

Abstract
BACKGROUND
Type 2 diabetes mellitus (T2D) confers a two- to three-fold increased risk of cardiovascular disease (CVD). However, the mechanisms underlying increased CVD risk among people with T2D are only partially understood. We hypothesized that a genetic association study among people with T2D at risk for developing incident cardiovascular complications could provide insights into molecular genetic aspects underlying CVD.
METHODS
From 16 studies of the Cohorts for Heart & Aging Research in Genomic Epidemiology (CHARGE) Consortium, we conducted a multi-ancestry time-to-event genome-wide association study (GWAS) for incident CVD among people with T2D using Cox proportional hazards models. Incident CVD was defined based on a composite of coronary artery disease (CAD), stroke, and cardiovascular death that occurred at least one year after the diagnosis of T2D. Cohort-level estimated effect sizes were combined using inverse variance weighted fixed effects meta-analysis. We also tested 204 known CAD variants for association with incident CVD among patients with T2D.
RESULTS
A total of 49,230 participants with T2D were included in the analyses (31,118 European ancestries and 18,112 non-European ancestries) which consisted of 8,956 incident CVD cases over a range of mean follow-up duration between 3.2 and 33.7 years (event rate 18.2%). We identified three novel, distinct genetic loci for incident CVD among individuals with T2D that reached the threshold for genome-wide significance (P<5.0×10−8): rs147138607 (intergenic variant between CACNA1E and ZNF648) with a hazard ratio (HR) 1.23, 95% confidence interval (CI) 1.15 – 1.32, P=3.6×10−9, rs11444867 (intergenic variant near HS3ST1) with HR 1.89, 95% CI 1.52 – 2.35, P=9.9×10−9, and rs335407 (intergenic variant between TFB1M and NOX3) HR 1.25, 95% CI 1.16 – 1.35, P=1.5×10−8. Among 204 known CAD loci, 32 were associated with incident CVD in people with T2D with P<0.05, and 5 were significant after Bonferroni correction (P<0.00024, 0.05/204). A polygenic score of these 204 variants was significantly associated with incident CVD with HR 1.14 (95% CI 1.12 – 1.16) per 1 standard deviation increase (P=1.0×10−16).
CONCLUSIONS
The data point to novel and known genomic regions associated with incident CVD among individuals with T2D.
INTRODUCTION
Type 2 diabetes mellitus (T2D) is a significant risk factor for cardiovascular disease (CVD), leading in a two- to three-fold higher likelihood of developing CVD. CVD is the leading cause of morbidity and mortality in people with diabetes1, 2 and previous studies suggest that life expectancy is reduced by up to eight years in people with T2D3. Although CVD mortality rates have declined substantially in the general population in recent decades, this improvement has been less substantial in people with T2D4. Various modifiable factors have been extensively investigated, with high blood pressure, hypercholesterolemia, smoking, and diabetes being well-validated risk factors of CVD that are used to estimate the 10-year risk of incident events5. However, diabetes itself may explain as much as 75 to 90% of the excess risk of coronary disease in people with T2D6, 7.
There are distinct clinical characteristics of CVD in people with T2D that result in less favorable outcomes. People with T2D have an earlier onset of extensive CVD that tends to progress more rapidly than among those without T2D. There is a greater number of atherosclerotic vessels involved and longer diseased vessel segments in people with T2D compared to those without8, even after accounting for standard CVD risk factors. The mortality risk at 30 days and one year after acute coronary syndrome was higher in people with T2D than those without T2D9. These features, as well as excess CVD risk in people with T2D not solely attributable to more aggregated risk factors, suggest there could be additional risk factors for CVD that are specific to T2D.
Other than traditional risk factors, recent genome-wide association studies (GWASs) have identified at least 204 genetic loci associated with CVD in the general population. These studies have been mostly conducted among European ancestry participants,10 which calls for ancestry-diverse studies. So far, identified CVD variants implicate pathways involved in cholesterol metabolism, insulin resistance, thrombosis, inflammation, endothelial function, and vascular remodeling11. Genetic risk factors of CVD in the general population are mostly thought to be relevant to people with T2D12, leading to the suggestion that genetic predisposition to CVD involves additional loci and/or stronger associations in this group. There are shared genetic loci between T2D and CVD, which include chromosome 9p21.3, IRS1, TCF7L2, HNF1A, and APOE13, 14. In addition, there is a significant genetic correlation between T2D and CVD15. Still, genetic risk factors for CVD in people with T2D have not been thoroughly investigated. Most studies have been underpowered given the stringent significance threshold required for a GWAS and were cross-sectional13, 16, 17. To identify genetic risk factors for incident CVD specifically in people with T2D, it is crucial to investigate T2D cases longitudinally and in follow-up studies where diabetes clearly precedes CVD.
We hypothesize that individuals with T2D share an increased CVD genetic burden and that a multi-ancestry GWAS for incident CVD among people with T2D could help identify these signals and infer biological mechanisms underlying the increased CVD risk among people with T2D. To test these hypotheses, we performed a time-to-event GWAS of incident CVD in a large, multi-ancestry sample of people with T2D ensuring that the occurrence of T2D preceded any CVD event.
METHODS
Study Design and Participating Cohorts
This is a meta-analysis of ancestry-specific cohort-level time-to-event GWAS for incident CVD in people with T2D, majority of which were from the Cohorts for Heart & Aging Research in Genomic Epidemiology (CHARGE) Consortium18. We studied 49,230 participants with T2D from 16 cohorts and of multiple ancestries. In the case of multi-ancestry cohorts, participants were grouped into major ancestries, resulting in 28 ancestry-specific cohort subgroups. Detailed information, including study design, study period, and a brief description of the participants of each cohort, is shown in Table S1. All human research was conducted according to the Declaration of Helsinki, and each cohort acquired institutional review board approval. Each participant provided written informed consent.
Definition of T2D
T2D was defined in each cohort by having one or more of the American Diabetes Association criteria19 (Table S2). T2D was defined as having at least one of the following conditions: fasting blood glucose (FBG) ≥ 126 mg/dL, hemoglobin A1c (HbA1c) ≥ 6.5%, 2-h glucose by 75-g oral glucose tolerance test ≥ 200 mg/dL, physician-diagnosed diabetes, or use of glucose-lowering medications. Participants with known type 1 diabetes mellitus (T1D) or other specific types of diabetes were excluded. To minimize contamination of T1D, we excluded people with age at diabetes diagnosis of diabetes below 40.
Definition of Cardiovascular Disease
CVD was defined as a composite of 1) coronary artery disease (CAD), 2) cerebrovascular disease, and 3) death from a cardiovascular cause. CAD included myocardial infarction, stable or unstable angina, percutaneous coronary intervention, coronary artery bypass grafting, and other cohort-defined events (Table S3). Cerebrovascular disease included ischemic stroke, hemorrhagic stroke, transient ischemic attack, carotid stenting, endarterectomy, and other cohort-defined events. Death from cardiovascular causes included death from myocardial infarction, stroke, unexpected death presumed to be from ischemic CVD, and other cohort-defined events. An incident CVD event was defined as the first CVD event occurring at least one year after T2D diagnosis. Participants with any CVD event prior to diagnosis of T2D or within one year after diagnosis of T2D were excluded from incident CVD analysis. If a participant had multiple CVD events, only the first event was considered. The source of CVD information varied by each cohort, which included a self-report of the participant, doctor’s notes, cohort visit examinations, linkage to primary care registers and secondary care registers, hospital admissions, and mortality data.
Cox Proportional Hazards Models
We applied Cox proportional hazards modeling for the time-to-event GWAS. Each single nucleotide variation (SNV) or short insertion-deletion was tested for its association with incident CVD considering observation time and adjusting for covariates. Observation time was defined as years between the age at diagnosis of T2D and age at incident CVD for cases or age at last follow-up for control participants who did not experience CVD event. Before running the Cox proportional hazards model, the model which did not include genotype information was evaluated to determine whether it met the proportional hazard assumption using the cox.zph() function in the ‘Survival’ R package20. If a variable violated the assumption, this was resolved either by including an interaction term or by stratifying the variables into 3 to 5 subgroups. Time-to-event GWAS was performed in each ancestry-specific cohort subgroup using either ‘GWASTools’ R package21 or ‘gwasurvivr’ R package22 (Table S4). The primary analysis included age at diagnosis of T2D and sex as covariates, and significant principal components (PCs) were used to adjust for population stratification (basic model). In the full model, body mass index (BMI), current smoking status, treatment for hypertension, systolic blood pressure, total cholesterol, and high-density lipoprotein (HDL) cholesterol level, all of which are used in 10-year risk estimation of CVD, were added to the basic model.
