

Abstract
The insulin receptor (IR) is a type II receptor tyrosine kinase (RTK) that plays essential roles in metabolism, growth, and proliferation. Dysregulation of IR signaling is linked to many human diseases, such as diabetes and cancers. A “resolution revolution” in cryo–electron microscopy (cryo-EM) has led to the determination of several structures of IR with different numbers of bound insulin molecules in recent years, which have tremendously improved our understanding of how IR is activated by insulin. Here, we review the insulin-induced activation mechanism of IR, including (1) the detailed binding modes and functions of site-1 and site-2 insulins, and (2) the insulin-induced structural transitions that are required for the IR activation. We highlight several other key aspects of the activation and regulation of IR signaling, and discuss the remaining gaps in our understanding of IR activation mechanism and potential avenues of future research.
Keywords: Insulin receptor, insulin, cryo-EM, activation mechanism, site-1, site-2
Introduction
The discovery of insulin in 1921 is one of the most significant medical breakthroughs that changed the world(1). In the past, children and adults who developed diabetes most often died within a few years, sometimes even within a few days or weeks of their diagnosis. Since Frederick Banting and Charles Best successfully isolated the hormone insulin, people suffering from diabetes have been treated with insulin, and diabetes is no longer considered fatal. Insulin was believed to play a role in the enzymatic process of glucose phosphorylation, for 30 years following its discovery. The concept of insulin action in facilitating glucose uptake into cells was proposed in 1950(2); however, how insulin initiates this process was unclear until the discovery of a membrane receptor for insulin, the insulin receptor (IR) in 1971(3). Kahn and colleagues discovered that insulin stimulates tyrosine phosphorylation of the IR by forming an IR/insulin complex (4, 5). IR cDNAs have been cloned in 1985(6, 7), and the concept of signal transduction by receptor tyrosine kinases (RTKs) has been established, making IR and aberrant RTK activation potential therapeutic targets.
In vertebrates, the IR belongs to the IR family that consists of three type II RTKs: IR(6, 7), insulin-like growth factor 1 receptor (IGF1R)(8), and IR-related receptor (IRR) (9). The IR is derived from a single polypeptide that is post-translationally cleaved into two subunits ❘ α-subunit and β-subunit, which are then cross-linked by multiple disulfide bridges to form a homodimer (α2β2)(10). The extracellular domains (ECD) of IR contain two leucine-rich-repeats (L1 and L2), a cysteine-rich region (CR), and three consecutive fibronectin type III (FnIII-1, −2, and −3) domains(11) (Figure 1a). The FnIII-2 domain contains a furin cleavage site. After cleavage by furin, the insulin proreceptor is converted into a mature receptor, consisting of α- and β-subunits(10). The C-terminal domain of α-subunit is referred to as α-CT motif, which is constitutively associated with the L1 domain. The remaining β-subunit passes through the cellular membrane via a single transmembrane (TM) domain that is linked to the intracellular domains (ICD) including juxtamembrane, tyrosine kinase and C-terminal domains. Different to all other RTKs, IR forms a stable dimer independent of ligand binding. The two protomers are covalently linked by multiple disulfide bonds between FnIII-1 domains and α-CTs(12, 13) (Figure 1a).
Figure 1.
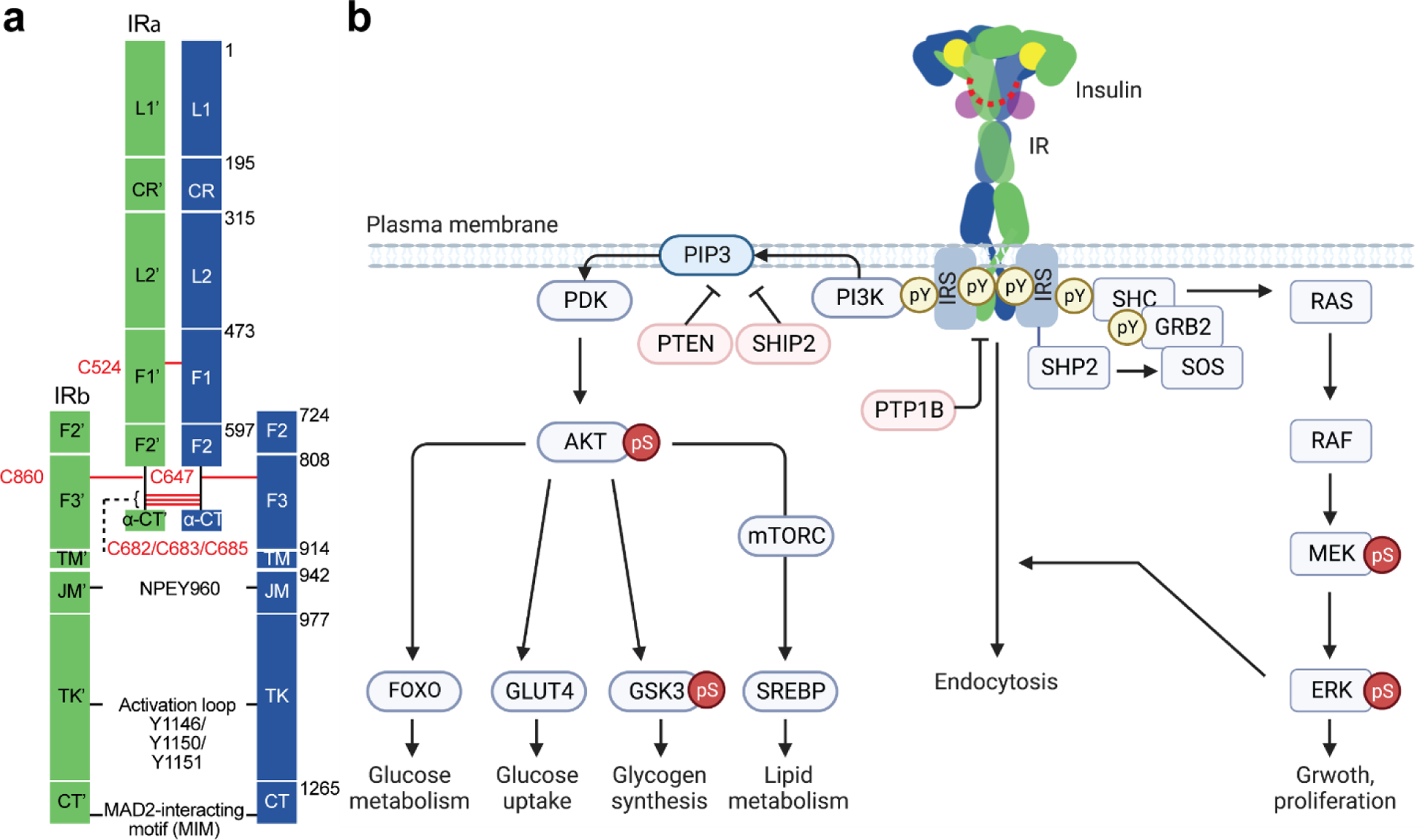
Structure and signaling of IR. (a) Schematic representation of the structure of the disulfide-linked IR. Each protomer is shown in green and blue. Disulfide bonds are shown in red. NPEY motif, MAD2-teracting motif (MIM), and tyrosine triplets in the activation loop are indicated. Leucine-rich-repeat (L1 and L2); cysteine-rich region (CR); fibronectin type III (F1, F2, and F3); C-terminal domain of α-subunit (α-CT); transmembrane (TM); juxtamembrane (JM); tyrosine kinase (TK); C-terminal region of β-subunit (CT). (b) Insulin-activated IR triggers two signaling cascades; PI3K-AKT pathway and MAPK pathway. The IR undergoes endocytosis, which redistributes and terminates the IR signaling. Protein nomenclatures are defined in the main text. Phosphor-Tyr (pY) and phosphor-Ser/Thr (pS). Arrows and blunt ends indicate activation and inhibition, respectively.
The mammalian IR exists two isoforms: isoform A (IR-A), lacking exon 11 and isoform B (IR-B), including exon 11(6, 7, 14). Exon 11 encodes 12 amino acids long segment at the α-CT. IR-A and IR-B bind to native insulin with similar affinity; however, the binding affinity of IR-A for IGF1 or IGF2 is significantly higher than that of IR-B(15), suggesting that the extension plays an important role in regulating the ligand binding process, and that IR-B is a more insulin-specific receptor. The molecular basis for the role of the 12 amino acids segments in the activation of insulin-bound or IGF-bound IR is still unclear.
Most mammalian cells express IR, but its expression levels are regulated depending on cell types, developmental stages, and disease states. The main target tissues of insulin’s metabolic action are liver, muscle, and adipose tissue(16–19). In liver, insulin-activated IR promotes glycogen and triglyceride synthesis, and inhibits glucose production(16, 18, 20). In muscle, insulin-activated IR promotes glucose uptake and glycogen synthesis, while in adipose tissue, insulin promotes glucose uptake and inhibits lipolysis(17, 20). Aside from controlling systemic metabolic homeostasis, IR in brain regulates cognitive behavior, food intake, and depression(21, 22). In highly proliferative T-cells, IR expression is increased, and loss of IR causes defects in proliferation and optimal immunity(23). Furthermore, IR plays a role in insulin clearance, while IR in endothelial cells transports insulin via transcytosis to brain, muscle and adipose tissue(24, 25). Because IR signaling affects all tissues in the body and controls diverse biological responses, IR signaling dysfunction leads to various diseases including diabetes, cancer, cardiovascular diseases, and even Alzheimer’s disease.
In this review, we mainly focus on the molecular basis of insulin-dependent IR activation and discuss how multiple insulin bindings to two distinct sites promotes optimal IR signaling. We also highlight key aspects of activation and regulation of IR signaling and provide insight into structure-function relationship of the IR. However, structural works on the IR ICD as well as its complex with downstream effector proteins will not be discussed here. For this topic, we refer the reader to references(26–30).
1. Insulin-induced IR signaling
Insulin activates two main signaling pathways (PI3K-AKT and MAPK) through IR on the plasma membrane by forming an IR-insulin high-affinity complex (16, 31–33) (Figure 1b). The binding of insulin to IR shows complex characteristics involving multiple binding events (i.e., insulin binds to two distinct types of sites on IR – site-1 and site-2) and negative cooperativity (34–36). Moreover, it has also been established that the binding of insulin to IR induces a large conformational change of IR that facilitates auto-phosphorylation and triggers downstream signaling cascades. The insulin-activated IR kinase trans auto-phosphorylates multiple tyrosine residues in the intracellular region: phospho-Y953 (pY953) and pY960 in the juxtamembrane domain, pY1146, pY1150, and pY1151 in the activation loop of the kinase domain, and pY1316 and pY1322 in the C-terminal domain(3–7, 37, 38) (note that we will use the numbering system of the mature form of human IR-A). The phosphorylated tyrosine residues provide docking sites for downstream effector and adaptor proteins, thus triggering phosphorylation-mediated signaling cascades(39) (Figure 1b).
The first discovered IR substrates (IRSs) are a family of adaptor proteins that convert the tyrosine phosphorylation signal into a lipid kinase signal through recruiting phosphoinositide 3-kinase (PI3K) to plasma membrane(40–45) (Figure 1b). In human, three homologous IRS proteins exist (IRS1, IRS2, and IRS4)(42). IRS1 and IRS2 are expressed widely, meanwhile the expression of IRS4 is tissue-specific. The activated IR auto-phosphorylates its motif named NPEY960 in the juxtamembrane domain, and IRS directly binds to the NPEpY960 motif. Subsequently, the IR phosphorylates multiple tyrosine residues on IRS, which in turn recruit SH2-domain containing downstream proteins including PI3K. The triphosphorylated inositol produced by PI3K activates the serine/threonine kinase AKT which initiates serine/threonine phosphorylation cascades(46, 47). The insulin dependent metabolic processes are largely governed by the PI3K-AKT pathway(48–52). More specifically, the PI3K-AKT pathway regulates glucose uptake by glucose transporter type 4 (GLUT4)(53–56), glycogen synthesis by glycogen synthase kinase 3 (GSK3)(57, 58), protein and lipid metabolism by mammalian target of rapamycin (mTOR)(59–61), and glucose metabolism by forkhead family box O (FOXO)(62–67).
Furthermore, the insulin-activated IR recruits and phosphorylates an adaptor protein❘ SH3-containing protein (SHC), which can interact with the SH2/SH3 adaptor protein GRB2 (growth factor receptor bound protein 2) and guanine nucleotide exchange factor SOS (son of sevenless)(68–71) (Figure 1b). The GRB2-SOS complex activates the mitogen-activated protein kinase (MAPK) pathway, which mainly controls cell growth and proliferation. IRS also recruit GRB2-SOS2 complex, contributing to the activation of the MAPK pathway as well. A non-receptor protein tyrosine phosphatase SHP2 facilitates the activation of MAPK pathway(72, 73). The complexities of IR signaling pathways are thoroughly reviewed. We hereby refer the reader to the references that provide more comprehensive summary of the IR signaling(16, 31, 33, 74).
