

Abstract
Cutaneous neurofibromas (cNFs) are the most common tumor in people with the rasopathy neurofibromatosis type 1. They number in hundreds or even thousands throughout the body, and currently, there are no effective interventions to prevent or treat these skin tumors. To facilitate the identification of novel and effective therapies, essential studies including a more refined understanding of cNF biology and the role of RAS signaling and downstream effector pathways responsible for cNF initiation, growth, and maintenance are needed. This review highlights the current state of knowledge of RAS signaling in cNF pathogenesis and therapeutic development for cNF treatment.
INTRODUCTION
The condition, neurofibromatosis type 1 (NF1) results from inactivating alterations in the NF1 gene on chromosome 17q11.2. NF1 was first cloned in 1990 and is one of the largest human genes (350 kb and 60 exons) (Viskochil et al., 1990; Wallace et al., 1990). More than 500 variants in the NF1 gene have been identified; most result in loss of function (Ars et al., 2003; Carey et al., 1986; Huson et al., 1989). Recent scientific advances have begun to shed light on the complex function and regulation of the NF1 gene. NF1 encodes neurofibromin, a RAS GTPase-activating protein (GAP) ubiquitously expressed in tissues but most abundant in the brain, spinal cord, and peripheral nervous system (Daston and Ratner, 1992; Shen et al., 1996; Upadhyaya et al., 1997). Neurofibromin negatively regulates RAS signaling by promoting the conversion of the active GTP-bound form of RAS (i.e., RAS-GTP) to the inactive guanosine diphosphate (GDP)-bound form. In the absence of neurofibromin, the active RAS-GTP form is stabilized, resulting in excessive stimulation of multiple progrowth pathways (Le and Parada, 2007; Ratner and Miller, 2015). Although a pathogenic alteration in one germline allele is sufficient for patients to present with features of NF1, tumor formation requires biallelic loss of NF1 (Skuse et al., 1989).
NF1 is a multisystem disorder that can present with a myriad of manifestations, including but not limited to neoplasia, hyperpigmentation, neurocognitive deficits, and skeletal disease; however, the most universal is the development of cutaneous neurofibromas (cNFs). cNFs are histologically benign skin tumors, comprising multiple cell types, including Schwann cells, fibroblasts, mast cells, and macrophages. These tumors involve the dermis, can be present in any region of the body, and can number in the hundreds or even thousands in an individual with NF1. Although histologically benign, these tumors can be a significant source of disfigurement, anxiety, itch, and pain (Page et al., 2006; Wolkenstein et al., 2003). There are currently no Food and Drug Administration (FDA)–approved medical therapies to prevent or treat cNF, and symptomatic lesions are primarily removed through surgical procedures, including excision, electrodesiccation, and ablative laser therapy (Kim et al., 2016; Levine et al., 2008; Lutterodt et al., 2016; Méni et al., 2015). Although effective for tumor removal in most instances, these procedures often fail to prevent tumor regrowth, may lead to scarring and additional disfigurement, and can be costly and time consuming given the number of tumors needing treatment.
The RAS pathway has emerged as a therapeutic target for cNFs. Similar to other NF1-related tumors, cNFs arise owing to biallelic inactivation of the NF1 gene (Storlazzi et al., 2005), leading to strongly elevated RAS signaling and activation of the canonical RAS/MAPK pathway (Figure 1). MAPK/extracellular signal–regulated kinase (ERK) kinase (MEK) is a kinase in the RAS–MAPK pathway, downstream of RAS that phosphorylates and activates ERK. Currently, the majority of approaches targeting the RAS–MAPK pathway in cNF focus on inhibiting MEK. MEK inhibitors (MEKis) have shown efficacy in shrinking neurofibromas in cNF mouse models (Mo et al., 2021) and clinically in plexiform neurofibromas (pNFs) in NF1 (Dombi et al., 2016; Gross et al., 2020). Despite promising data reporting that MEKis may be efficacious in treating NF1-related tumors, systemic dosing of MEKis commonly leads to adverse effects, including a high rate of skin toxicity, which can be dose limiting. In addition, it is unclear whether solely targeting the RAS–MAPK pathway with MEKis will be sufficient to have a meaningful clinical effect on cNF growth or development over prolonged periods of treatment (i.e., decades) or whether adaptive resistance may limit efficacy (Wang et al., 2022). Despite some concerns about MEKi as a singular approach for cNFs, the RAS pathway is a major driver of cNF emergence and progression. A first step toward the development of therapeutics that target the RAS pathway for cNF is understanding the impact of NF1 on RAS pathway activity relative to skin and cNF.
Figure 1. Signaling pathways of RAS isoforms.
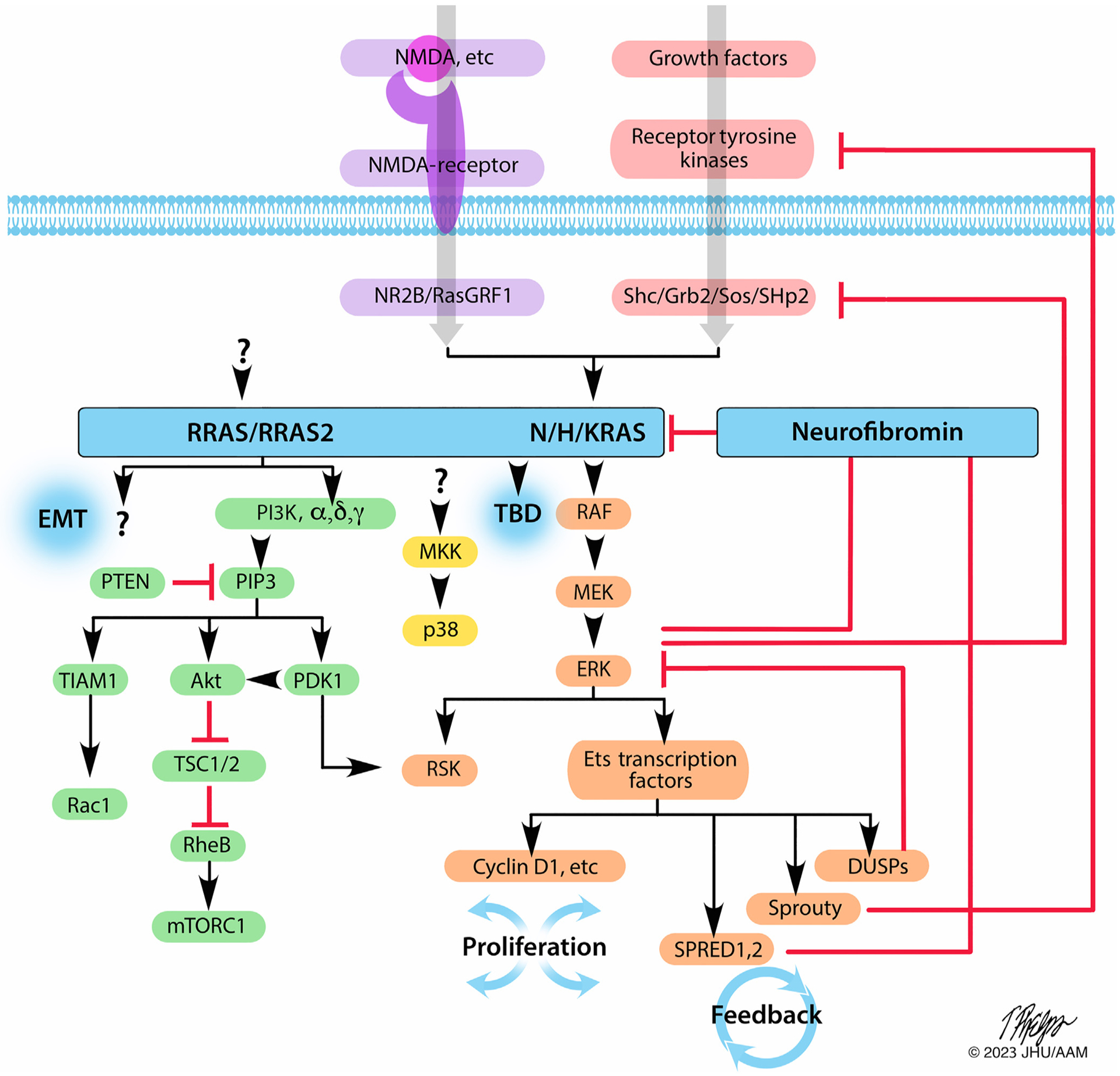
NMDA and GFs activate NMDA receptors and receptor tyrosine kinases, respectively, to activate canonical N/H/KRAS, which signal through the RAF/MEK/ERK cascade to activate downstream effector proteins, including ETS transcription factors, cyclin D1, SPRED1 and SPRED2, Sprouty, and DUSPs, which not only drive cellular proliferation but also complex feedback mechanisms depicted by the red inhibitory signals. MKK, which activates p38 to drive inflammatory cytokine production, represents another distinct signaling axis governed by RAS. Neurofibromin, SPRED1, and SPRED2 also function as GAPs for nonclassical RRAS/RRAS2 isoforms, which signal predominately through PI3K and other less-characterized pathways to modulate EMT and other cellular phenotypes. Undoubtedly, other downstream effectors of both canonical and noncanonical RAS isoforms have yet to be determined. Illustration: Tim Phelps © 2022 JHU AAM Department of Art as Applied to Medicine, Johns Hopkins University School of Medicine. AAM, Department of Art as Applied to Medicine; Akt, protein kinase B; EMT, epithelial-to-mesenchymal transition; ERK, extracellular signal–regulated kinase; GAP, GTPase-activating protein; JHU, Johns Hopkins University; MEK, MAPK/extracellular signal–regulated kinase kinase; MKK, MAPK kinase; NMDA, N-methyl D-aspartate; PI3K, phosphoinositide 3-kinase.
