

Abstract
While CD8+ T cells are critical for elimination of cancer cells, factors within the tumor microenvironment (TME) drive them to a hypofunctional state: exhaustion. The most terminally exhausted T (tTex) cells are not the targets of checkpoint blockade immunotherapy but rather may limit immunotherapeutic efficacy. As intratumoral CD8+ tTex cells bear phenotypic similarities to regulatory T (Treg) cells, we asked whether tTex cells may be directly anti-functional and constrain cancer immunity. Here we show intratumoral CD8+ tTex cells carried similar capacity to suppress T cell proliferation as CD4+Foxp3+ Treg cells. tTex cell suppression requires CD39, which generates immunosuppressive adenosine. Deletion of CD39 in CD8+ T cells slows tumor progression and improves immunotherapy response. CD39 is induced on tTex by tumor hypoxia, thus mitigation of hypoxia limits tTex suppression. Our data suggest tTex cells are a significant regulatory population in cancer: strategies to limit their generation, reprogram their immunosuppressive nature, or remove them from the tumor microenvironment may potentiate immunotherapy.
T cells are critical mediators of immunity, harboring a diverse repertoire of T cell receptors (TCR) that facilitate recognition and elimination of both foreign infectious agents and malignant host cells. Differentiation of a naïve T cell to an effector (Teff) cell is orchestrated by metabolic, epigenetic, and transcriptional reprogramming events required for effective disease control. Yet the activity of Teff cells is tightly regulated to prevent autoimmunity. Upon antigen clearance, Teff cells contract, maintaining a pool of long-lived memory T (Tmem) cells. In non-resolving inflammatory settings like cancer, however, T cell fate diverges from memory formation to a metabolically and epigenetically distinct effector lineage termed exhaustion1–3. T cell infiltration in cancer is a well reported positive prognostic marker, yet even patients with immunologically active tumors may progress despite therapeutic strategies targeting inhibitory pathways on exhausted T cells3–5. Immunotherapy via antibody-mediated blockade of ‘checkpoint’ receptors, like PD-1, have highlighted both strengths and limitations of reigniting anti-tumor immunity. While checkpoint blockade therapy has been a success, clinical efficacy of these agents can vary dramatically across tumor types, with most patients not achieving sustained remission. Beyond the simple number of infiltrating cells, the makeup of a patient’s tumor infiltrating lymphocytes (TIL) plays a significant role in the immunotherapy response3,5. Further, cell extrinsic factors like metabolic restriction and interaction with immunoregulatory populations also hinder effective TIL-mediated tumor control6,7. A deeper understanding of mechanisms of immune suppression in cancer is needed to advance development of better immunotherapies.
Exhaustion has broad, and somewhat contested, terminology, although it has now been well established that exhaustion in CD8+ T cells is not a singular state but is instead a progressive lineage that differentiates though (at least) two discrete stages. A pool of progenitor exhausted T (pTex) cells, maintained by the transcription factor TCF-1, is held in check by the PD-1 receptor and carries self-renewal capacity, which can seed the predominant TCF1− terminally exhausted T (tTex) cell pool1. Progression of the pTex lineage towards the tTex cell fate has been defined by progressive loss of effector cytokine production and proliferative potential, the upregulation of multiple coinhibitory and costimulatory receptors, and metabolic derangements that precipitate these impairments. Exhausted T cells have been further characterized by a distinct epigenic and transcriptional signature2,3,8. Persistent TCR signaling, exaggerated by metabolic and inflammatory stress, is believed to be the primary driver of T cell exhaustion9. Considerable efforts to deconvolute this state have proposed that diminished responsiveness in exhausted T cells is a compensatory mechanism to limit, or suppress, immunopathology in settings of persistent stimulation1,10. Indeed, tTex cells have been identified in numerous chronic pathologic settings but also in the context of homeostatic immune processes, such as in the placenta during gestation11. In both, T cell exhaustion serves to tolerize T cells experiencing continuous antigen exposure. Thus, exhaustion may be a developmentally advantageous program to control persistent T cell activation that arises in various pathologic states. A high proportion of terminally exhausted T (tTex) cells among the total tumor immune infiltrate, for instance, predicts resistance, not response, to therapeutic PD-1 blockade, despite these cells expressing the highest levels of PD-1 receptor3,5,12. Functionally, tTex cells are often characterized by what is lost: diminished polyfunctionality, proliferative potential, and stunted capacity to lyse target cells. Yet beyond the loss-of-function associated with terminal exhaustion, tTex cells have also been characterized by gain-of-functions in the upregulation of effector molecules involved in immune suppression and wound repair (e.g. IL-10, CD39).
As cancer progresses, increased secretion of angiogenic factors and heightened oxidative metabolism yield significant oxygen deprivation across the tumor bed and activate hypoxia and energetic stress responses6,7. In CD8+ T cells, sustained exposure to hypoxia rapidly accelerates differentiation to terminal exhaustion and represses effective anti-tumor immunity13. In both preclinical models and patients undergoing anti-PD-1 immunotherapy, heightened tumor oxidative metabolism and hypoxia staining correlates with poor immunotherapeutic efficacy driven, in part, by CD8+ T cell dysfunction14,15. T cells sense low oxygen tensions through hypoxia-sensitive elements (e.g. HIF1𝛼) that orchestrate transcriptional and post-translational programs. One such gene is CD39 (Entpd1), a key member of the adenosine pathway and an ectoenzyme regularly cited as a faithful marker of tumor antigen experienced tTex cells in murine and human disease16. CD39 rapidly catabolizes extracellular proinflammatory adenosine tri- and diphosphate (eATP, eADP) to the mono-phosphorylated form. CD73 then acts on AMP to produce adenosine, inhibiting local signaling cascades. It remains unclear, however, whether expression of such regulatory molecules like CD39 on CD8+ T cells engenders significant consequence to anti-tumor immunity.
Here we reveal CD8+ tTex cells as a significant regulatory population in solid tumors which carry similar suppressive capacity to neighboring CD4+Foxp3+ Treg cells. However, unlike Treg cells, which utilize many nonredundant suppressive mechanisms, tTex cells elicit their inhibitory function solely through CD39. Deletion of CD39 in CD8+ tTex cells augments immunotherapy, and its expression on tTex cells is reduced by pharmacologically or genetically elevating intratumoral oxygen tension, dampening the suppressor potential of tTex cells. Thus, we define tTex cells not solely as a hypofunctional state, but one capable of conditionally possessing potent anti-functional characteristics that suppress local T cell immunity.
Intratumoral exhausted T cells are functionally suppressive
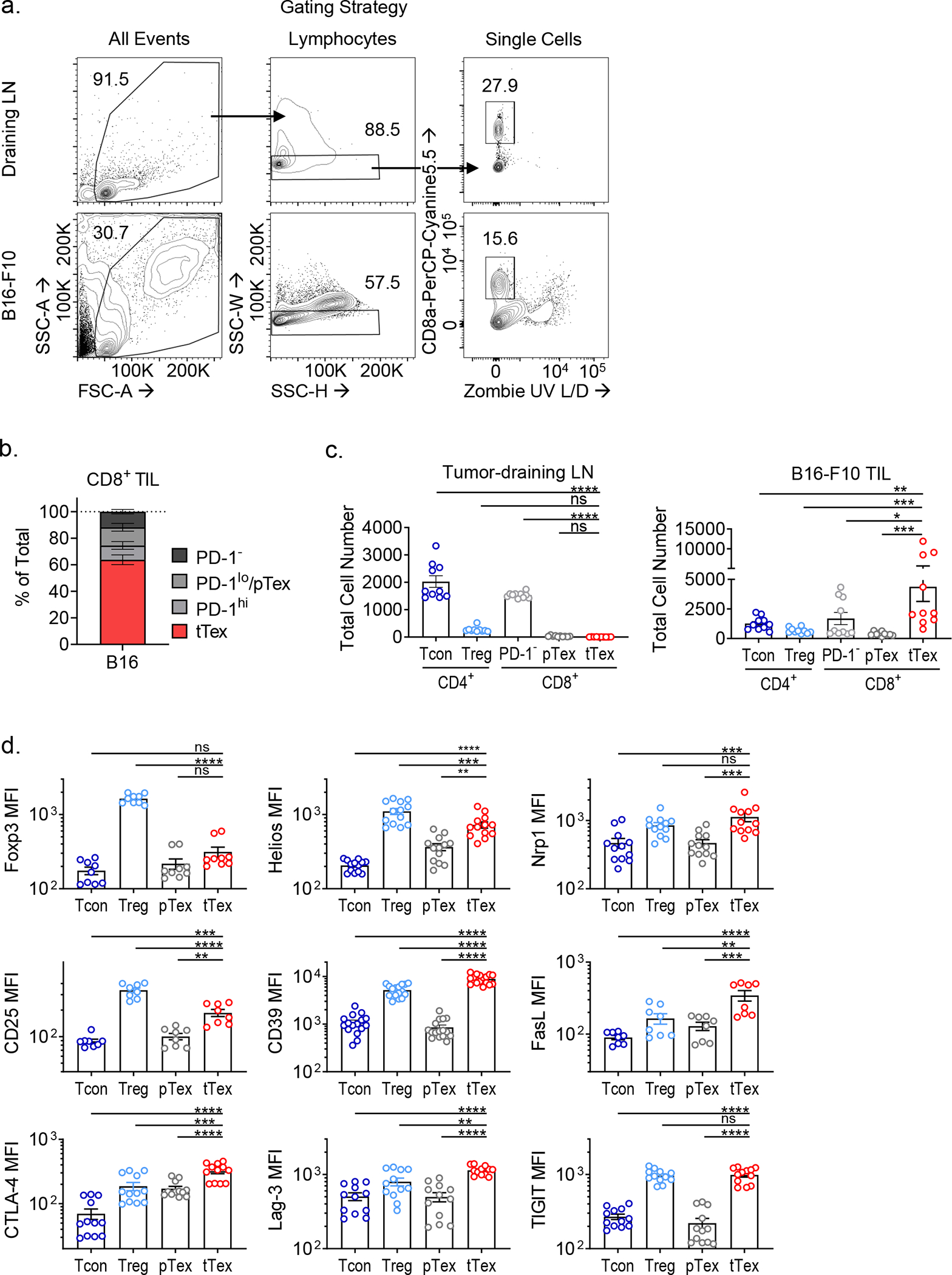
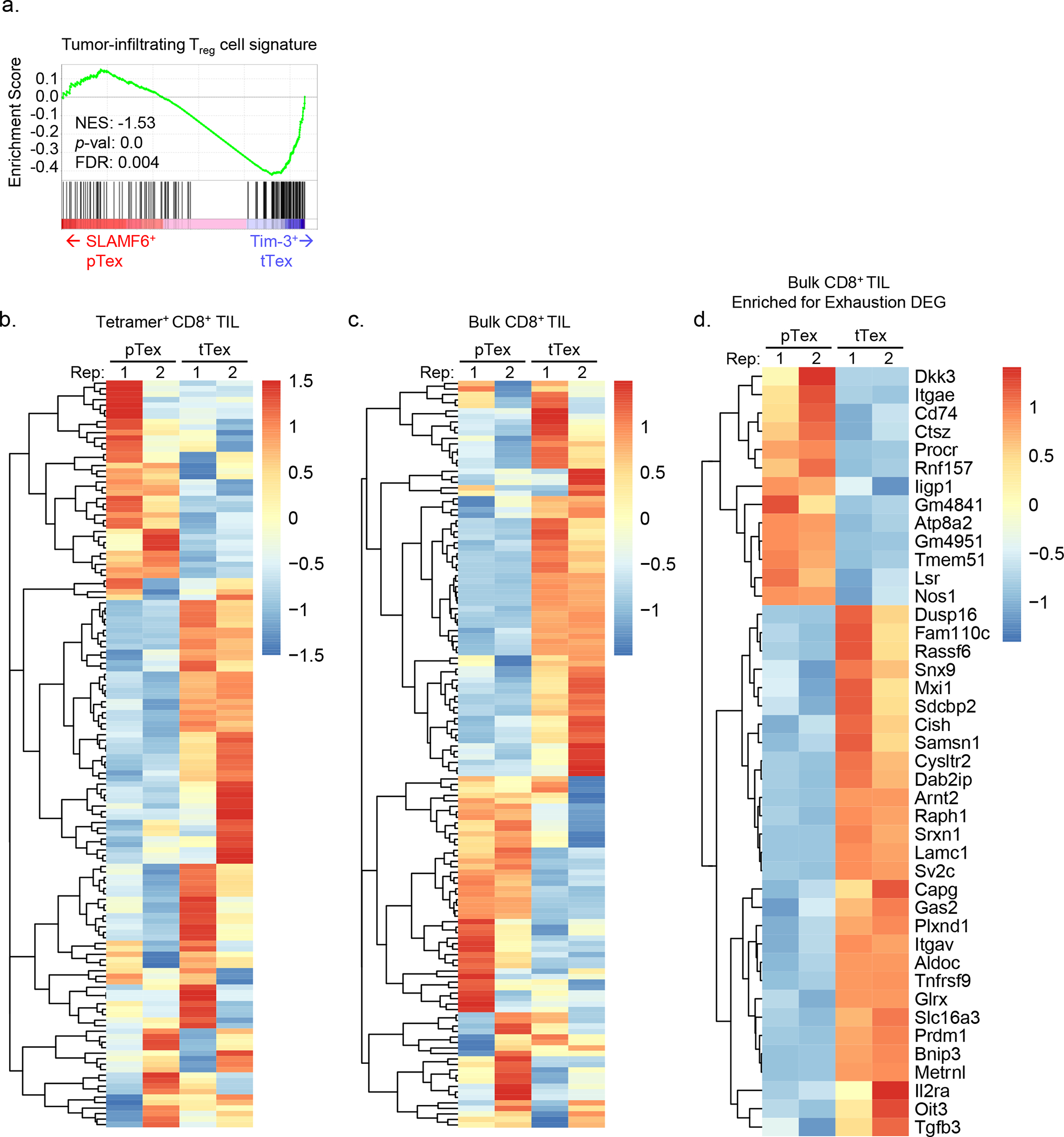
Enrichment of terminally differentiated exhausted CD8+ T cells (tTex cells) in solid tumors has been shown to correlate with poor clinical responses to checkpoint blockade therapy3,5. However, it remains unclear whether ineffective restimulation of anti-tumor immunity is simply the result of a loss of T cell functionality, or whether tTex cells themselves contribute to the tolerogenic tumor microenvironment. Thus, we sought to determine whether tTex cells were directly immunosuppressive. To begin to delineate alternative effector roles in terminal exhaustion, we employed the aggressive, immunotherapy resistant B16-F10 melanoma line, orthotopically implanted intradermally in C57BL6 mice. On day 14 (tumor area, 8 to 10 mm2), PD-1hiTim-3+ tTex cells are the plurality both among CD8+ T cell populations and among total CD3+ T cells (Fig. 1A,B, Extended Data Fig. 1A–C). At day 14, infiltrating CD8+ T cells were sorted into four groups, stratified by the expression of inhibitory receptors, PD-1 and Tim-3 (PD-1−, PD-1int, PD-1hiTim-3−, and PD-1hiTim-3+;)8,13. Co-expression of PD-1 and Tim-3 has been shown to reliably identify tTex cells with tumor antigen specificity and classic hallmarks of exhaustion–loss of polyfunctional cytokine production, poor proliferation, and elevated TOX expression. Effector pTex cells with anti-tumor potential are identified by isolated expression of PD-1 (PD-1−, PD-1int) concomitant with Tcf1, Slamf6 (Ly108), and CXCR5. As CD8+ T cells progress to terminal exhaustion and proinflammatory functions are repressed, tTex cells tend to upregulate genes associated with Foxp3+CD4+ regulatory T (Treg) cells (Fig. 1C; Extended Data Fig. 1D): these include the transcription factor Helios (Ikzf2) and molecules driving Treg cell suppressor functions: neuropillin 1 (Nrp1), interleukin 2 receptor alpha (Il2ra, CD25), Fas ligand (Fasl), CD39 (Entpd1), and numerous co-inhibitory and co-stimulatory receptors (cytotoxic T lymphocyte antigen 4 [Ctla4], lymphocyte activating 3 [Lag3], and T cell immunoreceptor with Ig and ITIM domains [Tigit]). Remarkably, by mining well-studied RNAseq datasets3,8,17, we discovered that beyond upregulation of immunosuppression-related effector molecules, tTex cells upregulate a broader transcriptional signature in common with intratumoral Treg cells. Tumor infiltrating CD8+ T cell transcriptomes expectedly cluster separately from CD4+Foxp3+ Treg cells and Foxp3− conventional CD4+ T (Tcon) cells from the same environment (Fig. 1D). However, if we utilize the Magnuson et al. tumor infiltrating Treg cell signature as a template, which defines genes uniquely upregulated in B16-infiltrating Treg cells compared to their LN-resident counterparts, we observe significant gene set enrichment in CD8+ tTex cells versus their progenitor counterparts (Fig. 1E; Extended Data Fig. 2A–F). These data suggest CD4+ Treg cells and CD8+ tTex cells possess some overlap in transcriptional programming in the TME which may support exhausted CD8+ T cells possessing a suppressive phenotype upon terminal differentiation.
Figure 1. PD-1hiTim-3+Foxp3− terminally exhausted CD8+ T cells predominate in solid tumors and adopt a suppressive signature that correlates with immunotherapy response.
