screens identify genomic ribonucleotides as a source of PARP‐trapping lesions. Nature 559: 285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]