

Abstract
The use of optogenetics in metabolic engineering for light-controlled microbial chemical production raises the prospect of utilizing control and optimization techniques routinely deployed in traditional chemical manufacturing. However, such mechanisms require well-characterized, customizable tools that respond fast enough to be used as real-time inputs during fermentations. Here, we present OptoINVRT7, a new rapid optogenetic inverter circuit to control gene expression in Saccharomyces cerevisiae. The circuit induces gene expression in only 0.6 h after switching cells from light to darkness, which is at least 6 times faster than previous OptoINVRT optogenetic circuits used for chemical production. In addition, we introduce an engineered inducible GAL1 promoter (PGAL1-S), which is stronger than any constitutive or inducible promoter commonly used in yeast. Combining OptoINVRT7 with PGAL1-S achieves strong and light-tunable levels of gene expression with as much as 132.9 ± 22.6-fold induction in darkness. The high performance of this new optogenetic circuit in controlling metabolic enzymes boosts production of lactic acid and isobutanol by more than 50% and 15%, respectively. The strength and controllability of OptoINVRT7 and PGAL1-S open the door to applying process control tools to engineered metabolisms to improve robustness and yields in microbial fermentations for chemical production.
Graphical Abstract
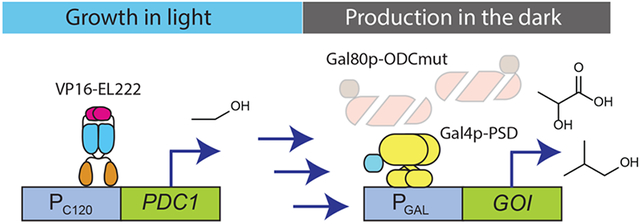
Dynamic control of metabolic pathways is an effective way to improve the efficiency of chemical production in engineered microbial hosts.1-3 Microbial fermentations are often separated temporally into two phases: an initial “growth phase”, in which metabolism is devoted to replicating cells (growing the cell culture), and a subsequent “production phase”, in which metabolism is shifted to maximize flux toward a product of interest. This helps balance the allocation of media and cellular resources between making the product of interest and providing the cell with what it needs to survive and grow, resulting in an overall increase in chemical production. The transition between the growth phase and production phase is most often controlled by adding a chemical inducer, such as isopropyl β-d-1-thiogalactopyranoside (IPTG) or doxycycline, or making a change in the carbon source or a nutrient, such as galactose or methionine, which activate or repress specific promoters used to regulate the metabolic enzymes. Autonomous induction is another strategy to achieve dynamic control, in which transcription of the metabolic enzymes is regulated by promoters or biosensors that respond to specific metabolites or quorum cues that automatically change as the fermentation progresses.1,4 We recently showed that it is also possible to dynamically control microbial fermentations for chemical production using light signals by linking metabolic enzyme expression to optogenetic transcriptional circuits,5,6 or enzyme colocalization to light-responsive synthetic organelles.7
Using light for dynamic control in metabolic engineering offers several advantages. Light, unlike chemical signals, is easy to reverse any number of times and at any frequency (light schedule) during the fermentation.5,8-11 In addition, light signals are easily tuned by intensity or frequency of exposure, and using light avoids the need for expensive chemical inducers or complex media changes (e.g., of carbon source) that are difficult to implement. Critically, the challenge of light penetration in high cell density fermentations can be overcome, at least in lab-scale bioreactors, by using inverter circuits that induce gene expression in the dark instead of light,5,6 as well as by using highly light-sensitive transcription factors such as VP16-EL222,8 which allows robust gene expression induced by light in bioreactors of up to at least 5 L at cell densities of 50 OD.5 These features enable dynamic control of fermentations not only in triggering their transition from growth to production, but also during the production phase using blue light pulses to boost chemical production.
Another unique advantage of using optogenetics to control microbial fermentations for chemical production is the ease with which light inputs can interface with computers to implement control systems in living cells, the emerging area of cybergenetics.12,13 While such a light-enabled in silico-bound control system has not yet been developed for metabolic engineering, it has been elegantly demonstrated for dynamic control of protein production.14,15 Light is thus an attractive agent to implement computer-assisted control of engineered metabolisms to enable new open- and closed-loop modalities of dynamic control of microbial fermentations to optimize chemical production.1 However, this goal requires new optogenetic systems of rapid response that reach induction levels necessary for effectively controlling metabolism.
In this study, we present OptoINVRT7, which is at least 6 times faster than previous OptoINVRT circuits developed for yeast metabolic engineering.5 As an inverter circuit, it represses gene expression in blue light, and strongly activates it in the dark. Combining OptoINVRT7 with a redesigned GAL1 promoter, which we call PGAL1-S, we developed light-controlled system variants that cover and exceed the gamut of strength, sensitivity, and fold-change of induction in existing inducible and constitutive promoters commonly used in yeast metabolic engineering. Comparing the performance of OptoINVRT7 to that of previous circuits when controlling the biosynthesis of lactic acid and isobutanol, we found that enhanced activation kinetics lead to increased and more robust chemical production. This technology is an important step toward developing real-time computer-assisted dynamic control systems for metabolic engineering.
RESULTS AND DISCUSSION
Development and Characterization of OptoINVRT5 and OptoINVRT7.
Robust controls for metabolic engineering using light require optogenetic circuits with a wide dynamic range of gene expression and fast kinetics. We recently described optogenetic circuits OptoINVRT1, OptoINVRT2, and OptoINVRT3, which are designed to invert the transcriptional response to light, allowing genes to be repressed by blue light and expressed by darkness5 (Figure 1a,b). These circuits harness the yeast galactose regulon, involving Gal4p (a transcriptional activator of PGAL1) and Gal80p (a repressor of Gal4p) (Supplementary Figure S1). By controlling GAL80 expression with the blue light-responsive VP16-EL222 transcription factor,8 OptoINVRT circuits effectively repress genes transcribed by the GAL1 promoter (PGAL1) when yeast is exposed to blue light. However, the already translated Gal80p continues to repress Gal4p even after blue light stimulation is removed, causing a delay in the response of OptoINVRT circuits. Gal4p is only able to induce gene expression in the dark when Gal80p is sufficiently degraded or diluted by mitotic division (Figure 1a,b).5,16 Therefore, the constraints placed by Gal80p clearance on the rates of activation of OptoINVRT1, OptoINVRT2, and OptoINVRT3 limit their use in tight real-time control of metabolic pathways.
Figure 1.

OptoINVRT circuit engineering. (a) Schematic of the OptoINVRT circuit design. The OptoINVRT circuits (1, 2, 3, 5, or 7) using each element are specified in parentheses. Blue light-activated steps, including VP16-EL222-activation of PC120 and PSD-mediated degradation of Gal4p, are marked with blue arrows. Constitutive expression of GAL4 by PADH1 or PPGK1 is shown with a purple arrow. Gal80p-repression of Gal4p is shown with a red blunt arrow. Enhanced degradation of Gal80p by CLN2 or ODCmut is shown with a brown arrow. (b) OptoINVRT circuits use VP16-EL222 to express Gal80p (orange) in the light, which represses Gal4p (yellow) to control GFP (green) expression; GAL4 is expressed with PADH1 (OptoINVRT1) or PPGK1 (OptoINVRT2, 3, 5, and 7). A photosensitive degron domain (blue) fused to Gal4p increases its degradation rate in the light (OptoINVRT3, 5, and 7). Gal80p is fused to constitutive degron domains (brown) such as CLN2-PEST (OptoINVRT5) or ODCmut (OptoINVRT7). (c) Specific GFP expression of OptoINVRT circuits driving expression from PGAL1, characterized in CENPK.2–1C-derived strains YEZ100 (OptoINVRT1), YEZ101 (OptoINVRT2), YEZ102 (OptoINVRT3), YEZ230–5 (OptoINVRT5), and YEZ230C (OptoINVRT7); constitutive PTEF1 control (YEZ186) is shown. Light-sensitive strains are shown in full light (light blue), 8 s ON/72 s OFF (dark blue), and full darkness (black). All data are shown as mean values; dots represent individual data points; error bars represent the s.d. of four biologically independent 1-mL sample replicates exposed to the same conditions. All experiments were repeated at least three times.