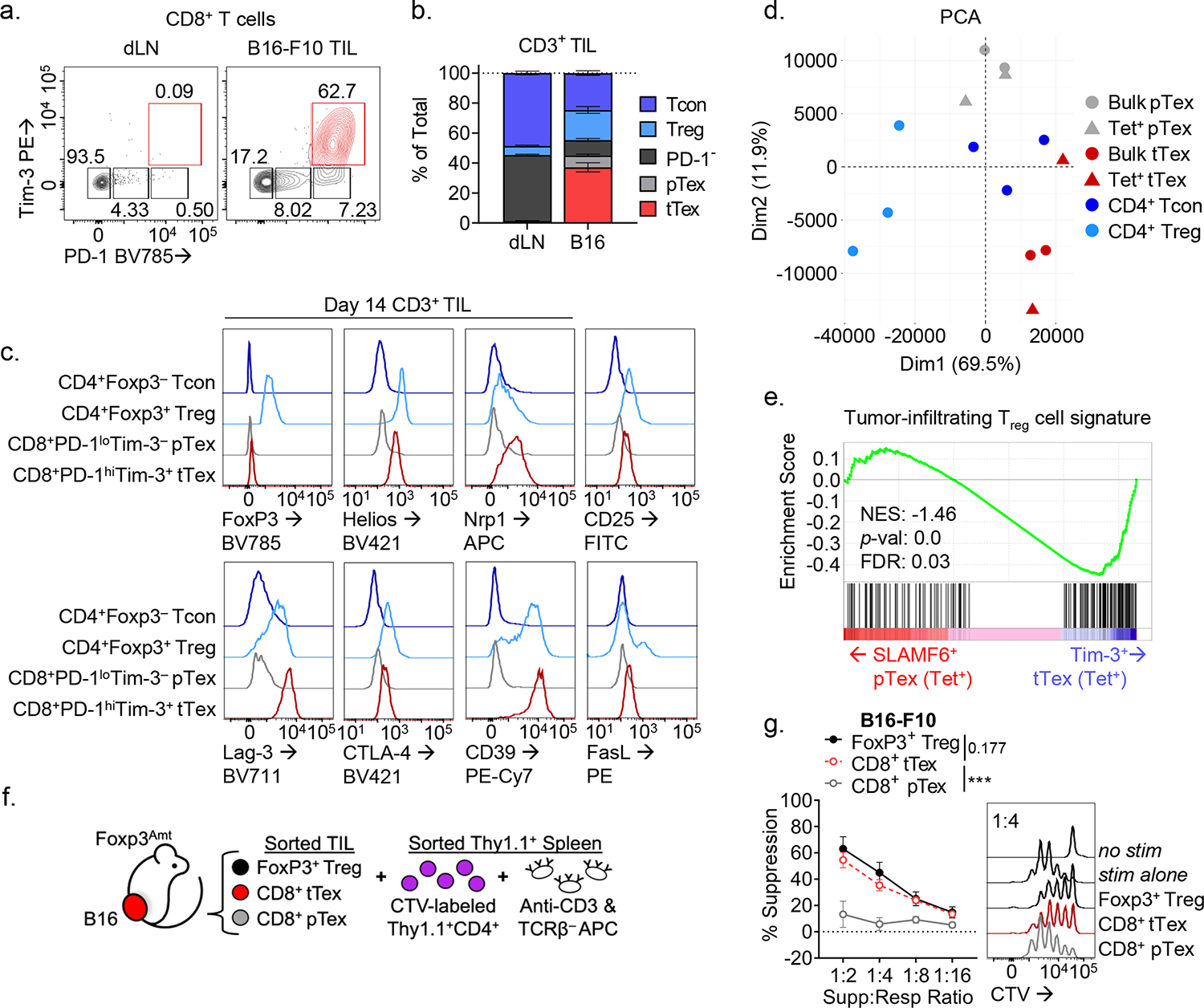
(A) CD8+ tumor-infiltrating T cells from day 14 B16-F10 murine melanoma tumor and tumor draining lymph nodes (dLN). (B) Total CD3+ T cells from B16-F10 TIL and dLN with quantified fold change. (C) Expression of CD4+Foxp3+ Treg cell-associated genes among tumor infiltrating T cell populations. (D) Principal component analysis (PCA) plot of tumor-infiltrating T cells from RNAseq mined from Magnuson et al. (2018) and Miller et al. (2019). (E) Gene set enrichment analysis (GSEA) of tumor infiltrating Treg cell signature on (B) tetramer+ SLAMF6+ progenitor and Tim-3+ terminally exhausted CD8+ T cell transcripts from Miller et al. (F) Model of suppression assay and (G) results from sorted B16-F10 melanoma TIL. Statistics are linear regression (G) with *p<0.05, **p<0.01, and ***p<0.001.
To determine if intratumoral tTex cells indeed possess intrinsic suppressive functions, we next compared tumor infiltrating CD8+ T cell populations with CD4+ Treg cells directly ex vivo in miniaturized T cell suppression assays18. B16-F10 tumors were intradermally injected into Treg cell reporter mice (FlpO and mAmetrine concomitant with Foxp3; hereafter, Foxp3Ametrine-Flpo) and T cell populations were isolated at day 14. These putative ‘suppressor’ populations were then co-cultured at diminishing ratios with proliferation dye-labeled, congenically mismatched naïve CD4+ T cells (responder T cells) and T cell-depleted splenocytes coated with anti-CD3 (Fig. 1F). Strikingly, on a per-cell basis, CD8+ tTex cells and CD4+Foxp3+ Treg cells, sorted from the same B16-F10 tumor, displayed equivalent suppression of co-cultured responding T cells (Fig. 1G). Progenitor exhausted T cells carried no suppressor capacity; in fact, no other tumor infiltrating CD4+ or CD8+ T cell population assayed, regardless of their inhibitory receptor expression, displayed detectable suppressive function (Fig. 2A). While suppression assays typically utilize naïve CD4+ or CD8+ T cells as responders, in repeat experiments, we found tTex cells were also capable of suppressing memory or effector CD8+ T cells isolated from the tumor draining lymph nodes (dLN) of B16-F10-bearing mice (Fig. 2B) or their intratumoral pTex cell counterparts (Fig. 2C).
Figure 2. Suppressive functionality in tTex cells can act on multiple target cells and is environment-dependent.
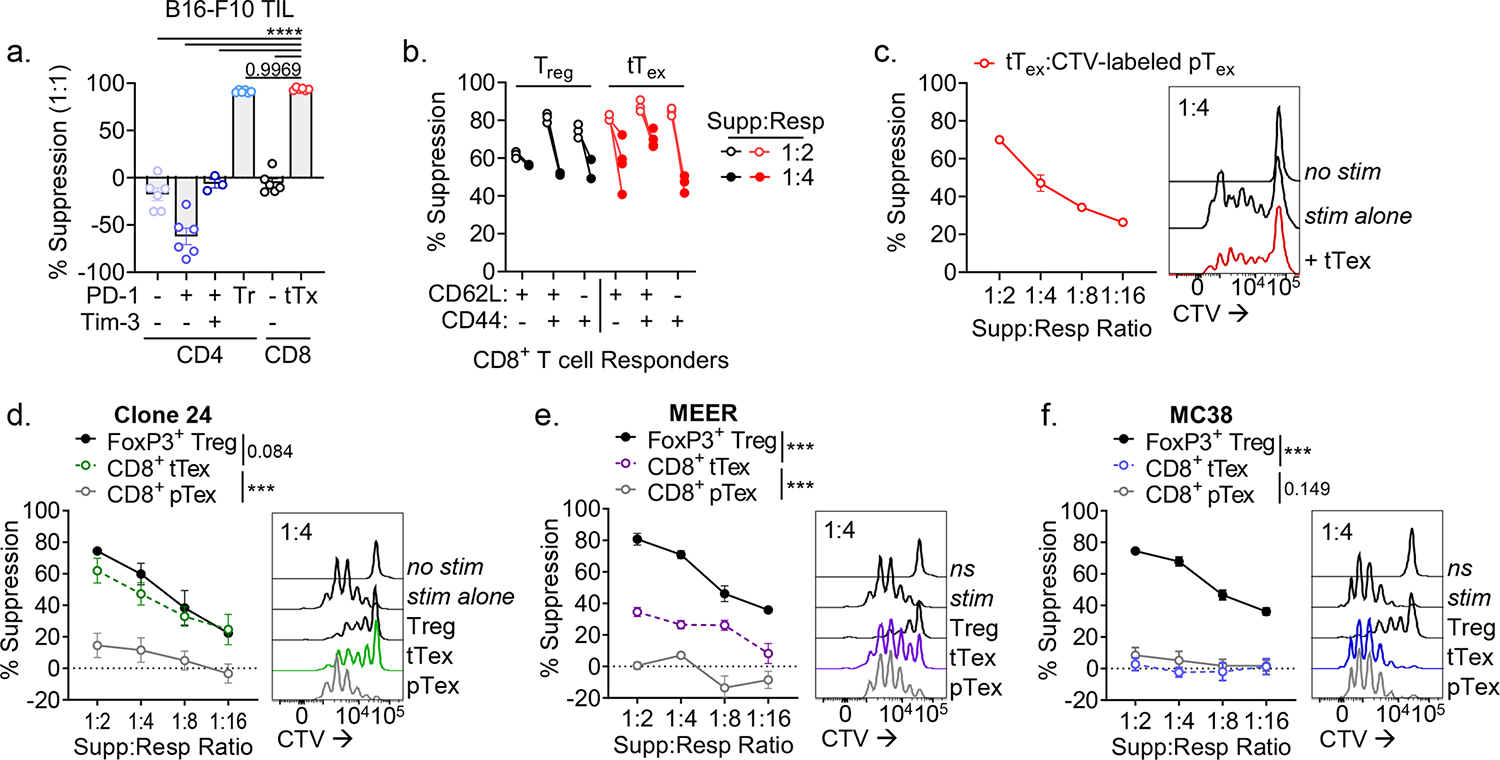
(A) CD4+ and CD8+ TIL populations from B16-F10 tumors stratified by inhibitory receptor expression and utilized as the ‘suppressor’ group in ex vivo suppression assay. (B) Suppression assay utilizing diverse responder populations from tumor draining lymph nodes and co-cultured with PD-1hiTim-3+ tTex cells or Foxp3+ Treg cells. (C) Proliferation dye labeled pTex cells co-cultured with tTex cells in repeat suppression assays. (D-F) Suppression assay with TIL from (D) Ptenf/fBRafLSL.V600ETyr2Cre.ER–derived melanoma clone, dubbed Clone 24, (E) PD-1-resistant MEER head and neck carcinoma, or (F) PD-1-sensitive MC38 adenocarcinoma. Statistics are one-way ANOVA (A) and linear regression (D-F) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
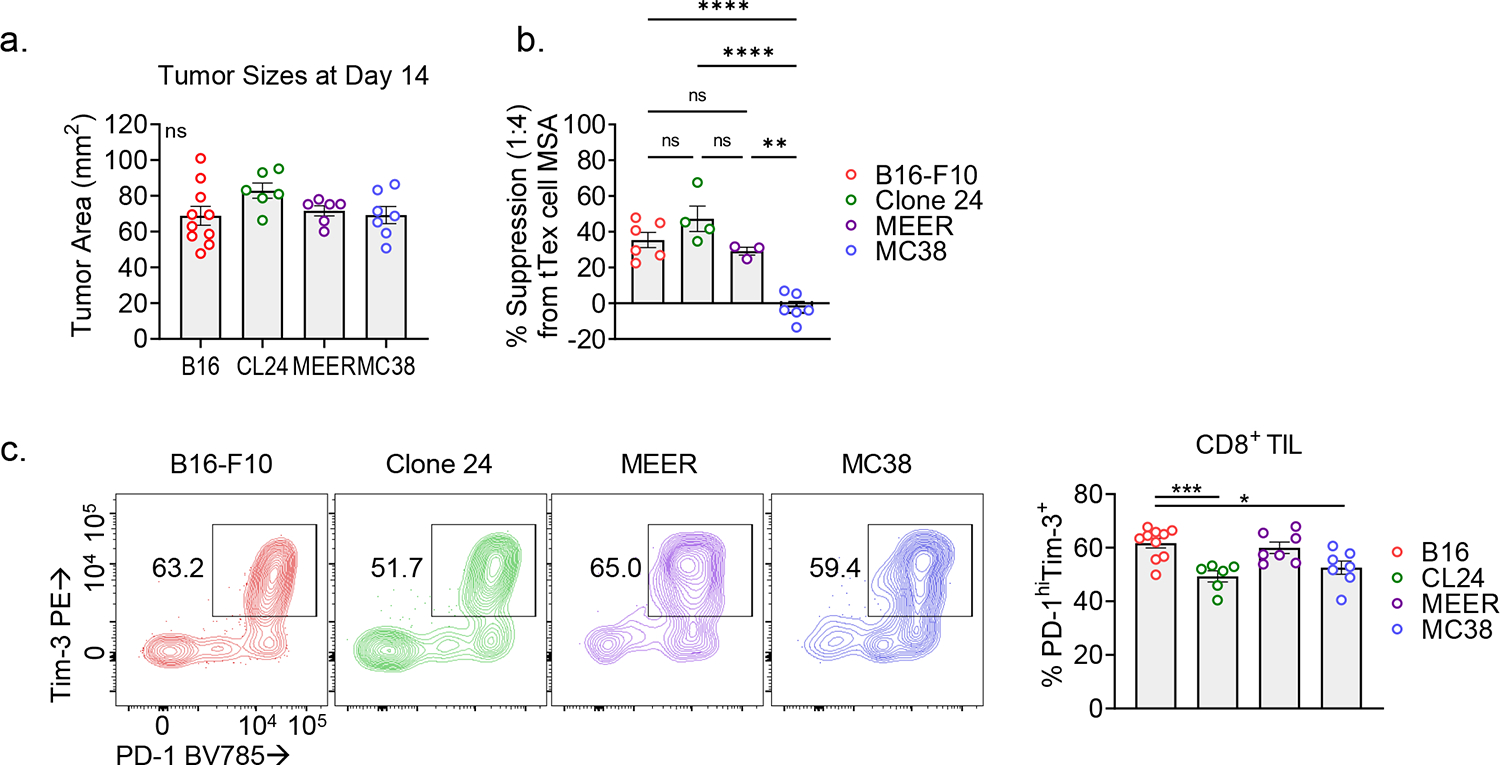
Taken together, these data suggest that tTex cells harbor a regulatory function unique among CD8+ TIL and, given tTex cells are significantly enriched the TME, represent an unappreciated suppressor population within solid tumors. We next validated our ex vivo findings in other size-matched immunotherapy resistant tumor models including: clone 24, a poorly infiltrated melanoma line derived from the autochthonous Ptenf/fBrafLSL.V600ETyr2Cre.ER mouse model15 (Fig. 2D; Extended Data Fig. 3A) and MEER, an immunotherapy-resistant head and neck carcinoma line14,18 (Fig. 2E). Isolated from these diverse environments, tTex cells consistently displayed measurable ex vivo immune suppression, while progenitor cells did not. However, not every tumor microenvironment yielded PD-1hiTim-3+ tTex cells with functional suppressive character. CD8+PD-1hiTim-3+ TIL isolated from anti-PD-1-sensitive adenocarcinoma MC38 failed to suppress T cell proliferation (Fig. 2F; Extended Data Fig. 3B). Notably, we recently reported that the regulatory capacity of Foxp3+ Treg cells is mediated by their local metabolic milieu18. Here we observe the suppressive features of tTex cells, too, are tuned by their environment, but within a greater dynamic range. These data suggest there is some plasticity in this anti-functional suppressor program that may be targeted for therapeutic benefit. Further, these data highlight inhibitory receptor expression is not requisite for suppressive functionality, as PD-1hiTim-3+ tTex cells were similarly enriched across all tumors assayed (Extended Data Fig. 3C). We thus sought to understand the mechanism of tTex cell suppression in order to delineate the environmental cues that enforce regulatory functions in CD8+ TIL.
tTex cell apoptosis enhances suppressive function ex vivo
CD4+Foxp3+ Treg cells are potent suppressors that employ multiple, sometimes redundant means to suppress immune function. We dissected various potential inhibitory mechanisms employed by terminally exhausted CD8+ T cells. Several reports have identified a subpopulation of CD8+ T cells that secrete IL-10 and play far-reaching regulatory roles in chronic disease states: protecting against autoimmune disease19, orchestrating chronicity in viral infection20, and supporting cancer progression21. However, germline deletion of IL-10 did not perturb tumor growth versus wild-type C57/BL6 mice (Extended Data Fig. 4A,B) and sorted Il10−/− tTex cells had similar suppressive function to wild-type tTex cells (Fig. 3A). A well-characterized IL-10 neutralizing antibody22 also failed to neutralize tTex cell-mediated suppression (Extended Data Fig. 4C). These experiments suggest while IL-10 has classically been defined by its suppressive character in situ (and increasing by its influence on T cell metabolism as well21,23), in this context, IL-10 does not contribute to observed suppression by tTex cells.
Figure 3. CD8+ tTex cell ex vivo suppression is associated by tTex cell-intrinsic apoptosis.