In this review, we present what is known about the NF1 gene and its product as a modulator of RAS and address key knowledge gaps. We dissect the complex circuitry of RAS signaling as it governs not only oncogenesis and survival but also cellular differentiation and the role that aberrant RAS activity plays as a primary regulator in cNF biology and pathogenesis.
NEUROFIBROMIN AS A RAS MODULATOR
Neurofibromin exists as a homodimer and forms a lemniscate-shaped molecule as a consequence of a head-to-tail dimerization. Each monomer comprises an N-terminal HEAT domain, a GAP-related domain (GRD) required for RAS interaction, a Sec14-PH module necessary for membrane binding, and a C-terminal HEAT domain (Lupton et al., 2021; Naschberger et al., 2021; Sherekar et al., 2020). Neurofibromin exhibits different functional states, such as closed, self-inhibited, Zinc stabilized, and open (Lupton et al., 2021; Naschberger et al., 2021). The closed conformation is marked by self-occlusion of the GRD interface by the N-HEAT domain, with the SPRED1-binding site exposed on the surface (Lupton et al., 2021; Naschberger et al., 2021). SPRED1 is known to recruit neurofibromin from the cytosol to the plasma membrane where RAS resides (Stowe et al., 2012; Yan et al., 2020). Conformational rearrangements of the GRD modules are required for RAS binding; they are likely initiated through a complex interaction of SPRED1 with GRD that reorients Sec14-PH (Figure 2a). This results in interaction with the cellular membrane to access and bind RAS (Lupton et al., 2021; Naschberger et al., 2021). Zinc reduces RAS–GAP activity by promoting a self-inhibited, closed conformation through binding by N-HEAT and the GRD–Sec14-PH linker (Naschberger et al., 2021). The neurofibromin scaffold interacts with many proteins that may also play a role in its multifaceted presentation. Of the many proteins interacting with neurofibromin, the interactions with RAS and SPRED1 are the best understood (Lorenzo and McCormick, 2020; Yan et al., 2020).
Figure 2. Structural and functional domains of neurofibromin.
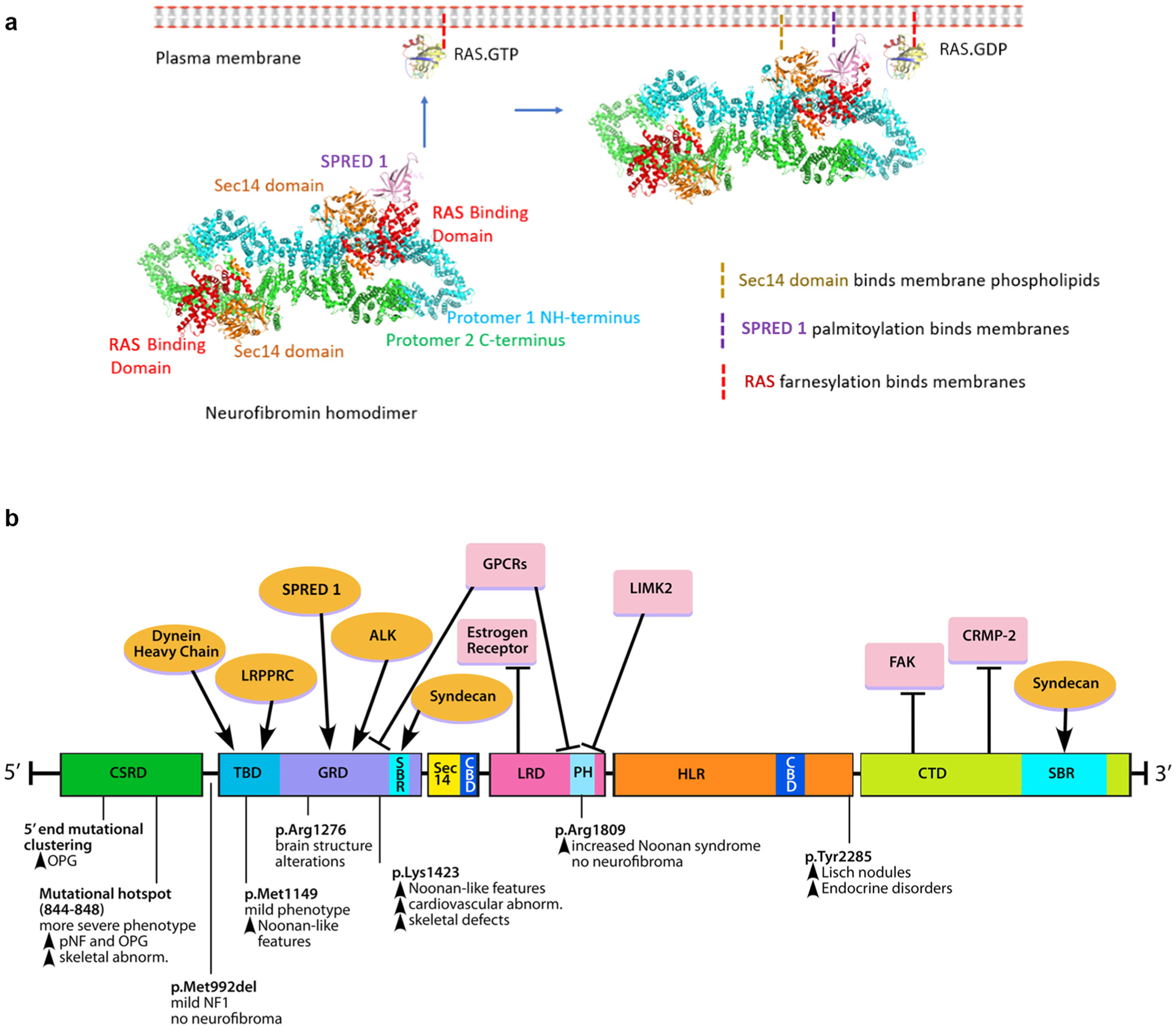
(a) Neurofibromin exists as a lemniscate-shaped homodimer as a result of head-to-tail dimerization between the NH- and C-terminal domains. Recruitment of neurofibromin from the cytosol to associate with RAS at the plasma membrane is shown. Complex interactions between SPRED1 and the GRD are thought to reorient the Sec14-PH to allow for RAS binding at the plasma membrane. (b) Functional domains of neurofibromin and their putative protein–protein interacting partners (top). Amino acid residues spanning each domain are denoted in the figure. The domains include CBD, CSRD, CTD, GRD, HLR, LRD, PH, SBR, and TBD. The proteins include ALK, CELF, CRMP-2, FAK, GPCRs, LIMK2, LRPPRC, SPRED1, and TTIA-1. Reported genotype–phenotype correlations associated with recurrent NF1 variant hotspots (bottom) are shown. The figure is adapted from Mo et al. (2022). Illustration (for Figure 2b): Tim Phelps © 2022 JHU AAM Department of Art as Applied to Medicine the Johns Hopkins University School of Medicine. AAM, Department of Art as Applied to Medicine; CBD, caveolin-binding domain; CSRD, cysteine/serine-rich domain; CTD, C-terminal domain; FAK, focal adhesion kinase; GPCR, G-proteine–coupled receptor; GRD, GTPase-activating protein–related domain; HLR, HEAT-like repeat; JHU, Johns Hopkins University; LRD, leucine-rich domain; NF1, neurofibromatosis type 1; PH, pleckstrin homology; SBR, syndecan-binding region; TBD, tubulin-binding domain.
Loss of the neurofibromin GTPase activity and consequent RAS pathway activation is considered the canonical pathway through which tumors develop in people with NF1. RAS represents a family of proto-oncogenes that can be transformed into oncogenes implicated in both common, often aggressive cancers (i.e., melanoma) and in NF1-associated benign tumors (Le and Parada, 2007). In healthy cells, RAS regulates proliferation, differentiation, transformation, and apoptosis. RAS is most often maintained in the inactive (GDP-bound) conformation. When stimulated, RAS releases GDP and binds GTP. RAS-GTP is the activated form that stimulates several progrowth pathways, including the RAF/MEK/ERK and phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mTOR pathways (Khosravi-Far and Der, 1994; Weiss et al., 1999). Activating variants and/or overexpression of RAS genes (HRAS, KRAS, and NRAS) are commonly found in sporadic solid tumors and have been identified in breast, thyroid, prostate, lung, colorectal, and brain cancers (Barbacid, 1987; Bos, 1989; Harris and McCormick, 2010). Moreover, somatic pathogenic variants in the NF1 gene have been linked to several malignancies that occur independent of NF1, including melanoma, breast cancer, glioblastoma, and primary lung adenocarcinoma (Bowman et al., 2021; Furukawa et al., 2003; Gutmann et al., 1995; Gutzmer et al., 2000; Hölzel et al., 2010; Iyengar et al., 1999; Johnson et al., 1993; Sangha et al., 2008; Side et al., 1998); NF1 is a driver variant in an estimated 26–27% of newly diagnosed melanomas (Ascierto et al., 2017; Jour et al., 2023; Luo et al., 2022). Therefore, therapeutics developed to address neurofibromin dysfunction may affect both NF1-related tumors (caused by germline and somatic variants) and other treatment-resistant cancers (caused by somatic variants).