Cohort-level Analysis
Participants were genotyped using GWAS SNV genotyping arrays (Table S5). Each cohort had its specific pre-imputation genotype quality control criteria for call rate, minor allele frequency (MAF), and deviation from Hardy-Weinberg equilibrium. Genotype imputation was performed using either TOPMed reference panel (GRCh38), Haplotype Reference Consortium reference panel (GRCh37), 1,000 Genomes phase 3 reference panel (GRCh37), or population-specific reference panel. All the genotypes aligned on the positive strand of the reference genome. SNVs with poor imputation quality were removed from the analysis in each cohort (INFO score < 0.4 or R2 score < 0.3). Within each cohort, analysis was performed separately for four major ancestries: African American (AFR), East Asian (EAS), European (EUR), and Hispanic (HIS). Time-to-event GWAS for incident CVD was performed under the additive genetic model. Familial relationships were handled either by excluding related individuals or by using a robust estimate of variance when relationships were known for the family-based cohort. Cohort-level summary statistics were collected for meta-analysis.
Meta-Analysis
Summary statistics of the cohort level results underwent standard quality control procedures using EasyQC software23. First, the genetic coordinates of GRCh38 were converted to GRCh37/hg19 using LiftOver24 software. Then variants with unreliably large effect size (β≥10) or large standard error (≥10) were excluded in each cohort. Variants with minor allele count ≤ 6, or variants with a frequency difference > 0.20 compared to the corresponding ancestry in the 1,000 Genomes phase 3 data were also removed. Allele coding and marker names were harmonized. Meta-analysis of cohort-level summary statistics was conducted using an inverse-variance-weighted fixed-effect method as implemented in METAL25. Genomic control was applied at the cohort level to control for possible inflation in the type I error due to residual population stratification. Both overall meta-analysis and ancestry-level meta-analysis were performed. A genome-wide significance threshold was set as P < 5.0 × 10−8.
Approximate Conditional Analysis
Approximate conditional analysis using summary statistics was carried out using GCTA-Cojo26 (version 1.93.2). Variants within a 120 kilobase (Kb) region (+/− 60 Kb) of the lead signal from the meta-analysis were selected to be included in the conditional analysis. Conditional analyses were approximated using ancestry-specific summary statistics from our association models and then meta-analyzed using a fixed-effect, inverse variance weighted approach, excluding lead variants that only occurred in one ancestry. Linkage disequilibrium was estimated using GCTA by providing the 1000-Genomes reference panel using super populations ‘AFR’ to represent our African ancestry, ‘EUR’ to represent our European ancestry, ‘EAS’ to represent our East Asian ancestry, and the union of ‘AMR’, ‘CEU’ and ‘YRI’ populations to represent our Hispanic ancestry individuals. We applied a multiple testing threshold at each locus when considering variants for distinct secondary signals by dividing 0.05 by the number of variants in the 120 Kb region.
Fine-Mapping of Distinct Association Signals to Identify 95% Credible Sets
To fine-map distinct association signals, we computed credible sets with 95% confidence. For each variant within +/− 90 Kb of a lead variant with a minor allele count greater than 40, i.e., the most associated variants in a region, we computed ancestry-specific Bayes factors in favor of association using estimated allelic effect sizes and standard errors from each available ancestry-specific meta-analysis. For example, a variant present only in AFR and EUR ancestries would have two Bayes factors corresponding to the AFR and EUR ancestry-specific analyses. To find the Bayes factor for the variant in a particular ancestry, we considered
where and represent the effect estimate and variance, respectively, from the ancestry-specific meta-analysis. The constant describes the prior variance in allelic effects, which we set to 0.0462.
After computing the Bayes factors, we then calculated the posterior probability that the variant drives the association signal in a particular ancestry using
where n denotes the total number of variants within +/− 90 kilobases of the lead variant. This was repeated for all variants in each region.
Finally, ancestry-specific 95% credible sets for each locus were constructed by sorting the variants in a particular locus and ancestry in descending order of probability and finding the smallest subset of the top variants whose sum of probabilities exceeded 0.95.
Functional Annotations
A genome-wide map of 18 distinct human umbilical vein endothelial cell (HUVEC) chromatin state annotations was retrieved from the Common Metabolic Diseases Genome Atlas (CMDGA, https://cmdga.org/). These chromatin states were characterized from ENCODE27 ChIP-seq data using ChromHMM28 v1.18. Each variant in each ancestry-specific credible set was matched to its corresponding chromatin state annotation.
Colocalization of GWAS and eQTL
To estimate the posterior probability of our genome-wide significant variants and eQTL sharing the identical causal variants, we performed a Bayesian colocalization method as implemented in R package ‘coloc’29 (cran.r-project.org/web/packages/coloc). eQTL data were obtained from the eQTLGen Consortium30 (31,684 whole blood samples), and GWAS variants were extracted from the summary statistics for variants located within one megabase (Mb) of the lead GWAS variants. We defined the variants as colocalized when the posterior probability of a colocalized signal (PP4) was >0.8 as generally recommended.
Phenome-Wide Association Analysis
For the genome-wide significant variants, we performed additional analyses to gain further insight into how these variants are linked to the pathophysiology of CVD using phenome-wide association analysis. The Common Metabolic Disease Knowledge Portal was used to investigate phenome-wide association (https://hugeamp.org/). As there were 388 phenotypic traits (some of which are interrelated) included in the portal, the significance threshold was conservatively set as P < 1.2×10−4 (0.05/388).
Association of known 204 CAD variants
We tested previously reported 204 CAD variants identified in the general population for association with incident CVD in people with T2D10, 12. These variants represent genetic risk factors for prevalent CAD in the general population. Adjusting for the multiple comparisons, the association’s significance threshold was set as P < 0.00024 (0.05/204). The summary statistics, including effect size (odds ratio) and the P value of these variants, were reported previously12. In addition, a weighted polygenic score based on these 204 known CAD variants was constructed as previously described12 and tested for its association with incident CVD in people with T2D.
RESULTS
Study Overview
A total of 49,230 people with T2D who did not have CVD at diagnosis of T2D or within one year of diagnosis were included in the analysis (Table 1). There were 16 participating cohorts which were divided into 28 ancestry-specific cohort subgroups (EUR: 14 cohorts, AFR: 8 cohorts, HIS: 3 cohorts, EAS: 3 cohorts). Participants with European ancestry consisted of about 63.2% (N=31,118) of the participants, and the remaining 36.8% (N=18,112) consisted of participants with non-European ancestry (AFR 22.6%, 11,124; HIS 8.8%, 4,325; EAS 5.4%, 2,663). UK Biobank was the largest cohort, representing about 31.8% (N=15,643) of the participants. Among 49,230 participants with T2D, 8,956 developed an incident CVD (event rate 18.2%) over a range of mean follow-up duration between 3.2 and 33.7 years. The CVD event rate by ancestry group was highest in those of African ancestry (25.6%). The detailed clinical characteristics of the participants for each cohort, including mean age at diagnosis of T2D, BMI, smoking status, blood pressure, lipid level, are shown in Table S6. In general, and in UK Biobank, those who developed incident CVD had an earlier age at diagnosis of T2D, higher BMI, higher rate of smoking and dyslipidemia (Table S6).
Table 1.
Sample size and event rate according to ancestry and cohort.