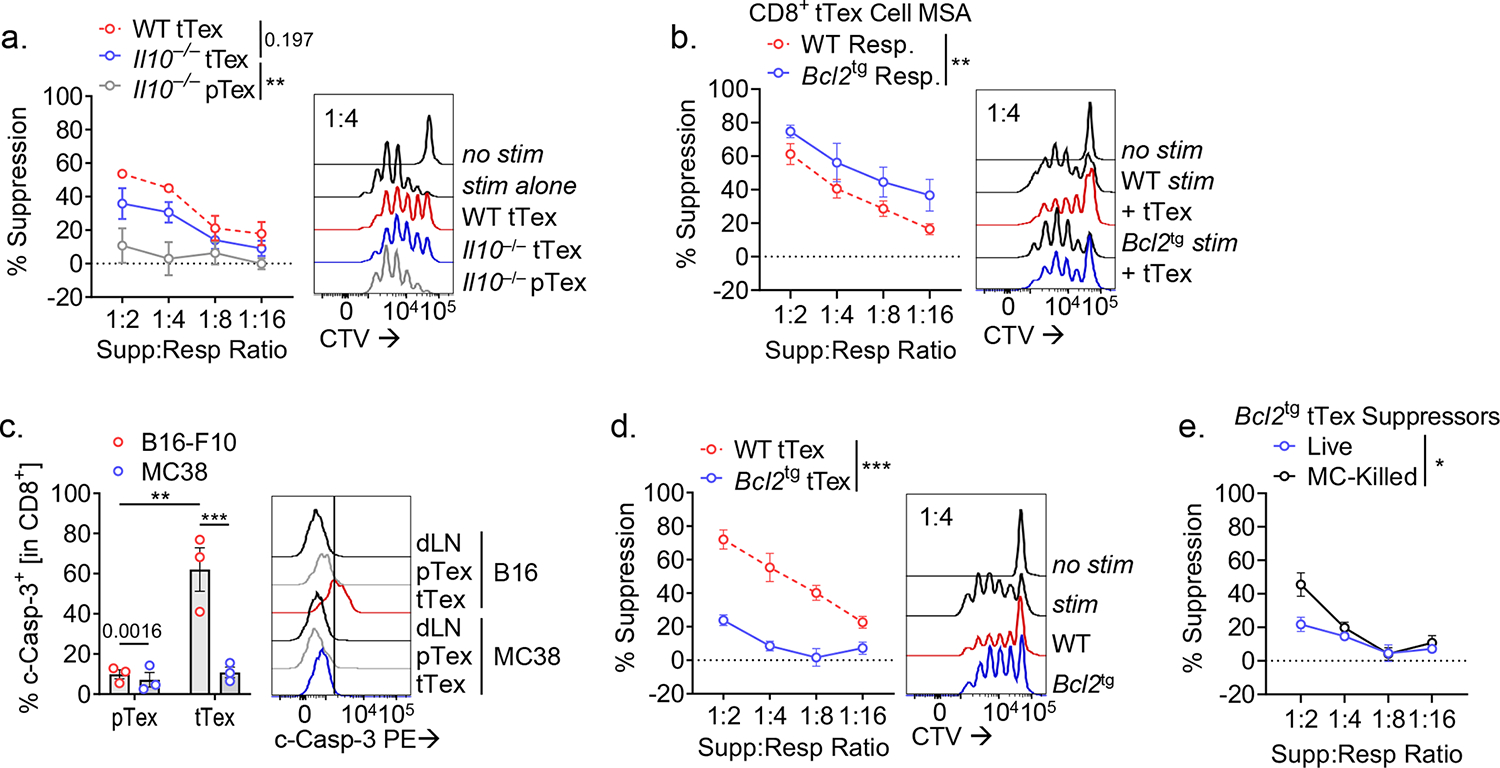
(A) Suppression assay of B16-F10 TIL from wild-type C57/BL6 or Il10−/− mice. (B) Suppression assay of B16-F10-derived tTex cells co-cultured with with activated C57/BL6 or Bcl2tg responding T cells. (C) Cleaved caspase-3 staining in CD8+ T cells from B16-F10 or MC38 tumors and draining lymph nodes (dLN). (D) Suppression assay of B16-F10 TIL from C57/BL6 or Bcl2tg mice. (E) Repeat experiments from Fig. 4D with live or Mytomycin C (MC)-killed tTex cells. Statistics are linear regression (A,B,D,E) and one-way ANOVA (C) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
We next investigated whether tTex cell-mediated cytotoxicity could explain suppressor function in ex vivo assays. In both mice and humans, CD8+ T cells have repeatedly been shown to induce tolerance in autoimmune disease through lysis of autoreactive CD4 T cells24. Indeed, cytotoxicity also contributes to CD4+Foxp3+ Treg cell suppression25. However, in ex vivo suppression assays, we do not observe meaningful changes in cell viability among responding CD4+ T cells or APCs regardless of the co-cultured putative ‘suppressor’ population, or the ratio of suppressors to responders (Extended Data Fig. 4D,E). These data suggest CD8+ tTex cells are not enforcing immune suppression by direct cell lysis ex vivo. Despite little evidence that tTex cell cytotoxicity was responsible for the observed immunosuppression, we sought a more robust study to validate our initial observations. To accomplish this, we derived the responding T cell and APC pools from the well-established model of apoptosis resistance via BCL-2 overexpression (Bcl2tg). Here too, we observed no diminishing of tTex cell-mediated suppression and, in fact, by improving cell viability in the responding population (Extended Data Fig. 4F), tTex cells appeared more suppressive (Fig. 3B). Interestingly, viability of antigen-presenting cells was dispensable for the assay: if apoptosis was directly induced in APC by mitomycin C treatment prior to their addition in suppression assay, tTex cells retained suppressive capacity (Extended Data Fig. 4G). However, tTex cell suppression was overcome by replacing the typically used T cell depleted-splenocyte APCs with anti-CD3/anti-CD28-coated microbeads, suggesting local immune cells may be directly influencing the capacity of tTex cells to suppress T cell responses.
While direct cytotoxicity by tTex cells was not apparent in our ex vivo experiments, numerous studies have suggested tTex cells themselves are apoptosis prone2,13,20. Indeed, directly ex vivo, suppressive tTex cells from B16-F10 display elevated cleaved caspase 3 staining versus progenitor counterparts or non-suppressive MC38 tTex cells (Fig. 3C). In ex vivo co-culture too, tTex cells are significantly less viable than progenitors after three days, despite initiating the assay with sorted live populations (Extended Data Fig. 4H). Intriguingly, apoptosis-resistant Bcl2tg tTex cells from B16-F10 tumors displayed markedly reduced suppressive capacity ex vivo (Fig. 3D; Extended Data Fig. 4I), suggesting that there may be some role for cell death in mediating tTex cell control of local immune functions. Further, when we triggered apoptosis in Bcl2tg tTex cells with mitomycin C treatment prior to the assay, we could augment their suppressive function (Fig. 3E). Evidence suggests CD4+Foxp3+ Treg cells too, retain suppressive functions as they undergo apoptosis26. Thus, tTex cells may not suppress local immunity by killing, but rather further exert their regulatory capabilities though dying.
CD39 is required for tTex cell suppressor function
Apoptosis bolsters Treg cell suppressive function through the release and subsequent CD39/CD73-mediated hydrolysis of extracellular ATP (eATP) to adenosine27. CD39 faithfully marks the most terminally exhausted CD8+ T cells16,28 both in human (Extended Data Fig. 5A,B) and murine (Fig. 1C) cancers. Interestingly, in our ex vivo system, higher CD39 expression among tTex correlated with increased suppression (9.64% increase per 0.1 log CD39 MFI, p < 0.001; Fig. 4A). To determine the importance of CD39 density in suppression, we flow cytometrically purifed CD39hi or CD39lo tTex cells from either B16 (harbors highly suppressive tTex) or MC38 (which harbors poorly suppressive tTex cells). This revealed that within MC38-derived tTex, CD39hi carried some suppressor function, compared to CD39lo cells (Fig. 4B; Extended Data Fig. 5C). These data highlight that density of surface CD39 expression (rather than simply whether it is expressed) likely plays a role in observed ex vivo suppressive function.
Figure 4. tTex cells suppress through CD39-mediated extracellular ATP depletion and adenosine production.
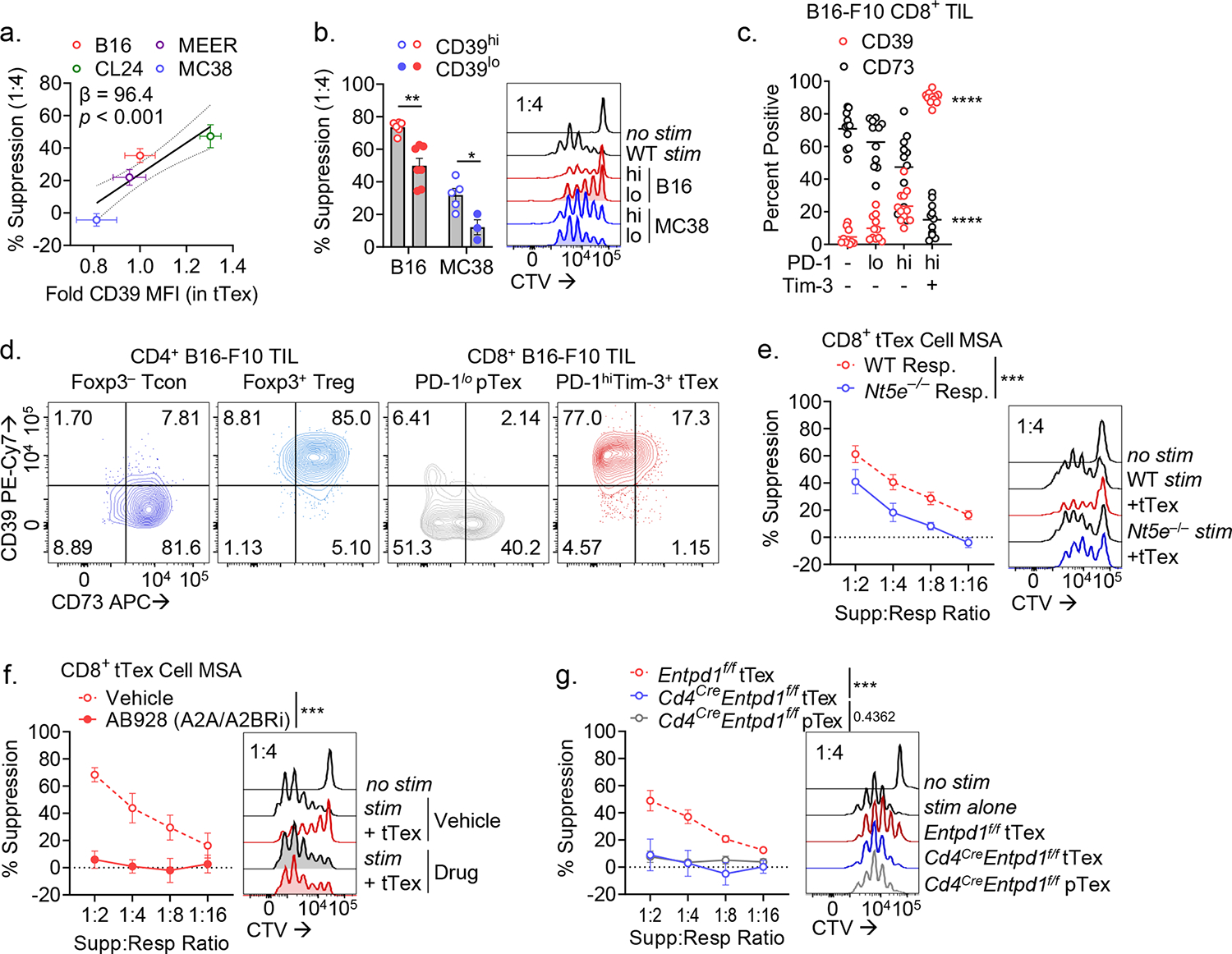
(A) Pearson correlation of percent tTex cell-mediated suppression cell in 1:4 ratio in suppression assay versus fold change in CD39 MFI in tTex cells between murine tumor lines. (B) Repeat experiments from Fig. 1G and 2F, in which tTex cells from B16-F10 and MC38 tumors were stratified by CD39 expression. (C) Percent CD8+ T cell populations from B16-F10 expressing CD39 or CD73. (D) Representative bivariant plots of B16-F10 infiltrating T cell populations. (E) Suppression assay of B16-F10 TIL co-cultured with with activated CTV-labeled C57/BL or Nt5e– ‘responding’ T cells. (F) Suppression assay of B16-F10 infiltrating CD8+ tTex cells co-cultured with DMSO vehicle or A2AR/A2BR small-molecule inhibitor AB928 at 3 μg/mL. (F) MSA of B16-F10 TIL from Entpd1f/f or Cd4CreEntpd1f/f mice. Statistics are linear regression (A,E-G) and one-way ANOVA (B,C) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Foxp3+ Treg cells generate adenosine through coordinate action of CD39 and CD73, which they coexpress. Notably, unlike Treg cells, CD8+ tTex cells largely do not co-express CD73, and during differentiation to exhaustion ‘switch’ from being CD73hiCD39− to CD73loCD39+ (Fig. 4C,D). Analysis of CD8+ T cells across numerous lymphoid and non-lymphoid organs in a tumor-bearing mouse reveals that CD39 expression is essentially restricted to intratumoral tTex cells and undetectable in cells from non-inflamed healthy tissues (Extended Data Fig. 5D). Thus, while tTex cells may be poorly capable of producing adenosine in isolation, as they are CD73lo, elevated CD39 expression may contribute to immune suppression by supporting the generation of an adenosine rich environment with local CD73+ cells. Indeed, circulating human CD73loCD39+ Treg cells have been shown to associate with CD73-expressing cells or exosomes to elicit their suppressive function29, supporting the notion that CD39+ tTex cells may be suppressive only when in proximity to CD73+ cells.
To model the influence of CD73+ cells in tTex cell-mediated suppression, we next derived responder T cell and APC populations from CD73-deficient (Nt5e−/−) mice. Indeed, wild-type tTex cells were significantly less suppressive when APCs and responding T cells were CD73-deficient (Fig. 4E). However, tTex cell suppressive capacity was not wholly absent in this setting, suggesting that the residual CD73 expression on tTex cells may mediate some control of local immune activation via the eATP:adenosine axis. To further elucidate the role of adenosine signaling in tTex cell-mediated suppression, we treated cells with a small molecule inhibitor specific to the adenosine receptors, A2AR and A2BR (AB928; Arcus Biosciences), which showed blockade of adenosine receptor signaling is sufficient to completely ablate tTex cell suppression ex vivo (Fig. 4F). Interestingly, neither CD73 deficiency (Extended Data Fig. 5E) nor adenosine receptor blockade (Extended Data Fig. 5F) prevented CD4+Foxp3+ Treg cell-mediated suppression in ex vivo co-culture, revealing that unlike the numerous inhibitory mechanisms employed by Treg cells, tTex cells employ a single dominant method of constraining local immune responses–via the generation of a local environment primed for generating adenosine.
Next, to specifically interrogate the consequences of CD39 activity on tTex cells, we bred Entpd1f/f (encoding CD39) mice to Cd4Cre mice (hereafter, Cd4CreEntpd1f/f)30, generating T cell specific deletion of CD39 (Extended Data Fig. 6A). In large B16-F10 tumors (8 to 10 mm; day 14), CD39 expression was dispensable for expression of inhibitory receptors, PD-1 and Tim-3 (Extended Data Fig. 6B). While the percent of PD-1+Tim-3+ tTex among CD8+ TIL did not change with CD39 deletion in T cells, tTex cells were slightly but significantly reduced by cell number in Cd4CreEntpd1f/f mice (Extended Fig. 6C). However CD39-deficient tTex cells displayed a complete loss of inhibitory capacity compared to CD39-competent counterparts (Fig. 4F). T cell specific deletion of CD39 was also sufficient to slow tumor growth (Extended Data Fig. 6D); however, in this Cre-lox system, we cannot distinguish the contributions of CD39 deletion in CD4+ Treg cells versus CD8+ tTex cells to the improved tumor response.
It is important to note that CD39, like many other T cell exhaustion-associated markers (e.g. PD-1 and Tim-3), is rapidly and transiently upregulated in the immediate response to antigen recognition. Acute infection models have revealed the temporal nature of CD39 upregulation in activated CD4+ and CD8+ T cells. To determine whether activation induced CD39 expression, or activated Teff cells in general, were capable of suppressing local T cell responses, we generated Teff cells from a model of acute infection, transferring 1 × 105 naïve Thy1.1+ OT-I TCR transgenic CD8+ T from Cd4CreEntpd1f/f or Entpd1f/f mice into wild-type Thy1.2+ hosts infected with OVA-expressing Vaccinia virus (VacciniaOVA). Regardless of genotype, Thy1.1+CD44+ OT-I Teff cells isolated at 8 days after transfer failed to suppress co-cultured responding T cell proliferation, (Extended Data Fig. 6E) and displayed minimal CD39 expression in ex vivo culture (Extended Data Fig. 6F).
We next assessed the requirement for TCR stimulation in tTex cell suppression. When OT-I CD8+ T cells were used as responders, polyclonal intratumoral tTex cell-mediated suppression was observed when cultures were stimulated with anti-CD3 (engaging the TCR of both tTex cells and responder cells), typically performed in suppression assay protocols. However, when TCR signaling in tTex cell was bypassed by activating OT-I responders with cognate peptide, tTex cell-mediated suppression was lost (Extended Data Fig. 6G). Notably, while PD-1 and Tim-3 expression remained elevated in tTex cells between these two stimulation conditions (Extended Data Fig. 6H–I), CD39 was absent in tTex cells in SIINFEKL co-cultures, suggesting TCR signaling was required to maintain CD39 expression on tTex cells (Extended Data Fig. 6J).
CD39 overexpression restricts local T cell activation
We next sought to better understand the mechanism by which CD39 may be limiting anti-tumor immunity with a gain-of-function approach. We retrovirally overexpressed CD39 concurrent with mCherry (pMSCV-Entpd1-IRES-mCherry; Fig. 5A). Transduced polyclonal CD8+ T cells bore CD39 density similar to B16-F10-derived tTex cells in situ (Fig. 5B) and possessed functional enzymatic activity (Fig. 5C). As eATP has been defined as a crucial autocrine signaling molecule in activated T cells, we first investigated the effects of CD39 overexpression on the overexpressing cell itself. Compared to empty vector control CD8+ T cells (pMSCV T cells), CD39-overexpressing T cells displayed significant reduction in TCR signaling via phosphorylated ZAP-70 (Y319) and Akt (S473) (Fig. 5D,E), consistent with the role of adenosine on T cell activation. CD39-overexpressing T cells also displayed both diminished and delayed switch to glycolysis when acutely stimulated during extracellular flux assays as previously described31 (Extended Data Fig. 7A–D). Reduced TCR signaling in CD39-overexpressing T cells was also evident in diminution of downstream calcium flux (Extended Data Fig. 7E) and cytokine assays (Extended Data Fig. 7F,G). These data highlighted CD39-mediated autoregulation, yet did not reveal the ability of CD39 to impose suppression of nearby T cells. We next investigated the effect of CD39 overexpression on neighboring cells, as presence of the ectonucleotidase would presumably generate an adenosine rich microenvironment. We repeated restimulation experiments in 1:1 co-culture using congenically mismatched CD39-overexpressing and empty vector control T cells. Compared to control T cells cultured alone, CD39-overexpressing T cells again displayed a significant reduction in cytokine production (Fig. 5F) with correspondingly reduced proliferation (Fig. 5G). However, when control transduced T cells were cultured in the presence of CD39-overexpressing T cells, the control T cells suffered an equivalent loss of cytokine production and proliferation compared to the CD39-overexpressing cells alone (Fig. 5F,G), suggesting enforced CD39 expression is sufficient for suppressor functions even in vitro generated Teff cells.
Figure 5. Enforced CD39 expression in Teff cells inhibits effector functions in neighboring CD39− T cells.