Although neurofibromin’s GRD is a major potential therapeutic target, the protein contains additional functional domains that may have therapeutic implications (Figure 2b). Among other pathways regulated by neurofibromin, the best understood is the cAMP pathway, a ubiquitous mediator of intracellular signaling activated by a wide variety of pathways through G protein–coupled receptors. Intracellular cAMP activity has been linked to both growth and senescence in NF1 tumors (Warrington et al., 2010). Interestingly, data obtained to date suggest that the impact of cAMP in the setting of an Nf1 variant is dependent on cell type. For example, cAMP is reduced in astrocytes in the setting of Nf1 inactivation (Dasgupta et al., 2003). In contrast, Schwann cells display increased cAMP levels in the absence of Nf1, which likely promote cell growth via cyclin D1 by allowing continual activation after exposure to growth factors (Dang and De Vries, 2011). This mechanism may be directly related to the development of neurofibromas independent of the RAS pathway. The significance of the remaining neurofibromin domains is not well-understood. The pleckstrin homology and Sec14-homology (Sec14) domains form a lipid-binding module; their impact on tumorigenesis in NF1 is not yet known, but they may alter the protein–protein interactions between neurofibromin and other proteins that influence its interaction with RAS (D’Angelo et al., 2006; Welti et al., 2007). Other described domains of neurofibromin are the tubulin-binding domain, a nuclear localization sequence, and a closely related focal adhesion kinase (Arun et al., 2013; Kweh et al., 2009; Li et al., 2001). The influence of these domains in mutated neurofibromin is not yet known.
Nf1/RAS-DEPENDENT SIGNALING AS A MASTER REGULATOR OF cNF PATHOGENESIS
Second-hit somatic inactivating variants in NF1 resulting in loss of heterozygosity (LOH) are required for the genesis of cNFs (Sawada et al., 1996; Serra et al., 1997) and are frequently observed in other NF1 tumors, such as pNF (Colman et al., 1995) and low-grade astrocytomas (Gutmann et al., 2003; Kluwe et al., 2001). Studies in genetically engineered mouse models (GEMMs) suggest that Nf1 LOH most likely occurs in Schwann cell precursors (SCPs) expressing primitive neural crest markers, which represent the cells of origin for neurofibromas (Chen et al., 2014). Cre-mediated recombination of Nf1 in boundary cap cells—driven by either the HoxB7 (Chen et al., 2019) or Prss56 (Radomska et al., 2019) promoter—spontaneously gives rise to both cNF and pNF in mice.
Although cNFs and pNFs have similar histological appearances, they are characterized by distinctive growth patterns. pNFs are likely to present at birth and grow most rapidly throughout early childhood (Akshintala et al., 2020). Upon entering adulthood, growth largely ceases or occurs at an indolent rate (Akshintala et al., 2020; Nguyen et al., 2012). In contrast, cNFs may be present in childhood but are increasingly clinically apparent in adolescence and early adulthood and progressively increase in number throughout life in an unpredictable manner (Cannon et al., 2018; Ehara et al., 2018; Guiraud et al., 2019). Furthermore, cNF growth can be highly variable, with some reporting periods of emergence and rapid and significant growth followed by terminal growth arrest and long-term stability and others showing slow growth incrementally over decades and yet others with barely any activity.
Notably, in contrast to pNFs or diffuse infiltrating neurofibromas—which may involve deep nerve, soft tissue, and skin and are associated with an ~10% lifetime incidence of malignant transformation (Evans et al., 2002)—cNFs do not progress to malignancy (Ortonne et al., 2020, 2018). Although the reasons for this phenomenon remain poorly understood, emerging data suggest that Nf1 haploinsufficiency within the tumor field may serve as a double-edged sword enhancing the growth of benign NF1-associated tumors while impeding malignant transformation (Brosseau et al., 2018). Supporting this paradigm, Krox20-Cre–mediated ablation of Nf1 in SCPs requires a superimposed Nf1+/− background for the genesis of pNF (Zhu et al., 2002), whereas adoptive transfer of wild-type bone marrow abolishes pNF formation in an Nf1+/− background (Yang et al., 2008). Similarly, optic nerve glioma requires biallelic Nf1 gene inactivation in astrocytes coupled with heterozygosity of Nf1 in surrounding brain tissue (Bajenaru et al., 2003).
Although an Nf1+/− background is not required for pNF genesis in some models (Wu et al., 2008), PLPCreERT2-mediated targeting of myelinating SCPs in Nf1flox/− mice (Nf1+/− background) led to pNF more rapidly than their Nf1flox/flox counterparts (wild-type background) (Brosseau et al., 2018). Intriguingly, the Nf1flox/flox mice showed a spontaneous transformation of pNFs to malignant peripheral nerve sheath tumors in 10% of experimental mice but never in Nf1flox/− mice, suggesting that an Nf1+/− background has the capacity to restrain the outgrowth of malignant disease in mice with benign pNF. Enhanced immune surveillance is one potential mechanism for this phenotype, that is, T cells from Nf1+/− mice exhibit enhanced proliferation in response to CD3 stimulation, and increased fractions of activated CD8 cytotoxic T cells in response to delayed-type hypersensitivity assays were also observed in vivo (Brosseau et al., 2018). These findings challenge the traditional conception of a strictly protumorigenic role for germline NF1 variants and may serve to reconcile apparent discrepancies as to why the NF1 gene is somatically mutated with high frequency in a number of sporadic cancers, including approximately one quarter of all melanomas (Jour et al., 2023; Luo et al., 2022) and remarkably in 93% of desmoplastic melanomas (Wiesner et al., 2015). Although desmoplastic melanoma has rarely been reported in persons with NF1, a recent large retrospective cohort study did reveal an increased incidence of melanoma in persons with NF1 compared with that in matched controls (OR = 2.27) (Trinh et al., 2022). Comparatively, ORs for basal cell carcinoma and squamous cell carcinoma in patients with NF1 were 1.30 and 1.32, respectively. Thus, possible selection bias of patients with NF1 being evaluated more frequently by dermatologists and thereby potentially increasing the chances of diagnosing skin cancer is unlikely to fully account for these relative risk differences. These data highlight the complex and conflicting roles of NF1 in promoting malignancy.
Dueling roles of RAS activation in cNF cells of origin
The nuanced mechanisms through which NF1-dependent hyperactivation of RAS signaling differentially orchestrates both proliferation and growth arrest/quiescence phenotypes within distinct phases of the cNF life cycle remain poorly understood (Figure 3). Although the role of oncogenic RAS in promoting growth and survival in cancer and immortalized cell lines is well-established (Barbacid, 1987), the effects of chronic RAS hyperactivation and primary cell lines are far more complex. In human-induced pluripotent stem cells, Mo et al. (2021) found that homozygous NF1 deletion increased the pools of SCPs by impeding Schwann cell lineage maturation. In contrast, loss of Nf1 in the CNS resulted in enhanced astrocytic differentiation (Dasgupta and Gutmann, 2005). Similarly, in murine Schwann cells, biallelic inactivation of Nf1 resulted in a transient proliferative burst, followed by induction of Cdkn2a (Ink4a/Arf)-mediated senescence growth arrest (Rhodes et al., 2019). Loss of the Cdkn2a alternate reading frame resulted in the development of atypical neurofibromas and malignant transformation in vivo, a key secondary genetic driver event that has been observed in humans (Beert et al., 2011; Brohl et al., 2017; Carrió et al., 2018; Lee et al., 2014; Pemov et al., 2019). Collectively, these findings suggest that LOH-mediated NF1 hyperactivation of RAS pathway activity produces distinct phenotypes that depend on the lineage and differentiative state at which the variants are introduced (Figure 2): either proliferation and survival of SCPs or terminal differentiation and senescence-induced growth arrest, in a context-dependent manner.
Figure 3. Dueling roles of NF1/RAS-dependent signaling in governing the fate and function of cNF cells of origin.
