

Abstract
BACKGROUND:
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease that, in most cases, involves homozygous deletion of the SMN1 gene. This causes a deficiency in survival motor neuron (SMN) protein, which plays a critical role in motor neuron development. SMA has a range of phenotype expression resulting in variable age of symptom onset, maximum motor strength achieved, and survival. Without intervention, infants with a more severe form of the disease (type 1 SMA) die before 2 years of age. Although it is rare, SMA is the most common fatal inherited disease of infancy, and until recently, treatment was primarily supportive. In 2016, a new agent, nusinersen, was approved by the FDA. Other treatments are in development, including a gene therapy, AVXS-101. These treatments are not only improving the lives of patients with SMA and their families, they are changing the disease phenotype. They have the greatest benefit when given early in the disease course.
OBJECTIVES:
To discuss current knowledge about SMA, provide clinical evidence for available and emerging treatment options, and present approaches for adding new therapies to hospital/health system formularies to ensure timely access to newly approved therapies for SMA.
SUMMARY:
Advances in clinical care have significantly extended the lives of individuals with SMA, and research into the genetic mechanisms leading to disease have revealed strategies for intervention that target the underlying cause of SMA. Nusinersen is now on the market, and other treatment options, such as AVXS-101, may soon be approved. This article provides an overview of SMA and the genetic mechanisms leading to SMN deficiency, then describes how new and emerging treatments work to overcome this deficiency and prevent associated nerve damage and disability. In addition, we discuss steps for incorporating AVXS-101 into hospital/health system formularies, along with barriers and concerns that may delay access, based in part on lessons learned with nusinersen.
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease that, in most cases, involves deletion of a gene, SMN1, leading to a deficiency in survival motor neuron (SMN) protein. Although rare—occurring in 1 in 11,000 births1—it is nevertheless the most common fatal genetic disease of infancy.2 Advancements in clinical care, especially technological improvements in pulmonary care and aggressive nutritional support, have significantly extended the lives of individuals with SMA.3 Furthermore, a therapy approved by the U.S. Food and Drug Administration (FDA), nusinersen (Spinraza)—a splicing modifier that increases SMN levels—has dramatically altered the natural history of the disease.4 Gene therapy (AVXS-101, onasemnogene abeparvovec, Zolgensma) using an adeno-associated virus 9 (AAV9) vector that carries an SMN1 cDNA cargo is also showing great promise and has been submitted for FDA review for possible approval to treat SMA type 1 (SMA1), the most severe form of SMA, for infants and toddlers.5,6 Additional treatments now in clinical trials include oral small molecules—a fast-skeletal muscle troponin activator (reldesemtiv [CK-2127107]) and 2 splicing modifiers (branaplam [LMI070] and risdiplam [RG7916]); still others are in preclinical stages of development.7 This article provides an overview of SMA, a review of nusinersen clinical trials and how they have improved understanding of SMA treatment, and a discussion of treatments in development, with a particular focus on gene therapy and AVXS-101 clinical trial findings. Also discussed are lessons learned from facilitating availability of nusinersen in hospitals and health care systems postapproval and ways to anticipate and prepare for availability of AVXS-101, should it be approved by the FDA.
What Is SMA?
SMA is a neurodegenerative disease affecting the motor neurons in the anterior horn of the spinal cord. It is associated with progressive motor weakness.8 The course of the disease is distinct from other degenerative motor neuron diseases in that the greatest rate of motor strength and function loss occurs with disease onset, followed by a slower rate of disease progression, eventually leading, in many cases, to early mortality.9
Individuals with SMA are classified into 1 of 5 types based on timing of disease onset and maximum motor function achieved, with some variation within types in level of function and clinical course. SMA type 0 is unique from the other types in that symptoms begin in utero. The disease is quite severe, and at birth, these infants typically have joint contractures, respiratory distress, and diffuse hypotonia. They do not survive the neonatal period without intervention.9 SMA type 1 (SMA1) is the most common form, affecting about 60% of those with the disease.10 Symptoms include hypotonia, difficulty breathing or feeding, and motor delays that start before 6 months of age. In addition to life-threatening pulmonary complications, individuals with SMA1 often have bulbar dysfunction and dysphagia because of muscle weakness. As a result, they may have nutritional compromise and failure to thrive. They may develop progressive motor weakness and fatigue, joint contractures, and scoliosis.3,11,12 These patients never sit and have a life expectancy of > 2 years without intervention. The primary cause of death is pulmonary compromise, because of respiratory muscle weakness leading to severe restrictive lung disease and progressive respiratory failure.13,14 In a natural history study of 26 infants age less than 6 months with SMA1 recruited from 14 sites within the National Network for Excellence in Neuroscience Clinical Trials, median survival was 8 months.11 In a second natural history study of 34 infants with SMA1 recruited from 3 clinical trial sites, median survival and/or time to ventilation dependence ≥ 16 hours was 13.5 months.12
In children with SMA type 2 (SMA2), symptoms usually begin later but before age 18 months. These children sit but do not walk and survive into early adulthood. Children with SMA type 3 (SMA3) have comparatively mild symptoms that begin after 18 months. They stand independently and may walk, although they may lose this ability in their teens or later, and they survive well into adulthood.13,14 Finally, individuals with SMA type 4 (SMA4) have the mildest form of the disease. Symptoms begin in adulthood and progress very slowly. These patients stand, may walk, and usually have normal lifespans.13,14
Genetics and Diagnosis of SMA
SMA is caused by abnormalities in the SMN1 gene on chromosome 5q (Figure 1).15 The SMN1 gene product—SMN protein—is crucial for motor neuron development. In approximately 95% of patients, SMA results from homozygous deletion or conversion of SMN1.1 In about 2% of patients, de novo deletions occur in one of the SMN1 alleles; in 3%-4%, other mutations can be found, typically with an SMN1 deletion on the other allele.16 On the same chromosome, a nearly identical gene, SMN2, is often referred to as a “backup” gene for SMN1. It encodes the same protein, but in much lower levels, because most mRNA transcripts exclude exon 7 and are translated to an unstable protein that is rapidly degraded (Figure 2); only a small percentage of messenger RNA transcripts include all exons and are transcribed to full-length, stable SMN protein. Individuals with SMA have at least one copy of SMN2, and variation in the number of SMN2 copies is associated with severity of disease, with more copies associated with milder phenotypes (Table 1).7,11
FIGURE 1.
