

Abstract
Several homeoprotein transcription factors transfer between cells and regulate gene expression, protein translation, and chromatin organization in recipient cells. ENGRAILED‐1 is one such homeoprotein expressed in spinal V1 interneurons that synapse on α‐motoneurons. Neutralizing extracellular ENGRAILED‐1 by expressing a secreted single‐chain antibody blocks its capture by spinal motoneurons resulting in α‐motoneuron loss and limb weakness. A similar but stronger phenotype is observed in the Engrailed‐1 heterozygote mouse, confirming that ENGRAILED‐1 exerts a paracrine neurotrophic activity on spinal cord α‐motoneurons. Intrathecal injection of ENGRAILED‐1 leads to its specific internalization by spinal motoneurons and has long‐lasting protective effects against neurodegeneration and weakness. Midbrain dopaminergic neurons express Engrailed‐1 and, similarly to spinal cord α‐motoneurons, degenerate in the heterozygote. We identify genes expressed in spinal cord motoneurons whose expression changes in mouse Engrailed‐1 heterozygote midbrain neurons. Among these, p62/SQSTM1 shows increased expression during aging in spinal cord motoneurons in the Engrailed‐1 heterozygote and upon extracellular ENGRAILED‐1 neutralization. We conclude that ENGRAILED‐1 might regulate motoneuron aging and has non‐cell‐autonomous neurotrophic activity.
Keywords: ALS, ENGRAILED1, Motoneurons, Neurodegeneration, Paracrine
Subject Categories: Neuroscience
Reducing ENGRAILED‐1 expression in V1 interneurons of the ventral spinal cord or blocking its transfer from interneurons to motoneurons leads to α‐motoneuron retrograde degeneration. Exogenous ENGRAILED‐1 addressed to motoneurons restores a wild‐type phenotype in ENGRAILED‐1 heterozygotes.
Introduction
Homeoprotein (HP) transcription factors are key transcriptional regulators with well‐established developmental functions, including control of morphogenesis, lineage decisions, and cell differentiation (Gehring, 1987; Holland & Takahashi, 2005). In addition to their classical cell‐autonomous activities, non‐cell‐autonomous developmental functions based on HP intercellular transfer were identified for ENGRAILED‐1 (EN1), EN2, VAX1, OTX2, and PAX6. Early in development, paracrine ENGRAILED induces anterior cross‐vein formation in the fly wing disk and shapes the zebrafish optic tectum (Layalle et al, 2011; Rampon et al, 2015; Amblard et al, 2020b). ENGRAILED‐2 and VAX1, respectively, regulate retinal ganglion cell (RGC) axon guidance in the tectum and decussation at the optic chiasma (Brunet et al, 2005; Wizenmann et al, 2009; Yoon et al, 2012; Kim et al, 2014), while PAX6 regulates the size of the zebrafish eye anlagen, enhances oligodendrocyte progenitor cell migration in the chick neural tube, and guides Cajal–Retzius cell migration in the mouse neuroepithelium (Lesaffre et al, 2007; Di Lullo et al, 2011; Kaddour et al, 2019). At late developmental stages and in the adult, OTX2 non‐cell‐autonomous activity regulates cerebral cortex plasticity (Sugiyama et al, 2008; Di Nardo et al, 2020), inner retinal physiological functions and RGC survival (Torero‐Ibad et al, 2020; Pensieri et al, 2021; Torero‐Ibad et al, 2011). This novel signaling pathway, involving unconventional secretion and internalization with direct access to the cytoplasm, is likely common to a larger number of HPs since the transfer domains are highly conserved between most HPs (Prochiantz & Joliot, 2003). The finding that more than 150 HPs can transfer between cells, in vitro and in the embryonic brain, even though without identified functions for most of them, lends weight to this hypothesis (Lee et al, 2019).
Several vertebrate HPs remain expressed throughout life, but their adult physiological functions are still poorly understood. In the context of neurodegenerative diseases, EN1 is of particular interest since it is expressed in the mesencephalic dopaminergic (mDA) neurons of the ventral tegmental area (VTA) and substantia nigra pars compacta (SNpc) (Sgadò et al, 2006; Di Nardo et al, 2007), two cell populations that degenerate in Parkinson's Disease (PD), and promotes their survival in PD animal models (Di Nardo et al, 2018). In the En1 +/− mouse (En1‐Het), mDA neurons from the VTA and SNpc undergo progressive retrograde degeneration leading to a loss of 20 and 40% of their initial number, respectively (Sgadò et al, 2006; Sonnier et al, 2007; Nordström et al, 2015). Conversely, EN1 injection in the midbrain and its subsequent internalization by mDA neurons, thanks to its transduction properties, prevents their degeneration in rodent and macaque PD models (Alvarez‐Fischer et al, 2011; Thomasson et al, 2019). Since mDA neurons express EN1, the latter observations reflect a cell‐autonomous pro‐survival activity of EN1. This incited us to search for other neurons that may require EN1 for their survival, either expressing En1 or in the vicinity of En1‐expressing cells and thus potentially exposed to secreted EN1. In the spinal cord, inhibitory V1 interneurons, including Renshaw cells (RCs), have been shown to express En1, and α‐motoneurons (αMNs) are candidate non‐cell‐autonomous targets owing to their direct contact with En1‐expressing V1 interneurons (Wenner et al, 2000; Higashijima et al, 2004; Alvarez et al, 2005).
We confirm here that En1 is expressed in adult spinal cord V1 interneurons and now report that these interneurons do not degenerate in the En1‐Het mouse. In contrast, αMNs of En1‐Het mice show retrograde degeneration associated with progressive limb strength loss. This phenotype is in part the consequence of local EN1 non‐cell‐autonomous activity since it can be partially mimicked in wild‐type (WT) mice following EN1 neutralization in the spinal cord extracellular space. Furthermore, EN1 intrathecally injected at 3 months gains direct access to MNs and, in En1‐Het mice, a single injection restores limb strength and prevents αMN death for at least 2.5 months. This demonstrates a novel non‐cell‐autonomous function of EN1 in the adult CNS and suggests that this HP may be of interest for developing novel therapeutic strategies and targets for diseases involving αMN degeneration.
Results
ENGRAILED‐1 is transferred from V1 interneurons to motoneurons in the ventral spinal cord
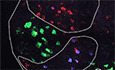
Previous studies reported En1 expression in embryonic and early postnatal spinal cord V1 interneurons but, to our knowledge, no study has directly visualized EN1 in adult mouse V1 interneurons. The available studies identify En1‐expressing cells in the adult mouse spinal cord using EN1‐Cre‐inducible reporter genes activated during development that maintain reporter gene expression throughout life (Sapir, 2004; Alvarez et al, 2005; Lane et al, 2021). In contrast with classical in situ hybridization (ISH) that gave no or very low signal in our hands, RNAscope ISH clearly reveals En1 expression in adult 4.5‐month‐old mice V1 interneurons, including Renshaw cells (RCs) identified by Calbindin‐1 expression (Fig 1A). These En1‐expressing cells are localized dorsally (V1P) and ventrally (V1R, RCs) to a population of En1‐negative neurons identified by a choline acetyltransferase (ChAT) ISH (RNAscope) probe, thus MNs. En1 expression, quantified by RT–qPCR, is fully maintained between 4.5 and 16 months and reduced by twofold in the En1‐Het mouse (Fig 1B).
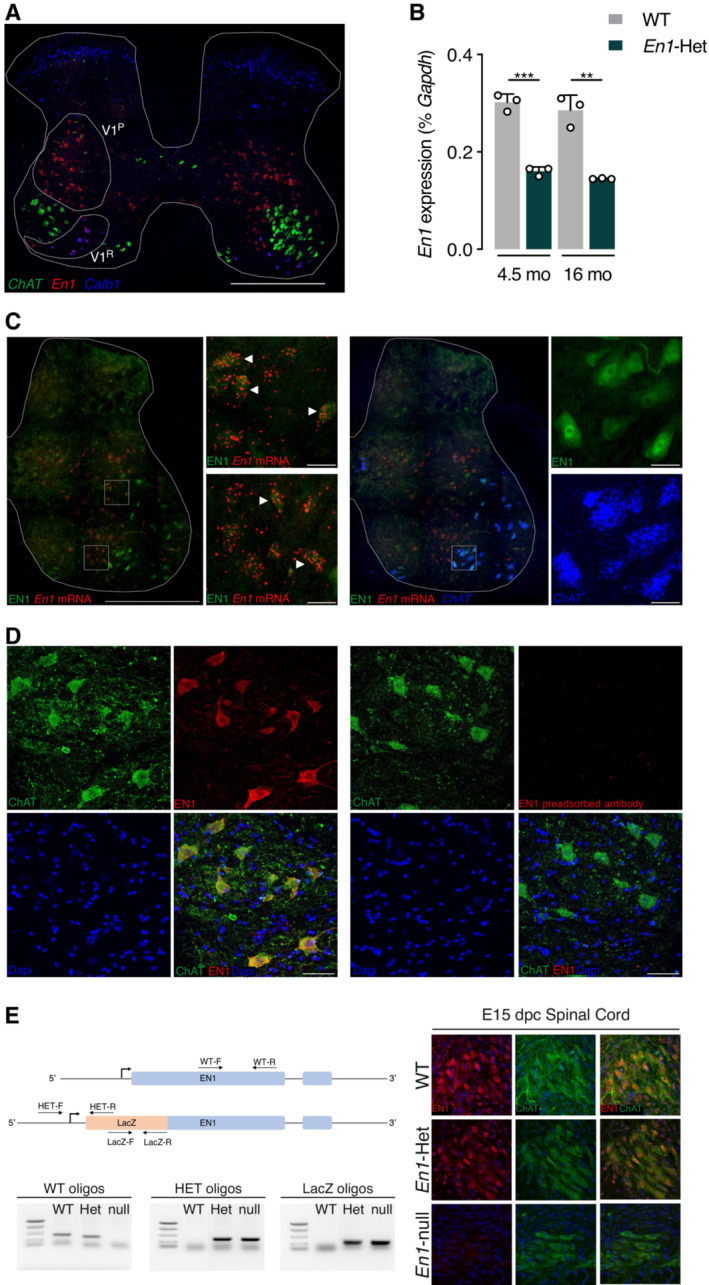
Figure 1. En1 expression in the adult spinal cord.
- Triple RNAscope in situ hybridization showing Engrailed‐1 (En1), Choline acetyltransferase (ChAT), and Calbindin‐1 (Calb1) expression in the lumbar spinal cord. En1 is expressed in V1 interneurons, dorsal (V1P) and ventral (V1R) to the main ChAT‐expressing motoneuron pool. Ventral interneurons correspond to Renshaw cells as shown by Calb1 expression. Scale bar: 500 μm.
- RT–qPCR of RNA from the lumbar enlargement at 4.5 and 16 months of age shows stable En1 expression in WT at both ages and a twofold reduction of expression in heterozygous mice. Unpaired two‐sided t‐test. **P < 0.005; ***P < 0.0005. n = 3. Values are mean ± SD.
- Triple staining EN1 IHC (green), En1 RNAscope ISH (red), and ChAT RNAscope (blue) demonstrating the double staining of En1 mRNA and protein (EN1) in the V1 interneuron population (left panel insets, arrowheads point toward examples of double‐stained V1 interneurons), and the presence of EN1 protein in large cells not expressing En1 mRNA (left panel) but expressing ChAT (right panel insets). Scale bar: 500, 30 μm for high magnification insets.
- Left: EN1 (red) detected with the LSBio antibody is localized in ChAT‐expressing neurons (green) in the ventral horns of the spinal cord. Right: EN1 signal is lost upon preincubation of the antibody with 1.5 M excess of recombinant hEN1. Scale bar: 50 μm.
- Left: Relative positions of the different oligonucleotides selected to genotype the E15 embryos and examples of the genotyping based on the combination of PCRs with the different pairs of primers. Right: Double ChAT/EN1 immunostaining demonstrating the co‐localization of the two proteins in the WT and the En1‐Het ventral cord and the absence of EN1 staining in MNs from En1‐KO embryos. Note that the staining is reduced in En1‐Het embryos, compared with WT embryos.
Data information: These experiments were performed once.
Source data are available online for this figure.
To examine En1 expression at the protein level, we tested several antibodies and found that LS‐B9070 from Lifespan Biosciences (LSBio) directed against EN1 amino acids 1 to 30 gives a clear and consistent staining of both V1 interneurons (Fig 1C, left panels) and of ChAT‐expressing (ChAT+) MNs (Fig 1C, right panels). Antibody specificity was verified in several ways. First, in spinal cord and ventral midbrain extracts, the LSBio antibody and a previously characterized 86/8 antibody (Sonnier et al, 2007) recognize a major band that migrates at the same position as recombinant EN1 (Fig EV1A). Second, immunostaining of ChAT+ cells by the LSBio antibody (Fig 1D, left panel) is totally lost upon preincubation of the antibody with recombinant hEN1 (right panel). Third, in En1‐KO mice at embryonic day 15 (the KO is embryonic lethal), the staining is entirely absent in ChAT+ neurons (Fig 1E) demonstrating, in addition to antibody specificity, that EN1 transfers also at embryonic stages. Finally, serial antibody dilutions demonstrate that the intensity of ChAT‐expressing (ChAT+) MN staining is decreased in adult En1‐Het mouse compared with WT (Fig EV1B).
Figure EV1. Characterization of the anti‐EN1 LSBio antibody.
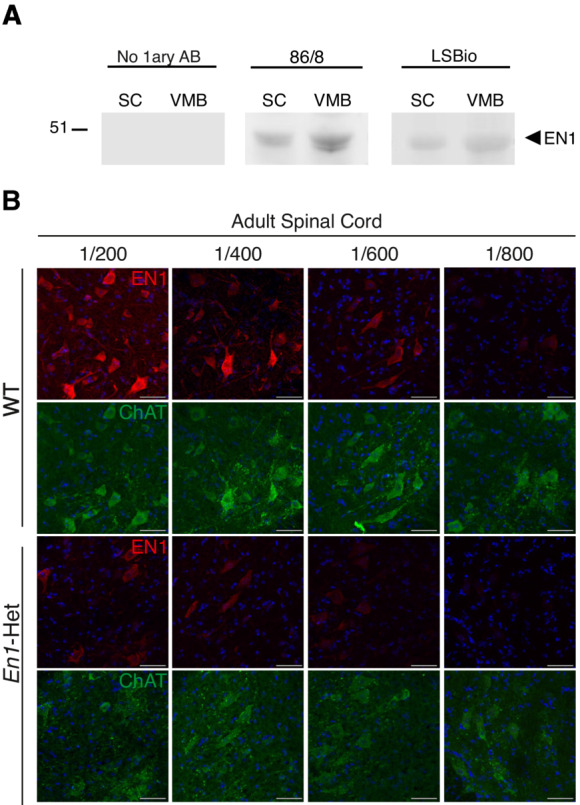
- Western blots of spinal cord (SC) and ventral midbrain (VMB) extracts demonstrating that the 86/8 and LSBio antibodies recognize in both structures the same protein migrating with recombinant EN1 velocity. No staining is observed in the absence of primary antibody (left panel). This experiment was performed twice.
- Double staining of 3‐month‐old WT and En1‐Het ventral MNs with the anti‐ChAT antibody and the anti‐EN1 LSBio antibody at various dilutions. EN1 staining decreases with increasing dilutions of the antibody. The loss of staining is more rapid in En1‐Het than in WT mice. Scale bar = 50 μm.
Data information: This experiment was performed once.
The presence of EN1 protein but not mRNA in MNs suggests that EN1 produced by the interneurons is efficiently secreted and internalized by adult spinal MNs. As will be described below, EN1 intercellular transfer is further demonstrated by its extracellular neutralization through the expression of a secreted single‐chain antibody (scFvEN1) directed against EN1 (Wizenmann et al, 2009), resulting in an important reduction of MN staining by the LSBio antibody (see below).
Engrailed1‐heterozygote mice show alpha‐motoneuron retrograde degeneration
Because En1‐expressing mDA neurons degenerate in the En1‐Het midbrain (Sonnier et al, 2007), we examined whether ventral spinal cord neuronal populations, in particular interneurons that express En1, also degenerate in the En1‐Het mouse. We used RNAscope labeling to compare the number of En1‐expressing neurons in the spinal cord ventral horns of En1‐Het mice and their WT littermates at 4.5 months of age. Figure 2A shows no loss of V1 interneurons, including RCs (Calbindin1‐expressing cells). Given the absence of a change in En1 expression between 4.5 and 16 months quantified by RT–qPCR (Fig 1B), these results demonstrate that V1 interneuron survival is not affected by reduced En1 expression in the En1‐Het mouse mutant. We also quantified the total number of ChAT+ motoneurons in 4.5‐month‐old mice and did not observe a reduction in the En1‐Het mouse (Fig 2A).