2. The structure of unliganded apo-IR
The structure of the complete ECD of IR in the unliganded, apo-state was first determined at 3.8 Å resolution by X-ray crystallography in 2006 (75), and it was further improved to 3.3 Å resolution in 2016 (PDB: 4ZXB) (76) (Figure 2). These structures represent major breakthroughs in the structural biology of IR family receptors. The overall structure of IR in the unliganded, apo-state displays a ‘Λ’-shape (Figure 2). Two IR protomers, each of which adopts a ‘7’-shaped conformation, pack tightly together through the interaction between the L1 and L2 domains of one protomer and FnIII-2’ and FnIII-1’ domains of another. The L1 domain interacts with the FnIII-2’ domain in the middle part of the ‘Λ’, through highly complementary surfaces (Figure 2). The α-CT’ as well as the long linker between the α-CT’ and FnIII-2’ domain also contribute to this interaction. In the top region of the ‘Λ’, the L2 domain packs against the FnIII-1’ domain using a number of hydrophobic and polar residues, such as L403, H429, and Y430 (L2 domain) and L569, T571, and F572 (FnIII-1’ domain). At the same interface, the two L2 domains also make strong homotypic interactions mainly via hydrogen bonds. With such interaction patterns, the two membrane-proximal FnIII-3 domains are separated by a long distance (Figure 2), which prevents the intracellular kinase domains from undergoing efficient auto-phosphorylation, suggesting that such a ‘Λ’-shaped structure represents the auto-inhibited state of IR. It is, therefore, reasonable to imagine that a large conformational rearrangement, induced by insulin binding, is required for IR activation.
Figure 2.
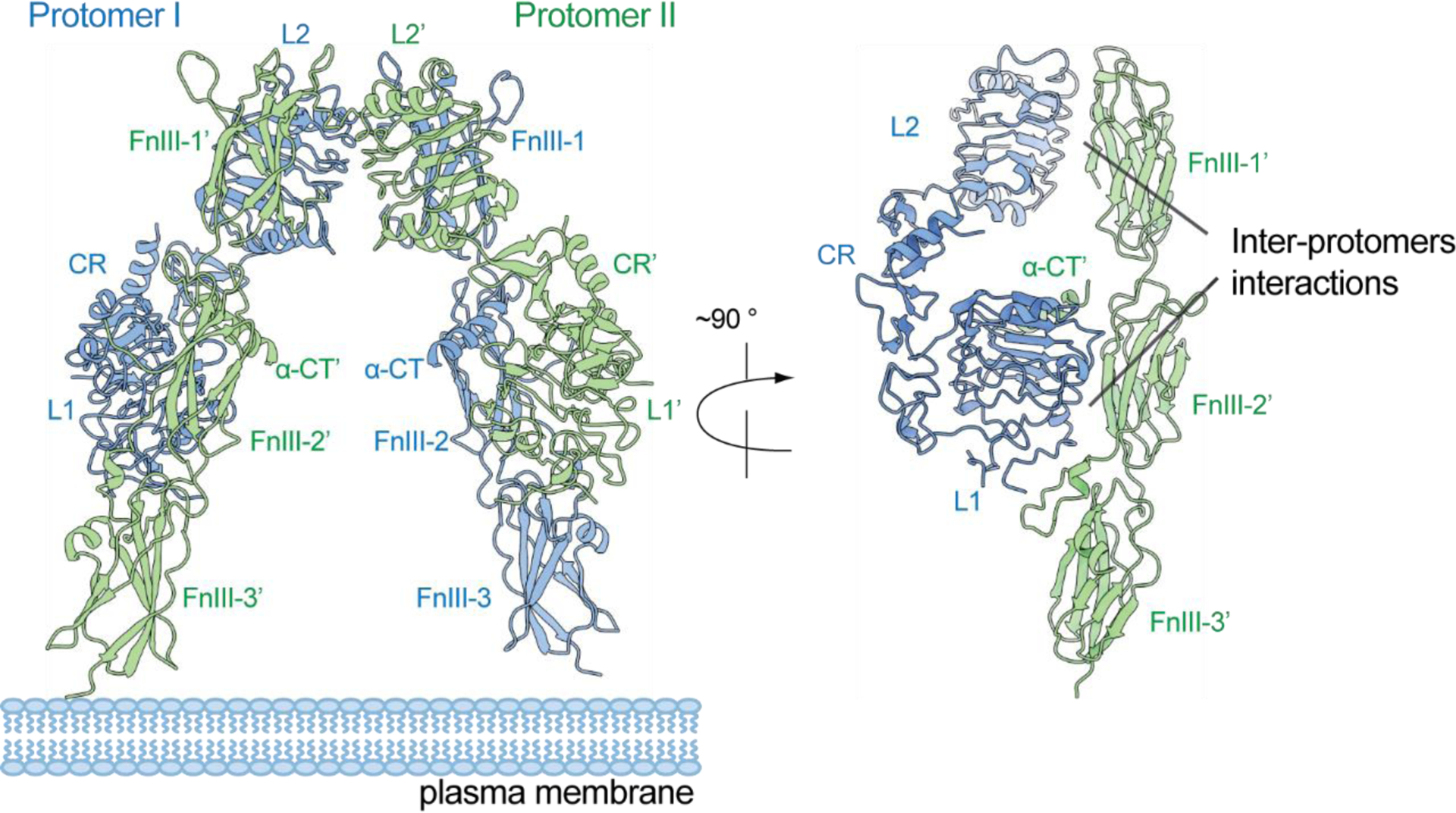
The X-ray crystal structure of IR ECD in the apo-, unliganded state, showing a ‘Λ’-shaped conformation (PDB: 4ZXB).
3. The structure of ligand-bound, active state of IR
In 2013, the first structure of insulin-bound IR was determined by X-ray crystallography, using a truncated ECD of IR which only contains the L1 and CR domains and a α-CT peptide, revealing how insulin engages site-1 of IR (PDB: 3W11) (77). Nevertheless, the crystallization of the insulin-bound intact IR-ECD or full-length IR is a challenge, due to the difficulties in large scale of protein purification and the nature of structural dynamics. In 2011, a combination of hardware and software advances led to the “resolution revolution” in single-particle cryo-electron microscopy (cryo-EM) (78). Sooner than later, cryo-EM has become the major tool for structural studies of single-pass transmembrane receptors, including IR, as it requires a much smaller amount of protein and can tolerate certain levels of impurity and structural heterogeneity (79). In 2018, two different groups determined the cryo-EM structures of the complete ECD of IR bound with insulin, at approximately 4 Å resolution(80, 81). Despite some differences in construct design, these two structures of the IR-ECD/insulin complex share similar structural characteristics. Specifically, upon insulin binding to IR site-1, the overall architecture of the IR changes from an auto-inhibited ‘Λ’-shape to a ‘T’-shape. The large structural change brings the two kinase domains of the IR into close proximity, allowing efficient auto-phosphorylation, which leads to the activation of IR signaling.
Although these two cryo-EM structures represented another major advance in the structural studies of IR, they were both resolved at relatively low resolution, suggesting the intrinsic instability of the truncated IR-ECD samples. In addition, none of them could uncover the second insulin binding site. As insulin binds to full-length IR more strongly than the truncated IR-ECD, structural studies of full-length IR in complex with insulin are essential for capturing a stable, fully liganded state of IR(82–84).
The cryo-EM structure of full-length IR in the presence of saturating insulin concentrations was determined at 3.2 Å resolution (1:4, IR: insulin; PDB: 6PXV) in 2019(85), and the structure of IR-ECD was determined at oversaturated insulin concentration at 4.3 Å resolution (1:28, IR: insulin; PDB: 6SOF) in 2020(86) (Figure 3a). To obtain sufficient full-length IR for cryo-EM analysis, several new strategies were applied during the expression and purification of full-length IR. These include (1) several mutations in the IR-ICD were introduced to reduce IR endocytosis(87); (2) a new affinity purification approach based on the strong interaction between T6SS effector Tse3 and its immunity protein Tsi3 was developed and applied(88). In the full-length IR/insulin complex, the dimerized TM domains of IR were resolved at the secondary structural level, showing a crossover at the N-terminus of the TM helices(85). This structural observation suggests that the TM–TM interaction plays a role in stabilizing the active conformation of IR, which explains in part why the cryo-EM structure of the full-length IR/insulin complex was resolved at a higher resolution than the IR-ECD/insulin complex. Strikingly, these structures reveal that IR in the presence of saturating insulin concentration adopts a ‘T’-shaped symmetric conformation, with four insulins bound at four sites (named sites-1,−1’, −2 and −2’) that are related by a C2 symmetry (Figure 3a). More importantly, for the first time, the binding mode between IR and site-2 insulin has been revealed in atomic detail, representing another major milestone in the structural biology of IR. In the next section, we will describe in detail the binding mode and the function of the binding at site-1 and site-2.
Figure 3.
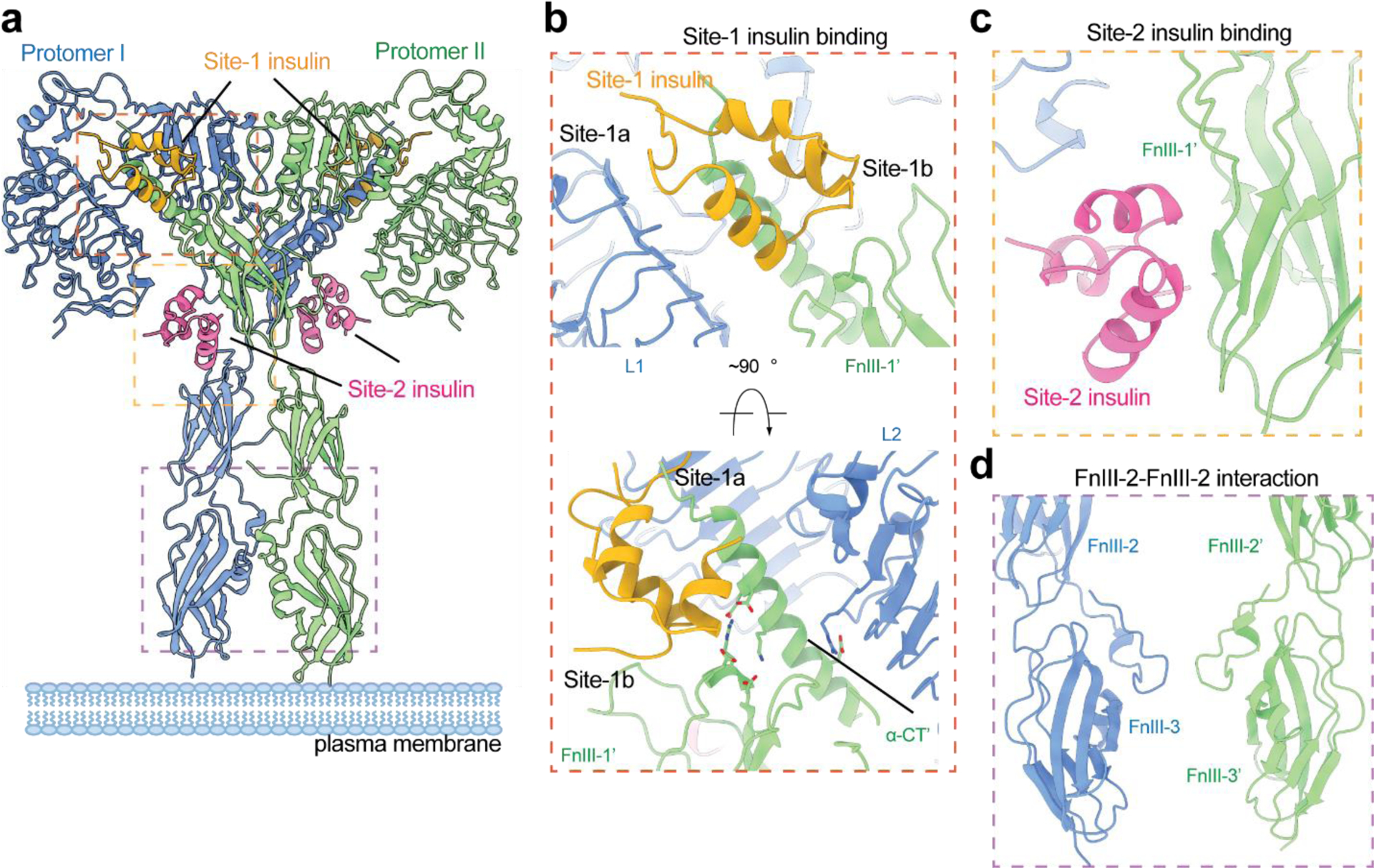
The cryo-EM structure of full-length IR in the active, fully liganded state, showing a ‘T’-shaped symmetric conformation (PDB: 6PXV). (a) ‘T’-shaped IR dimer bound with 4 insulin at site-1, site-1’, site-2, and site-2’. (b) The detailed binding mode of site-1 insulin in the ‘T’-shaped IR. The site-1 insulin crosslinks the L1 of one protomer and α-CT’ and FnIII-1’ of another. The α-CT’ contacts both L2 and FnIII-1’ domains in the ‘T’-shaped IR. (c) The detailed binding mode of site-2 insulin in the ‘T’-shaped IR. Site-2 insulin interacts with a side surface of FnIII-1 domain. (d) The homotypic FnIII-2-FnIII-2’ interaction in the ‘T’-shaped IR.