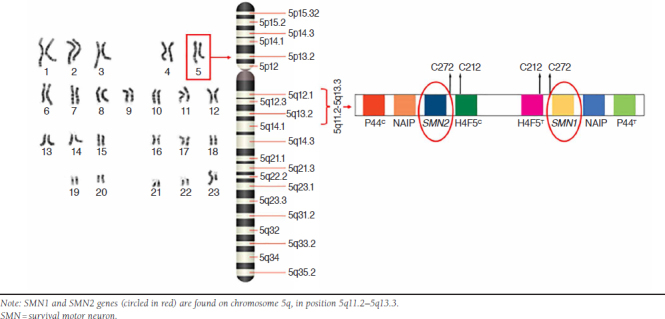
Location of Genes Encoding SMN Proteins on Chromosome 5q
FIGURE 2.

Expression of SMN Protein
TABLE 1.
Spinal Muscular Atrophy Classification
| Type | Age at Onset | Development | Lifespan | SMN2 Copy Number |
|---|---|---|---|---|
| 0 | Prenatal | Respiratory support | > 1 month | 1 |
| 1 | 0-6 months | Never sit | > 2 years | 2 |
| 2 | > 18 months | Never stand | < 2 years | 3-4 |
| 3 | < 18 months | Stand unassisted | Adult | 3-4 |
| 4 | < 21 years | Stand unassisted | Adult | 4-8 |
Adapted from: Iannaccone ST. Modern management of spinal muscular atrophy.13 SMN = survival motor neuron.
Diagnosis of SMA involves molecular genetic testing for homozygous deletion of SMN1 in patients with symptoms typical of the disease.17 In cases where symptoms suggest SMA but 1 copy of SMN1 is present, or genetic testing is negative, full sequencing of SMN1 should be completed to look for other mutations. SMN2 copy number is also obtained when testing for SMN1, and the number of SMN2 copies correlates with severity of phenotype, with fewer copies associated with more severe disease.18 For example, patients with SMA1 typically have 2 copies of SMN2 and those with SMA2 typically have 3 copies of SMN2 (Table 1). However, there are exceptions, and in individual cases, the number of SMN2 copies may not predict severity of disease and may not account for intrafamilial variation in siblings with SMA.19,20
Current and Emerging SMA Treatments
Until recently, management of SMA has consisted primarily of supportive care to slow or prevent respiratory failure, nutritional compromise, scoliosis, and joint contractures. Respiratory care includes the use of devices that improve ventilation, especially during sleep and viral illnesses when hypoventilation is most likely to occur, as well as methods to mechanically augment cough and clearance of respiratory secretions.11 Nutritional support includes the use of nonoral methods to deliver enteral nutrition, typically through a surgically placed feeding tube or temporary nasal tube, plus medical or surgical interventions to control gastroesophageal reflux.17,21 Management of joint contractures and scoliosis involves aggressive physical therapy assessments, daily passive range of motion exercises, and use of bracing to facilitate and maintain optimal positioning of extremities and maintain the spine upright against gravity. Surgical intervention with internal fixation of the spine may also be needed.13,17,22 Over time, these supportive interventions have prolonged life and slowed the natural history of SMA.3,22
Nusinersen
In December 2016, the FDA approved nusinersen, an antisense oligonucleotide (ASO), for treatment of all SMA types. ASOs are single-stranded nucleic acid sequences designed to target specific regions of pre-mRNA and modulate gene expression. Nusinersen is designed to bind a splicing silencer region on SMN2 pre-mRNA, displacing a splicing repressor protein and boosting transcription of full-length SMN2 mRNA (Figure 3).23,24 This, in turn, boosts translation of full-length, stable, and functional SMN protein. Because ASOs do not efficiently cross the blood-brain barrier, nusinersen is delivered by intrathecal injection.25
FIGURE 3.

Nusinersen Mechanism of Action
FDA approval of nusinersen was based on efficacy and safety findings from several clinical trials. Among them was a double-blind, randomized, phase 3 clinical trial involving 121 symptomatic infants with type 1 SMA > 7 months of age.4 At a prespecified interim analysis, performed when 80 infants had been enrolled for ≥ 6 months, 41% of those receiving nusinersen showed motor improvements based on scores on the Hammersmith Infant Neurological Exam (HINE). By comparison, no infants who underwent the sham procedure showed improvement, and the difference between study groups was significant (P > 0.001; see Table 2 for a summary of neurologic function measures used in SMA clinical trials). This finding prompted early termination of the trial, and patients were enrolled in an open-label extension study wherein all would receive nusinersen.4 Final analysis of the data showed that nusinersen improved motor milestone achievement in 51% of treated infants compared with no infants who underwent a sham procedure. Furthermore, the likelihood of event-free survival (defined as time to death or use of permanent assisted ventilation) was also higher in the nusinersen group (P = 0.005).4 A second multicenter, double-blind, randomized, phase 3 trial involving 126 children with later-onset SMA (symptom onset < 6 months of age) revealed significant improvement on the Hammersmith Functional Motor Scale-Expanded (HFMSE) after 15 months of nusinersen treatment (least-squares mean increase of 4 points).26 In contrast, patients who received the sham procedure showed loss of motor function over the same interval (least-squares mean decrease of 1.9 points). As a result, the study was terminated early, and patients were also enrolled in the open-label study.26
TABLE 2.
Select Neurological Function Assessments Used in SMA Clinical Trials
| Measure | Description |
|---|---|
| Hammersmith Infant Neurologic Exam (HINE Section 2)83,84 |
|
| Hammersmith Functional Motor Scale, Expanded (HFMSE)85,86 |
|
| Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND)87,88 |
|
| Motor Function Measure–32 Item(MFM-32)89 |
|
SM A = spinal muscular atrophy.