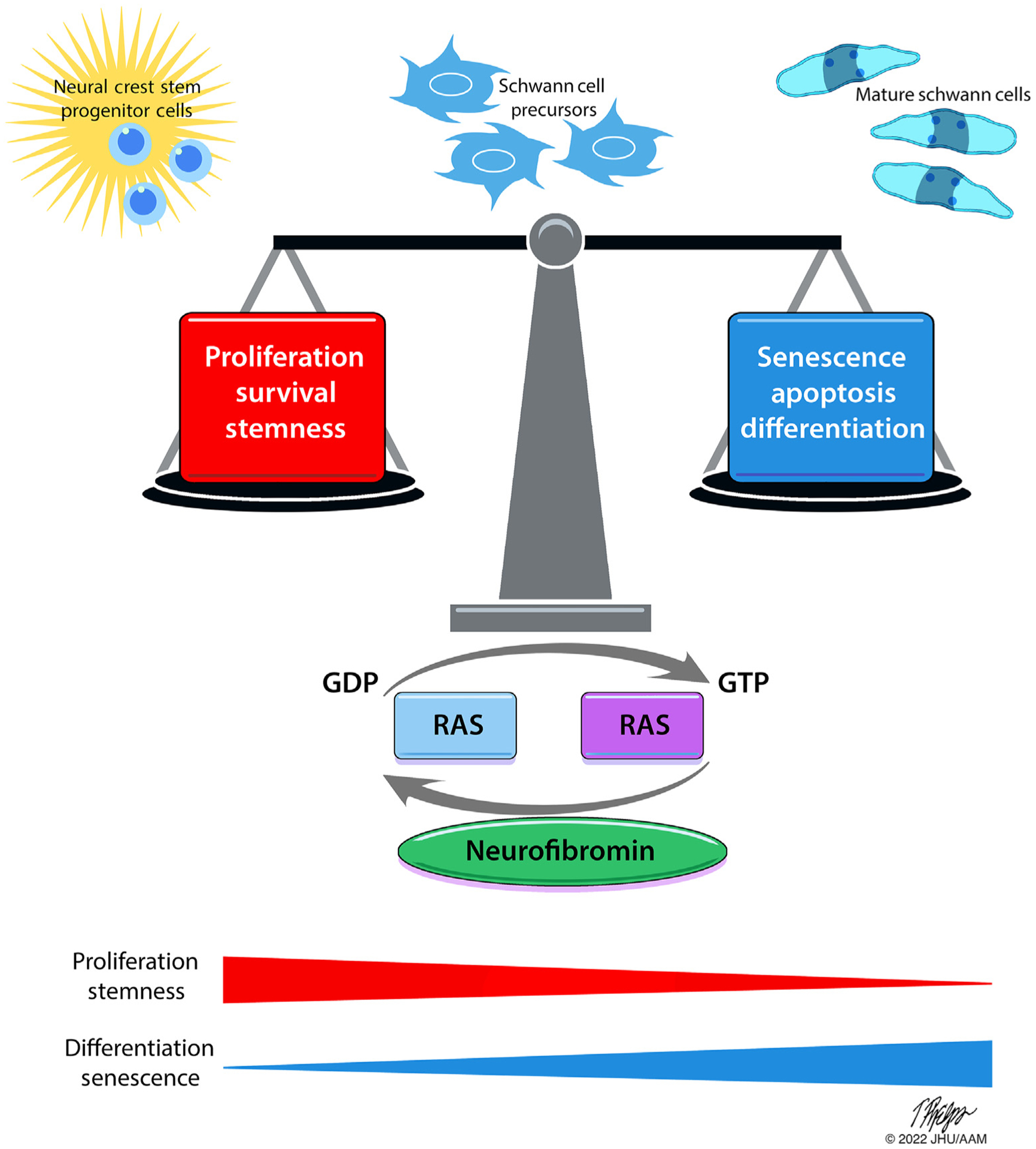
Schematic depicting the complex cellular phenotypes invoked by chronic RAS hyperactivation in Schwann cell precursors, promoting proliferation, survival, and cell stemness on one hand while driving senescence, growth arrest, apoptosis, and cellular differentiation on the other hand. Emerging data suggest that these phenotypes are highly context dependent upon the lineage and cellular differentiation state in which the NF1 inactivation occurs and may further be influenced by epigenetic programs as well. Illustration: Tim Phelps © 2022 JHU AAM Department of Art as Applied to Medicine the Johns Hopkins University School of Medicine. AAM, Department of Art as Applied to Medicine; cNF, cutaneous neurofibroma; GDP, guanosine diphosphate; GTP, guanosine triphosphate; JHU, Johns Hopkins University; NF1, neurofibromatosis type 1.
Epigenetic signatures may be responsible at least in part for endowing distinct RAS-dependent phenotypes invoked by NF1 LOH in the Schwann cell lineage. NF1 LOH in Schwann cells is associated with distinct epigenetic alterations that influence RAS signaling outputs (Grit et al., 2021). Steensma and colleagues recently compared the methylation profiles of cNFs with those of pNFs (Grit et al., 2021). They observed consistent site-specific methylation events in MAP2K3 (MKK3) and an upstream regulatory site for MAPK14 (p38) in a large cohort of cNFs. These alterations were associated with increased MKK3/p38-dependent signaling in cNFs, a critical pathway linking RAS-dependent signaling with inflammatory cytokine production. In contrast, epigenetic reinforcement of canonical RAS/MEK/ERK signaling was observed in pNFs where unchecked growth and proliferation typically pre-dominate. These emerging insights in neurofibroma epigenetics may thus provide a molecular basis for the distinct growth kinetics, pathophysiological paradigms, and responses to MEKi therapy observed between cNF and pNF. Further investigation of how epigenetic programs modulate RAS signaling in neurofibromas is needed, including how differential activation of RAS/MKK/p38 and RAS/MEK/ERK effector pathways influence growth, tumor configuration, and pain phenotypes in cNF.
The specific RAS isoforms that show preferential activity in cNF remain ill defined and represent another area where additional study is needed. In murine models, optic pathway glioma genesis is driven by a proclivity for KRAS activation as opposed to NRAS or HRAS (Dasgupta et al., 2005). In Nf1+/− mice with cognitive phenotypes, heterozygous KRAS or NRAS inactivation normalized RAS-dependent signaling and ameliorated learning deficits (Costa et al., 2002; Cui et al., 2008). In addition to the classical RAS proteins, HRAS, NRAS, and KRAS, whose major function is the activation of the MAPK pathway, neurofibromin is a GAP for RRAS proteins (Patmore et al., 2012). These proteins regulate PI3K activity, among other less well-characterized pathways. It is likely that activation of these pathways after loss of NF1 contributes to the cNF phenotype (Figure 1).
The role of certain NF1 variants in the phenotype of cNF is another area of active investigation. Recently, missense variants affecting NF1 codons 844–848 have been associated with a severe NF1 phenotype, including a high burden of cNF and a high rate of malignancies (Koczkowska et al., 2018). Intriguingly, these variants reside outside the GRD, within a highly conserved region of the cysteine/serine-rich domain of NF1, and it is unclear precisely how variants in this domain alter interactions between neurofibromin and RAS (Koczkowska et al., 2018). Germline NF1 micro-deletions are also associated with increased severity of cNF and other manifestations of the NF1 condition, which has been attributed to loss of neighboring modifier genes such as CRLF3, ATAD5, OMG, RAB11FIP4, SUZ12, and ILRRC37B among others. Notably, cNFs arising in the context of NF1 microdeletion do not exhibit somatic LOH of the second NF1 allele but instead typically harbor NF1 single nucleotide variants (De Raedt et al., 2006).
Nf1 gene dose and RAS-dependent signaling in the cNF microenvironment
Interactions between Schwann cells and the tumor micro-environment (including mast cells, macrophages, fibroblasts, and neuronal elements) are critical for the genesis of benign tumors in NF1, including cNF (Bui et al., 2021) (Figure 4). Beyond the requirement of LOH in tumor-initiating Schwann cells, haploinsufficiency of Nf1 has also been shown to elevate RAS activity in multiple cell lineages (Staser et al., 2010). In response to stem cell factor (SCF)–mediated stimulation of the c-kit receptor, Nf1+/− mast cells exhibit increased levels of RAS-GTP and enhanced the activity of downstream effector pathways, including the MAPK, p38, PI3K/Akt, and RAC GTPases (Ingram et al., 2001, 2000; Khalaf et al., 2007; McDaniel et al., 2008). Genetic and pharmacologic disruption of the SCF receptor c-Kit prevented pNF formation in Nf1flox/−;Krox20-Cre mice (Yang et al., 2008), and a subset of patients, particularly young children with airway-associated pNF, showed clinical response to imatinib mesylate in a phase 2 trial (Robertson et al., 2012; Yang et al., 2008). A case report of ketotifen, a mast cell–stabilizing agent started early in life and continued for decades, suggested prevention or stabilization of cNF (Riccardi, 2015). However, in spontaneous GEMMs, ketotifen did not affect pNF initiation or growth (Burks et al., 2019). Concordantly, disruption of SCF production by SCPs in PlpCre-ERT2;Nf1 floxed mice effectively disrupted the recruitment of mast cells into the tumor microenvironment but did not improve tumor burden (Liao et al., 2018), suggesting that mast cells were not required for pNF initiation and progression in this model.
Figure 4. Schwann cell–microenvironment interactions shape cNF development.