| Ancestry | Cohort | Total (N) | Event (N) | Event Rate (%) |
|---|---|---|---|---|
|
| ||||
| European/European American | ARIC | 2,184 | 543 | 24.9 |
| BIOME | 851 | 160 | 18.8 | |
| CHS | 379 | 225 | 59.4 | |
| FHS | 547 | 185 | 33.8 | |
| MESA | 392 | 64 | 16.3 | |
| MGB | 1,161 | 389 | 33.5 | |
| PMBB | 603 | 244 | 40.5 | |
| PROSPER | 452 | 39 | 8.6 | |
| REGARDS | 303 | 195 | 64.4 | |
| ROTTERDAM | 611 | 98 | 16 | |
| SANFORD | 2,062 | 174 | 8.4 | |
| UKBB | 15,643 | 1,499 | 9.6 | |
| WGHS | 2,043 | 105 | 5.1 | |
| WHI | 3,887 | 998 | 25.7 | |
|
| ||||
| Subtotal | 31,118 (63.2%) | 4,918 (54.9%) | 15.8 | |
|
| ||||
| African American | ARIC | 1,142 | 310 | 27.1 |
| BIOME | 1,764 | 467 | 26.5 | |
| CHS | 130 | 76 | 58.5 | |
| JHS | 726 | 92 | 12.7 | |
| MESA | 508 | 85 | 16.7 | |
| PMBB | 1,242 | 388 | 31.2 | |
| REGARDS | 2,483 | 733 | 29.5 | |
| WHI | 3,129 | 693 | 22.1 | |
|
| ||||
| Subtotal | 11,124 (22.6%) | 2,844 (31.8%) | 25.6 | |
|
| ||||
| Hispanic/Latino | BIOME | 2,681 | 668 | 24.9 |
| MESA | 482 | 89 | 18.5 | |
| WHI | 1,162 | 177 | 15.2 | |
|
| ||||
| Subtotal | 4,325 (8.8%) | 934 (10.4%) | 21.6 | |
|
| ||||
| East Asian | KOGES | 2,317 | 185 | 8.0 |
| MESA | 194 | 42 | 21.6 | |
| WHI | 152 | 33 | 21.7 | |
|
| ||||
| Subtotal | 2,663 (5.4%) | 260 (2.9%) | 9.8 | |
|
| ||||
| Total | 49,230 | 8,956 | 18.2 | |
A total of 49,230 participants with T2D from 16 cohorts and of multiple ancestries were included in this study: 31,118 (63.2%) of European, 11,124 (22.6%) of African American, 4,325 (8.8%) of Hispanic/Latino, and 2,663 (5.4%) of East Asian ancestry. The total incident CVD event rate was 18.2% with those of African ancestry having the highest event rate of 25.6%. ARIC, Atherosclerosis Risk in Communities Study; BIOME, BioMe BioBank; CHS, Cardiovascular Health Study; FHS, Framingham Heart Study; JHS, Jackson Heart Study; KOGES, Korean Genome and Epidemiology Study; MESA, Multi-Ethnic Study of Atherosclerosis; MGB, Mass General Brigham Biobank; PMBB, Penn Medicine BioBank; PROSPER, Prospective Study of Pravastatin in the Elderly at Risk; REGARDS, Reasons for Geographic and Racial Differences in Stroke Study; ROTTERDAM, Rotterdam Study; SANFORD, Sanford Health; UKBB, UK Biobank; WGHS, Women's Genome Health Study; and WHI, Women's Health Initiative.
Loci for Incident CVD in People with T2D
After genotype level quality control, we tested 15,471,776 variants with overall MAF ≥ 1% for association with incident CVD. A plot of expected-by-observed association statistics showed minimal inflation (λGC = 1.093 for variants with MAF ≥ 1%, λGC = 1.058 for variants with MAF ≥ 5%) (Figure 1A). An association plot of variants with CVD in T2D by chromosomal location identified three variants associated with incident CVD in people with T2D in genome-wide significance (P < 5.0×10−8) (Figure 1B, Table 2). The variant rs147138607 (chr1:181855562:G>C, MAF 10.7%) had an HR for incident CVD in T2D of 1.23 (95% CI 1.15 – 1.32, P = 3.6×10−9) and resides in an intergenic region between the genes CACNA1E and ZNF648 (Figure 2A). This variant was most significant in those of African ancestry and was at least nominally significant in the three other ancestry groups (Table 2). The second most significant variant rs77142250 (chr4:11444867:T>C, MAF 1.3%) was present at low frequency (1.3%) only in those of African ancestry, had an HR 1.89 (95% CI 1.52 – 2.35, P = 9.9×10−9), and resides near the gene HS3ST1 (Figure 2B). The third variant rs335407 (chr6:155665441:C>T, MAF 5.5%) had an HR of 1.25 (95% CI 1.16 – 1.35, P = 1.58×10−8), resides in an intergenic region between the genes TFB1M and NOX3 (Figure 2C). This variant was significantly associated with increased risk of CVD in those of European or African ancestry and showed a consistent direction of effects across all ancestry groups. An approximate conditional analysis of these three index variants at each of the three regions of interest showed no evidence of secondary signals (Figure S1).
Figure 1. QQ plot and Manhattan plot of the time-to-event GWAS for incident CVD in people with T2D.

A, QQ plot showing the distribution of the observed P values from the meta-analysis of GWAS result against the expected distribution under the null hypothesis. The gray zone indicates the 95% CI. λGC was 1.093 for variants with MAF ≥ 1% and λGC was 1.058 for variants with MAF ≥ 5%. B, Manhattan plot depicting the significance of all the variants after meta-analysis of GWAS results. SNP locations are plotted on the x-axis according to their chromosomal position. The negative log10 of P values of time-to-event analysis based on Cox proportional hazard model under the additive model are plotted on the y-axis. Dark dots highlight the significance of association for the previously known 204 CAD variants identified in the general population.
Table 2.
Genetic variants significantly associated with incident CVD in people with T2D in basic model.
| CHR | POS (rsID) | Effect Allele | Ancestry | Frequency | HR (95% CI) | P | Het P | Sample Size |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| 1 | 181855562 (rs147138607) | G>C | European/European American | 0.018 | 1.20 (1.00–1.44) | 0.047 | 0.756 | 24,457 |
| African American | 0.127 | 1.22 (1.12–1.33) | 2.3×10−6 | 0.381 | 8,929 | |||
| Hispanic/Latinx | 0.065 | 1.26 (1.03–1.55) | 0.027 | 0.198 | 3,163 | |||
| East Asian | 0.050 | 1.55 (1.07–2.25) | 0.021 | 0.469 | 2,511 | |||
| Combined | 0.107 | 1.23 (1.15–1.32) | 3.6×10−9 | 0.713 | 39,060 | |||
|
| ||||||||
| 4 | 11444867 (rs77142250) | T>C | African American | 0.013 | 1.89 (1.52–2.35) | 9.9×10−9 | 0.363 | 9,748 |
|
| ||||||||
| 6 | 155665441 (rs335407) | C>T | European/European American | 0.027 | 1.33 (1.19–1.50) | 1.4×10−3 | 0.664 | 29,910 |
| African American | 0.084 | 1.18 (1.05–1.31) | 3.8×10−3 | 0.891 | 7,765 | |||
| Hispanic/Latinx | 0.033 | 1.34 (0.99–1.81) | 0.055 | 0.596 | 3,163 | |||
| East Asian | 0.026 | 0.92 (0.45–1.88) | 0.810 | 0.541 | 2,511 | |||
| Combined | 0.055 | 1.25 (1.16–1.35) | 1.5×10−8 | 0.859 | 43,349 | |||
Three distinct genetic loci increased risk of incident CVD among individuals with T2D with genome-wide significance in time-to-event analysis (P<5.0×10−8). CHR, chromosome; CI, confidence interval; Het P, significance of heterogeneity; HR, hazard ratio; POS, position in GRCh37/hg19; rsID, reference SNP id.
Figure 2. Regional association plots for the three genome-wide significant variants.
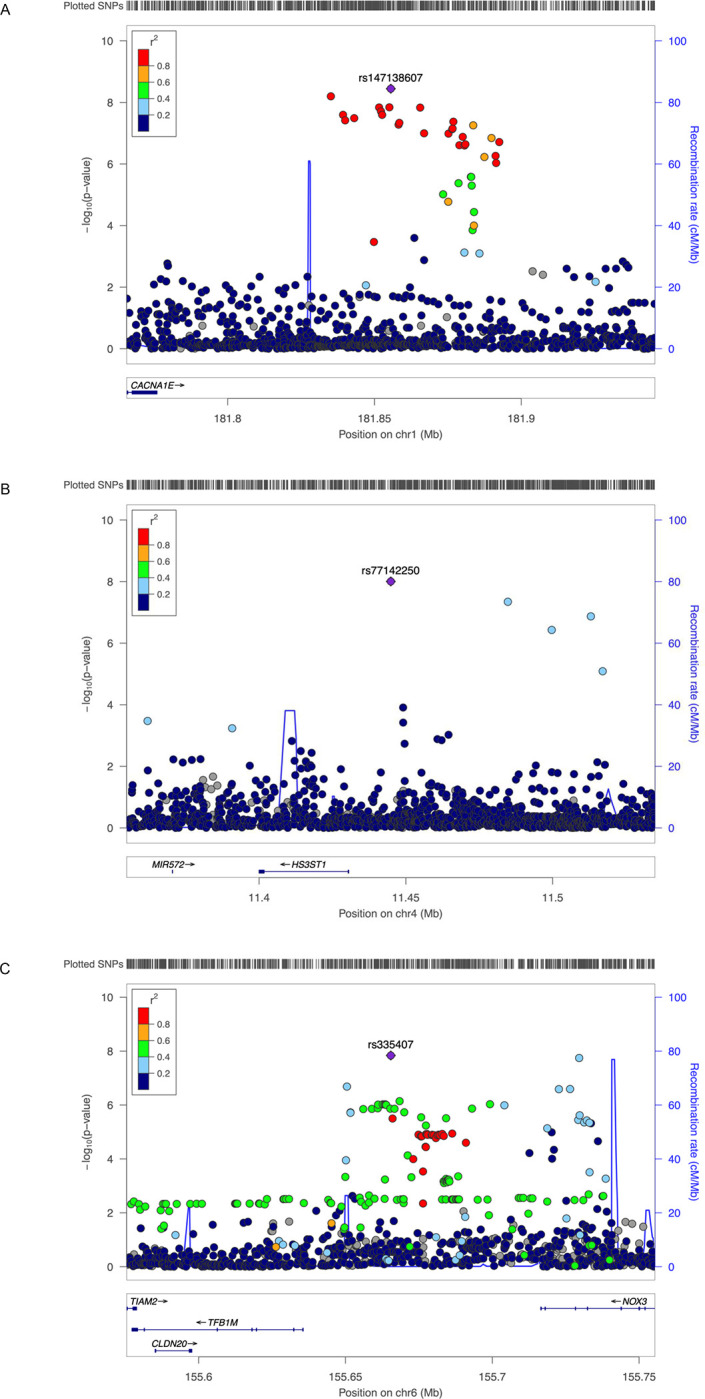
A, rs147138607 near CACNA1E and ZNF648. B, rs77142250 near HS3ST1. C, rs335407 near TFB1M and NOX3. The hash marks above the panel represent the position of each SNP that was genotyped or imputed. The negative log10 of P values from the Cox regression are shown in the y-axis. Estimated recombination rates are plotted to reflect recombination hot spots. The SNPs in LD with the most significant SNP are color coded to represent their strength of LD based on Europeans for A, and C, and Africans for B.