Figure 2. Progressive loss of αMNs and strength decrease in En1‐Het mice.
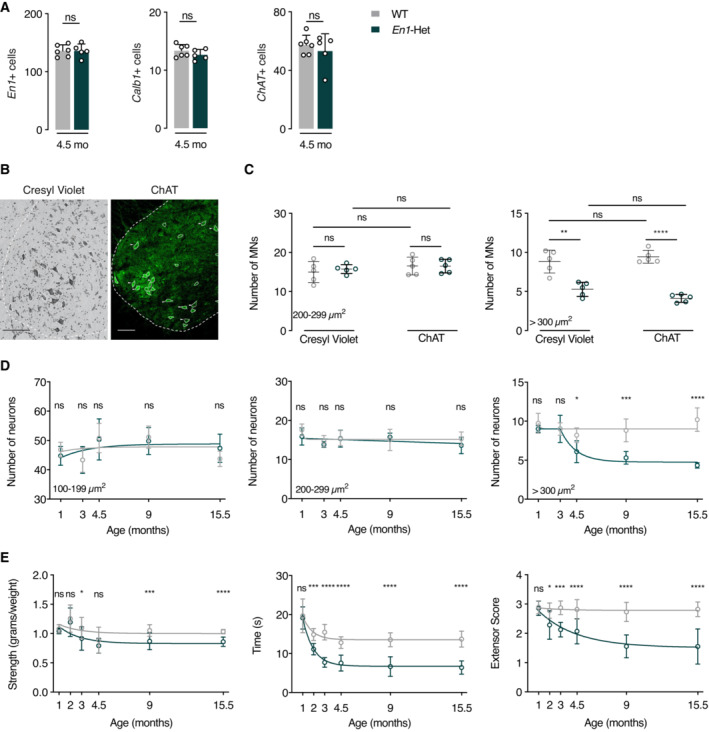
- Analysis of the number of En1+, Calb1+, and ChAT+ neurons (triple RNAscope). At 4.5 months, WT and En1‐Het mice show no difference in the number of cells expressing En1, Calb1, or ChAT. Unpaired two‐sided t‐test. n = 5–6.
- Cresyl violet and ChAT staining of a ventral WT spinal cord at the lumbar level. Scale bar: 100 μm.
- Cresyl violet and ChAT staining at the lumbar level show no medium‐size (200–299 μm2) cell loss (γMNs) and a decrease of about 50% in the number of large‐size (> 300 μm2) cells (αMNs). Unpaired two‐sided t‐test. **P < 0.005; ****P < 0.0001. n = 5.
- Lumbar level Cresyl violet staining shows that, in contrast with small‐ and medium‐size neurons (interneurons and γMNs, left and center panels), large‐size neurons (αMNs, right panel) undergo progressive death first measured at 4.5 months. The values represent the average number of cells per ventral horn. For the small neurons (100–199 μm2), there was no main effect, two‐way ANOVA for repeated measures for treatment group: F(1, 43) = 0.0017, P = 0.968, ns. For the medium‐sized neurons (200–299 μm2) two‐way ANOVA for repeated measures for treatment group: showed no main effect F(1, 43) = 2.085, ns. For the large neurons (> 300 μm2), two‐way ANOVA for repeated measures showed a significant main effect F(1, 43) = 59.99, P < 0.0001. Post hoc comparisons were performed by unpaired two‐sided t‐test with equal SD comparing WT with En1‐Het at each time point (*P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001). n = 5–6.
- Compared with WT mice, En1‐Het mice experience gradual strength loss. This loss is observed with the forepaw grip strength (left panel), the inverted grid test (center panel) and the hindlimb extensor reflex test (right panel). Strength loss is first observed between 2 and 3 months, thus before measurable αMN cell body loss. Two‐way ANOVA showed significant main effects for grip strength (F(1, 136) = 19.18, P < 0.0001), inverted grid test (F(1, 103) = 143.1, P < 0.0001), and extensor score (F(1, 103) = 10.1, P < 0.0001). Comparisons were made by unpaired two‐sided t‐test with equal SD comparing WT with En1‐Het at each time point (*P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001). n = 4–20.
Data information: The analysis of neuron number in (A), the direct comparison of Cresyl violet and ChAT in (C), and the longitudinal studies in (D and E) were performed once. Values are mean ± SD.
Source data are available online for this figure.
The ChAT‐expressing population is heterogeneous with a mixture of γMNs and αMNs and the global analysis of ChAT+ cells in Fig 2A may have missed degeneration, total or partial, of one of the two populations. Gamma and αMNs differ by size with the surface of γMNs ranging from 200 to 300 μm2 and that of αMNs equal or greater than 300 μm2 (Powis & Gillingwater, 2016). We counted MNs on Cresyl violet‐stained and ChAT‐labeled lumbar enlargement spinal cord sections (Fig 2B), taking soma size into account. We first performed this analysis at 9 months, reasoning that any degeneration would be more apparent at later ages. Cresyl violet and ChAT staining yield identical results and demonstrate a specific loss of large (> 300 μm2) neurons (Fig 2C). The concordance between data obtained from Cresyl violet and ChAT immunostaining precludes that the loss of ChAT+ cells in En1‐Het mice is a consequence of ChAT expression downregulation. We used Cresyl violet to quantify the number of small (100–199 μm2), medium (200–299 μm2), and large (> 300 μm2) neuronal populations as a function of age. Figure 2D demonstrates that, in the En1‐Het mutant, the number of small‐size (100–199 μm2) and intermediate (200–299 μm2) neurons does not decrease with age (Fig 2D, left and middle panels). In contrast, the number of αMNs strongly decreases between 3 and 4.5 months of age, with about 50% loss at 9 months and no further decrease between 9 and 15.5 months (Fig 2D, right panel). The plateau in αMN death is best explained by the fact that, with time and due to αMN death, each remaining MN receives a higher amount of EN1 (Fig EV2A–C) which may slow down neurodegeneration. We then determined the consequences of αMN degeneration on muscle strength by measuring forepaw grip strength, the time holding onto an inverted grid, and the hindlimb extensor reflex. Figure 2E shows that strength loss appears between 2 and 3 months and reaches a maximum between 4.5 and 9 months of age. The loss of strength between 2 and 3 months therefore precedes αMN loss first observable at 4.5 months (Fig 2D, right panel).
Figure EV2. Evolution of EN1 content per MN in WT and En1‐Het mice.
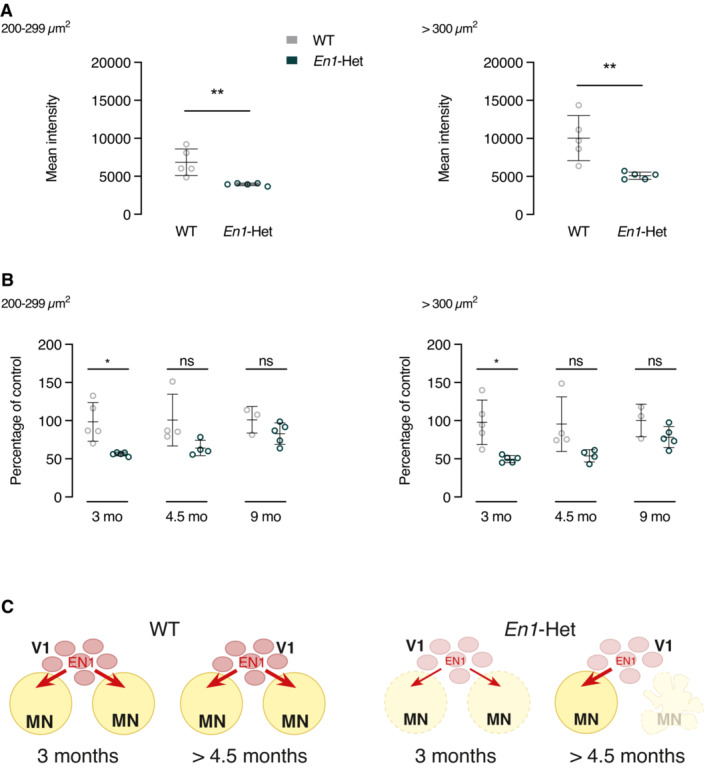
- Analysis of EN1 amount in MNs at 3 months in WT and En1‐Het mice. EN1 content is reduced by about half in γMNs (left panel) and αMNs (right panel). EN1 was revealed by the LSBio antibody allowing for the visualization of endogenous EN1. Unpaired two‐sided t‐test with equal SD (**P < 0.005; n = 5). Values are mean ± SD.
- Quantification of EN1 amount in γMNs and αMNs with the LSBio antibody at 3, 4.5 and 9 months in WT and En1‐Het mice. Values at 3 months correspond to the ones shown in panel A. At 4.5 and 9 months, the amount of EN1 in MNs is similar in En1‐Het and WT mice suggesting that, with time, each remaining MN receives a higher amount of EN1. Two‐way ANOVA showed a significant main effect for the γMNs (F(1, 20) = 16.93, P = 0.0005) and αMNs (F(1, 20) = 19.03, P = 0.0003). Unpaired two‐sided t‐test with equal SD (*P < 0.05; n = 4–5). Data information: This experiment was performed once. Values are mean ± SD.
- Hypothetical representation of EN1 availability to αMNs in WT and Het mice. In the En1‐Het mouse, each V1 interneurons only provides half as much EN1 to the full population of αMNs at 3 months of age. At 4.5 months of age and later, half of the αMNs have been lost allowing each remaining αMN to receive its full complement of EN1 from the V1 interneurons.
To further characterize αMN degeneration, we analyzed the organization of neuromuscular junctions (NMJs) by following the number of acetylcholine receptor (AChR) clusters, the endplate area, the percentage of endplates with perforations, and that of fully occupied endplates (Sleigh et al, 2014). The number of AChR clusters identified with fluorescent α‐bungarotoxin (α‐BTX) did not change with time (Fig 3A) and the endplate area remained stable for 9 months, with a modest 20–25% decrease at 15.5 months. A similar late‐occurring (15.5 months) and modest effect was also observed for the percentage of endplates with perforations, a maturation index that is decreased in the mutant mice only at 15.5 months (Comley et al, 2016). In contrast, a stronger and earlier difference was observed in the percentage of fully occupied endplates identified by the binding of fluorescent α‐BTX and immunostaining of the high molecular weight neurofilament (2H3) and the synaptic vesicle glycoprotein SV2A (Fig 3B, left panel). Fully occupied endplates were defined by an overlap between SV2A/2H3 and α‐BTX staining (pixel analysis) occupying more than 80% of the endplate (see Materials and Methods). Using this criterion, significant retrograde degeneration could already be measured in 3‐month‐old animals (Fig 3B, right panel). Quantitative RT–PCR (Fig EV3A) and EN1 immunohistochemistry (Fig EV3B) failed to reveal EN1 protein or mRNA at the level of the endplate, precluding that degeneration is a consequence of EN1 partial loss of function at the endplate level.
Figure 3. Neuromuscular junction (NMJ) morphology differences between WT and En1‐Het mice.
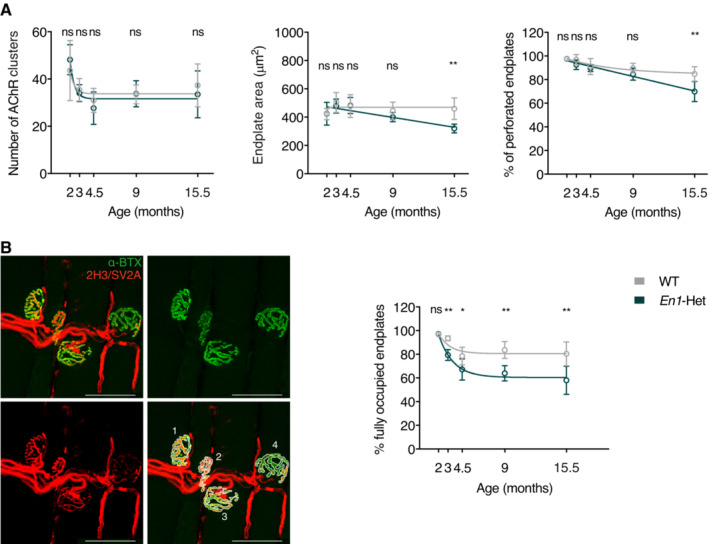
- NMJs of En1‐Het and WT mice show similar numbers of AChR clusters (left panel) and late‐occurring (15.5 months) decrease in endplate area (center panel) and percentage of perforated endplates (right panel). The number of AChR clusters seems to decrease between 2 and 3 months, but two‐way ANOVA showed no significant genotype effect (F(1, 47) = 0.1291, ns). There was a significant main effect for endplate area (F(1, 47) = 5.778, P = 0.0202) and for the percentage of perforated endplates (F(1, 47) = 13.82, P = 0.0005). Post hoc analysis revealed significant genotype differences at 15.5 month of age. Unpaired two‐sided t‐test with equal SD comparing WT with En1‐Het at each time point (**P < 0.005). n = 4–8.
- Left panel illustrates the use of Alexa Fluor 488‐conjugated α‐bungarotoxin (α‐BTX, in green) and of neurofilament and synaptic vesicle glycoprotein antibodies (2H3 and SV2A, in red) to evaluate the percentage of fully occupied endplates (> 80% occupancy). The right panel shows that the % of fully occupied endplates decreases progressively in the En1‐Het mouse, starting between 3 and 4.5 months of age. Scale bar: 50 μm. Two‐way ANOVA showed a significant main effect (F(1, 47) = 45.45, P < 0.0001). Unpaired two‐sided t‐test with equal SD comparing WT with En1‐Het at each time point (*P < 0.05; **P < 0.005). n = 4–8. See details of analysis in the Materials and Methods section.
Data information: The longitudinal study in (A and B) was performed once. Values are mean ± SD.
Source data are available online for this figure.
Figure EV3. Absence of En1 transcription or EN1 protein at endplate level.
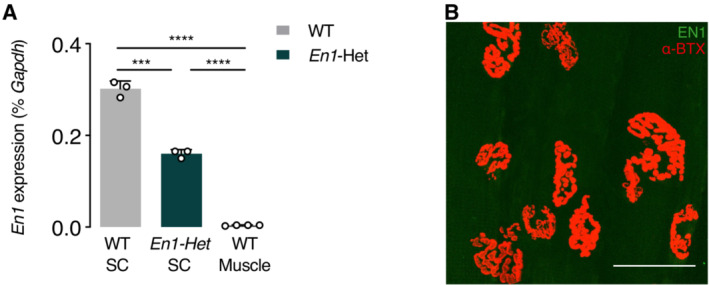
- RT–qPCR of RNA from the lumbar enlargement at 4.5 months of WT and En1‐Het mice and from WT muscle. En1 expression is absent from the muscle of WT mice. Unpaired two‐sided t‐test with equal SD (***P < 0.005; ****P < 0.0005; n = 4–5). Values are mean ± SD.
- Immunohistochemistry for EN1 protein (LSBio antibody, in green) shows its absence at the level of the NMJ (α‐BTX, in red). Scale bar = 50 μm.
Data information: This experiment was done once. Values are mean ± SD.
All in all, muscle weakness and NMJ changes preceding cell body loss demonstrate selective retrograde degeneration of αMNs in the En1‐Het mouse.
Extracellular neutralization of ENGRAILED‐1 partially reproduces the Engrailed1‐heterozygote phenotype
Since αMNs do not express En1, their degeneration in the En1‐het mouse must be a consequence of the En1 heterozygote status of the V1 interneurons with several mechanisms contributing, alone or in combination, to the phenotype. In light of the presence of EN1 in MNs (Fig 1), of its known direct non‐cell‐autonomous activity in several developmental phenomena (Brunet et al, 2005; Wizenmann et al, 2009; Layalle et al, 2011; Rampon et al, 2015; Amblard et al, 2020b), and of EN1/2 protective activity on mDA neurons (Sonnier et al, 2007; Alvarez‐Fischer et al, 2011; Rekaik et al, 2015; Blaudin de Thé et al, 2018; Thomasson et al, 2019), a parsimonious hypothesis is that EN1 exerts a direct non‐cell‐autonomous trophic activity on αMNs.