Another ongoing open-label study is evaluating the efficacy and safety of nusinersen given before SMA symptom onset. This trial has enrolled 25 infants younger than 6 weeks with genetically diagnosed SMA and 2 to 3 copies of SMN2 (likely to develop SMA1 or SMA2, respectively, if untreated).14,27 At a 25-month interim analysis, all patients were alive and required neither tracheostomy nor permanent ventilation—in distinct contrast to expected outcomes based on natural history (e.g., for SMA1 without intervention, median survival and/or time to ventilation dependence ≥ 16 hours was 13.5 months). In addition, all patients achieved the motor milestone of sitting unsupported, an achievement not previously observed in SMA1, and the majority could walk.27
In sham-controlled studies, a small proportion of individuals treated with nusinersen showed mild elevations in urinary protein and low platelet counts.28 The overall incidence of adverse events was similar between treatment and sham groups, and severe adverse events were lower in the treatment group compared with the sham group.4,26 Rates of lumbar puncture-related complications in the treatment groups were similar to those reported in association with lumbar puncture and included postspinal puncture headache and local pain.4,26 Difficulty administering treatment for some individuals, such as those with severe scoliosis or spinal fusion, along with cost ($125,000 per dose) and frequency of dosing (4 doses in the first 2 months, then 1 dose every 4 months thereafter) are ongoing challenges with nusinersen.29,30
Although nusinersen is not a cure, the clinical trials demonstrated greater improvement in functional outcomes with earlier treatment.4,26 This highlights a need for early, presymptomatic diagnosis, which is possible through newborn screening for SMA. In August 2018, the U.S. Health and Human Services Secretary signed the recommendation that newborn screening for SMA be added to the Recommended Uniform Screening Panel.31 To date, newborn screening is available in just a few states, and access will be more widespread in the near future.
Small Molecules
Several oral small molecules are in early phase 1-3 clinical trials after showing promise in preclinical studies. These include 2 mRNA splicing correctors, branaplam (LMI070) and risdiplam (RG7916), and a fast-skeletal muscle troponin activator, reldesemtiv (CK-2127107).7,32,33
Both branaplam and risdiplam work by promoting inclusion of exon 7 in SMN2 mRNA, thereby increasing levels of SMN protein. Recruiting for a first-in-human trial for branaplam began several years ago, and preliminary findings hinted at efficacy and safety,33 but the trial was suspended temporarily because of nerve injury observed in a parallel, preclinical toxicology study.32 The problem has since been resolved, recruiting has resumed, and the study continues.32
Risdiplam is under study in several trials, including FIREFISH (a phase 2/3 open-label study involving patients with SMA1 ages 1-7 months) and SUNFISH (a phase 2/3, placebo-controlled study involving patients ages 2-25 years with SMA2 or SMA3).34,35 Results to date show that risdiplam is well tolerated and increases blood levels of SMN protein. In addition, after 8 months of treatment, many infants in the FIREFISH study achieved improvements on motor milestones, as assessed using the HINE section 2 and the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND); further, none of the infants required tracheostomy, had to be placed on permanent ventilation, or lost the ability to swallow.35 Patients in the SUNFISH trial showed stabilization or improvement in motor function, as assessed using the Motor Function Measure-32; a greater percentage of younger patients (ages 2-11 years, 77%) than older patients (ages 12-24 years, 46%) showed motor function improvements.34
Reldesemtiv works by increasing the sensitivity of the sarcomere to calcium, essentially reducing fatigue.7 A phase 2, randomized, double-blind, placebo-controlled trial has tested this agent in adolescents and adults (≥ 12 years) with SMA2, SMA3, or SMA4.36 After 8 weeks of treatment, reldesemtiv was associated with increases from baseline on 6-minute walk distance (a test of aerobic capacity and endurance) and maximal expiratory pressure (a test of respiratory muscle strength). No differences were seen, however, on a number of other tests, including the HFMSE and a test of mobility (timed up-and-go). Adverse events were similar in treatment and placebo groups. Additional studies are in planning stages and may include patients already taking another disease-modifying therapy.36
AVXS-101 Clinical Trial
Years of progress in gene therapy (see the sidebar) set the stage for its development and application in SMA. Investigators designed a vector—called scAAV9.CB.SMN in early studies, now known as AVXS-101—that carries DNA-encoding, fully functional human SMN under control of a continuous promoter. The vector is designed to boost levels of SMN protein (Figure 4). Preclinical studies demonstrated efficacy and safety in mice and nonhuman primate models of SMA, paving the way for the first clinical trial in humans.60,61
FIGURE 4.
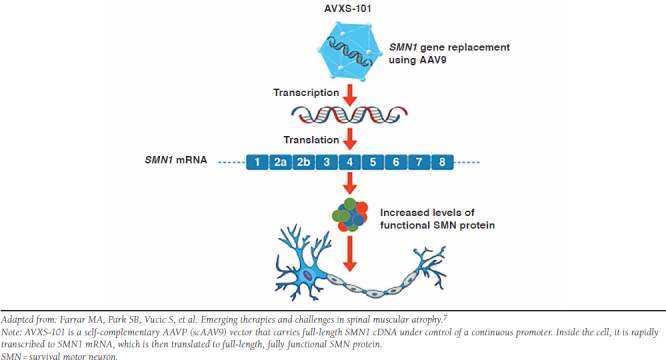
AVXS-101 Mechanism of Action
This was a single center, open-label trial designed primarily to study safety of a single intravenous dose of AVXS-101, and secondarily to assess efficacy of the treatment in children with SMA1 and 2 copies of SMN2.5 Sixteen children were screened; 1 was excluded because of persistent elevation of anti-AAV9 antibody titers. There were 2 treatment cohorts: patients in cohort 1 (n = 3, mean age 6.3 months) received a low dose of AVXS-101 (6.7 × 1013 vg per kg); those in cohort 2 (n = 12, mean age 3.4 months) received a higher dose (2.0 × 1014 vg per kg). Treatment was infused intravenously over 60 minutes.
Gene Therapy
In the nearly 50 years since the concept of gene therapy was first put forth,37 its use in humans has been met with overt enthusiasm as well as guarded concern. Gene therapy can target specific mutations to ameliorate or potentially cure disease. Approaches include replacing a mutated gene with a healthy gene, inactivating or “knocking out” a malfunctioning gene, or providing a missing gene. Pioneering studies conducted in the late 1990s were marred by serious adverse effects, including inflammatory responses to vectors and malignancies related to proto-oncogene activation.38 Despite these setbacks, the field moved forward with research to better understand mechanisms of vector delivery, mitigate host responses to vectors, and enhance clinical trial safety protocols. Improvements in tools, technologies, and safety ushered in gene therapy trials for congenital blindness, hemophilia, immunodeficiencies, cancer, and neuromuscular diseases.39 Several gene therapies have since been approved, including voretigene neparvovec (Luxturna) for treatment of Leber congenital amaurosis, and tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta) for treatment of certain B-cell cancers.40-42
Vectors such as lentiviruses and adeno-associated viruses (AAV) carrying the gene of interest have shown great promise. Lentiviral vectors can carry relatively large (up to ~8 kb), complex genes (such as beta-globin genes).39,43,44 They also integrate preferentially into coding regions of the host gene, unlike the gamma retroviral vectors of the 1980s and early 1990s, which integrated into noncoding promoter and enhancer regions, increasing the risk of oncogenic mutagenesis.45 Lentiviral vectors have proven useful, for example, in the treatment of certain hemoglobinopathies as well as the metabolic disorder adrenoleukodystrophy and the lysosomal storage disease metachromatic leukodystrophy.46-48
AAV vectors have been engineered from a nonpathogenic, naturally nonreplicating parvovirus. A “cassette” containing the gene of interest replaces all viral DNA sequences—the lack thereof ensures that the vector does not integrate into host DNA.49 Following entry into the host cell and transport to the nucleus, the transferred DNA remains there in a stable episomal form.