An effective way to shorten the response time of OptoINVRT circuits is to reduce the lingering activity of Gal80p after the light is turned off. To achieve this, we fused Gal80p to degradation tags to reduce its half-life, expecting it would increase the rate and maximum level of gene expression by Gal4p upon switching cells from light to darkness. Preempting a weaker activity of Gal80p due to the degradation tags, we also fused Gal4p to a light-sensitive degron domain to reduce the basal activity of Gal4p in the light, similar to OptoINVRT3.5 The fusion of Gal4p to a degron domain also reduces Gal4p activity, which lowers the maximum level of expression in the dark, but this can be mitigated by leaving the endogenous (untagged) copy of GAL4 in the strain background5 (Figure 1a,b). The combined intervention of reducing the protein stabilities of Gal80p (constitutively) and Gal4p (by light induction) constitutes the platform upon which two new inverter circuits, OptoINVRT5 and OptoINVRT7, are designed.
OptoINVRT5 and OptoINVRT7, like previous Opto-INVRT circuits, operate in a GAL80Δ strain background. They both use an integrated copy of VP16-EL222 driven by the PTEF1 promoter, GAL4 fused to a photosensitive degron domain driven by the PPGK1 promoter (PPGK1-GAL4-PSD), and two copies of GAL80 under the PC120 promoter (which is activated in blue light by VP16-EL222). In addition, GAL80 is fused to two possible degradation tags. In the case of OptoINVRT5 (strain YEZ230–5), GAL80 is fused to the CLN217 degradation tag, while in OptoINVRT7 (strain YEZ230C), GAL80 is fused to a mutated version of the murine ornithine decarboxylase degradation tag,18,19 (which we call ODCmut; Supplementary Sequence 1). Using green fluorescent protein (GFP) driven by PGAL1 as a reporter of gene expression, we observed that both circuits have higher dark state activation levels than any previous OptoINVRT circuit, about 82.1 ± 2.8% and 79.8 ± 3.1% the activity of PTEF1, respectively (Figure 1c). This can be explained by the reduced stability of Gal80p substantially increasing the activity of Gal4p in the dark. The relatively high basal activity of OptoINVRT5 in the light suggests that the CLN2 degradation tag is the stronger of the two tags, keeping Gal80p levels too low to fully repress Gal4p in the light. In contrast, the ODCmut degradation tag provides OptoINVRT7 with higher levels of light sensitivity (the percent reduction in GFP expression at 8/80 s pulse: 78% for OptoINVRT7 vs 64% for OptoINVRT5, see Methods) and equal maximum gene expression in the dark as OptoINVRT5, but a significantly lower basal gene expression in the light, resulting in the highest dark-to-light fold of induction of all OptoINVRT circuits: up to 132.9 ± 22.6. While toxicity of the VP16 domain in yeast has been previously suggested,20 we found that OptoINVRT7, like previous OptoINVRT circuits, only causes a mild increase in the lag phase, with no other observable growth defects (Supplementary Figure S2), instabilities, or off-target effects. The strength, sensitivity, tunability, and dynamic range of OptoINVRT7 makes it the most versatile optogenetic circuit we have developed for metabolic engineering.
Development of a Super GAL1 Promoter (PGAL1-S) for Enhanced Gene Expression.
The GAL1 promoter (PGAL1) is the strongest natural promoter known in yeast, including inducible and constitutive promoters.21 However, PGAL1 is natively regulated by glucose via the Multicopy Inhibitor of Galactose gene repressor Mig1p, which is translocated into the nucleus in the presence of glucose.22 By deleting the two Mig1p-binding sites in PGAL1,23 we developed PGAL1-M (Supplementary Sequence 2), which increases the level of darkness-induced gene expression of OptoINVRT7 in glucose by 37.9 ± 7.6% (YEZ230) relative to the level obtained with OptoINVRT7 controlling the wild type PGAL1 (YEZ230C) (Figure 2a). This modification is enough to make Opto-INVRT7 achieve the same levels of expression as PTEF1, but with the benefit of light-controlled tunability.
Figure 2.

PGAL1 promoter engineering. (a) Specific GFP expression by engineered GAL1 promoters controlled by OptoINVRT7: PGAL1 (YEZ230C), PGAL1-M (YEZ230) with deleted Mig1p binding sites; PGAL1-S (YEZ229) with four extra Gal4p-binding added to the PGAL1-M sequence. (b) Specific GFP expression controlled: optogenetically (blue and black) by darkness-induced OptoINVRT7 (YEZ229); with nutrients (green): galactose-induced PGAL1 (YEZ48), ethanol-induced PADH2 (YEZ294) and methionine-repressed PMET3 (YEZ295); with synthetic promoters: β-estradiol-induced (orange) LexABD-ER-B112AD31 (yMAL34), or LexABD-ER-VP16AD31 (yMAL35), and tetracycline-repressed (pink and purple) tTA32,33 (yMAL36); or constitutive promoters (periwinkle): PTDH3 (YEZ228) and PTEF1 (YEZ186). Light-sensitive strains in (a) and (b) are shown in full light (light blue), 8 s ON/72 s OFF (dark blue), and full darkness (black); yMAL34 and yMAL35 in 2000 nM, 15 nM, and 0 nM β-estradiol31 and yMAL36 in 0, 10, and 1000 ng/mL tetracycline.33 YEZ140 (no GFP control) was used to subtract background and autofluorescence. All data are shown as mean values; dots represent individual data points; error bars represent the s.d. of four biologically independent 1-mL sample replicates exposed to the same conditions. All experiments were repeated at least three times.
We next sought to increase the maximum level of gene expression achievable by OptoINVRT7 by further engineering PGAL1-M. A previous study showed that adding four extra copies of the Gal4p upstream activating sequence (UAS) to PGAL1 (at the 5′-end of the native UAS, and 200 base pairs apart) increases galactose-induced gene expression by 10–15%.24 Following a similar strategy, we added four copies of the Gal4p UAS to PGAL1-M, but only 93 base pairs apart, which corresponds to approximately 9 turns of a DNA helix between binding sites. This arrangement attempts to allow Gal4p dimers to bind on the same phase of the DNA. The resulting promoter, which we call the Super-GAL1 promoter PGAL1-S (Supplementary Sequence 3), increases the level of gene expression when using OptoINVRT7 (strain YEZ229) in the dark by 147.7 ± 4.0% (Figure 2a), relative to the same circuit using PGAL1-M (YEZ230). This achieves a 32-fold induction from full light to dark, with a maximum expression level 2.5 times stronger than PTEF1, one of the strongest promoters used in yeast metabolic engineering. Furthermore, this system allows light-tunable gene expression in glucose (Figure 2a), and other carbon sources, although OptoINVRT1 and PGAL1 still offer tighter control in the latter (Supplementary Figure S3). The higher leakiness of the OptoINVRT7-PGAL1-S combination in glucose is probably due, at least in part, to the endogenous GAL4 left in the strain. This is consistent with the substantially higher basal expression of this system in non-glucose containing media, in which the endogenous GAL4 is further derepressed. This effect may occur during fermentations, after glucose is consumed and yeast starts to consume ethanol, and should be considered when using this circuit in such regimes. OptoINVRT7 is also functional in the two different strain backgrounds we tested, CEN.PK2–1C (Figure 2a) and BY4741 (Supplementary Figure S4). This makes OptoINVRT7 and PGAL1-S exceptionally versatile for multiple metabolic engineering applications in different types of media and strains.
Comparison of PGAL1-S-OptoINVRT7 System with Other Yeast Promoters.
Several different promoters are commonly used to express genes in engineered yeast strains. Some are endogenous constitutive promoters, the strongest of which include PTDH3 and PTEF1.25 Other promoters are inducible, which may be controlled by specific nutrients, such as PMET3 repressed by methionine,26 or carbon sources, such as PGAL1 activated by galactose27,28 and PADH2 activated by ethanol.29 The limitations of controlling gene expression with changes in nutrient composition or carbon source (e.g., medium restrictions and poor tunability) have been addressed with the development of exogenous or synthetic systems, including systems based on the human estrogen receptor (hER),30,31 activated by β-estradiol, or the tetracycline transactivator protein (tTA),32,33 repressed by tetracycline or doxycycline. These systems represent the state of the art in yeast gene expression, widely used in basic and applied studies, including in yeast genetics, cell biology, and metabolic engineering.