To investigate this possibility, we used a strategy successfully developed in previous studies consisting in the expression of a secreted neutralizing anti‐EN1 single‐chain antibody (scFvEN1) (Wizenmann et al, 2009) in the spinal cord of WT mice. The antibody, or its inactive control harboring a single cysteine to serine mutation that prevents disulfide bond formation and EN1 recognition, was cloned into an AAV8 under the transcriptional control of the glial fibrillary acidic protein (GFAP) promoter (Fig 4A). This promoter directs scFvEN1 expression and secretion by astrocytes, thus allowing for the neutralization of EN1 in the extracellular space. Viruses were injected intrathecally (IT) at lumbar level 5 (L5) in 1‐month‐old WT mice, allowing for robust scFvEN1 expression 1 month later (Fig 4B). The amount of EN1 in MNs was quantified 6 months after virus injection, demonstrating a strong reduction in mice expressing scFvEN1 but not the inactive mutated form (Fig 4C).
Figure 4. Effects of extracellular EN1 neutralization on strength, MN survival, and NMJ morphology.
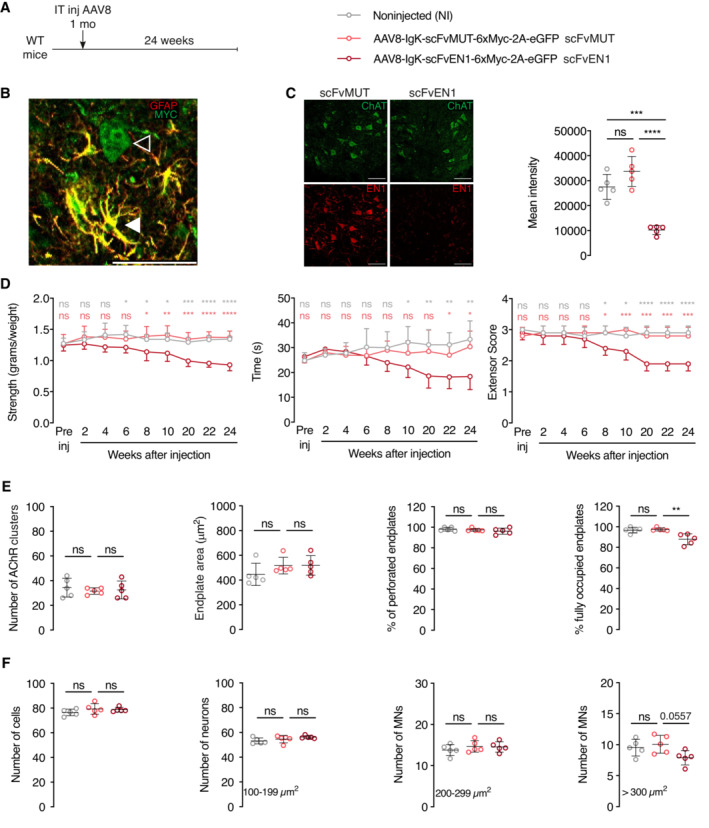
- Experimental paradigm and structure of AAV8‐encoded constructs containing glial fibrillary acidic protein (GFAP) promoter for expression in astrocytes, Immunoglobulin K (IgK) signal peptide for secretion, anti‐ENGRAILED single‐chain antibody (scFvEN1), 6 myc tags (6xMyc), skipping P2A peptide, and enhanced Green Fluorescent Protein (eGFP). An inactive control antibody (scFvMUT) contains a cysteine to serine mutation that prevents disulfide bond formation between IgG chains, thus epitope recognition. The AAV8 was injected in 1‐month‐old WT mice, and the strength phenotypes were followed for 6 months before anatomical analysis.
- Analysis of 1‐month postinjection showing that scFv antibodies are expressed in astrocytes (white arrowhead) double‐stained for GFAP and Myc and exported (empty arrowhead). Scale bar: 50 μm.
- Left panel illustrates that expressing the scFvEN1, but not scFvMUT, abolishes EN1 staining by LSBio anti‐EN1 antibody in ventral horn ChAT+ cells and right panel quantifies this inhibition 1‐way ANOVA followed by Tukey corrected post hoc comparisons (n = 5 mice per group, ***P < 0.0005, ****P < 0.0001). Scale bar: 100 μm.
- The three graphs illustrate how the WT antibody but not its mutated version leads to progressive strength decrease. Two‐way ANOVA showed significant main effects for grip strength (F(2, 12) = 15.88, P < 0.0005), inverted grid (F(2, 107) = 19.86, P < 0.0001), and extensor score (F(2, 12) = 30.22, P < 0.0001) followed by Tukey corrected post hoc comparisons (*P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001). n = 5 per treatment.
- Six months following infection (7‐month‐old mice), extracellular EN1 neutralization does not modify the number of AChR clusters, nor the endplate surface area, nor the percentage of perforated endplates. In contrast, the percentage of fully occupied endplates is diminished (right end panel). 1‐way ANOVA followed by Tukey corrected post hoc comparisons (**P < 0.005). n = 5 per treatment.
- Six months following infection, extracellular EN1 neutralization does not globally modify the total neuron number of cells at the lumbar level (left panel). A separate analysis of small (100–199 μm2), medium (200–299 μm2), and large (>300 μm2) neurons demonstrate a specific (P < 0.0557) loss of the latter category (αMNs). 1‐way ANOVA followed by Tukey corrected post hoc comparisons. N = 5 per treatment.
Data information: The extracellular neutralization study was performed twice. Values are mean ± SD.
Source data are available online for this figure.
In the three strength tests (anterior limb grasping, holding onto an inverted grid and hindlimb extensor reflex), mice expressing extracellular scFvEN1 experience gradual strength loss that begins between 6 and 8 weeks after virus injection (Fig 4D). In contrast, the performance of mice expressing the mutated inactive antibody is indistinguishable from that of WT animals. Endplate morphology and αMN survival were evaluated 6 months after injection of the antibody‐encoding viruses (at 7 months of age). The results demonstrate no effect of EN1 extracellular neutralization on the number of AChR clusters, endplate area, and percentage of endplates with perforations, but a modest effect on the percentage of fully occupied endplates (Fig 4E). The analysis of the total number of cells and, separately, of the number of small‐, medium‐, and large‐size neurons shows a small but specific loss of αMNs (Fig 4F).
Qualitatively, the latter results are similar to those obtained with the En1‐Het mice with a small decrease in the number of fully occupied endplates and the specific loss of large‐size αMNs. However, it is of note that the magnitude of the changes at 7 months was less than in the En1‐Het mouse, with a 15% loss in fully occupied endplates and a 25% reduction in the number of αMNs, compared to 30 and 50% in 7‐month‐old En1‐Het, respectively (Figs 2 and 3). This comparison, illustrated in Fig EV4, shows that secreted EN1 participates in the En1‐Het phenotype but may not fully explain its magnitude.
Figure EV4. Comparison of En1‐Het and scFvEN1‐expressing mice phenotypes.
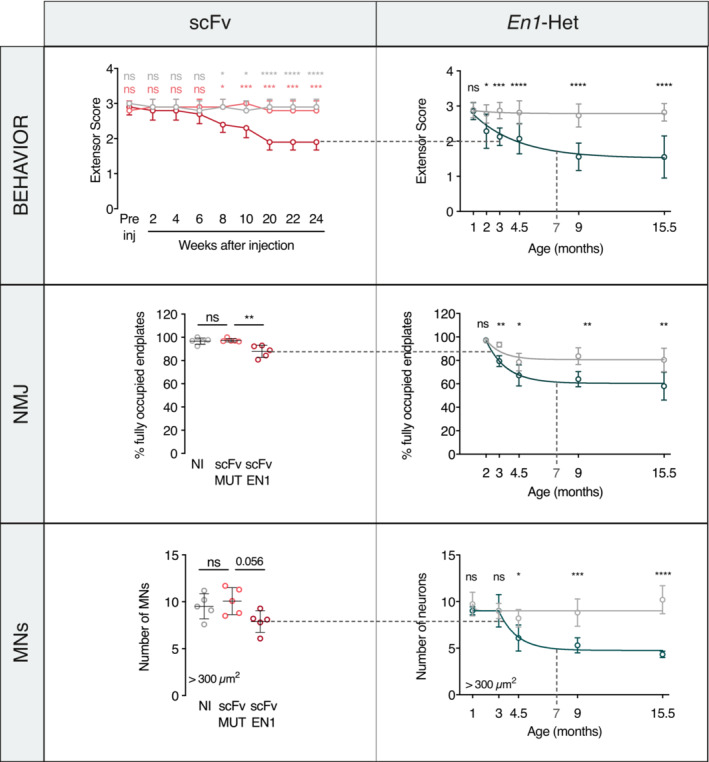
Comparison between En1‐het mouse and scFvEN1 models. Data and graphs are from main figures (primarily Figs 2 and 4). WT mice injected with scFvEN1 show similar results to those obtained in the En1‐Het mouse with a milder strength loss, a smaller decrease in the number of fully occupied endplates, and the specific loss of large‐size αMNs. At 7 months, scFvEN1 injected mice have a phenotype similar to 3‐month‐old En1‐Het mice.
Exogenous ENGRAILED‐1 rescues the Engrailed1‐heterozygote phenotype
We have previously demonstrated that EN1 administration in the ventral midbrain promotes mDA neuron survival in mouse and macaque PD models (Sonnier et al, 2007; Alvarez‐Fischer et al, 2011; Rekaik et al, 2015; Blaudin de Thé et al, 2018; Thomasson et al, 2019). Since extracellular EN1 exerts a trophic activity on αMNs, we evaluated whether exogenous administration of EN1 could prevent αMN death in En1‐Het mice. Human recombinant EN1 (hEN1) was intrathecally injected at the L5 level at a concentration of 1 μg in 5 μl in 3‐month‐old En1‐Het mice (Fig 5A), a time when all αMNs are still present but muscle weakness is already noticeable, a consequence of retrograde degeneration initiated at the endplate (Fig 3). One and a half months later, the single EN1 injection had restored strength as measured by the three tests of anterior limb grasping, holding onto an inverted grid, and hindlimb extensor reflex (Fig 5A). Accordingly, the percentage of fully occupied endplates was found to be back to WT levels (Fig 5B, left panel), in parallel with the prevention of αMN death (Fig 5B, right panel). This last series of experiments establishes that extracellular EN1 exerts a curative trophic activity on αMNs of already symptomatic mice allowing for endplate reinnervation and the arrest of αMN degeneration.
Figure 5. Intrathecal hEN1 injection in En1‐Het mice restores strength and prevents αMN death.
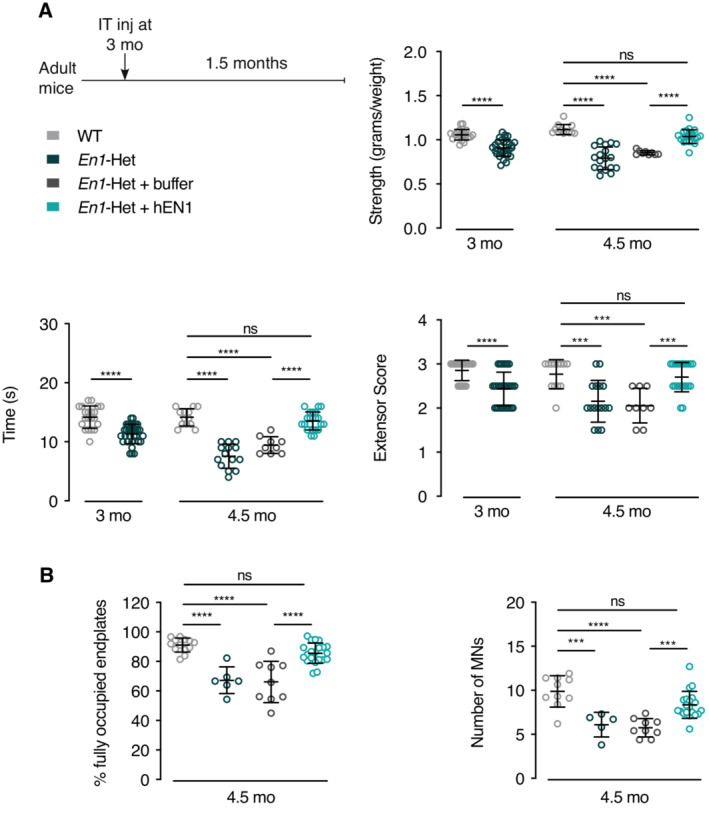
- Mice were tested for strength at 3 months of age, before the onset of αMN loss, but when strength has already decreased in En1‐het mice as measured in the forepaw grip strength, inverted grid, and extensor reflex tests (left side of each graph). The next day, the En1‐het mice were separated into two groups. One group received buffer and the other group recombinant hEN1 (1 μg in 5 μl), injected at the L5 level. One and a half months later (4.5 months of age) En1‐het mice injected with hEN1 have recovered normal strength, in contrast with noninjected mice or mice injected with buffer. Unpaired two‐sided t‐test used at 3 months of age. ****P < 0.0001. For 4.5‐month comparisons, 1‐way ANOVA followed by Tukey corrected post hoc comparisons. ***P < 0.0005; ****P < 0.0001. n = 9–31.
- At 4.5 months, following hEN1 injection at 3 months, the percentage of fully occupied endplates and the number of αMNs are not significantly different from control values. 1‐way ANOVA followed by Tukey corrected post hoc comparisons. ***P < 0.0005; ****P < 0.0001. n = 5–21.
Data information: The weakness and reversal with hEN1 and the neuroprotection were replicated in five independent experiments. Values are mean ± SD.
Source data are available online for this figure.
The duration of the effect of EN1 injection on strength was evaluated over time. Figure 6A summarizes an experimental design consisting of a single EN1 injection at 3 months. Strength analysis (anterior grip, holding time on the inverted grid, and extensor reflex score) demonstrates that the effect of a single injection is maintained until approximately Week 12 postinjection and decreases thereafter. In spite of this decrease, the strength of EN1‐injected En1‐Het mice is still superior to that of age‐matched noninjected controls 24‐week postinjection. Analysis of the % of fully occupied endplates and αMN number at 24 weeks confirms the long‐lasting effect of a single injection. This led us to test whether a second injection 12 weeks after the first one could prolong the protective effect. Figure 6B summarizes this experimental protocol and illustrates that a second injection prolongs strength for another 8–12 weeks, depending on the test. Moreover, the percentage of fully occupied endplates and the number of αMNs at 24 weeks were identical to those observed in WT siblings.
Figure 6. EN1 injection at 3 months has long‐lasting effects on strength, endplate occupancy, and αMN survival and the latter recoveries are prolonged by a second injection.
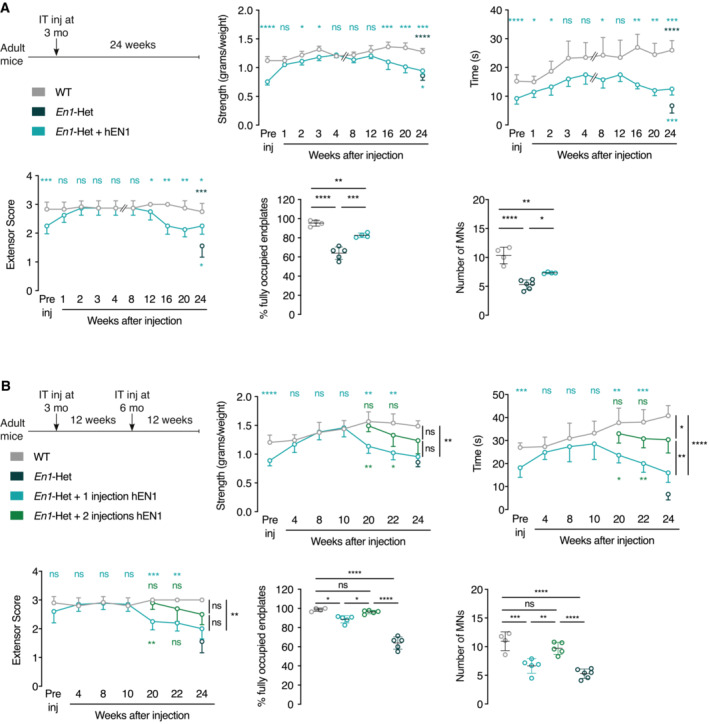
- Top left panel shows the single injection protocol whereby hEN1 is injected at 3 months (1 μg in 5 μl) intrathecally at the L5 level and mouse behavior followed for 24 weeks. The 3 time‐course graphs demonstrate that a single injection restores strength measured by the tests of grip strength, time on the inverted grid and extensor reflex and that this effect lasts for 12 weeks. After 12 weeks, strength decreases progressively but, even after 24 weeks, remains superior to that of untreated En1‐Het mice. At 24 weeks, the % of fully occupied endplates is inferior to that of WT mice, but superior to that of noninjected En1‐Het mice. The same holds true for the number of αMNs. Two‐way ANOVA revealed significant main effects for grip strength (F(1, 76) = 143.6, P < 0.0001), inverted grid (F(1, 76) = 128.6, P < 0.0001), and extensor score (F(1, 76) = 34.91, P < 0.0001). At the different times, the groups were compared by unpaired t‐test with equal variances comparing WT with En1‐Het injected at each time point. (*P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001). For the endplate analysis and αMNs, groups were compared by 1‐way ANOVA followed by Tukey corrected post hoc comparisons (*P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001). n = 4–10.