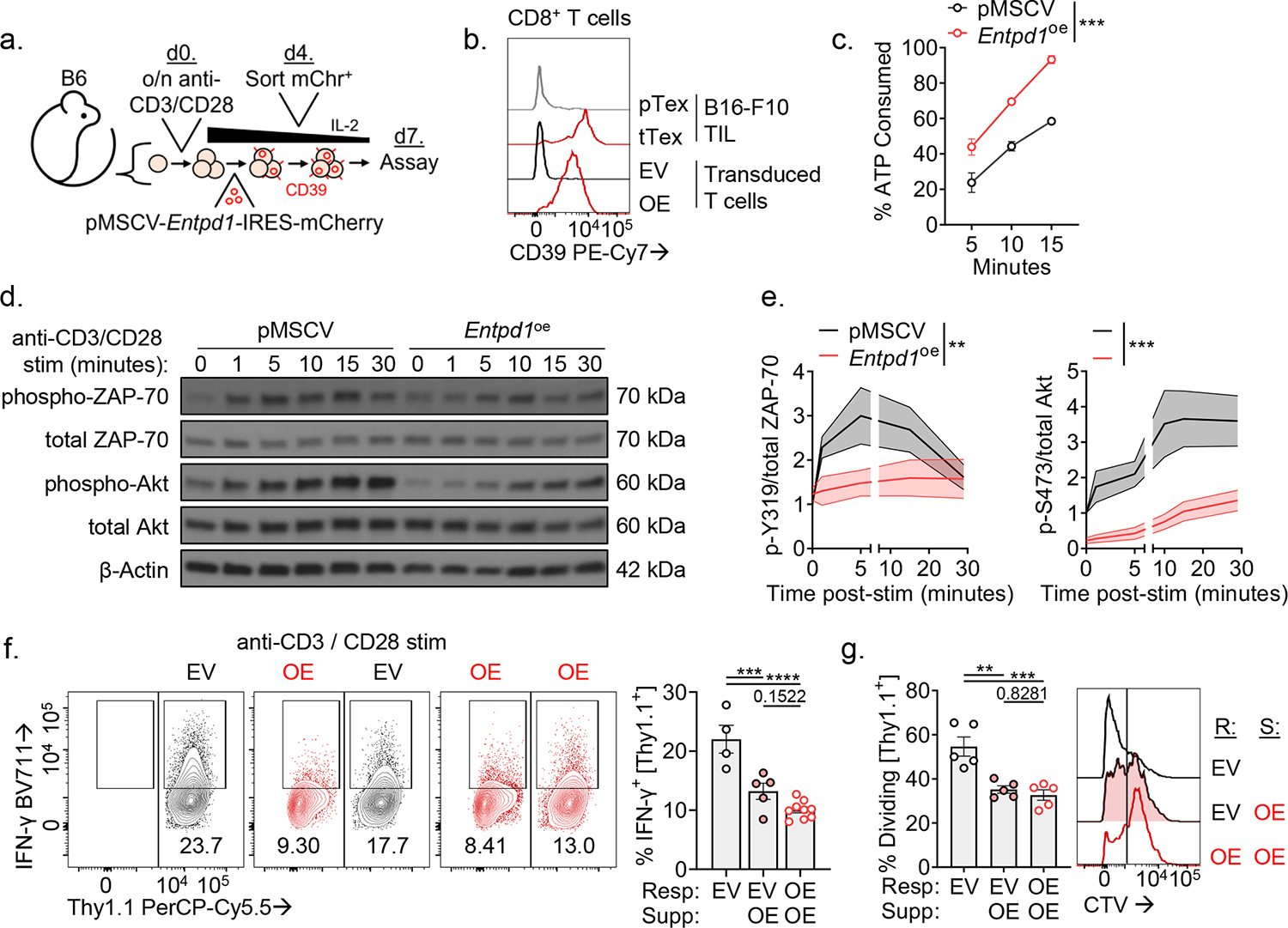
(A) Model of activated T cells transduced with pMSCV-Entpd1-IRES-mCherry and then rested for seven days prior to assay. (B) CD39 expression in day 7 transduced T cells or day 14 B16-F10 TIL. (C) Percent of spiked in ATP consumed in transduced T cells. (D) Western Blot of phosphorylation events following of anti-CD3/anti-CD28-coated microbead stimulation in transduced T cells and (E) quantified band intensity normalized to t = 0 in empty vector T cells. (F) Cytokine production of transduced T cells in 24-hour co-culture experiments following anti-CD3/anti-CD28-coated microbead stimulation. (G) Division on CTV-labeled transduced T cells over 72 hours. Statistics are linear regression (C) and one-way ANOVA (E,F,G) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
tTex cells are functionally significant suppressors in tumors
Several immune cells within the TME, as well as some tumor cells and stroma, can express some level of CD39. Both extracellular and intracellular factors regulate the expression of CD39, including activation signals and hypoxia-sensitive programs. So, we next sought to determine the specific contribution of tTex cell-mediated immunosuppression to the tumor microenvironment. Within the CD8+ compartment of a tumor-bearing mouse, CD39 expression is essentially restricted to tTex cells (Extended Data Fig. 5D); therefore, to determine the significance of CD39 expression of tTex cells alone, we employed a tamoxifen (TAM)-inducible, CD8-specific mouse, in which transgenic Cre-ERT2 is under control of the CD8a enhancer (E8i), and a Rosa26-driven recombination reporter, crossed with Entpd1f/f (E8iGFP−Cre−ERT2−Rosa26LSL-TdTomatoEntpd1f/f; hereafter, E8iCre-ERT2Entpd1f/f). This mouse was used to induce Entpd1 deletion specifically in CD8+ T cells prior to B16-F10 tumor engraftment (Extended Data Fig. 8A). Indeed, similar to our initial findings in the Cd4Cre system, inducible CD8-specific deletion of CD39 resulted in significantly enrichment of Slamf6+Tim-3− pTex cells and fewer in Slamf6−Tim-3+ tTex cells in B16 tumors compared to TAM-treated controls (Fig. 6A). Notably, in these advanced tumors, we did not observe significant changes in ex vivo cytokine production in TIL (Extended Data Fig. 8B).
Figure 6. Restricted deletion of CD39 on endogenous CD8+ tTex cells reduces suppression of newly infiltrating T cells.
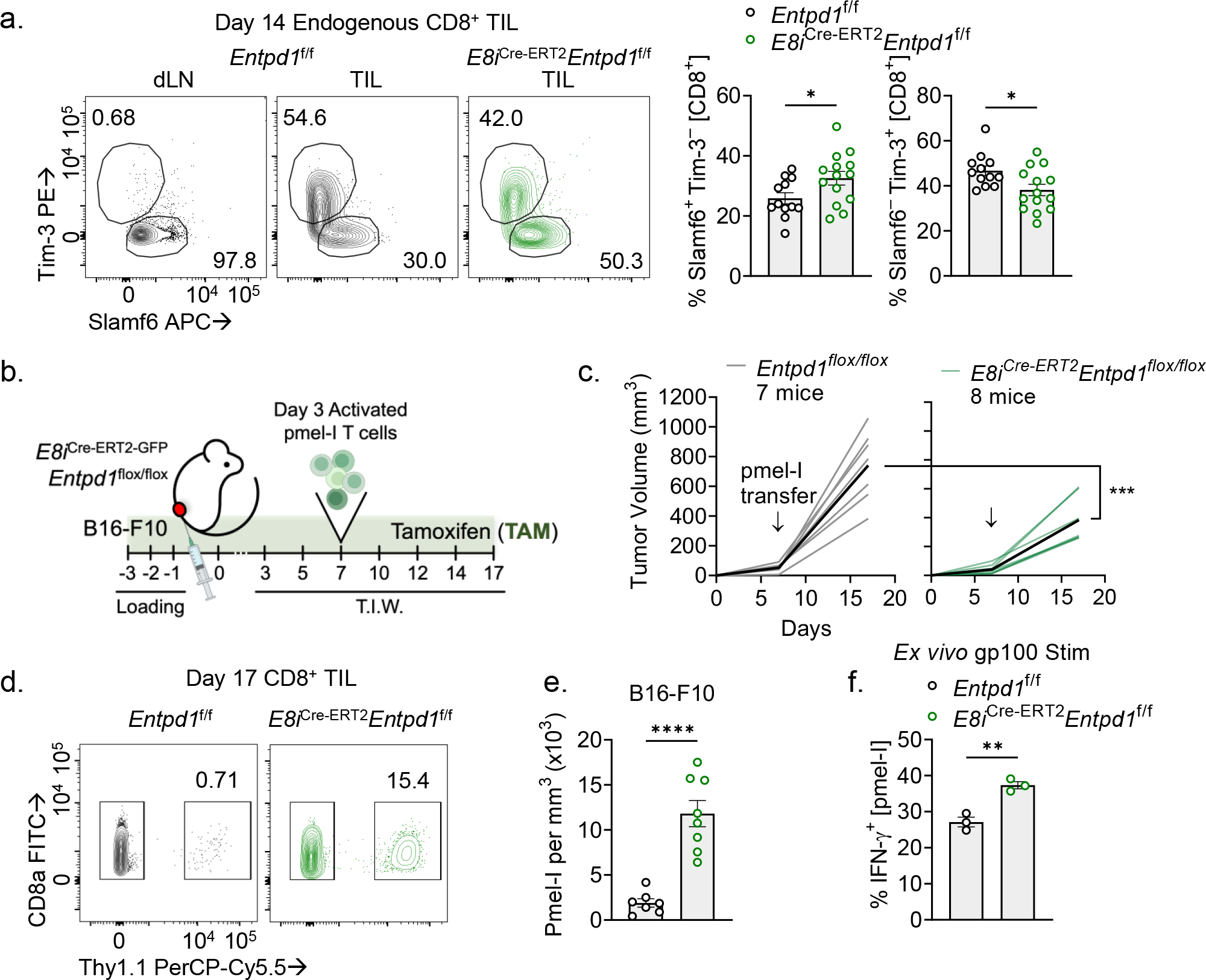
(A) Tim-3 and Slamf6 staining on CD8+ T cells from day 14 B16-F10 tumors. (B) Schematic of adoptive cell therapy protocol. Entpd1f/f or E8iCre-ERT2Entpd1f/f mice were treated with tamoxifen (TAM) starting three days prior to B16-F10 tumor inoculation then continued thrice weekly thereafter. 3–5 million activated pmel-I T cells were transferred retro-orbitally on day 7. (C) Tumor growth from adoptive T cell transfer experiment. (D,E) Percent infiltration and absolute numbers of adoptively transferred pmel-I T cells into B16-F10 tumor. (F) IFN-gamma production in pmel-I T cells restimulated ex vivo with gp100. Statistics are Mann-Whitney (A,E,F), and two-way ANOVA (C) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
We next sought to determine, using the E8iCre-ERT2Entpd1f/f system, whether CD39+ tTex cells suppressed tumor-infiltrating T cells in vivo. To do this, we inoculated B16-F10 cells into TAM-treated E8iCre-ERT2Entpd1f/f or control mice, and at day 7, adoptively transferred a subtherapeutic dose of activated, congenically marked pmel-I T cells (carrying a transgenic TCR specific for the melanocyte antigen, gp100, expressed in B16-F10) (Fig. 6B), allowing us to determine if CD39 expression on tTex cells mediated suppression of a wild-type T cell population in tumors. Ten days after pmel-I T cell transfer, B16-F10 tumors on E8iCre-ERT2Entpd1f/f mice were significantly smaller compared to controls (Fig. 6C). Further, while the number of transferred T cells in tumor-draining lymph nodes was equivalent between groups (Extended Data Fig. 8C), pmel-I T cells within the tumor were on average 4 times more numerous in E8iCre-ERT2Entpd1f/f mice compared controls (Fig. 6D,E; Extended Data Fig. 8D). Moreover, these pmel-I T cells possessed significantly elevated IFN-γ production over controls (Fig. 6F; Extended Data Fig. 8E). Thus, our data suggest CD39-expressing tTex cells are capable of suppressing tumor-specific T cell populations in vivo.
Evidence of in situ suppression by tTex cells encouraged further investigation of the therapeutic benefit of targeting CD39 expression. Thus, we orthotopically inoculated E8iCre-ERT2Entpd1f/f mice and controls with B16-F10 as before, but delayed TAM treatment–and thus CD39 deletion–in all groups to when tumors were palpable (~2 mm, day 5; Fig. 7A). After 3 days of tamoxifen treatment, we treated half of our cohort now carrying advanced tumors (~5 mm, day 8) with anti-PD-1 and anti-CTLA-4 immunotherapy. At this timepoint, checkpoint blockade immunotherapy is incapable of causing complete regressions in B16-F10 tumors and mice progress with little therapeutic benefit (Fig. 7B,C). As described earlier, CD8+ T cell-restricted deletion of CD39 itself was sufficient to significantly slow tumor progression (Fig. 7B,C) to a degree similar to combination checkpoint blockade alone. However, checkpoint blockade in the context of CD39 deletion further improved the therapeutic response, resulting in 37.5% of mice achieving a partial response (3/8; static or decreased tumor size after two days) and 12.5% with complete tumor remission (1/8; grossly undetectable disease; Fig. 7C).
Figure 7. tTex cell-restricted deletion of CD39 invigorates immune responses in an aggressive preclinical model.
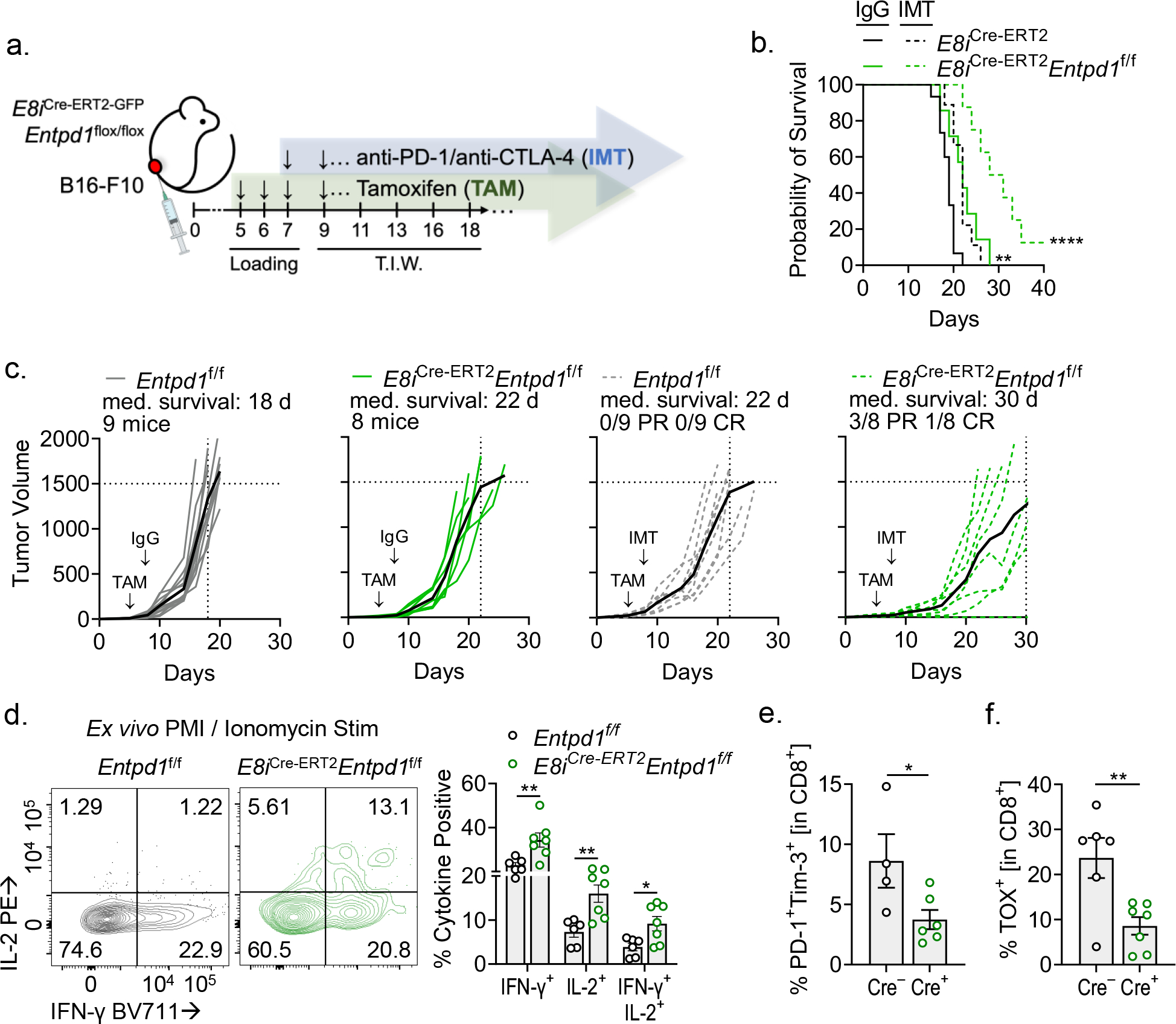
(A) Schematic of experimental design. TAM treatment began at day 5 post-tumor inoculation then continued thrice weekly thereafter; immunotherapy (IMT) treatment began on day 8 and was administered thrice weekly. (B,C) Tumor growth over time and survival in Entpd1f/f or E8iCre-ERT2Entpd1f/f mice bearing B16-F10 with or without combination immunotherapy. (D) CD8+ TIL restimulated with PMA and Ionomycin at day 8 of tumor growth, day 3 of TAM treatment. (E,F) PD-1 and Tim-3, or TOX expression in CD8+ T cells on day 8 of tumor growth, day 3 of TAM treatment. Statistics are Log-Rank test (B), one-way ANOVA (D), and Mann-Whitney (E,F) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Given the absence of improved cytokine production in intratumoral CD39-deficient T cells (Extended Data Fig. 8C) despite a diminished proportion of these being Slamf6−Tim-3+ tTex cells (Fig. 6A), we sought to interrogate the immune milieu early in tumor progression. Here, we treated E8iCre-ERT2Entpd1f/f mice with a three-day loading course of TAM beginning at day 5 post-tumor engraftment as before, then isolated CD8+ TIL when tumors between groups were still similarly sized (~5 mm, day 8). At this timepoint, CD39 expression was significantly depleted in E8iCre-ERT2Entpd1f/f TIL as compared to controls (~60% decrease from control; Extended Data Fig. 8F). Strikingly, B16-F10-infiltrating CD39-deficient CD8+ T cells displayed significantly improved cytokine competency when restimulated ex vivo as compared to control mice, with ~2-fold increase IFN-γ+ and IL-2+ T cells by flow cytometry (Fig. 7D). Further, at this early time point, CD8+ T cell-specific deletion of CD39 also resulted in delayed accumulation of hallmark features of exhaustion, including inhibitory receptor (PD-1 and Tim-3) expression as well as expression of the exhaustion-associated transcriptional repressor, Tox (Fig. 7E,F; Extended Data Fig. 8G,H). Thus, preventing suppressor activity in tTex cells creates an environment favorable for immunotherapy response.