We compared OptoINVRT7-controlled PGAL1-S with the yeast gold standard systems mentioned above because they include the most widely used promoters in yeast metabolic engineering (Figure 2b). Using GFP as a reporter, we found that PGAL1-S, when fully activated by OptoINVRT7, is 3.4 times stronger than the best estrogen receptor system we tested and 5.3 times stronger than the Dox repressed system, while still maintaining a comparable fold of induction. It is also 34.1% stronger than the wild type PGAL1 in galactose, the strongest inducible promoter in S. cerevisiae, and 152.3% stronger than PTEF1, one of the strongest constitutive promoters (as a control, we tested PTEF in different media conditions, Supplementary Figure S5). In GAL80 strains, PGAL1-S is also induced by galactose and repressed by glucose, akin to the natural PGAL1, with 36% higher level of gene expression and 80-fold induction (Supplementary Figure S6a). It remains to be seen how much PGAL1-S enhances gene expression control when combined with other synthetic inducible systems that utilize PGAL1, such as those based on hER,30,31 or other blue-light responsive proteins including cryptochrome 2 (CRY2)34-36 or VIVID/WC-1.37 Nevertheless, controlling PGAL1-S or PGAL1 with OptoINVRT circuits makes these promoters readily tunable in glucose (Figures 1c, 2a, and Supplementary Figure S6b), using light instead of chemicals or complex media changes for induction. Therefore, the combination of OptoINVRT7 with PGAL1-S adds enhanced strength, tunability, and versatility to the existing set of yeast promoters.
Kinetic Characterization of OptoINVRT7.
To test how much the OptoINVRT7 design improves its activation kinetics, we compared it with earlier OptoINVRT circuit designs. We used flow cytometry to measure the rate of GFP expression from each circuit upon switching from full light to dark conditions during the early exponential phase (OD600 = 0.5) of fermentations (see Methods). We found that OptoINVRT7 shows significant activation by the first time point measurement (2 h), reaching close to maximum activation after only 6 h in the dark (Figure 3). In contrast, OptoINVRT1 and OptoINVRT2 show only slight activation after 6 h in the dark, and even after 9 h only achieve 36.4% and 13.3% of the maximum activation observed experimentally in complete darkness. Additionally, OptoINVRT7 achieves more homogeneous gene induction compared to OptoINVRT1 and OptoINVRT2 (Supplementary Figure S7). Thus, the increased turnover rate of Gal80p, due to the fused degradation tag, and the light activated degradation of Gal4p confer a clear kinetic advantage to OptoINVRT7 activation.
Figure 3.

Activation kinetics (light-to-dark) of OptoINVRT circuits. Time-course expression of GFP using OptoINVRT1 (YEZ100, dark blue line), OptoINVRT2 (YEZ101, red line), and OptoINVRT7 (YEZ230C, cyan line). Strains were grown under light to an OD600 = 0.5, and then switched to full darkness at t = 0. Samples were taken during the exponential growth phase only (up until OD600 = 5). A negative control containing no GFP (YEZ140, yellow line) and positive control containing PTEF1-GFP (YEZ186, gray line) are also shown. Individual points are median values of flow cytometry measurements (n = 4 biologically independent 1 mL samples).
To quantify the kinetics of the inverted circuits, we define an apparent time delay, which characterizes the time when a signal above background is first detected (see Methods). While the apparent time delay for OptoINVRT1 and OptoINVRT2 is approximately 5 to 5.5 h, the time delay for OptoINVRT7 is approximately 0.8 h. Therefore, OptoINVRT7 is between 6 and 7 times faster than OptoINVRT1 and OptoINVRT2 by this metric.
OptoINVRT7 Is Superior for Chemical Production in Dynamic Fermentations.
We previously developed an optogenetic method that uses light to control the transition from growth- to production-phase in fermentations.5 We used OptoEXP (Supplementary Figure S8a) to induce with blue light the expression of PDC1, a gene essential for growth in glucose, but also responsible for ethanol byproduct formation (Supplementary Figure S8b). In the same strain, we also used OptoINVRT circuits to induce the production of lactic acid (LA) or isobutanol in the dark (Supplementary Figure S8c,d). Thus, cells grow only under blue light when they are able to produce ethanol. At some measured cell density, ρs, cultures are moved to the dark, where cells switch from OptoEXP-mediated expression of PDC1 and OptoINVRT-mediated repression of LDH or ILV2 (for lactic acid or isobutanol production, respectively), to expressing the biosynthetic pathway of interest with reduced competition from ethanol production (Figure 4a). This strategy relies on decreasing concentrations of Pdc1p and Gal80p by degradation or mitotic dilution upon moving cells from light to dark, which allows them to switch from growth to production. Therefore, we expected to see higher chemical production when using OptoINVRT7 due to its enhanced rate of Gal80p degradation.
Figure 4.

Comparison of optogenetic circuits for lactic acid production. (a) Schematic of using OptoEXP and OptoINVRT circuits to control PDC1 and LDH, respectively for lactic acid (LA) production. Enhanced degradation of Gal80p in OptoINVRT7 leads to more rapid activation of LDH and increased LA production. (b,c) LA production at different ρs values using (b) OptoINVRT2 (YEZ212C) and OptoINVRT7 (YEZ212) with PGAL1-M or (c) OptoINVRT2 (YEZ597–5) and OptoINVRT7 (YEZ598–4) with PGAL1-S; full light samples were kept under continuous light throughout growth and production phases. **P < 0.01, ***P < 0.001. Statistics are derived using a one-sided t-test. All data are shown as mean values; dots represent individual data points; error bars represent the s.d. of four biologically independent 1-mL sample replicates exposed to the same conditions. All experiments were repeated at least three times.
To analyze OptoINVRT7 for LA production, we compared it to OptoINVRT2, which we previously found to achieve the highest LA titers among the original OptoINVRT circuits.5 We used OptoINVRT2 or OptoINVRT7 to control the expression of LDH for LA production in 48-h fermentations in the dark. We tested these circuits, initially using PGAL1-M to drive LDH expression, in strains carrying pdc1Δ, pdc5Δ, pdc6Δ, gpd1Δ, and gal80Δ deletions, as well as PC120-PDC1 controlled by OptoEXP5 (Supplementary Figure S8a,b), making strains YEZ212C with OptoINVRT2 and YEZ212 with OptoINVRT7 (see Methods, Supplementary Table S2). After screening for the most productive colonies, we optimized the cell density, ρs, at which cultures are switched into the dark (measured as optical density units, OD600). When using OptoINVRT2 (YEZ212C), the maximum LA production occurs at the relatively low ρs of 3.5 (Figure 4b). This is likely caused by the slower turnover rate of Gal80p in OptoINVRT2, and thus the overreliance on time and mitotic dilution to reduce the intracellular concentrations of Gal80p upon moving cells into the dark. In contrast, OptoINVRT7 (YEZ212) reaches maximum LA production when ρs = 7.0 (Figure 4b), achieving 180.2% higher LA production compared to OptoINVRT2 (YEZ212C) at the same ρs. The design of OptoINVRT7 is consistent with the higher optimal ρs obtained because the degron domain decreases the stability of Gal80p, making its depletion faster and less dependent on mitotic dilution, which allows for strong expression of LDH and thus LA production when shifting light conditions later in the fermentation at higher cell densities.
The effect of these circuits on the relative expression of LDH and PDC1 can be indirectly observed by the ratios of LA and ethanol production (Supplementary Figure S9). For Opto-INVRT2, this LA:ethanol ratio reaches as much as 1.1 only when ρs is 3.5 (Supplementary Figure S9a). On the other hand, OptoINVRT7 can reach higher ratios, of up to around 1.5, in a wider range of ρs values (from 3.5 to at least 7.0). Comparing ethanol production between full light and light turned off at different cell densities, it is apparent that the levels of LDH expressed are able to effectively compete with Pdc1p at all ρs values when using OptoINVRT7, which for OptoINVRT2 only occurs when ρs is 3.5 or lower (Supplementary Figure S9b). By allowing higher values of ρs, fermentations regulated by OptoINVRT7 achieve higher final cell densities (Supplementary Discussion, Supplementary Figure S9c), which, combined with faster expression of LDH, results in more productive fermentations (Figure 4b). These results demonstrate how the faster overexpression kinetics afforded by OptoINVRT7 improve chemical production.