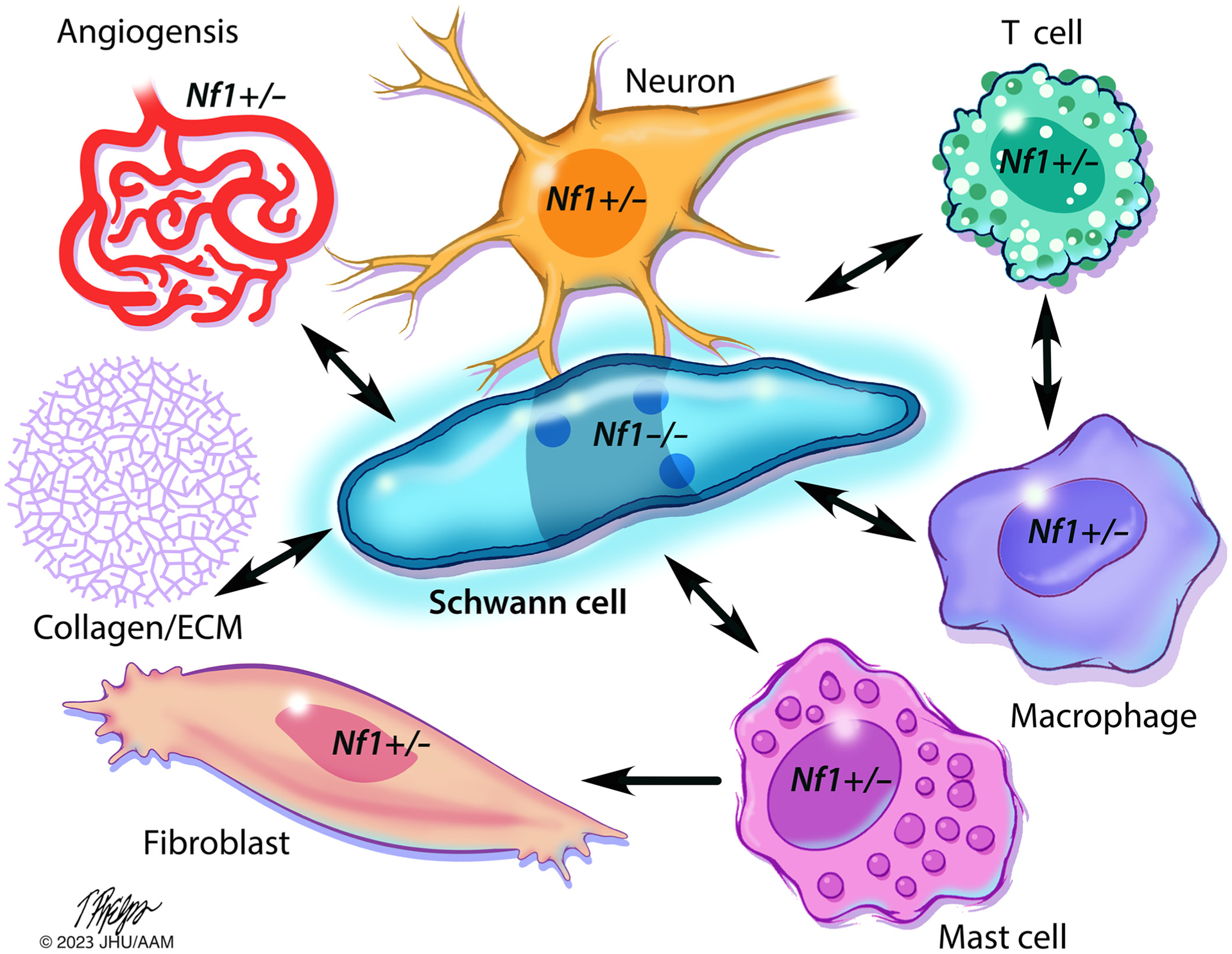
Nf1 LOH leads to aberrant proliferation of Schwann cells and their precursors, the tumorigenic cells of origin for cNF. Paracrine and cell–cell contact interactions between (Nf1−/−) Schwann cells and other Nf1 heterozygous (Nf1+/−) components of the tumor microenvironment, including neurons, mast cells, macrophages, T cells, fibroblasts, and endothelial cells, further affect cNF development. These Nf1+/− heterozygous lineages exhibit multiple RAS-dependent gain in functions in response to inflammatory cytokines and growth factors that further perpetuate cNF initiation and growth. Fibroblasts deposit abundant ECM and collagen, which comprises a significant proportion of the tumor’s dry weight. Illustration: Tim Phelps © 2022 JHU AAM Department of Art as Applied to Medicine the Johns Hopkins University School of Medicine. AAM, Department of Art as Applied to Medicine; cNF, cutaneous neurofibroma; ECM, extracellular matrix; JHU, Johns Hopkins University; LOH, loss of heterozygosity; NF1, neurofibromatosis type 1.
Fibroblasts and secreted collagen are abundant components of the neurofibroma microenvironment, with collagen itself comprising an estimated 50% of the tumor’s dry weight (Peltonen et al., 1986). Until recently, the particular subtypes of collagen that predominate within the extracellular matrix of cNFs had yet to be defined. Single-cell RNA sequencing of a series of human cNFs revealed that neurofibroma-associated fibroblasts (NFAFs) express increased amounts of collagen types I, III, VI, and XV, with a notable abundance of collagen VI (Brosseau et al., 2021). In triple-negative breast cancer, collagen VI binds directly with membrane glycoprotein NG2 to drive invasion through EGFR–MAPK–dependent signaling (Wishart et al., 2020), thus implicating a role for NF1 haploinsufficiency in NFAFs in amplifying collagen VI responses in a RAS-dependent manner. Inflammatory and profibrotic factors such as TGFβ, secreted by Nf1+/− mast cells, have also been shown to exert paracrine effects on Nf1+/− fibroblasts within the tumor microenvironment, resulting in enhanced RAS-c-abl–dependent proliferation and collagen synthesis (Yang et al., 2006).
Although the contribution of Nf1 haploinsufficiency in macrophages has not directly been interrogated in cNFs to date, Nf1+/− monocytes exhibit increased RAS-dependent chemotaxis and functions in response to monocyte chemotactic protein-1 stimulation of the CCR2 receptor, resulting in enhanced neointima formation in response to carotid artery ligation (Bessler et al., 2016). In addition, macrophages from Nf1+/− mice exhibited enhanced RAS/protein kinase C-delta–mediated p47Phox phosphorylation, resulting in enhanced macropinocytosis and polarization of macrophages toward a proinflammatory M1 phenotype, with increased cytokine secretion (Ghoshal et al., 2019). pNFs arising in PlpCre-ERT2;Nf1–floxed mice also exhibited a preponderance of M1 proinflammatory macrophages (compared with that of M2 protumorigenic macrophages), although macrophage levels were not affected by Nf1 heterozygosity status (Liao et al., 2018).
The contribution of enhanced Nf1/RAS-dependent signaling in neurons themselves is another field of study that merits further attention, specifically with respect to the pathogenesis of cNF. Nf1 haploinsufficiency in peripheral nervous system neurons resulted in increased RAS/Akt-dependent neurite lengths and survival (Brown et al., 2012). Nf1+/− mice with conditional biallelic Nf1 inactivation in neurons exhibit increased GABAergic interneural excitability resulting from attenuated activity of HTCN1. The N-terminal domain of neurofibromin binds directly to HTCN1 to modulate cationic currents (Omrani et al., 2015), thus representing a key RAS-independent function of neurofibromin that modulates neuronal excitability.
CONCLUSIONS
cNFs are one of the most prevalent, uniform, and burden-some aspects of NF1 for which there are currently no FDA-approved therapies. There is an immediate need for improved awareness of these tumors and options for therapeutics in clinical dermatology. In addition, there are rich opportunities for collaboration across dermatology, neuroscience, oncology, molecular biology, and genetics to understand cNF development and progression to not only develop needed therapies for cNF but also to improve treatment approaches for the many common conditions such as melanoma driven by NF1 variants.
As a modulator of RAS, neurofibromin regulates the development of both benign and malignant tumors of the skin, with somatic NF1 variants identified in approximately one fourth of all melanoma diagnoses. Although both tumors share a neural crest–derived cell of origin, with cNF arising from SCPs and melanoma from melanocytes, cNFs are uniformly benign and never progress to malignancy. Moreover, despite the high frequency of somatic NF1 variants in melanoma, persons with NF1 rarely develop melanoma (Uusitalo et al., 2016). Further study is needed to understand how the contributions of the germline NF1 loss and RAS activity within the tumor microenvironment influence tumor initiation and malignant transformation in the skin. The role of epigenetics and differentiative states within the respective cells of origin for cNFs and pNFs that acquire NF1 LOH also merits further exploration because this may further help to explain the distinct growth patterns and natural histories of these tumors.