In the fully adjusted model, where covariates of cardiovascular risk factors were included, the statistical significance of the above three variants decreased, which was expected because of the reduced sample size. However, the effect size remained similar. We found two additional genome-wide significant variants that were significant in the full model (Table S7). One was an insertion/deletion variant, rs140159474 (chr1:181836968:T>TA, MAF 11.5%) which had an HR 1.23 (95% CI 1.14 – 1.33, P = 5.0×10−8). This variant was in linkage disequilibrium with the rs147138607 variant, which was significant in the basic model. The second variant was rs76919663 (chr12:61625023:A>G) and was present only in those of African ancestry or Hispanic ancestry, had an HR 1.48 (95% CI 1.30 – 1.67, P = 9.8×10−10). The nearest gene to this variant was TAFA2.
Insights from Downstream Analysis
We performed fine-mapping analysis for each of the three regions to narrow the number of potentially causal variants using credible set analysis31. We constructed a total of nine credible sets: four for the region on chromosome 1, one for chromosome 4, and four for chromosome 6. Each credible set accounted for ≥ 95% of the posterior probability of association with T2D in CVD in its corresponding ancestry-specific analysis. The median credible set size (i.e., number of variants included) for the chromosome 1 locus was 468, with a minimum size of 28 (AFR), and a maximum size of 816 (HIS). Likewise, the median size for the chromosome 6 locus was 637, with a minimum size of 42 (EUR), and a maximum size of 867 (AFR). However, the chromosome 4 credible set only contained 4 variants: rs77142250, rs114281229, rs7677123, and rs77129258. Small credible sets like this are favorable for prioritizing variants for functional follow-up.
Functional interrogation for the three loci included chromatin state annotations from HUVEC cell lines, a system relevant to endothelial function, atherosclerosis, and CVD (Figure S2). The chromosome 4 locus contained narrow bands of transcription and enhancer annotations near HS3ST1 and wider bands dispersed upstream. One of the four variants in the credible set, rs114281229, falls within a region containing an active enhancer annotation. Additionally, rs77129258 resides in a region of weak transcription. Given their proximity, rs114281229 and rs77129258 may affect expression of HS3ST1, but further investigation is required to substantiate this. The chromosome 6 locus contained a wide region of active annotations near TFB1M and a few smaller regions near NOX3, but the size of each credible set in this region was too large to make meaningful functional prioritizations. The chromosome 1 locus contained mostly inaccessible quiescent annotations, with narrow regions of zinc finger protein (ZNF) annotations dispersed throughout.
We used colocalization with expression QTL data from the eQTLGen Consortium to further characterize the three new loci29. We found chromosome 6 rs335407 to be a significant eQTL for TIAM2 in peripheral leukocytes P = 7.71×10−238. TIAM2 is essential for endothelial barrier and cell-cell contact maintenance32. However, posterior probabilities from colocalization analysis did not support rs335407 as causal for both incident CVD and expression of TIAM2. Using a phenome-wide association analysis, we surveyed other phenotypes associated with the three novel loci by interrogating the Common Metabolic Disease Knowledge Portal. Metabolic phenotypes nominally (P < 0.05) associated included, for chromosome 1 rs147138607, small artery occlusion and stroke; for chromosome 4 rs7714225, sleep with oxyhemoglobin saturation under 90% and BMI; and for chromosome 6 rs335407, BMI and snoring (Table S8).
Role of Known CAD Variants in People with T2D
We investigated 204 variants of CAD identified in the general population CAD10, 12 for its association with CAD in people with T2D (Table S9). Among these variants, we observed nominally significant associations with CAD in people with T2D for 38 variants, which included five that were significant after Bonferroni correction (Figure 3A). These include rs9349379 at PHACTR1 locus, rs2891168 at CDKN2A/2B locus, rs111245230 at SVEP1 locus, rs11057830 at SCARB1 locus, and rs11838776 at COL4A1/A2. Among the nominally significant 38 variants, 35 had consistent dirrection of association for CAD in people with T2D and in the general population (binomial P=3.1×10−8). For the 204 variants, we further observed consistency in the direction of association for risk of CVD between the general population and people with T2D with Spearman coefficient 0.51, P=7.2×10−15 (Figure 3B). Next, we modeled a polygenic score composed of these 204 CAD variants and used this in a model for incident CVD in T2D (Table 3). The CAD polygenic score was associated with increased CVD in people with T2D, with an estimated HR of 1.14 (95% CI 1.12 – 1.16) per 1 SD increase. We showed that the association between the CAD polygenic score and CVD differed by ancestry groups (non-significant in East Asians if European derived summary statistics was used). Overall, one standard deviation increase in polygenic score was associated with a 14% increased risk (HR 1.14, 95% CI 1.12 – 1.16) of CVD in people with T2D.
Figure 3. Association of previously identified 204 CAD variants with incident CVD in people with T2D.

A, QQ plot showing the distribution of the observed P values for the 204 CAD variants with risk of incident CVD in people with T2D against the expected distribution under the null hypothesis. The red dots highlight five variants that were significantly associated with incident CVD after Bonferroni correction. B, Comparison of the effect size of known 204 CAD variants in the general population and incident CVD in people with T2D. Effect size of the known 204 CAD variants for prevalent CAD in the general population (x-axis, β-coefficient from logistic regression analysis) and incident CVD in people with T2D (y-axis, β-coefficient from Cox regression analysis) are plotted. There was a significant correlation between the effect sizes (Spearman coefficient 0.509, P=7.2×10−15). The red dots highlight 32 variants that were nominally (P<0.05) associated with incident CVD and had same direction of association in the general population.
Table 3.
Association of polygenic score of 204 known CAD variants and incident CVD in people with T2D.
| Ancestry | HR | 95% CI | Het P | P |
|---|---|---|---|---|
|
| ||||
| European/European American | 1.18 | 1.14 – 1.21 | 0.010 | <1.0×10−16 |
| African American | 1.10 | 1.05 – 1.15 | 0.255 | 8.3×10−5 |
| Hispanic/Latinx | 1.10 | 1.03 – 1.18 | 0.474 | 0.0031 |
| East Asian | 0.99 | 0.88 – 1.13 | 0.586 | 0.982 |
|
| ||||
| Overall | 1.14 | 1.12 – 1.16 | 0.002 | <1.0×10−16 |
Polygenic score of 204 CAD variants discovered from the general population was associated with increased risk of incident CVD in people with T2D. CI, confidence interval; Het P, significance of heterogeneity; HR, hazard ratio.
DISCUSSION
In this study, we sought to identify novel genetic loci associated with incident CVD in people with T2D by performing a time-to-event GWAS. We discovered three distinct variants that reached genome-wide significance: rs147138607 on chromosome 1 between CACNA1E and ZNF648, rs77142250 on chromosome 4 near HS3ST1, and rs335407 on chromosome 6 between TFB1M and NOX3. All were significant in African ancestry while rs147138607 and rs335407 were also at least nominally significant in European ancestry. None of these variants were significantly associated with CVD in the general population. We found that most CAD variants already known from cross-sectional GWAS in the general population were also associated with incident CVD events in people with T2D, and that a polygenic score composed of 204 CAD variants was associated with incident CVD. To the best of our knowledge, this is the first large-scale genetic association study to investigate genetic risk factors of incident CVD specifically in people with T2D.