To investigate the effect that promoter strength has on the chemical production response of OptoINVRT circuits, we carried out similar experiments using PGAL1-S to drive LDH expression (using strains YEZ597–5 with OptoINVRT2 and YEZ598–4 with OptoINVRT7). We found that the stronger PGAL1-S increases LA production, as well as LA to ethanol ratios in both circuits (Figure 4c and Supplementary Figure S10) relative to PGAL1-M (Figure 4b and Supplementary Figure S9). While using PGAL1-S with OptoINVRT7 results in only a 5% increase in the maximum level of LA production compared to using PGAL1-M, the stronger PGAL1-S substantially widens the range of ρs values (3.5–9.5 OD) at which LA titers surpass 6 g/L, which reflects an increase in the robustness of fermentations carried out with YEZ598–4 (Figure 4c). On the other hand, using PGAL1-S with OptoINVRT2 results in a more substantial increase, 37%, in the maximum levels of LA production compared to using PGAL1-M. The much higher effect of PGAL1-S in OptoINVRT2 suggests that slower circuits have more to gain from the strength of the promoter they control than more rapid ones. However, PGAL1-S does not increase the robustness of fermentations done with OptoINVRT2, as the maximum LA titer is still achieved only in a narrow window of ρs = 3.5. Furthermore, OptoINVRT7 controlling PGAL1-S boosts the ratios of LA to ethanol production to 2.5 (Supplementary Figure S10a), indicating a stronger shift of yeast metabolism toward LA fermentation, compared to other circuits or promoters. This suggests that the combination of a faster optogenetic circuit and a stronger inducible promoter results in overall enhanced production robustness.
As a second example of a biosynthetic pathway, we explored how much the faster OptoINVRT7 improves production of isobutanol, an advanced biofuel whose biosynthesis we have demonstrated can be controlled optogenetically.5 We previously found that isobutanol is best produced with OptoINVRT1, and that the maximum isobutanol titers are achieved at ρs values that are higher compared to the optimal ρs for lactic acid production.5 Thus, we compared OptoINVRT7 with OptoINVRT1 in their efficacy to produce isobutanol, using strains containing pdc1Δ, pdc5Δ, pdc6Δ, gpd1Δ, ald6Δ, bat1Δ, and gal80Δ, as well as multiple copies of PC120-PDC1 controlled by OptoEXP,5 and PGAL1-M-ILV2 controlled by OptoINVRT1 (YEZ535) or OptoINVRT7 (YEZ536). Next, we transformed these strains with a 2 μ plasmid containing the mitochondrial isobutanol pathway38 (including PGAL1-M-ILV2) and screened for high-producing colonies to find YEZ544–1 (with OptoINVRT1) and YEZ546–2 (with OptoINVRT7) (see Methods, Supplementary Table S2). We found that YEZ546–2 produced more isobutanol than YEZ544–1 at all ρs values tested (Supplementary Figure S11). Moreover, the highest increase in isobutanol production with OptoINVRT7 occurs at lower ρs values, obtaining as much as a 631% boost in isobutanol titers at ρs = 3.0. This also indicates an increase in process robustness and suggests that for isobutanol production, the rate of ILV2 induction is more important when inducing at lower cell densities.
The increase in process robustness that OptoINVRT7 brings to isobutanol production is more modest than what we observe for LA production, which is probably due to various factors that complicate isobutanol biosynthesis. This includes the mitochondrial compartmentalization of the isobutanol pathway, which sensitizes productivity to changes in mitochondrial activity.39 Isobutanol is also substantially more toxic than LA, which could negatively affect the health and productivity of strains when induced at lower cell densities. Finally, isobutanol biosynthesis, as engineered in our strains, can theoretically restore only as much as 50% of the NAD+ that ethanol or LA biosynthesis can restore, which may also impact how cells respond to earlier chemical induction (Supplementary Discussion). Nevertheless, these results demonstrate that the faster kinetics of OptoINVRT7 can help improve the productivity of different biosynthetic pathways.
CONCLUSIONS
In this study, we demonstrate the versatility with which OptoINVRT circuits can be designed to influence their activation kinetics and dynamic range. The key design parameters to manipulate the OFF-to-ON (light-to-dark) kinetics of these circuits are the protein stability and activity of the transcription factor (Gal4p) and the repressor (Gal80p). Much of the time delay in OptoINVRT circuit activation comes from the high stability of Gal80p, so tagging this repressor with a protein degradation tag enhances the light-to-dark system kinetics. However, if the degradation tag is too strong, as in OptoINVRT5, it increases the leakiness of the circuit. Therefore, finding a balanced level of constitutive Gal80p degradation, by fusing it to ODCmut, was key to achieving the high performance of OptoINVRT7. Combining this moderately destabilized Gal80p with light-dependent degradation of Gal4p, by fusing it to a photosensitive degron domain, provides OptoINVRT7 with a substantially enhanced activation kinetics, with more than a 6- to 7-fold reduction in apparent time delay relative to earlier OptoINVRT circuit designs, and low leakiness, reaching a light-to-dark induction fold of 132.9 ± 22.6. Thus, protein stability of Gal80p and Gal4p alone accounts for much of the kinetic differences between OptoINVRT circuits. Manipulating the stability of regulatory proteins could also change the kinetics of other gene circuits, such as negative autoregulation systems, which have also been shown to increase response rates and reduce noise.40-42 For yeast metabolic engineering, this establishes OptoINVRT7 as the most rapid OptoINVRT circuit to date, capable of offering robust control over engineered metabolic pathways for enhanced chemical production without observable harmful or off-target effects.
Promoter engineering to boost gene expression further enhances the performance of OptoINVRT circuits. By eliminating Mig1p-binding and introducing additional, evenly spaced, UAS in PGAL1, we developed PGAL1-S, one of the strongest promoters ever produced in S. cerevisiae. Using OptoINVRT7 to control PGAL1-S achieves light-tunable levels of gene expression, which in full darkness surpasses the activities of the most commonly used constitutive and inducible promoters in yeast (Figure 2b), including the activity of PGAL1 in galactose, the strongest native transcriptional activity known in yeast. While the maximum fold of induction attainable with these circuits is not as high as what has been reported for other systems,37 the combination of flexibility, tunability, dynamic range, and maximum levels of gene expression afforded by OptoINVRT-controlled PGAL1-S is particularly useful for metabolic engineering applications.
The rapid response of OptoINVRT7 enhances microbial chemical production and the robustness of fermentations. Strains using OptoINVRT7 produce 53.6% more LA than strains using OptoINVRT2 (the previous best optogenetic circuit for lactic acid production5) and 16.3% more isobutanol than strains using OptoINVRT1 (the previous best optogenetic circuit for isobutanol production5). The improved light-to-dark activation kinetics of OptoINVRT7, due to rapid degradation of Gal80p, allows higher induction levels of biosynthetic enzymes at potentially higher optimal ρs values, relative to slower OptoINVRT circuits, resulting in biocatalysts of higher activity and density. This explains in part why production of LA benefits more from OptoINVRT7 than isobutanol production. For lactic acid production, Opto-INVRT7 doubles the optimal ρs from 3.5 to 7.0, resulting in a more active and plentiful biocatalyst. The biocatalyst activity is further enhanced when using PGAL1-S, boosting lactic acid production at any ρs, but the optimal ρs is the same. In contrast, the optimal ρs for isobutanol production with OptoINVRT1 is already high (ρs = 8.75) and remains unchanged with OptoINVRT7, resulting only in a more active (but not more plentiful) biocatalyst. Furthermore, the inability of isobutanol biosynthesis in this strain to replenish NAD+ to the same extent as LDH limits the amount of isobutanol produced when the light is switched off at cell densities outside the optimal ρs range (see Supplementary Discussion). Nevertheless, OptoINVRT7 improves the productivity and robustness of fermentations for both products by improving chemical production at any ρs and, in the case of lactic acid, widening the range of ρs that achieve high levels of production. Therefore, metabolic pathways that compete with cell growth can benefit from the fast kinetics of OptoINVRT7, especially when it increases the optimal ρs values.
OptoINVRT circuits may be used to control gene expression with light in other settings beyond metabolic engineering. These circuits could make it possible to exploit the tunability, reversibility, and orthogonality of light for basic research, using their fast kinetics and high-fold levels of induction. Additionally, OptoINVRT circuits may be applicable in other organisms in which the functions of GAL4 and GAL80 are transferable; for example, Drosophila melanogaster,43 or zebrafish.44 This would require maintaining the intact Gal4p activation domain for proper interaction with Gal80p. Therefore, OptoINVRT circuits can be used to address fundamental questions in cell biology in yeast and potentially other organisms.
Moving forward, we envision a modular technological platform of optogenetic gene expression systems with high dynamic ranges and different activation kinetics. We anticipate that the combination of rapid optogenetic circuits, such as OptoINVRT7, with accurate dynamics models augmented by live feedback from genetically encoded biosensors that monitor metabolic activity or cell physiology, as well as cell density measurements, and other real-time data, will enable tighter closed-loop control systems to improve yields from microbial fermentations.1,12 The technology in this study represents a critical step in bringing us closer to such computer-assisted automatic control and optimization of engineered metabolisms for microbial chemical production.