Development of new and effective treatments for cNFs will require a more refined understanding of cNF biology and the role of RAS signaling and downstream effector pathways responsible for cNF initiation, growth, and maintenance. RAS exhibits a complex and dual role by not only perpetuating the growth and survival of SCPs that give rise to cNF but also driving Schwann cell differentiation and senescence-mediated growth arrest. The contribution of specific RAS isoforms that show preferential activity in cNF remains ill defined. Integrated multiomic approaches to further define epigenetic mechanisms that modulate the transmission of RAS-dependent signals through various downstream effector pathways across multiple stages of the cNF lifecycle will also be critical in informing the utility of putative strategies for cNF treatment and prevention. The revolution of single-cell and spatial biology will undoubtedly continue to provide unprecedented insights into how NF1/RAS-dependent signaling orchestrates the cNF microenvironment at various stages along the continuum from emergence and proliferation to quiescence and stability. Addressing these knowledge gaps will allow us to accelerate the identification and clinical translation of novel therapeutic agents to ameliorate a significant and life-long source of morbidity for persons with NF1 and potentially other diseases of the skin.
ACKNOWLEDGMENTS
This publication was supported by funding from the Neurofibromatosis Therapeutic Acceleration Program (NTAP) at the Johns Hopkins University School of Medicine. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of The Johns Hopkins University School of Medicine. The authors would like to thank Tim Phelps for illustration assistance.
CONFLICT OF INTEREST
FM is a consultant to BridgeBio, Quanta, Amgen, and Pfizer; all have RAS inhibitor programs, but none work on neurofibromatosis type 1. SDR, RLC, VS, SYL, CGR, JOB, and KYS receive support from the Neurofibromatosis Therapeutic Acceleration Progam (NTAP) at Johns Hopkins University. SDR, VS, MRS, and JOB receive funding from the Department of Defense. IL and JOB are consultants for SpringWorks Therapeutics. The remaining authors state no conflict of interest.
Abbreviations:
- Akt
protein kinase B
- cNF
cutaneous neurofibroma
- ERK
extracellular signal–related kinase
- FDA
Food and Drug Administration
- GAP
GTPase-activating protein
- GDP
guanosine diphosphate
- GEMM
genetically engineered mouse model
- GRD
GTPase-activating protein–related domain
- LOH
loss of heterozygosity
- MEK
MAPK/extracellular signal–regulated kinase syndical
- MEKi
MAPK/extracellular signal–regulated kinase syndical inhibitor
- NFAF
neurofibroma-associated fibroblast
- NF1
neurofibromatosis type 1
- PI3K
phosphoinositide 3-kinase
- pNF
plexiform neurofibroma
- SCF
stem cell factor
- SCP
Schwann cell precursor
Footnotes
Disclaimer
The contents of this paper are solely the responsibilities of the authors and do not necessarily represent the official views of the Johns Hopkins University School of Medicine.
REFERENCES
- Akshintala S, Baldwin A, Liewehr DJ, Goodwin A, Blakeley JO, Gross AM, et al. Longitudinal evaluation of peripheral nerve sheath tumors in neurofibromatosis type 1: growth analysis of plexiform neurofibromas and distinct nodular lesions. Neuro Oncol 2020;22:1368–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ars E, Kruyer H, Morell M, Pros E, Serra E, Ravella A, et al. Recurrent mutations in the NF1 gene are common among neurofibromatosis type 1 patients. J Med Genet 2003;40:e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun V, Worrell L, Wiley JC, Kaplan DR, Guha A. Neurofibromin interacts with the cytoplasmic Dynein Heavy Chain 1 in melanosomes of human melanocytes. FEBS Lett 2013;587:1466–73. [DOI] [PubMed] [Google Scholar]
- Ascierto ML, Makohon-Moore A, Lipson EJ, Taube JM, McMiller TL, Berger AE, et al. Transcriptional mechanisms of resistance to anti-PD-1 therapy. Clin Cancer Res 2017;23:3168–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajenaru ML, Hernandez MR, Perry A, Zhu Y, Parada LF, Garbow JR, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res 2003;63:8573–7. [PubMed] [Google Scholar]
- Barbacid M RAS genes. Annu Rev Biochem 1987;56:779–827. [DOI] [PubMed] [Google Scholar]
- Beert E, Brems H, Daniëls B, De Wever I, Van Calenbergh F, Schoenaers J, et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer 2011;50:1021–32. [DOI] [PubMed] [Google Scholar]
- Bessler WK, Kim G, Hudson FZ, Mund JA, Mali R, Menon K, et al. Nf1+/− monocytes/macrophages induce neointima formation via CCR2 activation. Hum Mol Genet 2016;25:1129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL. RAS oncogenes in human cancer: a review. Cancer Res 1989;49:4682–9. [PubMed] [Google Scholar]
- Bowman L, Tiu R, Smyth EN, Willard MD, Li L, Beyrer J, et al. Clinical characteristics, treatments, and concurrent mutations in non-small cell lung cancer patients with NF1 mutations. Clin Lung Cancer 2021;22:32–41.e1. [DOI] [PubMed] [Google Scholar]
- Brohl AS, Kahen E, Yoder SJ, Teer JK, Reed DR. The genomic landscape of malignant peripheral nerve sheath tumors: diverse drivers of Ras pathway activation. Sci Rep 2017;7:14992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosseau JP, Liao CP, Wang Y, Ramani V, Vandergriff T, Lee M, et al. NF1 heterozygosity fosters de novo tumorigenesis but impairs malignant transformation. Nat Commun 2018;9:5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosseau JP, Sathe AA, Wang Y, Nguyen T, Glass DA 2nd, Xing C, et al. Human cutaneous neurofibroma matrisome revealed by single-cell RNA sequencing. Acta Neuropathol Commun 2021;9:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Diggs-Andrews KA, Gianino SM, Gutmann DH. Neurofibromatosis-1 heterozygosity impairs CNS neuronal morphology in a cAMP/PKA/ROCK-dependent manner. Mol Cell Neurosci 2012;49:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui A, Jiang C, McKay RM, Klesse LJ, Le LQ. Insights into the pathogenesis of NF1-associated neoplasms. JID Innov 2021;1:100044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burks CA, Rhodes SD, Bessler WK, Chen S, Smith A, Gehlhausen JR, et al. Ketotifen modulates mast cell chemotaxis to kit-ligand, but does not impact mast cell numbers, degranulation, or tumor behavior in neurofibromas of Nf1-deficient mice. Mol Cancer Ther 2019;18:2321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon A, Chen MJ, Li P, Boyd KP, Theos A, Redden DT, et al. Cutaneous neurofibromas in neurofibromatosis type I: a quantitative natural history study. Orphanet J Rare Dis 2018;13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey JC, Baty BJ, Johnson JP, Morrison T, Skolnick M, Kivlin J. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci 1986;486:45–56. [DOI] [PubMed] [Google Scholar]
- Carrió M, Gel B, Terribas E, Zucchiatti AC, Moliné T, Rosas I, et al. Analysis of intratumor heterogeneity in Neurofibromatosis type 1 plexiform neurofibromas and neurofibromas with atypical features: correlating histological and genomic findings. Hum Mutat 2018;39:1112–25. [DOI] [PubMed] [Google Scholar]
- Chen Z, Liu C, Patel AJ, Liao CP, Wang Y, Le LQ. Cells of origin in the embryonic nerve roots for NF1-associated plexiform neurofibroma. Cancer Cell 2014;26:695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Mo J, Brosseau JP, Shipman T, Wang Y, Liao CP, et al. Spatiotemporal loss of NF1 in Schwann cell lineage leads to different types of cutaneous neurofibroma susceptible to modification by the hippo pathway. Cancer Discov 2019;9:114–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman SD, Williams CA, Wallace MR. Benign neurofibromas in type 1 neurofibromatosis (NF1) show somatic deletions of the NF1 gene. Nat Genet 1995;11:90–2. [DOI] [PubMed] [Google Scholar]
- Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, et al. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 2002;415:526–30. [DOI] [PubMed] [Google Scholar]
- Cui Y, Costa RM, Murphy GG, Elgersma Y, Zhu Y, Gutmann DH, et al. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 2008;135:549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang I, De Vries GH. Aberrant cAMP metabolism in NF1 malignant peripheral nerve sheath tumor cells. Neurochem Res 2011;36:1697–705. [DOI] [PubMed] [Google Scholar]
- D’Angelo I, Welti S, Bonneau F, Scheffzek K. A novel bipartite phospholipid-binding module in the neurofibromatosis type 1 protein. EMBO Rep 2006;7:174–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Dugan LL, Gutmann DH. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J Neurosci 2003;23:8949–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Gutmann DH. Neurofibromin regulates neural stem cell proliferation, survival, and astroglial differentiation in vitro and in vivo. J Neurosci 2005;25:5584–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Li W, Perry A, Gutmann DH. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. Cancer Res 2005;65:236–45. [PubMed] [Google Scholar]