Hypoxia mitigation weakens suppressor capacity of tTex cells
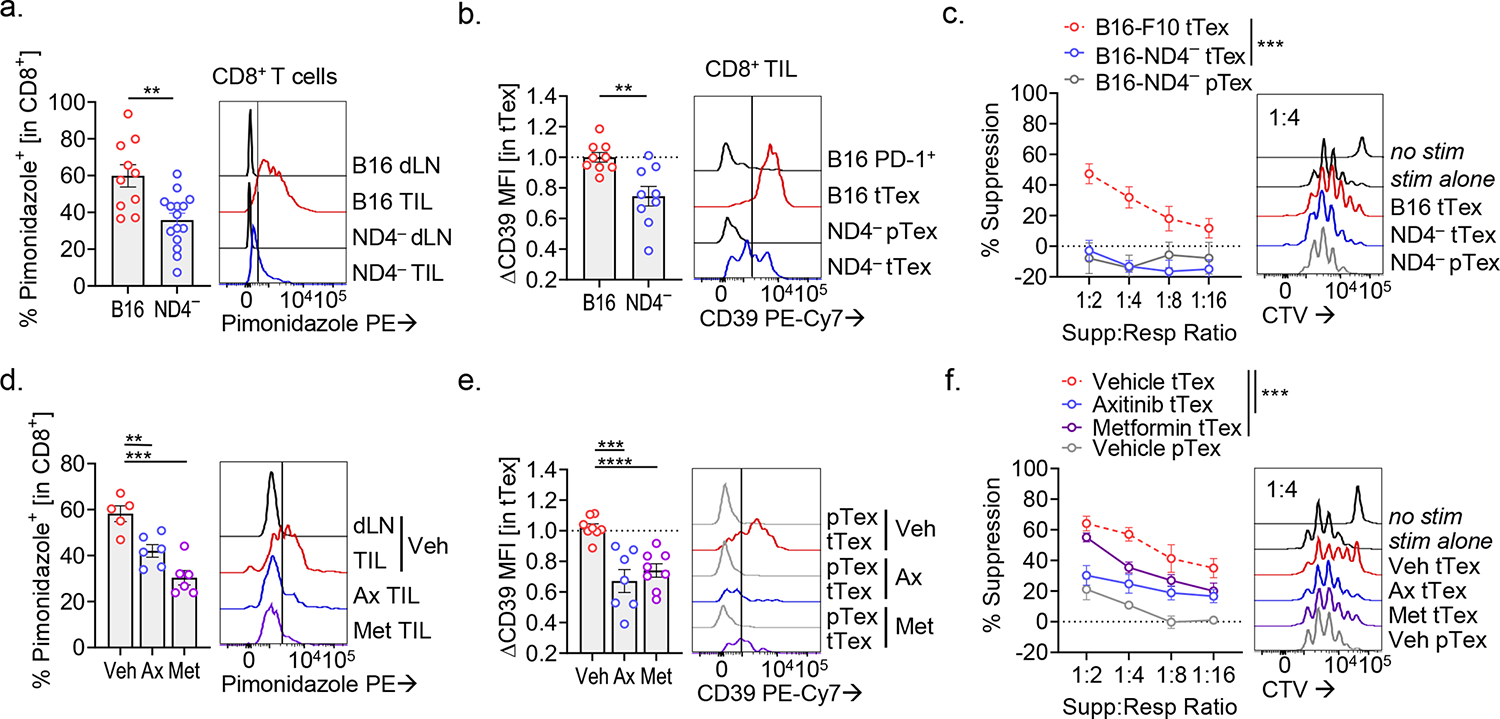
While the hypoxia-Hif1α-VHL axis has been implicated in T cell activation and proinflammatory effector functions32, the repressive role that hypoxia plays in the tumor microenvironment has been well documented by our group and others7,13–15. Disruption of this pathway through genetic or pharmacologic approaches reinvigorates anti-tumor immunity and sensitizes immunotherapy-resistant tumors. Hypoxia, oxidative stress, and proinflammatory mediators have all been shown to induce CD39 expression7. Hypoxia-selective pimonidazole (commercially known as Hypoxyprobe) labeling15, expectedly shows CD8+ TIL experiencing low oxygen tension compared to dLN (Extended Data Fig. 9A). As we have previously shown13, tTex cells experience significant hypoxia (pimonidazolehi) relative to progenitors (Fig. 8A), and exposure to hypoxia is associated with enrichment of surface CD39 staining (Fig. 8B). Thus, we asked if expression of CD39 could be driven by hypoxia in isolation, via in vitro culture at oxygen tensions similar to what is observed in tumors33 (~1.5% O2). Indeed, CD8+ T cells cultured in tumor hypoxic conditions displayed significantly higher CD39 staining compared to those maintained in atmospheric oxygen tensions (~20%), in a Hif1α-dependent manner (via Cd4CreHif1αf/f; Fig. 8C), supporting the hypoxia-Hif1α-VHL axis as a key contributor to the suppressor fate of CD39+ tTex cells. MC38 tumor-infiltrating tTex cells, which we show are largely non-suppressive due to being CD39lo (Extended Data Fig. 9B), experience significantly lower hypoxia exposure versus their B16-F10-resident counterparts34 (Extended Data Fig. 9C).
Figure 8. In vitro exposure to continuous TCR stimulation under hypoxic conditions yields exhausted-like, suppressive CD8+ T cells.
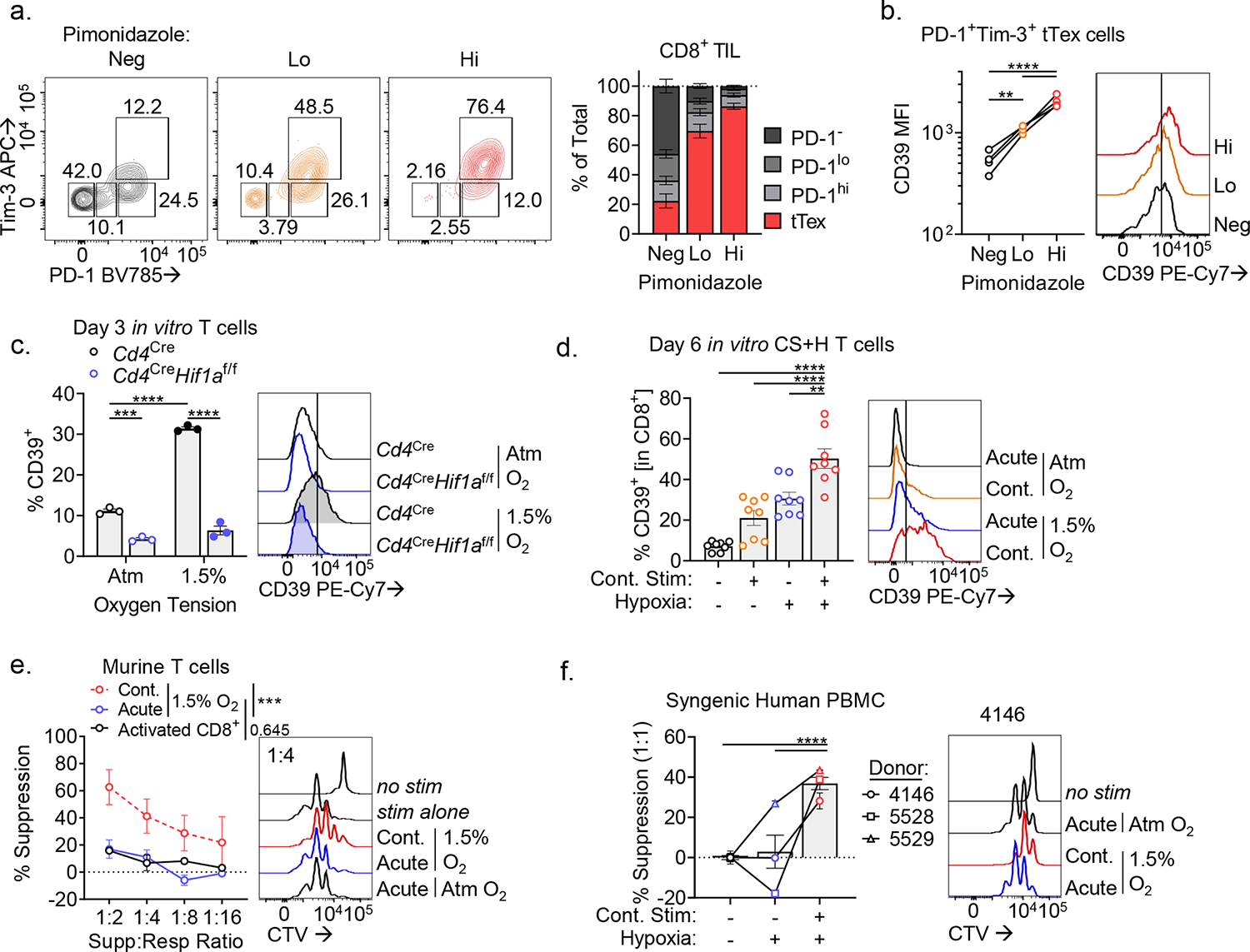
(A) PD-1 and Tim-3 staining in Pimonidazole fractions–negative, low, and high. (B) CD39 staining in PD-1+Tim-3+ tTex cells from Pimonidazole fractions. (C) Percent of CD8+ T cells expressing CD39 following 72-hour stimulation at atmospheric or tumor hypoxic conditions (1.5% O2). (D) CD39 staining of day 6 CS+H cells. (E) Proliferation of CTV-labeled murine CD4+ T cells in MSA experiments with CS+H cells. (F) Proliferation of three human healthy donor CTV-labeled CD8+ T cells in MSA experiments with syngeneic human CS+H cells. Statistics are one-way ANOVA (B-D,F), and linear regression (E) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
To further examine the role of hypoxia in the generation of suppressor tTex cells, we next asked whether our recently published in vitro protocol (Extended Data Fig. 9D,E) for generating tTex-like cells de novo could also recapitulate our observed suppressor functions in co-culture assay. As we have previously reported13, the combination of continuous TCR stimulation and hypoxia exposure resulted in significantly higher CD39 expression than either signal alone (Fig. 8D). Thus, we can leverage this system to generate T cells with sustained CD39 expression and to test their capacity to suppress effector responses in co-culture assay. CD39hi T cells that had previously experienced continuous TCR stimulation under tumor hypoxic conditions displayed potent suppressive capacity in suppression assay relative to CD39lo T cells acutely stimulated under hypoxia (Fig. 8E). Further, these findings could be recapitulated in human healthy donor derived CD8+ T cells cultured under similar conditions ex vivo (Extended Data Fig. 9F–H): continuous TCR signaling under hypoxia was sufficient to generate human CD39hi T cells that were capable of suppressing co-cultured activated syngeneic T cells (Fig. 8F).
We have previously demonstrated that disruption of tumor oxygen consumption through deletion of mitochondrial complex I subunit, NADH:ubiquinone oxidoreductase subunit S4 (Ndufs4; B16-F10ND4−) significantly reduces tumor hypoxia13,15 (Fig. 9A, Extended Data Fig. 10A). Further, B16-F10ND4−-infiltrating CD8+ T cells display a more activated and less exhausted signature compared to those derived from the parental B16-F10 line (Extended Data Fig. 10B). In direct comparison, PD-1+Tim-3+ tTex cells that differentiate in the less hypoxic B16-F10ND4− tumor display significantly reduced surface CD39 staining (Fig. 9B) and, strikingly, when assayed in ex vivo co-culture, B16-F10ND4− tTex cells failed to display any functional suppression (Fig. 9C). Taken together, these data propose tumor hypoxia as an enforcer of tTex cell suppressor functions via elevated expression of CD39. However, within this system we cannot assess whether this anti-functional state is irreversible, or if alleviation of tumor hypoxia may rescue tTex cells from a suppressive state.
Figure 9. Genetic or pharmacologic mitigation of hypoxia reduces the CD39-dependent suppressive phenotype in tTex cells.
(A) Histogram overlay displaying Pimonidazole staining in intratumoral CD8+ T cells from parental B16-F10 or Ndufs4− (B16ND4−) tumors and draining lymph nodes (dLN). (B) CD39 staining intratumoral T cells B16-F10 or B16ND4− tumors. (C) Biological replicate suppression assays utilizing B16-F10 or B16ND4− TIL. (D) Pimonidazole staining in intratumoral CD8+ T cells infiltrating B16-F10 tumors or draining lymph nodes following two treatments of anti-hypoxia agents Axitinib (Ax) and metformin (Met). (E) CD39 staining in intratumoral T cells from various treatment groups. (F) Biological replicate suppression assays of B16-F10 TIL following two treatments of either vehicle, Axitinib, or metformin. Statistics are Mann-Whitney (A,B), linear regression (C,F), and one-way ANOVA (D,E), with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
To assess the plasticity of tTex cell functions in vivo, we treated Foxp3Ametrine-Flpo mice bearing advanced B16-F10 tumors (~6 mm; day 10;) with one of two tumor hypoxia-targeting agents, axitinib or metformin (Extended Data Fig. 10C). We have previous shown that when treated early, both axitinib and metformin display a mild therapeutic benefit and sensitize hypoxic tumors to checkpoint blockade immunotherapy13,34. Two treatments of axitinib (2 mg kg−1) or metformin (50 mg kg−1), delivered intraperitoneally and late in B16-F10 melanoma disease course, were sufficient to significantly mitigate tumor hypoxia (Fig. 9D, Extended Data Fig. 10D). As with our B16-F10ND4− model, infiltrating CD8+ T cells from either axitinib or metformin treatment groups displayed lower inhibitory receptor expression (Extended Data Fig. 10E) and further, when subset by PD-1 and Tim-3 expression, tTex cells from treatment groups expressed lower levels of CD39 (Fig. 9E). Moreover, axitinib or metformin treated tTex cells were reliably less suppressive ex vivo than their vehicle-treated counterparts (Fig. 9F). These findings further support tumor hypoxia as a critical mediator of suppressor function in tTex cells and reveal a degree of reversibility of this state. In contrast, while tTex cell suppressor functions were significantly compromised by targeting tumor hypoxia, Foxp3+ Treg cells from the same environments–B16-F10ND4− or axitinib/metformin-treated tumors–maintained their potent inhibitory character ex vivo (Extended Data Fig. 10F,G). These data reveal that the therapeutic benefit derived from anti-hypoxia agents in combination with checkpoint blockade is not primarily derived from altered suppressive capabilities of Foxp3+ Treg cells, but may result from changes in CD8+ T cell functionality, by (1) limiting metabolic stress experienced by tumor-specific T cells and (2) preventing progression of tTex cells to an anti-functional state that directly counteracts anti-tumor immunity.
DISCUSSION
T cell exhaustion has been described in numerous inflammatory contexts, in both pathological settings such as cancer, chronic viral infection, and autoimmunity1,10, but also in states requiring immune homeostasis and tolerance, such as in gestation11. The commonality within these inflammatory contexts is that the differentiation program of exhaustion is dependent upon chronic exposure of T cells to their cognate antigen. While the impact and consequence of T cells exhaustion in pathologic or homeostatic settings are still being untangled, it is likely that the evolutionary advantages of this functional state and how this program is co-opted in disease settings such as cancer is context dependent10. Indeed, recent studies have highlighted a critical role for suppressor CD8+ T cells in mitigating pathologic autoreactive CD4+ T cells via perforin-mediated lysis24. In tumor-infiltrating tTex cells however, this pathway has been shown to be defective35,36. Interestingly, therapeutic strategies that enforce terminal exhaustion in T cells have also been shown to dampen autoimmune disease progression37. In this study, using gold-standard assays, we clearly demonstrate CD8+ tTex cells, a common cellular fate of CD8+ T cells in solid tumors, upregulate the ectonucleotidase CD39 to levels sufficient to create a suppressive microenvironment. CD39 is a well characterized immunosuppressive molecule, and indeed recent efforts to therapeutically block CD39 and CD73 in combination with checkpoint blockade have yielded encouraging results38,39.
We have described a transcriptional and post-translational circuit that allows terminally exhausted T cells to suppress conditionally. Specifically, tTex cells mediate their suppressor function through (1) CD39 expression driven principally through the TCR and hypoxic sensing, (2) apoptotic generation of eATP, and (3) CD73 expression on the responding cell population. This triad likely evolved to prevent immunopathology in inflamed and damaged tissues, promoting immune tolerance, but only to newly activated, CD73hi T cells. However, in the deregulated tumor microenvironment, this drives immune dysfunction and immunotherapy resistance. While tTex cells have been characterized by high cell turnover via apoptosis2,20, several groups have reported that maintenance of T cell viability through overexpression of BCL-2 not only augments anti-tumor responses, but also lessens immune tolerance in the tumor microenvironment40,41. Indeed, enforced expression of BCL-2 in CAR-T cells improved persistence and response in both preclinical models and in human trials. These data are supported by our findings of maintaining tTex cell viability through BCL-2 expression restricts their suppressor functionality. In several models tested, the suppressive capacity of apoptosis prone tTex cells was equivalent to Foxp3+ Treg cells isolated from the same tumor environment. These suppressive functions can be inhibited upon BCL-2 overexpression, genetic deletion of CD39, or pharmacologic blockade of adenosine signaling. Further, we delineate a specific in vivo role of CD39 expression on tTex cells, revealing that CD8+ T cell-restricted deletion of CD39 results in retained polyfunctionality in both the endogenous tumor infiltrating T cell compartment as well as transferred wild-type tumor antigen specific T cells. This improvement to CD8+ TIL effector functionality via endogenous CD39 deletion resulted in better control of tumor progression and enhanced therapeutic efficacy of adoptive T cell transfer and checkpoint blockade strategies. Notably, targeting this pathway is insufficient to prevent T cell exhaustion in cancer, but we do show evidence that CD39 deletion can slow terminal differentiation. These data not only improve our understanding of the breadth of functions attributed to exhausted T cells, but also supports the eATP:adenosine axis as attractive targets to subvert resistance to checkpoint blockade by exhausted T cells.
Adenosine rapidly accumulates in response to tissue injury and ischemia, subverting effective T cell activation and pushing the balance to tissue repair42. In cancer, pioneering studies have identified many pathways upregulated in immune and stromal cells generating adenosine through consumption of immunostimulatory metabolites. Among these, the ectonucleotidase CD39, is increasing reported as a faithful marker of tumor antigen experienced CD8+ T cells16,28, particularly terminal exhaustion3,5. An increasing number of preclinical studies have revealed the therapeutic potential of CD39 inhibition–in addition to other targets in adenosine metabolism–spurring several phase I/II trials in solid tumors and lymphoma with antibody or inhibitor therapy43. In solid tumors, the metabolic features of tumor cells15, and the resulting hypoxic13,14 and nutrient dearth microenvironment6 accelerates CD8+ T cell dysfunction and diminish efficacy of checkpoint blockade therapy. We demonstrate that tTex cells encountering tumor hypoxia gain a functional suppressor program principally via expression of CD39, and can limit effective tumor control in a manner independent of CD39 expression on other infiltrating immune cells, tumor cells, or stroma. Our study supports a model in which chronically stimulated tumor resident CD8+ T cells sensing their hypoxic microenvironment respond by rapidly progressing to a terminally differentiated exhausted fate and upregulating immunosuppressive programs to repress local anti-tumor immunity.