METHODS
Assembly of DNA Constructs.
We cloned promotergene-terminator sequences into previously described standardized vector series (pJLA vectors).38 This allows for easy manipulation and generation of multigene plasmids. All genes were designed to have NheI and XhoI restriction sites at the 5′ and 3′ ends, respectively, which were used to insert the genes into pJLA vectors. Each promoter-gene-terminator construct is flanked by XmaI and AgeI restriction sites at their 5′ ends, and MreI, AscI, and BspEI sites at their 3′ ends, which we used for easy assembly of multigene plasmids, as previously described5,38 (Supplementary Figure S12; see Supplementary Table S1).
Qiagen Miniprep, Qiagen Gel Extraction, and Qiagen PCR purification kits were used to extract and purify plasmids and DNA fragments, following manufacturer’s instructions. Most genes and promoters (GAL4, GAL80, GFP, PGAL1, PTEF, PTDH3, PPGK1, PCYC1, PADH1, CLN2-PEST) were amplified from yeast genomic DNA or lab plasmids, using Phusion Polymerase from NEB, following manufacturer’s instructions. Other genes were amplified from plasmids kindly provided by other groups: PsLDH from plasmid pET28a, LDH from Dr. Jinsuk J. Lee; and the photosensitive degron derived from the fusion of phototropin1 LOV2-V19L domain from Arabidopsis thaliana and a synthetic degradation sequence derived from the murine ornithine decarboxylase (ODC) from plasmid pDS143 from Dr. Christof Taxis.45 The codon-optimized sequence for ODCmut was purchased as a gBlock from IDT. The sequence for the PGAL1-M promoter was synthesized as a gBlock from IDT. The sequence for the PGAL1-S promoter was synthesized by Bio Basic’s gene synthesis service. When pJLA vectors were not available, we used Gibson isothermal assembly to produce our constructs, based on the protocols of the Megason lab at Harvard. Enzymes were purchased from NEB (XmaI, AscI, NheI, XhoI, BspEI, AgeI, T4 DNA ligase, Phusion Polymerase) and Thermo Fisher (MreI).
Gene constructs in pYZ12-B were integrated into the HIS3 locus of the genome by linearizing the plasmid with PmeI. Gene constructs in pYZ23 were integrated into the YARCdelta546 sites of the genome by linearizing the plasmid with PmeI.
All vectors were sequenced with Sanger Sequencing from GENEWIZ before using them to transform yeast. We avoid using tandem repeats to prevent recombination after yeast transformation, and thus do not observe instability of plasmids.
Yeast Transformations.
Yeast transformations were carried out using standard lithium acetate protocols, and the resulting strains are cataloged in Supplementary Table S2. Gene constructs derived from pYZ12-B and pYZ23 vectors were genomically integrated into the HIS3 locus and δ-sites (YARCdelta5), respectively. These vectors were first linearized with PmeI, followed by purification of the DNA fragments using the Qiagen PCR purification kit before using them for yeast transformation. Gene deletions were carried out by homologous recombination. DNA fragments containing antibiotic resistance cassettes flanked with Lox-P sites (located in the original plasmids) were amplified with PCR from pAG2647 (containing the hygromycin resistance gene HygB-PT) or pUG6 (containing the G418 resistance gene KanMX), using primers with 40 base pairs of homology to the promoter and terminator regions of the gene targeted for deletion. After transformation, cells were plated on synthetic complete (SC) drop out media depending on the autotrophy restored by the construct. In the case of antibiotic selection, cells were plated onto nonselective YPD plates for 16 h, then replica plated onto YPD plates with 300 μ/mL hygromycin (purchased from Invitrogen), 200 μg/mL nourseothricin (purchased from WERNER BioAgents), or 200 μg/mL G418, purchased from Gibco by Life Technologies). Zeocin was used to select for δ-integration ranging from 800 to 1200 μg/mL (purchased from Thermo Fisher Scientific).
All strains with genomic integrations or gene deletions were genotyped with PCR to confirm their accuracy. We integrated constructs in the HIS3 locus or δ-sites to promote strain stability.
Yeast Cell Culture Growth, Centrifugation, and Optical Measurements.
Unless otherwise specified, liquid yeast cultures were grown in 24-well plates, at 30 °C and shaken at 200 rpm, in either YPD or SC-dropout media with 2% glucose. To stimulate cells with light, we used blue LED panels (HQRP New Square 12′′ Grow Light Blue LED 14W), placed 40 cm from cell cultures (Supplementary Figure S13). To control light duty cycles, the LED panels were regulated with a Nearpow Multifunctional Infinite Loop Programmable Plug-in Digital Timer Switch (purchased from Amazon). Cell cultures were centrifuged in a table-top centrifuge, with 24-well plate rotor adaptors. Unless otherwise specified, plates were centrifuged at 1000 rpm for 10 min.
Fluorescence and optical density (OD600) measurements were taken using a TECAN plate reader (infinite M200PRO). The excitation and emission wavelengths used for GFP fluorescence measurements were 485 and 535 nm, respectively, using an optimal gain for all measurements. To process fluorescence data, the background fluorescence from the media was first subtracted from values. Then, the GFP/OD600 values of cells lacking a GFP construct were subtracted from the fluorescence values (GFP/OD600) of each sample to correct for light bleaching of the media and cell contents (GFP bleaching was not observed under our conditions). Thus, reported values were calculated per the following formula, where GFP corresponds to fluorescence measurements and OD corresponds to OD600.
All fluorescence measurements were done at the end of experiments or on samples taken from experimental cultures, such that potential activation of VP16-EL222 by the light used to excite GFP did not affect our experiments or results. Controls using constitutive PTEF1 to express GFP showed that this fluorophore does not bleach under the light conditions tested (Figure 1b,c).
To measure cell concentration, optical density measurements were taken at 600 nm wavelength, using media (exposed to the same conditions as the yeast) as blank. Measurements were done with the TECAN plate reader (infinite M200PRO) or Eppendorf spectrophotometer (BioSpectrometer basic), from samples diluted to a range of OD600 of 0.1 to 1.0.
All experiments involving light-inducible strains were done under minimal ambient light, unless otherwise specified, to avoid unwanted activation of optogenetic systems.
Duty cycles used in experiments shown in Figure 3b were chosen based on cycles that are able to fine-tune gene expression of OptoEXP.5
OptoINVRT circuit sensitivities were calculated using the formula below, as previously described.5
Flow Cytometry Experiments.
GFP expression was quantified by flow cytometry using a BD LSR II flow cytometer (BD Biosciences, San Jose, CA, USA) with the excitation wavelength of 488 nm and the emission wavelength of 530 nm. The gating used in our analyses was defined to include positive (YEZ186) and negative (YEZ140) control cells based on GFP fluorescence, but exclude particles that are either too small or too large to be single living yeast cells, based on the side scatter (SSC-A) vs forward scatter (FSC-A) plots as previously described.5 Median fluorescence values were determined from 20,000 cells. Data were analyzed with the FlowJo Version 10 software (Tree Star, Ashland, OR, USA).
All fluorescence measurements were done at the end of experiments or on samples taken from experimental cultures, such that potential activation of VP16-EL222 by the light used to excite GFP did not affect our experiments or results.
Construction of OptoINVRT Systems.
OptoINVRT1–3 circuits were previously characterized in yeast strains YEZ44 (CENPK.2–1C, gal80-Δ, gal4-Δ) and Y202 (S288C, pdc1Δ, pdc5Δ, pdc6Δ, gal80-Δ containing pJLA121PDC10202). We chose to test the new OptoINVRT5 and 7 circuits in both Y202 and YEZ25 (CENPK.2–1C, gal80-Δ), because we previously observed that endogenous GAL4 improves the activity of GAL4-PSD.5
Gene circuits were assembled using restriction enzyme digests and ligations afforded by pJLA vectors, in which ORFs were inserted using NheI and XhoI sites, and multiple cassettes assembled using XmaI (or AgeI), MreI (or BspEI), and AscI.38 OptoINVRT5 and OptoINVRT7 were constructed by assembling five promoter-gene-terminator sequences into single integration vectors targeting the HIS3 locus (EZ-L400 and EZ-L437C; Supplementary Table S1). The photosensitive degron (PSD) used is derived from the fusion of phototropin1 LOV2 domain (V19L mutant) from Arabidopsis thaliana and a synthetic degradation sequence derived from the murine ornithine decarboxylase (ODC). This construct, EZ-L266, was published previously.5
The new promoters, PGAL1-M and PGAL1-S, were constructed in a similar method to create EZ-L437 and EZ-L436 (Supplementary Table S1).