- Based on the results shown in A, a new experiment was performed with a second injection 12 weeks after the first injection at 3 months of age. Again, the injection at 3 months of age restored strength and in mice receiving the second injection strength was maintained an additional 10 weeks or more compared with mice receiving a single injection. The three strength graphs demonstrate a positive effect of the second injection with values intermediate between those measured in WT mice and En1‐Het mice with a single injection. The percentage of fully occupied endplates and the number of αMNs are back to wild‐type values in En1‐Het mice injected twice. Two‐way ANOVA revealed significant main effects for grip strength (F(2, 81) = 25.47, P < 0.0001), inverted grid (F(2, 91) = 51.96, P < 0.0001), and extensor score (F(2, 104) = 30.42, P < 0.0001). At all times, groups were compared by unpaired t‐test with equal variances through 12 weeks. After, the groups were compared by Tukey corrected post hoc comparisons (*P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001). For the endplate analysis and αMNs, groups were compared by 1‐way ANOVA followed by Tukey corrected post hoc comparisons (*P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001). n = 4–10.
Data information: The single injection and two injection time‐course studies were performed once each. Values are mean ± SD.
Source data are available online for this figure.
Intrathecally injected EN1 preferentially accumulates in MNs
We then followed EN1 localization after intrathecal injection and observed that 24 h after injection EN1 is present in the perivascular space and in ventral horn cells, primarily in MNs characterized by ChAT expression (Fig 7A, upper row left panel). Figure 7A also illustrates that EN1 staining in MNs is transient, peaking between 6 and 24 h, still visible at 48 h and barely so at 72 h. The latter images were taken with the 86/8 antibody that poorly stains endogenous EN1 allowing us to see primarily the exogenous protein, but similar images were obtained with the LSBio antibody. Motoneurons identified by ChAT staining were separated by size to distinguish between αMNs and γMNs and the kinetics of internalization were quantified in the two subpopulations (Fig 7A, bottom row), using the 86/8 (left panels) and LSBio (right panels) antibodies. Indeed, the difference following internalization is much higher with the 86/8 antibody since the basal staining due to endogenous EN1 is very low, but the time courses obtained with the two antibodies are very similar. Furthermore, the data obtained with the LSBio antibody, which recognizes endogenous and exogenous EN1, suggest that exogenous EN1 at its peak (24 h) doubles the concentration of EN1 per MN (assuming that the antibody recognizes equally endogenous and exogenous EN1). All in all, the latter series of experiments demonstrates similar patterns for αMNs and γMNs, with a very rapid accumulation that peaks at 24 h and decays rapidly, reaching nearly basal levels at 72 h, indicating a short half‐life for the internalized protein.
Figure 7. Following intrathecal injection, hEN1 is addressed specifically to all MNs.
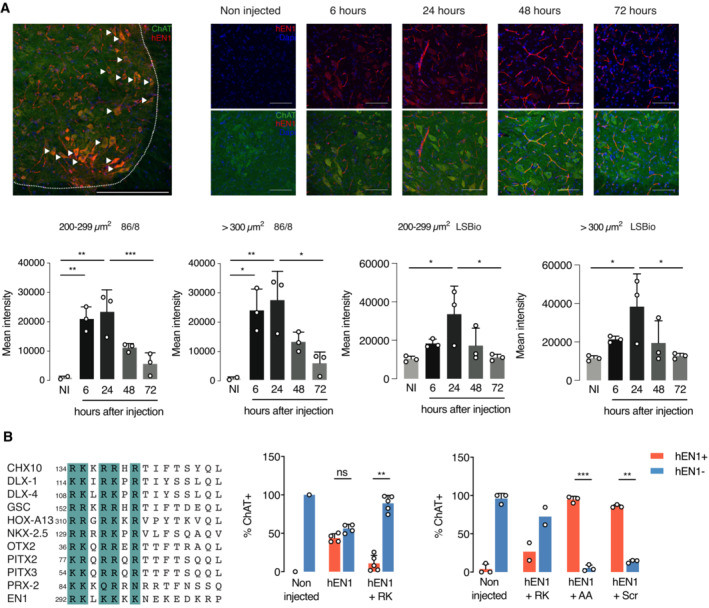
- Top left panel shows that 24 h after intrathecal injection (1 μg in 5 μl) at the L5 level of 2‐month‐old mice, hEN1 (red) can be primarily visualized in ventral horn ChAT+ cells (green). Arrowheads show hEN1 internalized by MNs. Scale bar: 100 μm. Top right panels show the progressive accumulation and clearance of hEN1 in ventral horn MNs, with a peak between 6 and 24 h. EN1 was revealed by the 86/8 antibody allowing for the visualization of exogenous EN1 only (scale bar: 100 μm). Bottom panels show the quantification of EN1 in γMNs and αMNs with the 86/8 (two left panels) and the LSBio (two right panels) antibodies. Since the LSBio sees both endogenous and exogenous EN1, the increase is only threefold, but qualitatively, the results are very similar, demonstrating rapid internalization and clearance of the exogenous protein and allowing one to calculate a half‐life of 24 h. One‐way ANOVA followed by Tukey corrected post hoc comparisons (*P < 0.05; **P < 0.005; ***P < 0.0005). When not significant, P‐values are shown. n = 3. Values are mean ± SD. Data information: The internalization experiments were performed once with each antibody.
- Left panel gives examples of putative glycosaminoglycan (GAG)‐binding domain in 11 homeoprotein transcription factors. Based on the alignment and on published work on OTX2 (Beurdeley et al, 2012) and EN2 (preprint: Cardon et al, 2023), a putative EN1 GAG‐binding domain (RK‐EN1) was designed. Middle panel quantifies the inhibitory effect of RK‐EN1 on hEN1 capture (86/8 antibody) by ChAT+ cells demonstrating that RK‐EN1 in a 1 to 20 ratio reduces the % of EN1‐positive MNs (ChAT+) from 50 to less than 10%. The right panel demonstrates that this inhibitory activity is not shared by the mutant AA peptide or by a scrambled (Scr) peptide. Unpaired two‐sided t‐test with equal SD (**P < 0.005; ***P < 0.0005). n = 2–5.
Data information: The GAG competition experiment was performed twice as described in the text. Values are mean ± SD except for conditions with two observations for which both data points are shown.
Source data are available online for this figure.
The preferential capture of EN1 by MNs might reflect the presence of EN1 binding sites at the MN surface. We previously identified a glycosaminoglycan (GAG) binding domain within the homeoprotein OTX2 that allows for its preferential capture by cerebral cortex parvalbumin (PV) neurons (Beurdeley et al, 2012). This RK‐OTX2 domain peptide (RKQRRERTTFTRAQL) competes for OTX2 binding to PV cells and antagonizes its specific internalization upon infusion or injection into the cerebral cortex (Beurdeley et al, 2012). A similar GAG‐binding domain was recently described in EN2 (Cardon et al, 2023) and sequence comparisons between OTX2, EN2, and several other HPs identified putative GAG‐binding sites in many of them (examples in Fig 7B, left panel). In the case of EN1, this domain (RKLKKKKNEKEDKRPRTAF), thereafter RK‐EN1, is present between residues 292 and 310 of hEN1.
To test its potential role in EN1 localization, RK‐EN1 was co‐injected with EN1 (in a 20:1 molar ratio) to competitively inhibit EN1 access to MNs. Twenty‐four hours after injection, EN1 gains access primarily to MNs, but the percentage of MNs that capture the protein is variable. In Fig 7B (central panel), EN1 gained access to 50% of all ventral horn MNs, characterized by ChAT expression, and this percentage dropped to 10% in the presence of the competing RK‐peptide, with most ChAT+ MNs devoid of EN1. In a separate experiment (Fig 7B, right panel), we tested peptide specificity by co‐injecting EN1 with a peptide (AKLKAKKNEKEDKAPRTAF) in which one R and one K were replaced by alanine residues (AA peptide) or with a scrambled (Scr) RK‐peptide (RLKNKEKPRDREKTKAKFK) composed of the same amino acids, but in a different order. For control of RK‐EN1 activity, two mice were injected with EN1 and the RK‐EN1 peptide, reproducing the inhibition of internalization observed in the first experiment (central panel). In contrast, the two variant peptides were devoid of inhibitory activity. The absence of inhibition by the scrambled peptide demonstrates that inhibition cannot be explained only by a positive charge effect, suggesting that RK‐EN1 is an addressing sequence for MNs in the ventral mouse spinal cord. It is of note that, even though only αMNs degenerate in the En1‐Het mouse, EN1 is specifically captured by both γMNs and αMNs.
Bioinformatic analyses reveal p62/SQSTM1 as a regulated ENGRAILED‐1 non‐cell‐autonomous target in motoneurons
Retrograde degeneration of αMNs in the En1‐Het and their long‐lasting (2 months at least) rescue after a single EN1 injection is reminiscent of the situation described for mDA neurons, with the difference that EN1 activity is non‐cell‐autonomous for αMNs and cell‐autonomous for mDA neurons (Rekaik et al, 2015). This similar protective effect led us to investigate whether genetic pathways interacting with En1 might be common to αMNs and mDA neurons. To that end, we compared previously acquired RNA sequencing data of the SNpc in WT and En1‐Het mice at 6 weeks of age, thus before the initiation of mDA neuron death but after the initiation of retrograde degeneration (Sonnier et al, 2007; Nordström et al, 2015). A significance threshold of P = 0.05 was used, generating a list of differentially expressed genes (Rekaik et al, 2015) that was crossed with a library of MN‐expressed genes (Bandyopadhyay et al, 2013). Several physiological pathways were selected (see Materials and Methods), resulting in a list of 402 genes. As in PD, there is ample evidence that the first steps in ALS occur in the distal motor neuron axon that retracts from the muscle in a “dying back” phenomenon (Fischer et al, 2004). To focus on relevant genes in the context of αMN retrograde degeneration, we used the STRING database to extract within the 402 genes those independently interacting with SOD1, FUS, TARDBP‐43, and C9ORF72, 4 genes mutated in a large majority of familial ALS (fALS). This resulted in a short list of 20 genes modified in mDA neurons following En1 loss of function, expressed in MNs and in interaction with one or several of the four fALS genes (Fig 8A).
Figure 8. SQSTM1/p62 expression in MNs increases with age.
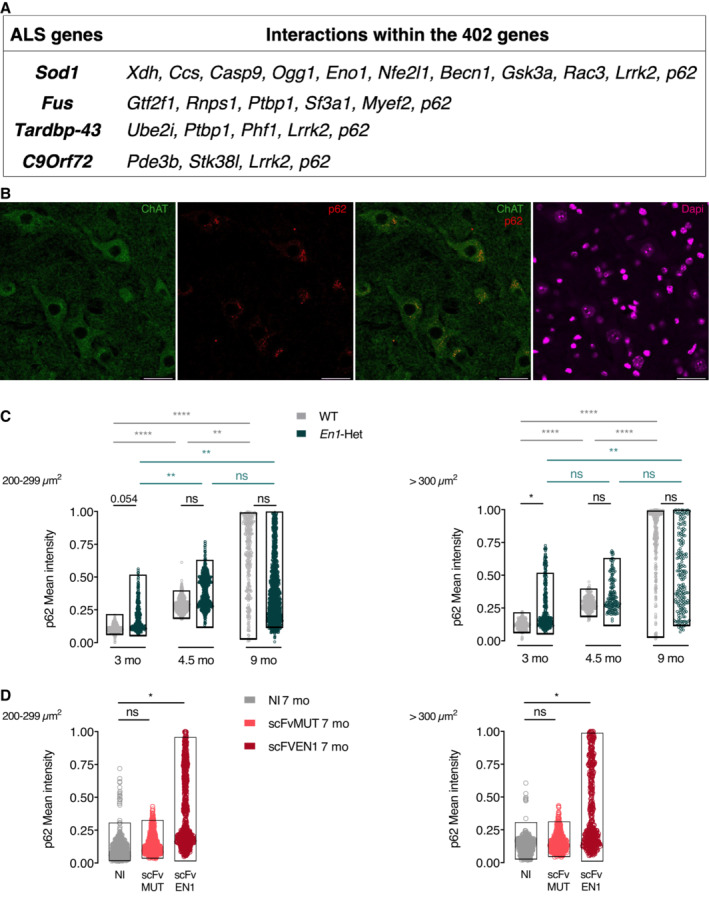
- The search for genes differentially expressed in WT and En1‐Het mDA neurons and expressed in MNs, thus putative non‐cell‐autonomous EN1 targets in MNs, allowed for the identification of 402 genes (after pathway selection). These genes were investigated for an interaction with genes mutated in the main 4 familial ALS forms. Among them, p62/SQSTM1 (p62) expression is upregulated in the SNpc (RNA‐seq) and in MNs of En1‐Het mice.
- Immunohistochemical staining shows the presence of high amounts of p62/SQSTM1 in ChAT+ cell bodies of 3‐month‐old mice. Scale bar: 50 μm.
- Intensity measurements demonstrate that the mean of p62/SQSTM1 expression increases with age in WT γMNs (left) and αMNs (right). However, comparing WT and En1‐Het shows a significant difference only at 3 months and not later. Unpaired two‐sided t‐test. (*P < 0.05; **P < 0.005; ***P < 0.0005, ****P < 0.0001). Data information: This experiment was done once. Values are mean ± SD. Between 151 and 1,215 neurons and 3–5 mice were analyzed for each condition.
- Mean intensity of p62/SQSTM1 expression is increased in γMNs (left) and αMNs (right) in mice expressing scFvEN1 6 months after virus injection (7‐month‐old mice) demonstrating that EN1 extracellular neutralization increases p62/SQSTM1 expression. Expression of the mutated antibody (scFvMUT) does not increase p62/SQSTM1 expression. Unpaired two‐sided t‐test. (*P < 0.05; **P < 0.005; ***P < 0.0005, ****P < 0.0001).
Data information: This experiment was performed once. Values are mean ± SD. Between 448 and 759 neurons and 5 mice were analyzed for each condition.
Source data are available online for this figure.
Among these 20 genes, p62 or p62/SQSTM1 interacts with the four fALS genes, is mutated in sporadic forms of the disease (Fecto et al, 2011; Shimizu et al, 2013; Chen et al, 2014; Yang et al, 2015), and was recently shown to have variants in 486 fALS patients (Yilmaz et al, 2019). p62/SQSTM1 is a multifunctional protein that regulates the degradation of ubiquitinated proteins by the proteasome and carries ubiquitinated cargoes to the LC3 receptor at the autophagosome surface, a key step in cargo engulfment and lysosomal hydrolysis (Doherty & Baehrecke, 2018). The importance of protein degradation and autophagy in protein homeostasis and age‐associated pathologies (Menzies et al, 2015; Leidal et al, 2018; Klionsky et al, 2021) led us to compare the immunostaining for p62/SQSTM1 in lumbar αMNs (> 300μm2) and γMNs (200–299 μm2) in different conditions. A first observation (Fig 8B) is that expression of p62/SQSTM1 in WT ventral spinal cord is very strong in MNs characterized by ChAT expression. A second one is that the intensity of p62/SQSTM1 expression increases with age in WT αMNs and γMNs and may thus be considered as a MN physiological age marker (Fig 8C).