In human cancer, CD39 expression in CD8+ T cells is associated with tumor antigen specificity and a terminally exhausted signature16,28, with some reports stating enrichment of CD39 prevents effective anti-tumor immunity44,45. Intriguingly, infiltration of CD39+CD103+ T cells–likely identifying tissue-resident memory populations–has also been shown to correlate with improved outcomes46,47. Taken together, these data suggest that while CD39 restricts inflammation, enrichment of tumor-specific T cells remains a dominant prognostic factor. Our data also suggest density of CD39, rather than its expression per se, is critical for suppressor function. Empowering those specific T cells through CD39 blockade may be an attractive target to circumvent the suppressor program enforced upon terminal exhaustion38,39.
A rather surprising result was that IL-10 was dispensable for tTex cell suppression, although the diversity of roles of IL-10 in cancer continues to widen. Numerous reports have highlighted a role for this pleiotropic cytokine both in the progression of cancer and restriction of anti-tumor immunity48, in enforcement of terminal exhaustion21,49, and in the cytotoxicity of CD8+ T cells21,50. We investigated whether exposure to IL-10 within the tumor or secretion of IL-10 by tTex cells19 could contribute to suppression of T cell responses. Contrary to studies highlighting a role for IL-10 in CD8+ T cell-mediated control of inflammation19,51, neither genetic deletion of IL-10 nor in vitro blockade diminished tTex cell-mediated suppression in our system. Thus, IL-10 does not appear to be a dominant tolerogenic mechanism utilized by tTex cells in cancer. Notably, recent reports have highlighted that engineered IL-10 molecules can metabolically reprogram tTex cells, improving their oxidative capacity and inflammatory functions21,52.
Exposure to hypoxia accelerates T cell exhaustion via metabolic byproducts like ROS, and alleviation of hypoxia sensitizes tumors otherwise resistant to immunotherapy13. We posit differentiation to terminal exhaustion results not only in dysfunctional but anti-functional cells, coordinated primarily through CD39 expression and adenosine-mediated suppression of T cell activation. However unlike many features of exhaustion, we show this suppressor state retains plasticity: while T cell exhaustion may not be capable of being fully reversed by checkpoint blockade immunotherapy, the inhibitory functions of tTex cells may be diminished through targeting tumor hypoxia or CD39 enzymatic activity. We propose multiple tenable therapeutic avenues to restrict tTex cell-mediated immune regulation and propose a mechanism to explain how anti-hypoxia therapies may promote improved CD8+ T cell effector functions–by dampening tumor hypoxia, CD39 expression is diminished on tTex cells and their regulatory potential is limited. Sequencing checkpoint blockade after tumor hypoxia mitigation and/or blockade of the eATP-adenosine axis may bring the curative promise of immunotherapy to additional patients.
METHODS
Mice
All experiments requiring live animals or ex vivo immune cells were performed in accordance with the Institutional Animal Care and Use Committee of the University of Pittsburgh. All mice were housed in specific pathogen-free conditions prior to use. Mice were maintained at room temperature on a 12h:12h light-dark cycle in boxes containing five (for female) or four (for male) mice. Experiental mice were received Purina Prolab Isopro RMH 3000 (5P75, 5P76) chow ad libitum. Both male and female mice were used, and mice were 6–8 weeks old at the start of experimentation. C57BL/6, Cd4Cre, Hif1αf/f, Nt5e−/−, Il10−/−, Bcl2tg mice were obtained from The Jackson Laboratory. Entpd1f/f mice were obtained from SC Robson (Beth Israel Deaconess Medical Center). E8iGFP−Cre−ERT2-Rosa26-LSL-TdTomato and Foxp3mAmetrine-Flpo mice were generated by DAA Vignali (University of Pittsburgh). Tumor experiments for ex vivo functional assays and analysis were typically completed on day 14, when tumors were 8 to 10 mm in diameter. For immunotherapy experiments, mice were sacrificed when tumors reached 15 mm in diameter or 1500 mm3, or were moribund.
In vivo treatments and immunotherapy were performed as previously described13,34 with Bio X Cell-supplied anti-PD-1 (clone J43), anti-CTLA-4 (clone 9H10), and host-appropriate isotype controls. Cayman Chemical supplied Axitinib (2 mg/kg IP; suspended in DMSO (10%), Tween 80 (10%), and 30% Captisol (80%)) and Metformin (50 mg/kg IP; saline (100%)). Sigma supplied tamoxifen (50 mg/kg PO; 100% EtOH (5%), corn oil (95%)).
Tissue Culture and T cell Isolation
B16-F10, MC38, and MEER were originally purchased from American Type Culture Collection (ATCC). B16-F0OVA was obtained from P Basse and L Falo (University of Pittsburgh). Platinum-E (Plat-E) was obtained from L Kane (University of Pittsburgh). Some immortalized tumor lines were generated in-house as previously described; B16-F10ND4– 13, Clone 24 (CL24)15, MEERPD−1res 14. All cell lines were maintained in complete DMEM (10% heat-inactivated fetal bovine serum, 1 mM sodium pyruvate, 2 mM L-glutamine, 5 mM HEPES Buffer, 100 U penicillin, 100 μg/mL streptomycin, 500 μg/mL gentamycin) at no greater than 80% confluence.
Tumor, tumor-draining lymph nodes, and spleens were surgically resected, then enzymatically and mechanically digested to a single cell suspension. Cells were purified by either magnetic bead isolation as previously described13, or fluorescence-activated cell sorting (FACS; on Beckman Coulter Mo-Flo Astrios High Speed Cell Sorter or Sony MA900 Cell Sorter), or a combination of the two. All T cells were cultured in complete RPMI (10% heat-inactivated fetal bovine serum, 1 mM sodium pyruvate, 2 mM L-glutamine, 5 mM HEPES Buffer, 100 U penicillin, 100 μg/mL streptomycin, 500 μg/mL gentamycin, 5 nM 2-mercaptoethanol) plus 100–25 U of murine IL-2 (PeproTech).
Ex vivo miniaturized suppression assays were performed as previously described18. To measure cytokine production, cells were incubated at 37°C for 6 hours or overnight in GolgiPlug Protein Transport Inhibitor (BD) with either 2:1 bead:cell ratio of Dynabeads T-Activator CD3/CD28 beads (ThermoFisher) or phorbol 12-myristate 13-acetate (PMA) and ionomycin (Sigma). Metabolic assays were performed as previously described13,31. In vitro continuous stimulation under hypoxia protocol was performed as previously described13.
Human Peripheral Blood or Tumor Infiltrating Lymphocyte Isolation
Human blood was obtained from Vitalant; tumor preparations were obtained via UPMC Hillman Cancer Center IRB protocol 96–099. Each patient was consented as outlined in our protocol, then subject to peripheral blood collection and tumor biopsy. Tumor samples were mechanically digested to a single cell suspension, then passaged through 70 μm cell strainers (BD). Lymphocytes were purified from bulk suspension by density centrifugation and/or cell sorting.
Flow Cytometry and FACS
Viability staining with Zombie dyes (Biolegend) and surface staining (see below) were performed in PBS at 4°C for 20 minutes. Samples were then washed and immediately analyzed on a BD LSRFortessa Flow Cytometer. Fixation of samples for intracellular hypoxia/transcription factor staining: following viability/surface staining, cells were incubated in 4% paraformaldehyde at room temperature for 5 minutes, washed in PBS, permeabilized with eBioscience Foxp3/Transcription Factor Staining Kit (ThermoFisher) per manufacturers protocol, washed in permeabilization buffer, then stained in permeabilization buffer at 4°C overnight. Fixation of samples for intracellular cytokine staining: following viability/surface staining, cells were fixed in BD Cytofix/Cytoperm Fixation/Permeablization Kit per manufacturers protocol, washed in permeabilization buffer, then stained in permeabilization buffer at 4°C overnight.
Biolegend supplied: Zombie Fixable Viability Dyes (UV, Aqua, Green, NIR); anti-mouse antibodies including, anti-CD8a (clone 53.6.7), anti-CD4 (GK1.5), anti-PD-1 (29F.1A12), anti-Tim-3 (RMT3–23), anti-Helios (22F6), anti-Nrp1 (3E12), anti-CD25 (PC61), anti-CTLA-4 (UC10–4B9), anti-Lag-3 (C9B7W), anti-CD39 (Duha59), anti-FasL (MFL3), anti-TIGIT (1G9), anti-ICOS (C398.4A), anti-CD73 (TY/11.8), anti-IL-2 (JES6–5H4), anti-IFN-γ (XMG1.2), anti-TNFα (MP6-XT22), anti-Thy1.1 (OX-7); anti-human antibodies including, anti-CD8a (RPA-T8), anti-CD4 (RPA-T4), anti-PD-1 (EH12.2H7), anti-Tim-3 (F38–2E2), anti-CD73 (AD2), anti-CD39 (TU66), anti-IL-2 (MQ1–17H12), anti-IFN-γ (4S.B3). Invitrogen supplied: anti-mouse antibody, anti-Foxp3 (FJK-16s); anti-human antibodies, anti-Lag-3 (3DS223H), anti-TNFα (Mab11). R&D Systems supplied: anti-Hif1α (241812). Thermo Fisher supplied: CellTrace Violet Cell Proliferation Kit, anti-Tox (TXRX10), anti-Blimp-1 (5E7).
Immunoblotting analysis
Immunoblotting was performed as previously described13. Santa Cruz Biotechnology supplied: anti-actin (clone C4); Cell Signaling supplied: Zap70 (D1C10E), phospho-Zap70 Y319/Y352 (65E4), Akt (C67E7), phosphor-Akt S473 (D9E). Immunoblots were detected via standard secondary detection and chemiluminescent exposure to film. Digitally captured films were analyzed densitometrically using ImageJ software.
Retroviral Vector Construction and T cell Transduction
Entpd1 (encoding CD39) was originally generated by and cloned into a murine stem cell virus (MSCV)-driven retroviral expression vector encoding an internal ribosome entry site (IRES)–GFP cassette, from DAA Vignali (Addgene plasmid # 52114; http://n2t.net/addgene:52114; RRID:Addgene_52114). For T cell transductions, T cells were activated with plate-bound anti-CD3 (5 μg/mL), soluble anti-CD28 (1 μg/mL), and IL-2 (100 U) for 24-hours. Retroviral supernatants were harvested, filtered, and supplemented with 6 μg/ml of polybrene. Prepared retrovirus was spun onto activated T cells (2,200 rpm, 2 hours, 37°C) then the cell/virus culture was rested in a tissue culture incubator for another 2 hours. Cells were then washed and cultured in fresh complete RPMI plus IL-2 (50 U) for three days to allow for expansion and expression of vector cassette. After 3 days, cells were purified via FACS by mCherry expression. CD39 expression was validated by flow cytometry and enzymatic activity verified by ENLIGHTEN ATP Assay System (Promega).
Statistics
If the data appeared to follow a normal distribution, we calculated the P values in GraphPad Prism using one-way analysis of variation (ANOVA) with Dunnett’s multiple comparison test, two-way ANOVA with Sidak’s multiple comparison test, unpaired Student’s t-test or paired Student’s t-test. For data that did not follow a normal distribution, we used the non-parametric Kruskal-Wallis and Mann-Whitney tests as indicated. The ex vivo suppression assays were analyzed using linear regression, treating the percent suppression as the dependent continuous variable, the type of cells as a categorical independent variable and the dilution as a continuous independent variable. Interactions were tested and included in the linear regression model if statistically significant. Values of p < 0.05 were considered significant and ranked as: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Extended Data
Extended Data Figure 1. PD-1hiTim-3+ terminally exhausted CD8+ T cells are a numerically dominant in tumor and express numerous Treg cell-associated effector molecules.
(A) Gating strategy for isolating CD8+ T cells in B16-F10 tumor or tumor-draining lymph nodes. (B) Quantified distribution of inhibitory receptor expression on CD8+ TIL from Fig. 1A. (C) Total cell numbers per size-matched B16-F10 tumor or draining lymph node. (D) Quantification from Fig. 1C. Mean fluorescence intensity (MFI) of CD4+Foxp3+ Treg cell-associated genes among tumor infiltrating T cell populations. Statistics are one-way ANOVA (C,D) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001
Extended Data Figure 2. Tumor infiltrating CD8+ T cells upregulate Treg cell signature upon terminal differentiation.
(A) Gene set enrichment analysis (GSEA) of tumor infiltrating Treg cell signature on bulk SLAMF6+ progenitor and Tim-3+ terminally exhausted CD8+ T cell transcripts from Miller et al. (B,C) Heatmap displaying DESeq2 of log2 normalized transcript expression of genes from the tumor infiltrating Treg cell signature gene set in (B) tetramer+ or (C) bulk progenitor and terminally exhausted CD8+ T cells. Values are transformed log2 (TPM) scaled to row. (D) Heatmap of log2 normalized DESeq2-defined differentially expressed genes (DEG) between progenitor and terminally exhausted CD8+ T cells.
Extended Data Figure 3. Quantification of tumor sizes and TIL populations from preclinical models with diverse sensitivity to immunotherapy.
(A) Tumor sizes from MSA experiments in Fig. 1 and 2 (B) (F) Average calculated percent suppression at 1:4 suppressor to responder ratio from all replicate experiments in Fig. 1 and supplementary Fig. 3R. (G) Percent of CD8+PD-1+Tim-3+ tTexh cells in various murine tumor models. Bivariant plots and histograms are representative of ≥3 experiments. Statistics are one-way ANOVA (A-C) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Extended Data Figure 4. tTex cells do not suppress via IL-10 secretion or direct cytotoxicity.
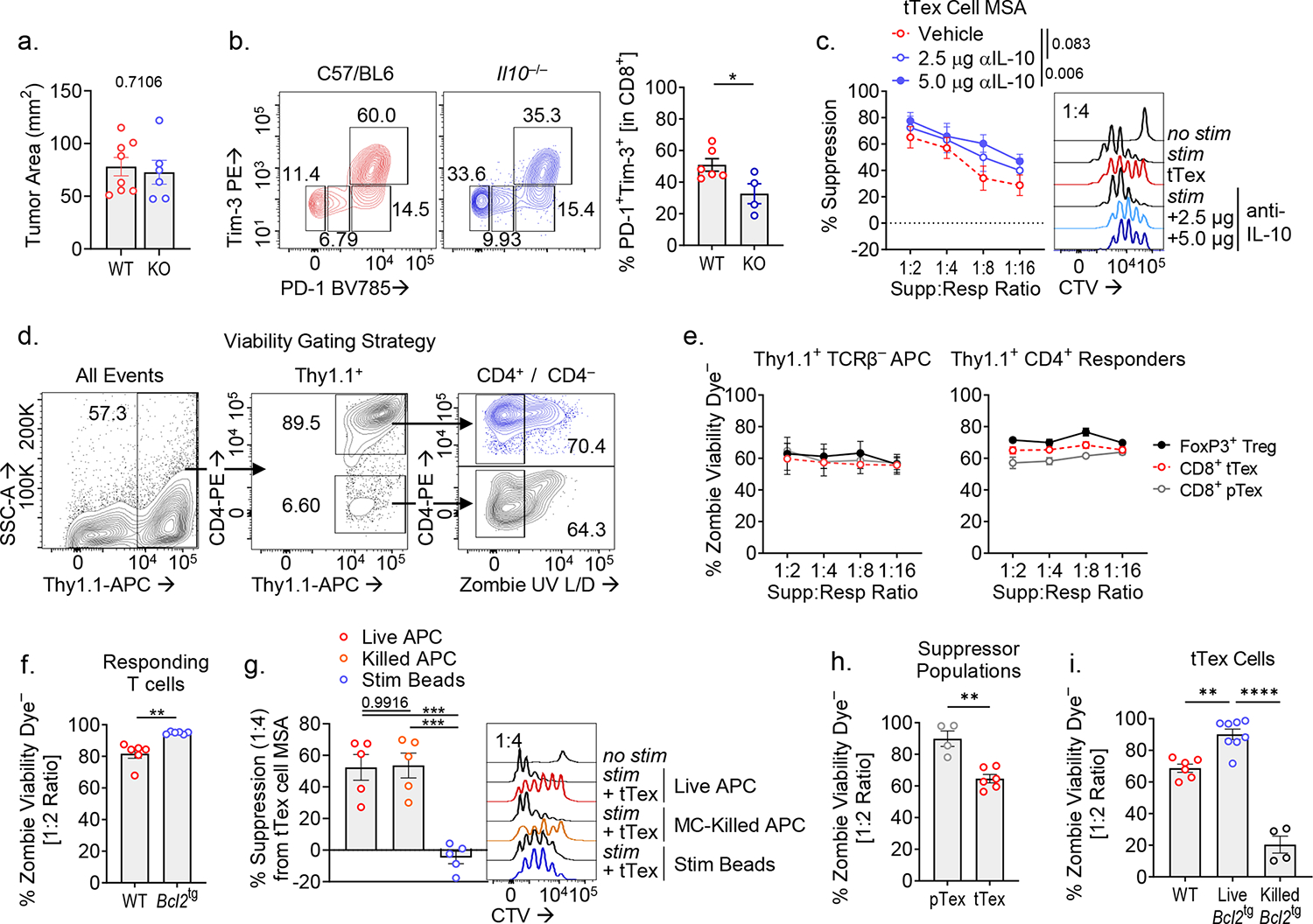
(A,B) Day 14 B16-F10 tumor sizes from Il10−/− experiments and matched CD8+ T cell infiltrate. (C) Suppression assay of B16-F10 infiltrating tTex cells co-cultured with isotype IgG or neutralizing IL-10 antibody at 2.5 or 5.0 μg/mL. (D) Gating strategy for calculation of cell viability in suppression assay populations. (E) Calculated averages of ‘responder’ or ‘APC’ population cell viability per dilution series of ‘suppressor’ T cells from Fig. 1G. (F) Cell viability of responder T cell populations from Fig. 3B. (G) Suppression assay of B16-F10 infiltrating tTex cells co-cultured with CTV-labeled responding T cells with either live or Mytomycin C (MC)-killed T cell-depleted splenocytes, or anti-CD3/anti-CD28 bound microbeads. (H) Cell viability of ‘suppressor’ populations from Fig. 1G. (I) Cell viability of ‘suppressor’ populations from Fig. 3D,E. Statistics are Mann-Whitney (A,B,F,H), linear regression (C), and one-way ANOVA (G,I) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Extended Data Figure 5. tTex cells suppress through CD39-mediated extracellular ATP depletion and adenosine production.