Characterization of OptoINVRT Systems.
OptoINVRT5 and OptoINVRT7, controlling GFP expression, were transformed in YEZ25 (CENPK.2–1C, gal80-Δ) to make YEZ230–5 (EZ-L400) and YEZ230C (EZ-L437C), respectively. OptoINVRT circuits were characterized in strains YEZ100 (OptoINVRT1), YEZ101 (OptoINVRT2), YEZ102 (OptoINVRT3), YEZ230–5 (OptoINVRT5), and YEZ230C (OptoINVRT7), using YEZ140 and YEZ186 as controls. We monitored GFP expression as previously described,5 except in this case we exposed cells to full light, complete darkness, or light pulses of 8 s ON/72 s OFF.
We tested PGAL1-M and PGAL1-S in an S288C background, Y202 (S288C, gal80Δ, pdc1Δ, pdc5Δ, and pdc6Δ, containing pJLA121-PDC10202), with strains YEZ210 (EZ-L437) and YEZ209 (EZ-L436), (Supplementary Table S2), using YEZ94 and YEZ171 as controls. The new promoters were tested in a CEN.PK2–1C background with YEZ229 (EZ-L436) and YEZ230 (EZ-L437), using YEZ140 and YEZ186 as controls (Supplementary Table S2). We monitored GFP expression, as previously described.5
To investigate the effects of OptoINVRT circuits on cell growth, we grew cultures of OptoINVRT1 (YEZ100), OptoINVRT2 (YEZ101), OptoINVRT3 (YEZ102), and OptoINVRT7 (YEZ230C), as well as a control strain (YEZ140), overnight in SC-his + 2% glucose under blue light (485 nm). The next day, cultures were diluted in triplicates into fresh media to an OD600 of 0.1, and grown under full 485 nm light or in the darkness at 200 rpm and 30 °C. OD600 values were taken 10 h after inoculation, then taken every 2–3 h until stationary phase was reached.
Comparison of OptoINVRT7 to Other Yeast Promoters.
The previously described artificial transcription factors activated by β-estradiol were made by fusing the bacterial LexA DNA binding domain, hER, and various transactivation domains.31 From that study, we chose the two transcription factors achieving the highest levels of gene expression, LexA-ER-B112 and LexA-ER-VP16, to compare them to OptoINVRT7. We obtained constructs containing these systems (AddGene (#58430 and #58437)), as well as a minimal PCYC1 promoter containing 8 lexAop binding sites (AddGene #58435). We subcloned LexA-ER-B112 and LexA-ER-VP16 using Gibson Assembly into pJLA1210301 under the strong constitutive promoter PPGK1.48 We then placed the synthetic promoter upstream of GFP using Gibson Assembly; during cloning, an additional binding site was generated, resulting in 9 total lexAop binding sites. This P9xlexAop-GFP cassette was then inserted downstream of LexA-ER-B112 and LexA-ER-VP16 using sequential gene insertion,38 generating pMAL81 and pMAL125. We integrated these constructs into CEN.PK2–1C to produce YMAL34 and YMAL35.
In a separate study, the Tetracycline Off system was used to induce transcriptional repression in yeast with doxycycline.33 The tTA transcription factor driven by a minimal PCYC1 promoter containing 7 tetO binding sites (P7xtetO) was inserted into pJLA1210202, which contains the terminator TACT1. The tTa and P7xtetO were respectively amplified from plasmids pRS304 and Eze-kIura3mb-7xtetOpr (generously provided by Kristala Prather). We then inserted P7xtetO upstream of GFP using Gibson Assembly and placed the P7xtetO-GFP cassette downstream of P7xtetO_tTA using sequential gene insertion, generating pMAL140. We integrated this construct into CEN.PK2–1C to produce YMAL36.
OptoINVRT Light-to-Dark Kinetic Evaluation.
Cultures of YEZ100, YEZ230C, YEZ140, and YEZ186 were grown overnight in SC-his + 2% glucose and blue light (485 nm). The next day, cultures were diluted in quadruplicates into fresh media to an OD600 of 0.1, and grown under full 485 nm light for 3 h at 200 rpm and 30 °C. Cultures were then switched to complete darkness (wrapped in aluminum foil) maintaining their incubation under the same agitation and temperature conditions. Samples were taken at 0, 2, 4, 6, and 9 h, diluted in PBS (to an OD600 of 0.1), and kept in ice until fluorescence measurements were taken with flow cytometry.
Percent of maximum activation was calculated using the following equation:
To calculate the half activation time of each circuit, we constructed a trendline using the linear portion of each activation curve and used it to calculate the time at the x-intercept of the trendline (i.e., calculated the time (x) value at which GFP (y) = background fluorescence).
Screening of Lactic Acid Producing Strains.
Starting from Y202, a triple PDC deletion S288C strain, we deleted GPD1 using HygB to produce YEZ200, from which we removed the antibiotic resistance markers with Cre recombinase to produce YEZ203. We then integrated OptoEXP and either OptoINVRT2 (EZ-L260) or OptoINVRT7 (EZ-L439) into YEZ203 to produce YEZ207C and YEZ207, respectively. We integrated multiple copies of a construct containing LDH from Pelodiscus sinensis5 controlled by either PGAL1-M or PGAL1-S and PC120-PDC1 (EZ-L353 or EZ-L827, respectively) into delta sites of YEZ207C and YEZ207. Then we counter-selected these strains against plasmid pJLA121-PDC10202 containing PDC1 using 5FOA, resulting in strains YEZ212C (OptoINVRT2) and YEZ212 (OptoINVRT7), respectively for PGAL1-M constructs, and YEZ597–5 (OptoINVRT2) and YEZ598–4 (OptoINVRT7) for PGAL1-S constructs (see Supplementary Table S2). These strains induce PDC1 and repress LDH expression in the light, while in the dark they stop inducing PDC1 and induce LDH instead.
Colonies from each transformation plate (grown in glucose and under blue light) were screened for lactic acid (8 from each strain). Each colony was used to inoculate 1 mL of SC-his + 2% media in 24 well plates and grown overnight at 30 °C, 200 rpm, and blue light. The next morning, each culture was diluted to 0.1 OD600 and grown for 12 h at 30 °C, 200 rpm, and blue light. After these incubation periods, the cultures reached OD600 values of 5. The cultures were then moved into the dark for 6 h. Then the cultures were resuspended in fresh media, in plates that were then sealed with Nunc Sealing Tape (Thermo Scientific) to begin the fermentations. The plates were incubated in the dark at 30 °C and shaken at 200 rpm for 48 h during fermentation. Subsequently, the cultures were centrifuged, and the supernatants collected for HPLC analysis.
The highest lactic acid-producing colonies of each strain were used to optimize the pregrowth parameters of fermentation. To find the optimal cell density at which to switch cultures from light to dark (ρs), we diluted the overnight cultures into 1 mL of the SC dropout medium to different OD600 values, ranging from 0.04 to 0.32. The lactic acid-producing strains were then grown for 16 h under continuous 15 s ON/65 s OFF blue light (18.75% dose), which is sufficient to achieve robust growth. By not using 100% light dose during cell growth, we avoid maximal production of Gal80p and Pdc1p, which need to be degraded or diluted during the production phase. We then measured the final OD600 values, which correspond to ρs, and incubated the cultures in the dark for 6 h optimized as previously described.5 After this dark incubation period, the cultures were centrifuged at 1000 rpm for 5 min and suspended in fresh SC dropout media containing glucose at 20.2 g/L. The plates were sealed with Nunc Sealing Tape, and incubated in the dark for fermentation at 30 °C and 200 rpm. The “Full-Light” control cultures were grown under constant blue light throughout the procedure (during the growth phase, the “dark” incubation period, and the fermentation). Cultures were harvested after 48 h and centrifuged at 1000 rpm for 10 min. Supernatants were analyzed with HPLC.
Construction and Screening of Isobutanol Producing Strains.
We integrated OptoEXP and either OptoINVRT1 (EZ-L571) or OptoINVRT7 (EZ-L439) into YEZ235 (CEN.PK2–1C with BAT1, GPD1, GAL80, ALD6, PDC1, PDC5, and PDC6 deleted; and with all antibiotic resistance markers removed using Cre recombinase, except HygB-PT marker used to delete GPD1) resulting in YEZ531 and YEZ532, respectively. We integrated multiple copies of a construct containing PGAL1-M-ILV2 and PC120-PDC1 (from EZ-L352) into δ-sites of YEZ531 and YEZ532 and then counter-selected against plasmid pJLA121-PDC10202 containing PDC1 using 5FOA, resulting in strains YEZ535 and YEZ2536, respectively (see Supplementary Table S2). These strains induce PDC1 and repress ILV2 expression in the light; while in the dark they stop inducing PDC1 and induce ILV2 instead. After confirming that these strains grow in the light but not in the dark on SC–his + 2% glucose plates, we transformed YEZ535 and YEZ536 with EZ-L390 to make YEZ544 and YEZ546, respectively.