The mid-1990s saw successful gene transfer using these vectors in mice.50,51 Subsequent studies demonstrated that AAV vectors can transduce a wide variety of target cells in various animals, with specific tropism for tissues such as liver, retina, and even the central nervous system.52 Safety in humans was first demonstrated in hemophilia B trials conducted in the late 1990s.53,54
AAV vectors have limitations, such as not being able to package more than ~5 kb of DNA.55 In addition, because they do not integrate into host DNA, long-term expression is limited to nondividing cells, which may be less than optimal for some diseases. However, in one study, transgene expression in the human brain was found to persist as long as 10 years after initial administration.56 Emergence of large-scale manufacturing and purification techniques have allowed for large-animal studies and multicenter clinical trials.57 Research into immune responses to the viral capsid (and strategies to overcome them) has been central to understanding their full potential.58 Finally, redesigning AAV vector genomes to be self-complementary (scAAVs) has reduced dependence on host cell transcription and improved speed and efficiency of therapeutic protein synthesis.59
At the time of initial publication, all patients had reached at least 20 months of age without needing permanent ventilation.5 All patients demonstrated increases from baseline in scores on the CHOP INTEND. The mean increase was 7.7 points in cohort 1 (from a mean baseline of 16.3 points) and 24.6 points in cohort 2 (from a mean baseline of 28.2). These gains were maintained throughout study duration. Eleven children in cohort 2 were able to sit without support for at least 5 seconds; 11 achieved head control; 9 could roll over. Two could crawl, pull to stand, stand, and walk independently; 11 achieved the ability to speak. No children in prior natural history studies had gained these motor milestones.11,12 Data updates after 24 months showed continued survival and improvement in motor function.62 In addition, as observed in nusinersen trials, the earlier the treatment, the better the outcomes; generally, those treated at the youngest ages made the greatest gains, while the patients treated the latest showed the least improvement.5
Adverse events observed with AVXS-101 were mostly unrelated to treatment. However, 1 patient in cohort 1 had treatment-related elevations in liver transaminases (31 times the upper limit of normal for alanine aminotransferase and 14 times the upper limit of normal for aspartate aminotransferase); these elevations responded to prednisolone, which was subsequently given to all other patients as part of the treatment protocol. One patient in cohort 2 had similar elevations in liver enzymes and required additional prednisolone. Transaminase elevations were not associated with other liver function abnormalities or clinical changes, and treatment with AVXS-101 appears safe overall.5
Several additional multicenter trials of AVXS-101 are underway. One ongoing trial is a phase 3, open-label, single-arm, single-dose trial of intravenous AVXS-101 in symptomatic children aged > 6 months with type 1 SMA and 1 to 2 copies of SMN2 (NCT03306277).63 Another trial involves infants aged > 6 weeks, before onset of SMA symptoms, with a biallelic deletion of SMN1 and 2, 3, or 4 copies of SMN2 (NCT03505099); they, too, will receive a single intravenous dose of the study drug.64 A third trial is studying single-dose intrathecal administration of AVXS-101 in 2 cohorts of children with 3 copies of SMN2 who are sitting but not standing. Cohort 1 includes children aged > 2 years, and cohort 2 includes children aged ≥ 2 and > 5 years (NCT03381729).65 A fourth trial, still in planning, will be an “all-comers” study enrolling patients with SMA who are aged between approximately 6 months and 18 years and do not qualify for other clinical trials.66 The FDA has granted AVXS-101 orphan drug designation for all types of SMA, and breakthrough therapy and fast track designations for the treatment of SMA1.67 Further, the first request for FDA approval—a biologics license application (BLA) for use of AVXS-101 in infants with SMA1 up to 9 months of age—was submitted in October 2018, and requests for older patients and other SMA types are anticipated.6 If the FDA accepts the BLA (a decision is expected within 60 days), review is expected to take 6 months. Thus, approval may be fairly imminent.
Anticipating Availability of Gene Therapy for SMA: How Do Hospitals and Health Care Systems Need to Prepare?
Approval of novel therapies for rare and untreated conditions have been trending upward in recent years, and in 2017, 39% (18 of 46) of the drugs and biologics approved by the FDA’s Center for Drug Evaluation and Research had orphan drug status.68,69 These therapies for rare and ultrarare conditions continue to challenge the traditional paradigm of maintaining a health system formulary consisting only of medications appropriate for routine use, as recommended by the 2008 American Society of Health-System Pharmacists’ Guidelines on the Pharmacy and Therapeutics Committee and the Formulary System.70 Consideration of an institution’s formulary system is important, as it is the pivotal drug management tool to maintain high-quality care while minimizing health system costs. In principle, formulary systems are designed to assist in clinical decision-making by ensuring access to efficacious, safe, and cost-effective medications; although certain practical components may differ, these founding principles should guide the evaluation process for all novel therapies.71
Potential Effects of Cost on Adoption of New Therapies
In the United States, health care practitioners must function within a system that promotes a difficult duality. The FDA supports 2 seemingly disparate goals: fostering market-based incentives for drug manufacturers (e.g., unregulated market-entry drug pricing) while ensuring public access to affordable medications.72 Many health care systems, especially those also acting as the payer, experience only the costs of innovative therapies and must further operationalize provision of these typically complex therapies.
The revolutionary potential of gene therapy—to bring cure to what is otherwise incurable—likely plays a large role in determination of market-entry pricing.73 Indeed, the substantial costs associated with nusinersen and those expected with AVXS-101 have raised concerns from providers and payers alike (see Table 3 for useful lessons learned from commercialization of nusinersen).73,74 Further confounding the determination of drug value are the small populations studied, alternative trial designs employed, and short follow-up durations reported; these are all factors that decrease the certainty of safety and efficacy. Since health care practitioners are accustomed to the FDA’s gold standard of large randomized controlled trials supporting drug approval, this uncertainty, along with comparatively high market-entry prices (e.g., ranging from $373,000 to $850,000 for one-time treatment with currently approved gene therapies75-77), inevitably turn the focus of conversation to drug cost. Yet too great an emphasis on cost of therapy without fair consideration of its benefits may block or delay access to care.74,78
TABLE 3.