The main objective of this study was to identify genetic variants that could explain the excess risk of CVD in people with T2D. We show that people with T2D are enriched with genetic risk factors of CAD observed in the general population: 1) there was an excess number of common single variants known to be associated with CAD in people with T2D, and 2) polygenic score composed of these variants were significantly associated with incident CVD in people with T2D. Furthermore, we identified genetic loci associated with incident CVD, specifically in people with T2D. These variants were not identified as genetic risk factors of CVD in the general population. Taken together, we show that the excess CVD risk in people with T2D is conferred at least in part by excess of known CAD variants and variants that specifically exert their effect in T2D.
The locus on chromosome 1 contained narrow regions of ZNF annotations in a largely quiescent region of inaccessible chromatin. This region has previously been shown to contain a SNP (rs10911021, 226 Kb distal to rs181855562 with R2 0.015) with significant gene-by-diabetes synergism on CVD risk33 and all-cause mortality in people with T2D34. Located between ZNF648 and GLUL, the rs10911021 variant has been characterized by decreased expression of the GLUL gene in human endothelial cells and lower pyroglutamic-to-glutamic acid ratio compared to the protective allele homozygotes, suggesting a mechanistic link between glutamic acid metabolism and CVD risk among people with diabetes35. By contrast, not much is known about the role of ZNF648 itself on T2D and CVD risk, although it has been shown to be a highly conserved gene across species and is essential for erythroid and megakaryocyte differentiation36. Further studies are needed to provide mechanistic insights on the potential link between this locus and CVD risk in T2D.
On the chromosome 4 locus, we identified two variants (rs114281229 and rs77129258) in 95% credible set that may play a role in the expression of HS3ST1 (Heparan Sulfate-Glucosamine 3-Sulfotransferase 1), a sulfated glycosaminoglycan involved in mediating the activities of leukocytes during inflammation through cellular differentiation, proliferation, and homeostasis37. HS3ST1 plays a significant role in the insulin secretory pathway through its effects on membrane depolarization of pancreatic β-cells38. An intronic SNP of HS3ST1 (rs16881446) has previously been implicated in CAD severity and CV events in a candidate gene analysis37. One of the variants included in the credible set in our study, rs114281229, fell within a region containing an active enhancer annotation, while the other variant, rs77129258, fell within a region of weak transcription. These findings increase the likelihood that these variants play an important role in the expression of HS3ST1, which would need to be verified with functional assays and in vivo models of T2D.
Although the size of the credible sets near the TFB1M and NOX3 genes on chromosome 6 was too large to identify specific variants or make functional prioritizations, the rs335407 SNP identified in our study is a known significant eQTL for the TIAM2 gene in peripheral leukocytes30. TIAM2 (T-cell lymphoma invasion and metastasis 2) is a RAC1 guanine nucleotide exchange factor essential for endothelial barrier and cell-cell contact maintenance32. Given its importance in maintaining endothelial cell integrity, TIAM2 may play an important role in mediating the effects of CVD risk in T2D. Additional studies are needed to validate these findings.
The strengths of this study include the use of time-to-event GWAS for incident CVD rather than performing conventional case-control analysis. This allows us to dissect the temporal relationship between T2D and CVD and assures that T2D qualifies as an exposure variable for CVD and capture CVD events that occur specifically after T2D diagnosis. As far as we are aware of, we have gathered the largest number of T2D samples (N=49,230) with information on incident CVD events (overall event rate 18.2%, N=8,956). We leveraged large-scale biobanks and used age at diagnosis of T2D and CVD event to construct Cox proportional hazards models with observation time defined as years between age at diagnosis of T2D and CVD or last follow-up. This study also benefits from the fact that we included samples from different ancestries and performed a multi-ancestry meta-analysis. As many as 36.8% of the participants were from non-European ancestry. Multi-ancestry meta-analysis is known to increase power where association signal is shared across ancestry groups and improves fine-mapping resolution39.
There are certain limitations in this study. First, although we compiled a large number of T2D cases with information on incident CVD, the sample size was still modest. We had limited statistical power compared to the latest GWAS with a case-control design that included more than one million participants40. Beyond the three robust association signals we identified, there are likely to be variants lying below the significance threshold. The summary results of our study, which are publicly available on the Common Metabolic Disease Knowledge Portal (https://hugeamp.org), provide a useful resource for further validation and replication. Second, we excluded participants having CVD prior to or within one year of T2D diagnosis. Participants with early onset CVD might have been excluded, and the role of genetic risk factors of CVD discovered in the general population would have been underestimated. However, our focus was to identify genetic risk factors that are specific in people with T2D which could explain the excess risk of CVD, which is the main cause of morbidity and mortality. Finally, there were limitations in performing downstream analysis with our multi-ancestry GWAS results. Most of the publicly available resources, such as GTEx, were developed from the genetic information from European samples, and many of the low-frequency variants in African Americans were absent. In addition, statistical methods for fine mapping, eQTL analysis, and colocalization were not optimized to account for the different LD patterns in multi-ancestry analysis.
In conclusion, we conducted a time-to-event GWAS to identify genetic risk factors of CVD in people with T2D using multi-ancestry cohorts and biobanks. We discovered three loci that are robustly associated with incident CVD and show that known CAD variants identified in the general population are also enriched in people with T2D. These genetic findings might partially explain the excess risk of CVD in people with T2D. Even though the three loci warrant further replication and validation, they point to novel targets for early prevention and treatment of CVD in people with T2D.
CLINICAL PERSPECTIVE.
What is new?
We conducted a large-scale multi-ancestry time-to-event GWAS to identify genetic variants associated with CVD among people with T2D.
Three variants were significantly associated with incident CVD in people with T2D: rs147138607 (intergenic variant between CACNA1E and ZNF648), rs11444867 (intergenic variant near HS3ST1), and rs335407 (intergenic variant between TFB1M and NOX3).
A polygenic score composed of known CAD variants identified in the general population was significantly associated with the risk of CVD in people with T2D.
What are the clinical implications?
There are genetic risk factors specific to T2D that could at least partially explain the excess risk of CVD in people with T2D.
In addition, we show that people with T2D have enrichment of known CAD association signals which could also explain the excess risk of CVD.
Sources of Funding
J.M. was supported by American Diabetes Association grant No. 7-21-JDFM-005. J.M.M. is supported by American Diabetes Association Innovative and Clinical Translational Award 1-19-ICTS-068, American Diabetes Association grant #11-22-ICTSPM-16, and by NHGRI U01HG011723. M.O.G. was supported by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Disease (R01-DK109588 and P30-DK063491), the National Center for Advancing Translational Sciences (UL1TR001420, UL1TR001881), and by the Eris M. Field Chair in Diabetes Research. R.H.C. was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number T32GM100842. S.M.D. receives research support from RenalytixAI, outside the current work and is supported by the US Department of Veterans Affairs Clinical Research and Development award IK2-CX001780. This publication does not represent the views of the Department of Veterans Affairs or the United States Government. R.M. was supported by the National Heart, Lung, and Blood Institute (R01HL142809 and R01HL159514), the American Heart Association (22TPA969625), and the Wild Family Foundation.
Cardiovascular Health Study: This CHS research was supported by NHLBI contracts HHSN268201200036C, HHSN268200800007C, HHSN268201800001C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, 75N92021D00006; and NHLBI grants U01HL080295, R01HL085251, R01HL087652, R01HL105756, R01HL103612, R01HL120393, and U01HL130114 with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided through R01AG023629 from the National Institute on Aging (NIA). A full list of principal CHS investigators and institutions can be found at CHS-NHLBI.org. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The Framingham Heart Study (FHS) is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with Boston University (Contract No. N01-HC-25195, HHSN268201500001I and 75N92019D00031).
The Jackson Heart Study (JHS) is supported and conducted in collaboration with Jackson State University (HHSN268201800013I), Tougaloo College (HHSN268201800014I), the Mississippi State Department of Health (HHSN268201800015I), and the University of Mississippi Medical Center (HHSN268201800010I, HHSN268201800011I and HHSN268201800012I) contracts from the National Heart, Lung, and Blood Institute and the National Institute for Minority Health and Health Disparities.
The KoGES analysis was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare (grant number: HI15C3131).
MESA and the MESA SHARe projects are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, and UL1-TR-001420, UL1TR001881, DK063491, and R01HL105756. Funding for SHARe genotyping was provided by NHLBI Contract N02-HL-64278. The authors thank the other investigators, the staff, and the participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutes can be found at http://www.mesa-nhlbi.org. This study was also supported in part by the NHLBI contracts R01HL151855 and R01HL146860 and the National Institute of Diabetes and Digestive and Kidney Diseases contract UM1DK078616.