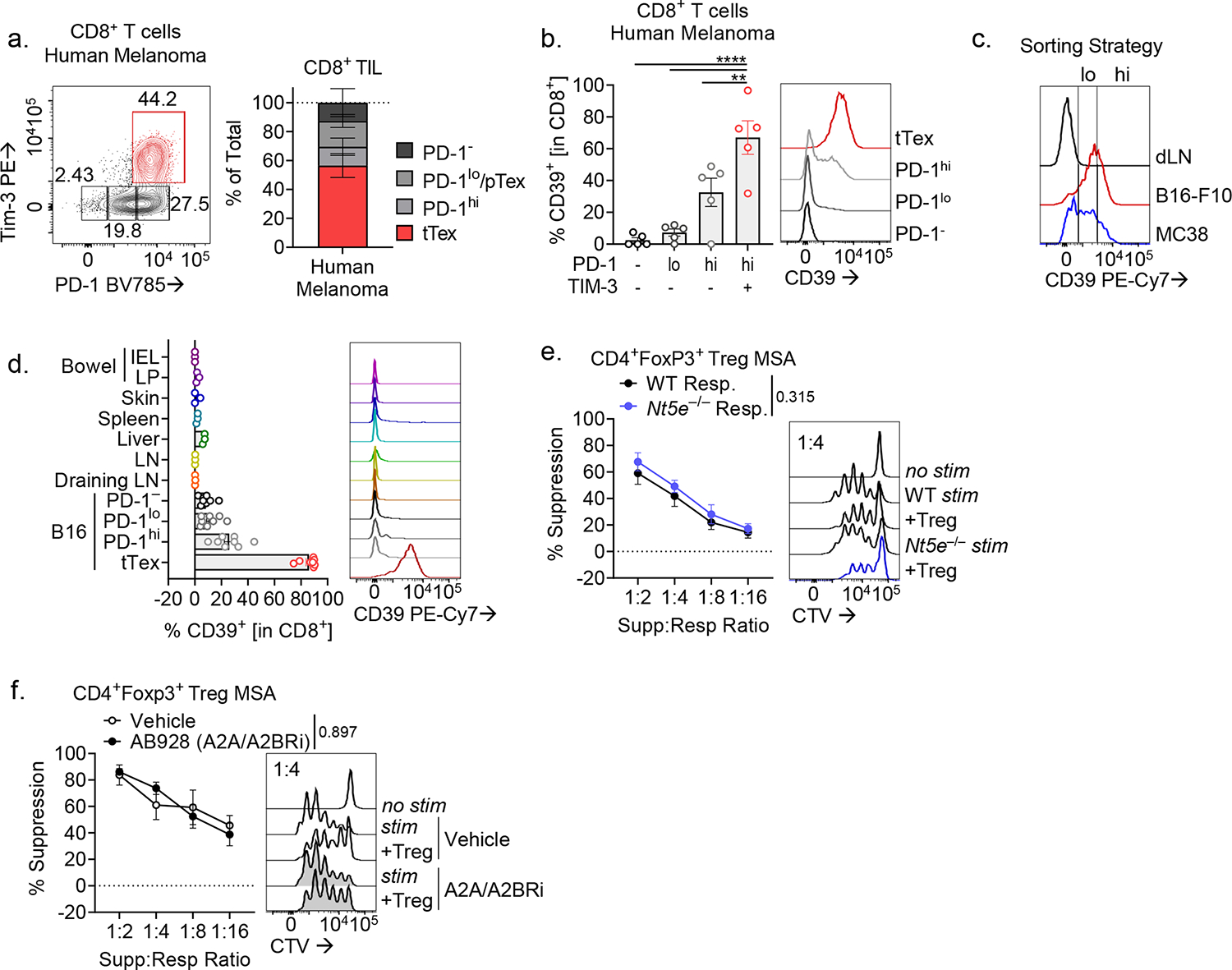
(A,B) Human CD8+ T cell populations from five melanoma biopsies from treatment-naïve patients and corresponding CD39 expression. (C) Sorting strategy for Fig. 4B. (D) Percent CD8+ T cells expressing CD39 in various tissues in a B16-F10 tumor-bearing C57/BL6 mice. (E,F) Suppression assays of B16-F10 infiltrating CD4+ Treg cells, co-cultured (E) with activated CTV-labeled C57/BL or Nt5e− responding T cells or (F) with DMSO vehicle or A2AR/A2BR small-molecule inhibitor AB928 at 3 μg/mL. Statistics are one-way ANOVA (B), and linear regression (E,F) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Extended Data Figure 6. Cd4-driven Cre recombinase expression efficiently deletes CD39 on CD8+ TIL.
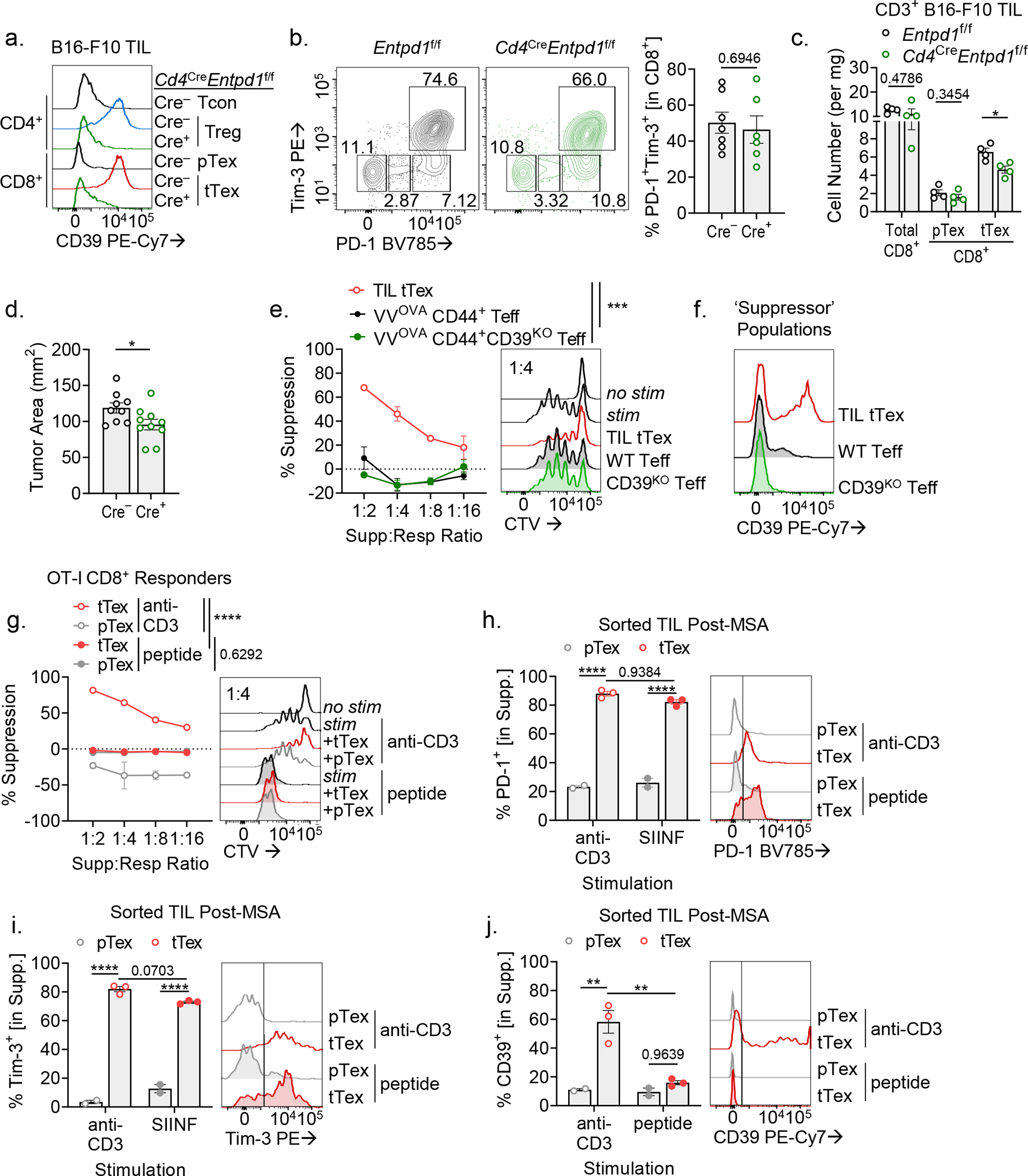
(A,B) TIL analysis of CD39 and inhibitory expression in CD8+ T cells from Cd4CreEntpd1f/f mice. (C) Cell counts per milligram of tumor mass from experiments in Extended Fig. 6A,B. (D) Tumor areas at day 14 from experiments in Fig. 2F. (E) Suppression assay of Cd4CreEntpd1f/f Thy1.1+CD44+ OT-I Teff cells isolated from day 8 of acute VacciniaOVA infection. (F) CD39 expression on TIL tTex cells or VacciniaOVA OT-I T cells in suppression assay co-cultures. (G) Suppression assay of B16-F10-derived CD8+ tTex cells co-cultured with OT-I TCR transgenic CD8+ T cells. Culture were stimulated with either anti-CD3 antibodies as before, or OT-I specific peptide, SIIFEKL. (H-J) Flow cytometric analysis of TIL following MSA from Extended Data Fig. 6G. Statistics are Mann-Whitney (B-D,H-J) and linear regression (E,G) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Extended Data Figure 7. Enforced CD39 expression on CD8+ T cells limits metabolic reprogramming via disruption of TCR signaling.
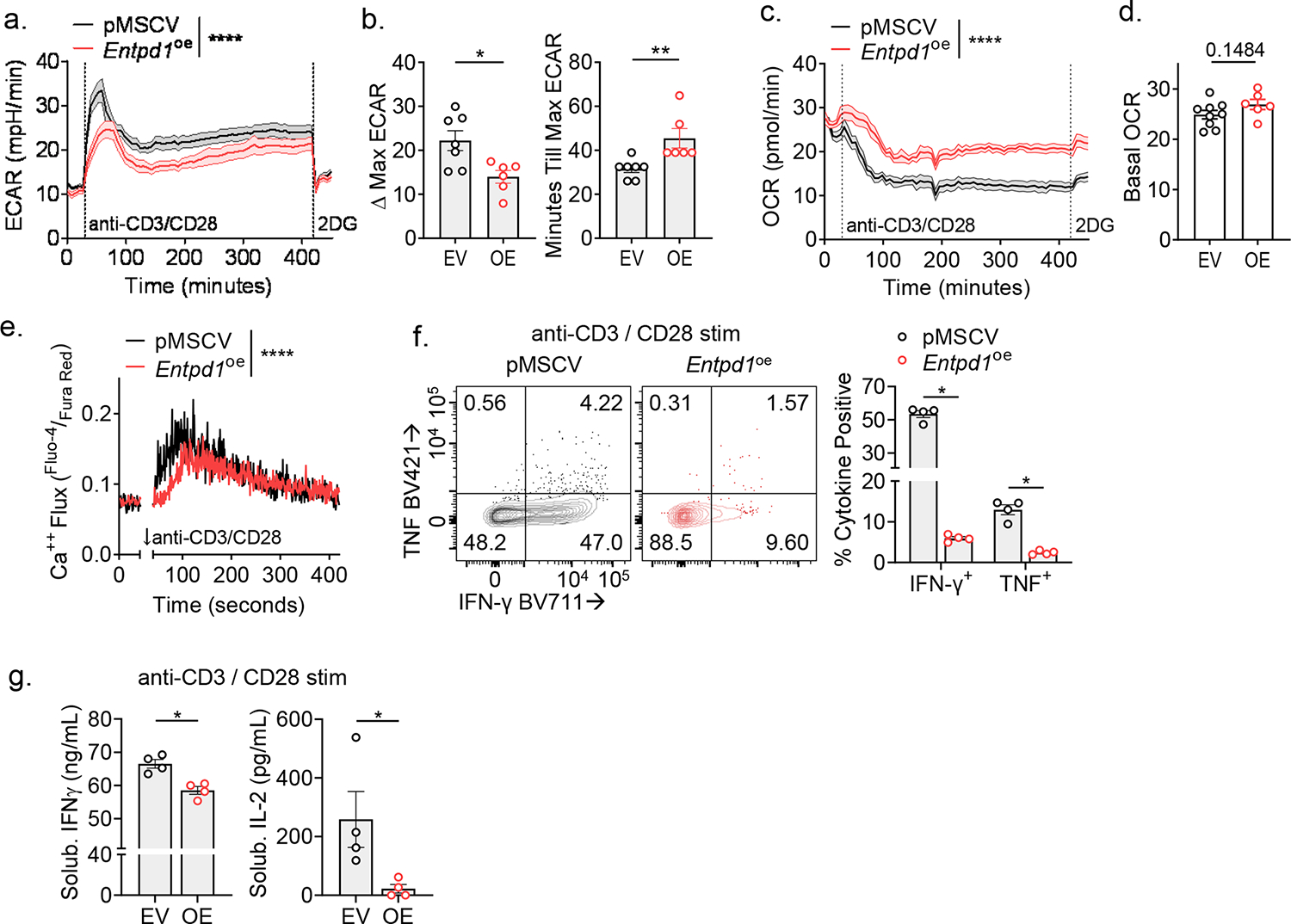
(A) Extracellular acidification rate (ECAR) in in-Seahorse activation of rested CD8+ T cells stability transduced with pMSCV or pMSCV-Entpd1. (B) Delta-maximal ECAR quantified by basal ECAR – maximal ECAR; minutes till maximal ECAR per sample well. (C) Oxygen consumption rate (OCR) of cells from Extended Data Fig. 7C with (D) quantified basal OCR. (E) Intracellular calcium flux in day 7 transduced T cells restimulated in a calcium-buffered solution with anti-CD3/anti-CD28-bound microbeads in the presence of calcium indicators, Fluo-4 and Fura Red. (F) Cytokine production of transduced T cells following 24-hour anti-CD3/anti-CD28-bound microbeads stimulation. (G) ELISA of supernatants from repeat experiments of Extended Data Fig. 7F. Statistics are Mann-Whitney (B,D,G) and one-way ANOVA (E,F) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Extended Data Figure 8. E8i-driven Cre recombinase expression efficiently deletes CD39 on CD8+ TIL.
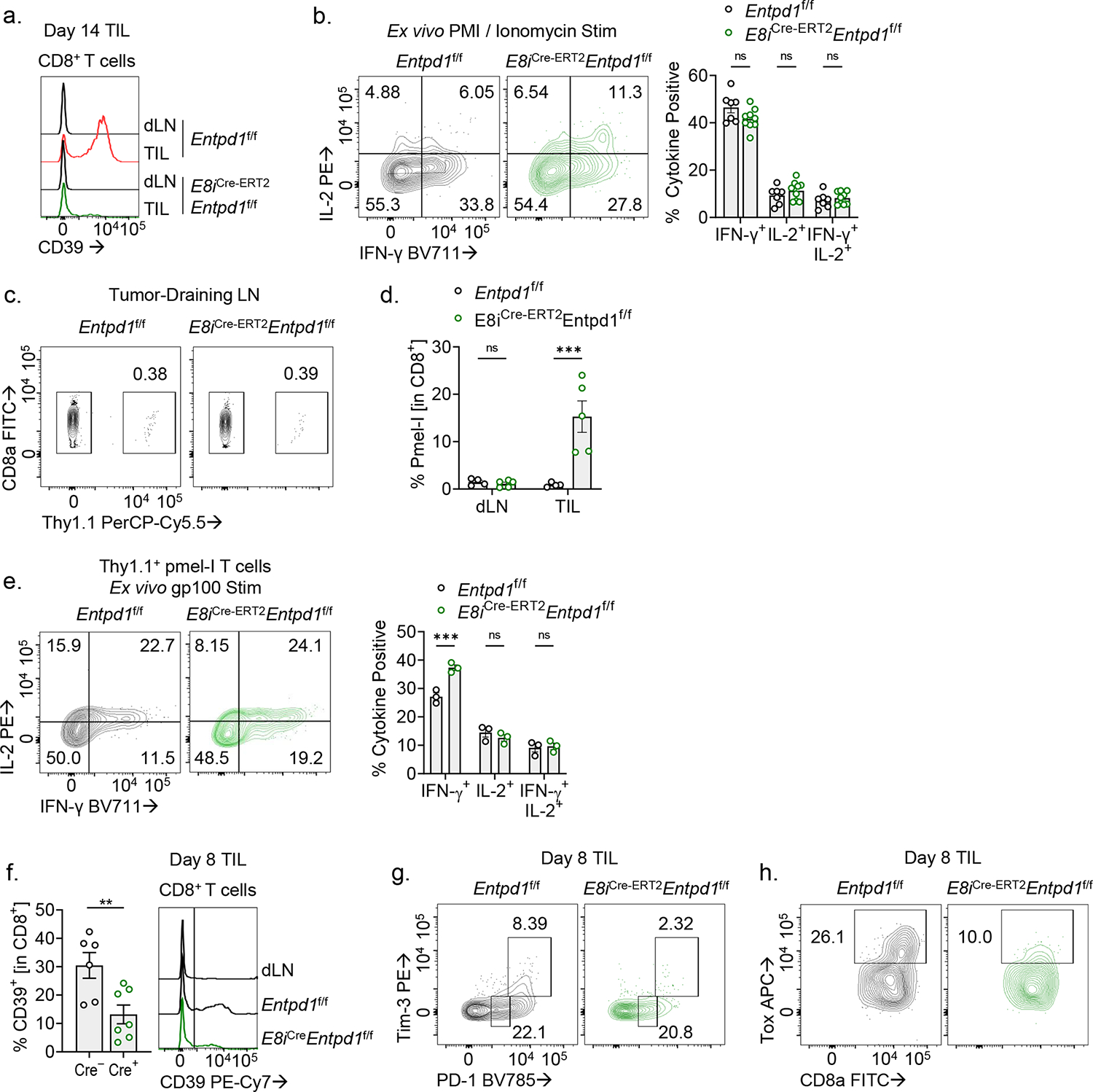
(A) CD39 expression in day 14 B16-F10 tumor infiltrating CD8+ T cells from Entpd1f/f or E8iCre-ERT2Entpd1f/f mice. (B) Cytokine production following PMA/Ionomycin re-simulation of day 14 CD8+ TIL. (C,D) Infiltration of adoptively transferred pmel-I T cells into tumor-draining lymph nodes (dLN; C) and (D) quantification of pmel-I percentages in a representative experiment. (E) Ex vivo cytokine production of pmel-I T cells following gp100 re-stimulation. (F) TIL analysis of CD39 expression in CD8+ T cells from E8iCre-ERT2Entpd1f/f mice treated for three consecutive days with tamoxifen. (G,H) Representative flow cytometry plots from Fig. 7E,F. Statistics are one-way ANOVA (B,D,E) and Mann-Whitney (F) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Extended Data Figure 9. Tumor hypoxia enforces CD39 expression on tTex cells.