4. Insulin-binding site-1
The primary insulin-binding site of IR is composed of the L1 domain and the α-CT (namely site-1a) (Figure 3b). Structural comparison between the insulin-free and insulin-bound L1/α-CTs reveals that insulin binding to this site induces a remarkable relocation of the α-CT in relative to the L1 domain, suggesting that certain level of flexibility of α-CT is required for the insulin binding. Strikingly, 24 of the 51 insulin residues are involved in binding to the site-1a(77, 85). The residues from the A chain of insulin predominantly contact the α-CT, while the residues from the B chain of insulin interact with both the L1 domain and α-CT, through a combination of Van der Waals and hydrogen bonding interactions (Figure 3b). As many other review papers have described the insulin binding to site-1a in detail(89, 90), we will not go into detail here. In addition to this primary interface, the site-1a-bound insulin in the ‘T’-shaped IR form simultaneous contacts with the loops in the top region of FnIII-1’ domain from the adjacent protomer (namely site-1b)(85) (Figure 3b). Several residues from the B chain of insulin, including His-B5, Ser-B9, and His-B10, contribute to this interaction. With such binding mode, one insulin concurrently engages two protomers, thereby stabilizing the ‘T’-shaped active conformation of IR (Figure 3b).
5. Insulin-binding site-2
The high-resolution cryo-EM structure of full-length IR/insulin in the fully-liganded state identified the location of insulin-binding site-2 in IR and revealed the detailed binding mode of insulin at site-2(85). The newly discovered site is located at the side surface of a β-sheet in the FnIII-1 domain of IR (Figure 3c). A total of 14 residues from both A and B chains of insulin, such as Leu-A13, Glu-A17, His-B10, Glu-B13, and Leu-B17, are critical for the insulin binding to site-2. As the site-2 insulin only contacts one surface of the ‘T’-shaped IR, it is non-essential for maintaining the active conformation (Figure 3c). Nevertheless, the significance of insulin-binding at site-2 in IR activation has been demonstrated by mutagenesis, binding and cell-based assays(85, 91). The following structural studies have revealed the function of insulin binding at site-2 and will be discussed in the section 9.
6. Receptor-receptor interfaces for maintaining the ‘T’-shaped active IR dimer
The cryo-EM structure of the full-length IR/insulin complex also revealed several receptor-receptor interfaces critical to the structural stability of the ‘T’-shaped IR dimer(85). Firstly, in the top-part of the ‘T’, each α-CT adopts a long α-helical conformation and contacts both the FnIII-1 domain of the same protomer and the L2’ domain of another (Figure 3b). These interactions exclusively exist in the ‘T’-shaped IR dimer and are critical for maintaining the active conformation. These interfaces involve a cluster of charged residues, including E697, K703, E706 and D707 (α-CT); D496, R498 and D499 (FnIII-1 domain), and R345 (L2 domain), forming multiple salt bridges (Figure 3b). Secondly, in the bottom-part of the ‘T’, the two FnIII-2 domains form a homotypic interaction, further stabilizing the membrane-proximal domains of IR (Figure 3d). The FnIII-2-FnIII-2’ interaction may also facilitate dimerization of the TM domains, which place the two intracellular kinase domains in optimal relative positions for auto-phosphorylation.
7. Structures of IR at sub-saturating insulin concentration
In two recent studies, cryo-EM structures of a series of partially liganded full-length IR with one or two insulins bound were determined, using the insulin site-specific mutants or sub-saturating insulin concentrations(92, 93) (Figure 4a, b). The structure of one insulin bound full-length IR assumes a ‘Γ’-shaped conformation, which is almost identical to that of a single-IGF1 bound full-length IGF1R(94) (Figure 4a). This single insulin is bound at one of the site-1s in the top-part of the ‘Γ’, forming extensive interactions with L1’/α-CT and FnIII-1 domains. In the bottom-part of the ‘Γ’, the unliganded L1 domain contacts both membrane-proximal domains, which further stabilizes the ‘Γ’-shaped conformation. This structural configuration places both intracellular kinase domains close together, enabling auto-phosphorylation to occur. Thus, the ‘Γ’-shaped IR dimer represents an (partially) active state of IR (Figure 4a).
Figure 4.
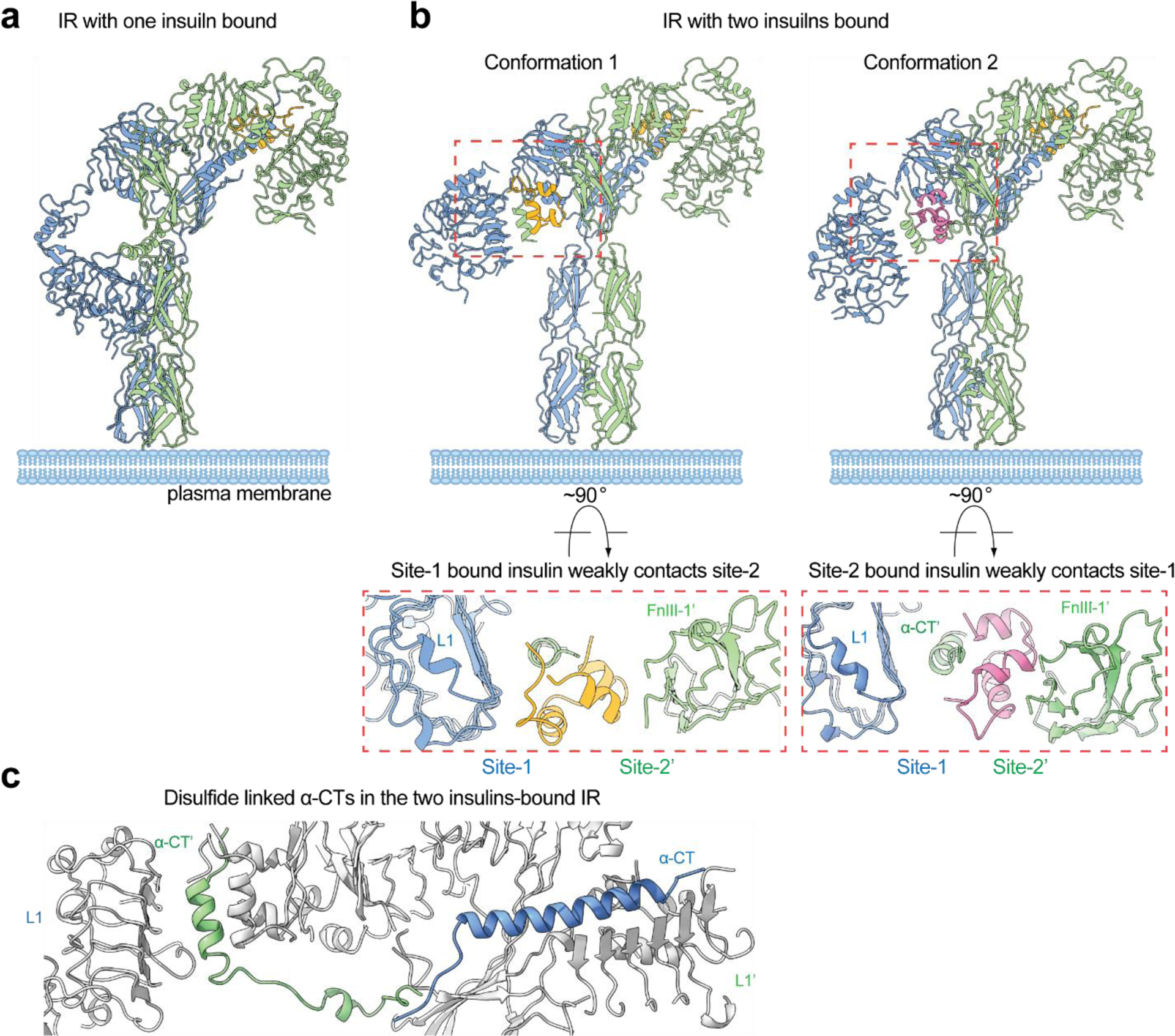
The cryo-EM structures of full-length IR bound with sub-saturated insulins. (a) The cryo-EM structure of IR in one insulin-bound state, showing a ‘Γ’-shape. (b) The cryo-EM structure of IR in two insulins-bound state, showing two different types of ‘Ƭ’-shape. In the middle region of conformation 1, site-1-bound insulin also weakly contacts site-2. In the middle region of conformation 2, site-2-bound insulin also weakly contacts site-1. (c) The disulfide-linked α-CTs adopt stretched conformation in the ‘Ƭ’-shaped IR with two insulins bound.
Strikingly, in response to the binding of two insulins, IR predominantly adopts a ‘Ƭ’-shaped asymmetric conformation (~80% of particles) (Figure 4b), while only a small population of IR forms a ‘T’-shaped symmetric dimer (~20% of particles)(92, 93). The conformation of the ‘T’-shaped IR with two insulins bound at site-1s is almost identical to that of 2:4 IR/insulin complex. In the ‘Ƭ’-shaped asymmetric IR, one insulin is bound at site-1 in the top-part of the ‘Ƭ’ in the same fashion as the site-1 insulin binding in the ‘T’-shaped symmetric IR, meanwhile another insulin is bound in the middle region of the ‘Ƭ’, simultaneously contacting sites-1 and −2’ from two adjacent protomers (named hybrid site) (Figure 4b). Intriguingly, the cryo-EM analyses of the full-length IR show that the hybrid site can be crosslinked and stabilized by one insulin in two different ways(92) (Figure 4b): (1) insulin binds primarily to site-1 and contacts a side surface of FnIII-1’ (site-2’) at the same time (Conformation 1); (2) The site-1 bound insulin rotates approximately 60° around the α-CT. As a result, this insulin binds at site-2’, while also contacting the α-CT of the adjacent site-1 (Conformation 2).
8. The function of site-1 insulin binding
8.1. Break the auto-inhibited apo-state of IR
The ‘Λ’-shaped IR dimer in the absence of insulin represents a stable, auto-inhibited state of IR. It is, therefore, conceivable that insulin binding is required to destabilize the auto-inhibited conformation of IR. Indeed, the superimposition of the L1/α-CT’ (site-1a)-bound insulin onto the structure of apo-IR reveals that the insulin at site-1a of apo-IR sterically clashes with the linker between the FnIII-1’ and FnIII-2’ domains from the adjacent protomer(85) (Figure 5a). Thus, insulin binding to site-1a of apo-IR will push the FnIII-2’ domain away from the L1 domain, thereby partially disrupting the protomer-protomer interactions that are responsible for the auto-inhibition of IR. The relaxed apo-IR dimer would allow structural rearrangement between the two protomers, leading to a ‘T’-shaped active conformation. Similar mechanism has been proposed in the IGF1-induced activation of IGF1R (95).
Figure 5.