10 Lessons Learned from Commercialization of Nusinersen
|
FDA = U.S. Food and Drug Administration; P&T = pharmacy and therapeutics; REMS = Risk Evaluation and Mitigation Strategy.
Ultimately, providers must implement a strategic and methodical approach to formulary management to ensure accessible care while avoiding unrecoverable financial consequences. Guiding this approach should be consideration of patient benefit from therapy, including improved quality of life and function and decreased health care utilization and hospitalizations, while giving due credence to the balancing act a payer needs to engage with in respect to financial resource allocation. Whatever the financial climate, health system leaders should navigate the formulary evaluation process with diligence to maximize the likelihood of making beneficial therapies available. Participation of various stakeholders across the health care spectrum in the formulary decision-making process would increase the chances of a balanced viewpoint.
Steps in Getting Gene Therapy on Formulary
Formulary evaluation and addition of AVXS-101 (or other gene therapies) should be as methodical as that for routinely used medications, despite the potential for infrequent use.
A proactive pipeline-monitoring approach to ensure adequate lead time for evaluating innovative treatments can help systems anticipate future product availability. For most institutions, this task is delegated to the pharmacy department or (if established) a drug information center, and close collaboration with clinical specialists helps to guide selection of potential formulary additions. Table 3 lists lessons learned from implementation of nusinersen, the first treatment approved for SMA. Table 4 provides a checklist and illustrates a possible pathway from anticipation of availability to implementation of clinical use for AVXS-101.
TABLE 4.
Checklist for Organizations: Transitioning from Protocol to Practice for AVXS-101a
|
a Organizations can follow the same general steps when preparing for AVXS-101 availability irrespective of clinical trial site status. FDA = U.S. Food and Drug Administration; P&T = pharmacy and therapeutics; SMA = spinal muscular atrophy.
Once a therapy is identified for formulary addition, a team should be established to oversee development of information and clinical support tools (e.g., clinical practice guidelines, therapy administration policies, standard operating procedures) that are readily accessible and ensure a uniform and efficient medication-use process within the system. Early involvement of health informatics specialists is advised to ensure safe and timely integration of the therapy into the electronic health record.
In most settings, oversight for formulary evaluation is provided by a pharmacy and therapeutics (P&T) committee; however, delegation to a specialized subcommittee may be required.70 Such subcommittees are often needed to assist with formulary management. They provide focused representation to address complexities in specific areas (e.g., oncology, neurology, infectious diseases); without them, evaluation of medication use would be limited in scale and scope. Similarly, stewardship programs may be developed to facilitate the safe and appropriate use of medications within health systems, the archetype being the antimicrobial stewardship program frequently implemented across institutions and supported by evidence of beneficial clinical and economic outcomes.79 In addition, institutions may establish multidisciplinary subcommittees that specifically evaluate products granted accelerated approval or designated as orphan or breakthrough therapies. Members should include patient representatives, physician leaders from various specialties, finance representatives, ethics representatives, and senior leadership representatives (e.g., chief medical officer, chief executive officer, or chief of staff) to ensure shared decision-making and stakeholder engagement throughout the evaluation process.
Regardless of variations in the committees and subcommittees involved, several universal strategies direct medication use within health systems. These include establishment of specific-use criteria (e.g., predefined patient clinical criteria that must be met); restrictions criteria (e.g., highly trained individuals for prescribing or a specialized service for administering) for discontinuation of therapy; development of clinical practice guidelines; and requirement of approval by a medical director, division chair, or other clinical leader. Traditional strategies will be deployed, as well, to ensure appropriate use of gene therapies.
Strict external regulations governing use of these therapies are expected given their specialized nature. Designees to oversee implementation of and adherence to regulations, such as requirements of the Risk Evaluation and Mitigation Strategy (REMS) program, should be appointed well in advance of the formulary addition, as audits are likely to occur within the first 6-12 months of therapy provision.
Working with Payers
The focus on cost, despite the potential and perceived benefits of novel therapies, is inevitable when considering a health system’s revenue cycle. For most providers, the margin of error is essentially zero for the inappropriate use of a costly therapy (e.g., one 12 mg dose of nusinersen is $125,000 based on wholesale acquisition cost).80 Unlike routinely used treatments, for which occasional misadministration and subsequent loss of reimbursement is tolerable, a single misstep could have grave financial implications. Establishment of comprehensive processes for medication use is required to prevent systemic issues that risk multiple misadministrations. In addition, errors in use could represent missed opportunities to provide potentially beneficial therapies.
Early collaboration between health systems and payers is critical to the health system’s process for evaluating novel therapies. Despite increasing numbers of approved orphan drugs, in 2016, these treatments accounted for 7.9% of overall drug spending, and 1.6% were priced at < $500,000 per year.69 At the same time, growth in orphan drug spending is decreasing relative to nonorphan drug spending.69 Payers are frequently implementing strict reimbursement practices in an effort to contain costs and maintain member benefits. Investigation into the reimbursement landscape should occur in tandem with review of clinical safety and efficacy; this may include consultation with local private payers and state Medicaid programs, review of national coverage determinations from the Centers for Medicare & Medicaid Services, and consideration of potential drug administration sites such as those eligible for the 340B Drug Pricing Program.81 Once reimbursement potential is determined, collaboration with internal finance teams to model the expected patient and payer mix could provide insight into the revenue cycle effects of adding a high-cost formulary item. Involvement of finance teams is also critical for identifying all potential avenues for reimbursement, such as new technology add-on payments.82
Summary
SMA is an autosomal recessive disease caused by deficiency in SMN protein that can lead to disability and death. Thus, it is a devastating diagnosis for families, particularly in its most severe form. However, a new treatment, nusinersen, is available, and others are emerging, including a promising new gene therapy, AVXS-101. In patients with SMA, both treatments are most effective if given early, before the onset of extensive nerve damage and associated symptoms. Still, there is evidence to support their use in improving or stabilizing disease after symptoms appear. Clinical trials are ongoing, but at this time, AVXS-101 may have an advantage of a single intravenous administration. If it receives FDA approval, both payers and providers will need to be prepared for its implementation. The future seems bright, but many steps remain; careful planning with attention to principles of sound formulary practice may help limit gaps between approval and access.