Colonies from each transformation plate (grown in SC-ura + 2% glucose and under blue light) were screened for isobutanol (16 colonies from each plate). Each colony was used to inoculate 1 mL of SC-ura + 2% media in 24 well plates and grown overnight at 30 °C, 200 rpm, and continuous blue light (100% dose). The next morning, each culture was diluted to 0.1 OD600 and grown for 20 h at 30 °C, 200 rpm, and 15 s ON, 65 s OFF blue light (18.75% dose). After these incubation periods, the cultures reached cell densities between 6 and 9 OD600. The cultures were then incubated in the dark for 4 h at 30 °C, 200 rpm. Then the cultures were centrifuged at 1000 rpm for 5 min and resuspended in fresh SC-ura + 4% glucose media, in plates that were then sealed with Nunc Sealing Tape (Thermo Scientific) to begin the fermentations. The plates were incubated in the dark at 30 °C and shaken at 200 rpm for 48 h during fermentation. Subsequently, the cultures were centrifuged, and the supernatants collected for HPLC analysis.
After finding the highest producing colonies of YEZ544 (YEZ544–1) and YEZ546 (YEZ546–2), from the screens above, we optimized their pregrowth parameters of fermentation to maximize isobutanol production. To find the optimal cell density at which to switch cultures from light to dark, ρs, we grew overnight cultures in SC-ura + 2% glucose and continuous blue light (100% dose). The next day we diluted these cultures into 1 mL of the SC-ura + 2% glucose to different initial OD600 values, ranging from 0.04 to 0.32. The isobutanol-producing strains were then grown for 20 h under continuous 15 s ON/65 s OFF (18.75% dose) blue light. We then incubated the cultures in the dark for 6 h at 30 °C and shaken at 200 rpm. After this dark incubation period, the cultures were centrifuged at 1000 rpm for 5 min and suspended in fresh SC dropout media containing glucose at 20.4 g/L. The plates were sealed with Nunc Sealing Tape and incubated in the dark for fermentation at 30 °C and 200 rpm. “Full-Light” control cultures were grown under constant blue light during the growth phase, the dark incubation period, and the fermentation. Cultures were harvested after 48 h and centrifuged at 1000 rpm for 10 min. Supernatants were analyzed with HPLC.
Detection of Small Molecules.
The concentrations of glucose, lactic acid, isobutanol, and ethanol were quantified with high-performance liquid chromatography (HPLC), using an Agilent 1260 Infinity instrument (Agilent Technologies, Santa Clara, CA, USA). Samples were centrifuged at 1000 rpm for 10 min to remove cells and other solid debris and analyzed using an Aminex HPX-87H ion-exchange column (Bio-Rad, Richmond, CA, USA). The column was eluted with a mobile phase of 5 mM sulfuric acid at 55 °C and a flow rate of 0.6 mL/min. Glucose, lactic acid, isobutanol, and ethanol were monitored with a refractive index detector (RID). To determine their concentration, the peak areas were measured and compared to those of standard solutions for quantification.
Supplementary Material
ACKNOWLEDGMENTS
We thank all members of the Avalos, Kevrekidis, and Toettcher laboratories for helpful comments. We thank Dr. Christina DeCoste, Dr. Katherine Rittenbach, and the Princeton Molecular Biology Flow Cytometry Resource Center for assistance with flow cytometry experiments. We thank Dr. Kris Prather for pRS304 and kIura3mb-7xtetOpr, as well as Dr. Joerg Stelling for FRP467-PACT1(−1-520)-LexA-ER-haVP16, FRP880_PACT1(−1-520)-LexA-ER-haB112-TCYC1, and FRP795_insul-(lexA-box)8-PminCYC1-Citrine-TCYC1. This work was supported by the Maeder Graduate Fellowship in Energy and the Environment (to EMZ), The Pew Charitable Trusts, the U.S. DOE Office of Biological and Environmental Research, Genomic Science Program Award DE-SC0019363, and NSF CAREER Award CBET-1751840 (to JLA), the NIH grant DP2EB024247 (to JET) and a Schmidt Transformative Technology grant (to JET, IGK and JLA), and the DARPA Lagrange Program, Contract no. N66001-18-C-4031 (RJL and IGK).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.0c00305.
Supplementary discussion on lactic acid fermentations; sequences of engineered degradation tags and promoters; tables of plasmids and strains; schematics of yeast GAL regulon, chemical production pathways, and plasmid assemblies; effects of circuits on cell growth; performance of circuits and promoters in different media and background strains; effects of different circuits on lactic acid, ethanol, and isobutanol production (PDF)
The authors declare no competing financial interest.
The authors declare that all data supporting the findings of this study are available within the paper (and Supporting Information), but original data that supports the findings are available from the corresponding authors upon reasonable request.
Contributor Information
Evan M. Zhao, Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey 08544, United States
Makoto A. Lalwani, Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey 08544, United States
Robert J. Lovelett, Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey 08544, United States; Department of Chemical and Biomolecular Engineering, Johns Hopkins University, Baltimore, Maryland 21218, United States
Sergio A. García-Echauri, Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey 08544, United States
Shannon M. Hoffman, Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey 08544, United States
Christopher L. Gonzalez, Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey 08544, United States
Jared E. Toettcher, Department of Molecular Biology, Princeton University, Princeton, New Jersey 08544, United States
Ioannis G. Kevrekidis, Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey 08544, United States; Department of Chemical and Biomolecular Engineering, Johns Hopkins University, Baltimore, Maryland 21218, United States
José L. Avalos, Department of Chemical and Biological Engineering, Department of Molecular Biology, and The Andlinger Center for Energy and the Environment, Princeton University, Princeton, New Jersey 08544, United States
REFERENCES
- (1).Lalwani MA, Zhao EM, and Avalos JL (2018) Current and future modalities of dynamic control in metabolic engineering. Curr. Opin. Biotechnol 52, 56–65. [DOI] [PubMed] [Google Scholar]
- (2).Tan SZ, and Prather KLJ (2017) Dynamic pathway regulation: recent advances and methods of construction. Curr. Opin. Chem. Biol 41, 28–35. [DOI] [PubMed] [Google Scholar]
- (3).Venayak N, Anesiadis N, Cluett WR, and Mahadevan R (2015) Engineering metabolism through dynamic control. Curr. Opin. Biotechnol 34, 142–152. [DOI] [PubMed] [Google Scholar]
- (4).Gupta A, Reizman IMBB, Reisch CR, and Prather KLJJ (2017) Dynamic regulation of metabolic flux in engineered bacteria using a pathway-independent quorum-sensing circuit. Nat. Biotechnol 35, 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zhao EM, Zhang Y, Mehl J, Park H, Lalwani MA, Toettcher JE, and Avalos JL (2018) Optogenetic regulation of engineered cellular metabolism for microbial chemical production. Nature 555, 683–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lalwani MA, Ip SS, Carrasco-López C, Day C, Zhao EM, Kawabe H, and Avalos JL (2020) Optogenetic control of the lac operon for bacterial chemical and protein production. Nat. Chem. Biol, DOI: 10.1038/s41589-020-0639-1. [DOI] [PubMed] [Google Scholar]
- (7).Zhao EM, Suek N, Wilson MZ, Dine E, Pannucci NL, Gitai Z, Avalos JL, and Toettcher JE (2019) Light-based control of metabolic flux through assembly of synthetic organelles. Nat. Chem. Biol 15, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Motta-Mena LB, Reade A, Mallory MJ, Glantz S, Weiner OD, Lynch KW, and Gardner KH (2014) An optogenetic gene expression system with rapid activation and deactivation kinetics. Nat. Chem. Biol 10, 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pathak GP, Strickland D, Vrana JD, and Tucker CL (2014) Benchmarking of optical dimerizer systems. ACS Synth. Biol 3, 832–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Fiore G, Perrino G, Di Bernardo M, and Di Bernardo D (2016) In Vivo Real-Time Control of Gene Expression: A Comparative Analysis of Feedback Control Strategies in Yeast. ACS Synth. Biol 5, 154–162. [DOI] [PubMed] [Google Scholar]