- Daston MM, Ratner N. Neurofibromin, a predominantly neuronal GTPase activating protein in the adult, is ubiquitously expressed during development. Dev Dyn 1992;195:216–26. [DOI] [PubMed] [Google Scholar]
- De Raedt T, Maertens O, Chmara M, Brems H, Heyns I, Sciot R, et al. Somatic loss of wild type NF1 allele in neurofibromas: comparison of NF1 micro-deletion and non-microdeletion patients. Genes Chromosomes Cancer 2006;45:893–904. [DOI] [PubMed] [Google Scholar]
- Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, et al. Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N Engl J Med 2016;375:2550–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehara Y, Yamamoto O, Kosaki K, Yoshida Y. Natural course and characteristics of cutaneous neurofibromas in neurofibromatosis 1. J Dermatol 2018;45:53–7. [DOI] [PubMed] [Google Scholar]
- Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 2002;39:311–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, Yanai N, Fujita M, Harada Y. Novel mutations of neurofibromatosis type 1 gene in small cell lung cancers. Surg Today 2003;33:323–7. [DOI] [PubMed] [Google Scholar]
- Ghoshal P, Singla B, Lin H, Cherian-Shaw M, Tritz R, Padgett CA, et al. Loss of GTPase activating protein neurofibromin stimulates paracrine cell communication via macropinocytosis. Redox Biol 2019;27:101224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grit JL, Johnson BK, Dischinger PS, J Essenburg C, Adams M, Campbell S, et al. Distinctive epigenomic alterations in NF1-deficient cutaneous and plexiform neurofibromas drive differential MKK/p38 signaling. Epigenetics Chromatin 2021;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ, et al. Selumetinib in children with inoperable plexiform neurofibromas [published correction appears in N Eng J Med 2020;383:1290] N Engl J Med 2020;382:1430–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiraud M, Bouroubi A, Beauchamp R, Bocquet A, Grégoire JM, Rauly-Lestienne I, et al. Cutaneous neurofibromas: patients’ medical burden, current management and therapeutic expectations: results from an online European patient community survey. Orphanet J Rare Dis 2019;14:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann DH, Geist RT, Rose K, Wallin G, Moley JF. Loss of neurofibromatosis type I (NF1) gene expression in pheochromocytomas from patients without NF1. Genes Chromosomes Cancer 1995;13:104–9. [DOI] [PubMed] [Google Scholar]
- Gutmann DH, James CD, Poyhonen M, Louis DN, Ferner R, Guha A, et al. Molecular analysis of astrocytomas presenting after age 10 in individuals with NF1. Neurology 2003;61:1397–400. [DOI] [PubMed] [Google Scholar]
- Gutzmer R, Herbst RA, Mommert S, Kiehl P, Matiaske F, Rütten A, et al. Allelic loss at the neurofibromatosis type 1 (NF1) gene locus is frequent in desmoplastic neurotropic melanoma. Hum Genet 2000;107:357–61. [DOI] [PubMed] [Google Scholar]
- Harris TJ, McCormick F. The molecular pathology of cancer. Nat Rev Clin Oncol 2010;7:251–65. [DOI] [PubMed] [Google Scholar]
- Hölzel M, Huang S, Koster J, Ora I, Lakeman A, Caron H, et al. NF1 is a tumor suppressor in neuroblastoma that determines retinoic acid response and disease outcome. Cell 2010;142:218–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson SM, Compston DA, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. II. Guidelines for genetic counselling. J Med Genet 1989;26:712–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram DA, Hiatt K, King AJ, Fisher L, Shivakumar R, Derstine C, et al. Hyperactivation of p21(ras) and the hematopoietic-specific Rho GTPase, Rac2, cooperate to alter the proliferation of neurofibromin-deficient mast cells in vivo and in vitro. J Exp Med 2001;194:57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram DA, Yang FC, Travers JB, Wenning MJ, Hiatt K, New S, et al. Genetic and biochemical evidence that haploinsufficiency of the Nf1 tumor suppressor gene modulates melanocyte and mast cell fates in vivo. J Exp Med 2000;191:181–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyengar TD, Ng S, Lau CC, Welch WR, Bell DA, Berkowitz RS, et al. Differential expression of NF1 type I and type II isoforms in sporadic borderline and invasive epithelial ovarian tumors. Oncogene 1999;18:257–62. [DOI] [PubMed] [Google Scholar]
- Johnson MR, Look AT, DeClue JE, Valentine MB, Lowy DR. Inactivation of the NF1 gene in human melanoma and neuroblastoma cell lines without impaired regulation of GTP. Ras. Proc Natl Acad Sci USA 1993;90: 5539–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jour G, Illa-Bochaca I, Ibrahim M, Donnelly D, Zhu K, Miera EV, et al. Genomic and transcriptomic analyses of NF1-mutant melanoma identify potential targeted approach for treatment. J Invest Dermatol 2023;143: 444–55.e8. [DOI] [PubMed] [Google Scholar]
- Khalaf WF, Yang FC, Chen S, White H, Bessler W, Ingram DA, et al. K-ras is critical for modulating multiple c-kit-mediated cellular functions in wild-type and Nf1+/− mast cells. J Immunol 2007;178:2527–34. [DOI] [PubMed] [Google Scholar]
- Khosravi-Far R, Der CJ. The Ras signal transduction pathway. Cancer Metastasis Rev 1994;13:67–89. [DOI] [PubMed] [Google Scholar]
- Kim DH, Hyun DJ, Piquette R, Beaumont C, Germain L, Larouche D. 27.12 MHz radiofrequency ablation for benign cutaneous lesions. BioMed Res Int 2016;2016:6016943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluwe L, Hagel C, Tatagiba M, Thomas S, Stavrou D, Ostertag H, et al. Loss of NF1 alleles distinguish sporadic from NF1-associated pilocytic astrocy-tomas. J Neuropathol Exp Neurol 2001;60:917–20. [DOI] [PubMed] [Google Scholar]
- Koczkowska M, Chen Y, Callens T, Gomes A, Sharp A, Johnson S, et al. Genotype-phenotype correlation in NF1: evidence for a more severe phenotype associated with missense mutations affecting NF1 codons 844–848. Am J Hum Genet 2018;102:69–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kweh F, Zheng M, Kurenova E, Wallace M, Golubovskaya V, Cance WG. Neurofibromin physically interacts with the N-terminal domain of focal adhesion kinase. Mol Carcinog 2009;48:1005–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le LQ, Parada LF. Tumor microenvironment and neurofibromatosis type I: connecting the GAPs. Oncogene 2007;26:4609–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet 2014;46:1227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine SM, Levine E, Taub PJ, Weinberg H. Electrosurgical excision technique for the treatment of multiple cutaneous lesions in neurofibromatosis type I. J Plast Reconstr Aesthet Surg 2008;61:958–62. [DOI] [PubMed] [Google Scholar]
- Li C, Cheng Y, Gutmann DA, Mangoura D. Differential localization of the neurofibromatosis 1 (NF1) gene product, neurofibromin, with the F-actin or microtubule cytoskeleton during differentiation of telencephalic neurons. Brain Res Dev Brain Res 2001;130:231–48. [DOI] [PubMed] [Google Scholar]
- Liao CP, Booker RC, Brosseau JP, Chen Z, Mo J, Tchegnon E, et al. Contributions of inflammation and tumor microenvironment to neurofibroma tumorigenesis. J Clin Invest 2018;128:2848–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo C, McCormick F. SPRED proteins and their roles in signal transduction, development, and malignancy. Genes Dev 2020;34:1410–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Shen R, Arora A, Orlow I, Busam KJ, Lezcano C, et al. Landscape of mutations in early stage primary cutaneous melanoma: an InterMEL study. Pigment Cell Melanoma Res 2022;35:605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupton CJ, Bayly-Jones C, D’Andrea L, Huang C, Schittenhelm RB, Venugopal H, et al. The cryo-EM structure of the human neurofibromin dimer reveals the molecular basis for neurofibromatosis type 1. Nat Struct Mol Biol 2021;28:982–8. [DOI] [PubMed] [Google Scholar]
- Lutterodt CG, Mohan A, Kirkpatrick N. The use of electrodessication in the treatment of cutaneous neurofibromatosis: A retrospective patient satisfaction outcome assessment. J Plast Reconstr Aesthet Surg 2016;69:765–9. [DOI] [PubMed] [Google Scholar]
- McDaniel AS, Allen JD, Park SJ, Jaffer ZM, Michels EG, Burgin SJ, et al. Pak1 regulates multiple c-Kit mediated Ras-MAPK gain-in-function phenotypes in Nf1+/− mast cells. Blood 2008;112:4646–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méni C, Sbidian E, Moreno JC, Lafaye S, Buffard V, Goldzal S, et al. Treatment of neurofibromas with a carbon dioxide laser: a retrospective cross-sectional study of 106 patients. Dermatology 2015;230:263–8. [DOI] [PubMed] [Google Scholar]