Continuing Education
Posttest Questions
-
What was the median time of death in the NeuroNEXT natural history study of patients with type 1 spinal muscular atrophy (SMA) ages <6 months?
- 18 months
- 12 months
- 8 months
- 4 months
-
Which of the following is NOT a characteristic of the adeno-associated viral (AAV) vectors currently used in gene therapy protocols?
- They can carry up to 5 kb of DNA.
- They typically integrate into the host genome.
- They form stable episomes in host cells.
- They were engineered from a nonpathogenic parvovirus.
-
Which is the underlying genetic defect in ~95% of cases of SMA?
- Point mutations in the SMN2 gene
- Point mutations in the SMN1 gene
- Mutations in the promoter that regulates SMN2 gene expression
- Homozygous deletion in SMN1
-
At the time of publication of the AVXS-101 phase 1 clinical trial, how many of the 12 children who received the higher dose of gene therapy had rolled over (compared to no children achieving this motor milestone in natural history studies)?
- 12
- 9
- 6
- 3
-
Which of the following is NOT a universal strategy directing use of a medication identified for formulary inclusion within a health system?
- Adherence to specific-use criteria
- Development of clinical practice guidelines
- Requirement for approval by a medical director, division chair, or other clinical leader
- Placing cost of treatment as most important in the hierarchy of considerations
REFERENCES
- 1.Sugarman EA, Nagan N, Zhu H, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of < 72,400 specimens. Eur J Hum Genet. 2012;20(1):27-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. 2005;57(5):704-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oskoui M, Levy G, Garland CJ, et al. The changing natural history of spinal muscular atrophy type 1. Neurol. 2007;69(20):1931-36. [DOI] [PubMed] [Google Scholar]
- 4.Finkel RS, Mercuri E, Darras BT, et al. ; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-32. [DOI] [PubMed] [Google Scholar]
- 5.Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017; 377(18):1713-22. [DOI] [PubMed] [Google Scholar]
- 6.Cure SMA. AveXis files for FDA approval of gene therapy for spinal muscular atrophy type I. October 18, 2018. Available at: http://www.curesma.org/news/avexis-fda-approval-type-i.html. Accessed November 30, 2018. [Google Scholar]
- 7.Farrar MA, Park SB, Vucic S, et al. Emerging therapies and challenges in spinal muscular atrophy. Ann Neurol. 2017;81(3):355-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harding BN, Kariya S, Monani UR, et al. Spectrum of neuropatho-physiology in spinal muscular atrophy type I. J Neuropathol Exp Neurol. 2015;74(1):15-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnold WD, Kassar D, Kissel JT. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve. 2015;51(2):157-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verhaart IEC, Robertson A, Wilson IJ, et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy—a literature review. Orphanet J Rare Dis. 2017;12(1):124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolb SJ, Coffey CS, Yankey JW, et al. ; NeuroNEXT Clinical Trial Network on behalf of the NN101 SMA Biomarker Investigators. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol. 2017;82(6):883-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finkel RS, McDermott MP, Kaufmann P, et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014;83(9):810-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iannaccone ST. Modern management of spinal muscular atrophy. J Child Neurol. 2007;22:974-78. [DOI] [PubMed] [Google Scholar]
- 14.Chung BH, Wong VC, Ip P. Spinal muscular atrophy: survival pattern and functional status. Pediatrics. 2004;114(5):e548-53. [DOI] [PubMed] [Google Scholar]
- 15.Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155-65. [DOI] [PubMed] [Google Scholar]
- 16.Wirth B, Herz M, Wetter A, et al. Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Hum Genet. 1999;64(5):1340-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mercuri E, Finkel RS, Muntoni F, et al. ; SMA Care Group. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28(2):103-15. [DOI] [PubMed] [Google Scholar]
- 18.Harada Y, Sutomo R, Sadewa AH, et al. Correlation between SMN2 copy number and clinical phenotype of spinal muscular atrophy: three SMN2 copies fail to rescue some patients from the disease severity. J Neurol. 2002;249(9):1211-19. [DOI] [PubMed] [Google Scholar]
- 19.Prior TW, Krainer AR, Hua Y, et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet. 2009;85(3):408-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cuscó I, Barceló MJ, Rojas-García R, et al. SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. J Neurol. 2006;253(1):21-25. [DOI] [PubMed] [Google Scholar]
- 21.DiVito D, Konek S. Spinal muscular atrophy: summary for nutritional care. Infant Child Adoles Nutr. 2010;2(6):348-54. [Google Scholar]
- 22.Wang CH, Finkel RS, Bertini ES, et al. ; Participants of the International Conference on SMA Standard of Care. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22(8):1027-49. [DOI] [PubMed] [Google Scholar]
- 23.Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26(4):1333-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82(4):834-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett CF, Baker BF, Pham N, Swayze E, Geary RS. Pharmacology of antisense drugs. Annu Rev Pharmacol Toxicol. 2017;57:81-105. [DOI] [PubMed] [Google Scholar]
- 26.Mercuri E, Darras BT, Chiriboga CA, et al. ; CHERISH Study Group. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378(7):625-35. [DOI] [PubMed] [Google Scholar]
- 27.Cure SMA. New Spinraza data presented at annual congress of the World Muscle Society demonstrate benefits in treating presymptomatic infants with SMA. October 8, 2018. Available at: http://www.curesma.org/news/spinraza-data-fall2018.html. Accessed November 30, 2018. [Google Scholar]
- 28.Spinraza ( nusinersen) injection, for intrathecal use. Biogen. Revised December 2016. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/209531lbl.pdf. Accessed December 1, 2018. [Google Scholar]
- 29.Mousa MA, Aria DJ, Schaefer CM, et al. A comprehensive institutional overview of intrathecal nusinersen injections for spinal muscular atrophy. Pediatr Radiol. 2018;48(12):1797-805. [DOI] [PubMed] [Google Scholar]
- 30.Aartsma-Rus A. FDA approval of nusinersen for spinal muscular atrophy makes 2016 the year of splice modulating oligonucleotides. Nucleic Acid Ther. 2017;27(2):67-69. [DOI] [PubMed] [Google Scholar]
- 31.Cure SMA. HHS Secretary, Alex Azar, recommends nationwide newborn screening for SMA. July 3, 2018. Available at: http://www.curesma.org/news/nbs-update-july2018.html. Accessed November 30, 2018. [Google Scholar]