- (11).Benzinger D, and Khammash M (2018) Pulsatile inputs achieve tunable attenuation of gene expression variability and graded multi-gene regulation. Nat. Commun, DOI: 10.1038/s41467-018-05882-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Carrasco-López C, García-Echaurí SA, Kichuk T, and Avalos JL (2020) Optogenetics and biosensors set the stage for metabolic cybergenetics. Curr. Opin. Biotechnol 65, 1–14. [DOI] [PubMed] [Google Scholar]
- (13).Lugagne J-B, and Dunlop MJ (2019) Cell-machine interfaces for characterizing gene regulatory network dynamics. Curr. Opin. Syst. Biol 14, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Olson EJ, Hartsough LA, Landry BP, Shroff R, and Tabor JJ (2014) Characterizing bacterial gene circuit dynamics with optically programmed gene expression signals. Nat. Methods 11, 449–455. [DOI] [PubMed] [Google Scholar]
- (15).Milias-Argeitis A, Rullan M, Aoki SK, Buchmann P, and Khammash M (2016) Automated optogenetic feedback control for precise and robust regulation of gene expression and cell growth. Nat. Commun 7, 12546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Egriboz O, Goswami S, Tao X, Dotts K, Schaeffer C, Pilauri V, and Hopper JE (2013) Self-association of the Gal4 inhibitor protein Gal80 is impaired by Gal3: evidence for a new mechanism in the GAL gene switch. Mol. Cell. Biol 33, 3667–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mateus C, and Avery SV (2000) Destabilized green fluorescent protein for monitoring dynamic changes in yeast gene expression with flow cytometry. Yeast 16, 1313–1323. [DOI] [PubMed] [Google Scholar]
- (18).Takeuchi J, Chen H, Hoyt MA, and Coffino P (2008) Structural elements of the ubiquitin-independent proteasome degron of ornithine decarboxylase. Biochem. J 410, 401–407. [DOI] [PubMed] [Google Scholar]
- (19).Miyazaki Y, Matsufuji S, Murakami Y, and Hayashi S (1993) Single amino-acid replacement is responsible for the stabilization of ornithine decarboxylase in HMOAcells. Eur. J. Biochem 214, 837–844. [DOI] [PubMed] [Google Scholar]
- (20).González C, Ray JCJ, Manhart M, Adams RM, Nevozhay D, Morozov AV, and Balázsi G (2015) Stress-response balance drives the evolution of a network module and its host genome. Mol. Syst. Biol 11, 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Da Silva NA, and Srikrishnan S (2012) Introduction and expression of genes for metabolic engineering applications in Saccharomyces cerevisiae. FEMS Yeast Res. 12, 197–214. [DOI] [PubMed] [Google Scholar]
- (22).Schüller H-J (2003) Transcriptional control of non-fermentative metabolism in the yeast Saccharomyces cerevisiae. Curr. Genet 43, 139–160. [DOI] [PubMed] [Google Scholar]
- (23).Griggs DW, and Johnston M (1991) Regulated expression of the GAL4 activator gene in yeast provides a sensitive genetic switch for glucose repression. Proc. Natl. Acad. Sci. U. S. A 88, 8597–8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Blazeck J, Garg R, Reed B, and Alper HS (2012) Controlling promoter strength and regulation in Saccharomyces cerevisiae using synthetic hybrid promoters. Biotechnol. Bioeng 109, 2884–2895. [DOI] [PubMed] [Google Scholar]
- (25).Mumberg D, Müller R, and Funk M (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–22. [DOI] [PubMed] [Google Scholar]
- (26).Mao X, Hu Y, Liang C, and Lu C (2002) MET3 promoter: A tightly regulated promoter and its application in construction of conditional lethal strain. Curr. Microbiol 45, 37–40. [DOI] [PubMed] [Google Scholar]
- (27).Bhat PJ, and Murthy TVS (2001) Saccharomyces cerevisiae: mechanism of galactose-mediated signal transduction. Mol. Microbiol 40, 1059–1066. [DOI] [PubMed] [Google Scholar]
- (28).Platt A, and Reece RJ (1998) The yeast galactose genetic switch is mediated by the formation of a Gal4p-Gal80p-Gal3p complex. EMBO J. 17, 4086–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lee KM, and DaSilva NA (2005) Evaluation of the Saccharomyces cerevisiae ADH2 promoter for protein synthesis. Yeast 22, 431–440. [DOI] [PubMed] [Google Scholar]
- (30).McIsaac RS, Gibney PA, Chandran SS, Benjamin KR, and Botstein D (2014) Synthetic biology tools for programming gene expression without nutritional perturbations in Saccharomyces cerevisiae. Nucleic Acids Res. 42, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ottoz DSM, Rudolf F, and Stelling J (2014) Inducible, tightly regulated and growth condition-independent transcription factor in Saccharomyces cerevisiae. Nucleic Acids Res. 42, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Garí E, Piedrafita L, Aldea M, and Herrero E (1997) A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cervisiae. Yeast 13, 837–848. [DOI] [PubMed] [Google Scholar]
- (33).Tan SZ, Manchester S, and Prather KLJ (2016) Controlling Central Carbon Metabolism for Improved Pathway Yields in Saccharomyces cerevisiae. ACS Synth. Biol 5, 116–124. [DOI] [PubMed] [Google Scholar]
- (34).Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, and Tucker CL (2010) Rapid blue light induction of protein interactions in living cells. Nat. Methods 7, 973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Taslimi A, Zoltowski B, Miranda JG, Pathak GP, Hughes RM, and Tucker CL (2016) Optimized second-generation CRY2-CIB dimerizers and photoactivatable Cre recombinase. Nat. Chem. Biol 12, 425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Melendez J, Patel M, Oakes BL, Xu P, Morton P, and McClean MN (2014) Real-time optogenetic control of intracellular protein concentration in microbial cell cultures. Integr. Biol 6, 366–372. [DOI] [PubMed] [Google Scholar]
- (37).Salinas F, Rojas V, Delgado V, López J, Agosin E, and Larrondo LF (2018) Fungal light-oxygen-voltage domains for optogenetic control of gene expression and flocculation in yeast. mBio, DOI: 10.1128/mBio.00626-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Avalos JL, Fink GR, and Stephanopoulos G (2013) Compartmentalization of metabolic pathways in yeast mitochondria improves the production of branched-chain alcohols. Nat. Biotechnol 31, 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zhang Y, Lane S, Chen J-M, Hammer SK, Luttinger J, Yang L, Jin Y-S, and Avalos JL (2019) Xylose utilization stimulates mitochondrial production of isobutanol and 2-methyl-1-butanol in Saccharomyces cerevisiae. Biotechnol. Biofuels 12, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Nevozhay D, Adams RM, Murphy KF, Josić K, and Balázsi G (2009) Negative autoregulation linearizes the dose-response and suppresses the heterogeneity of gene expression. Proc. Natl. Acad. Sci. U. S. A 106, 5123–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Guinn MT, and Balázsi G (2019) Noise-reducing optogenetic negative-feedback gene circuits in human cells. Nucleic Acids Res. 47, 7703–7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Rosenfeld N, Elowitz MB, and Alon U (2002) Negative autoregulation speeds the response times of transcription networks. J. Mol. Biol 323, 785–793. [DOI] [PubMed] [Google Scholar]
- (43).McGuire SE, Le PT, Osborn AJ, Matsumoto K, and Davis RL (2003) Spatiotemporal Rescue of Memory Dysfunction in Drosophila. Science (Washington, DC, U. S.) 302, 1765–1768. [DOI] [PubMed] [Google Scholar]
- (44).Faucherre A, and López-Schier H (2011) Delaying Gal4-driven gene expression in the zebrafish with morpholinos and Gal80. PLoS One 6, e16587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Usherenko S, Stibbe H, Muscò M, Essen L-OO, Kostina EA, and Taxis C (2014) Photo-sensitive degron variants for tuning protein stability by light. BMC Syst. Biol 8, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Yuan J, and Ching CB (2015) Combinatorial Assembly of Large Biochemical Pathways into Yeast Chromosomes for Improved Production of Value-added Compounds. ACS Synth. Biol. 4, 23–31. [DOI] [PubMed] [Google Scholar]
- (47).Goldstein AL, and McCusker JH (1999) Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15, 1541–1553. [DOI] [PubMed] [Google Scholar]
- (48).Gibson DG, Young L, Chuang R-YY, Venter JC, Hutchison CA, and Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.