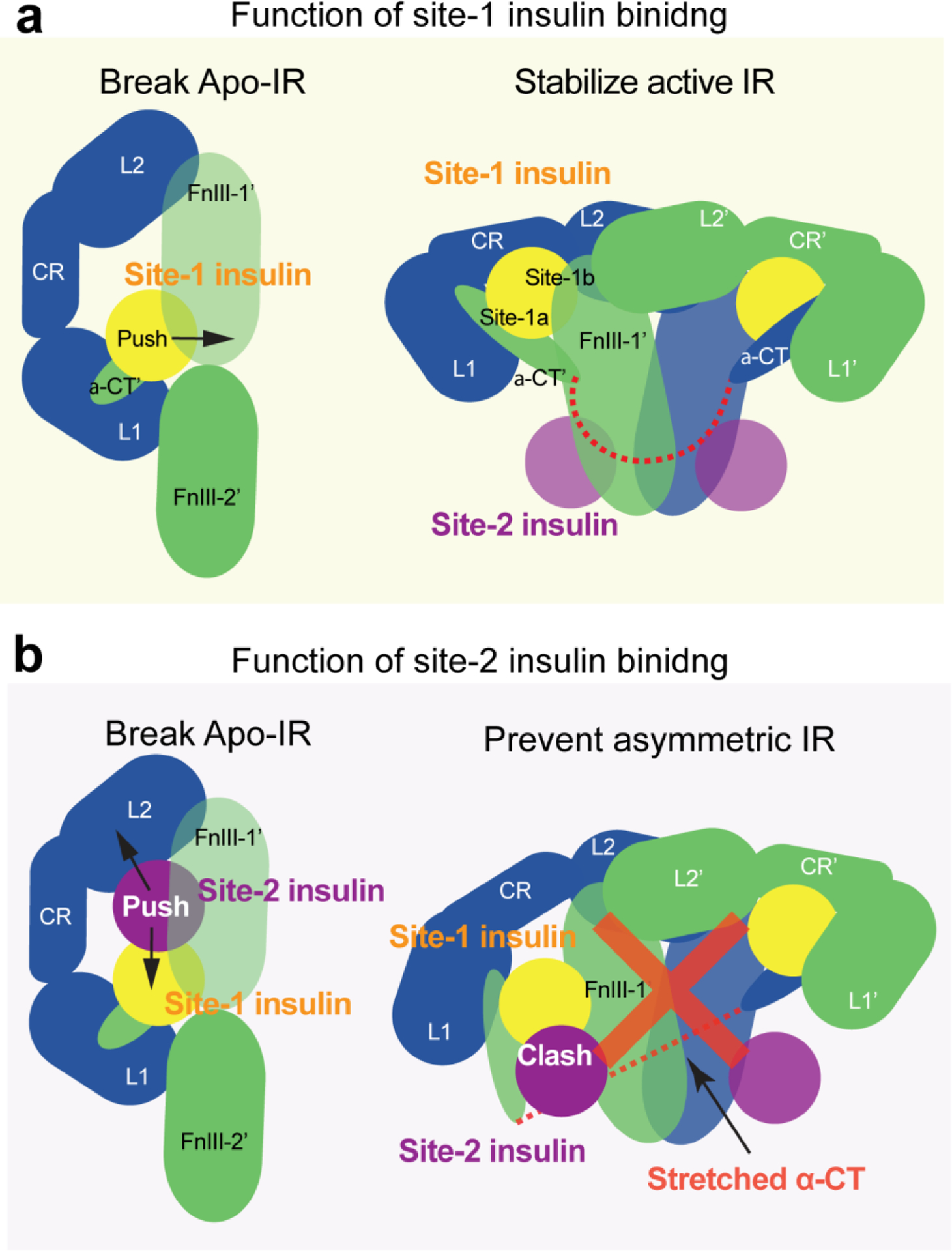
The functions of sites-1 and −2 insulin binding. (a) Site-1 insulin plays a critical role in insulin-induced IR activation by breaking the auto-inhibited conformation of IR and by stabilizing the active conformation. (b) Site-2 insulin plays an auxiliary role in insulin-induced IR activation by assisting site-1 insulin to break the auto-inhibited conformation of IR and by preventing the formation of asymmetric IR.
8.2. Stabilize the active state of IR
The structural comparison between the ‘Λ’-shaped (apo state) and ‘T’-shaped (active state) IR indicates that, once site-1 bound insulin disrupts the auto-inhibited IR, receptor activation is achieved by dramatic conformational changes that include both inter-protomer rotation and intra-protomer hinge motion. In the ‘T’-shaped active IR dimer, insulins at site-1 stabilize the active conformation by simultaneously contacting several domains between the two protomers, including the L1 of one protomer, and α-CT’ and FnIII-1’ of another (Figure 5a). Thus, site-1 insulins tightly “crosslink” the two protomers in the ‘T’-shaped IR.
9. The function of site-2 insulin binding
9.1. Assist site-1 insulin for breaking the auto-inhibited IR
Insulin binding to site-1a of apo-IR separates the L1 domain of one protomer from the FnIII-2’ domain of the another in the middle-part of the ‘Λ’-shaped IR dimer. Nevertheless, due to the long inter-domain linkers in each protomer, the interactions between the L2 and FnIII-1’ domains in the top-part of the ‘Λ’ remain intact, even with insulin bound at site-1a. This suggests that site-1 insulins may not be able to completely disrupt the auto-inhibited IR. The cryo-EM analysis of full-length IR with insulin bound only at site-2 demonstrates that insulin can bind to site-2 in the apo-IR without changing the conformation of IR(92). In addition, the two insulins bound at sites-1a and −2 in one-half of the apo-IR would clash(85) (Figure 5b). Such steric clash may push the site-2 insulin and its bound FnIII-1’ domain away from the L2 domain of adjacent protomer, thereby further fracturing the two protomers (Figure 5b). Thus, it is likely that the sites-1 and −2 insulins cooperatively disrupt the auto-inhibited state of IR.
9.2. Prevent the formation of asymmetric conformations of IR
IR predominantly forms asymmetric conformations when two insulins are bound at the IR site-1s (Figure 4b), in contrast to the ‘T’-shaped symmetric conformation induced by the binding of four insulins to both sites-1 and −2(92, 93). This structural observation suggests that insulin binding to site-2 prevents the formation of asymmetric conformations of IR. In other words, the insulin binding to site-2 is necessary to overcome the energetic hurdle associated with the asymmetric state of IR, allowing IR to reach a more stable, symmetric ‘T’-shape.
The cryo-EM results on the full-length IR obtained at sub-saturating insulin concentrations provide a structural explanation for why IR cannot form an asymmetric conformation when four insulins are bound to both sites-1 and −2(92). In the middle-part of the ‘Ƭ’-shaped asymmetric IR with two insulins at site-1s, one insulin primarily binds to site-1 but also weakly contacts site-2’ (hybrid site) (Figure 4b). The binding of another insulin to the site-2’ within the hybrid site would require an outward movement of the lower L1/α-CT domain to prevent steric clashes between the two insulins. However, the two disulfide-linked α-CTs in the ‘Ƭ’-shaped IR adopt a kinked and extended conformation (Figure 4c). Such stretched α-CT restricts the outward movement of the lower L1 domain, preventing the binding of another insulin to the hybrid site in the ‘Ƭ’-shaped IR (Figure 5b). Given these structural observations, we propose that, upon the binding of four insulins to both sites-1 and −2, the L1/α-CT’ domain together with bound insulin (i.e. site-1a) must move upward, ultimately reaching the top loop of the FnIII-1’ domain (i.e., site-1b), to prevent both (1) the clash with the site-2-bound insulin and (2) the overstretching of the two disulfide-linked α-CTs. As a result, the ‘T’-shaped symmetric dimer would be exclusively formed. Collectively, these structural results and analyses explain how the binding of insulin to site-2 prevents the formation of asymmetric IR as well as why only the ‘T’-shaped IR has been observed in cryo-EM datasets of IR at saturating insulin concentrations(85, 92).
Furthermore, a recent study using site-specific insulin mutants supports the functional significance of site-2 insulin binding(92): (1) Insulin mutants that bind only to IR site-2 (insulin site-1 mutants) fail to disrupt the apo-IR. Conversely, insulin mutants that bind only to IR site-1 (insulin site-2 mutants) break the apo-IR, but most of the insulin site-2 mutants-bound IR forms asymmetric conformations. (2) Insulin site-2 mutants displayed greatly reduced potency for triggering IR auto-phosphorylation as compared with native insulin. (3) Neither insulin site-1 nor site-2 mutants alone reduced blood glucose levels in mice. However, cotreatment with 1:1 stoichiometry of the insulin site-1 and site-2 mutants activated IR signaling and lowered blood glucose levels in mice. (4) Consistent with the functional results, the structure of IR with both insulin site-1 and site-2 mutants bound exhibits a symmetric ‘T’-shape, identical to the ‘T’-shaped IR with four native insulins. These structural and functional data together suggest that site-2 insulin binding plays a critical role in regulating the conformation of IR, i.e., converting IR from asymmetric to symmetric, and clearly demonstrates that the binding of multiple insulins to both site-1 and site-2 facilitates optimal IR signaling by promoting ‘T’-shaped symmetric conformation.
10. Insulin-dependent IR activation
10.1. Proposed activation mechanism of IR in response to one insulin binding
In principle, insulin should prefer to bind to IR site-1, as site-1 has a more than 10-fold higher affinity for insulin than site-2. The binding of one insulin to one of the two site-1s of apo-IR breaks one of the two L1-FnIII-2’ interactions, which is critical for the maintenance of the apo-IR, thus partially disrupting the auto-inhibited conformation, similar as observed in the IGF1R activation induced by IGF1 (Figure 6a). The released L1’/α-CT domains (site-1a), along with bound insulin, undergo a hinge motion and move upward, making interactions with the top loop of FnIII-1 domain (site-1b) from the neighboring protomer, forming the top-part of the ‘Γ’-shaped IR dimer (Figure 6a). The released FnIII-1−3 domains of another protomer swing toward to the neighboring leg and make close contact with the L1 domain of the same protomer in the bottom-part of the ‘Γ’ (Figure 6a). This swing motion of FnIII-1−3 reduces the distance between the two membrane-proximal domains, thus facilitating the auto-phosphorylation. The L1-FnIII-2’ interaction in the unliganded half of the IR remains unchanged in comparison to the apo-IR. In addition, the two covalently linked α-CTs form a rigid beam-like structure, further contributing to the structural stability of one insulin-bound ‘Γ’-shaped asymmetric IR(92).
Figure 6.
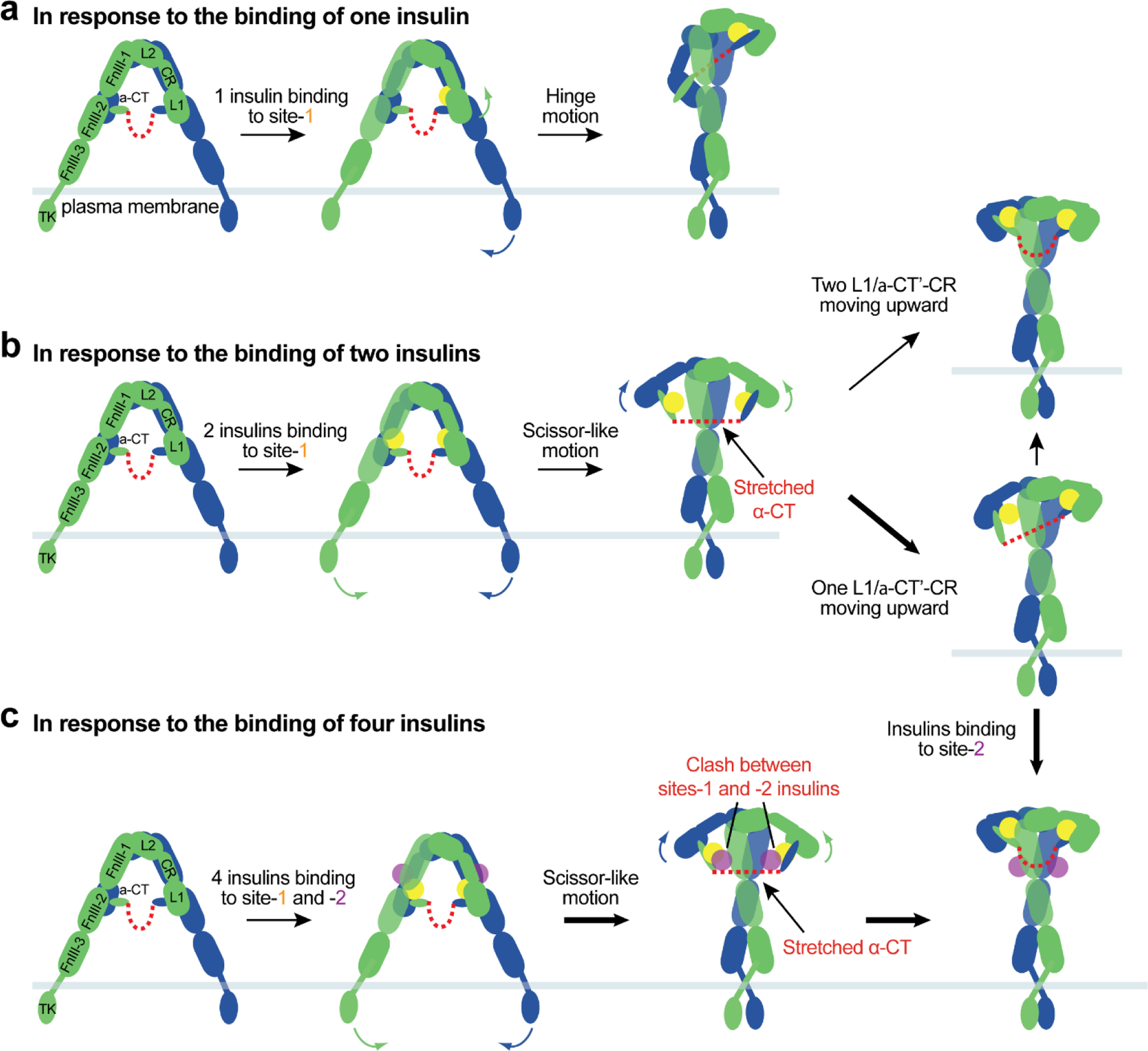
The proposed model of insulin-induced IR activation. At unsaturated insulin concentrations, one or two insulins bind to IR site-1(s) and disrupt the auto-inhibited conformation. (a) The released L1/α-CT’ domains with a site-1a insulin move upward to the top-loop of FnIII-1’ (site-1b), crosslinking two protomers. (b) Two insulins binding at both IR site-1s facilitates a scissor-like rotation of two protomers. We speculate that these conformational changes lead to an instable, intermediate state of IR. We further propose that hinge motions between the L1/α-CT’-CR-L2 domains within one or two protomers reduce the structural instability, resulting in ‘Ƭ’-shaped asymmetric or ‘T’-shaped symmetric IR dimers. (c) At saturating insulin concentrations, four insulins bind to IR sites-1 and −2, breaking the auto-inhibited conformation effectively and promoting a scissor-like rotation of two protomers. The collision between two insulins at sites-1 and −2 in combination with the tension generated by disulfide-linked α-CTs would promote the formation of the fully active, ‘T’-shaped IR.