- Mo J, Anastasaki C, Chen Z, Shipman T, Papke J, Yin K, et al. Humanized neurofibroma model from induced pluripotent stem cells delineates tumor pathogenesis and developmental origins. J Clin Invest 2021;131:e139807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo J, Moye SL, McKay RM, Le LQ. Neurofibromin and suppression of tumorigenesis: beyond the GAP. Oncogene 2022;41:1235–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naschberger A, Baradaran R, Rupp B, Carroni M. The structure of neurofibromin isoform 2 reveals different functional states. Nature 2021;599: 315–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen R, Dombi E, Widemann BC, Solomon J, Fuensterer C, Kluwe L, et al. Growth dynamics of plexiform neurofibromas: a retrospective cohort study of 201 patients with neurofibromatosis 1. Orphanet J Rare Dis 2012;7:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omrani A, van der Vaart T, Mientjes E, van Woerden GM, Hojjati MR, Li KW, et al. HCN channels are a novel therapeutic target for cognitive dysfunction in Neurofibromatosis type 1. Mol Psychiatry 2015;20:1311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortonne N, Carroll SL, Rodriguez FJ, Miller DC, Nazarian RM, Blakeley JO, et al. Assessing interobserver variability and accuracy in the histological diagnosis and classification of cutaneous neurofibromass. Neurooncol Adv 2020;2:i117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortonne N, Wolkenstein P, Blakeley JO, Korf B, Plotkin SR, Riccardi VM, et al. Cutaneous neurofibromas: current clinical and pathologic issues. Neurology 2018;91:S5–13. [DOI] [PubMed] [Google Scholar]
- Page PZ, Page GP, Ecosse E, Korf BR, Leplege A, Wolkenstein P. Impact of neurofibromatosis 1 on Quality of Life: a cross-sectional study of 176 American cases. Am J Med Genet A 2006;140:1893–8. [DOI] [PubMed] [Google Scholar]
- Patmore DM, Welch S, Fulkerson PC, Wu J, Choi K, Eaves D, et al. In vivo regulation of TGF-beta by R-Ras2 revealed through loss of the rasgap protein NF1. Cancer Res 2012;72:5317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltonen J, Penttinen R, Larjava H, Aho HJ. Collagens in neurofibromas and neurofibroma cell cultures. Ann N Y Acad Sci 1986;486:260–70. [DOI] [PubMed] [Google Scholar]
- Pemov A, Hansen NF, Sindiri S, Patidar R, Higham CS, Dombi E, et al. Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define premalignant neurofibromatosis type 1-associated atypical neurofibromas. Neuro Oncol 2019;21:981–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomska KJ, Coulpier F, Gresset A, Schmitt A, Debbiche A, Lemoine S, et al. Cellular origin, tumor progression, and pathogenic mechanisms of cutaneous neurofibromas revealed by mice with Nf1 knockout in boundary cap cells. Cancer Discov 2019;9:130–47. [DOI] [PubMed] [Google Scholar]
- Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer 2015;15: 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes SD, He Y, Smith A, Jiang L, Lu Q, Mund J, et al. Cdkn2a (Arf) loss drives NF1-associated atypical neurofibroma and malignant transformation. Hum Mol Genet 2019;28:2752–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi VM. Ketotifen suppression of NF1 neurofibroma growth over 30 years. Am J Med Genet A 2015;167:1570–7. [DOI] [PubMed] [Google Scholar]
- Robertson KA, Nalepa G, Yang FC, Bowers DC, Ho CY, Hutchins GD, et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: a phase 2 trial. Lancet Oncol 2012;13:1218–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangha N, Wu R, Kuick R, Powers S, Mu D, Fiander D, et al. Neurofibromin 1 (NF1) defects are common in human ovarian serous carcinomas and co-occur with TP53 mutations. Neoplasia 2008;10:1362–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada S, Florell S, Purandare SM, Ota M, Stephens K, Viskochil D. Identification of NF1 mutations in both alleles of a dermal neurofibroma. Nat Genet 1996;14:110–2. [DOI] [PubMed] [Google Scholar]
- Serra E, Puig S, Otero D, Gaona A, Kruyer H, Ars E, et al. Confirmation of a double-hit model for the NF1 gene in benign neurofibromas. Am J Hum Genet 1997;61:512–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MH, Harper PS, Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1). J Med Genet 1996;33:2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherekar M, Han SW, Ghirlando R, Messing S, Drew M, Rabara D, et al. Biochemical and structural analyses reveal that the tumor suppressor neurofibromin (NF1) forms a high-affinity dimer. J Biol Chem 2020;295:1105–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Side LE, Emanuel PD, Taylor B, Franklin J, Thompson P, Castleberry RP, et al. Mutations of the NF1 gene in children with juvenile myelomonocytic leukemia without clinical evidence of neurofibromatosis, type 1. Blood 1998;92:267–72. [PubMed] [Google Scholar]
- Skuse GR, Kosciolek BA, Rowley PT. Molecular genetic analysis of tumors in von Recklinghausen neurofibromatosis: loss of heterozygosity for chromosome 17. Genes Chromosomes Cancer 1989;1:36–41. [DOI] [PubMed] [Google Scholar]
- Staser K, Yang FC, Clapp DW. Plexiform neurofibroma genesis: questions of Nf1 gene dose and hyperactive mast cells. Curr Opin Hematol 2010;17: 287–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storlazzi CT, Von Steyern FV, Domanski HA, Mandahl N, Mertens F. Biallelic somatic inactivation of the NF1 gene through chromosomal translocations in a sporadic neurofibroma. Int J Cancer 2005;117:1055–7. [DOI] [PubMed] [Google Scholar]
- Stowe IB, Mercado EL, Stowe TR, Bell EL, Oses-Prieto JA, Hernández H, et al. A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Genes Dev 2012;26:1421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh P, Li S, Sarin KY. Neurofibromatosis Type 1 and risk of skin cancer. JAMA Dermatol 2022;158:1214–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyaya M, Osborn MJ, Maynard J, Kim MR, Tamanoi F, Cooper DN. Mutational and functional analysis of the neurofibromatosis type 1 (NF1) gene. Hum Genet 1997;99:88–92. [DOI] [PubMed] [Google Scholar]
- Uusitalo E, Rantanen M, Kallionpää RA, Pöyhönen M, Leppävirta J, Ylä-Outinen H, et al. Distinctive cancer associations in patients with neurofibromatosis Type 1. J Clin Oncol 2016;34:1978–86. [DOI] [PubMed] [Google Scholar]
- Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK, et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 1990;62:187–92. [DOI] [PubMed] [Google Scholar]
- Wallace MR, Marchuk DA, Andersen LB, Letcher R, Odeh HM, Saulino AM, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients [published correction appears in Science 1990;250:1749] Science 1990;249:181–6. [DOI] [PubMed] [Google Scholar]
- Wang W, Cui XW, Gu YH, Wei CJ, Li YH, Ren JY, et al. Combined cyclin-dependent kinase inhibition overcomes MAPK/extracellular signal-regulated kinase kinase inhibitor resistance in plexiform neurofibroma of neurofibromatosis Type I. J Invest Dermatol 2022;142(3):613–23.e7. [DOI] [PubMed] [Google Scholar]
- Warrington NM, Gianino SM, Jackson E, Goldhoff P, Garbow JR, Piwnica-Worms D, et al. Cyclic AMP suppression is sufficient to induce glioma-genesis in a mouse model of neurofibromatosis-1. Cancer Res 2010;70: 5717–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss B, Bollag G, Shannon K. Hyperactive Ras as a therapeutic target in neurofibromatosis type 1. Am J Med Genet 1999;89:14–22. [PubMed] [Google Scholar]
- Welti S, Fraterman S, D’Angelo I, Wilm M, Scheffzek K. The sec14 homology module of neurofibromin binds cellular glycerophospholipids: mass spectrometry and structure of a lipid complex. J Mol Biol 2007;366: 551–62. [DOI] [PubMed] [Google Scholar]
- Wiesner T, Kiuru M, Scott SN, Arcila M, Halpern AC, Hollmann T, et al. NF1 mutations are common in desmoplastic melanoma. Am J Surg Pathol 2015;39:1357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart AL, Conner SJ, Guarin JR, Fatherree JP, Peng Y, McGinn RA, et al. Decellularized extracellular matrix scaffolds identify full-length collagen VI as a driver of breast cancer cell invasion in obesity and metastasis. Sci Adv 2020;6:eabc3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolkenstein P, Zeller J, Revuz J, Ecosse E, Leplège A. Visibility of neurofibromatosis 1 and psychiatric morbidity. Arch Dermatol 2003;139: 103–4. [DOI] [PubMed] [Google Scholar]
- Wu J, Williams JP, Rizvi TA, Kordich JJ, Witte D, Meijer D, et al. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in Desert hedgehog-expressing cells. Cancer Cell 2008;13:105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Markegard E, Dharmaiah S, Urisman A, Drew M, Esposito D, et al. Structural insights into the SPRED1-neurofibromin-KRAS complex and disruption of SPRED1-neurofibromin interaction by oncogenic EGFR. Cell Rep 2020;32:107909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang FC, Chen S, Clegg T, Li X, Morgan T, Estwick SA, et al. Nf1 +/− mast cells induce neurofibroma like phenotypes through secreted TGF-beta signaling. Hum Mol Genet 2006;15:2421–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang FC, Ingram DA, Chen S, Zhu Y, Yuan J, Li X, et al. Nf1-dependent tumors require a microenvironment containing Nf1+/− and c-kit-dependent bone marrow. Cell 2008;135:437–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science 2002;296: 920–2. [DOI] [PMC free article] [PubMed] [Google Scholar]