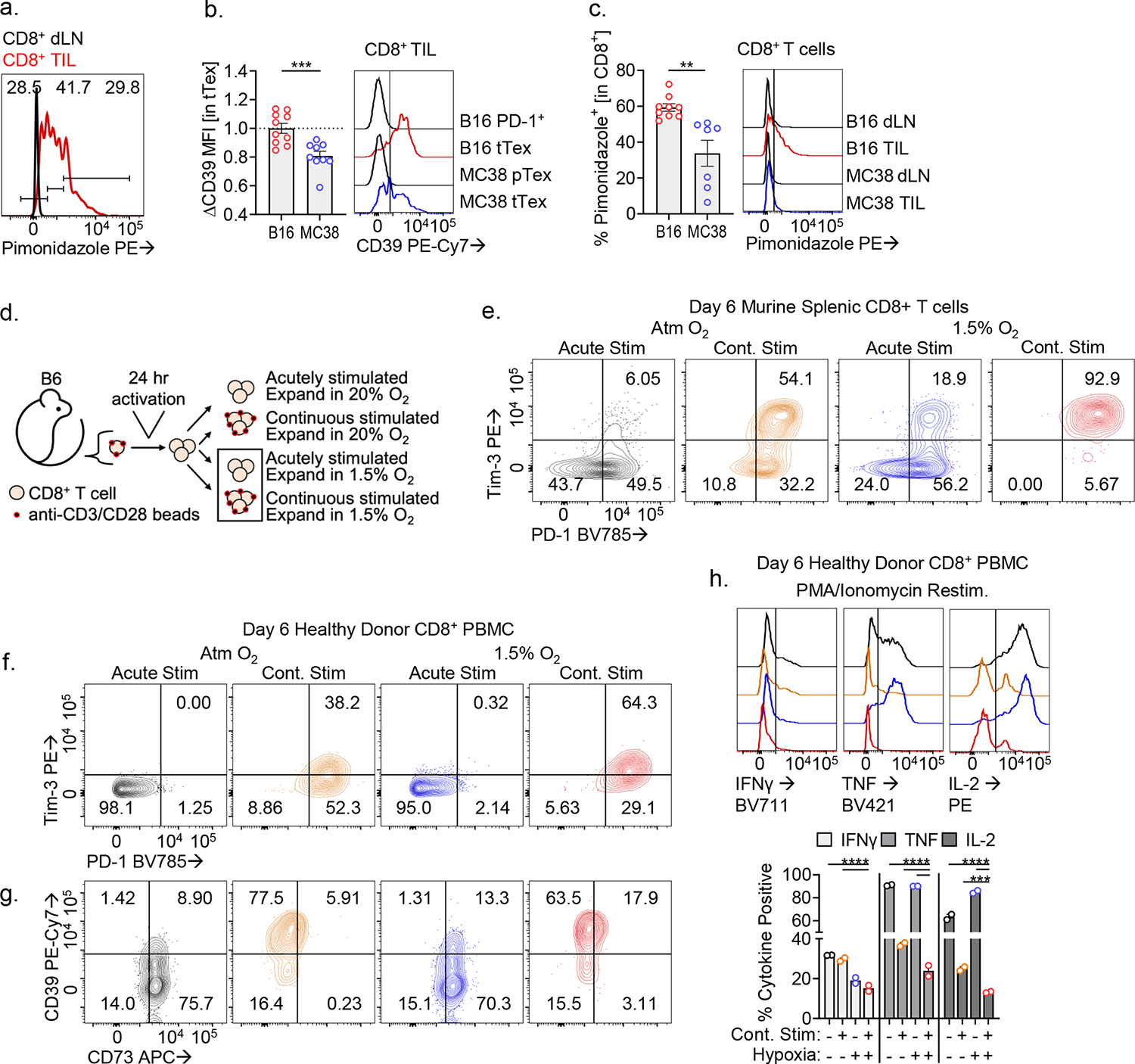
(A) Histogram overlay displaying hypoxia exposure in CD8+ dLN and TIL from B16-F10 tumors. (B) CD39 staining in exhausted T cells from B16-F10 or MC38, and (C) Pimonidazole staining of bulk TIL. (D) Continuous Activation under Hypoxia (CS+H) assay. In brief, naïve T cells are activated for 24 hours, then split into treatment groups of removal (acute stimulation) or continued presence (continuous stimulation) of anti-CD3/anti-CD28-bound microbeads and cultured under atmospheric oxygen tensions (~20% O2) or tumor hypoxic conditions (1.5% O2) for 5 days. (E) Inhibitory receptor staining in murine day 6 CS+H cells. (F-H) Validation of humanized CS+H assay with healthy donor PBMC-derived CD8+ T cells via staining of (F) inhibitory receptors and (G) enzymes of adenosine metabolism. (H) 24-hour PMA/Ionomycin restimulation of human CS+H cells. Statistics are Mann-Whitney (B,C), one-way ANOVA (H) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Extended Data Figure 10. Tumor hypoxia mitigation as a therapeutic target to lessen tTex cell-mediated suppression.
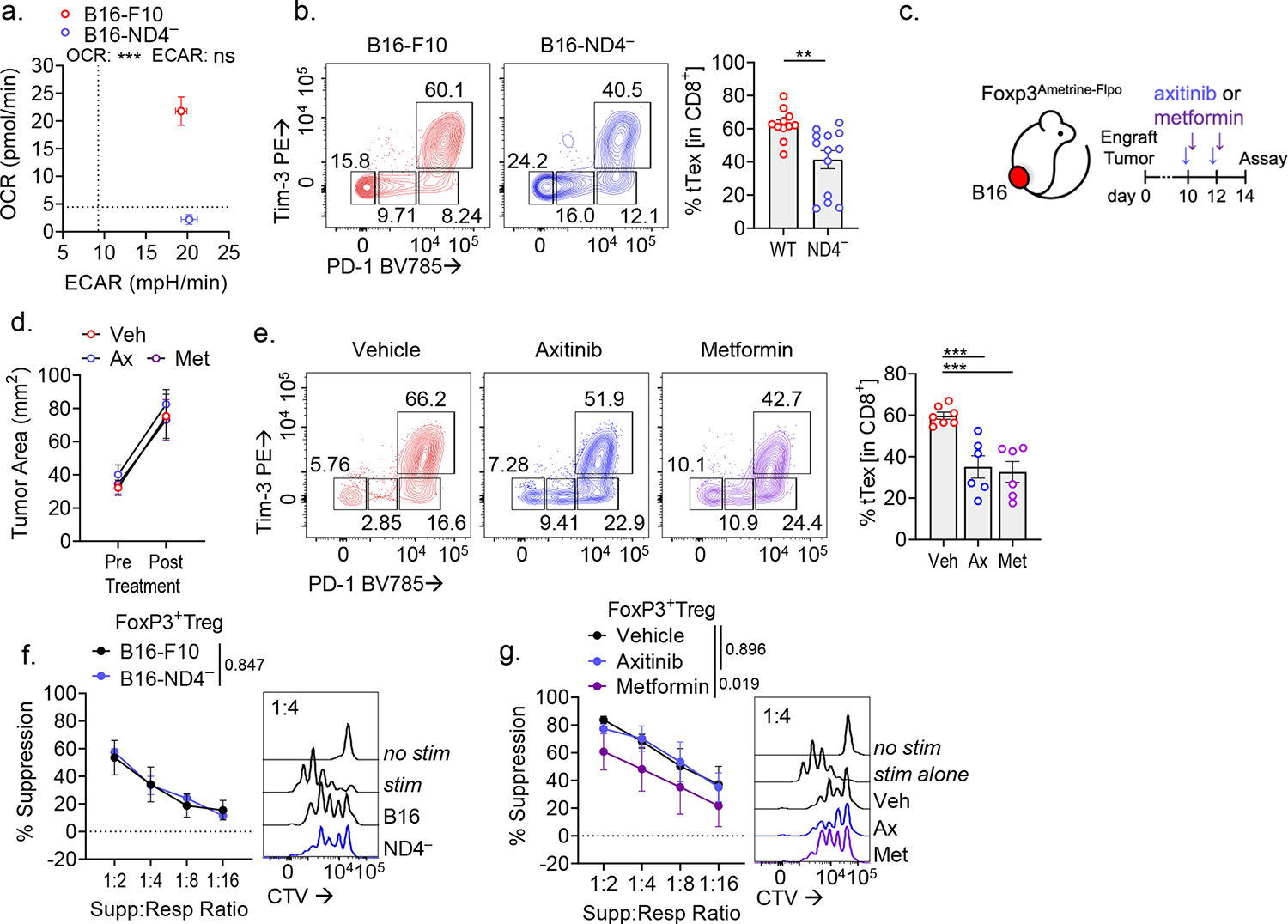
(A) Extracellular flux analysis showing validation of mitochondrial respiration knockdown (via OCR measurement) in B16ND4− tumor cells versus parental B16-F10. (B) PD-1 and Tim-3 staining in TIL from day 14 wild-type B16-F10 or B16ND4− tumors. (C) Schematic of treatment plan for therapeutic alleviation of tumor hypoxia. (D) Tumor sizes at treatment initiation and sacrifice in Axitinib/metformin experiments. (E) PD-1 and Tim-3 staining in TIL from Axitinib/metformin experiments in Extended Data Fig. 9D. (F) Suppression assays of CD4+Foxp3+ Treg cells from Fig. 9C. (G) Suppression assays of CD4+Foxp3+ Treg cells from Fig. 9F. Statistics are Mann-Whitney (B), one-way ANOVA (E) and linear regression (F,G) with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001
ACKNOWLEDGMENTS
The authors would like to thank all members of the Delgoffe lab for helpful and invigorating discussions. We would also like to thank friends and collaborators at the University of Pittsburgh, especially Ansen Burr and the Hand lab. This work was supported by a National Institutes of Health (NIH) Director’s New Innovator Award (DP2AI136598); the Hillman Fellows for Innovative Cancer Research Program; a Stand Up to Cancer–American Association for Cancer Research Innovative Research Grant (SU2C-AACR-IRG-04-16); the Alliance for Cancer Gene Therapy; the UPMC Hillman Cancer Center Skin Cancer and Head and Neck Cancer SPOREs (P50CA121973 and P50CA097190; NIH); the Mark Foundation for Cancer Research’s Emerging Leader Award; and a Cancer Research Institute-Lloyd J. Old STAR Award; and the Sy Holzer Endowed Immunotherapy Fund (all to G.M.D.). Trainees on this manuscript were supported by grants T32CA082084 (NIH) (to P.D.A.V., M.J.W., K.D., and B.R.F.), F30CA247034 (NIH) (to P.D.A.V.), F31AI149971 (NIH) (to M.J.W.), F31CA247129 (NIH) (to K.D.). This work used the UPMC Hillman Cancer Center Flow Cytometry and Animal Facilities, supported in part by grant P30CA047904 (NIH). This work was supported by the Health Sciences Sequencing Core (HSSC) at UPMC Children’s Research Hospital of Pittsburgh and the University of Pittsburgh Center for Research Computing. Some images were derived from Biorender.com. We also would like to acknowledge the authors of important papers that we could not cite due to space.
Footnotes
COMPETING INTERESTS STATEMENT
A.V.M. is currently an employee of Novasenta. G.M.D. declares competing financial interests and has submitted patents targeting exhausted T cells that are licensed or pending and is entitled to a share in net income generated from licensing of these patent rights for commercial development. G.M.D. consults for and/or is on the scientific advisory board of BlueSphere Bio, Century Therapeutics, Nanna Therapeutics, Novasenta, Pieris Pharmaceuticals, and Western Oncolytics/Kalivir; has grants from bluebird bio, Novasenta, Pfizer, Pieris Pharmaceuticals, TCR2, and Western Oncolytics/Kalivir; G.M.D. owns stock in Novasenta, BlueSphere Bio, and RemplirBio. The remaining authors have no competing interests.
REFERENCES
- 1.Blank CU et al. Defining “T cell exhaustion”. Nat. Rev. Immunol. 19, 665–674 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Im SJ et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller BC et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruni D, Angell HK & Galon J The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 20, 662–680 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Sade-Feldman M et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DePeaux K & Delgoffe GM Metabolic barriers to cancer immunotherapy. Nat. Rev. Immunol. 21, 785–797 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jayaprakash P, Vignali PDA, Delgoffe GM & Curran MA Hypoxia reduction sensitizes refractory cancers to immunotherapy. Annu. Rev. Med. 73, 251–265 (2022). [DOI] [PubMed] [Google Scholar]
- 8.Ford BR et al. Tumor microenvironmental signals reshape chromatin landscapes to limit the functional potential of exhausted T cells. Sci. Immunol. 7, eabj9123 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wherry EJ & Kurachi M Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collier JL, Weiss SA, Pauken KE, Sen DR & Sharpe AH Not-so-opposite ends of the spectrum: CD8+ T cell dysfunction across chronic infection, cancer and autoimmunity. Nat. Immunol. 22, 809–819 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardardottir L et al. The new old CD8+ T cells in the immune paradox of pregnancy. Front. Immunol. 12, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pagès F et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 391, 2128–2139 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Scharping NE et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 22, 205–215 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zandberg DP et al. Tumor hypoxia is associated with resistance to PD-1 blockade in squamous cell carcinoma of the head and neck. J. Immunother. Cancer 9, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Najjar YG et al. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canale FP et al. CD39 Expression Defines Cell Exhaustion in Tumor-Infiltrating CD8+ T Cells. Cancer Res. 78, 115–128 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Magnuson AM et al. Identification and validation of a tumor-infiltrating Treg transcriptional signature conserved across species and tumor types. Proc. Natl. Acad. Sci. USA 115, E10672–E10681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watson MJ et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 591, 645–651 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao Y et al. IL-4 induces a suppressive IL-10-producing CD8+ T cell population via a Cdkn2a-dependent mechanism. J. Leukoc. Biol. 94, 1103–1112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin H-T et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 107, 14733–14738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo Y et al. Metabolic reprogramming of terminally exhausted CD8+ T cells by IL-10 enhances anti-tumor immunity. Nat. Immunol. 22, 746–756 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seo N, Hayakawa S, Takigawa M & Tokura Y Interleukin-10 expressed at early tumour sites induces subsequent generation of CD4(+) T-regulatory cells and systemic collapse of antitumour immunity. Immunology 103, 449–457 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanna BS et al. Interleukin-10 receptor signaling promotes the maintenance of a PD-1int TCF-1+ CD8+ T cell population that sustains anti-tumor immunity. Immunity 54, 2825–2841.e10 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Li J et al. KIR+CD8+ T cells suppress pathogenic T cells and are active in autoimmune diseases and COVID-19. Science 376, eabi9591 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao X et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 27, 635–646 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Maj T et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 18, 1332–1341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deaglio S et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 204, 1257–1265 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta PK et al. CD39 expression identifies terminally exhausted CD8+ T cells. PLoS Pathog. 11, e1005177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuler PJ et al. Human CD4+ CD39+ regulatory T cells produce adenosine upon co-expression of surface CD73 or contact with CD73+ exosomes or CD73+ cells. Clin. Exp. Immunol. 177, 531–543 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rothweiler S et al. Selective deletion of ENTPD1/CD39 in macrophages exacerbates biliary fibrosis in a mouse model of sclerosing cholangitis. Purinergic Signal 15, 375–385 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Menk AV et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep. 22, 1509–1521 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doedens AL et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat. Immunol. 14, 1173–1182 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKeown SR Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 87, 20130676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scharping NE, Menk AV, Whetstone RD, Zeng X & Delgoffe GM Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol Res 5, 9–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wherry EJ et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Linsley PS & Long SA Enforcing the checkpoints: harnessing T-cell exhaustion for therapy of T1D. Curr Opin Endocrinol Diabetes Obes 26, 213–218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perrot I et al. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep. 27, 2411–2425.e9 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Allard D, Allard B & Stagg J On the mechanism of anti-CD39 immune checkpoint therapy. J. Immunother. Cancer 8, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee YG et al. Modulation of BCL-2 in both T Cells and Tumor Cells to Enhance Chimeric Antigen Receptor T cell Immunotherapy against Cancer. Cancer Discov. (2022). doi: 10.1158/2159-8290.CD-21-1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horton BL, Williams JB, Cabanov A, Spranger S & Gajewski TF Intratumoral CD8+ T-cell Apoptosis Is a Major Component of T-cell Dysfunction and Impedes Antitumor Immunity. Cancer Immunol Res 6, 14–24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boison D & Yegutkin GG Adenosine metabolism: emerging concepts for cancer therapy. Cancer Cell 36, 582–596 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo S, Han F & Zhu W CD39 - A bright target for cancer immunotherapy. Biomed. Pharmacother. 151, 113066 (2022). [DOI] [PubMed] [Google Scholar]
- 44.Zhang H et al. The role of NK cells and CD39 in the immunological control of tumor metastases. Oncoimmunology 8, e1593809 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tøndell A et al. Ectonucleotidase CD39 and Checkpoint Signalling Receptor Programmed Death 1 are Highly Elevated in Intratumoral Immune Cells in Non-small-cell Lung Cancer. Transl Oncol 13, 17–24 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duhen T et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 9, 2724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gallerano D et al. Genetically driven CD39 expression shapes human tumor-infiltrating CD8+ T-cell functions. Int. J. Cancer 147, 2597–2610 (2020). [DOI] [PubMed] [Google Scholar]
- 48.Rivas JR et al. Interleukin-10 suppression enhances T-cell antitumor immunity and responses to checkpoint blockade in chronic lymphocytic leukemia. Leukemia (2021). doi: 10.1038/s41375-021-01217-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sawant DV et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 20, 724–735 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oft M IL-10: master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol Res 2, 194–199 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Pfannenstiel LW et al. Immune-Checkpoint Blockade Opposes CD8+ T-cell Suppression in Human and Murine Cancer. Cancer Immunol Res 7, 510–525 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Naing A et al. PEGylated IL-10 (Pegilodecakin) Induces Systemic Immune Activation, CD8+ T Cell Invigoration and Polyclonal T Cell Expansion in Cancer Patients. Cancer Cell 34, 775–791.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]