10.2. Proposed activation mechanism of IR by two insulins binding only to site-1s
IR with two insulins bound at site-1s predominately assumes an asymmetric conformation(92). In this asymmetric IR, protomer I has a ‘7’-shaped conformation almost identical to that in the ‘Λ’-shaped IR (Figure 7a, b); while protomer II retains a similar ‘leg’ to that of protomer I with a different conformation of the ‘head’ part – extended versus compacted. This structural analysis hints that, after the apo-IR is disrupted by the insulin binding to two site-1s, a certain degree of IR activation could be achieved by a scissor-like rotation between the two protomers as rigid-bodies, using the interface between L2 and FnIII-1’ domains as a pivot point (Figure 6b). In principle, this would result in an extended ‘T’-shaped IR with membrane proximal stalks placed in close proximity. During the structural transition, however, the two L1 domains and disulfide-linked α-CTs would be separated by a long distance. As a consequence, the two disulfide-linked α-CTs would adopt a straightened and stretched conformation (similar to the game of tug-of-war), which may be energetically unfavorable (Figure 6b). This indicates that the extended ‘T’-shaped IR must be unstable, and may represent a transition state during IR activation.
Figure 7.
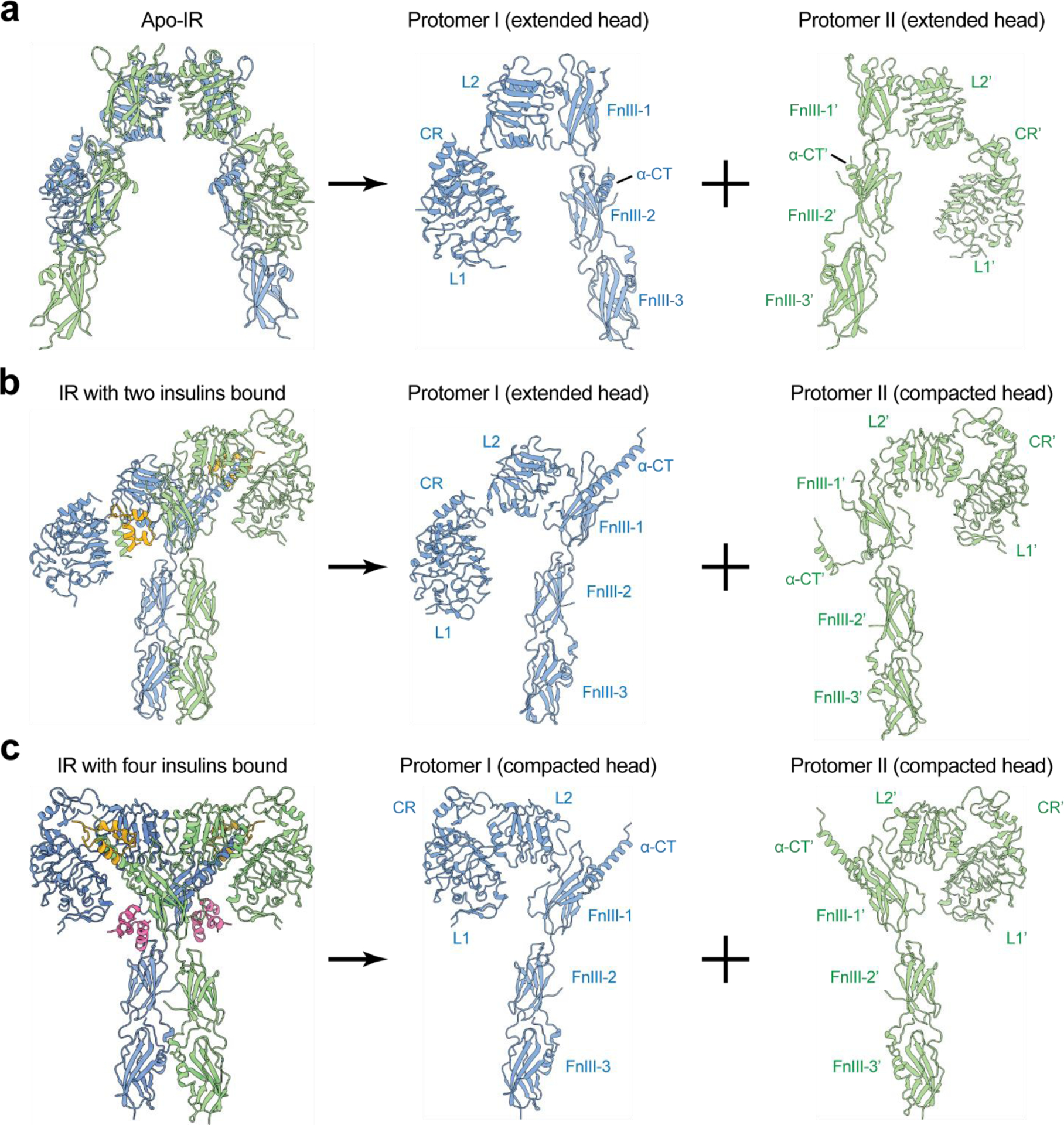
The protomer conformations of IR with different numbers of insulin bound. Two different types of IR protomer conformations exist – compacted and extended. (a) The apo-IR consists of two extended protomers. (b) The ‘Ƭ’-shaped IR with two insulins bound consists of one extended protomer and one compacted protomer. (c) The ‘T’-shaped IR with four insulins bound consists of two compacted protomers.
We speculate that the structural instability of extended ‘T’-shaped IR would be reduced through spontaneous hinge motions of the L1/α-CT’-CR-L2 domains within one or two protomers, which convert one or two protomers from an extended to a compacted conformation, leading to ‘Ƭ’-shaped asymmetric or ‘T’-shaped symmetric architecture. (Figure 6b). Indeed, these two distinct IR conformations were observed in cryo-EM data of full-length IR obtained at sub-saturating insulin concentrations(92) (Figure 4b). In the ‘Ƭ’- or compacted ‘T’-shaped IR, the distance between the two L1 domains was reduced, allowing the disulfide-linked α-CTs to bridge them more facilely. On the other hand, it is reasonable to imagine that the straightened disulfide-linked α-CTs in the extended ‘T’-shaped IR would act as a stretched spring that generates large contraction forces to promote the conformational rearrangement of IR from extended ‘T’-shape to ‘Ƭ’- or compacted ‘T’-shape (Figure 6b).
Two different types of site-1 insulin bindings in the top and middle regions of ‘Ƭ’ stabilize the ‘Ƭ’-shaped asymmetric IR (Figure 4b). However, the ‘Ƭ’-shaped IR is not likely to be very stable, as insulin bound at site-1 of one protomer in the middle part of the complex makes only weak contact with the site-2’ of another protomer. Thus, the ‘Ƭ’-shaped asymmetric IR may represent a local energy minimum and a partially active state. Consistently, IR exhibits lower activity in response to insulin mutants that can only bind to IR site-1(92).
10.3. Proposed activation mechanism of IR by four insulins binding to both sites-1 and −2
The surface of site-2 insulin binding in the apo-IR is exposed. Furthermore, the cryo-EM analysis of IR with insulin bound only at site-2 demonstrates that insulin can bind to site-2 in the apo-IR without altering its overall conformation(92). It is, therefore, tempting to speculate that four insulins are able to bind to all the sites (i.e. two site-1s and two site-2s) at saturating insulin concentrations (Figure 6c). Four insulins binding to two types of sites in apo-IR could more effectively disrupt the auto-inhibited conformation and trigger a scissor-like rotation between two rigid protomers, leading to an extended ‘T’-shaped conformation (Figure 6c). However, the extended ‘T’-shaped IR is unstable for the following reasons: (1) the disulfide-linked α-CTs would adopt an extended and stretched conformation, due to the long distance between the two L1 domains, and (2) insulins bound at sites-1 and −2’ are most likely to collide with each other on both sides of the extended ‘T’, owing to the limited space between the sites-1 and −2’ from two adjacent protomers, similar to that shown in the hybrid site of the ‘Ƭ’-shaped IR. Therefore, both the ‘tension’ generated by the over-stretched disulfide-linked α-CTs and the ‘repulsion’ force generated by the collision between two insulins at sites-1 and −2’ would trigger the rapid kinking movements of L1/α-CT’-CR-L2 domains in both protomers (Figure 6c). As a result, the two L1/α-CT’ domains (site-1a) together with bound insulins would move upward and ultimately touch the top-surface of FnIII-1’ domain (site-1b); consequently, the ‘head’ parts of two protomers would adopt a compacted conformation, and the IR is transformed from an extended ‘T’- to a compacted ‘T’-shaped conformation (Figures 6c and 7c). The ‘T’-shaped symmetric conformation was exclusively observed in the cryo-EM dataset of full-length IR obtained at saturating insulin concentration, supporting this activation model. Moreover, recent cryo-EM studies demonstrate that IR with physically decoupled α-CT predominantly adopts asymmetric conformations even at saturating insulin concentrations(96). This finding further supports the proposed activation model that both disulfide-linked α-CTs and insulin binding at two distinct sites are critical for the efficient formation of the fully active, ‘T’-shaped IR.
The ‘T’-shaped IR dimer is structurally stable due to the concurrent binding of insulin to both sites-1a and −1b, as well as extensive protomer-protomer interactions. In addition, the disulfide-linked α-CTs in this compacted ‘T’-shape assume a relaxed conformation due to the short distance between their two L1 domains. Collectively, these structural observations suggest that the ‘T’-shaped symmetric IR conformation has the highest structural stability, which may represent the most optimal state for IR activation.
11. The sources of negative cooperativity
Insulin binds to IR with a complex kinetics characterized by a curvilinear Scatchard plot, suggesting a negative cooperativity in the binding of insulin to IR(35). Recent cryo-EM studies of the full-length IR bound with different numbers of insulins provide a structural basis for understanding the source of negative cooperativity(92).
Structural plasticity of α-CT in IR is required for insulin binding. In the apo-IR, only the N-terminal part of α-CT is folded as a short α-helix, whereas the rest of this motif is disordered(76). This partially folded α-CT without any constraints in both N- and C-termini allows it to undergo the conformational change which is necessary for insulin binding. In the ‘Γ’-shaped IR dimer (with only one insulin bound at site-1), the disulfide-linked α-CTs adopt a rigid and elongated conformation(92) (Figure 4a). In this structural configuration, the conformational plasticity of the unliganded α-CT is restricted. Thus, the unliganded α-CT in the asymmetric IR dimer is likely to be less capable of binding insulin, suggesting a negative cooperativity between the two sites-1s. A similar molecular mechanism underlying the negative cooperativity in the binding of IGF1 to IGF1R has been proposed before(94). Furthermore, in the middle region of the ‘Ƭ’-shaped IR (with two insulins bound), sites-1 and −2’ from two neighbor protomers are located in close proximity, and largely overlap (Figure 4b). Therefore, the binding of insulin at site-1 will hinder the binding of another insulin to site-2’, or conversely, thereby underlying the potential negative cooperativity between sites-1 and −2’ in the ‘Ƭ’-shaped IR.