We acknowledge the Penn Medicine BioBank (PMBB) for providing data and thank the patient-participants of Penn Medicine who consented to participate in this research program. We would also like to thank the Penn Medicine BioBank team and Regeneron Genetics Center for providing genetic variant data for analysis. The PMBB is approved under IRB protocol# 813913 and supported by Perelman School of Medicine at University of Pennsylvania, a gift from the Smilow family, and the National Center for Advancing Translational Sciences of the National Institutes of Health under CTSA award number UL1TR001878.
The PROSPER study was supported by an investigator-initiated grant obtained from Bristol-Myers Squibb. Prof. Dr. J. W. Jukema is an Established Clinical Investigator of the Netherlands Heart Foundation (grant 2001 D 032). Support for genotyping was provided by the seventh framework program of the European commission (grant 223004) and by the Netherlands Genomics Initiative (Netherlands Consortium for Healthy Aging grant 050-060-810).
This REGARDS research was supported by the National Institutes of Health (NIH) National Heart, Lung, and Blood Institute R01HL136666 and T32HL007457. The REGARDS study is supported by a cooperative agreement U01 NS041588 from the National Institute of Neurological Disorders and Stroke, National Institutes of Health, U.S. Department of Health and Human Services. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke or the National Institutes of Health. Representatives of the funding agency have been involved in the review of the manuscript but not directly involved in the collection, management, analysis, or interpretation of the data. The authors thank the other investigators, the staff, and the participants of the REGARDS study for their valuable contributions. A full list of participating REGARDS investigators and institutions can be found at http://www.regardsstudy.org.
The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts 75N92021D00001,75N92021D00002, 75N92021D00003, 75N92021D00004, 75N92021D00005. Scientific Computing Infrastructure at Fred Hutch funded by ORIP grant S10OD028685.
The Women’s Genome Health Study (WGHS) is funded by the National Heart, Lung, and Blood Insitute (HL043851 and HL080467) and the National Cancer Institute (CA047988 and UM1CA182913), with funding for genotyping from Amgen.
Data Availability
Gentoype data and phenotypes for the participating cohorts of the CHARGE consortium are available via the database of Genotypes and Phenotypes (dbGAP) and the corresponding author upon reasonable request. Summary statistics of the study results will be available on the Common Metabolic Disease Knowledge Portal (https://hugeamp.org).
REFERENCES
- 1.Haffner SM, Lehto S, Ronnemaa T, Pyorala K and Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–34. [DOI] [PubMed] [Google Scholar]
- 2.Kannel WB and McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA. 1979;241:2035–8. [DOI] [PubMed] [Google Scholar]
- 3.Rosenquist KJ and Fox CS. Mortality Trends in Type 2 Diabetes. In: rd, Cowie C. C., Casagrande S. S, Menke A, Cissell M. A, Eberhardt M. S, Meigs J. B, Gregg E. W, Knowler W. C, Barrett-Connor E, Becker D. J., Brancati F. L, Boyko E. J, Herman W. H, Howard B. V, Narayan K. M. V, Rewers Mand Fradkin J. E, eds. Diabetes in America Bethesda (MD); 2018. [PubMed] [Google Scholar]
- 4.Rawshani A, Rawshani A, Franzen S, Eliasson B, Svensson AM, Miftaraj M, McGuire DK, Sattar N, Rosengren A and Gudbjornsdottir S. Mortality and Cardiovascular Disease in Type 1 and Type 2 Diabetes. N Engl J Med. 2017;376:1407–1418. [DOI] [PubMed] [Google Scholar]
- 5.Goff DC Jr., Lloyd-Jones DM, Bennett G, Coady S, D’Agostino RB, Gibbons R, Greenland P, Lackland DT, Levy D, O’Donnell CJ, Robinson JG, Schwartz JS, Shero ST, Smith SC Jr., Sorlie P, Stone NJ, Wilson PW, Jordan HS, Nevo L, Wnek J, Anderson JL, Halperin JL, Albert NM, Bozkurt B, Brindis RG, Curtis LH, DeMets D, Hochman JS, Kovacs RJ, Ohman EM, Pressler SJ, Sellke FW, Shen WK, Smith SC Jr, Tomaselli GF and American College of Cardiology/American Heart Association Task Force on Practice G. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129:S49–73. [DOI] [PubMed] [Google Scholar]
- 6.Fuller JH, Shipley MJ, Rose G, Jarrett RJ and Keen H. Coronary-heart-disease risk and impaired glucose tolerance. The Whitehall study. Lancet. 1980;1:1373–6. [DOI] [PubMed] [Google Scholar]
- 7.Rosengren A, Welin L, Tsipogianni A and Wilhelmsen L. Impact of cardiovascular risk factors on coronary heart disease and mortality among middle aged diabetic men: a general population study. BMJ. 1989;299:1127–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elezi S, Kastrati A, Pache J, Wehinger A, Hadamitzky M, Dirschinger J, Neumann FJ and Schomig A. Diabetes mellitus and the clinical and angiographic outcome after coronary stent placement. J Am Coll Cardiol. 1998;32:1866–73. [DOI] [PubMed] [Google Scholar]
- 9.Donahoe SM, Stewart GC, McCabe CH, Mohanavelu S, Murphy SA, Cannon CP and Antman EM. Diabetes and mortality following acute coronary syndromes. JAMA. 2007;298:765–75. [DOI] [PubMed] [Google Scholar]
- 10.van der Harst P and Verweij N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ Res. 2018;122:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Musunuru K and Kathiresan S. Genetics of Common, Complex Coronary Artery Disease. Cell. 2019;177:132–145. [DOI] [PubMed] [Google Scholar]
- 12.Morieri ML, Gao H, Pigeyre M, Shah HS, Sjaarda J, Mendonca C, Hastings T, Buranasupkajorn P, Motsinger-Reif AA, Rotroff DM, Sigal RJ, Marcovina SM, Kraft P, Buse JB, Wagner MJ, Gerstein HC, Mychaleckyj JC, Pare G and Doria A. Genetic Tools for Coronary Risk Assessment in Type 2 Diabetes: A Cohort Study From the ACCORD Clinical Trial. Diabetes Care. 2018;41:2404–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao W, Rasheed A, Tikkanen E, Lee JJ, Butterworth AS, Howson JMM, Assimes TL, Chowdhury R, Orho-Melander M, Damrauer S, Small A, Asma S, Imamura M, Yamauch T, Chambers JC, Chen P, Sapkota BR, Shah N, Jabeen S, Surendran P, Lu Y, Zhang W, Imran A, Abbas S, Majeed F, Trindade K, Qamar N, Mallick NH, Yaqoob Z, Saghir T, Rizvi SNH, Memon A, Rasheed SZ, Memon FU, Mehmood K, Ahmed N, Qureshi IH, Tanveer Us S, Iqbal W, Malik U, Mehra N, Kuo JZ, Sheu WH, Guo X, Hsiung CA, Juang JJ, Taylor KD, Hung YJ, Lee WJ, Quertermous T, Lee IT, Hsu CC, Bottinger EP, Ralhan S, Teo YY, Wang TD, Alam DS, Di Angelantonio E, Epstein S, Nielsen SF, Nordestgaard BG, Tybjaerg-Hansen A, Young R, Consortium CHDE, Benn M, Frikke-Schmidt R, Kamstrup PR, Consortium E-C, Consortium EP-I, Michigan B, Jukema JW, Sattar N, Smit R, Chung RH, Liang KW, Anand S, Sanghera DK, Ripatti S, Loos RJF, Kooner JS, Tai ES, Rotter JI, Chen YI, Frossard P, Maeda S, Kadowaki T, Reilly M, Pare G, Melander O, Salomaa V, Rader DJ, Danesh J, Voight BF and Saleheen D. Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nat Genet. 2017;49:1450–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodarzi MO and Rotter JI. Genetics Insights in the Relationship Between Type 2 Diabetes and Coronary Heart Disease. Circ Res. 2020;126:1526–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, ReproGen C, Psychiatric Genomics C, Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control C, Duncan L, Perry JR, Patterson N, Robinson EB, Daly MJ, Price AL and Neale BM. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fall T, Gustafsson S, Orho-Melander M and Ingelsson E. Genome-wide association study of coronary artery disease among individuals with diabetes: the UK Biobank. Diabetologia. 2018;61:2174–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shah HS, Gao H, Morieri ML, Skupien J, Marvel S, Pare G, Mannino GC, Buranasupkajorn P, Mendonca C, Hastings T, Marcovina SM, Sigal RJ, Gerstein HC, Wagner MJ, Motsinger-Reif AA, Buse JB, Kraft P, Mychaleckyj JC and Doria A. Genetic Predictors of Cardiovascular Mortality During Intensive Glycemic Control in Type 2 Diabetes: Findings From the ACCORD Clinical Trial. Diabetes Care. 2016;39:1915–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Psaty BM, O’Donnell CJ, Gudnason V, Lunetta KL, Folsom AR, Rotter JI, Uitterlinden AG, Harris TB, Witteman JC, Boerwinkle E and Consortium C. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.ElSayed NA, Aleppo G, Aroda VR, Bannuru RR, Brown FM, Bruemmer D, Collins BS, Hilliard ME, Isaacs D, Johnson EL, Kahan S, Khunti K, Leon J, Lyons SK, Perry ML, Prahalad P, Pratley RE, Seley JJ, Stanton RC, Gabbay RA and on behalf of the American Diabetes A. 2. Classification and Diagnosis of Diabetes: Standards of Care in Diabetes-2023. Diabetes Care. 2023;46:S19–S40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Therneau TM. A Package for Survival Analysis in R. 2022. [Google Scholar]
- 21.Gogarten SM, Bhangale T, Conomos MP, Laurie CA, McHugh CP, Painter I, Zheng X, Crosslin DR, Levine D, Lumley T, Nelson SC, Rice K, Shen J, Swarnkar R, Weir BS and Laurie CC. GWASTools: an R/Bioconductor package for quality control and analysis of genome-wide association studies. Bioinformatics. 2012;28:3329–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rizvi AA, Karaesmen E, Morgan M, Preus L, Wang J, Sovic M, Hahn T and Sucheston-Campbell LE. gwasurvivr: an R package for genome-wide survival analysis. Bioinformatics. 2019;35:1968–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winkler TW, Day FR, Croteau-Chonka DC, Wood AR, Locke AE, Magi R, Ferreira T, Fall T, Graff M, Justice AE, Luan J, Gustafsson S, Randall JC, Vedantam S, Workalemahu T, Kilpelainen TO, Scherag A, Esko T, Kutalik Z, Heid IM, Loos RJ and Genetic Investigation of Anthropometric Traits C. Quality control and conduct of genome-wide association meta-analyses. Nat Protoc. 2014;9:1192–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, Hillman-Jackson J, Kuhn RM, Pedersen JS, Pohl A, Raney BJ, Rosenbloom KR, Siepel A, Smith KE, Sugnet CW, Sultan-Qurraie A, Thomas DJ, Trumbower H, Weber RJ, Weirauch M, Zweig AS, Haussler D and Kent WJ. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 2006;34:D590–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Willer CJ, Li Y and Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang J, Ferreira T, Morris AP, Medland SE, Genetic Investigation of ATC, Replication DIG, Meta-analysis C, Madden PA, Heath AC, Martin NG, Montgomery GW, Weedon MN, Loos RJ, Frayling TM, McCarthy MI, Hirschhorn JN, Goddard ME and Visscher PM. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet. 2012;44:369–75, S1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ernst J and Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 2012;9:215–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallace C. Eliciting priors and relaxing the single causal variant assumption in colocalisation analyses. PLoS Genet. 2020;16:e1008720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vosa U, Claringbould A, Westra HJ, Bonder MJ, Deelen P, Zeng B, Kirsten H, Saha A, Kreuzhuber R, Yazar S, Brugge H, Oelen R, de Vries DH, van der Wijst MGP, Kasela S, Pervjakova N, Alves I, Fave MJ, Agbessi M, Christiansen MW, Jansen R, Seppala I, Tong L, Teumer A, Schramm K, Hemani G, Verlouw J, Yaghootkar H, Sonmez Flitman R, Brown A, Kukushkina V, Kalnapenkis A, Rueger S, Porcu E, Kronberg J, Kettunen J, Lee B, Zhang F, Qi T, Hernandez JA, Arindrarto W, Beutner F, Consortium B, i QTLC, Dmitrieva J, Elansary M, Fairfax BP, Georges M, Heijmans BT, Hewitt AW, Kahonen M, Kim Y, Knight JC, Kovacs P, Krohn K, Li S, Loeffler M, Marigorta UM, Mei H, Momozawa Y, Muller-Nurasyid M, Nauck M, Nivard MG, Penninx B, Pritchard JK, Raitakari OT, Rotzschke O, Slagboom EP, Stehouwer CDA, Stumvoll M, Sullivan P, t Hoen PAC, Thiery J, Tonjes A, van Dongen J, van Iterson M, Veldink JH, Volker U, Warmerdam R, Wijmenga C, Swertz M, Andiappan A, Montgomery GW, Ripatti S, Perola M, Kutalik Z, Dermitzakis E, Bergmann S, Frayling T, van Meurs J, Prokisch H, Ahsan H, Pierce BL, Lehtimaki T, Boomsma DI, Psaty BM, Gharib SA, Awadalla P, Milani L, Ouwehand WH, Downes K, Stegle O, Battle A, Visscher PM, Yang J, Scholz M, Powell J, Gibson G, Esko T and Franke L. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet. 2021;53:1300–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, Cook JP, Schmidt EM, Wuttke M, Sarnowski C, Magi R, Nano J, Gieger C, Trompet S, Lecoeur C, Preuss MH, Prins BP, Guo X, Bielak LF, Below JE, Bowden DW, Chambers JC, Kim YJ, Ng MCY, Petty LE, Sim X, Zhang W, Bennett AJ, Bork-Jensen J, Brummett CM, Canouil M, Ec Kardt KU, Fischer K, Kardia SLR, Kronenberg F, Lall K, Liu CT, Locke AE, Luan J, Ntalla I, Nylander V, Schonherr S, Schurmann C, Yengo L, Bottinger EP, Brandslund I, Christensen C, Dedoussis G, Florez JC, Ford I, Franco OH, Frayling TM, Giedraitis V, Hackinger S, Hattersley AT, Herder C, Ikram MA, Ingelsson M, Jorgensen ME, Jorgensen T, Kriebel J, Kuusisto J, Ligthart S, Lindgren CM, Linneberg A, Lyssenko V, Mamakou V, Meitinger T, Mohlke KL, Morris AD, Nadkarni G, Pankow JS, Peters A, Sattar N, Stancakova A, Strauch K, Taylor KD, Thorand B, Thorleifsson G, Thorsteinsdottir U, Tuomilehto J, Witte DR, Dupuis J, Peyser PA, Zeggini E, Loos RJF, Froguel P, Ingelsson E, Lind L, Groop L, Laakso M, Collins FS, Jukema JW, Palmer CNA, Grallert H, Metspalu A, Dehghan A, Kottgen A, Abecasis GR, Meigs JB, Rotter JI, Marchini J, Pedersen O, Hansen T, Langenberg C, Wareham NJ, Stefansson K, Gloyn AL, Morris AP, Boehnke M and McCarthy MI. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50:1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amado-Azevedo J, Reinhard NR, van Bezu J, de Menezes RX, van Beusechem VW, van Nieuw Amerongen GP, van Hinsbergh VWM and Hordijk PL. A CDC42-centered signaling unit is a dominant positive regulator of endothelial integrity. Sci Rep. 2017;7:10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qi L, Qi Q, Prudente S, Mendonca C, Andreozzi F, di Pietro N, Sturma M, Novelli V, Mannino GC, Formoso G, Gervino EV, Hauser TH, Muehlschlegel JD, Niewczas MA, Krolewski AS, Biolo G, Pandolfi A, Rimm E, Sesti G, Trischitta V, Hu F and Doria A. Association between a genetic variant related to glutamic acid metabolism and coronary heart disease in individuals with type 2 diabetes. JAMA. 2013;310:821–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prudente S, Shah H, Bailetti D, Pezzolesi M, Buranasupkajorn P, Mercuri L, Mendonca C, De Cosmo S, Niewczas M, Trischitta V and Doria A. Genetic Variant at the GLUL Locus Predicts All-Cause Mortality in Patients With Type 2 Diabetes. Diabetes. 