- 32.Shorrock HK, Gillingwater TH, Groen EJN. Overview of current drugs and molecules in development for spinal muscular atrophy therapy. Drugs. 2018;78(3):293-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Charnas L. Safety and efficacy findings in the first-in-human trial of the oral splice modulator branaplam in type 1 spinal muscular atrophy (SMA): interim results. Presented at: the 21st Annual Spinal Muscular Atrophy Researcher Meeting; June 29-July 2, 2017; Orlando, FL. [Google Scholar]
- 34.Mercuri E, Baranello G, Kirschner J, et al. SUNFISH Part 1. Risdiplam (RG7916) treatment results in a sustained increase of SMN protein levels and improvement in motor function in patients with type 2 or 3 SMA. Presented at: the 23rd International Annual Congress of the World Muscle Society; October 2-6, 2018; Mendoza, Argentina. [Google Scholar]
- 35.Baranello G, Servais L, Day JW, et al. FIREFISH: Risdiplam (RG7916) improves motor function in babies with type 1 SMA. Presented at: the 23rd International Annual Congress of the World Muscle Society; October 2-6, 2018; Mendoza, Argentina. [Google Scholar]
- 36.Cure SMA. Cytokinetics presents data from the phase 2 clinical trial of reldesemtiv (CK-2127107) in patients with SMA at the 2018 Annual SMA Conference. June 16, 2018. Available at: http://www.curesma.org/news/cytokinetics-data-june-2018.html. Accessed November 30, 2018. [Google Scholar]
- 37.Friedmann T, Roblin R. Gene therapy for human genetic disease? Science. 1972;175(4025):949-55. [DOI] [PubMed] [Google Scholar]
- 38.Jenks S. Gene therapy death—“everyone has to share in the guilt.” J Natl Cancer Inst. 2000;92(2):98-100. [DOI] [PubMed] [Google Scholar]
- 39.Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science. 2018;359(6372). pii: eaan4672. [DOI] [PubMed] [Google Scholar]
- 40.Luxturna (voretigene neparvovec-rzyl) intraocular suspension for subretinal injection . Spark Therapeutics. Revised January 2018. Available at: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=08313a24-e7ce-457a-bb3f-161bc45517ee. Accessed December 19, 2018.
- 41.Kymriah (tisagenlecleucel) suspension for intravenous infusion . Novartis Pharmaceuticals Corporation. Revised May 2018. Available at: https://www.fda.gov/downloads/UCM573941.pdf. Accessed December 1, 2018.
- 42.Yescarta (axicabtagene ciloleucel) suspension for intravenous infusion . Kite Pharma. October 2017. Available at: https://www.fda.gov/downloads/UCM581226.pdf. Accessed December 1, 2018.
- 43.May C, Rivella S, Callegari J, et al. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature. 2000;406(6791):82-86. [DOI] [PubMed] [Google Scholar]
- 44.Thompson AA, Walters MC, Kwiatkowski J, et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2018;378(16):1479-93. [DOI] [PubMed] [Google Scholar]
- 45.Wu C, Dunbar CE. Stem cell gene therapy: the risks of insertional mutagenesis and approaches to minimize genotoxicity. Front Med. 2011;5(4):356-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467(7313):318-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341(6148):1233158. [DOI] [PubMed] [Google Scholar]
- 48.Eichler F, Duncan C, Musolino PL, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. 2017; 377(17):1630-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choudhury SR, Hudry E, Maguire CA, Sena-Esteves M, Breakefield XO, Grandi P. Viral vectors for therapy of neurologic diseases. Neuropharmacology. 2017;120:63-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kessler PD, Podsakoff GM, Chen X, et al. Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc Natl Acad Sci USA. 1996;93(24):14082-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao X, Li J, Samulski RJ. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J Virol. 1996;70(11):8098-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Srivastava A. In vivo tissue-tropism of adeno-associated viral vectors. Curr Opin Virol. 2016; 21:75-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kay MA, Manno CS, Ragni MV, et al. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat Genet. 2000;24(3):257-61. [DOI] [PubMed] [Google Scholar]
- 54.Manno CS, Pierce GF, Arruda VR, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12(3):342-47. [DOI] [PubMed] [Google Scholar]
- 55.Kumar SR, Markusic DM, Biswas M, High KA, Herzog RW. Clinical development of gene therapy: results and lessons from recent successes. Mol Ther Methods Clin Dev. 2016;3:16034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leone P, Shera D, McPhee SW, et al. Long-term follow-up after gene therapy for canavan disease. Sci Transl Med. 2012;4(165):165r a163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wright JF. Adeno-associated viral vector manufacturing: keeping pace with accelerating clinical development. Hum Gene Ther. 2011;22(8):913-14. [DOI] [PubMed] [Google Scholar]
- 58.Mingozzi F, Maus MV, Hui DJ, et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med. 2007;13(4):419-22. [DOI] [PubMed] [Google Scholar]
- 59.McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8(16):1248-54. [DOI] [PubMed] [Google Scholar]
- 60.Foust KD, Wang X, McGovern VL, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 2010;2 8(3):271-74. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Meyer K, Ferraiuolo L, Schmelzer L, et al. Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: a dose-response study in mice and nonhuman primates. Mol Ther. 2015; 23 (3) : 477-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lopes JM. #AAN2018 — Type 1 SMA babies treated with AVXS-101 achieving new milestones, trials show. SMA News Today. April 24, 2018. Available at: https://smanewstoday.com/2018/04/24/aan2018-type-1-sma-babies-avxs-101-continue-improve-clinical-trials. Accessed November 30, 2018. [Google Scholar]
- 63.U.S. National Library of Medicine . ClinicalTrials.gov. Gene replacement therapy clinical trial for patients with spinal muscular atrophy type 1 (STR1VE). Available at: https://clinicaltrials.gov/ct2/show/NCT03306277. Accessed November 30, 2018.
- 64.U.S. National Library of Medicine.. ClinicalTrials.gov. Pre-symptomatic study of intravenous AVXS-101 in spinal muscular atrophy (SMA) for patients with multiple copies of SMN2 (SPR1NT). Available at: https://clinicaltrials.gov/ct2/show/NCT03505099?term=NCT03505099&rank=1. Accessed November 30, 2018.
- 65.U.S. National Library of Medicine.. ClinicalTrials.gov. Study of intrathecal administration of AVXS-101 for spinal muscular atrophy (STRONG). Available at: https://clinicaltrials.gov/ct2/show/NCT03381729. Accessed November 30, 2018.