12. The functional importance of α-CT
The cryo-EM structure of insulin bound full-length IR-3CS (cysteine triplets in α-CT substituted with serine, C682S/C683S/C685S) demonstrated that the IR with non-covalently linked α-CTs forms predominantly asymmetric conformations, despite insulins occupying all four binding sites(96). In one-half of these asymmetric conformations, insulins bind at site-1 and site-2’, similar to the ‘T’-shaped IR, while in the other half, two insulins bind to the hybrid site without stretching the α-CTs. The fact that fully liganded IR with physically decoupled α-CTs adopts asymmetric conformations supports the idea that disulfide-linked a-CTs plays a critical role in promoting the ‘T’-shaped symmetric IR. In addition, the levels of phosphorylation of AKT and ERK were markedly reduced in cells expressing IR-3CS, further suggesting that disulfide linkages between two α-CTs are essential for the formation of the ‘T’-shaped IR dimer and the optimal IR function(96).
13. Comparing the activation mechanisms of IR and IGF1R
Despite the fact that IR and IGF1R share high structural similarities, the characteristics of ligand binding are different between these two receptors(97). Recent cryo-EM studies have demonstrated that the activation mechanism of IR differs significantly from that of the IGF1R. In contrast to the maximum 2:4 stoichiometry of active IR/insulin complex, IGF1 is undetectable in the side surface of FnIII-1 in IGF1R (that is equivalent to the site-2 of IR), and only one IGF1 binding to the IGF1R is sufficient to fully activate the receptor(94).
Additionally, in the fully liganded, active state of IR, the stalks of the dimer that contain FnIII-2 and FnIII-3 domains are closely associated by homotypic interactions between two loops in FnIII-2 domain(85). In the fully active state of IGF1R, however, the unliganded L1 domain bridges the two stalks of the dimer into close proximity and further stabilizes the active conformation(94). It is possible that the different arrangements of membrane-proximal stalk regions between IR and IGF1R may provide a mechanism for defining their signaling specificity, allowing these two closely related receptors to generate distinct signaling outcomes.
Notably, to facilitate timely metabolic activity, the circulating levels of insulin are rapidly fluctuating in response to food intake and excise. In contrast, the IGF1 levels remain unchanged during the daytime and are responsible for long-term actions such as cell growth and differentiation(98–100). As IR has different insulin binding occupancy under different blood insulin concentrations, such a unique multi-sites system will allow IR to respond to a wide range of insulin concentrations at different metabolic states. In contrast, the one-site system in IGF1R will allow it to response to the IGF1 binding with high sensitivity. In part, this explains why these two related receptors use remarkably different mechanisms for activation.
14. Negative regulators of IR signaling
As insulin has multiple functions and is crucial to systemic homeostasis, the IR signaling should be tightly fine-tuned. Insulin itself contributes to the termination of the IR signaling by promoting IR endocytosis, and groups of phosphatases suppress the action of insulin in multiple steps. In addition, the levels of IR on the cell surface are regulated by ubiquitin-mediated degradation. These mechanisms are largely redundant, and vary depending on the tissue, developmental stage, and health condition.
14.1. IR endocytosis
Insulin-activated IR undergoes clathrin- or caveolae-mediated endocytosis, which controls spatiotemporal IR signaling, insulin clearance, and insulin delivery (25, 74, 101–104). The IR auto-phosphorylation and downstream signaling proteins control the clathrin-mediated IR endocytosis, which allows activated IR to be preferentially internalized(38, 87, 105–107). The internalized IR undergoes lysosomal degradation or recycles back to the plasma membrane. Thus, persistent hyperinsulinemia, which is closely related to insulin resistance, promotes IR endocytosis, and inhibits IR signaling. Consistent with this idea, IR levels on the cell surface are reduced in the livers of type 2 diabetes patients(108, 109) and insulin resistant mice(110). The assembly polypeptide 2 (AP2) links IR to clathrin, thus promoting the IR endocytosis. The SHP2-MAPK axis controls the phosphorylation of IRS, thus facilitating the association of IRS-AP2 with IR(74, 108). In addition, the cell division regulators, mitotic arrest deficient 2 (MAD2), budding uninhibited by benzimidazole-related 1 (BUBR1), cell division cycle protein 20 homolog (CDC20), and p31comet directly control the association of the AP2 with IR, thus regulating IR endocytosis(87, 102).
14.2. Ubiquitination
Several E3 ubiquitin ligases negatively regulate IR signaling by targeting IR and IRS. The canonical model of IR ubiquitination is that E3 ubiquitin-protein ligases (e.g. NEDD4 and CBL) ubiquitinate IR, facilitating the IR endocytosis and endosomal trafficking, thereby downregulating IR signaling(101). In addition to the canonical model, SOCS family proteins also inhibit IR signaling by recruiting E3 ubiquitin ligase to IR or IRS and promote proteasomal degradation(111). Moreover, Mitsugumin 53 (MG53) has been proposed to ubiquitinate IR and IRS1 in skeletal muscle (112, 113). Recently, RNAi screening identified MARCH1 as an E3 ligase of IR, demonstrating that MARCH1 regulates basal levels of IR in liver(114).
14.3. Phosphatases
Two types of phosphatases terminate IR signaling: lipid and protein phosphatases. PTEN and SH2-containing inositol 5’-phosphatase 2 (SHIP2) dephosphorylate lipid second messenger phosphatidylinositol 3,4,5-triphosphate (PI(3,4,5)P3), thereby attenuating IR signaling (Figure 1b). The PTEN converts PI(3,4,5)P3 to PI (4,5)P2, whereas the SHIP2 converts PI(3,4,5)P3 to PI(3,4)P2, terminating PI3K signaling(115–117). In addition, protein-tyrosine phosphatase 1B (PTP1B) is the well-characterized tyrosine phosphatase localized at endoplasmic reticulum (ER), negatively regulating IR signaling through direct binding to IR(118–120).
15. IR and IGF1R hybrid
Previous studies have demonstrated that a hybrid receptor may be composed of one half from IR and the other from IGF1R, thus forming a heterotetramer(121, 122). A combined binding assay with labeled IGF1 and monoclonal antibodies specific to IR or IGF1R demonstrated that these hybrid receptors could bind both insulin and IGF1 with high binding affinity, showing potential functional properties(123). It has been proposed that there is a variation in the expression levels of the IR/IGF1R hybrid in different tissues, and the IR/IGF1R hybrid is the major form of IGF1R in muscle(124). The two isoforms of IR can form hybrid receptors with IGF1R with similar efficiency; however, IR-A/IGF1R and IR-B/IGF1R hybrid receptors have different ligand binding affinities and activation potencies in response to insulin and IGF2(125). The IR-A/IGF1R hybrid binds to IGF2 with high affinity compared with the IR-B/IGF1R.
Given the distinct activation mechanisms of IR and IGF1R, the mechanism for ligand binding and activation of the hybrid receptor will be complicated. Moreover, the factors and mechanisms that regulate the formation of IR/IGF1R hybrids, as well as the physiological functions of the hybrid receptors, are unknown. The recent structure of IGF1-bound of ECD of IR-B/IGF1R hybrid receptor demonstrate that IGF1 binds to the L1/CR domains of IGF1R and the FnIII-1 domain of IR(126). The overall structure of the IGF1-bound IR-B/IGF1R hybrid receptors resembled the asymmetric conformation shown in IGF1R/IGF1(94), IGF1Rzip/IGF2(127) (zipper stabilized IGF1R ECD), IGF1R/insulin(128), and IRzip/insulin(81) (zipper stabilized IR ECD). Future studies are required to determine the structure of the apo-IR/IGF1R as well as the full-length structures of the IR/IGF1R hybrid with different types of ligands bound.
Conclusions and perspectives
The recent advances in structural and functional studies of IR have significantly contributed to our understanding of the IR activation. In addition to long-term studies of this important receptor using biochemistry and X-ray crystallography, recent developments in single-particle cryo-EM allow high-resolution structural determinations of dynamic IR/insulin complex(129). It is clear, however, that more remains to be learned about the activation mechanism of IR and its downstream signaling.
The TM domains in IR are critical for receptor activation and downstream signaling(130, 131). Visualizing the TMs in their dimerized active form and revealing the dimerization interface will be an important step towards fully understanding the activation mechanism of IR. Additionally, because the FnIII-3 domain is connected to the TM domain through a short linker (4 residues), it is reasonable to speculate that ECD, TM domain and ICD are coupled allosterically, and that the differences in the arrangements of the membrane proximal regions in the asymmetric and symmetric IR may cause differential dimeric assembly of TM domain and ICD. However, as of now, no cryo-EM structures of full-length IR/insulin complexes have been able to resolve a complete structure of IR, due to the flexible linkages of the TM helix with both the ECD and ICD. It may be necessary to stabilize the junction at the two ends of the TM domains using accessory factors such as conformationally selective nanobodies and TM-associating peptides(132). The cryo-EM density of the TM domains could also be improved by the application of advanced focused image classification techniques to distinguish different conformational states of the TM of IR(133). In addition, the reconstitution of full-length IR into a lipid bilayer, such as nanodisc, saposin or peptics(134, 135), may stabilize the TM domains by lipid-TM interaction and further improve its resolution. It has been suggested that certain types of lipids, such as phosphatidylinositol, play a key role in stabilizing the ICD of a variety of RTKs(136), including IR. Therefore, integrating such lipids into the nanodisc during the sample preparation may facilitate determining the entire structure of full-length IR/insulin complex.
Furthermore, IR have a large number of protein partners that play critical roles in promoting and regulating IR signaling(137). It remains challenging to determine the structure of full-length IR bound with downstream signaling proteins or other binding partners, due to the following reasons: (1) those binding partners are associated with IR transiently; (2) the binding of most of signaling proteins to IR requires receptor phosphorylation; and (3) the relative orientation between IR and its binding partners may not be fixed. It is important to note that all of the previously purified full-length IR for structural studies are kinase-dead, and cannot undergo auto-phosphorylation. In addition, a key IRS binding region of IR (NPEY960 motif) was mutated to prevent IR endocytosis. Thus, it would be essential to purify the over-expressed full-length IR wild-type or directly isolate IR from native source to reconstitute the IR/insulin in complex with their binding partners in vitro. Subsequently, the combination of biochemical, mass spectrometry, X-ray crystallography, single-particle cryo-EM, cryo-electron tomography (cryo-ET)(138) and computational modelling (Alpha fold(139)) will make it possible to visualize the entire structure of the IR signaling complex, thus providing a comprehensive understanding of the mechanism of IR signaling. The understanding of the structural nature of IR and IR signaling complex will assist in the development of novel therapeutics for diseases associated with dysregulation of IR signaling.
Acknowledgments
This work is supported in part by grants from the National Institutes Health (R01GM136976 to X.-C.B., and R01DK132361 to E.C.), the Welch foundation (I-1944 to X.-C.B.), and the Alice Bohmfalk Charitable (to E.C.). X.-C.B. is Virginia Murchison Linthicum Scholars in Medical Research at UTSW.
Footnotes
Disclosure statement
All authors declare no conflicts of interest.