2015;64:2658–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X, Zheng Y, Guasch-Ferre M, Ruiz-Canela M, Toledo E, Clish C, Liang L, Razquin C, Corella D, Estruch R, Fito M, Gomez-Gracia E, Aros F, Ros E, Lapetra J, Fiol M, Serra-Majem L, Papandreou C, Martinez-Gonzalez MA, Hu FB and Salas-Salvado J. High plasma glutamate and low glutamine-to-glutamate ratio are associated with type 2 diabetes: Case-cohort study within the PREDIMED trial. Nutr Metab Cardiovasc Dis. 2019;29:1040–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferguson DCJ, Mokim JH, Meinders M, Moody ERR, Williams TA, Cooke S, Trakarnsanga K, Daniels DE, Ferrer-Vicens I, Shoemark D, Tipgomut C, Macinnes KA, Wilson MC, Singleton BK and Frayne J. Characterization and evolutionary origin of novel C2H2 zinc finger protein (ZNF648) required for both erythroid and megakaryocyte differentiation in humans. Haematologica. 2021;106:2859–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smits NC, Kobayashi T, Srivastava PK, Skopelja S, Ivy JA, Elwood DJ, Stan RV, Tsongalis GJ, Sellke FW, Gross PL, Cole MD, DeVries JT, Kaplan AV, Robb JF, Williams SM and Shworak NW. HS3ST1 genotype regulates antithrombin’s inflammomodulatory tone and associates with atherosclerosis. Matrix Biol. 2017;63:69–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takahashi I, Ohashi K and Nata K. Involvement of heparan sulfate 3-O-sulfotransferase isoform-1 in the insulin secretion pathway. J Diabetes Investig. 2012;3:362–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mahajan A, Spracklen CN, Zhang W, Ng MCY, Petty LE, Kitajima H, Yu GZ, Rueger S, Speidel L, Kim YJ, Horikoshi M, Mercader JM, Taliun D, Moon S, Kwak SH, Robertson NR, Rayner NW, Loh M, Kim BJ, Chiou J, Miguel-Escalada I, Della Briotta Parolo P, Lin K, Bragg F, Preuss MH, Takeuchi F, Nano J, Guo X, Lamri A, Nakatochi M, Scott RA, Lee JJ, Huerta-Chagoya A, Graff M, Chai JF, Parra EJ, Yao J, Bielak LF, Tabara Y, Hai Y, Steinthorsdottir V, Cook JP, Kals M, Grarup N, Schmidt EM, Pan I, Sofer T, Wuttke M, Sarnowski C, Gieger C, Nousome D, Trompet S, Long J, Sun M, Tong L, Chen WM, Ahmad M, Noordam R, Lim VJY, Tam CHT, Joo YY, Chen CH, Raffield LM, Lecoeur C, Prins BP, Nicolas A, Yanek LR, Chen G, Jensen RA, Tajuddin S, Kabagambe EK, An P, Xiang AH, Choi HS, Cade BE, Tan J, Flanagan J, Abaitua F, Adair LS, Adeyemo A, Aguilar-Salinas CA, Akiyama M, Anand SS, Bertoni A, Bian Z, Bork-Jensen J, Brandslund I, Brody JA, Brummett CM, Buchanan TA, Canouil M, Chan JCN, Chang LC, Chee ML, Chen J, Chen SH, Chen YT, Chen Z, Chuang LM, Cushman M, Das SK, de Silva HJ, Dedoussis G, Dimitrov L, Doumatey AP, Du S, Duan Q, Eckardt KU, Emery LS, Evans DS, Evans MK, Fischer K, Floyd JS, Ford I, Fornage M, Franco OH, Frayling TM, Freedman BI, Fuchsberger C, Genter P, Gerstein HC, Giedraitis V, Gonzalez-Villalpando C, Gonzalez-Villalpando ME, Goodarzi MO, Gordon-Larsen P, Gorkin D, Gross M, Guo Y, Hackinger S, Han S, Hattersley AT, Herder C, Howard AG, Hsueh W, Huang M, Huang W, Hung YJ, Hwang MY, Hwu CM, Ichihara S, Ikram MA, Ingelsson M, Islam MT, Isono M, Jang HM, Jasmine F, Jiang G, Jonas JB, Jorgensen ME, Jorgensen T, Kamatani Y, Kandeel FR, Kasturiratne A, Katsuya T, Kaur V, Kawaguchi T, Keaton JM, Kho AN, Khor CC, Kibriya MG, Kim DH, Kohara K, Kriebel J, Kronenberg F, Kuusisto J, Lall K, Lange LA, Lee MS, Lee NR, Leong A, Li L, Li Y, Li-Gao R, Ligthart S, Lindgren CM, Linneberg A, Liu CT, Liu J, Locke AE, Louie T, Luan J, Luk AO, Luo X, Lv J, Lyssenko V, Mamakou V, Mani KR, Meitinger T, Metspalu A, Morris AD, Nadkarni GN, Nadler JL, Nalls MA, Nayak U, Nongmaithem SS, Ntalla I, Okada Y, Orozco L, Patel SR, Pereira MA, Peters A, Pirie FJ, Porneala B, Prasad G, Preissl S, Rasmussen-Torvik LJ, Reiner AP, Roden M, Rohde R, Roll K, Sabanayagam C, Sander M, Sandow K, Sattar N, Schonherr S, Schurmann C, Shahriar M, Shi J, Shin DM, Shriner D, Smith JA, So WY, Stancakova A, Stilp AM, Strauch K, Suzuki K, Takahashi A, Taylor KD, Thorand B, Thorleifsson G, Thorsteinsdottir U, Tomlinson B, Torres JM, Tsai FJ, Tuomilehto J, Tusie-Luna T, Udler MS, Valladares-Salgado A, van Dam RM, van Klinken JB, Varma R, Vujkovic M, Wacher-Rodarte N, Wheeler E, Whitsel EA, Wickremasinghe AR, van Dijk KW, Witte DR, Yajnik CS, Yamamoto K, Yamauchi T, Yengo L, Yoon K, Yu C, Yuan JM, Yusuf S, Zhang L, Zheng W, FinnGen e MC, Raffel LJ, Igase M, Ipp E, Redline S, Cho YS, Lind L, Province MA, Hanis CL, Peyser PA, Ingelsson E, Zonderman AB, Psaty BM, Wang YX, Rotimi CN, Becker DM, Matsuda F, Liu Y, Zeggini E, Yokota M, Rich SS, Kooperberg C, Pankow JS, Engert JC, Chen YI, Froguel P, Wilson JG, Sheu WHH, Kardia SLR, Wu JY, Hayes MG, Ma RCW, Wong TY, Groop L, Mook-Kanamori DO, Chandak GR, Collins FS, Bharadwaj D, Pare G, Sale MM, Ahsan H, Motala AA, Shu XO, Park KS, Jukema JW, Cruz M, McKean-Cowdin R, Grallert H, Cheng CY, Bottinger EP, Dehghan A, Tai ES, Dupuis J, Kato N, Laakso M, Kottgen A, Koh WP, Palmer CNA, Liu S, Abecasis G, Kooner JS, Loos RJF, North KE, Haiman CA, Florez JC, Saleheen D, Hansen T, Pedersen O, Magi R, Langenberg C, Wareham NJ, Maeda S, Kadowaki T, Lee J, Millwood IY, Walters RG, Stefansson K, Myers SR, Ferrer J, Gaulton KJ, Meigs JB, Mohlke KL, Gloyn AL, Bowden DW, Below JE, Chambers JC, Sim X, Boehnke M, Rotter JI, McCarthy MI and Morris AP. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat Genet. 2022;54:560–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aragam KG, Jiang T, Goel A, Kanoni S, Wolford BN, Atri DS, Weeks EM, Wang M, Hindy G, Zhou W, Grace C, Roselli C, Marston NA, Kamanu FK, Surakka I, Venegas LM, Sherliker P, Koyama S, Ishigaki K, Asvold BO, Brown MR, Brumpton B, de Vries PS, Giannakopoulou O, Giardoglou P, Gudbjartsson DF, Guldener U, Haider SMI, Helgadottir A, Ibrahim M, Kastrati A, Kessler T, Kyriakou T, Konopka T, Li L, Ma L, Meitinger T, Mucha S, Munz M, Murgia F, Nielsen JB, Nothen MM, Pang S, Reinberger T, Schnitzler G, Smedley D, Thorleifsson G, von Scheidt M, Ulirsch JC, Biobank J, Epic CVD, Arnar DO, Burtt NP, Costanzo MC, Flannick J, Ito K, Jang DK, Kamatani Y, Khera AV, Komuro I, Kullo IJ, Lotta LA, Nelson CP, Roberts R, Thorgeirsson G, Thorsteinsdottir U, Webb TR, Baras A, Bjorkegren JLM, Boerwinkle E, Dedoussis G, Holm H, Hveem K, Melander O, Morrison AC, Orho-Melander M, Rallidis LS, Ruusalepp A, Sabatine MS, Stefansson K, Zalloua P, Ellinor PT, Farrall M, Danesh J, Ruff CT, Finucane HK, Hopewell JC, Clarke R, Gupta RM, Erdmann J, Samani NJ, Schunkert H, Watkins H, Willer CJ, Deloukas P, Kathiresan S, Butterworth AS and Consortium CAD. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat Genet. 2022;54:1803–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Gentoype data and phenotypes for the participating cohorts of the CHARGE consortium are available via the database of Genotypes and Phenotypes (dbGAP) and the corresponding author upon reasonable request. Summary statistics of the study results will be available on the Common Metabolic Disease Knowledge Portal (https://hugeamp.org).