Comparison of p62/SQSTM1 expression in WT and En1‐Het mice (Fig 8C) reveals a statistically significant or close to significant difference at 3 months in αMNs and γMNs (P = 0.0499 and 0.0536, respectively), before αMNs begin to degenerate, even though strength loss is already measurable (Fig 2C and D). Taking p62/SQSTM1 as an age marker, its increased expression at 3 months suggests accelerated aging of αMNs and γMNs in En1‐Het mice during this period, thus before αMN death and the ensuing reestablishment, at 4.5 and 9 months of age, of nearly normal EN1 concentration per MN (Fig EV2B). This led us to follow p62/SQSTM1 expression after extracellular EN1 neutralization in WT mice. Figure 8D shows that p62/SQSTM1 is significantly upregulated 6 months after injection of the AAV8‐scFvEN1 in one‐month‐old mice, but not of its mutated inactive variant. It is of note that, due to a ceiling effect in measurements at 9 months, we used a ß distribution adjusted between 0 and 1 for all calculations in Fig 8C and D (see Materials and Methods).
At 3 months of age, all MNs (αMNs and γMNs) are still present in the En1‐Het mouse but the enhanced p62/SQSTM1 expression suggests that mutant mice experience accelerated aging. We thus investigated whether hEN1 injection at 1 month could antagonize MN premature aging at 3 months. Figure 9A describes the injection protocols and demonstrates that hEN1 injection prevents strength loss normally observed at 3 months using the three strength measurements (anterior limb grip strength, holding time on the inverted grid, and extensor reflex). Figure 9B illustrates and quantifies p62/SQSTM1 expression, demonstrating that hEN1 intrathecal injection at 1 month antagonizes p62/SQSTM1 increase taking place at 3 months of age.
Figure 9. Human recombinant EN1 injection at 1 month prevents p62/SQSTM1 overexpression and strength loss in 3‐month‐old En1‐Het mice.
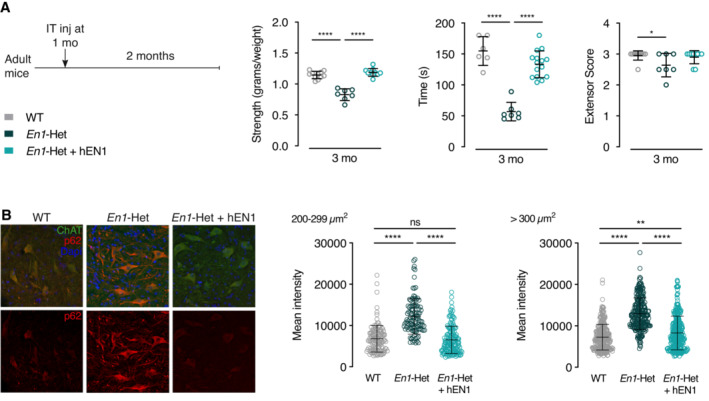
- Left panel: Injection and analysis protocol. Right panel: muscle strength analysis demonstrating that hEN1 injection at 1 month prevents muscular strength decrease observed in 3‐month‐old En1‐Het mice. One‐way ANOVA followed by two‐sided t‐test. (*P < 0.05, ****P < 0.0001). N = 6–10. Data information: This experiment was performed once. Values are mean ± SD.
- Left panel: Increased p62/SQSTM1 staining in 3‐month‐old ChAT+ MNs from control En1‐Het is abolished by hEN1 injection at 1 month. Right panel: quantification of p62/SQSTM1 staining in γMNs and αMNs of control and hEN1‐injected 3‐month‐old En1‐het mice. One‐way ANOVA followed by two‐sided t‐test. (**P < 0.01, ****P < 0.0001).
Data information: This experiment was performed once. Values are mean ± SD. Between 111 and 303 neurons and 4–5 mice were analyzed for each condition.
Source data are available online for this figure.
Together, these results suggest that p62/SQSTM1 expression is a MN aging marker, that the loss of an En1 allele or EN1 neutralization in adult WT animals accelerates MN aging, and that hEN1 injection at 1 month of age prevents accelerated aging and strength loss observed in 3‐month‐old En1‐Het mice.
Discussion
This study establishes a novel adult function of EN1 and the importance of its expression in adult spinal cord V1 interneurons for αMN survival and muscle strength. This non‐cell‐autonomous EN1 activity involves, at least in part, EN1 secretion as shown by the deleterious effect on the neuromuscular phenotype of extracellular EN1 neutralization. Moreover, hEN1 injected intrathecally accumulates in MNs. In the En1‐Het mouse, this extracellular gain of function, following a single intrathecal injection of hEN1, antagonizes premature aging, reverses muscle denervation, arrests αMN degeneration, and restores normal strength for several weeks. While EN1 injections were done using the human recombinant protein, human and mouse EN1 are 91.1% identical and show similar efficacy in mDA neuron survival experiments (Alvarez‐Fischer et al, 2011; Rekaik et al, 2015; Blaudin de Thé et al, 2018; Thomasson et al, 2019).
One day after injection, hEN1 is present around vascular structures probably in the perivascular space as well as within the parenchyma where it is observed within cells. Indeed, it has been shown previously that EN1 and EN2, similarly to many HPs (Lee et al, 2019), translocate across the plasma membrane and gain access to the cell cytoplasm and nucleus by a mechanism distinct from endocytosis (Brunet et al, 2007; Prochiantz & Di Nardo, 2015; Di Nardo et al, 2018, 2020; Amblard et al, 2020a). Injected hEN1 shows preferential accumulation in ChAT‐positive ventral MNs, although this does not preclude that some EN1 is present in the extracellular space and that a minority of other cell types may also capture the protein, albeit at lower levels. The deleterious effect of blocking extracellular EN1 with an anti‐EN1 scFv expressed in, and secreted by, astrocytes establishes that EN1 secreted by the V1 interneurons is internalized by αMNs and γMNs expressing EN1‐binding sites and participates in several aspects of MN physiology, including αMN survival.
The latter finding is in line with the non‐cell‐autonomous functions of EN1/2 reported in the fly wing disk and in the frog, fish, and chick optic tectum (Brunet et al, 2005; Wizenmann et al, 2009; Layalle et al, 2011; Rampon et al, 2015; Amblard et al, 2020b). It is also reminiscent of OTX2, another HP that specifically accumulates in PV interneurons of the cerebral cortex (Sugiyama et al, 2008). Prior to internalization, OTX2 binds to GAGs present at the surface of these interneurons thanks to a GAG‐binding domain (RKQRRERTTFTRAQL) that overlaps with the first helix of the homeodomain (Beurdeley et al, 2012; Miyata et al, 2012). Interestingly, similar putative GAG‐binding domains are present upstream of the homeodomain in many HPs (examples in Fig 7B), including EN2 (Cardon et al, 2023) and EN1 for which this putative GAG‐binding sequence is RKLKKKKNEKEDKRP. The role of this domain in the specific targeting of EN1 and the presence of EN1 binding sites at the MN surface are both illustrated by the ability of the peptide to abolish EN1‐specific uptake by ventral spinal cord MNs. Although we have not directly demonstrated that EN1 binds GAGs, the conservation of the GAG‐binding domain and the analogy with OTX2 (Beurdeley et al, 2012) and EN2 (Cardon et al, 2023) support a specific association of EN1 with MN‐expressed GAGs. If this is confirmed by future studies, it will be important to identify the molecular nature of this GAG as done for OTX2, EN2, and also VAX1 (Kim et al, 2014). Given the existence of a putative GAG‐binding domain in many HPs, this study and those on OTX2 and VAX1 raise the issue of the existence of a sugar code for the specific recognition of their target cells by transferring HPs.
In the En1‐Het mouse, deficits in strength begin before 3 months of age and might be a consequence of accelerated MN aging, as suggested by the enhanced p62/SQSTM1 expression at 3 months of age in mutant mice (discussed below). They coincide with a decrease in the percentage of fully occupied endplates first observed at 3 months when EN1 levels in MNs are about half of the WT levels (Fig EV2A) and precede αMN loss which is initiated between 3 and 4.5 months. This temporal delay likely reflects a retrograde degeneration of αMNs that starts at the terminals, whereby peripheral strength is affected before αMN cell body loss. Retrograde degeneration also occurs in En1‐expressing mDA neurons of En1‐Het mice, with their terminals showing signs of degeneration at 4 weeks, whereas they only start dying at 6 weeks postnatal (Nordström et al, 2015). Future studies will be necessary to elucidate the molecular mechanisms that operate at the presynaptic and postsynaptic sites and might be in part regulated by EN1 transfer into MNs.
Retrograde degeneration of αMNs is also a prominent feature of human motoneuron diseases (Fischer et al, 2004). In this context, it is of note that hEN1 injection at 3 months not only prevents lumbar αMN death for months but also restores normal endplate innervation and muscle strength, suggesting reinnervation. The neurodegenerative process is slowed or halted on a time scale of months and a second injection 3 months after the first one prolongs rescue for another 2–3 months. The time course of αMN degeneration in En1‐Het animals is rapid with approximately 70% of final αMNs death taking place between months 3 and 4.5. Then, the process slows down to reach a plateau with ~ 40% of the αMNs remaining at 15.5 months. It is thus possible that αMNs are heterogeneous in terms of their dependency on EN1. A more likely possibility is that when αMNs death gets close to 50%, the survivors receive sufficient secreted EN1 to halt accelerated aging and the ensuing degeneration. This hypothesis is supported by the data in Fig EV2B and schematized in Fig EV2C, and the fact that the difference in p62/SQSTM1 expression is only visible at 3 months when MNs receive half of the normal EN1 dose but not at 4.5 months or later when almost half of the αMNs have disappeared. A strict evaluation of αMN response to decreasing EN1 doses will be necessary to fully explore this hypothesis.
Although EN1 activity is cell‐autonomous in mDA neurons and non‐cell‐autonomous in αMNs, the similarities of their responses to En1 hypomorphism, such as progressive retrograde degeneration preceding cell body loss, led us to interrogate the repertoire of differentially expressed genes in WT and En1‐Het mDA neurons and to compare it with the MN transcriptome. This produced a short list of genes and one of them, p62/SQSTM1, interacts with SOD1, FUS, TARDBP‐43, and C9ORF72, 4 genes responsible for a majority of fALS. p62/SQSTM1 encodes p62/SQSTM1, a known regulator of degradation by the proteasome and autophagy through its role in bringing ubiquitinated proteins to the proteasome or to LC3 at the autophagosome surface (Klionsky et al, 2021). This unbiased identification of p62/SQSTM1 in the En1 “pathway” is particularly interesting because, in addition to interacting with the 4 main fALS mutations, it presents variants in 486 patients with familial ALS and is also mutated in sporadic ALS cases (Fecto et al, 2011; Shimizu et al, 2013; Chen et al, 2014; Yang et al, 2015). As already mentioned, p62/SQSTM1 expression in WT mice increases during aging and can be taken as an age marker. Compared with WT, we observe an upregulation of p62/SQSTM1 in the En1‐Het mouse at 3 months suggesting accelerated aging. This accelerated aging, antagonized by hEN1 injection at 1 month, is best explained, in the absence of αMN death at this age, by a dilution of EN1 content per MN. Similar upregulation, suggesting accelerated aging, is also measured in the scFvEN1 injected WT mice at 7 months when αMN death is limited to 20%, resulting in a significant decrease in EN1 content per MN (Fig 4C).
These series of observations can be interpreted in two non–mutually exclusive hypotheses. A first one is that p62/SQSTM1 plays a homeostatic role and that its increase represents a reaction against degeneration in aging neurons. A second, based on the fact that p62/SQSTM1 itself is degraded through autophagosomes, is that aging reduces autophagy and fosters p62/SQSTM1 accumulation (Bjørkøy et al, 2009; Jaakkola & Pursiheimo, 2009; Tai et al, 2016; Jakobi et al, 2020; Sun et al, 2023). P62/SQSTM1 is a multifunctional protein with distinct domains interacting with different co‐factors (Ma et al, 2019). Thus, if a protective activity is demonstrated in further studies, specific deletions will be necessary to identify the pathways involved, in particular within the N‐terminal domain, which presents a pro‐survival activity through the N‐kB pathway, and the C‐terminal domain, which regulates proteostasis through the activation of proteasome and autophagosome activities (Foster & Rea, 2020).
Although EN1 is internalized by all MNs, which show similar increases in p62/SQSTM1 expression in both the En1‐Het mouse or following EN1 extracellular neutralization, only αMNs and not γMNs degenerate in both models. This demonstrates that γMNs, in contrast with αMNs, are EN1‐independent for their survival. The survival of approximately half of the αMNs may reflect a differential sensitivity of αMN subpopulations and/or the fact that, as discussed above, the surviving neurons receive higher and sufficient amounts of EN1. The absence of dependency on EN1 for γMN survival also applies to En1‐expressing V1 interneurons that do not degenerate in the En1‐Het mouse. This differs with mDA neurons that express En1 and degenerate in the En1‐Het mouse. EN1 survival activity is therefore independent of its cell‐autonomous or non‐cell‐autonomous activity, but rather may reflect a higher sensitivity to stress of mDA neurons and αMNs, compared with V1 interneurons and γMNs.
The differential susceptibility of αMNs and γMNs to reduced En1 expression in the En1‐Het mouse or to EN1 extracellular availability following scFvEN1 viral expression may be explained by their different functions and the underlying anatomy (Kanning et al, 2010). Alpha‐MNs not only receive excitatory inputs from local spinal circuits but also receive an important contribution from descending upper motor neurons. They innervate extrafusal fibers and the innervation ratio (i.e., the number of muscle fibers per αMN axon) in humans can reach up to 2,000 (Feinstein et al, 1954; Masson et al, 2014). Gamma‐MNs innervate muscle spindles and receive their excitatory inputs from the reticular formation and their inhibitory ones from relay neurons mainly found in the dorsal horn that does not express EN1. They fire after αMNs, at a slower rate and, in humans, the innervation ratio is close to parity (i.e., 1–3 muscle fibers per axon), suggesting that their metabolic demand is less than that of αMNs. Finally, while γMNs can take up exogenous EN1 similarly to αMNs, they do not receive contacts from EN1‐expressing interneurons, and thus, the functions of EN1 might differ between these two MN types. A full understanding of these distinct functions requires further studies.
The deleterious phenotype is stronger in the En1‐Het mouse than following extracellular EN1 neutralization in WT mice. This may be due to the fact that EN1 neurotrophic activity exerted on αMNs is purely non‐cell‐autonomous and that scFvEN1 neutralizes less than 50% of extracellular EN1 (due to affinity, levels of expression and/or localization of the antibody). However, the more than twofold reduction in EN1 content in MNs, following scFvEN1 expression (Fig 4), rather suggests a more complex explanation whereby, while scFvEN1 only targets pure non‐cell‐autonomous EN1 activity, the En1‐Het phenotype may result from a 50% loss in secreted EN1 and, in addition, from cell‐autonomous dysfunctions affecting V1 interneurons but not inducing their death. V1 interneurons are an essential component of the reciprocal inhibitory αMN circuit and loss of their inhibitory input places αMNs at risk (Wang et al, 2008; Quinlan, 2011; Ramírez‐Jarquín et al, 2014; Ramírez‐Jarquín & Tapia, 2018). Furthermore, deficits in inhibitory interneurons in the spinal cord of motoneuron disease models have been reported (Hossaini et al, 2011; McGown et al, 2013; Allodi et al, 2021) and interneuron pathology has been observed before αMN loss (Chang & Martin, 2009; Allodi et al, 2021). One possibility is that EN1 regulates the expression of classical trophic factors in spinal V1 interneurons that are secreted and support αMN survival, in conjunction with EN1 signaling (Fig 10). Such synergy between classical secreted factors and HP signaling has been reported for ENGRAILED and DPP in the fly wing disk (Layalle et al, 2011), for EN2, adenosine, and EphrinA5 in chick RGC growth cone navigation (Wizenmann et al, 2009; Stettler et al, 2012), and for PAX6 and Netrin1 in the migration of chick oligodendrocyte precursors (Di Lullo et al, 2011).
Figure 10. Proposed schema of EN1 cell‐autonomous and non‐cell‐autonomous activities.