References
- 1.Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA. 1922. Pancreatic Extracts in the Treatment of Diabetes Mellitus. Can Med Assoc J 12: 141–6 [PMC free article] [PubMed] [Google Scholar]
- 2.Levine R, Goldstein MS, Huddlestun B, Klein SP. 1950. Action of insulin on the ‘permeability’ of cells to free hexoses, as studied by its effect on the distribution of galactose. Am J Physiol 163: 70–6 [DOI] [PubMed] [Google Scholar]
- 3.Freychet P, Roth J, Neville DM Jr., 1971. Insulin receptors in the liver: specific binding of (125 I)insulin to the plasma membrane and its relation to insulin bioactivity. Proc Natl Acad Sci U S A 68: 1833–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kasuga M, Karlsson FA, Kahn CR. 1982. Insulin stimulates the phosphorylation of the 95,000-dalton subunit of its own receptor. Science 215: 185–7 [DOI] [PubMed] [Google Scholar]
- 5.Kasuga M, Zick Y, Blithe DL, Crettaz M, Kahn CR. 1982. Insulin stimulates tyrosine phosphorylation of the insulin receptor in a cell-free system. Nature 298: 667–9 [DOI] [PubMed] [Google Scholar]
- 6.Ullrich A, Bell JR, Chen EY, Herrera R, Petruzzelli LM, et al. 1985. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 313: 756–61 [DOI] [PubMed] [Google Scholar]
- 7.Ebina Y, Ellis L, Jarnagin K, Edery M, Graf L, et al. 1985. The human insulin receptor cDNA: the structural basis for hormone-activated transmembrane signalling. Cell 40: 747–58 [DOI] [PubMed] [Google Scholar]
- 8.Ullrich A, Gray A, Tam AW, Yang-Feng T, Tsubokawa M, et al. 1986. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J 5: 2503–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shier P, Watt VM. 1989. Primary structure of a putative receptor for a ligand of the insulin family. J Biol Chem 264: 14605–8 [PubMed] [Google Scholar]
- 10.Bravo DA, Gleason JB, Sanchez RI, Roth RA, Fuller RS. 1994. Accurate and efficient cleavage of the human insulin proreceptor by the human proprotein-processing protease furin. Characterization and kinetic parameters using the purified, secreted soluble protease expressed by a recombinant baculovirus. J Biol Chem 269: 25830–7 [PubMed] [Google Scholar]
- 11.Bajaj M, Waterfield MD, Schlessinger J, Taylor WR, Blundell T. 1987. On the tertiary structure of the extracellular domains of the epidermal growth factor and insulin receptors. Biochim Biophys Acta 916: 220–6 [DOI] [PubMed] [Google Scholar]
- 12.Schaffer L, Ljungqvist L. 1992. Identification of a disulfide bridge connecting the alpha-subunits of the extracellular domain of the insulin receptor. Biochem Biophys Res Commun 189: 650–3 [DOI] [PubMed] [Google Scholar]
- 13.Sparrow LG, McKern NM, Gorman JJ, Strike PM, Robinson CP, et al. 1997. The disulfide bonds in the C-terminal domains of the human insulin receptor ectodomain. J Biol Chem 272: 29460–7 [DOI] [PubMed] [Google Scholar]
- 14.Seino S, Bell GI. 1989. Alternative splicing of human insulin receptor messenger RNA. Biochem Biophys Res Commun 159: 312–6 [DOI] [PubMed] [Google Scholar]
- 15.Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, et al. 1999. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol 19: 3278–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petersen MC, Shulman GI. 2018. Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev 98: 2133–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santoro A, McGraw TE, Kahn BB. 2021. Insulin action in adipocytes, adipose remodeling, and systemic effects. Cell Metab 33: 748–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rask-Madsen C, Kahn CR. 2012. Tissue-specific insulin signaling, metabolic syndrome, and cardiovascular disease. Arterioscler Thromb Vasc Biol 32: 2052–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nandi A, Kitamura Y, Kahn CR, Accili D. 2004. Mouse models of insulin resistance. Physiol Rev 84: 623–47 [DOI] [PubMed] [Google Scholar]
- 20.Samuel VT, Shulman GI. 2016. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest 126: 12–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kullmann S, Heni M, Hallschmid M, Fritsche A, Preissl H, Haring HU. 2016. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol Rev 96: 1169–209 [DOI] [PubMed] [Google Scholar]
- 22.Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang HY, et al. 2018. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol 14: 168–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai S, Clemente-Casares X, Zhou AC, Lei H, Ahn JJ, et al. 2018. Insulin Receptor-Mediated Stimulation Boosts T Cell Immunity during Inflammation and Infection. Cell Metab 28: 922–34 e4 [DOI] [PubMed] [Google Scholar]
- 24.Pulgar VM. 2018. Transcytosis to Cross the Blood Brain Barrier, New Advancements and Challenges. Front Neurosci 12: 1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee WL, Klip A. 2016. Endothelial Transcytosis of Insulin: Does It Contribute to Insulin Resistance? Physiology (Bethesda) 31: 336–45 [DOI] [PubMed] [Google Scholar]
- 26.Hubbard SR, Wei L, Ellis L, Hendrickson WA. 1994. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 372: 746–54 [DOI] [PubMed] [Google Scholar]
- 27.Eck MJ, Dhe-Paganon S, Trub T, Nolte RT, Shoelson SE. 1996. Structure of the IRS-1 PTB domain bound to the juxtamembrane region of the insulin receptor. Cell 85: 695–705 [DOI] [PubMed] [Google Scholar]
- 28.Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D. 2000. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell 6: 1401–12 [DOI] [PubMed] [Google Scholar]
- 29.Hu J, Liu J, Ghirlando R, Saltiel AR, Hubbard SR. 2003. Structural basis for recruitment of the adaptor protein APS to the activated insulin receptor. Mol Cell 12: 1379–89 [DOI] [PubMed] [Google Scholar]
- 30.Depetris RS, Hu J, Gimpelevich I, Holt LJ, Daly RJ, Hubbard SR. 2005. Structural basis for inhibition of the insulin receptor by the adaptor protein Grb14. Mol Cell 20: 325–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haeusler RA, McGraw TE, Accili D. 2018. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol 19: 31–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boucher J, Kleinridders A, Kahn CR. 2014. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.White MF, Kahn CR. 2021. Insulin action at a molecular level - 100 years of progress. Mol Metab 52: 101304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gavin JR 3rd, Gorden P, Roth J, Archer JA, Buell DN 1973. Characteristics of the human lymphocyte insulin receptor. J Biol Chem 248: 2202–7 [PubMed] [Google Scholar]
- 35.de Meyts P, Roth J, Neville DM Jr., Gavin JR 3rd, Lesniak MA 1973. Insulin interactions with its receptors: experimental evidence for negative cooperativity. Biochem Biophys Res Commun 55: 154–61 [DOI] [PubMed] [Google Scholar]
- 36.DeMeyts P, Bainco AR, Roth J. 1976. Site-site interactions among insulin receptors. Characterization of the negative cooperativity. J Biol Chem 251: 1877–88 [PubMed] [Google Scholar]
- 37.White MF, Shoelson SE, Keutmann H, Kahn CR. 1988. A cascade of tyrosine autophosphorylation in the beta-subunit activates the phosphotransferase of the insulin receptor. J Biol Chem 263: 2969–80 [PubMed] [Google Scholar]
- 38.Rajagopalan M, Neidigh JL, McClain DA. 1991. Amino acid sequences Gly-Pro-Leu-Tyr and Asn-Pro-Glu-Tyr in the submembranous domain of the insulin receptor are required for normal endocytosis. J Biol Chem 266: 23068–73 [PubMed] [Google Scholar]
- 39.Kahn CR, White MF. 1988. The insulin receptor and the molecular mechanism of insulin action. J Clin Invest 82: 1151–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun XJ, Rothenberg P, Kahn CR, Backer JM, Araki E, et al. 1991. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 352: 73–7 [DOI] [PubMed] [Google Scholar]
- 41.White MF, Maron R, Kahn CR. 1985. Insulin rapidly stimulates tyrosine phosphorylation of a Mr-185,000 protein in intact cells. Nature 318: 183–6 [DOI] [PubMed] [Google Scholar]
- 42.Bjornholm M, He AR, Attersand A, Lake S, Liu SC, et al. 2002. Absence of functional insulin receptor substrate-3 (IRS-3) gene in humans. Diabetologia 45: 1697–702 [DOI] [PubMed] [Google Scholar]
- 43.Sadagurski M, Dong XC, Myers MG Jr., White MF. 2014. Irs2 and Irs4 synergize in non-LepRb neurons to control energy balance and glucose homeostasis. Mol Metab 3: 55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.White MF, Livingston JN, Backer JM, Lauris V, Dull TJ, et al. 1988. Mutation of the insulin receptor at tyrosine 960 inhibits signal transmission but does not affect its tyrosine kinase activity. Cell 54: 641–9 [DOI] [PubMed] [Google Scholar]
- 45.Backer JM, Myers MG Jr., Shoelson SE, Chin DJ, Sun XJ, et al. 1992. Phosphatidylinositol 3’-kinase is activated by association with IRS-1 during insulin stimulation. EMBO J 11: 3469–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kohn AD, Kovacina KS, Roth RA. 1995. Insulin stimulates the kinase activity of RAC-PK, a pleckstrin homology domain containing ser/thr kinase. EMBO J 14: 4288–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cantley LC. 2002. The phosphoinositide 3-kinase pathway. Science 296: 1655–7 [DOI] [PubMed] [Google Scholar]
- 48.Vanhaesebroeck B, Stephens L, Hawkins P. 2012. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol 13: 195–203 [DOI] [PubMed] [Google Scholar]
- 49.Cheatham B, Vlahos CJ, Cheatham L, Wang L, Blenis J, Kahn CR. 1994. Phosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis, and glucose transporter translocation. Mol Cell Biol 14: 4902–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hopkins BD, Pauli C, Du X, Wang DG, Li X, et al. 2018. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560: 499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu M, Wan M, Leavens KF, Chu Q, Monks BR, et al. 2012. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med 18: 388–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manning BD, Toker A. 2017. AKT/PKB Signaling: Navigating the Network. Cell 169: 381–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suzuki K, Kono T. 1980. Evidence that insulin causes translocation of glucose transport activity to the plasma membrane from an intracellular storage site. Proc Natl Acad Sci U S A 77: 2542–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Foley K, Boguslavsky S, Klip A. 2011. Endocytosis, recycling, and regulated exocytosis of glucose transporter 4. Biochemistry 50: 3048–61 [DOI] [PubMed] [Google Scholar]
- 55.Huang S, Czech MP. 2007. The GLUT4 glucose transporter. Cell Metab 5: 237–52 [DOI] [PubMed] [Google Scholar]
- 56.Leto D, Saltiel AR. 2012. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol 13: 383–96 [DOI] [PubMed] [Google Scholar]
- 57.Cohen P 1999. The Croonian Lecture 1998. Identification of a protein kinase cascade of major importance in insulin signal transduction. Philos Trans R Soc Lond B Biol Sci 354: 485–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. 1995. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378: 785–9 [DOI] [PubMed] [Google Scholar]
- 59.Li S, Brown MS, Goldstein JL. 2010. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A 107: 3441–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, et al. 2011. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146: 408–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laplante M, Sabatini DM. 2012. mTOR signaling in growth control and disease. Cell 149: 274–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin HV, Accili D. 2011. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab 14: 9–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. 2007. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab 6: 208–16 [DOI] [PubMed] [Google Scholar]
- 64.Nakae J, Kitamura T, Silver DL, Accili D. 2001. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest 108: 1359–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haeusler RA, Hartil K, Vaitheesvaran B, Arrieta-Cruz I, Knight CM, et al. 2014. Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nat Commun 5: 5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakae J, Park BC, Accili D. 1999. Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J Biol Chem 274: 15982–5 [DOI] [PubMed] [Google Scholar]
- 67.Biggs WH 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC 1999. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A 96: 7421–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gehart H, Kumpf S, Ittner A, Ricci R. 2010. MAPK signalling in cellular metabolism: stress or wellness? EMBO Rep 11: 834–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pronk GJ, McGlade J, Pelicci G, Pawson T, Bos JL. 1993. Insulin-induced phosphorylation of the 46- and 52-kDa Shc proteins. J Biol Chem 268: 5748–53 [PubMed] [Google Scholar]
- 70.English J, Pearson G, Wilsbacher J, Swantek J, Karandikar M, et al. 1999. New insights into the control of MAP kinase pathways. Exp Cell Res 253: 255–70 [DOI] [PubMed] [Google Scholar]
- 71.Cobb MH. 1999. MAP kinase pathways. Prog Biophys Mol Biol 71: 479–500 [DOI] [PubMed] [Google Scholar]
- 72.Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, et al. 2016. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535: 148–52 [DOI] [PubMed] [Google Scholar]