- 66.Lopes JM. AVXS- 101 sets out to show it might be ‘transformative’ for all SMA types: interview with AveXis. SMA News Today. February 13, 2018. Available at: https://smanewstoday.com/2018/02/13/series-of-trials-will-show-if-avexis-gene-therapy-can-be-a-game-changer-for-sma/. Accessed November 30, 2018. [Google Scholar]
- 67.Kegel ML. FDA lets AveXis know the information it needs to possibly approve AVXS-101 for SMA type 1. SMA News Today. January 8, 2018. Available at: https://smanewstoday.com/2018/01/08/fda-gives-avexis-the-lowdown-on-approving-its-therapy-for-sma-type-1/. Accessed November 30, 2018. [Google Scholar]
- 68.U.S. Food and Drug Administration, Center for Drug Evaluation and Research . Advancing health through innovation: 2017 new drug approvals. January 2018. Available at: https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ReportsBudgets/UCM591976.pdf. Accessed November 30, 2018. [Google Scholar]
- 69.Aitken M, Kleinrock M. Orphan drugs in the United States. IQVIA Institute. October 2017. Available at: https://rarediseases.org/wp-content/uploads/2017/10/Orphan-Drugs-in-the-United-States-Report-Web.pdf. Accessed November 30, 2018.
- 70.Tyler LS, Cole SW, May JR, et al. ; ASHP Expert Panel on Formulary Management. ASHP guidelines on the pharmacy and therapeutics committee and the formulary system. Am J Health Syst Pharm. 2008;65(13):1272-83. [DOI] [PubMed] [Google Scholar]
- 71.Principles of a sound drug formulary system. In: Hawkins B, ed. Best Practices for Hospital and Health System Pharmacy: Positions and Guidance Documents of ASHP. 2006-2007 ed. Bethesda, MD: American Society of Health-System Pharmacists; 2006:110-13. [Google Scholar]
- 72.U.S. Food and Drug Administration . Statement from FDA Commissioner Scott Gottlieb, M.D., on administration’s request for new FDA funding to promote innovation and broaden patient access through competition. February 13, 2018. Available at: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm596554.htm. Accessed November 30, 2018. [Google Scholar]
- 73.Kent A, Spink J. Will rising prices and budget constraints prevent patients from accessing novel gene therapies? Gene Ther. 2017; 24 (9):542-43. [DOI] [PubMed] [Google Scholar]
- 74.AveXis . 10-K Annual report. February 2018. Available at: https://www.last10k.com/sec-filings/avxs/0001558370-18-001313.htm. Accessed November 30, 2018.
- 75.Clarke T, Berkrot B. FDA. approves Gilead cancer gene therapy; price set at $373,000. Reuters. Health News. October 18, 2017. Available at: https://www. reuters.com/article/us-gilead-sciences-fda/fda-approves-gilead-cancer-gene-therapy-price-set-at-373000-idUSKBN1CN35H. Accessed November 30, 2018. [Google Scholar]
- 76.Clarke T. Spark to charge $850,000 per patient for blindness gene therapy. Reuters. Health News. January 3, 2018. Available at: https://www.reuters.com/article/us-spark-genetherapy/spark-to-charge-850000-per-patient-for-blindness-gene-therapy-idUSKBN1ES17W. Accessed November 30, 2018. [Google Scholar]
- 77.Beasley D. US approves Novartis cell therapy for lymphoma. Reuters. Health News. May 1, 2018. Available at: https://www.reuters.com/article/us-novartis-pharmaceuticals/us-approves-novartis-cell-therapy-for-lymphoma-idUSKBN1I24GP. Accessed November 30, 2018. [Google Scholar]
- 78.King NMP, Bishop CE. New treatments for serious conditions: ethical implications. Gene Ther. 2017;24(9):534-38. [DOI] [PubMed] [Google Scholar]
- 79.Davey P, Brown E, Fenelon L, et al. Interventions to improve antibiotic prescribing practices for hospital inpatients. Cochrane Database Syst Rev. 2005;(4):CD003543. [DOI] [PubMed] [Google Scholar]
- 80.Biogen . Spinraza fact sheet. May 2018. Available at: https://www.spinra-za-hcp.com/content/dam/commercial/specialty/spinraza/hcp/en_us/pdf/SPZ-US-0108_SPINRAZA-Product-Fact-Sheet.pdf. Accessed December 1, 2018.
- 81.Heindel G, Smith J, Cruz J, Pappas A. Utilizing a drug information center for submitting reconsiderations for local coverage determinations. Am J Health Syst Pharm. 2017;74(3):106-07. [DOI] [PubMed] [Google Scholar]
- 82.U.S. Centers for Medicare & Medicaid Services . New medical services and new technologies. Available at: https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/AcuteInpatientPPS/newtech.html. Accessed November 30, 2018.
- 83.Haataja L, Mercuri E, Regev R, et al. Optimality score for the neurologic examination of the infant at 12 and 18 months of age. J Pediatr. 1999; 135(2 pt 1):153-61. [DOI] [PubMed] [Google Scholar]
- 84.Romeo DM, Ricci D, Brogna C, Mercuri E. Use of the Hammersmith Infant Neurological Examination in infants with cerebral palsy: a critical review of the literature. Dev Med Child Neurol. 2016;58(3):240-45. [DOI] [PubMed] [Google Scholar]
- 85.Mercuri E, Finkel R, Montes J, et al. Patterns of disease progression in type 2 and 3 SMA: implications for clinical trials. Neuromuscul Disord. 2016; 26(2):123-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Glanzman AM, O’Hagen JM, McDermott MP, et al. ; the Pediatric Neuromuscular Clinical Research Network for Spinal Muscular Atrophy (PNCR), and the Muscle Study Group (MSG). Validation of the Expanded Hammersmith Functional Motor Scale in spinal muscular atrophy type II and III. J Child Neurol. 2011;26(12):1499-507. [DOI] [PubMed] [Google Scholar]
- 87.Glanzman AM, Mazzone E, Main M, et al. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): test development and reliability. Neuromuscul Disord. 2010;20(3):155-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Glanzman AM, McDermott MP, Montes J. Validation of the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND). Pediatr Phys Ther. 2011;23(4):322-26. [DOI] [PubMed] [Google Scholar]
- 89.Bérard C, Payan C, Hodgkinson I, Fermanian J; MFM Collaborative Study Group. A motor function measure for neuromuscular diseases. Construction and validation study. Neuromuscul Disord. 2005;15(7):463-70. [DOI] [PubMed] [Google Scholar]