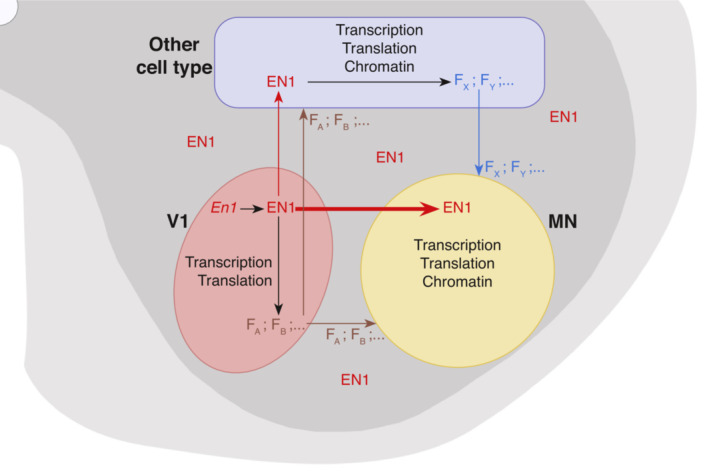
EN1 transcribed and synthesized in V1 (pink) has a cell‐autonomous activity in these cells contributing to the expression of several V1 factors (FA, FB, …) that may signal to MNs (yellow) and other cell types (blue). All the latter F factors do not necessarily require EN1 activity. EN1 after its secretion by V1 interneurons is preferentially internalized by MNs (fat red arrow) and less by other cell types (thin red arrow), if at all. These other cell types express other F Factors (FX, FY, …) which are secreted and are, or not, under non‐cell‐autonomous EN1 activity. As a result, MNs experience the signaling activity of F factors (possibly EN1‐dependent as in the scheme, but not necessarily so) and internalize EN1. Following internalization, EN1 can work alone or in synergy with the F factors to regulate transcription, translation, and chromatin conformation within MNs.
It remains that the scFvEN1 experiment demonstrates an EN1 non‐cell‐autonomous activity. This activity could be not only through extracellular EN1 acting directly on αMNs but also indirectly through other cell types, such as astrocytes, oligodendrocytes, or microglial cells (Fig 10). However, the internalization preference of injected EN1 by MNs and the fact that injected EN1 totally restores a healthy phenotype in the En1‐Het mouse strongly suggest that EN1 exerts its non‐cell‐autonomous activity primarily on αMNs. This activity is reminiscent of the direct non‐cell‐autonomous protective activity of OTX2 for RGCs upon NMDA excitotoxic insult (Torero‐Ibad et al, 2011). Moreover, when comparing the phenotypes of the En1‐Het and scFvEN1 mice, one must be aware of the fact that, in the En1‐Het, EN1 levels are lower very early during development. We thus cannot exclude that extracellular EN1 acts in conjunction with compromised interneuron physiological properties in En1‐Het animals, even if the interneurons are present in the appropriate number.
EN1 may protect αMNs through one or several of the mechanisms previously characterized for mDA neurons of the SNpc, which can be protected from oxidative stress by a single EN1 injection (Rekaik et al, 2015; Blaudin de Thé et al, 2018; Thomasson et al, 2019). Among these mechanisms are the regulation of Complex I mitochondrial activity, DNA break repair, heterochromatin maintenance, and the repression of long interspersed nuclear elements (LINE‐1) expression (Alvarez‐Fischer et al, 2011; Rekaik et al, 2015; Blaudin de Thé et al, 2018). The evaluation of these possibilities in the spinal cord will be the objective of future studies but the long‐lasting effect of a transient twofold increase in EN1 concentration within MNs strongly suggests an activity taking place at the chromatin level.
The En1‐Het mouse presents muscle weaknesses, abnormal spinal reflex, NMJ denervation, and αMN loss, all of which are phenotypes reminiscent of changes in ALS patients and in some ALS mouse models. This raises the question as to whether the results reported here are relevant for αMN diseases. To our knowledge, although they may exist in some sporadic cases, as for PD (Haubenberger et al, 2011), no studies so far have associated ALS with mutations affecting En1 integrity or En1 expression. It is thus clear that the En1‐het mouse cannot, as yet, be considered as an ALS mouse model. However, En1 genetic interactions with pathways leading to ALS, and its therapeutic activity in classical mouse and nonhuman primate models of PD (Alvarez‐Fischer et al, 2011; Rekaik et al, 2015; Blaudin de Thé et al, 2018; Thomasson et al, 2019), allows us to envisage its use as a long‐lasting therapeutic protein in ALS mouse models or iPSC‐derived human MNs.
One main mechanism possibly linking EN1 expression to ALS is that, in ALS as in many neurodegenerative diseases, aging is a major risk factor. Indeed, our results indicate that SQSTM1/p62 expression is a potential marker of age, both in αMNs and in γMNs, in agreement with its interaction with redox regulation pathways (Hensley & Harris‐White, 2015). Since En1‐Het mDA neurons are more sensitive than WT to oxidative stress and given that EN1 protects them against experimental oxidative stress (Rekaik et al, 2015), we speculate that the upregulation of SQSTM1/p62 expression, either in the mutant or following extracellular loss due to scFvEN1 expression, reveals an EN1 anti‐aging activity that may explain the ALS‐like phenotype of En1 hypomorphs. This hypothesis is supported by the finding that hEN1 injection at 1 month prevents the upregulation of p62/SQSTM1 in the MNs of 3‐month‐old En1‐Het mice and prevents their loss of strength. Indeed, we now need to follow the effects of gain and loss of EN1 functions in bona fide ALS animal models.
In conclusion, we have demonstrated a novel non‐cell‐autonomous activity for EN1 in adult spinal cord. Constitutive reduction in EN1 expression or local neutralization of extracellular EN1 causes motor endplate degeneration, muscle weakness, and αMN loss. This is an example of an important non‐cell‐autonomous activity for this transcription factor as is also the case for EN2, OTX2, PAX6, and VAX1 (Brunet et al, 2007; Prochiantz & Di Nardo, 2015; Di Nardo et al, 2018, 2020). Given that the sequences necessary for intercellular transfer are conserved in most HPs (Prochiantz & Joliot, 2003; Joliot & Prochiantz, 2004) and that such transfer has been demonstrated for about 150 of them (Lee et al, 2019), it is not unreasonable to predict that the developmental and physiological non‐cell‐autonomous functions demonstrated for a handful of HPs only represent the tip of the iceberg.
Materials and Methods
Reagents and Tools table
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Sheep anti‐ChAT | ABCAM | ab18736; RRID:AB_2244867 |
| Goat anti‐ChAT | Millipore | AB‐144P; RRID:AB_2079751 |
| Rabbit anti‐EN1 86/8 | Home made | Sonnier et al (2007) |
| Rabbit anti‐EN1 LSBio | CliniSciences | LS‐B9070 |
| Mouse anti‐neurofilament 165kDa | DSHB | 2H3; RRID:AB_531793 |
| Mouse anti‐SV2A | DSHB | SV2; RRID:AB_2315387 |
| Rabbit anti‐p62/SQSTM1 | ABCAM | ab109012; RRID:AB_2810880 |
| Alpha bungarotoxin, Alexa fluor 488 conjugate | Thermo Fisher | B13422 |
| Chemicals | ||
| PBS | Thermo Fisher | 14200075 |
| Triton X‐100 | Sigma‐ Aldrich | 9036‐19‐5 |
| Normal Goat Serum | Thermo Fisher | 31873 |
| Fetal Bovine Serum | Gibco | 26140087 |
| DAPI Fluoromount‐G | Southern Biotech | 0100‐20 |
| O‐Xylene | CARLO‐HERBA | 403926 |
| Cresyl Violet acetate | Sigma Aldrich | 105235 |
| Eukitt Quick‐hardening mounting medium | Sigma Aldrich | 03989 |
| Sucrose | Sigma Aldrich | 84097 |
| SYBR‐Green | Roche | 04887352001 |
| EDTA‐free Complete Proteases inhibitor cocktail | Roche | 11873580001 |
| 250U/ml of benzonase endonuclease | Sigma Aldrich | E1014‐25kU |
| Nu‐PAGE 4‐12% acrylamide 1 mm SDS‐PAGE | Invitrogen | NP0321BOX |
| LDS‐DTT Sample Buffer | Invitrogen | NP0007 |
| Tween‐20 | Sigma Aldrich | 9005‐64‐5 |
| Commercial assays | ||
| RNAscope Multiplex Fluorescent V2 Assay | ACD | 323100 |
| RNA‐Protein Co‐detection Ancillary Kit | ACD | 323180 |
| RNA Extraction RNeasy Kit | QIAGEN | 74104 |
| Chemiluminescence substrate CLARITY ECL | BIORAD | 170‐5061 |
| Pierce BCA assay | 23225 | |
| Oligonucleotides | ||
| qPCR Engrailed‐1 sense | This study | CCTGGGTCTACTGCACACG |
| qPCR Engrailed‐1 antisens | This study | CGCTTGTTTTGGAACCAGAT |
| qPCR Gapdh sense | This study | TGACGTGCCGCCTGGAGAAAC |
| qPCR Gapdh antisense | This study | CCGGCATCGAAGGTGGAAGAG |
| RNAscope Mm‐En1 | ACD | 442651 |
| RNAscope Mm‐ChAT | ACD | 408731‐C2 |
| RNAscope Mm‐Calb1 | ACD | 428431‐C3 |
| Viruses | ||
| scFvMUT | Vector Biolabs | AAV8‐IgK‐scFvMUT‐6xMyc‐2A‐eGFP |
| scFvEN1 | Vector Biolabs | AAV8‐IgK‐scFvEN1‐6xMyc‐2A‐eGFP |
| Experimental models | ||
| En1 lacZ (En1‐Heterozygous; M. musculus) | Jackson Lab | 00712 |
| Software | ||
| Image Studio Lite | Li‐Cor | https://www.licor.com/bio/image-studio-lite/ |
| GraphPad Prism | GraphPad | https://www.graphpad.com |
| Fiji | ImageJ | https://imagej.net/Fiji |
| Other | ||
| Grip Strength Meter | AHLBORN | ALMEMO 2450 |
Methods and Protocols
Animal management
All animal treatments followed the guidelines for the care and use of laboratory animals (US National Institutes of Health), the European Directive number 86/609 (EEC Council for Animal Protection in Experimental Research and Other Scientific Utilization), and French authorizations no 00703.01 “Therapeutic homeoproteins in Parkinson Disease” and no APAFIS #6034–2016071110167703 v2, “Spinal cord motoneuron neuroprotection” delivered by the Ministry of higher education, research, and innovation. Adult En1‐Het mice and WT littermates (available Jackson Labs strain #:007912) were housed two to five per cage with ad libitum access to food and water and under a 12‐h light/dark cycle. Transgenic mouse strain En1‐Het was bred by the Rodent Breeding Services provided by the Animal Care Services at College de France. Females and males were included in all studies. The endpoint limit for euthanasia was a 15% or greater loss of bodyweight or signs of paralysis; in all experiments, no mouse reached these endpoints.
Behavior analyses
Mice were habituated to the behavioral room and to the experimenter 24 h before the day of testing and again before each behavioral test. All tests were performed on the same day and behavioral assessment was carried out by evaluators blind to genotype and treatment.
Forepaw grip strength
The Transducer (IITC Life Science Grip Strength Meter, ALMEMO 2450 AHLBORN, World Precision Instruments) was calibrated and the scale of values set to grams. Each mouse was lifted by the tail to position the front paws at the height of the bar (about 15 cm) and then moved toward the bar. When a symmetric firm grip with both paws was established, the mouse was pulled backward at a constant speed until the grasp was broken and the maximal value was recorded. The test was repeated five times per animal with a minimal resting time of 5 min between tests and the mean of all values was normalized to the weight of each animal.
Inverted grid test
The mouse was placed on a wire grid (15 × 10 cm) and allowed to explore it. After 3–5 min, the grid was raised 30 cm above a soft surface and slowly inverted. Latency to release was recorded three times per mouse with a minimum resting time of 5 min between trials. The longest latency was used for analysis.
Hindlimb extensor reflex
Mice were suspended by the tail at a constant height (about 15 cm) and scored for hindlimb extension reflex. The scores were assigned from 0 to 3 as follows: 3 for normal symmetric extension in both hind limbs without visible tremors; 2.5 for normal extension in both hind limbs with tremor in one or both paws; 2.0 for unequal extension of the hind limbs without visible tremors; 1.5 for unequal extension in the hind limbs with tremors in one or both paws, 1.0 for extension reflex in only one hindlimb, 0.5 for minimum extension of both hindlimbs, and 0 for the absence of any hindlimb extension.
Tissue preparation
Spinal cord
Adult mice were euthanized by a 1 μl/g body weight dose of Dolethal (Euthasol: 4 μg/μl). Spinal cords were dissected and placed in phosphate buffer saline (PBS) to remove meninges and surrounding connective tissues. Cervical and lumbar enlargements were separately placed in paraformaldehyde 4% (PFA, Thermo Scientific) for 1 h at room temperature (RT) with gentle mixing, washed in PBS three times for 30 min at RT, and placed in PBS with 20% sucrose overnight at 4°C. After cryoprotection, the tissue was embedded in Tissue Freezing Medium (TFM, Microm Microtech) and frozen on dry ice and 30 μm sections were prepared using an HM 560 Microm cryostat (Thermo Scientific).
Muscle
The lumbrical muscles were dissected in cold PBS, fixed at RT in 4% PFA for 10 min, and washed in PBS. Muscle whole mounts were processed in toto to visualize the entire innervation pattern and allow for a detailed NMJ analysis (Sleigh et al, 2014).
Cresyl violet staining
Slides with 30 μm spinal cord sections were washed in PBS 3 times, cleared in O‐Xylene (CARLO‐HERBA) for 5 min, then hydrated in graded alcohol with increasing water, and placed in Cresyl Violet acetate (MERCK). Sections were then dehydrated in increasing alcohol and mounted in Eukitt® Quick‐hardening mounting medium (Sigma).
Spinal cord and muscle immunofluorescence labeling
Slides with 30 μm spinal cord sections or whole‐mount muscles were washed in PBS and permeabilized with 2% Triton. After 30 min at RT in 100 μM glycine buffer, 10% normal goat serum (NGS, Invitrogen) or fetal bovine serum (FBS, Gibco) was added in the presence of 1% Triton before incubation with primary antibodies (sheep anti‐Choline Acetyltransferase [ChAT] ABCAM 1:1,000, goat anti‐ChAT Millipore 1:500, rabbit anti‐EN1 86/8 1:300 [Sonnier et al, 2007], rabbit anti‐EN1 LSBio [CliniSciences] 1:200, mouse anti‐neurofilament 165 kDa Developmental Studies Hybridoma Bank 1:50, mouse anti‐synaptic vesicle glycoprotein 2A DSHB 1:100 and rabbit anti‐p62 ABCAM 1:1,000) overnight at 4°C, washed and further incubated with secondary antibodies for 2 h at RT. Where indicated, anti‐EN1 LSBio activity was neutralized by a 1‐h (RT) preincubation with hEN1 (1.5 EN1/LSBio molar ratio). For muscle staining, α‐bungarotoxin (Alexa Fluor 488 conjugate) was included at the same time as the secondary antibodies. Slides were washed and mounted with DAPI Fluoromount‐G® (Southern Biotech). Controls without primary antibodies were systematically included.
RT‐qPCR
Spinal cords were removed as above, and lumbar enlargements were rapidly frozen on dry ice. Total RNA was extracted (RNeasy Mini kit, Qiagen) and reverse transcribed using the QuantiTect Reverse Transcription kit (Qiagen). RT–qPCR was done using SYBR‐Green (Roche Applied Science) and a Light Cycler 480 (Roche Applied Science). Data were analyzed using the “2‐ddCt” method and values were normalized to Glyceraldehyde 3‐phosphate dehydrogenase (Gapdh). The following primers were used: Engrailed‐1 sense: CCTGGGTCTACTGCACACG, antisense: CGCTTGTTTTGGAACCAGAT; Gapdh sense: TGACGTGCCGCCTGGAGAAAC, antisense: CCGGCATCGAAGGTGGAAGAG.
Protein and single‐chain antibodies
Protein
Human EN1 (hEN1) was produced as described (Torero‐Ibad et al, 2011), and endotoxins were removed by Triton X‐144 phase separation. In brief, precondensed 1% Triton X‐144 (Sigma) was added to the protein preparation. The solution was incubated for 30 min at 4°C with constant stirring, transferred to 37°C for 10 min, and centrifuged at 1,500 g for 10 min at 25°C. The endotoxin‐free protein was aliquoted and kept at −80°C.
Single‐chain antibodies
The anti‐EN1 single‐chain antibody plasmid was prepared from the anti‐ENGRAILED 4G11 hybridoma (Developmental Hybridoma Bank, Iowa City, IA, USA). Cloning was as described (Wizenmann et al, 2009) with or without signal peptide (Lesaffre et al, 2007) with 6 myc tags at the C terminus of the antibody followed by a GFP downstream of a P2A skipping peptide. Addition of a GFAP promoter, insertion in an AAV8 backbone, and production of the AAV8 were done by Vector Biolabs.