- 73.Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, et al. 2018. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol 20: 1064–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hall C, Yu H, Choi E. 2020. Insulin receptor endocytosis in the pathophysiology of insulin resistance. Exp Mol Med [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McKern NM, Lawrence MC, Streltsov VA, Lou MZ, Adams TE, et al. 2006. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature 443: 218–21 [DOI] [PubMed] [Google Scholar]
- 76.Croll TI, Smith BJ, Margetts MB, Whittaker J, Weiss MA, et al. 2016. Higher-Resolution Structure of the Human Insulin Receptor Ectodomain: Multi-Modal Inclusion of the Insert Domain. Structure 24: 469–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Menting JG, Whittaker J, Margetts MB, Whittaker LJ, Kong GK, et al. 2013. How insulin engages its primary binding site on the insulin receptor. Nature 493: 241–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bai XC, McMullan G, Scheres SH. 2015. How cryo-EM is revolutionizing structural biology. Trends Biochem Sci 40: 49–57 [DOI] [PubMed] [Google Scholar]
- 79.Cai K, Zhang X, Bai XC. 2022. Cryo-electron Microscopic Analysis of Single-Pass Transmembrane Receptors. Chem Rev [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scapin G, Dandey VP, Zhang Z, Prosise W, Hruza A, et al. 2018. Structure of the insulin receptor-insulin complex by single-particle cryo-EM analysis. Nature 556: 122–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weis F, Menting JG, Margetts MB, Chan SJ, Xu Y, et al. 2018. The signalling conformation of the insulin receptor ectodomain. Nat Commun 9: 4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schaffer L 1994. A model for insulin binding to the insulin receptor. Eur J Biochem 221: 1127–32 [DOI] [PubMed] [Google Scholar]
- 83.De Meyts P, Whittaker J. 2002. Structural biology of insulin and IGF1 receptors: implications for drug design. Nat Rev Drug Discov 1: 769–83 [DOI] [PubMed] [Google Scholar]
- 84.Whittaker J, Garcia P, Yu GQ, Mynarcik DC. 1994. Transmembrane domain interactions are necessary for negative cooperativity of the insulin receptor. Mol Endocrinol 8: 1521–7 [DOI] [PubMed] [Google Scholar]
- 85.Uchikawa E, Choi E, Shang G, Yu H, Bai XC. 2019. Activation mechanism of the insulin receptor revealed by cryo-EM structure of the fully liganded receptor-ligand complex. Elife 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gutmann T, Schafer IB, Poojari C, Brankatschk B, Vattulainen I, et al. 2020. Cryo-EM structure of the complete and ligand-saturated insulin receptor ectodomain. J Cell Biol 219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Choi E, Zhang X, Xing C, Yu H. 2016. Mitotic Checkpoint Regulators Control Insulin Signaling and Metabolic Homeostasis. Cell 166: 567–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lu D, Shang G, Zhang H, Yu Q, Cong X, et al. 2014. Structural insights into the T6SS effector protein Tse3 and the Tse3-Tsi3 complex from Pseudomonas aeruginosa reveal a calcium-dependent membrane-binding mechanism. Mol Microbiol 92: 1092–112 [DOI] [PubMed] [Google Scholar]
- 89.De Meyts P 2015. Insulin/receptor binding: the last piece of the puzzle? What recent progress on the structure of the insulin/receptor complex tells us (or not) about negative cooperativity and activation. Bioessays 37: 389–97 [DOI] [PubMed] [Google Scholar]
- 90.Lawrence MC. 2021. Understanding insulin and its receptor from their three-dimensional structures. Mol Metab 52: 101255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kristensen C, Kjeldsen T, Wiberg FC, Schaffer L, Hach M, et al. 1997. Alanine scanning mutagenesis of insulin. J Biol Chem 272: 12978–83 [DOI] [PubMed] [Google Scholar]
- 92.Li J, Park J, Mayer JP, Webb KJ, Uchikawa E, et al. 2022. Synergistic activation of the insulin receptor via two distinct sites. Nat Struct Mol Biol 29: 357–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nielsen J, Brandt J, Boesen T, Hummelshoj T, Slaaby R, et al. 2022. Structural Investigations of Full-Length Insulin Receptor Dynamics and Signalling. J Mol Biol 434: 167458. [DOI] [PubMed] [Google Scholar]
- 94.Li J, Choi E, Yu H, Bai XC. 2019. Structural basis of the activation of type 1 insulin-like growth factor receptor. Nat Commun 10: 4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kavran JM, McCabe JM, Byrne PO, Connacher MK, Wang Z, et al. 2014. How IGF-1 activates its receptor. Elife 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li J, Wu J, Bai X-c, Choi E 2022. Molecular basis for the role of disulfide-linked αCTs in the activation of insulin-like growth factor 1 receptor and insulin receptor. bioRxiv: 2022.07.06.498988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nakae J, Kido Y, Accili D. 2001. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr Rev 22: 818–35 [DOI] [PubMed] [Google Scholar]
- 98.Tengholm A, Gylfe E. 2009. Oscillatory control of insulin secretion. Mol Cell Endocrinol 297: 58–72 [DOI] [PubMed] [Google Scholar]
- 99.Siddle K 2012. Molecular basis of signaling specificity of insulin and IGF receptors: neglected corners and recent advances. Front Endocrinol (Lausanne) 3: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim JJ, Accili D. 2002. Signalling through IGF-I and insulin receptors: where is the specificity? Growth Horm IGF Res 12: 84–90 [DOI] [PubMed] [Google Scholar]
- 101.Goh LK, Sorkin A. 2013. Endocytosis of receptor tyrosine kinases. Cold Spring Harb Perspect Biol 5: a017459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Choi E, Yu H. 2018. Spindle Checkpoint Regulators in Insulin Signaling. Front Cell Dev Biol 6: 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Carpentier JL. 1994. Insulin receptor internalization: molecular mechanisms and physiopathological implications. Diabetologia 37 Suppl 2: S117–24 [DOI] [PubMed] [Google Scholar]
- 104.Fagerholm S, Ortegren U, Karlsson M, Ruishalme I, Stralfors P. 2009. Rapid insulin-dependent endocytosis of the insulin receptor by caveolae in primary adipocytes. PLoS One 4: e5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Backer JM, Kahn CR, Cahill DA, Ullrich A, White MF. 1990. Receptor-mediated internalization of insulin requires a 12-amino acid sequence in the juxtamembrane region of the insulin receptor beta-subunit. J Biol Chem 265: 16450–4 [PubMed] [Google Scholar]
- 106.Backer JM, Shoelson SE, Haring E, White MF. 1991. Insulin receptors internalize by a rapid, saturable pathway requiring receptor autophosphorylation and an intact juxtamembrane region. J Cell Biol 115: 1535–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tang R, Jiang Z, Chen F, Yu W, Fan K, et al. 2020. The Kinase Activity of Drosophila BubR1 Is Required for Insulin Signaling-Dependent Stem Cell Maintenance. Cell Rep 31: 107794. [DOI] [PubMed] [Google Scholar]
- 108.Choi E, Kikuchi S, Gao H, Brodzik K, Nassour I, et al. 2019. Mitotic regulators and the SHP2-MAPK pathway promote IR endocytosis and feedback regulation of insulin signaling. Nat Commun 10: 1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Caro JF, Ittoop O, Pories WJ, Meelheim D, Flickinger EG, et al. 1986. Studies on the mechanism of insulin resistance in the liver from humans with noninsulin-dependent diabetes. Insulin action and binding in isolated hepatocytes, insulin receptor structure, and kinase activity. J Clin Invest 78: 249–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Soll AH, Kahn CR, Neville DM Jr., 1975. Insulin binding to liver plasm membranes in the obese hyperglycemic (ob/ob) mouse. Demonstration of a decreased number of functionally normal receptors. J Biol Chem 250: 4702–7 [PubMed] [Google Scholar]
- 111.Howard JK, Flier JS. 2006. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab 17: 365–71 [DOI] [PubMed] [Google Scholar]
- 112.Song R, Peng W, Zhang Y, Lv F, Wu HK, et al. 2013. Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature 494: 375–9 [DOI] [PubMed] [Google Scholar]
- 113.Yi JS, Park JS, Ham YM, Nguyen N, Lee NR, et al. 2013. MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat Commun 4: 2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nagarajan A, Petersen MC, Nasiri AR, Butrico G, Fung A, et al. 2016. MARCH1 regulates insulin sensitivity by controlling cell surface insulin receptor levels. Nat Commun 7: 12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Carracedo A, Pandolfi PP 2008. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 27: 5527–41 [DOI] [PubMed] [Google Scholar]
- 116.Clement S, Krause U, Desmedt F, Tanti JF, Behrends J, et al. 2001. The lipid phosphatase SHIP2 controls insulin sensitivity. Nature 409: 92–7 [DOI] [PubMed] [Google Scholar]
- 117.Lazar DF, Saltiel AR. 2006. Lipid phosphatases as drug discovery targets for type 2 diabetes. Nat Rev Drug Discov 5: 333–42 [DOI] [PubMed] [Google Scholar]
- 118.Anderie I, Schulz I, Schmid A. 2007. Direct interaction between ER membrane-bound PTP1B and its plasma membrane-anchored targets. Cell Signal 19: 582–92 [DOI] [PubMed] [Google Scholar]
- 119.Popov D 2012. Endoplasmic reticulum stress and the on site function of resident PTP1B. Biochem Biophys Res Commun 422: 535–8 [DOI] [PubMed] [Google Scholar]
- 120.Issad T, Boute N, Boubekeur S, Lacasa D. 2005. Interaction of PTPB with the insulin receptor precursor during its biosynthesis in the endoplasmic reticulum. Biochimie 87: 111–6 [DOI] [PubMed] [Google Scholar]
- 121.Soos MA, Siddle K. 1989. Immunological relationships between receptors for insulin and insulin-like growth factor I. Evidence for structural heterogeneity of insulin-like growth factor I receptors involving hybrids with insulin receptors. Biochem J 263: 553–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Moxham CP, Duronio V, Jacobs S. 1989. Insulin-like growth factor I receptor beta-subunit heterogeneity. Evidence for hybrid tetramers composed of insulin-like growth factor I and insulin receptor heterodimers. J Biol Chem 264: 13238–44 [PubMed] [Google Scholar]
- 123.Soos MA, Whittaker J, Lammers R, Ullrich A, Siddle K. 1990. Receptors for insulin and insulin-like growth factor-I can form hybrid dimers. Characterisation of hybrid receptors in transfected cells. Biochem J 270: 383–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bailyes EM, Nave BT, Soos MA, Orr SR, Hayward AC, Siddle K. 1997. Insulin receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues: quantification of individual receptor species by selective immunoprecipitation and immunoblotting. Biochem J 327 ( Pt 1): 209–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, Belfiore A. 2002. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem 277: 39684–95 [DOI] [PubMed] [Google Scholar]
- 126.Xu Y, Margetts MB, Venugopal H, Menting JG, Kirk NS, et al. 2022. How insulin-like growth factor I binds to a hybrid insulin receptor type 1 insulin-like growth factor receptor. Structure [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xu Y, Kirk NS, Venugopal H, Margetts MB, Croll TI, et al. 2020. How IGF-II Binds to the Human Type 1 Insulin-like Growth Factor Receptor. Structure 28: 786–98 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang X, Yu D, Sun J, Wu Y, Gong J, et al. 2020. Visualization of Ligand-Bound Ectodomain Assembly in the Full-Length Human IGF-1 Receptor by Cryo-EM Single-Particle Analysis. Structure 28: 555–61 e4 [DOI] [PubMed] [Google Scholar]
- 129.Bai XC. 2021. Seeing Atoms by Single-Particle Cryo-EM. Trends Biochem Sci 46: 253–54 [DOI] [PubMed] [Google Scholar]
- 130.Yamada K, Goncalves E, Kahn CR, Shoelson SE. 1992. Substitution of the insulin receptor transmembrane domain with the c-neu/erbB2 transmembrane domain constitutively activates the insulin receptor kinase in vitro. J Biol Chem 267: 12452–61 [PubMed] [Google Scholar]
- 131.Cheatham B, Shoelson SE, Yamada K, Goncalves E, Kahn CR. 1993. Substitution of the erbB-2 oncoprotein transmembrane domain activates the insulin receptor and modulates the action of insulin and insulin-receptor substrate 1. Proc Natl Acad Sci U S A 90: 7336–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.McMahon C, Baier AS, Pascolutti R, Wegrecki M, Zheng S, et al. 2018. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat Struct Mol Biol 25: 289–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bai XC, Rajendra E, Yang G, Shi Y, Scheres SH. 2015. Sampling the conformational space of the catalytic subunit of human gamma-secretase. Elife 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Denisov IG, Sligar SG. 2016. Nanodiscs for structural and functional studies of membrane proteins. Nat Struct Mol Biol 23: 481–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Frauenfeld J, Loving R, Armache JP, Sonnen AF, Guettou F, et al. 2016. A saposin-lipoprotein nanoparticle system for membrane proteins. Nat Methods 13: 345–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Michailidis IE, Rusinova R, Georgakopoulos A, Chen Y, Iyengar R, et al. 2011. Phosphatidylinositol-4,5-bisphosphate regulates epidermal growth factor receptor activation. Pflugers Arch 461: 387–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Virkamaki A, Ueki K, Kahn CR. 1999. Protein-protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J Clin Invest 103: 931–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Turk M, Baumeister W. 2020. The promise and the challenges of cryo-electron tomography. FEBS Lett 594: 3243–61 [DOI] [PubMed] [Google Scholar]
- 139.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, et al. 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–89 [DOI] [PMC free article] [PubMed] [Google Scholar]