Intrathecal injections
Mice were anesthetized with 1 μl/g ketamine‐xylazine (Imalgene1000: 10 μg/μl, Rompur 2%: 0.8 μg/μl) and placed on the injection platform. The tail of the animal was taken between two fingers, and the spinal column was gently flattened against a padded surface with the other hand. The L3 vertebral spine was identified by palpation, and a 23G × 1″ needle (0.6 × 25 mm Terumo tip) was placed at the L1 and T13 groove and inserted through the skin at an angle of 20° (Hylden & Wilcox, 1980). The needle was slowly advanced into the intervertebral space until it reached the injection point, provoking a strong tail‐flick reflex. Injections of 5 μl were done at a rate of 1 μl/min with 1 μg recombinant protein, AAV8‐IgK‐scFvMUT‐6xMyc‐2A‐eGFP (scFvMUT), or AAV8‐IgK‐scFvEN1‐6xMyc‐2A‐eGFP (scFvEN1) (Vector Biolabs, 1012 GC/ml). The peptides (ProteoGenix) were co‐injected with hEN1 at a 20‐fold molar excess of peptides. The needle was left in place for 2 min after injection and then slowly removed. Animals were placed in a warmed chamber until recovery from anesthesia. Extensor reflex and gait analysis were examined 2 and 24 h after injection to verify the absence of spinal cord damage.
RNAscope fluorescent in situ hybridization
Mice aged 4.5 months were euthanized with Dolethal and transcardially perfused with PBS followed by 4% PFA in PBS. The lumbar region of the spinal cord was dissected, fixed overnight in 4% PFA at 4°C, and cryoprotected in PBS 30% sucrose for 24 h at 4°C. Spinal cord sections 40 μm thick were prepared using a Leitz (1400) sliding microtome. Sections were washed in PBS, incubated with RNAscope hydrogen peroxide solution from Advanced Cell Diagnostics (ACD) for 10 min at RT, rinsed in Tris‐buffered saline with Tween (50 mM Tris‐Cl, pH 7.6; 150 mM NaCl, 0.1% Tween 20) at RT, collected on Super Frost plus microscope slides (Thermo Scientific), dried at RT for 1 h, rapidly immersed in ultrapure water, dried again at RT for 1 h, heated for 1 h at 60°C, and dried at RT overnight. The next day, sections were immersed in ultrapure water, rapidly dehydrated with 100% ethanol, incubated at 100°C for 15 min in RNAscope 1X Target Retrieval Reagent (ACD), washed in ultrapure water, and dehydrated with 100% ethanol for 3 min at RT. Protease treatment was carried out using RNAscope Protease Plus solution (ACD) for 30 min at 40°C in a HybEZ oven (ACD). Sections were then washed in PBS before in situ hybridization using the RNAscope Multiplex Fluorescent V2 Assay (ACD). Probes were hybridized for 2 h at 40°C in HybEZ oven (ACD), followed by incubation with signal amplification reagents according to the manufacturer's instructions. Probes were purchased from ACD: Mm‐En1‐C1 (catalog #442651), Mm‐Chat‐C2 (catalog #408731‐C2), and Mm‐Calb1‐C3 (catalog #428431‐C3). The hybridized probe signal was visualized and captured on an upright Leica CFS SP5 confocal microscope with a 40× oil objective.
RNAscope and immunofluorescence co‐detection
Sections prepared as described above were collected on Super Frost plus microscope slides and heated for 30 min at 60°C. RNAscope was performed according to the manufacturer's instructions (Multiplex Fluorescent V2 Assay and RNA‐Protein Co‐detection Ancillary Kit Cat No. 323180). In brief, sections were postfixed in 4% PFA for 15 min at 4°C, dehydrated in 50, 70, and 100% (twice) ethanol solutions for 5 min each, incubated with RNAscope hydrogen peroxide solution for 10 min at RT, rinsed in ultrapure water, incubated at 100°C for 5 min in RNAscope 1X Co‐detection Target Retrieval, rinsed in ultrapure water, and washed in Phosphate buffer, 0.1% tween 20 pH7.2 (PBS‐T). Sections were incubated overnight at 4°C with anti‐EN1 LSBio diluted (1:200) in Co‐detection Antibody Diluent, washed in PBS‐T, fixed in 4% PFA for 30 min at RT, and washed twice in PBS‐T. Sections were treated with protease 30 min at 40°C in a HybEZ oven and washed in PBS before in situ hybridization using the RNAscope Multiplex Fluorescent V2 Assay. Probes were hybridized for 2 h at 40°C in HybEZ oven, followed by incubation with signal amplification reagents according to manufacturer instructions. Finally, sections were incubated with the secondary antibody diluted in the Co‐detection Antibody Diluent for 30 min at RT.
Western blots
Tissue extracts
Spinal cords and ventral midbrains dissected from adult mice were kept frozen at −80°C. After thawing on ice and 3 rinses in PBS, tissues were resuspended in 500 μl dissociation buffer (DB: 50 mM Tris–HCl pH8.0, 0.3 M NaCl, Triton X‐100 1%, EDTA‐free Complete Proteases inhibitor cocktail from Roche and 250 U/ml of benzonase endonuclease from Sigma). After homogenization in a Dounce tissue homogenizer, 10 times with the A piston and 10 times with the B piston, lysates were incubated for 30 min on ice, sonicated for 5 min in Diagenode Bioruptor set with 30/30 s ON/OFF at high power, incubated on ice for 30 min, and centrifuged for 10 min at 12,000 g. Supernatants were harvested and their protein contents measured (Pierce BCA assay) and frozen at −80°C.
Western blots
Invitrogen Nu‐PAGE 4–12% acrylamide 1 mm SDS–PAGE gels were used to run spinal cord extracts or the hEN1 recombinant protein. Extracts (2–3 μg total proteins) and 2 ng of purified EN1 were diluted in 10 μl Invitrogen LDS‐DTT Sample Buffer, heated for 10 min at 70°C, briefly centrifuged at 10,000 g and loaded. For the immunoprecipitation followed by western blot, 1/200 of total inputs or flow‐throughs and 1/20 of the total protein A–sepharose E2 elution were used per well. SeeBlue 2 and molecular weight markers were from Invitrogen. Gels were run in MOPS buffer at 150 V for 1 h and 15 min and the proteins transferred for 30 min at 100 V on Millipore PVDF membranes (BIORAD Criterion Blotter apparatus). Membranes were blocked for 1 h at RT with 4% nonfat dry milk (NFDM) diluted in TBS containing 0.2% Tween‐20 (TBST). Antibodies to EN1 86/8 and LSBio were diluted 1/200 in TBST 4% NFDM and incubated overnight at 4°C. Blots were rinsed 3 times for 15 min each with TBST and incubated with HRP‐labeled anti‐rabbit antibody diluted 1/2,500 for 1 h at RT. When preceded by immunoprecipitations, revelation was with HRP‐protein G diluted 1/5,000 for 1 h at RT followed by 3 TBST rinses of 15 min each. HRP activity was revealed with chemiluminescence substrate CLARITY from BIORAD, and the light was quantified with the FUJI LAS 4000 imager. Signals were quantified with the Image Studio Lite software from Li‐Cor.
Bioinformatic analysis
Differentially expressed genes (cutoff P < 0.05) from the RNA‐seq performed on microdissected six‐week‐old SNpc from WT and En1‐Het mice (GSE72321; Rekaik et al, 2015) were compared with genes identified from an RNA‐seq of microdissected MNs from WT mice (GSE38820) (Bandyopadhyay et al, 2013). Transcripts present in the two lists were retained. Using STRING database, 10 biological processes of interest were selected including regulation of gene expression, cellular response to stress, cell communication, regulation of I‐kappaB kinase/NF‐kappaB signaling, cellular response to DNA damage stimulus, response to oxygen‐containing compounds, locomotion, DNA repair, modulation of chemical synaptic transmission, and brain development. Using these pathways reduced the list of selected genes to 402. To the 402 genes, we used the STRING database to identify known interactions with the four main genes involved in familial ALS (fALS): SOD1, FUS, C9Orf72, and TARDBP.
Image analyses
Cresyl violet‐stained spinal cord section images were acquired with a Nikon‐i90 microscope under bright‐field conditions at 10× with a Z‐stack step of 0.5 μm. RNAscope FISH images were acquired using a Yokogawa X1 Spinning Disk confocal microscope at 20× (Nikon) and acquisitions of 3D z‐stack (1 μm) using 491, 561, and 642 nm laser beams. For cell quantification, at least five lumbar spinal cord sections (10 ventral horns) through levels L1 to L5 were analyzed for each animal. Immunofluorescence‐stained spinal cord sections images were acquired with a Leica SP5 inverted confocal microscope at 20× (Leica DMI6000) and acquisitions of 3D z‐stack (0.5 μm) were made using the UV (405 nm, 50 mW), Argon (488 nm, 200 mW), and DPSS (561 nm, 15 mW) lasers. For cell counting, at least five 30‐μm‐thick spinal cord sections separated by ≥ 900 μm were analyzed for each animal. ChAT+ cells and Cresyl violet‐stained cells with a soma area above 100 μm2 were manually outlined using ImageJ software (NIH, Bethesda, MD) and their area determined. Analyses were carried out on Z‐stacks through the entire 30 μm thickness of the section. Cells were classified as small (100–199 μm2 cross‐sectional area), intermediate (200–299 μm2), or large (> 300 μm2). For example, in a WT mouse 4.5 months of age 558, 158, and 112 cells were counted in the 100–199, 200–299, and > 300 μm2 classes, respectively (5 sections). In a 4.5‐month‐old En1‐Het mouse, the values were 562, 149, and 66, respectively. The data in the graphs are the average number of each cell category in one ventral horn. Thus, the WT mouse had an average of 11.2 large MNs and the En1‐Het mouse had an average of 6.6 large MNs. ChAT and Cresyl violet gave similar results for medium‐size (200–299 μm2) and large (> 300 μm2) neurons thus allowing us to characterize αMNs based on size (> 300 μm2), with or without additional ChAT staining. For p62 staining analysis, the motoneuron region of five lumbar sections was acquired at 40× for each animal. Mean intensity was measured in ChAT+ cells with a soma greater than 150 μm2 using ImageJ software. For each experiment, image acquisition was performed with the same parameter settings of the confocal microscope to allow for comparison between experimental conditions.
For endplate analysis, lumbrical muscles were imaged with a Leica SP5 inverted confocal microscope (Leica DMI6000) with a motorized XY stage. 3D z‐stack (0.5 μm) acquisitions were made as above (UV, Argon and DPSS) and images analyzed using ImageJ software. Analyses were performed blind to the genotype and treatment. The Z‐stack with maximum overall intensity was determined. Red (SV2A/NF) and green channels (α‐BGT) were separated and images were filtered for better segmentation using a Median Filter with a radius of 2 pixels. Each channel image was then converted to a gray LUT, and an automatic threshold was established for both channels to outline the areas of analysis. For postsynaptic analysis, each endplate was identified by α‐BGT signal. The integrated density of one channel overlapping the other channel was determined using the AND function in the ROI manager for each pair of measurements. Measurements were performed in the original images, and the overlap was quantified based on integrated density as a percentage (overlap values/total values in the α‐BGT channel ×100). Endplates were categorized as fully innervated, partially innervated, or denervated. A fully innervated endplate is defined as an endplate in which 80% or more of the green pixels (α‐BGT) are covered by a red pixel (SV2A/NF), a partially innervated one is between 20 and 80% and a denervated one below 20% coverage. Endplate morphology was evaluated by counting the number of endplates with perforations (areas lacking α‐bungarotoxin staining). All analyses were performed on the entire Z‐stacks through the NMJ.
Statistical analyses
Sample size estimate was based on our previous observations. Results are expressed as mean ± SD. Statistical significance was determined as follows. For RT–qPCR, WT and En1‐Het mice were compared by unpaired t‐test with equal SD. For behavioral and NMJ analyses and αMN counting in the time‐course studies, a 2‐way ANOVA was performed and if the main effect was significant groups were compared by unpaired t‐test with equal SD comparing WT with En1‐Het for each time point. For intrathecal injections, behavioral and NMJ analyses, and αMN counting, experimental data were compared by one‐way ANOVA followed by a post hoc Tukey test for comparisons with WT. For behavioral analysis in the time‐course protection of injected hEN1, a 2‐way ANOVA was performed and if the main effect was significant groups were compared by unpaired t‐test with equal variances comparing WT with En1‐Het injected at each time point.
The p62 measures are bounded between 0 and an upper quantification limit (ceiling effect), and classical normally distributed models are ill‐suited to account for these two constraints. We solicited PharmaLex Belgium to develop a suitable analysis. Since it can take a wide variety of shapes and is naturally bounded in the [0;1] interval, beta‐distribution was fitted on the p62/SQSTM1 measure, for all MNs, γMNs, and αMNs separately, after rescaling the original data to the [0;1] interval. The shape and scale parameters account for time, group, and time*group interaction as fixed effect. It models the variance observed in the data by time, group, and time*group interaction, meaning each combination has its own variability. Finally, the mouse‐to‐mouse variability (or between‐mouse variability) is modeled by time and group.
Author contributions
Mélanie Leboeuf: Formal analysis; investigation; methodology; writing – review and editing. Stephanie E Vargas‐Abonce: Formal analysis; investigation; methodology; writing – review and editing. Eugénie Pezé‐Hedsieck: Resources. Edmond Dupont: Investigation. Lucia Jimenez‐Alonso: Investigation. Kenneth L Moya: Conceptualization; formal analysis; supervision; methodology; writing – original draft; project administration; writing – review and editing. Alain Prochiantz: Conceptualization; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
AP and KLM are co‐founders and hold shares in BrainEver, a company developing HPs for therapeutic use.
Supporting information
Expanded View Figures PDF
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Source Data for Figure 9
Acknowledgments
We would like to thank Anne Bousseau, Ariel Di Nardo, Lizzie di Lullo, and Julien Spatazza for reading the manuscript and discussing experiments and results with the authors. We are grateful for the help brought by the CIRB Imaging and animal facilities. Memolife Labex PhD fellowship for SEVA, Association Nationale de la Recherche et de la Technologie (Cifre/ANRT no 2017/0488) to ML, BrainEver, Homeosign ERC‐AdG no 339379, Fondation Schueller‐Bettencourt.
EMBO reports (2023) 24: e56525
Contributor Information
Kenneth L Moya, Email: ken.moya@college-de-france.fr.
Alain Prochiantz, Email: alain.prochiantz@college-de-france.fr.
Data availability
All source images are available at: https://www.ebi.ac.uk/biostudies/studies/S-BSST961 (S‐BSST961).
References
- Allodi I, Montañana‐Rosell R, Selvan R, Löw P, Kiehn O (2021) Locomotor deficits in a mouse model of ALS are paralleled by loss of V1‐interneuron connections onto fast motor neurons. Nat Commun 12: 3251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez FJ, Jonas PC, Sapir T, Hartley R, Berrocal MC, Geiman EJ, Todd AJ, Goulding M (2005) Postnatal phenotype and localization of spinal cord V1 derived interneurons. J Comp Neurol 493: 177–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Fischer D, Fuchs J, Castagner F, Stettler O, Massiani‐Beaudoin O, Moya KL, Bouillot C, Oertel WH, Lombès A, Faigle W et al (2011) Engrailed protects mouse midbrain dopaminergic neurons against mitochondrial complex I insults. Nat Neurosci 14: 1260–1266 [DOI] [PubMed] [Google Scholar]
- Amblard I, Dupont E, Alves I, Miralvès J, Queguiner I, Joliot A (2020a) Bidirectional transfer of homeoprotein EN2 across the plasma membrane requires PIP2. J Cell Sci 133: jcs244327 [DOI] [PubMed] [Google Scholar]
- Amblard I, Thauvin M, Rampon C, Queguiner I, Pak VV, Belousov V, Prochiantz A, Volovitch M, Joliot A, Vriz S (2020b) H2O2 and engrailed 2 paracrine activity synergize to shape the zebrafish optic tectum. Commun Biol 3: 536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay U, Cotney J, Nagy M, Oh S, Leng J, Mahajan M, Mane S, Fenton WA, Noonan JP, Horwich AL (2013) RNA‐seq profiling of spinal cord motor neurons from a Presymptomatic SOD1 ALS mouse. Plos One 8: e53575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurdeley M, Spatazza J, Lee HHC, Sugiyama S, Bernard C, Di Nardo AA, Hensch TK, Prochiantz A (2012) Otx2 binding to perineuronal nets persistently regulates plasticity in the mature visual cortex. J Neurosci 32: 9429–9437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T (2009) Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 452: 181–197 [DOI] [PubMed] [Google Scholar]
- Blaudin de Thé XB, Rekaik H, Heidsieck EP, Massiani‐Beaudoin O, Joshi RL, Fuchs J, Prochiantz A (2018) Engrailed homeoprotein blocks degeneration in adult dopaminergic neurons through LINE‐1 repression. EMBO J 37: e97374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet I, Weinl C, Piper M, Trembleau A, Volovitch M, Harris W, Prochiantz A, Holt C (2005) The transcription factor Engrailed‐2 guides retinal axons. Nature 438: 94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet I, Di Nardo AA, Sonnier L, Beurdeley M, Prochiantz A (2007) The topological role of homeoproteins in the developing central nervous system. Trends Neurosci 30: 260–267 [DOI] [PubMed] [Google Scholar]
- Cardon S, Hervis YP, Bolbach G, Lopin‐Bon C, Jacquinet J‐C, Illien F, Walrant A, Ravault D, He B, Molina L et al (2023) A cationic motif upstream Engrailed2 homeodomain controls cell internalization through selective interaction with heparan sulfates. Nat Commun 14: 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Martin LJ (2009) Glycinergic innervation of motoneurons is deficient in amyotrophic lateral sclerosis mice: a quantitative confocal analysis. Am J Pathol 174: 574–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zheng Z‐Z, Chen X, Huang R, Yang Y, Yuan L, Pan L, Hadano S, Shang H‐F (2014) SQSTM1 mutations in Han Chinese populations with sporadic amyotrophic lateral sclerosis. Neurobiol Aging 35: 726.e7–726.e9 [DOI] [PubMed] [Google Scholar]
- Comley LH, Nijssen J, Frost‐Nylen J, Hedlund E (2016) Cross‐disease comparison of amyotrophic lateral sclerosis and spinal muscular atrophy reveals conservation of selective vulnerability but differential neuromuscular junction pathology. J Comp Neurol 524: 1424–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lullo E, Haton C, Poupon CL, Volovitch M, Joliot A, Thomas JL, Prochiantz A (2011) Paracrine Pax6 activity regulates oligodendrocyte precursor cell migration in the chick embryonic neural tube. Development 138: 4991–5001 [DOI] [PubMed] [Google Scholar]
- Di Nardo AA, Nedelec S, Trembleau A, Volovitch M, Prochiantz A, Montesinos ML (2007) Dendritic localization and activity‐dependent translation of Engrailed1 transcription factor. Mol Cell Neurosci 35: 230–236 [DOI] [PubMed] [Google Scholar]
- Di Nardo AA, Fuchs J, Joshi RL, Moya KL, Prochiantz A (2018) The physiology of homeoprotein transduction. Physiol Rev 98: 1943–1982 [DOI] [PubMed] [Google Scholar]
- Di Nardo AA, Joliot A, Prochiantz A (2020) Homeoprotein transduction in neurodevelopment and physiopathology. Sci Adv 6: eabc6374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty J, Baehrecke EH (2018) Life, death and autophagy. Nat Cell Biol 20: 1110–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H et al (2011) SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 68: 1440–1446 [DOI] [PubMed] [Google Scholar]
- Feinstein B, Lindegard B, Nyman E, Wohlfart G (1954) Studies on action potentials in normal human muscles. Acta Psych Neurol 29: 189–195 [DOI] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano‐Sanchez A, Khan J, Polak MA, Glass JD (2004) Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol 185: 232–240 [DOI] [PubMed] [Google Scholar]
- Foster A, Rea S (2020) The role of sequestosome 1/p62 protein in amyotrophic lateral sclerosis and frontotemporal dementia pathogenesis. Neural Regen Res 15: 2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring W (1987) Homeo boxes in the study of development. Science 236: 1245–1252 [DOI] [PubMed] [Google Scholar]
- Haubenberger D, Reinthaler E, Mueller JC, Pirker W, Katzenschlager R, Froehlich R, Bruecke T, Daniel G, Auff E, Zimprich A (2011) Association of transcription factor polymorphisms PITX3 and EN1 with Parkinson's disease. Neurobiol Aging 32: 302–307 [DOI] [PubMed] [Google Scholar]
- Hensley K, Harris‐White ME (2015) Redox regulation of autophagy in healthy brain and neurodegeneration. Neurobiol Dis 84: 50–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashijima S, Masino MA, Mandel G, Fetcho JR (2004) Engrailed‐1 expression marks a primitive class of inhibitory spinal interneuron. J Neurosci 24: 5827–5839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland PWH, Takahashi T (2005) The evolution of homeobox genes: implications for the study of brain development. Brain Res Bull 66: 484–490 [DOI] [PubMed] [Google Scholar]
- Hossaini M, Cano SC, vsn Dis V, Haasdijk ED, Hoogenraad CC, Holstege JC, Jaarsma D (2011) Spinal inhibitory interneuron pathology follows motor neuron degeneration independent of glial mutant superoxide dismutase 1 expression in SOD1‐ALS mice. J Neuropathol Exp Neurol 70: 662–677 [DOI] [PubMed] [Google Scholar]
- Hylden JLK, Wilcox GL (1980) Intrathecal morphine in mice: a new technique. Eur J Pharmacol 67: 313–316 [DOI] [PubMed] [Google Scholar]
- Jaakkola PM, Pursiheimo J‐P (2009) p62 degradation by autophagy: another way for cancer cells to survive under hypoxia. Autophagy 5: 410–412 [DOI] [PubMed] [Google Scholar]
- Jakobi AJ, Huber ST, Mortensen SA, Schultz SW, Palara A, Kuhm T, Shrestha BK, Lamark T, Hagen WJH, Wilmanns M et al (2020) Structural basis of p62/SQSTM1 helical filaments and their role in cellular cargo uptake. Nat Commun 11: 440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joliot A, Prochiantz A (2004) Transduction peptides: from technology to physiology. Nat Cell Biol 6: 189–196 [DOI] [PubMed] [Google Scholar]
- Kaddour H, Coppola E, Di Nardo AA, Poupon CL, Mailly P, Wizenmann A, Volovitch M, Prochiantz A, Pierani A (2019) Extracellular Pax6 regulates tangential Cajal–Retzius cell migration in the developing mouse neocortex. Cereb Cortex 30: 465–475 [DOI] [PubMed] [Google Scholar]
- Kanning KC, Kaplan A, Henderson CE (2010) Motor neuron diversity in development and disease. Annu Rev Neurosci 33: 409–440 [DOI] [PubMed] [Google Scholar]
- Kim N, Min KW, Kang KH, Lee EJ, Kim H‐T, Moon K, Choi J, Le D, Lee S‐H, Kim JW (2014) Regulation of retinal axon growth by secreted Vax1 homeodomain protein. Elife 3: e02671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, Pedro JMB, Cadwell K, Cecconi F, Choi AMK et al (2021) Autophagy in major human diseases. EMBO J 40: e108863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AR, Cogdell IC, Jessell TM, Bikoff JB, Alvarez FJ (2021) Genetic targeting of adult Renshaw cells using a calbindin 1 destabilized Cre allele for intersection with parvalbumin or Engrailed1. Sci Rep 11: 19861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layalle S, Volovitch M, Mugat B, Bonneaud N, Parmentier ML, Prochiantz A, Joliot A, Maschat F (2011) Engrailed homeoprotein acts as a signaling molecule in the developing fly. Development 138: 2315–2323 [DOI] [PubMed] [Google Scholar]
- Lee EJ, Kim N, Park JW, Kang KH, Kim W, Sim NS, Jeong C‐S, Blackshaw S, Vidal M, Huh S‐O et al (2019) Global analysis of intercellular homeodomain protein transfer. Cell Rep 28: 712–722.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidal AM, Levine B, Debnath J (2018) Autophagy and the cell biology of age‐related disease. Nat Cell Biol 20: 1338–1348 [DOI] [PubMed] [Google Scholar]
- Lesaffre B, Joliot A, Prochiantz A, Volovitch M (2007) Direct non‐cell autonomous Pax6 activity regulates eye development in the zebrafish. Neural Dev 2: 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Attarwala IY, Xie X‐Q (2019) SQSTM1/p62: a potential target for neurodegenerative disease. ACS Chem Nerosci 10: 2094–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson GL, Przedborski S, Abbott LF (2014) A computational model of motor neuron degeneration. Neuron 83: 975–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGown A, McDearmid JR, Panagiotaki N, Tong H, Mashhadi SA, Redhead N, Lyon AN, Beattie CE, Shaw PJ, Ramesh TM (2013) Early interneuron dysfunction in ALS: insights from a mutant sod1 zebrafish model. Ann Neurol 73: 246–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies FM, Fleming A, Rubinsztein DC (2015) Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci 16: 345–357 [DOI] [PubMed] [Google Scholar]
- Miyata S, Komatsu Y, Yoshimura Y, Taya C, Kitagawa H (2012) Persistent cortical plasticity by upregulation of chondroitin 6‐sulfation. Nat Neurosci 15: 414–422 [DOI] [PubMed] [Google Scholar]
- Nordström U, Beauvais G, Ghosh A, Sasidharan BCP, Lundblad M, Fuchs J, Joshi RL, Lipton JW, Roholt A, Medicetty S et al (2015) Progressive nigrostriatal terminal dysfunction and degeneration in the engrailed1 heterozygous mouse model of Parkinson's disease. Neurobiol Dis 73: 70–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensieri P, Mantilleri A, Plassard D, Furukawa T, Moya KL, Prochiantz A, Lamonerie T (2021) Photoreceptor cKO of OTX2 enhances OTX2 intercellular transfer in the retina and causes photophobia. ENeuro 8: 10.1523/ENEURO.0229-21.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powis RA, Gillingwater TH (2016) Selective loss of alpha motor neurons with sparing of gamma motor neurons and spinal cord cholinergic neurons in a mouse model of spinal muscular atrophy. J Anat 228: 443–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochiantz A, Di Nardo AA (2015) Homeoprotein signaling in the developing and adult nervous system. Neuron 85: 911–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochiantz A, Joliot A (2003) Can transcription factors function as cell–cell signalling molecules? Nat Rev Mol Cell Bio 4: 814–819 [DOI] [PubMed] [Google Scholar]
- Quinlan KA (2011) Links between electrophysiological and molecular pathology of amyotrophic lateral sclerosis. Integr Comp Biol 51: 913–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez‐Jarquín UN, Tapia R (2018) Excitatory and inhibitory neuronal circuits in the spinal cord and their role in the control of motor neuron function and degeneration. ACS Chem Nerosci 9: 211–216 [DOI] [PubMed] [Google Scholar]
- Ramírez‐Jarquín UN, Lazo‐Gómez R, Tovar‐y‐Romo LB, Tapia R (2014) Spinal inhibitory circuits and their role in motor neuron degeneration. Neuropharmacology 82: 101–107 [DOI] [PubMed] [Google Scholar]
- Rampon C, Gauron C, Lin T, Meda F, Dupont E, Cosson A, Ipendey E, Frerot A, Aujard I, Saux TL et al (2015) Control of brain patterning by engrailed paracrine transfer: a new function of the Pbx interaction domain. Development 142: 1840–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekaik H, Blaudin de Thé F‐X, Fuchs J, Massiani‐Beaudoin O, Prochiantz A, Joshi RL (2015) Engrailed homeoprotein protects mesencephalic dopaminergic neurons from oxidative stress. Cell Rep 13: 242–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapir T (2004) Pax6 and engrailed 1 regulate two distinct aspects of Renshaw cell development. J Neurosci 24: 1255–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgadò P, Alberi L, Gherbassi D, Galasso SL, Ramakers GMJ, Alavian KN, Smidt MP, Dyck RH, Simon HH (2006) Slow progressive degeneration of nigral dopaminergic neurons in postnatal engrailed mutant mice. Proc Natl Acad Sci USA 103: 15242–15247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Toyoshima Y, Shiga A, Yokoseki A, Arakawa K, Sekine Y, Shimohata T, Ikeuchi T, Nishizawa M, Kakita A et al (2013) Sporadic ALS with compound heterozygous mutations in the SQSTM1 gene. Acta Neuropathol 126: 453–459 [DOI] [PubMed] [Google Scholar]
- Sleigh JN, Burgess RW, Gillingwater TH, Cader MZ (2014) Morphological analysis of neuromuscular junction development and degeneration in rodent lumbrical muscles. J Neurosci Meth 227: 159–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnier L, Pen GL, Hartmann A, Bizot J‐C, Trovero F, Krebs M‐O, Prochiantz A (2007) Progressive loss of dopaminergic neurons in the ventral midbrain of adult mice heterozygote for Engrailed1. J Neurosci 27: 1063–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stettler O, Joshi RL, Wizenmann A, Reingruber J, Holcman D, Bouillot C, Castagner F, Prochiantz A, Moya KL (2012) Engrailed homeoprotein recruits the adenosine A1 receptor to potentiate ephrin A5 function in retinal growth cones. Development 139: 215–224 [DOI] [PubMed] [Google Scholar]
- Sugiyama S, Di Nardo AA, Aizawa S, Matsuo I, Volovitch M, Prochiantz A, Hensch TK (2008) Experience‐dependent transfer of Otx2 homeoprotein into the visual cortex activates postnatal plasticity. Cell 134: 508–520 [DOI] [PubMed] [Google Scholar]
- Sun C, Seranova E, Cohen MA, Chipara M, Roberts J, Astuti D, Palhegyi AM, Acharjee A, Sedlackova L, Kataura T et al (2023) NAD depletion mediates cytotoxicity in human neurons with autophagy deficiency. Cell Rep 42: 112372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai H, Wang Z, Gong H, Han X, Zhou J, Wang X, Wei X, Ding Y, Huang N, Qin J et al (2016) Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress‐induced senescence. Autophagy 13: 99–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomasson N, Pioli E, Friedel C, Monseur A, Lavaur J, Moya KL, Bezard E, Bousseau A, Prochiantz A (2019) Engrailed‐1 induces long‐lasting behavior benefit in an experimental Parkinson primate model. Mov Disord 34: 1082–1084 [DOI] [PubMed] [Google Scholar]
- Torero‐Ibad R, Mazhar B, Vincent C, Bernard C, Dégardin J, Simonutti M, Lamonerie T, Nardo AD, Prochiantz A, Moya KL (2020) OTX2 non‐cell autonomous activity regulates inner retinal function. eNeuro 7: ENEURO.0012‐19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torero-Ibad R, Rheey J, Mrejen S, Forster V, Picaud S, Prochiantz A, Moya KL (2011) Otx2 promotes the survival of damaged adult retinal ganglion cells and protects against excitotoxic loss of visual acuity in vivo . J Neurosci 31: 5495–5503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Li L, Goulding M, Frank E (2008) Early postnatal development of reciprocal Ia inhibition in the murine spinal cord. J Neurophysiol 100: 185–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenner P, O'Donovan MJ, Matise MP (2000) Topographical and physiological characterization of interneurons that express engrailed‐1 in the embryonic chick spinal cord. J Neurophysiol 84: 2651–2657 [DOI] [PubMed] [Google Scholar]
- Wizenmann A, Brunet I, Lam JSY, Sonnier L, Beurdeley M, Zarbalis K, Weisenhorn‐Vogt D, Weinl C, Dwivedy A, Joliot A et al (2009) Extracellular engrailed participates in the topographic guidance of retinal axons in vivo. Neuron 64: 355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Tang L, Zhang N, Pan L, Hadano S, Fan D (2015) Six SQSTM1 mutations in a Chinese amyotrophic lateral sclerosis cohort. Amyotroph Lateral Scler Frontotemporal Degener 16: 378–384 [DOI] [PubMed] [Google Scholar]
- Yilmaz R, Müller K, Brenner D, Volk AE, Borck G, Hermann A, Meitinger T, Strom TM, Danzer KM, Ludolph AC et al (2019) SQSTM1/p62 variants in 486 familial ALS patients from Germany and Sweden. Neurobiol Aging 87: 139.e9–139.e15 [DOI] [PubMed] [Google Scholar]
- Yoon BC, Jung H, Dwivedy A, O'Hare CM, Zivraj KH, Holt CE (2012) Local translation of extranuclear Lamin B promotes axon maintenance. Cell 148: 752–764 [DOI] [PMC free article] [PubMed] [Google Scholar]